Abstract
Proteins of the plakin family are predominantly expressed in the epidermis and play a crucial role in cytoskeletal assembly by crosslinking intracellular structural components with cell–cell junctions and the plasma membrane. While most plakins are critical for maintaining epidermal integrity, the role of epiplakin (EPPK1) in inflammatory skin disorders has not been thoroughly investigated. We therefore used single-cell RNA sequencing (scRNAseq) analysis, immunofluorescence, and ex vivo cytokine treatment of human skin explants to investigate EPPK1 regulation in psoriasis. ScRNAseq analysis of psoriatic and healthy skin revealed that EPPK1 was the only member of the plakin family showing specific downregulation in the epidermis of psoriatic lesions. This finding was corroborated at the protein level by immunostaining of human psoriasis samples showing a specific downregulation of EPPK1 in the suprabasal granular layer of psoriatic epidermis. Transcriptomic profiling of Eppk1−/− murine epidermis revealed reduced expression of genes involved in epithelial adhesion and lipid metabolism, partially overlapping with the psoriatic keratinocyte signature, suggesting that EPPK1 loss may predispose the skin to barrier dysfunction under inflammatory stress. Investigation of the mechanism underlying the EPPK1 regulation in psoriasis revealed that interferon-γ (IFN-γ) was the main cytokine involved in its downregulation in human ex vivo skin. Collectively, our findings demonstrate a specific IFN-γ-dependent downregulation of EPPK1 in psoriasis, suggesting that lack of EPPK1 might contribute to the epithelial defects observed in this inflammatory skin condition.
1 Introduction
Plakins are high-molecular-weight proteins which are also referred to as cytolinkers that play a crucial role in cytoskeletal assembly in various cells by crosslinking intracellular structural components with junctional complexes and the plasma membrane (Bouameur et al., 2014). The plakin family comprises seven members: plectin (PLEC), periplakin (PPL), microtubule-actin cross-linking factor 1 (MACF1), epiplakin (EPPK1), envoplakin (EVPL), desmoplakin (DSP), and bullous pemphigoid antigen 1 (DST) (Bouameur et al., 2014). These proteins are all expressed in the epidermis, and most have been demonstrated to be essential for epidermal integrity in knockout animal studies, with many members implicated in blistering skin diseases (Bouameur et al., 2014). One notable exception is EPPK1, the ablation of which did not result in any obvious alterations in the epidermal architecture in mice (Spazierer et al., 2006).
EPPK1 is distinct from all other plakins because it consists exclusively of plakin repeat domains (PRDs), whereas other plakin family members contain additional structural domains, such as actin and microtubule-binding domains (Spazierer et al., 2003; Bouameur et al., 2014). A detailed analysis of the EPPK1 genomic locus in humans and mice has revealed the presence of multiple variants containing 9 to 16 PRDs, caused by varying numbers of identical PRD copies at the C-terminus (Ishikawa et al., 2018; Ueo et al., 2021). The expression of EPPK1 is restricted to epithelial tissues, with the highest levels found in the epidermis (Fujiwara et al., 2001; Spazierer et al., 2003). Co-localization of EPPK1 with keratin filaments has been demonstrated in cultured cell lines (Jang et al., 2005). Specifically, PRDs of mouse Eppk1 have been shown to bind to simple keratins 8 and 18, as well as epidermal keratins 5 and 14 (Spazierer et al., 2003; Szabo et al., 2015).
Interestingly, although the epidermis of Eppk1−/− mice did not show any ultrastructural abnormalities, faster keratinocyte migration was observed during wound healing, indicating a subtle role for Eppk1 in keratinocyte dynamics (Goto et al., 2006; Spazierer et al., 2006). A similar phenotype was observed in the cornea of Eppk1−/− animals, characterized by increased fragility, impaired intraepithelial cell differentiation, and accelerated cell migration of epithelial cells (Kokado et al., 2013). These findings suggest that while EPPK1 is not strictly required for epidermal integrity at least under non-stressed conditions, it may play a regulatory role in keratinocyte migration and wound repair. Recent live microscopy studies have shown that EPPK1 is diffusely distributed in the cytoplasm under homeostatic conditions, a finding that contrasts with previous reports describing its association with keratin filaments in fixed cells. It was further observed that under conditions of elevated cytoplasmic Ca2+ levels, EPPK1 associates with keratin filaments, leading to reduced keratin dynamics (Ratajczyk et al., 2022). In addition, EPPK1 presence in vertebrate genomes for over 450 million years, except in whales and dolphins, indicates an evolutionarily conserved role (Fuchs et al., 2022). However, it remains unclear whether EPPK1 mutations are associated with skin pathologies, as no reports have described human EPPK1 mutations linked to disease thus far.
As little is known about plakins in inflammatory skin conditions, we conducted the present study to investigate their expression and regulation in human and imiquimod induced murine psoriasis as well as atopic dermatitis, two chronic inflammatory skin diseases with different pathogenesis.
2 Materials and methods
2.1 Sample dissociation and preparation of the single-cell suspension
Wildtype and Eppk1−/− mice were bred and maintained as previously described (Spazierer et al., 2006). Mice were sacrificed, and dorsal skin was excised under sterile conditions. Tissue samples were washed in Dulbecco’s phosphate-buffered saline (PBS, without Ca2+/Mg2+; Gibco™, Thermo Fisher Scientific, Waltham, MA, United States, cat#: 14190144) under laminar airflow. Six mm punch biopsies were taken from intact dorsal skin of each a wild type C57BL/6J and a Eppk1−/− mouse backcrossed into the C57BL/6J background, mechanically minced, and enzymatically dissociated using the Whole Skin Dissociation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany, cat#: 130-101-540) in gentleMACS™ C Tubes (Miltenyi Biotec, cat#: 130-093-237) for 2.5 h at 37°C. Dissociation was performed using the gentleMACS™ Octo Dissociator with Heaters (Miltenyi Biotec, cat#: 130-096-427), following the manufacturer’s instructions. This protocol yielded single-cell suspensions with >90% viability. Cell suspensions were filtered sequentially through 100 μm and 40 µm strainers (Corning Inc., Corning, NY, United States, cat#: 352360 and cat#: 352340, respectively), then washed twice with 0.04% bovine serum albumin (BSA; Sigma-Aldrich, St. Louis, MO, United States, cat#: A9418) in PBS. Cell concentration and viability were assessed using the Acridine Orange/Propidium Iodide (AO/PI) Cell Viability Kit (Logos Biosystems, Anyang-si, Gyeonggi-do, South Korea, cat#: F23001) and counted with the LUNA-FL™ Dual Fluorescence Cell Counter (Logos Biosystems, cat#: L20001).
2.2 Single-cell processing and library preparation
Single-cell RNA sequencing was performed using the Chromium Single Cell Fixed RNA Profiling Kit (10x Genomics, Pleasanton, CA, United States) according to the manufacturer’s instructions (CG000527, protocol version F). Approximately 10,000 cells per sample were targeted for recovery. Cells were hybridized with the Mouse Whole Transcriptome Probe Set (10x Genomics, Pleasanton, CA, United States, cat#: 1000496) and barcoded using the Chromium Next GEM Dual Index Kit TS Set A (10x Genomics, Pleasanton, CA, United States, cat#: 1000215) as part of the Pooled Wash Workflow. Library construction was performed using the Next GEM Single Cell Fixed RNA Hybridization and Library Kit (10x Genomics, Pleasanton, CA, United States, cat#: 1000415) and Gel Bead Kit (10x Genomics, Pleasanton, CA, United States, cat#: 1000421). Libraries were amplified using 9 cycles of PCR, followed by quality control using the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, United States), which showed clean profiles with expected fragment distributions.
2.3 Sequencing and data processing
Final libraries were sequenced on an Illumina NextSeq 2000 platform (Illumina Inc., San Diego, CA, United States) using a P4 100-cycle flow cell in paired-end mode, following 10x Genomics recommendations for the Fixed RNA Profiling workflow. Raw base call (BCL) files were demultiplexed using Cell Ranger mkfastq with the SI-TS-G2 dual index set (10x Genomics, Pleasanton, CA, United States). Reads were aligned to the mm10 mouse reference genome provided by 10x Genomics, and gene expression matrices were generated based on unique molecular identifier (UMI) counts.
2.4 scRNAseq analysis
Publicly available scRNAseq data sets GSE173706 (Ma et al., 2023), GSE165021,GSE150361,GSE193350 (Guo et al., 2024) and GSE153760 (Bangert et al., 2021) were retrieved from the NCBI Gene Expression Omnibus (GEO) database. Processing and analysis were performed using R (version 4.3.1, The R Foundation, Vienna, Austria) and RStudio (version 2023.6.2.0 + 561). Computational scRNAseq analyses were conducted using the Seurat package (version 4.9.9.9059) as per the developer’s vignettes (Hao et al., 2023). For each individual dataset, UMI count matrices were generated and converted to Seurat objects which were subsequently merged into a single object and subjected to quality control. For the psoriasis dataset (GSE173706) (Ma et al., 2023), cells were filtered based on the following criteria: fewer than 500 UMIs, fewer than 500 or more than 6,000 expressed genes, and more than 10% mitochondrial reads. Similarly, for the GSE153760 (Bangert et al., 2021) atopic dermatitis, the imiquimod dataset GSE165021,GSE150361,GSE193350 (Guo et al., 2024), as well as the data generated from Eppk1−/− mice (GSE300052) cells with fewer than 200 or more than 45,000 UMIs, fewer than 100 or more than 7,500 expressed genes, and more than 10% mitochondrial reads were excluded from further analysis. Additionally, all mitochondrial genes were removed from the UMI count matrices. Normalization and variance stabilization were performed using the ‘SCTransform’ function with v2 regularization, regressing out the percentage of mitochondrial reads. Dimensionality reduction was performed via principal component analysis (PCA) with 50 principal components, after which initial clustering was conducted using the ‘RunUMAP’, ‘FindNeighbors’, and ‘FindClusters’ functions. To correct for batch effects, the merged Seurat object was split by sample, and each subset was normalized. For data integration, the merged dataset was divided into ‘count’ and ‘data’ layers, followed by normalization and identification of variable features, per Seurat v5 integrative analysis guidelines. Features were scaled and centered before conducting PCA. Data integration was performed using the “IntegrateLayers” function with an anchor-based RPCA approach. The integrated, batch-corrected expression matrix was used for further analyses. Clustering was performed following the standard Seurat workflow, using UMAP and Louvain clustering with the “RunUMAP,” “FindNeighbors,” and “FindClusters” functions. Validation was performed by examining known marker panels to ensure the plausibility of initial clustering. For sub-clustering analysis, clusters of interest were subset, and the processes of normalization, variance stabilization, and integration were reiterated. PCA, UMAP, and Louvain clustering were then performed on the reintegrated subsets to derive sub-clusters. Differentially expressed genes (DEGs) were identified using the MAST statistical framework (Finak et al., 2015), implemented in Seurat’s ‘FindMarkers’ and ‘FindAllMarkers’ functions, focusing on genes expressed in a minimum of 25% of cells in one group. The “AddModuleScore” function within the Seurat environment was used for module score calculation of gene sets.
To functionally annotate DEGs, enrichment analysis was performed using the Enrichr R package (Kuleshov et al., 2016), querying the “GO_Biological_Process_2025”, “GO_Cellular_Component_2025”, and “WikiPathways_2024_Human” databases. Only significantly regulated genes (adjusted p-value < 0.05 and log2FC > 0.5) were included in the analysis. Enriched terms were ranked according to Enrichr’s “combined score,” which integrates the Fisher’s exact test p-value with the deviation from expected rank (z-score). The top enriched terms were visualized as dot plots indicating both combined score and gene ratio.
2.5 Data visualization
Data visualization was conducted using Seurat v. 4.9.9.9059 (Hao et al., 2023), ggplot2 v.3.4.2 (Wickham, 2016), EnhancedVolcano v.1.18.0 (Blighe et al., 2023), and scCustomize v.1.1.1 (Marsh et al., 2023).
2.6 Sampling of psoriatic and healthy skin
Lesional skin of psoriatic patients was collected at the Department of Dermatology, Medical University of Vienna via 6 mm punch biopsies. Healthy skin samples were obtained from abdominal tissue post plastic surgery at the Division of Plastic and Reconstructive Surgery, Department of Surgery, Medical University of Vienna.
2.7 Ex-vivo treatment of skin biopsies
The ex-vivo maintenance of the skin samples was carried out using KBM™-2 basal medium (Lonza Group AG, Basel, Switzerland, cat#: CC-3103) supplemented with KGM™-2 SingleQuots™ (Lonza Group AG, Basel, Switzerland, cat#: CC-4152), ensuring that the epidermis was not submerged in culture medium. Samples were treated with either 10 ng/mL recombinant IL-17A (R&D Systems®, cat#: 7955-IL/CF; n = 6), 10 ng/mL recombinant human IL-22 (R&D Systems®, cat#: 782-IL-010/CF; n = 4), 10 ng/mL recombinant human IL-1β (BioLegend®, cat#: 579402; n = 6), 10 ng/mL recombinant human IFN- γ (PeproTech®, cat#: 300-02-20UG; n = 6), a combination of the cytokines (n = 6), or medium alone (n = 6). Treatments were carried out for durations of 2 days, 4 days, or 8 days, with medium and cytokine changes performed every other day. After the treatment period, samples were fixed in 7.5% buffered neutral formaldehyde solution for further processing.
2.8 Immunofluorescence staining and microscopy
Paraffin-embedded sections from healthy donors, psoriasis patients, and ex-vivo treated skin samples were deparaffinized using a heater fan until the paraffin melted, followed by incubation in xylene. Sections were sequentially rehydrated through ethanol solutions (96%, 80%, 30%) and then rinsed in deionized water (dH2O). Antigen retrieval was performed using a 10 mM sodium citrate buffer (pH 6) prepared by diluting 25 mL of Target Retrieval Solution 10x (Dako, cat#: S2369) in 225 mL dH2O. Slides were placed in a jar containing 80 mL buffer and heated in a pressure cooker, followed by cooling to room temperature. The primary anti-EPPK1 antibody (Abcam, cat#: ab247172) was diluted 1:50 in 2% PBS/BSA, and 100 µL was added to each pap pen encircled section, followed by overnight incubation at 4°C. The sections were washed in PBS and incubated for 30 min at room temperature in the dark with Goat anti-Rabbit IgG (H+L), Alexa Fluor™ 546 (Invitrogen, ThermoFisher Scientific, cat#: A-11035), at a dilution of 1:500 in 2% PBS/BSA. The mixture included 10% goat serum and DAPI (1:1,000) (ThermoFisher Scientific, cat#: 62248). Sections were washed in PBS and dH2O and subsequently mounted with aqua polymount medium (Polysciences, cat#: 18606) and stored at 4°C. Fluorescence microscopy was conducted using an OLYMPUS BX63 fluorescence microscope, together with Olympus cellSens software (Olympus, Shinjuku, Tokyo, Japan).
2.9 Statistical analysis
Immunofluorescence quantification was performed using ImageJ (version 1.53t, National Institutes of Health, Bethesda, MD, United States). EPPK1 expression was assessed by calculating the Corrected Total Cell Fluorescence (CTCF), defined as: Integrated Density – (Area × Mean Background Fluorescence), providing a background-adjusted measure of total fluorescence signal over the EPPK1-positive area. All statistical analyses and data visualization were conducted using GraphPad Prism version 10.0.3 (GraphPad Software, San Diego, CA, United States). Comparisons involving more than two groups were analyzed using one-way ANOVA followed by Dunnett’s multiple comparisons test. Differences between two groups were assessed using unpaired two-tailed t-tests. Associations between continuous variables were examined using Pearson’s correlation coefficient. Relationships between categorical and continuous variables were analyzed by simple logistic regression. A p-value < 0.05 was considered statistically significant. For multiple comparisons, adjusted p-values were applied where indicated.
3 Results
3.1 EPPK1 mRNA-expression is significantly reduced in psoriatic lesions
To assess the expression levels of plakin family members, we re-analyzed a previously published scRNAseq dataset (Ma et al., 2023) from lesional and non-lesional psoriatic skin, alongside healthy controls. Following initial clustering of cell types (Figure 1A; Supplementary Figure S1A), we calculated a psoriasis module score based on key psoriasis-associated genes (S100A7A, S100A8, S100A9, IL17A, IL1B, IL22, IFNG, KRT6A, KRT16, MKI67, and IL23A), which revealed a strong increase in lesional psoriatic skin and a moderate increase in non-lesional skin compared to healthy controls (Figures 1B–D), confirming accuracy of the disease-state and providing a reliable platform for further analysis. Notably, the module score also incorporated proinflammatory cytokines (IL17A, IL1B, IL22, IL23, and IFNG) which were upregulated in lesional psoriasis, providing context for the regulation of plakin family members (Figures 1B–D). Subsequent keratinocyte subsetting (Figure 1E) and clustering into differentiation stages, using well-established marker genes (Supplementary Figures S1B, S2), identified proliferative basal keratinocytes and differentiated keratinocyte stages. A plakin gene module score, derived from the expression levels of all plakin members, including DSP, DST, EPPK1, EVPL, MACF1, PLEC, and PPL, was subsequently calculated. This revealed a marked reduction in the overall expression of plakin family members in lesional psoriatic skin (Figures 1F–H), while expression levels in non-lesional skin remained largely similar to healthy controls. Further dissection of individual plakin family members expression revealed that EPPK1 stood out as the most significantly downregulated plakin in psoriasis (Figure 1I). Violin plots illustrated that EPPK1 expression was substantially decreased across all epidermal layers of lesional skin, with a pronounced reduction observed in the late differentiation stages of keratinocytes (Figures 1J–L). Non-lesional psoriatic skin showed a moderate reduction in EPPK1 expression compared to lesional skin, though its levels remained lower than those observed in healthy controls. Interestingly, all other plakin family members showed less, or no deviating expression levels in psoriasis (Supplementary Figure S3). Importantly, the significant downregulation of EPPK1 contrasts with the relatively stable expression of EVPL and PPL, two plakins highly expressed in differentiated keratinocytes (Figure 1I; Supplementary Figure S3). Taken together with the reduced but largely preserved expression of key terminal differentiation markers FLG and CDSN (Supplementary Figures S2E, F), this pattern indicates that EPPK1 downregulation is unlikely to be solely a secondary consequence of impaired keratinocyte differentiation in lesional psoriasis. These findings underscore the distinct downregulation of EPPK1 mRNA expression in psoriasis, particularly in relation to keratinocyte differentiation, with non-lesional skin retaining EPPK1 levels closer to healthy controls.
FIGURE 1
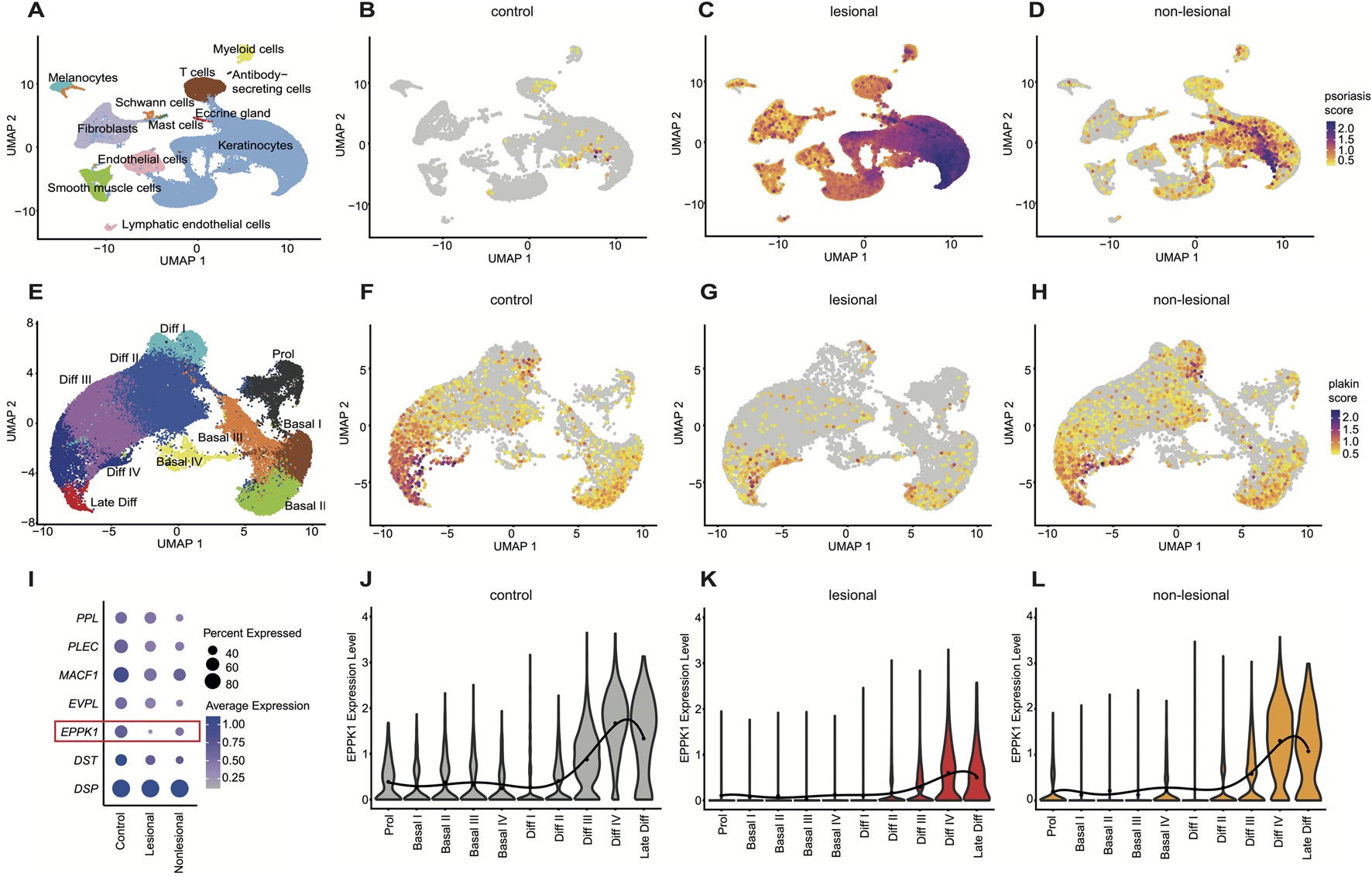
Comprehensive scRNAseq analysis of plakin regulation in psoriasis (A) UMAP visualization from a comprehensive scRNAseq analysis, revealing 12 distinct cell clusters. (B–D) Feature plots illustrating a psoriasis module score, including the expression levels of S100A7A, S100A8, S100A9, IL17A, IL1B, IL22, IFNG, KRT6A, KRT16, MKI67, and IL23A genes. Feature plots are split by condition into (B) (F) control, (C) lesional, and (D) non-lesional psoriasis. (E) UMAP of a subset from the keratinocyte cluster, showing 10 distinct clusters representing stages of keratinocyte differentiation. (F–H) Feature plots showing a plakin module score, including expression levels of DSP, DST, EPPK1, EVPL, MACF1, PLEC, and PPL genes, split by condition into (F) control, (G) lesional, and (H) non-lesional. (I) Dot plot showing expression levels of epidermal plakins in the keratinocyte subset split by condition. Dot size represents the expression percentage for each gene with color intensity reflecting average gene expression levels. (J–L)EPPK1 expression levels as violin plots, split by condition. The mean expression level for each keratinocyte cluster is indicated by a black dot, with the solid black line representing a smooth sixth-degree polynomial regression fit through the means. (J) Grey violins show EPPK1 expression levels in healthy skin, (K) red violins illustrate EPPK1 expression in lesional psoriasis, and (L) orange violins show EPPK1 expression in non-lesional psoriasis.
3.2 Eppk1−/− keratinocytes downregulate adhesion and lipid genes, partly mirroring psoriatic profiles
To investigate the role of EPPK1 in epidermal homeostasis and its potential involvement in psoriasis-associated transcriptional changes, we performed scRNA-seq of skin isolated from wildtype and Eppk1−/− mice. Following initial clustering of epidermal populations (Figure 2A), we subsetted the keratinocyte compartment (Figure 2B) and assessed the expression of plakin family members. As expected, Eppk1 expression was selectively lost in knockout keratinocytes. In contrast, expression of other plakins, including Macf1, Ppl, Evpl, Dsp, Plec, and Dst remained largely unchanged between wildtype and Eppk1−/− mice (Figure 2C), indicating that loss of Eppk1 is not compensated by transcriptional upregulation of related plakin genes. Differential gene expression analysis within the keratinocyte subset identified 29 significantly upregulated and 62 significantly downregulated genes in Eppk1−/− skin compared to wildtype controls (log2FC > 0.5, adjusted p < 0.05; Figure 2D). This transcriptional shift was relatively modest compared to the broader dysregulation observed in lesional psoriatic keratinocytes versus healthy skin (Figure 2E). Strikingly, however, nearly half (48.4%) of the significantly downregulated genes in Eppk1−/− keratinocytes were also suppressed in lesional psoriatic keratinocytes (Figure 2F), suggesting transcriptional convergence in epidermal dysregulation. Visualization of a module score based on the overlapping downregulated gene set revealed pronounced suppression in lesional keratinocytes, with an intermediate reduction in non-lesional psoriasis (Figure 2G), a pattern that was also evident at the individual gene level (Figure 2H). Enrichment analysis revealed that the shared downregulated genes are significantly associated with pathways essential to epidermal function, including tight and apical junction assembly, hemidesmosome organization, focal adhesion dynamics, intermediate filament bundling, and lipid metabolic processes (Figure 2I). These data indicate that Eppk1 loss partially recapitulates psoriasis-like transcriptional changes in keratinocytes and suggest a role for Eppk1 in maintaining epidermal integrity, promoting barrier formation, and supporting resilience during stress.
FIGURE 2

Comparative scRNA-seq analysis of Eppk1−/− and psoriatic keratinocytes (A) UMAP visualization of single-cell RNA-seq data from murine wildtype and Eppk1−/− skin, revealing 8 distinct clusters. (B) UMAP of the keratinocyte subset showing seven subclusters corresponding to stages of keratinocyte differentiation. (C) Dot plot displaying expression of plakin family genes, split by condition into wildtype and Eppk1−/−. Dot size reflects the percentage of expressing cells, and color intensity indicates average expression levels. (D) Volcano plot showing differentially expressed genes (DEGs) between wildtype and Eppk1−/− keratinocytes. Genes significantly upregulated in Eppk1−/− (log2FC > 0.5, adjusted p < 0.05) are shown with positive fold changes, while downregulated genes exhibit negative fold changes. (E) Volcano plot of DEGs between healthy skin and lesional psoriasis within the keratinocyte subset (based on data shown in Figure 1). Significantly upregulated genes in psoriasis compared to healthy skin have positive log2FC values, and significantly downregulated genes have negative log2FC values. (F) Venn diagrams illustrating the overlap of significantly regulated genes in keratinocytes between Eppk1−/−, lesional psoriasis, and non-lesional psoriasis, compared to healthy controls/wildtype. (G) Feature plot showing a module score for genes that are consistently downregulated across Eppk1−/− and human psoriasis datasets (as identified in F), split by condition. (H) Dot plot of individual overlapping downregulated genes (from F) across conditions in the human psoriasis dataset. Dot size reflects the percentage of expressing cells; color intensity reflects average expression. (I) Enrichment analysis of overlapping downregulated genes in Eppk1−/− and psoriatic keratinocytes. Top 13 enriched pathways are shown with corresponding combined scores and gene ratios.
3.3 IMQ-induced psoriasis mouse model exhibits generalized downregulation of plakins including Eppk1
To explore whether EPPK1 downregulation is conserved in mouse models of psoriasis, we performed scRNA-seq analysis of skin from wild-type and imiquimod (IMQ)-treated mice (Guo et al., 2024). Unsupervised clustering identified 12 cell populations (Figure 3A), with cluster identities defined by established marker gene expression (Supplementary Figure S1D). Consistent with findings in human psoriasis, the psoriasis-associated gene module score was markedly elevated in IMQ-treated skin compared to controls (Figures 3B,C), confirming disease induction. Most genes within the module were upregulated in the IMQ condition, including Il23a, Il17a, Il22, Il1b, Ifng, and S100a8/9, whereas Mki67 exhibited higher expression in control samples (Figure 3D), possibly reflecting reduced proliferative drive or differing kinetics of proliferation in the IMQ model compared to human psoriasis. Focusing on keratinocytes, we subsetted and reclustered this population into four differentiation stages (Figure 3E), corroborated through canonical marker genes (Supplementary Figure S1E). Calculation of a plakin gene module score, comprising Dsp, Dst, Eppk1, Evpl, Macf1, Plec, and Ppl, revealed a broad reduction in the IMQ-treated condition, partially recapitulating the findings observed in human lesional psoriasis (Figures 3F,G). Interestingly, whereas Eppk1 was markedly and specifically downregulated in human lesional psoriatic skin, the IMQ mouse model exhibited a broader and less selectively reduced expression of plakins, including Ppl, Evpl, Plec, and Dst (Figure 3H). These differences might indicate species-specific responses or distinct inflammatory mechanisms underlying epidermal structural disruptions in the IMQ model compared to human psoriasis.
FIGURE 3
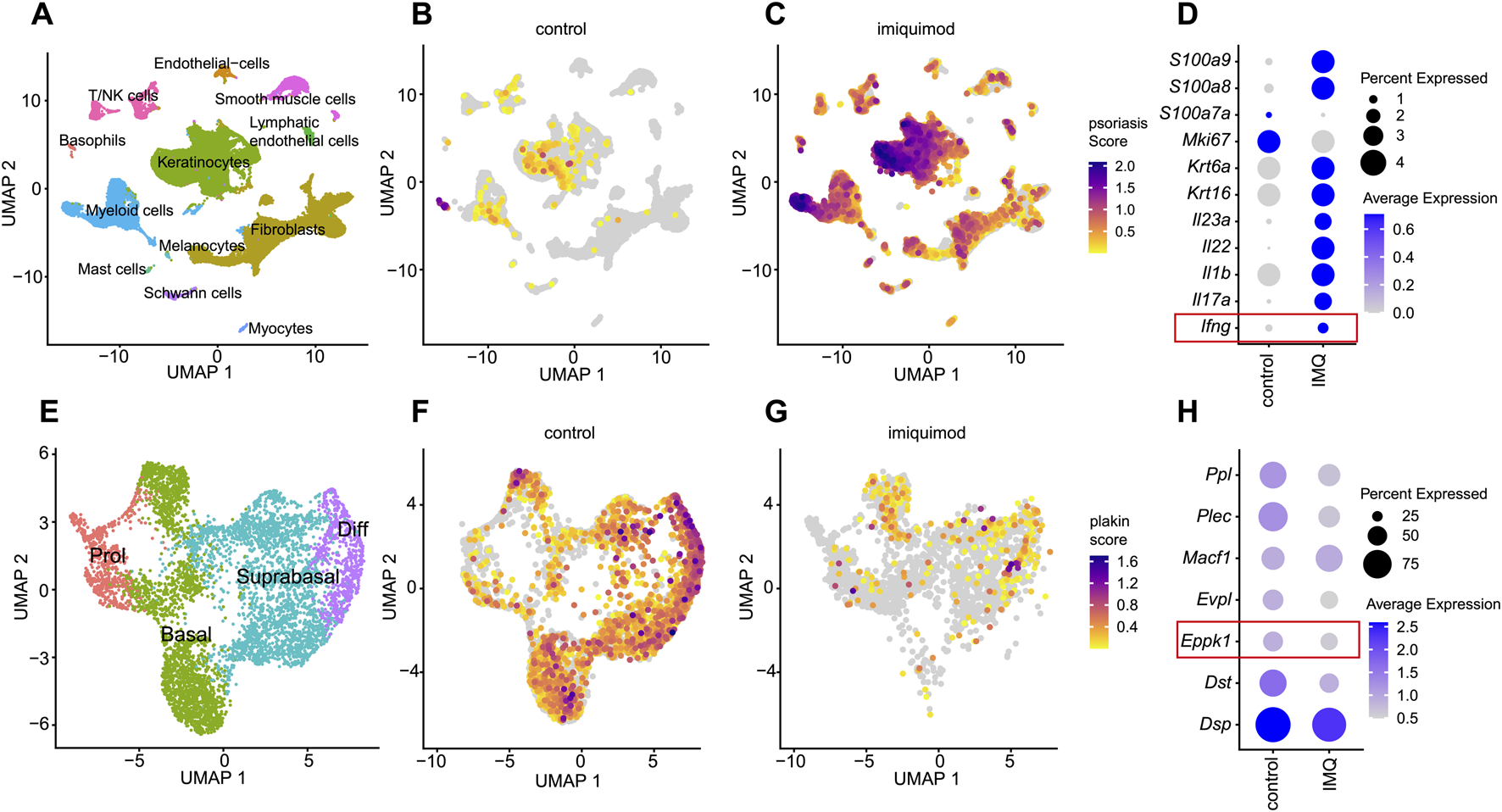
Comprehensive scRNAseq analysis of plakin regulation in IMQ-induced murine psoriasis (A) UMAP visualization from a scRNAseq analysis, revealing 12 distinct cell clusters. (B,C) Feature plots illustrate a psoriasis module score, including the expression levels of Ifng, Il17a, Il1b, Il22, Il23a, Krt16, Krt6a, Mki67, S100a7a, S100a8, S100a9 genes. Feature plots are split by condition into (B) healthy skin and (C) imiquimod (IMQ) conditions. (D) Dot plot of psoriasis related genes split by condition. Dot size represents the expression percentage for each gene with color intensity reflecting average gene expression levels. (E) UMAP of a subset from the keratinocyte cluster, showing 4 clusters representing stages of keratinocyte differentiation. (F,G) Feature plots showing a plakin module score, including expression levels of Dsp, Dst, Eppk1, Evpl, Macf1, Plec, and Ppl genes, split by condition into (F) healthy controls and (G) imiquimod (IMQ). (H) Dot plot of epidermal plakins in the keratinocyte subset split by condition. Dot size represents the expression percentage for each gene with color intensity reflecting average gene expression levels.
3.4 EPPK1 shows consistent expression in atopic dermatitis
To evaluate whether the observed downregulation of EPPK1 is specific to psoriasis or also occurs in other chronic inflammatory skin diseases, we performed a comparative analysis using atopic dermatitis (AD) another chronic inflammatory skin disorder to contextualize the psoriasis findings. We thus re-analyzed our previously published dataset (Bangert et al., 2021) to assess plakin expression in skin lesions of AD. Initial clustering of cell types identified five distinct clusters, including keratinocytes, melanocytes, T cells, Langerhans cells, and myeloid cells (Figure 4A). An AD module score was then calculated based on the expression levels of key AD-related genes (CCL27, CD74, IFITM3, IFNG, KRT6A, S100A7, S100A8, S100A9, and SERPINB4), revealing a clear distinction between control and AD samples (Figures 4B,C). Notably, our analysis revealed that in contrast to psoriasis, IFNG expression showed little to no variation between healthy skin and AD (Figure 4D), whereas increased levels of IFNG have been observed in psoriasis, particularly in lesional skin within the T-cell cluster (Supplementary Figure S4). Next, keratinocytes were subsetted and clustered into four distinct differentiation stages: proliferative, basal, differentiating, and late differentiating keratinocytes (Figure 4E). A plakin gene module score was calculated to assess overall plakin expression, revealing only minor differences between AD and control samples, with a slight overall increase in plakin expression observed in AD (Figures 4F,G). Upon further analysis of individual plakin family members, little or no difference in expression levels was found between AD and control conditions. Interestingly, EPPK1 exhibited no marked differences in mRNA expression between AD and controls, with a slight increase observed in AD, contrasting with the significant downregulation as seen in psoriatic lesions (Figure 4H). These findings suggest that while minor alterations in overall plakin expression were observed in AD, with a slight increase in the plakin module score, reduced EPPK1 expression appears to be more characteristic of psoriasis.
FIGURE 4
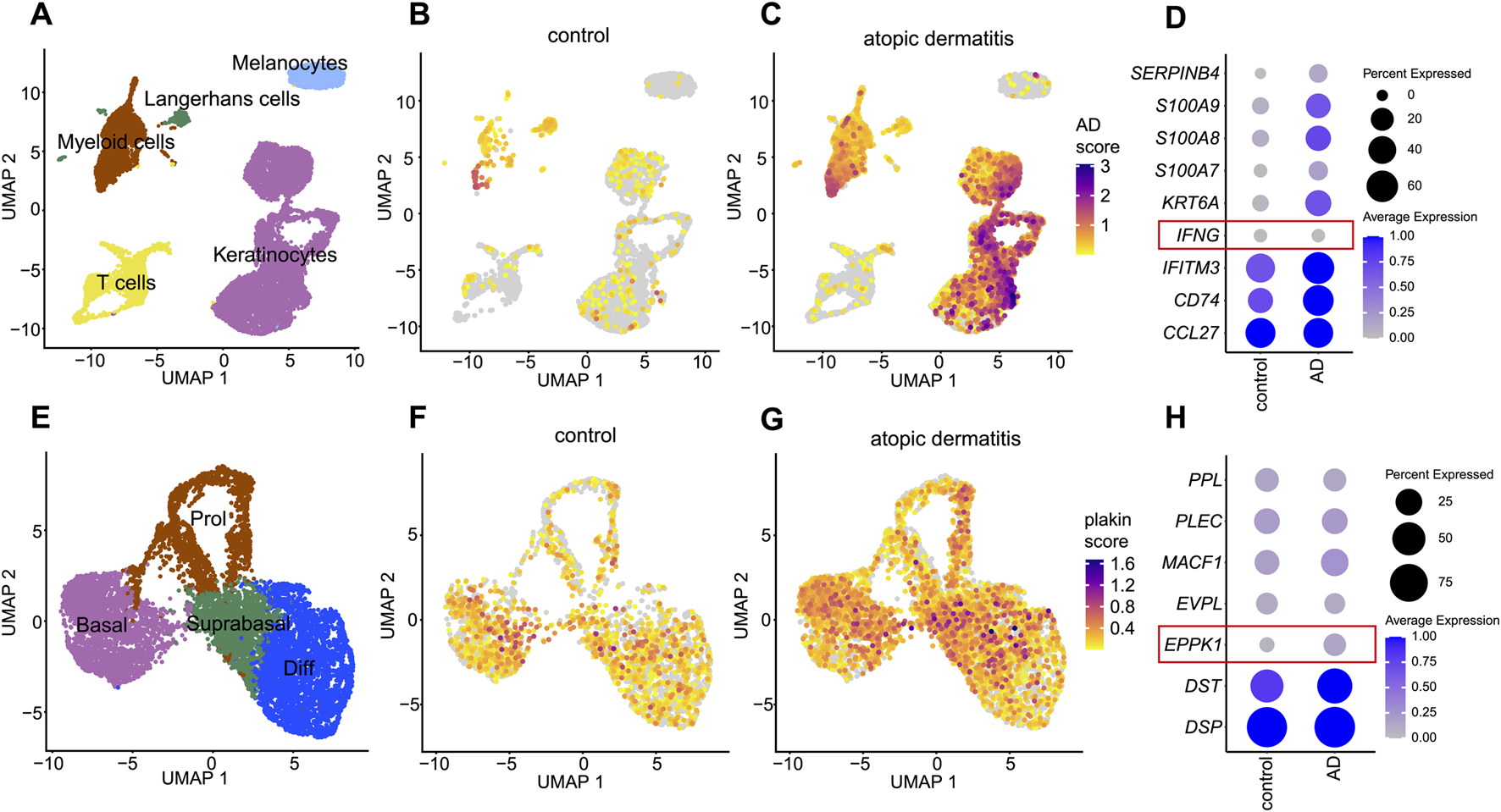
Comprehensive scRNAseq analysis of plakin regulation in atopic dermatitis (A) UMAP visualization from a scRNAseq analysis, revealing 5 distinct cell clusters. (B,C) Feature plots illustrate an atopic dermatitis module score, including the expression levels of CCL27, CD74, IFITM3, IFNG, KRT6A, S100A7, S100A8, S100A9 and SERPINB4 genes. Feature plots are split by condition into (B) healthy skin and (C) atopic dermatitis (AD). (D) Dot plot of AD-related genes split by condition. Dot size represents the expression percentage for each gene with color intensity reflecting average gene expression levels. (E) UMAP of a subset from the keratinocyte cluster, showing 4 clusters representing stages of keratinocyte differentiation. (F,G) Feature plots showing a plakin module score, including expression levels of DSP, DST, EPPK1, EVPL, MACF1, PLEC, and PPL genes, split by condition into (F) healthy skin and (G) atopic dermatitis (AD). (H) Dot plot of epidermal plakins in the keratinocyte subset split by condition. Dot size represents the expression percentage for each gene with color intensity reflecting average gene expression levels.
3.5 Immunostaining confirms EPPK1 downregulation in psoriatic lesions
Consequently, we further concentrated our study on EPPK1 in psoriasis, using immunostaining to validate the mRNA expression results from the scRNAseq analysis. Immunofluorescence staining in healthy skin revealed strong EPPK1 protein expression, specifically localized to the suprabasal granular layer of the epidermis, particularly in late-stage differentiated keratinocytes (Figure 5A). This pattern is consistent with the elevated expression levels observed in the “Diff III”, “Diff IV”, and “Late Diff” keratinocyte clusters from the scRNA-seq analysis (Figures 1F,J). In sharp contrast, virtually no EPPK1 staining was observed in lesional psoriatic skin (Figure 5B), consistent with the significant downregulation of EPPK1 mRNA observed in our earlier transcriptomic analysis (Figures 1G,K). In line with these observations, semi-quantitative analysis revealed a significantly lower corrected total cell fluorescence (CTCF) of EPPK1 in psoriatic lesions compared to healthy skin (p < 0.0001; Figure 5C). These findings were corroborated across all psoriasis donors analyzed (n = 12), with a marked absence of EPPK1 staining in the majority of cases, while EPPK1 expression was consistently detected in healthy skin samples (Supplementary Figure S5). Interestingly, EPPK1 staining intensity in psoriatic lesions showed no significant association with sex (Tjur’s R2 = 0.022, p = 0.6204), disease duration (R2 = 0.2025, p = 0.1421), or PASI scores (R2 = 0.0039, p = 0.8477), indicating that EPPK1 downregulation occurs independently of these clinical parameters (Supplementary Table S1; Supplementary Figure S6).
FIGURE 5

EPPK1 immunostaining and ex-vivo cytokine treatment. Immunofluorescence imaging of (A) healthy skin and (B) psoriatic skin. Images at ×40 magnification display DAPI-stained nuclei (blue) and EPPK1 fluorescence (red). (C) Quantification of EPPK1 signal intensity based on corrected total cell fluorescence (CTCF) normalized to the mean of healthy control samples. Data are presented as box plots displaying all individual data points. Red indicates statistically significant differences (p < 0.05). (D–F) Immunofluorescence staining of human skin cultured ex vivo and treated with pro-inflammatory cytokines. Representative images at ×40 magnification showing EPPK1 (red) and DAPI (blue) at days 2, 4, and 8 post-treatment. (D) Untreated ex vivo skin. (E) IFN-γ treatment. (F) Combined treatment with IL-1β, IL-17A, IL-22, and IFN-γ. (G) Quantification of EPPK1 expression (CTCF) in cytokine-treated ex vivo skin, normalized to untreated control samples. Data are shown as box plots with all data points. Red highlights statistically significant changes (adjusted p < 0.05).
3.6 IFN-γ mediates EPPK1 downregulation in ex-vivo skin cultures
We further aimed to investigate the factors responsible for the observed downregulation of EPPK1 in psoriasis. To this end, we first examined the expression of IL1B, IL17, IL22 and IFNG, all well-known cytokines involved in the pathogenesis of psoriasis (Baliwag et al., 2015; He et al., 2023), in the scRNAseq dataset. As shown in Supplementary Figure S4, IL17A, IL22 and IFNG were strongly upregulated in specific subsets of T-cells, and IL1B expression was upregulated in myeloid cells. To explore the effects of these cytokines on EPPK1 expression, we stimulated biopsies of healthy skin ex vivo with each of these cytokines individually and in combination. After 2, 4, and 8 days, we harvested the samples and performed immunostaining for EPPK1 (Figures 5D–F). While stimulation with IL-1β, IL-17A, or IL-22 alone did not significantly alter EPPK1 expression in this setting (Supplementary Figure S7), treatment with IFN-γ led to a marked reduction in EPPK1 levels by day 4 (p = 0.0385) and an even more pronounced decrease by day 8 (p < 0.0001) (Figures 5E,G). Notably, IFN-γ–treated samples also displayed mild parakeratosis, reminiscent of psoriatic histopathology. Combined treatment with IL-1β, IL-17A, IL-22, and IFN-γ produced similar effects to IFN-γ alone (Figures 5F,G), suggesting that IFN-γ is the dominant effector in this cytokine mix. Quantitative analysis of EPPK1 CTCF corroborated these observations, showing statistically significant reductions in EPPK1 intensity upon IFN-γ exposure and combinatorial treatment (Figure 5G). These findings indicate that IFN-γ is a key negative regulator of EPPK1 expression in keratinocytes and may contribute to its downregulation in psoriatic lesions.
4 Discussion
In the present study we investigated the role of EPPK1 in inflammatory skin conditions. Our data demonstrate that EPPK1 is robustly expressed in the suprabasal granular layer of healthy human epidermis, yet markedly reduced at both the mRNA and protein levels in lesional psoriatic skin. This finding contrasts with previous reports suggesting increased EPPK1 expression in psoriasis (Tian et al., 2022). A plausible explanation for this discrepancy may lie in methodological differences, including antibody specificity and the use of immunohistochemistry in earlier studies versus scRNA-seq and immunofluorescence in our work. Notably, we found that EPPK1 shares an expression pattern with other plakins such as EVPL and PPL, both of which are well-established markers of epidermal differentiation in healthy skin. However, unlike EPPK1, EVPL and PPL were not downregulated in lesional psoriasis, indicating a unique role of EPPK1 in psoriatic pathogenesis.
Structurally, EPPK1 stands out among plakins due to its lack of a coiled-coil rod domain, functioning likely as a single-chain molecule (Fujiwara et al., 2001; Bouameur et al., 2014). Its conservation across nearly all vertebrate species, with the notable exception of cetaceans, emphasizes its evolutionary importance (Fuchs et al., 2022). The absence of EPPK1 in cetaceans may be attributed to their aquatic environment, which imposes lower mechanical stress on the skin. Interestingly, cetacean skin exhibits morphological parallels to psoriatic epidermis, including thickening, hyperkeratosis, and parakeratosis (Springer et al., 2021), raising intriguing questions about the evolutionary role of EPPK1 in barrier adaptation.
Given that EPPK1 protein expression is most prominent in late-stage differentiated keratinocytes, its reduction in psoriasis may contribute primarily to disrupted differentiation rather than the hyperproliferation that characterizes the disease. An effect on cell proliferation has previously been reported in cancer models showing that EPPK1 knockdown inhibits cell proliferation (Yang et al., 2023; Arimura et al., 2024), a phenotype opposite to that observed in psoriatic epidermis. The apparent contradiction could reflect context-specific differences in the drivers of proliferation. In psoriasis, chronic inflammation, particularly mediated by IFN-γ, IL-17A, and IL-22 (Niehues et al., 2021), may override any anti-proliferative effect of EPPK1 loss. Therefore, we propose that EPPK1 downregulation predominantly impairs keratinocyte differentiation and migration (Goto et al., 2006). However, it remains unclear whether EPPK1 contributes directly to the barrier defect observed in psoriasis or if its downregulation is merely a bystander effect of impaired keratinocyte differentiation. Importantly, several lines of evidence argue against the idea that EPPK1 downregulation is a mere secondary consequence of altered differentiation. For example, terminal differentiation markers like FLG and CDSN remain largely intact in psoriatic skin, and other suprabasal plakins with similar expression profiles are unaffected. Furthermore, we found that IFN-γ stimulation of ex vivo skin samples led to a significant reduction in EPPK1 expression within 4 days, without impacting overall skin architecture. This suggests that the regulation of EPPK1 is cytokine-driven and occurs independently of gross differentiation defects. Thus, IFN-γ may act as a direct regulator of EPPK1 in the context of inflammation. Nonetheless, additional research is required to definitively address this issue.
Our exploration of Eppk1-deficient mice revealed no overt epidermal abnormalities under steady-state conditions (Spazierer et al., 2006), reinforcing the notion that EPPK1 is not essential for maintaining basic epidermal morphology. However, scRNA-seq of Eppk1−/− murine skin revealed reduced expression of genes associated with epithelial cohesion and lipid barrier formation. Intriguingly, nearly half of these genes were also downregulated in human psoriatic keratinocytes, pointing to a shared transcriptional program that may undermine epidermal homeostasis and barrier function. While Eppk1 deficiency alone does not cause spontaneous skin pathology, the overlapping gene signature may sensitize the skin to inflammation-induced damage. Notably, Eppk1−/− keratinocytes did not exhibit compensatory upregulation of other plakin family members (Ppl, Evpl, Dst, etc.), key cytoskeletal genes such as Krt5, Krt14, or actin-associated linker proteins. A similar absence of compensation was observed in lesional psoriatic keratinocytes, suggesting that EPPK1 loss creates a structural void that is not buffered by alternative cytolinker proteins. This aligns with previous work in plectin-deficient cells, where keratin filament instability was not corrected by transcriptional adaptation but was instead shaped by post-translational mechanisms and MAPK signaling (Osmanagic-Myers et al., 2006). Similarly, EPPK1 loss does not appear to trigger a compensatory transcriptional program, though compensation at the post-transcriptional level, possibly involving ERM proteins or filamins, remains a possibility and warrants further study.
In support of our human findings, the IMQ-induced mouse model of psoriasis also showed a downregulation of Eppk1. However, in contrast to the selective downregulation observed in human psoriasis, IMQ treatment led to broader suppression of multiple plakins, including Ppl, Evpl, and Dst. These differences may reflect species-specific immune responses or distinct cytokine environments; the IMQ model primarily activates the IL-17/IL-23 axis and lacks the chronic inflammation and complex immune milieu characteristic of human disease (van der Fits et al., 2009; Swindell et al., 2017). While the IMQ model supports the relevance of EPPK1 loss in inflammatory skin conditions, these limitations underscore the need for more refined models, such as Eppk1-deficient mice crossed with genetic models of psoriasis, to fully dissect the functional role of Eppk1 in psoriasis in vivo.
Despite its downregulation in psoriasis, the contribution of EPPK1 to epidermal homeostasis appears to be context- and species-dependent. Although mice lacking Eppk1 do not show overt skin pathology under baseline conditions, several studies suggest that EPPK1 becomes functionally important during stress or injury. For example, Eppk1−/− mice show accelerated keratinocyte migration during wound healing (Goto et al., 2006), and Eppk1-deficient liver and pancreas tissues exhibit stress-induced defects (Wögenstein et al., 2014; Szabo et al., 2015). Although our study did not directly assess keratin filament architecture, several previous studies strongly support a role for EPPK1 in cytoskeletal organization under stress. EPPK1 colocalizes with keratins 5, 6, 10, and 17 during wound healing (Ishikawa et al., 2010), indicating a role in keratin network organization under mechanical stress. Mice lacking Eppk1 exhibit thinner keratin filaments, suggesting compromised structural resilience, an effect that may also contribute to impaired barrier function in psoriatic skin (Elango et al., 2018; Cohen et al., 2024). Recent work further supports a stress-responsive function for EPPK1. Under elevated cytoplasmic calcium levels, EPPK1 binds to keratin filaments and stabilizes them by reducing filament dynamics (Ratajczyk et al., 2022). In resting cells, however, EPPK1 remains diffusely cytoplasmic. This dynamic behavior suggests that EPPK1 reinforces cytoskeletal networks specifically during periods of stress or inflammation, a mechanism that may be disrupted in psoriatic skin due to its downregulation.
In summary, our findings highlight a previously underappreciated role for EPPK1 in maintaining epidermal integrity under inflammatory stress. We identify IFN-γ as a key upstream regulator of EPPK1 in psoriatic lesions and propose that EPPK1 downregulation contributes to disrupted differentiation and weakened barrier function in psoriasis. These results position EPPK1 as potential modulator of epithelial resilience in inflammatory skin diseases. Future research exploring how inflammatory signals intersect with cytoskeletal remodeling could open new therapeutic avenues aimed at reinforcing barrier function in psoriasis.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/, GSE300052.
Ethics statement
This study was conducted in accordance with the guidelines of the Council for International Organizations of Medical Sciences (CIOMS) and the World Health Organization (WHO). All patient samples were anonymized. Research has been approved by the ethics committee of the Medical University of Vienna (EK1969/2021 and EK1449/2016), adhering to the principles outlined in the Declaration of Helsinki and the “Good Scientific Practice” guidelines of the Medical University of Vienna. Written informed consent was obtained from all participants.
Author contributions
HK: Writing – original draft, Formal Analysis, Investigation, Data curation, Conceptualization. CD: Conceptualization, Investigation, Writing – original draft, Formal Analysis, Data curation. MS: Writing – original draft, Formal Analysis, Data curation. LA: Writing – original draft, Data curation, Formal Analysis. JG: Formal Analysis, Data curation, Writing – original draft. MM: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Supervision, Writing – original draft. PF: Supervision, Writing – original draft, Funding acquisition, Investigation, Conceptualization, Formal Analysis.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by the Federal Ministry of Education, Science and Research, Austria (BMBWF, Grant No. SPSC_02_002) and the Austrian Science Fund (FWF, Grant No. P30310). JG acknowledges funding from the LEO foundation (Grant LF-OC-23-001394).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Correction note
This article has been corrected with minor changes. These changes do not impact the scientific content of the article.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2025.1617737/full#supplementary-material
References
1
ArimuraK.KammerM.RahmanS. M. J.Sheau-ChiannC.ZhaoS.HeidiC.et al (2024). Elucidating the role of EPPK1 in lung adenocarcinoma development. BMC Cancer24, 441. 10.1186/s12885-024-12185-x
2
BaliwagJ.BarnesD. H.JohnstonA. (2015). Cytokines in psoriasis. Cytokine73, 342–350. 10.1016/j.cyto.2014.12.014
3
BangertC.RindlerK.KrausgruberT.AlkonN.ThalerF. M.KurzH.et al (2021). Persistence of mature dendritic cells, TH2A, and Tc2 cells characterize clinically resolved atopic dermatitis under IL-4Rα blockade. Sci. Immunol.6, eabe2749. 10.1126/sciimmunol.abe2749
4
BligheK.RanaS.LewisM. (2023). EnhancedVolcano: publication-ready volcano plots with enhanced colouring and labeling. Version 1.13.2. 10.18129/B9.bioc.EnhancedVolcano
5
BouameurJ.-E.FavreB.BorradoriL. (2014). Plakins, a versatile family of cytolinkers: roles in skin integrity and in human diseases. J. Invest Dermatol134, 885–894. 10.1038/jid.2013.498
6
CohenE.JohnsonC. N.WasikowskiR.BilliA. C.TsoiL. C.KahlenbergJ. M.et al (2024). Significance of stress keratin expression in normal and diseased epithelia. iScience27, 108805. 10.1016/j.isci.2024.108805
7
ElangoT.SunJ.ZhuC.ZhouF.ZhangY.SunL.et al (2018). Mutational analysis of epidermal and hyperproliferative type I keratins in mild and moderate psoriasis vulgaris patients: a possible role in the pathogenesis of psoriasis along with disease severity. Hum. Genomics12, 27. 10.1186/s40246-018-0158-2
8
FinakG.McDavidA.YajimaM.DengJ.GersukV.ShalekA. K.et al (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol.16, 278. 10.1186/s13059-015-0844-5
9
FuchsP.DrexlerC.RatajczykS.EckhartL. (2022). Comparative genomics reveals evolutionary loss of epiplakin in cetaceans. Sci. Rep.12, 1112. 10.1038/s41598-022-05087-0
10
FujiwaraS.TakeoN.OtaniY.ParryD. A.KunimatsuM.LuR.et al (2001). Epiplakin, a novel member of the plakin family originally identified as a 450-kDa human epidermal autoantigen. Structure and tissue localization. J. Biol. Chem.276, 13340–13347. 10.1074/jbc.M011386200
11
GotoM.SumiyoshiH.SakaiT.FässlerR.OhashiS.AdachiE.et al (2006). Elimination of epiplakin by gene targeting results in acceleration of keratinocyte migration in mice. Mol. Cell Biol.26, 548–558. 10.1128/MCB.26.2.548-558.2006
12
GuoD.LiX.WangJ.LiuX.WangY.HuangS.et al (2024). Single-cell RNA-seq reveals keratinocyte and fibroblast heterogeneity and their crosstalk via epithelial-mesenchymal transition in psoriasis. Cell Death Dis.15, 207–214. 10.1038/s41419-024-06583-z
13
HaoY.StuartT.KowalskiM. H.ChoudharyS.HoffmanP.HartmanA.et al (2023). Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol.42, 293–304. 10.1038/s41587-023-01767-y
14
HeJ.ZhaoM.MaX.LiD.KongJ.YangF. (2023). The role and application of three IFN-related reactions in psoriasis. Biomed. and Pharmacother.167, 115603. 10.1016/j.biopha.2023.115603
15
IshikawaK.FuruhashiM.SasakiT.KudohJ.TsuchisakaA.HashimotoT.et al (2018). Intragenic copy number variation within human epiplakin 1 (EPPK1) generates variation of molecular size of epiplakin. J. Dermatol Sci.S0923-1811 (18), 228–231. 10.1016/j.jdermsci.2018.05.008
16
IshikawaK.SumiyoshiH.MatsuoN.TakeoN.GotoM.OkamotoO.et al (2010). Epiplakin accelerates the lateral organization of keratin filaments during wound healing. J. Dermatol Sci.60, 95–104. 10.1016/j.jdermsci.2010.08.011
17
JangS.-I.KalininA.TakahashiK.MarekovL. N.SteinertP. M. (2005). Characterization of human epiplakin: RNAi-mediated epiplakin depletion leads to the disruption of keratin and vimentin IF networks. J. Cell Sci.118, 781–793. 10.1242/jcs.01647
18
KokadoM.OkadaY.GotoM.IshikawaK.MiyamotoT.YamanakaO.et al (2013). Increased fragility, impaired differentiation, and acceleration of migration of corneal epithelium of Epiplakin-Null mice. Investigative Ophthalmol. and Vis. Sci.54, 3780–3789. 10.1167/iovs.12-11077
19
KuleshovM. V.JonesM. R.RouillardA. D.FernandezN. F.DuanQ.WangZ.et al (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res.44, W90–W97. 10.1093/nar/gkw377
20
MaF.PlazyoO.BilliA. C.TsoiL. C.XingX.WasikowskiR.et al (2023). Single cell and spatial sequencing define processes by which keratinocytes and fibroblasts amplify inflammatory responses in psoriasis. Nat. Commun.14, 3455. 10.1038/s41467-023-39020-4
21
MarshS.TangM.KozarevaV.GraybuckL. (2023). scCustomize: custom visualizations and functions for streamlined analyses of single cell sequencing. Version 3.0.1. 10.5281/zenodo.10724532
22
NiehuesH.RikkenG.Vlijmen-WillemsI. M.Rodijk-OlthuisD.ErpP. E.ZeeuwenP. L.et al (2021). Identification of keratinocyte mitogens: implications for hyperproliferation in psoriasis and atopic dermatitis. JID Innov.2, 100066. 10.1016/j.xjidi.2021.100066
23
Osmanagic-MyersS.GregorM.WalkoG.BurgstallerG.ReipertS.WicheG. (2006). Plectin-controlled keratin cytoarchitecture affects MAP kinases involved in cellular stress response and migration. J. Cell Biol.174, 557–568. 10.1083/jcb.200605172
24
RatajczykS.DrexlerC.WindofferR.LeubeR. E.FuchsP. (2022). A Ca2+-Mediated switch of epiplakin from a diffuse to keratin-bound state affects keratin dynamics. Cells11, 3077. 10.3390/cells11193077
25
SpaziererD.FuchsP.PröllV.JandaL.OehlerS.FischerI.et al (2003). Epiplakin gene analysis in mouse reveals a single exon encoding a 725-kDa protein with expression restricted to epithelial tissues. J. Biol. Chem.278, 31657–31666. 10.1074/jbc.M303055200
26
SpaziererD.FuchsP.ReipertS.FischerI.SchmuthM.LassmannH.et al (2006). Epiplakin is dispensable for skin barrier function and for integrity of keratin network cytoarchitecture in simple and stratified epithelia. Mol. Cell Biol.26, 559–568. 10.1128/MCB.26.2.559-568.2006
27
SpringerM. S.Guerrero-JuarezC. F.HuelsmannM.CollinM. A.DanilK.McGowenM. R.et al (2021). Genomic and anatomical comparisons of skin support independent adaptation to life in water by cetaceans and hippos. Curr. Biol.31, 2124–2139.e3. 10.1016/j.cub.2021.02.057
28
SwindellW. R.MichaelsK. A.SutterA. J.DiaconuD.FritzY.XingX.et al (2017). Imiquimod has strain-dependent effects in mice and does not uniquely model human psoriasis. Genome Med.9, 24. 10.1186/s13073-017-0415-3
29
SzaboS.WögensteinK. L.ÖsterreicherC. H.GuldikenN.ChenY.DolerC.et al (2015). Epiplakin attenuates experimental mouse liver injury by chaperoning keratin reorganization. J. Hepatol.62, 1357–1366. 10.1016/j.jhep.2015.01.007
30
TianZ.WongW.WuQ.ZhouJ.YanK.ChenJ.et al (2022). Elevated expressions of BTN3A1 and RhoB in psoriasis vulgaris lesions by an immunohistochemical study. Appl. Immunohistochem. Mol. Morphol.30, 119–125. 10.1097/PAI.0000000000000977
31
UeoD.FuruhashiM.SasakiT.KudohJ.ParryD. A. D.WinterD. J.et al (2021). Intragenic copy number variation in mouse epiplakin 1 (Eppk1) and the conservation of the repeat structures in the lower vertebrates. J. Dermatol Sci.103, 186–189. 10.1016/j.jdermsci.2021.07.004
32
van der FitsL.MouritsS.VoermanJ. S. A.KantM.BoonL.LamanJ. D.et al (2009). Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J. Immunol.182, 5836–5845. 10.4049/jimmunol.0802999
33
WickhamH. (2016). ggplot2: elegant graphics for data analysis. Second edition. Switzerland: Springer. 10.1007/978-3-319-24277-4
34
WögensteinK. L.SzaboS.LunovaM.WicheG.HaybaeckJ.StrnadP.et al (2014). Epiplakin deficiency aggravates murine caerulein-induced acute pancreatitis and favors the formation of acinar keratin granules. PLOS ONE9, e108323. 10.1371/journal.pone.0108323
35
YangL.HuangW.BaiX.WangH.WangX.XiaoH.et al (2023). Androgen dihydrotestosterone promotes bladder cancer cell proliferation and invasion via EPPK1-mediated MAPK/JUP signalling. Cell Death Dis.14, 363–12. 10.1038/s41419-023-05882-1
Summary
Keywords
plakins, epiplakin, psoriasis, atopic dermatitis, scRNAseq
Citation
Kühtreiber H, Drexler C, Salek M, Auer L, Griss J, Mildner M and Fuchs P (2025) Epiplakin expression is lost in psoriatic skin lesions and is downregulated by IFN-γ in ex vivo skin cultures. Front. Cell Dev. Biol. 13:1617737. doi: 10.3389/fcell.2025.1617737
Received
24 April 2025
Accepted
29 June 2025
Published
17 July 2025
Corrected
07 August 2025
Volume
13 - 2025
Edited by
Giorgia Cardinali, San Gallicano Dermatological Institute, IRCCS, Italy
Reviewed by
Tara Chand Yadav, University of North Carolina at Chapel Hill, United States
Karin Vicente Greco, University College London, United Kingdom
Mirco Mastrangelo, University of L’Aquila, Italy
Updates
Copyright
© 2025 Kühtreiber, Drexler, Salek, Auer, Griss, Mildner and Fuchs.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Fuchs, peter.fuchs@univie.ac.at; Michael Mildner, michael.mildner@meduniwien.ac.at
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.