

- Department of Microbiology, Immunology, and Cancer Biology, University of Virginia Health System, Charlottesville, VA, USA
The Burkholderia cepacia complex (Bcc) is a group of Gram-negative bacteria that are ubiquitous in the environment and have emerged as opportunistic pathogens in immunocompromised patients. The primary patient populations infected with Bcc include individuals with cystic fibrosis (CF), as well as those with chronic granulomatous disease (CGD). While Bcc infection in CF is better characterized than in CGD, these two genetic diseases are not obviously similar and it is currently unknown if there is any commonality in host immune defects that is responsible for the susceptibility to Bcc. CF is caused by mutations in the CF transmembrane conductance regulator, resulting in manifestations in various organ systems, however the major cause of morbidity and mortality is currently due to bacterial respiratory infections. CGD, on the other hand, is a genetic disorder that is caused by defects in phagocyte NADPH oxidase. Because of the defect in CGD, phagocytes in these patients are unable to produce reactive oxygen species, which results in increased susceptibility to bacterial and fungal infections. Despite this significant defect in microbial clearance, the spectrum of pathogens frequently implicated in infections in CGD is relatively narrow and includes some bacterial species that are considered almost pathognomonic for this disorder. Very little is known about the cause of the specific susceptibility to Bcc over other potential pathogens more prevalent in the environment, and a better understanding of specific mechanisms required for bacterial virulence has become a high priority. This review will summarize both the current knowledge and future directions related to Bcc virulence in immunocompromised individuals with a focus on the roles of bacterial factors and neutrophil defects in pathogenesis.
Introduction
The Burkholderia cepacia complex (Bcc) is a group of Gram-negative bacilli that have emerged over the past 30 years as opportunistic pathogens in immunocompromised populations, specifically cystic fibrosis (CF) and chronic granulomatous disease (CGD). Bcc has recently been recognized as an emerging nosocomial pathogen in patients without CF and CGD, and its spread in the hospital setting has been associated with cross-transmission, frequent pulmonary procedures, and central venous access (Bressler et al., 2007). This complex of at least 17 distinct species was first identified as a plant pathogen in 1950s and was shown to be the causative agent of onion rot (Burkholder, 1950; Vanlaere et al., 2009; Lipuma, 2010). Bcc is also present in many non-pathogenic, ecologically beneficial relationships with plants, where it can be advantageous through the production of antimicrobial compounds, degradation of contaminants, and fixation of nitrogen (Mahenthiralingam et al., 2005; Lipuma, 2010).
Since B. cenocepacia and B. multivorans are the two most prevalent species in Bcc infections of both CGD and CF patients (Reik et al., 2005; Bressler et al., 2007), most investigations into the mechanisms of virulence have focused on these two species. Although we will attempt to more broadly apply these data to Bcc, it is currently unknown whether the recognized virulence factors always function similarly in all species of the complex during their interactions with the host innate immune system. It is also unknown whether there are unique and distinct bacterial factors that are required for disease in CGD versus CF, considering that these two diseases do not share any significant known immune defects. Due to the specific sensitivity of these two patient populations to Bcc, there may be unrecognized commonalities in host defects that allow for Bcc infection.
Bcc as Opportunistic Pathogens
Because Bcc infections are primarily associated with individuals with CGD and CF, it is interesting to compare these two not obviously related genetic diseases. Table 1 summarizes the disease manifestations and defects in phagocyte function associated with CGD and CF.
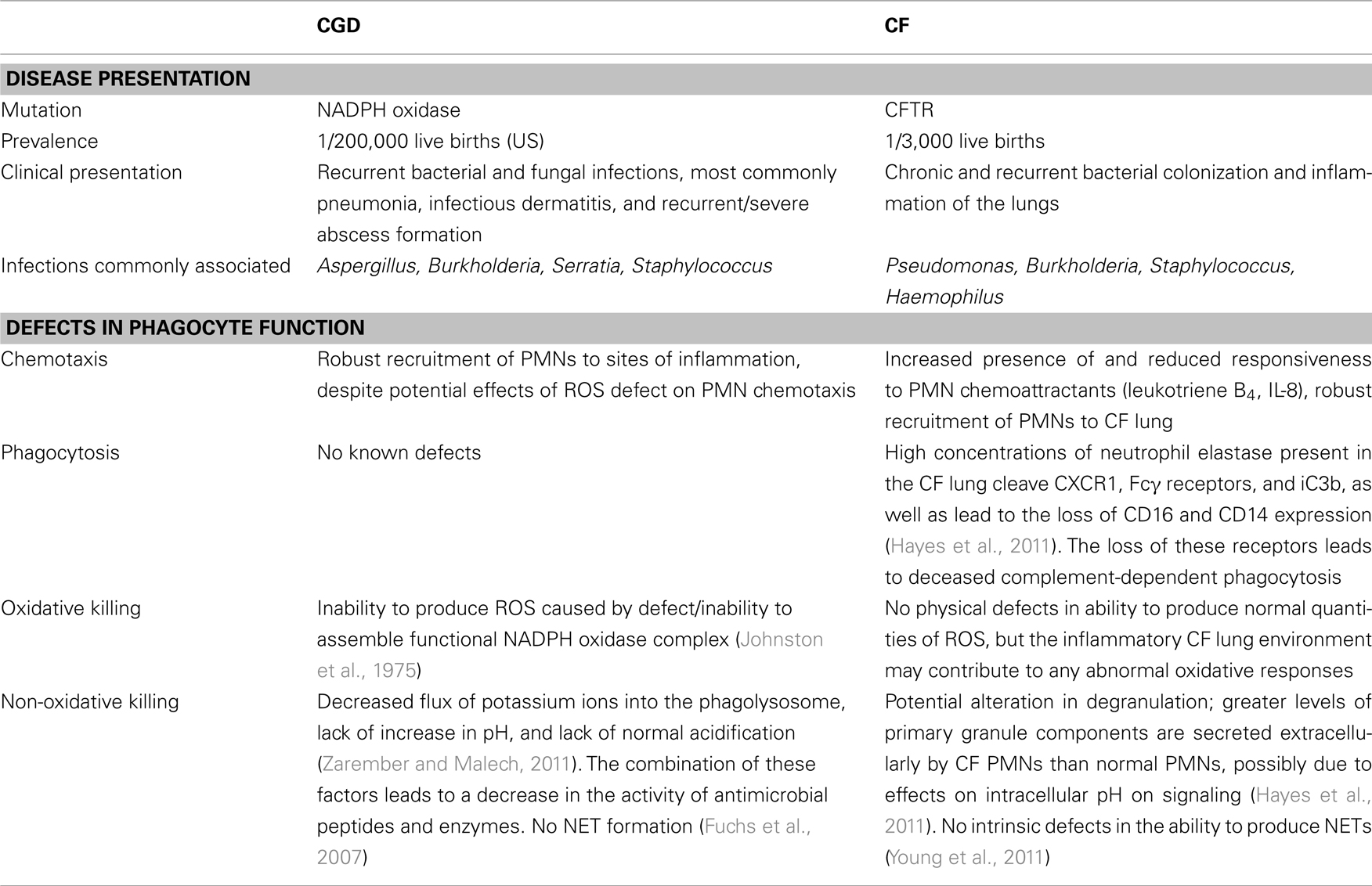
Table 1. Comparison of disease presentation and phagocyte function in CGD and CF.
Burkholderia cepacia (previously referred to as Pseudomonas cepacia) was recognized as an important pathogen in CGD in 1975 (Bottone et al., 1975). Bcc is now the leading cause of fatality from bacterial infections in CGD patients (Winkelstein et al., 2000). CGD is a genetic disorder caused by the inactivation of the phagocyte nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex. Because of this defect, some cells (neutrophils, mononuclear cells, macrophages, and eosinophils) in CGD patients are unable to produce reactive oxygen species (ROS), which results in ineffective clearance of some pathogens. These patients present with recurrent bacterial and fungal infections, which is frequently accompanied by uncontrolled inflammation and granuloma formation. The rate of incidence in the United States is 1/200,000 live births, or at least 20 patients per year (Holland, 2010).
Chronic granulomatous disease can be caused by mutations in any of the four components of NADPH oxidase. Flavocytochrome b558 is the catalytic center of the complex, and is a heterodimer composed of p22phox and gp91phox. It is contained on the membrane of specific granules and is incorporated into the phagosome membrane upon fusion (Segal, 2005). It provides a channel for electrons to be transferred through from NADPH in the cytosol to oxygen that is contained in the phagosome (Segal, 2005). The complex also contains two cytosolic components: p67phox and p47phox (Zarember and Malech, 2011). These cytoplasmic components are essential for stabilizing flavocytochrome b558 and facilitating the transfer of electrons. A defect in any one of these four components leads to inactivation of the complex and an inability to produce ROS. X-linked recessive CGD is caused by defects in gp91phox. Autosomal recessive CGD is caused by defects in p47phox, p67phox, or p22phox. Approximately 65% of CGD cases are X-linked (defects in gp91phox), 25% of cases are due to defects in p47phox, and the rest are split evenly between defects in p67phox and p22phox (Holland, 2010). There are two other genetic disorders with similar presentations to CGD that are not caused by mutations in NADPH oxidase components. Inhibitory mutations in Rac or deficiencies of glucose-6-phosphate dehydrogenase can also result in an inability to produce ROS (Johnston, 2001).
Overall mortality for CGD patients is around 2–5% per year, but morbidity remains a major issue for these patients and their families, as most patients have at least one severe infection every 3–4 years (Winkelstein et al., 2000; Marciano et al., 2004). In the initial cases of CGD diagnosed between 1957 and 1976, Staphylococcus was the most common infectious agent, followed by the common enteric pathogens Klebsiella species and Escherichia coli (Johnston, 2001). Now Aspergillus species are the most common causative infectious agents, followed by Bcc and Staphylococcus aureus (Winkelstein et al., 2000). The most common presentations of infection include pneumonia, infectious dermatitis, and recurrent/severe abscess formation beneath the skin and in organs (Winkelstein et al., 2000). Other symptoms have been described that are not caused directly by infection, including enteritis/colitis, discoid lupus, and chorioretinitis (Goldblatt et al., 1999; Segal et al., 2000; Winkelstein et al., 2000). The treatment regimen for these patients includes interferon-γ, antibacterial prophylaxis, and antifungal prophylaxis. Prophylactic treatment usually includes trimethoprim–sulfamethoxazole and itraconazole (Seger, 2008). Bone marrow transplantation can cure the disease if a histocompatible donor is available, which is not an option for most patients. As with other monogenetic diseases, gene therapy has the potential to be an effective curative treatment for CGD, however, to date, clinical trials have not been successful. The promise and problems associated with this approach have been recently reviewed (Grez et al., 2011).
Cystic fibrosis is caused by mutations in the CF transmembrane conductance regulator (CFTR), which functions as an ion channel. This defect results in the accumulation of mucus on epithelial surfaces, which contributes to ineffective mucociliary clearance in the lungs. CF is one of the most common inherited genetic diseases in the world with an annual frequency of approximately 1/3,000 live births (Hayes et al., 2011). In fact, one out of every 25 Caucasians is a heterozygous carrier of a mutated CFTR gene (Sheppard and Nicholson, 2002). The airway in CF patients is chronically colonized with potentially pathogenic bacteria, specifically Pseudomonas aeruginosa, S. aureus, and Haemophilus influenzae. Colonization and frequent infection of the CF airway leads to an excessive neutrophil (polymorphonuclear leukocyte, PMN) driven inflammatory response, which results in host tissue damage (McElvaney et al., 1992; Birrer et al., 1994). These patients experience recurrent respiratory infections and chronic lung inflammation that eventually culminates in respiratory failure (Dinwiddie, 2000). Pulmonary inflammation in CF is associated with high levels of inflammatory cytokines (IL-8, IL-6, TNF-alpha, and leukotriene B4), reduced levels of anti-inflammatory cytokines and antiproteases, and a rapid and substantial influx of PMNs (Rowe et al., 2005).
Burkholderia cepacia complex infections in both the CGD and CF patient populations initially present as a respiratory tract infection. An interesting distinction between infections in these two patient populations is the clinical presentation of this infection. In CF, pulmonary infection with Bcc generally causes endobronchial disease, and diagnosis is made by identification of bacteria in the sputum. In CGD, Bcc lung infection results in a pneumonic process associated with nodular infiltrates and diagnosis requires isolation following lung biopsy. Antibiotic treatment is generally able to clear Bcc in CGD, although recurrent re-infections do occur. In CF, Bcc infections are usually seen in older patients that are already chronically colonized with other CF pathogens, notably P. aeruginosa (Sheppard and Nicholson, 2002). According to the 2009 Cystic Fibrosis Foundation Registry Report, about 2.7% of CF patients are infected with Bcc, and these infections are associated with earlier fatality than non-Bcc-infected patients. Potentially due to the chronic nature of Bcc infections in CF and the sustained presence of the microbial community in the form of biofilms in the CF lung, Bcc isolates associated with CF show a higher degree of antibiotic resistance than Bcc isolates from CGD infections (Greenberg et al., 2009). Bcc infections in CF patients can result in a severe, necrotizing pneumonia, referred to as “cepacia syndrome,” that is not observed in Bcc infections in CGD patients. However, Bcc infections in CGD patients can become invasive and result in septicemia.
Chronic granulomatous disease and CF are both rare genetic diseases and, while infections by Bcc are prominent in these populations, Bcc infections are still relatively infrequent. Studies investigating Bcc virulence in CGD and CF aim to decrease morbidity and mortality in these patient populations, but they can also increase our understanding of how healthy individuals are able to fight off these rare infections. Since Bcc infections are very uncommon in immunocompetent individuals, the normal host immune response must be able to efficiently remove these pathogens, and the immune response or environment in CGD and CF, as well as other immunocompromised patients, would appear to be defective in this clearance. It is the investigation into the distinction between these normal and susceptible hosts that gives insight into normal and defective immune function in bacterial infections. Previous studies investigating CGD and other rare genetic diseases have contributed greatly to knowledge in the field on normal phagocyte function and bacteria–phagocyte interactions (Quie et al., 1967; Root et al., 1972; Lekstrom-Himes and Gallin, 2000). A better understanding of the infectious processes in patients with rare genetic defects in phagocyte function contributes to current knowledge on how normal, healthy individuals resist infections.
Functions of Normal PMNs and Defects in CGD and CF Patients
Chronic granulomatous disease manifests as a defect in all phagocytes normally capable of producing ROS. Macrophages and PMNs are normally the focus of study of CGD host phagocyte–pathogen interactions because of their essential roles as professional phagocytes. A recent review by Saldias and Valvano (2009) has summarized the current literature on the interactions between Bcc and macrophages, thus here we will focus specifically on Bcc interactions with PMNs. Bcc infections are characterized by a rapid and robust recruitment of PMNs to the site of infection. Normal PMNs have the ability to phagocytose and kill bacteria via oxidative and non-oxidative mechanisms, but CGD PMNs are only capable of utilizing non-oxidative killing mechanisms, leading to ineffective clearance of some pathogens. Because of the essential role of PMNs in normal host responses to bacteria and the substantial contribution of defective PMN function in CGD to pathogenesis, this review will focus on the defects associated with CGD PMNs and will provide comparison with potential defects in CF PMN function.
Polymorphonuclear leukocytes are highly motile phagocytes that comprise the first line of defense of the innate immune system against bacteria and play a crucial role in host defense in the lung. Four percent of the adult body mass is devoted to the production of PMNs, and as a result, PMNs are the most common nucleated cell in blood. Although PMNs are terminally differentiated once they leave bone marrow, they are still able to act autonomously as wandering phagocytes in all tissues, requiring highly specific and specialized responses (Zarember and Malech, 2011). They kill bacteria by fusing granules containing antimicrobial compounds with phagocytic compartments containing bacteria. These granules can also fuse with the plasma membrane to kill extracellular bacteria.
There are three types of granules that PMNs synthesize, and these different granule types contain molecules with different antimicrobial activities that often work synergistically to kill microbes once fused with the phagosome. Granules contain components that kill bacteria through both oxidative and non-oxidative mechanisms. Azurophil or primary granules contain myeloperoxidase, three neutral proteinases (cathepsin G, elastase, and proteinase 3), bactericidal permeability-increasing protein (BPI), and defensins. Specific or secondary granules contain lactoferrin, transcobalamin II, lysozyme, gelatinase-associated lipocalin, and flavocytochrome b558. Gelatinase or tertiary granules are classified by the presence of gelatinase but not lactoferrin and can contain any of the other components in specific granules. The matrix in granules contains negatively charged sulfated proteoglycans that sequester antimicrobial components until the granule is fused with the phagosome (Borregaard and Cowland, 1997; Segal, 2005).
Oxidative killing relies on the generation of ROS, primarily through NADPH oxidase. Electron transfer to oxygen through NADPH oxidase generates superoxide  in the phagosome. Superoxide is rapidly degraded into hydrogen peroxide (H2O2) by superoxide dismutase. Hydrogen peroxide can readily cross bacterial membranes and cause intracellular damage. CGD PMNs are defective in their ability to assemble functional NADPH oxidase complexes and therefore are unable to utilize oxidative killing mechanisms.
in the phagosome. Superoxide is rapidly degraded into hydrogen peroxide (H2O2) by superoxide dismutase. Hydrogen peroxide can readily cross bacterial membranes and cause intracellular damage. CGD PMNs are defective in their ability to assemble functional NADPH oxidase complexes and therefore are unable to utilize oxidative killing mechanisms.
In addition to oxidative killing mechanisms, normal PMNs can also use non-oxidative killing, which includes interactions between cationic regions of the granule proteins and the negatively charged bacterial membrane, as well as bacterial degradation by granule proteases. Examples of cationic peptides and proteins involved in non-oxidative killing of bacteria are BPI, LL-37, defensins, and azurocidin. These cationic granule peptides and proteins bind to and accumulate on the bacterial surface, leading to membrane permeabilization and osmotic lysis. These components are extremely important in non-oxidative killing of Gram-negative pathogens by PMNs (Hancock and Diamond, 2000). Granules also contain other components that possess different antimicrobial activities. Serine proteases degrade phagocytosed bacteria (Wiedow and Meyer-Hoffert, 2005). Lactoferrin sequesters iron away from pathogens, but also possesses direct antimicrobial activity (Ellison and Giehl, 1991). Many of the factors that function in non-oxidative killing strategies act synergistically or depend on each other for full activity after fusion or exocytosis. The lack of functional NADPH oxidase in CGD results in the reduced flux of potassium ions into the phagolysosome (Zarember and Malech, 2011). This influx plays a role in the release of cationic proteins from the matrix, and because of this CGD PMNs may also exhibit defects in non-oxidative killing. In CGD PMNs, the phagolysosome additionally does not increase pH and does not acidify as profoundly, which also has negative effects on the activity of antimicrobial peptides and enzymes (Dri et al., 2002).
Normal PMNs also use neutrophil extracellular traps (NETs) as a mechanism to kill extracellular microbes. These NETs are composed of PMN chromatin coated in granule components that have been expelled from the cell. The chromatin binds extracellular bacteria and fungi, trapping them in close contact with high concentrations of antimicrobial granule contents. NET formation is dependent on the generation of ROS by NADPH oxidase, and therefore CGD PMNs are also deficient in their ability to produce NETs (Fuchs et al., 2007). It has been shown that this defect also contributes to the inability of CGD PMNs to kill pathogens commonly associated with infections in CGD, specifically S. aureus and Aspergillus species (Brinkmann et al., 2004; Bianchi et al., 2009).
Most prior research has focused on the lung epithelial cell defects in CF (Conese and Assael, 2001; Machen, 2006). However more recently, there has been an emphasis in the field on defective immune cell function in the CF lung environment, particularly on potential defects in PMN function. These studies may assist in bridging the gap in understanding the specific susceptibilities of these two patient populations to Bcc infections. Defects in CF PMN function could contribute to ineffective clearance of lung pathogens in the presence of zealous PMN recruitment and inflammation (Hayes et al., 2011). It is already well established that PMN necrosis and release of proteolytic enzymes contribute significantly to the tissue damage in the CF lung. It has been shown that PMNs can become trapped in bacterial biofilms in the CF lung and undergo necrosis; after necrosis, their biomass, NETs, and intracellular leakage can contribute biofilm formation and lung injury (Jesaitis et al., 2003; Brinkmann et al., 2004; Walker et al., 2005). CF PMNs display excessive and prolonged superoxide production when stimulated, are constitutively primed (likely via the presence of IL-8, TNF-alpha, and P. aeruginosa alginate in the CF airway), and are unresponsive to anti-inflammatory signals through IL-10 (Pedersen et al., 1990; Koller et al., 1995; Taggart et al., 2000). Another observed potential defect is an alteration in degranulation; greater levels of primary granule components are secreted extracellularly by CF PMNs than by normal PMNs, possibly due to effects of intracellular pH on signaling (Koller et al., 1995; Coakley et al., 2000, 2002; Taggart et al., 2000). This release of toxic granule contents contributes to the extensive lung damage characteristic of CF. The absence of CFTR has also been shown to have direct effects on PMN function. The addition of chloride ion to hydrogen peroxide to produce hypochlorous acid (HOCl) through myeloperoxidase is another ROS-dependent killing mechanism, and CF PMNs display a decreased ability to chlorinate bacteria and a decreased ability to kill P. aeruginosa that is exacerbated by a chloride-deficient microenvironment (Painter et al., 2006, 2008).
There are also indications that the environment in the CF lung may significantly impair PMN function. High concentrations of neutrophil elastase present in the CF lung cleave CXCR1, Fcγ receptors, and iC3b, as well as lead to the loss of CD16 and CD14 expression (Tosi et al., 1990; Birrer et al., 1994; Alexis et al., 2006; Hartl et al., 2007; Tirouvanziam et al., 2008). The loss of these receptors leads to deceased complement-dependent phagocytosis. Hypersalinity of the airway surface fluid has been shown to inhibit the killing actions of epithelial cells in the CF lung through inactivation of β-defensin-1 and potentially other antimicrobials, although these findings are considered somewhat controversial in the CF field (Smith et al., 1996; Goldman et al., 1997; Krouse, 2001). Peripheral PMNs isolated from CF patients show comparable NET formation to normal PMNs, but there is also evidence that PMNs in the CF lung environment display increased NET formation that contributes to the pulmonary obstruction typical in CF (Marcos et al., 2010; Young et al., 2011). Further investigation into defective phagocyte function in CF could provide insight into links between the susceptibilities of both these patient populations to Bcc infections.
Although resident and migrating phagocytes play an essential role in bacterial clearance in the airway, there are other important components of the host defense at the mucosal airway surface. The airway epithelium protects the host from infection at the interface with the environment, where it is constantly exposed and bombarded with microbes. Epithelial cells produce cytokines, chemokines, and antimicrobial peptides, either constitutively or upon stimulation with microbial products (Bals and Hiemstra, 2004). Examples of antimicrobial products produced by airway epithelial cells include β-defensins, LL-37, and lactoferrin (Bals and Hiemstra, 2004). Dual oxidases (Duox) are also expressed by epithelial cells in the airways and produce hydrogen peroxide, which contributes to antimicrobial activities through the lactoperoxidase system (Geiszt et al., 2003). This theoretically allows for partial reconstitution of ROS production in the lung in response to bacterial infection, but interesting it has been shown that Bcc does not stimulate hydrogen peroxide production by bronchial epithelial cells through Duox (Rada and Leto, 2010).
Models Used to Assess Virulence of Bcc in CGD and CF
Many previous studies have focused on Bcc virulence in models of CF, and these models have been well described in other reviews (Scholte et al., 2004; Mahenthiralingam et al., 2005; Loutet and Valvano, 2010). Therefore, this section will focus on describing models of CGD and their potential for use in studies of Bcc virulence. Many in vitro and in vivo models of CGD are available, and some of these models have been adapted to assess microbial virulence in this patient population. There are two characterized, commercially available mouse models of CGD. The first is a model of X-linked CGD, which lacks the mouse equivalent of human gp91phox (Pollock et al., 1995). This strain of mouse exhibits a complete lack of functional protein, an inability to form a functional NADPH oxidase complex, and phagocytes that are unable to produce a respiratory burst. Bcc are virtually avirulent in wild-type mouse strains but are able to cause lethal infections in mouse models of CGD (Chiu et al., 2001; Sousa et al., 2007). Bcc CF isolates have been tested in this model, and the infecting bacteria multiplied five orders of magnitude in 3–6 days after intratracheal infection. The infected mice displayed neutrophil-dominated lung inflammation and abscesses, sepsis, and eventual death, which models a disease progression reflective of that observed in CGD human hosts. Isogenic mutants in some known virulence factors of B. cepacia and B. cenocepacia, in particular in quorum sensing and exopolysaccharide production, as well as a panel of less virulent isolates were also examined, and a loss or decrease in virulence was observed (Sousa et al., 2007).
The autosomal recessive mouse model of CGD lacks the mouse equivalent of p47phox, which is the complex component most commonly associated with human cases of autosomal recessive CGD. Characterization of this model showed a phenotype similar to human CGD; these mice spontaneously developed severe infections, usually by the same or related pathogens that are associated with infections in human CGD hosts (Jackson et al., 1995). Phagocytes from these animals display an inability to produce an oxidative burst and are unable to kill S. aureus and other pathogens characteristic of CGD (Jackson et al., 1995). This model has also been used to study the virulence of Bcc in CGD. B. multivorans clinical isolates from both CGD and CF patients were shown to establish lethal infections when injected intraperitoneally, while an environmental isolate was avirulent (Zelazny et al., 2009).
Mouse models allow analysis of Bcc virulence in a CGD host, but additional approaches are necessary to directly investigate Bcc interactions with host phagocytes. Both cell lines and primary cells have been used in assessment of bacterial virulence in interactions with CGD phagocytes. PMNs and macrophages can be derived from CGD patients, when available, or from CGD animals (Morgenstern et al., 1997; Kaneda et al., 1999; Zelazny et al., 2009). Because these cells are isolated directly from patients or animals with CGD, these cells most closely replicate the CGD defect, but unfortunately are unable to be genetically modified or adequately controlled experimentally. Also, the use of primary cells adds an additional level of variation to experiments due to donor-specific genetic and environmental factors. Cell lines that are genetically modified to lack functional NADPH oxidase proteins have also been constructed. PBL-985, a human myeloid leukemia cell line, is deficient in gp91phox and can be differentiated into monocytes or granulocytes (Zhen et al., 1993). These cells can be further genetically modified and lack the donor variability of primary cells. Unfortunately, PMNs are terminally differentiated cells, and precursor immortal cell lines must be differentiated in vitro into a PMN-like state. In a related promyelocytic cell line, HL-60, it has been demonstrated that in vitro differentiation from the promyelocytic stage results in an inability to synthesize secondary and tertiary granules (Le Cabec et al., 1997). Therefore, these precursor cell lines lack the full array of antimicrobial activities associated with mature, terminal PMNs. There are also variants of the J774.16 murine macrophage cell line that are deficient in gp91phox, and do not need to be differentiated (Goldberg et al., 1990). These immortal macrophage lines can be used to broadly study phagocyte interactions but are unable to utilize the full repertoire of killing mechanisms employed by PMNs.
Normal cell lines and primary cells can also be treated with inhibitors of NADPH oxidase function to create a CGD phenotype. Diphenyleneiodonium (DPI) is the most commonly used inhibitor and acts on flavoproteins, such as flavocytochrome b558 of NADPH oxidase. An electron is extracted from flavin adenine dinucleotide (FAD) by DPI to form a radical, which is then added back to protein groups in or near the active site of the flavoprotein to form covalent, phenylated adducts. Studies have also shown that DPI can also act on and inhibit nitric oxide synthase and xanthine oxidase (Riganti et al., 2004), while anaerobic killing, phagocytosis, and motility are not significantly affected by DPI-treatment (Ellis et al., 1988). There is a slight decrease in degranulation of azurophilic and specific granules after DPI-treatment, which is similar to what has been observed in PMNs isolated from CGD patients (Ellis et al., 1988). Another well-studied NADPH oxidase inhibitor is apocynin (4-hydroxy-3-methoxyacetophenone, also known as acetovanillone), which is a methoxy-substituted catechol that inhibits superoxide production by phagocytes while not affecting phagocytosis. It likely inhibits complex assembly by blocking sulfhydryl groups (Vejrazka et al., 2005). Although use of these inhibitors produces a CGD phenotype and creates an in vitro system amenable to controlled experimentation, the cells may exhibit additional defects due to non-specific action of these inhibitors or may not manifest completely the defects of CGD phagocytes. Also, in vitro experimentation with either primary cells or immortal cell lines can only be used to assess virulence factors that directly interact with host cells in comparison to factors that may indirectly affect cells through modulating the host environment.
Resistance of Bcc to Killing by PMNs
Burkholderia cepacia complex is a very diverse group of pathogens, both genetically and phenotypically. Studies have shown remarkable differences in virulence between clinical isolates and environmental isolates of Bcc, and even clinical strains of the same species have been observed to have varying virulence in vitro and in mouse models. In the context of Bcc infections in CF (Loutet and Valvano, 2010), factors have been shown to have direct effects on interactions with host innate immune cells. Both Figure 1 and Table 2 illustrate the contribution of Bcc factors to bacterial survival in interactions with phagocytes. Most of the factors described in this review have been investigated mainly in models of CF, but these factors may have broad roles for protection from host phagocyte killing and are likely also important in Bcc survival in interactions with CGD PMNs.
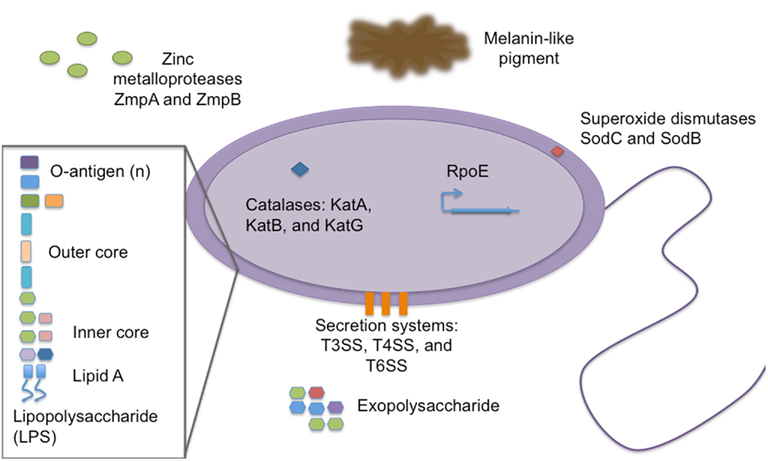
Figure 1. Burkholderia cepacia complex virulence factors important for survival in interactions with host phagocytes. Representation of known virulence factors utilized by members of Bcc in order to persist when confronting host phagocytes. The description and evidence in support of the roles of these factors is described in the text.
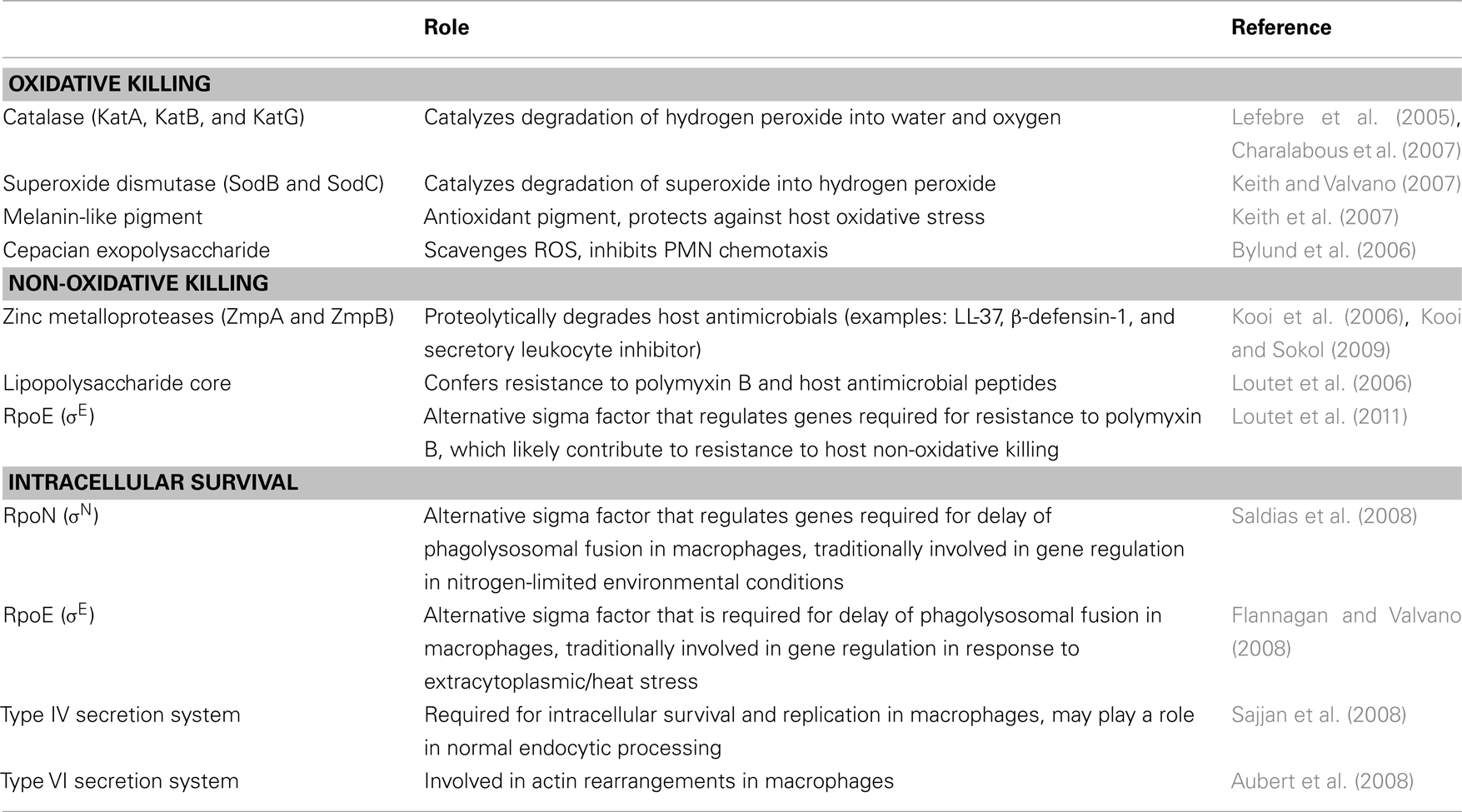
Table 2. Burkholderia cepacia complex virulence factors involved in interactions with host phagocytes.
B. cenocepacia has been shown to be readily ingested by normal PMNs and phagocytosis is associated with a robust oxidative burst. In PMN bactericidal assays, Bcc is killed by normal PMNs but is able to persist when incubated with CGD PMNs (Zelazny et al., 2009; Greenberg et al., 2010). Similarly B. cenocepacia has been found to induce PMN apoptosis, an effect dependent on viable bacteria. CGD PMNs were also equally apoptotic in comparison to normal PMNs after phagocytosis of B. cenocepacia, but B. cenocepacia also induced PMN necrosis in a subset of CGD PMNs, which could contribute to increased virulence in this population. This was true in both primary CGD PMNs and PMNs treated with DPI (Bylund et al., 2005). Another study found that a CGD clinical isolate of B. multivorans showed a higher association with CGD PMNs than with PMNs from normal, healthy donors, which suggests an additional distinction between the interactions of Bcc with normal and CGD PMNs (Zelazny et al., 2009).
Defenses Against Host Oxidative Killing
In the course of an infection, successful pathogens must evade the innate immune system in order to persist. Because of the prevalence of Bcc as CGD pathogens, oxidative killing mechanisms by host phagocytes, specifically neutrophils, are thought to be essential for Bcc clearance in healthy individuals. Superoxide dismutases and catalases protect bacteria from oxidative damage during normal respiration and in interactions with the host innate immune response. Bcc has been shown to be resistant to concentrations of ROS in vitro that are equivalent to an oxidative burst and this is due to periplasmic superoxide dismutase (Sod), catalase-peroxidases (Kat), and an antioxidant melanin-like pigment (Lefebre and Valvano, 2001; Saldias and Valvano, 2009). Both catalase and superoxide dismutase are universal mechanisms of bacterial resistance to host oxidative killing. A pigment produced by B. cenocepacia C5424 has been studied, and the absence of the pigment in a strain mutated in hppD (4-hydroxyphenylpyruvate dioxygenase) was shown to lead to an increase in sensitivity to extracellular superoxide and hydrogen peroxide in in vitro bacterial viability assays (Keith et al., 2007). This study also showed that the mutant co-localized to a higher extent than the wild-type strain with more degradative compartments in macrophages, and that this co-localization was dependent on NADPH oxidase activity. This finding suggested that wild-type bacteria that are able to produce the melanin-like pigment are more protected from oxidative killing in the phagosome, and that mutant bacteria that are unable to produce the pigment are associated with a loss of bacterial viability inside the phagosome and an inability to delay progression to a more degradative compartment. Thus, the melanin-like antioxidant pigment produced by B. cenocepacia has been shown to be protective against host oxidative killing (Keith et al., 2007). However not all strains of Bcc isolated from patients produce this pigment, suggesting that additional mechanisms of resisting host oxidative killing must exist.
Exopolysaccharides are bacterial virulence factors that have roles in evasion of host defenses, adhesion, and resistance to antimicrobials. Bcc produces at least four different exopolysaccharides, and the predominant exopolysaccharide is the heptasaccharide cepacian (Vinion-Dubiel and Goldberg, 2003; Zlosnik and Speert, 2010). Not all Bcc isolates make cepacian, but most clinical isolates do produce exopolysaccharide (Zlosnik and Speert, 2010). Cepacian produced by B. cenocepacia has been shown to scavenge ROS and inhibit neutrophil chemotaxis in in vitro migration assays (Bylund et al., 2006). Mutants that are unable to produce cepacian are less virulent than wild-type, cepacian-producing strains in a gp91phox−/− mouse model of CGD, which suggests that this exopolysaccharide is potentially an important bacterial factor for virulence in CGD (Sousa et al., 2007).
Defenses Against Host Non-Oxidative Killing
Burkholderia cepacia complex possesses tools to combat non-oxidative killing mechanisms, and these factors likely play an essential role in bacterial pathogenesis in CGD hosts that are defective in their ability to kill bacteria through oxidative killing mechanisms. Zinc metalloproteases are commonly utilized by bacteria to proteolytically degrade host antimicrobials. B. cenocepacia zinc metalloproteases ZmpA and ZmpB have been shown to act in resisting bacterial killing by host antimicrobial peptides in in vitro bacterial viability assays (Kooi and Sokol, 2009). ZmpB cleaves β-defensin-1, ZmpA cleaves LL-37, and both enzymes cleave elafin and secretory leukocyte inhibitor. All of these targets are components of the innate immune response to bacteria. Mutants in both zmpA and zmpB have been tested in a rat agar bead model, which is commonly used for assessing virulence of pathogens that chronically infect CF patients. Both mutant strains were able to persist and were present in the lung 14 days after infection, but they were less virulent than the wild-type strain. The zmpA mutant was characterized by a reduction in bacterial load in comparison to the wild-type strain, and the zmpB induced less lung tissue inflammation (Kooi et al., 2006). This indicates that ZmpA and ZmpB are required for full virulence but are not required for bacterial survival in this model. It would be interesting to evaluate these mutants in a mouse model of CGD, since such an acute model of infection could directly assess the importance of these zinc metalloproteases to infection.
Lipopolysaccharide (LPS) is dually important both in targeting of and protection from non-oxidative killing. LPS is a major component of Gram-negative membrane and is composed of lipid A, core oligosaccharides, and O-antigen. The negative charge of lipid A is an important binding target for cationic antimicrobial peptides. The lipid A of Bcc contains Kdo (3-deoxy-D-manno-oct-2-ulosinic acid), Ko monosaccharide (D-glycero-D-talo-oct-ulosonic acid), and Ara4N (4-amino-4-deoxy-L-arabinose; Silipo et al., 2005, 2007). Substitution of Ara4N on the lipid A and inner core oligosaccharide is protective against cationic antimicrobials, and synthesis of Ara4N is essential in B. cenocepacia for survival (Ortega et al., 2007). Inner core oligosaccharides are required for in vitro resistance to antimicrobial peptides and survival in a rat model of lung infection. Specifically, a deep rough mutant (lacking both O-antigen and the inner core) of B. cenocepacia was more sensitive to killing by polymyxin B, melittin, and HNP-1 in in vitro bacterial viability assays (Loutet et al., 2006).
In a signature-tagged mutagenesis screen of B. cenocepacia K56-2 in the rat agar bead model of chronic infection, several clones were identified as being avirulent that contained insertions in the genetic region responsible for O-antigen synthesis, indicating that under these conditions O-antigen was essential for virulence (Hunt et al., 2004). Interestingly, clinical isolates have been isolated with either an LPS-rough (with no O-antigen) or an LPS-smooth (with O-antigen) phenotype, indicating that O-antigen may not be required for virulence in all species of the complex under all conditions. For example, one of the best characterized B. cenocepacia isolates, the ET-12 lineage CF isolate J2315, which displays an LPS-rough phenotype. O-antigen has been shown to be essential for serum resistance but not in resistance to host antimicrobials (Ortega et al., 2005).
In addition to factors that have been recognized to act directly to protect against host defenses, genetic regulators can also contribute to virulence. The alternative sigma factor RpoE, a transcriptional regulator, has been shown to be required for lysosomal fusion delay, growth under high osmolarity, and growth under high temperature (Flannagan and Valvano, 2008). RpoE has also been shown to play a role in resistance to polymyxin B in in vitro bacterial viability assays, which suggests that expression of RpoE may protect the bacteria from non-oxidative killing as well (Loutet et al., 2011). Collectively, these two studies do implicate that regulation by RpoE is important for Bcc virulence, but the RpoE regulon has yet to be reported.
Since normal, healthy individuals do not typically contract clinical Bcc infections, the virulence associated with each of these factors likely relies partially on host susceptibility and bacterial factors, which may differ between these two genetic disorders. Although the virulence factors studied in CF likely also play important roles in Bcc pathogenesis in CGD, this is by no means a comprehensive list and research is currently being conducted to learn more about specific roles of both these molecules as well as currently unrecognized factors using models for assessing virulence in CGD.
Concluding Remarks and Future Directions
Despite the extensive amount of focus and new insight into Bcc virulence obtained in the past decade, there is still much to learn. Bcc is a very diverse group comprising of at least 17 distinct species, and the majority of studies have focused on only the most prevalent three species: B. cenocepacia, B. multivorans, and B. cepacia. Also, many clinical strains and available isogenic mutants within these species have only been investigated in subsets of the models available to assess virulence in either CGD or CF. A more comprehensive view of the virulence of this complex will be obtained through future studies characterizing the similarities and differences between each species and the role of each virulence factor in many models. It is already recognized that clinical isolates from the same species (example: B. cenocepacia J2315 and K56-2) display different virulence phenotypes, and differences between these related isolates could be characterized. In addition, most studies have focused on Bcc virulence in the context of CF. As presented in this review, many of these studies have contributed knowledge that can be applied to potential host–pathogen interactions in CGD. Further characterization of known virulence factors in models of CGD and high-throughput genetic screens to identify virulence factors that are essential specifically in CGD will generate a more complete understanding of Bcc pathogenesis in CGD. Also, as previously mentioned, there is no clear link between the immune defects in CF and CGD, and therefore the progression of disease and factors required could differ greatly between infections in these two patient populations.
A majority of the work investigating Bcc–host phagocyte interactions up to now has focused on macrophages, mainly due to the availability of immortal cell lines. Due to the large influx of PMNs to the site of infection in both CF and CGD, PMNs are playing an extremely important role in pathogenesis. The bacterial killing mechanisms used by PMNs are more numerous and varied than that of macrophages, and the process of phagosomal maturation also varies greatly between PMNs and macrophages. It is thought that Bcc exists in an intracellular Bcc-containing vacuole (BcCV) in macrophages, but it is unknown whether Bcc is able to persist in a similar compartment in PMNs (Lamothe et al., 2007). It has been shown that both the type IV and type VI secretion systems play a role in interactions with macrophages and may be important for intracellular survival, but it is unknown what their role is in interactions with PMNs (Aubert et al., 2008; Sajjan et al., 2008). It has been demonstrated that Bcc is ineffectively killed and cleared by CGD PMNs (Zelazny et al., 2009; Greenberg et al., 2010), but it is not clear whether the surviving Bcc are intracellular or associated extracellularly with PMNs. This distinction is very important for understanding the mechanism in which Bcc is able to persist in this patient population.
Another avenue for future research is to identify mechanisms of effective killing and clearance of Bcc in normal individuals. Normal PMNs are able to effectively kill Bcc while CGD PMNs are unable to do so. Because of the defect in oxidative killing in CGD, this was suggestive of oxidative killing being important for clearance by normal PMNs. However, in vitro studies have demonstrated high resistance of Bcc to both oxidative and non-oxidative killing mechanisms. There are also other defects in non-oxidative killing associated with CGD PMNs, specifically in granule component activation and NET formation. It could also be the synergistic effects of all of these defects together that allow for persistence in this patient population as well as in CF patients. These mechanistic studies are difficult in a terminally differentiated cell that is not amenable to genetic manipulation but could be feasible in vitro using purified components or in cell-based assays using combinations of inhibitors.
A more broad implication of understanding Bcc pathogenesis in CGD is to search for commonalities in virulence strategies employed by all CGD pathogens. Since the spectrum of potential pathogens causing infections in this population is very narrow and most infectious agents are opportunistic, there may be very specific factors or strategies utilized by this group of microbes to cause infections in CGD. Understanding these overarching themes of resistance to killing in the CGD host could lead to the development of both preventative care and treatment options.
Since Bcc infections are typically restricted to patients with CF or CGD, the comparison of the defects in these two genetic diseases remains an interesting venue for future research. As previously described in this review, Bcc infections in CF occur subsequent to colonization with P. aeruginosa, and the presence of bacterial biofilms and the effect of these biofilms on PMN function may contribute to the ability of Bcc to persist in CF patients. A recent study confirmed the contribution of the CF lung environment to susceptibility to Bcc by infecting wild-type, normally resistant mice with B. cenocepacia and alginate from P. aeruginosa; in this case the bacteria were able to persist and cause lung inflammation (Goldberg et al., 2011). It is also hypothesized that the CF lung environment may result in defective PMN function through anaerobic mucus-covered airways, high protease concentrations, and high levels of inflammatory signals (Boucher, 2004; Hayes et al., 2011). In addition to the lung environment, treatments for CF may also impact PMN function. Macrolide therapy has been shown to significantly improve lung function in CF, but these drugs also accelerate PMN apoptosis, inhibit ROS production by PMNs, and inactivate neutrophil elastase (Aoshiba et al., 1995; Gorrini et al., 2001; Shinkai and Rubin, 2005). The use of macrolides is largely ineffective in treatment and prevention of Bcc, and its effects on PMN function may actually contribute to the pathogenicity of Bcc in CF (Saiman et al., 2002). A more complete understanding of PMN defects in CF, whether attributed to treatment, the inflammatory environment, or the CFTR defect directly, could find a link between immune function in these two patient populations and better define the requirements for Bcc virulence in immunocompromised populations.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the members of the Goldberg laboratory and our collaborators for their intellectual contributions. Laura A. Porter is supported financially by the National Institutes of Health through the University of Virginia Infectious Diseases training grant AI07406.
References
Alexis, N. E., Muhlebach, M. S., Peden, D. B., and Noah, T. L. (2006). Attenuation of host defense function of lung phagocytes in young cystic fibrosis patients. J. Cyst. Fibros. 5, 17–25.
Aoshiba, K., Nagai, A., and Konno, K. (1995). Erythromycin shortens neutrophil survival by accelerating apoptosis. Antimicrob. Agents Chemother. 39, 872–877.
Aubert, D. F., Flannagan, R. S., and Valvano, M. A. (2008). A novel sensor kinase-response regulator hybrid controls biofilm formation and type VI secretion system activity in Burkholderia cenocepacia. Infect. Immun. 76, 1979–1991.
Bals, R., and Hiemstra, P. S. (2004). Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur. Respir. J. 23, 327–333.
Bianchi, M., Hakkim, A., Brinkmann, V., Siler, U., Seger, R. A., Zychlinsky, A., and Reichenbach, J. (2009). Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 114, 2619–2622.
Birrer, P., McElvaney, N. G., Rudeberg, A., Sommer, C. W., Liechti-Gallati, S., Kraemer, R., Hubbard, R., and Crystal, R. G. (1994). Protease-antiprotease imbalance in the lungs of children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 150, 207–213.
Borregaard, N., and Cowland, J. B. (1997). Granules of the human neutrophilic polymorphonuclear leukocyte. Blood 89, 3503–3521.
Bottone, E. J., Douglas, S. D., Rausen, A. R., and Keusch, G. T. (1975). Association of Pseudomonas cepacia with chronic granulomatous disease. J. Clin. Microbiol. 1, 425–428.
Boucher, R. C. (2004). New concepts of the pathogenesis of cystic fibrosis lung disease. Eur. Respir. J. 23, 146–158.
Bressler, A. M., Kaye, K. S., LiPuma, J. J., Alexander, B. D., Moore, C. M., Reller, L. B., and Woods, C. W. (2007). Risk factors for Burkholderia cepacia complex bacteremia among intensive care unit patients without cystic fibrosis: a case-control study. Infect. Control Hosp. Epidemiol. 28, 951–958.
Brinkmann, V., Reichard, U., Goosmann, C., Fauler, B., Uhlemann, Y., Weiss, D. S., Weinrauch, Y., and Zychlinsky, A. (2004). Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535.
Bylund, J., Burgess, L. A., Cescutti, P., Ernst, R. K., and Speert, D. P. (2006). Exopolysaccharides from Burkholderia cenocepacia inhibit neutrophil chemotaxis and scavenge reactive oxygen species. J. Biol. Chem. 281, 2526–2532.
Bylund, J., Campsall, P. A., Ma, R. C., Conway, B. A., and Speert, D. P. (2005). Burkholderia cenocepacia induces neutrophil necrosis in chronic granulomatous disease. J. Immunol. 174, 3562–3569.
Charalabous, P., Risk, J. M., Jenkins, R., Birss, A. J., Hart, C. A., and Smalley, J. W. (2007). Characterization of a bifunctional catalase-peroxidase of Burkholderia cenocepacia. FEMS Immunol. Med. Microbiol. 50, 37–44.
Chiu, C. H., Ostry, A., and Speert, D. P. (2001). Invasion of murine respiratory epithelial cells in vivo by Burkholderia cepacia. J. Med. Microbiol. 50, 594–601.
Coakley, R. J., Taggart, C., Canny, G., Greally, P., O’Neill, S. J., and McElvaney, N. G. (2000). Altered intracellular pH regulation in neutrophils from patients with cystic fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 279, L66–74.
Coakley, R. J., Taggart, C., McElvaney, N. G., and O’Neill, S. J. (2002). Cytosolic pH and the inflammatory microenvironment modulate cell death in human neutrophils after phagocytosis. Blood 100, 3383–3391.
Conese, M., and Assael, B. M. (2001). Bacterial infections and inflammation in the lungs of cystic fibrosis patients. Pediatr. Infect. Dis. J. 20, 207–213.
Dri, P., Presani, G., Perticarari, S., Alberi, L., Prodan, M., and Decleva, E. (2002). Measurement of phagosomal pH of normal and CGD-like human neutrophils by dual fluorescence flow cytometry. Cytometry 48, 159–166.
Ellis, J. A., Mayer, S. J., and Jones, O. T. (1988). The effect of the NADPH oxidase inhibitor diphenyleneiodonium on aerobic and anaerobic microbicidal activities of human neutrophils. Biochem. J. 251, 887–891.
Ellison, R. T. III, and Giehl, T. J. (1991). Killing of Gram-negative bacteria by lactoferrin and lysozyme. J. Clin. Invest. 88, 1080–1091.
Flannagan, R. S., and Valvano, M. A. (2008). Burkholderia cenocepacia requires RpoE for growth under stress conditions and delay of phagolysosomal fusion in macrophages. Microbiology 154, 643–653.
Fuchs, T. A., Abed, U., Goosmann, C., Hurwitz, R., Schulze, I., Wahn, V., Weinrauch, Y., Brinkmann, V., and Zychlinsky, A. (2007). Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 176, 231–241.
Geiszt, M., Witta, J., Baffi, J., Lekstrom, K., and Leto, T. (2003). Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J. 17, 1502–1504.
Goldberg, J. B., Ganesan, S., Comstock, A. T., Zhao, Y., and Sajjan, U. S. (2011). Cable pili and the associated 22 kda adhesin contribute to Burkholderia cenocepacia persistence in vivo. PLoS ONE 6, e22435. doi:10.1371/journal.pone.0022435
Goldberg, M., Belkowski, L. S., and Bloom, B. R. (1990). Regulation of macrophage function by interferon-gamma. Somatic cell genetic approaches in murine macrophage cell lines to mechanisms of growth inhibition, the oxidative burst, and expression of the chronic granulomatous disease gene. J. Clin. Invest. 85, 563–569.
Goldblatt, D., Butcher, J., Thrasher, A. J., and Russell-Eggitt, I. (1999). Chorioretinal lesions in patients and carriers of chronic granulomatous disease. J. Pediatr. 134, 780–783.
Goldman, M. J., Anderson, G. M., Stolzenberg, E. D., Kari, U. P., Zasloff, M., and Wilson, J. M. (1997). Human β-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 88, 553–560.
Gorrini, M., Lupi, A., Viglio, S., Pamparana, F., Cetta, G., Iadarola, P., Powers, J. C., and Luisetti, M. (2001). Inhibition of human neutrophil elastase by erythromycin and flurythromycin, two macrolide antibiotics. Am. J. Respir. Cell Mol. Biol. 25, 492–499.
Greenberg, D. E., Goldberg, J. B., Stock, F., Murray, P. R., Holland, S. M., and Lipuma, J. J. (2009). Recurrent Burkholderia infection in patients with chronic granulomatous disease: 11-year experience at a large referral center. Clin. Infect. Dis. 48, 1577–1579.
Greenberg, D. E., Marshall-Batty, K. R., Brinster, L. R., Zarember, K. A., Shaw, P. A., Mellbye, B. L., Iversen, P. L., Holland, S. M., and Geller, B. L. (2010). Antisense phosphorodiamidate morpholino oligomers targeted to an essential gene inhibit Burkholderia cepacia complex. J. Infect. Dis. 201, 1822–1830.
Grez, M., Reichenbach, J., Schwable, J., Seger, R., Dinauer, M. C., and Thrasher, A. J. (2011). Gene therapy of chronic granulomatous disease: the engraftment dilemma. Mol. Ther. 19, 28–35.
Hancock, R. E., and Diamond, G. (2000). The role of cationic antimicrobial peptides in innate host defenses. Trends Microbiol. 8, 402–410.
Hartl, D., Latzin, P., Hordijk, P., Marcos, V., Rudolph, C., Woischnik, M., Krauss-Etschmann, S., Koller, B., Reinhardt, D., Roscher, A. A., Roos, D., and Griese, M. (2007). Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat. Med. 13, 1423–1430.
Hayes, E., Pohl, K., McElvaney, N. G., and Reeves, E. P. (2011). The cystic fibrosis neutrophil: a specialized yet potentially defective cell. Arch. Immunol. Ther. Exp. (Warsz) 59, 97–112.
Hunt, T. A., Kooi, C., Sokol, P. A., and Valvano, M. A. (2004). Identification of Burkholderia cenocepacia genes required for bacterial survival in vivo. Infect. Immun. 72, 4010–4022.
Jackson, S. H., Gallin, J. I., and Holland, S. M. (1995). The p47phox mouse knock-out model of chronic granulomatous disease. J. Exp. Med. 182, 751–758.
Jesaitis, A. J., Franklin, M. J., Berglund, D., Sasaki, M., Lord, C. I., Bleazard, J. B., Duffy, J. E., Beyenal, H., and Lewandowski, Z. (2003). Compromised host defense on Pseudomonas aeruginosa biofilms: characterization of neutrophil and biofilm interactions. J. Immunol. 171, 4329–4339.
Johnston, R. B. Jr. (2001). Clinical aspects of chronic granulomatous disease. Curr. Opin. Hematol. 8, 17–22.
Johnston, R. B. Jr., Keele, B. B. Jr., Misra, H. P., Lehmeyer, J. E., Webb, L. S., Baehner, R. L., and RaJagopalan, K. V. (1975). The role of superoxide anion generation in phagocytic bactericidal activity. Studies with normal and chronic granulomatous disease leukocytes. J. Clin. Invest. 55, 1357–1372.
Kaneda, M., Sakuraba, H., Ohtake, A., Nishida, A., Kiryu, C., and Kakinuma, K. (1999). Missense mutations in the gp91-phox gene encoding cytochrome b558 in patients with cytochrome b positive and negative X-linked chronic granulomatous disease. Blood 93, 2098–2104.
Keith, K. E., Killip, L., He, P., Moran, G. R., and Valvano, M. A. (2007). Burkholderia cenocepacia C5424 produces a pigment with antioxidant properties using a homogentisate intermediate. J. Bacteriol. 189, 9057–9065.
Keith, K. E., and Valvano, M. A. (2007). Characterization of SodC, a periplasmic superoxide dismutase from Burkholderia cenocepacia. Infect. Immun. 75, 2451–2460.
Koller, D. Y., Urbanek, R., and Gotz, M. (1995). Increased degranulation of eosinophil and neutrophil granulocytes in cystic fibrosis. Am. J. Respir. Crit. Care Med. 152, 629–633.
Kooi, C., and Sokol, P. A. (2009). Burkholderia cenocepacia zinc metalloproteases influence resistance to antimicrobial peptides. Microbiology 155, 2818–2825.
Kooi, C., Subsin, B., Chen, R., Pohorelic, B., and Sokol, P. A. (2006). Burkholderia cenocepacia ZmpB is a broad-specificity zinc metalloprotease involved in virulence. Infect. Immun. 74, 4083–4093.
Krouse, M. E. (2001). Is cystic fibrosis lung disease caused by abnormal ion composition or abnormal volume? J. Gen. Physiol. 118, 219–222.
Lamothe, J., Huynh, K. K., Grinstein, S., and Valvano, M. A. (2007). Intracellular survival of Burkholderia cenocepacia in macrophages is associated with a delay in the maturation of bacteria-containing vacuoles. Cell. Microbiol. 9, 40–53.
Le Cabec, V., Calafat, J., and Borregaard, N. (1997). Sorting of the specific granule protein, NGAL, during granulocytic maturation of HL-60 cells. Blood 89, 2113–2121.
Lefebre, M., and Valvano, M. (2001). In vitro resistance of Burkholderia cepacia complex isolates to reactive oxygen species in relation to catalase and superoxide dismutase production. Microbiology 147, 97–109.
Lefebre, M. D., Flannagan, R. S., and Valvano, M. A. (2005). A minor catalase/peroxidase from Burkholderia cenocepacia is required for normal aconitase activity. Microbiology 151, 1975–1985.
Lekstrom-Himes, J. A., and Gallin, J. I. (2000). Immunodeficiency diseases caused by defects in phagocytes. N. Engl. J. Med. 343, 1703–1714.
Lipuma, J. J. (2010). The changing microbial epidemiology in cystic fibrosis. Clin. Microbiol. Rev. 23, 299–323.
Loutet, S. A., Flannagan, R. S., Kooi, C., Sokol, P. A., and Valvano, M. A. (2006). A complete lipopolysaccharide inner core oligosaccharide is required for resistance of Burkholderia cenocepacia to antimicrobial peptides and bacterial survival in vivo. J. Bacteriol. 188, 2073–2080.
Loutet, S. A., Mussen, L. E., Flannagan, R. S., and Valvano, M. A. (2011). A two-tier model of polymyxin B resistance in Burkholderia cenocepacia. Environ. Microbiol. Rep. 3, 278–285.
Loutet, S. A., and Valvano, M. A. (2010). A decade of Burkholderia cenocepacia virulence determinant research. Infect. Immun. 78, 4088–4100.
Machen, T. E. (2006). Innate immunity in the CF airway epithelia-hyperinflammatory? Am. J. Physiol. Cell Physiol. 291, 218–230.
Mahenthiralingam, E., Urban, T. A., and Goldberg, J. B. (2005). The multifarious, multireplicon Burkholderia cepacia complex. Nat. Rev. Microbiol. 3, 144–156.
Marciano, B. E., Wesley, R., De Carlo, E. S., Anderson, V. L., Barnhart, L. A., Darnell, D., Malech, H. L., Gallin, J. I., and Holland, S. M. (2004). Long-term interferon-gamma therapy for patients with chronic granulomatous disease. Clin. Infect. Dis. 39, 692–699.
Marcos, V., Zhou, Z., Yildirim, A. O., Bohla, A., Hector, A., Vitkov, L., Wiedenbauer, E. M., Krautgartner, W. D., Stoiber, W., Belohradsky, B. H., Rieber, N., Kormann, M., Koller, B., Roscher, A., Roos, D., Griese, M., Eickelberg, O., Doring, G., Mall, M. A., and Hartl, D. (2010). CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat. Med. 16, 1018–1023.
McElvaney, N. G., Nakamura, H., Birrer, P., Hebert, C. A., Wong, W. L., Alphonso, M., Baker, J. B., Catalano, M. A., and Crystal, R. G. (1992). Modulation of airway inflammation in cystic fibrosis. In vivo suppression of interleukin-8 levels on the respiratory epithelial surface by aerosolization of recombinant secretory leukoprotease inhibitor. J. Clin. Invest. 90, 1296–1301.
Morgenstern, D. E., Gifford, M. A., Li, L. L., Doerschuk, C. M., and Dinauer, M. C. (1997). Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J. Exp. Med. 185, 207–218.
Ortega, X. P., Cardona, S. T., Brown, A. R., Loutet, S. A., Flannagan, R. S., Campopiano, D. J., Govan, J. R., and Valvano, M. A. (2007). A putative gene cluster for aminoarabinose biosynthesis is essential for Burkholderia cenocepacia viability. J. Bacteriol. 189, 3639–3644.
Ortega, X. P., Hunt, T. A., Loutet, S., Vinion-Dubiel, A. D., Datta, A., Choudhury, B., Goldberg, J. B., Carlson, R., and Valvano, M. A. (2005). Reconstitution of O-specific lipopolysaccharide expression in Burkholderia cenocepacia strain J2315, which is associated with transmissible infections in patients with cystic fibrosis. J. Bacteriol. 187, 1324–1333.
Painter, R. G., Bonvillain, R. W., Valentine, V. G., Lombard, G. A., LaPlace, S. G., Nauseef, W. M., and Wang, G. (2008). The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J. Leukoc. Biol. 83, 1345–1353.
Painter, R. G., Valentine, V. G., Lanson, N. A., Leidal, K., Zhang, Q., Lombard, G. A., Thompson, C., Viswanathan, A., Nauseef, W. M., Wang, G., and Wang, G. (2006). CFTR expression in human neutrophils and the phagolysomal chlorination defect in cystic fibrosis. Biochemistry 45, 10260–10269.
Pedersen, S. S., Kharazmi, A., Espersen, F., and Hoiby, N. (1990). Pseudomonas aeruginosa alginate in cystic fibrosis sputum and the inflammatory response. Infect. Immun. 58, 3363–3368.
Pollock, J. D., Williams, D. A., Gifford, M. A., Li, L. L., Du, X., Fisherman, J., Orkin, S. H., Doerschuk, C. M., and Dinauer, M. C. (1995). Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat. Genet. 9, 202–209.
Quie, P. G., White, J. G., Holmes, B., and Good, R. A. (1967). In vitro bactericidal capacity of human polymorphonuclear leukocytes: diminished activity in chronic granulomatous disease of childhood. J. Clin. Invest. 46, 668–679.
Rada, B., and Leto, T. L. (2010). Characterization of hydrogen peroxide production by Duox in bronchial epithelial cells exposed to Pseudomonas aeruginosa. FEBS Lett. 584, 917–922.
Reik, R., Spilker, T., and Lipuma, J. J. (2005). Distribution of Burkholderia cepacia complex species among isolates recovered from persons with or without cystic fibrosis. J. Clin. Microbiol. 43, 2926–2928.
Riganti, C., Gazzano, E., Polimeni, M., Costamagna, C., Bosia, A., and Ghigo, D. (2004). Diphenyleneiodonium inhibits the cell redox metabolism and induces oxidative stress. J. Biol. Chem. 279, 47726–47731.
Root, R. K., Rosenthal, A. S., and Balestra, D. J. (1972). Abnormal bactericidal, metabolic, and lysosomal functions of Chediak-Higashi Syndrome leukocytes. J. Clin. Invest. 51, 649–665.
Rowe, S. M., Miller, S., and Sorscher, E. J. (2005). Cystic fibrosis. N. Engl. J. Med. 352, 1992–2001.
Saiman, L., Chen, Y., Gabriel, P. S., and Knirsch, C. (2002). Synergistic activities of macrolide antibiotics against Pseudomonas aeruginosa, Burkholderia cepacia, Stenotrophomonas maltophilia, and Alcaligenes xylosoxidans isolated from patients with cystic fibrosis. Antimicrob. Agents Chemother. 46, 1105–1107.
Sajjan, S. U., Carmody, L. A., Gonzalez, C. F., and LiPuma, J. J. (2008). A type IV secretion system contributes to intracellular survival and replication of Burkholderia cenocepacia. Infect. Immun. 76, 5447–5455.
Saldias, M. S., Lamothe, J., Wu, R., and Valvano, M. A. (2008). Burkholderia cenocepacia requires the RpoN sigma factor for biofilm formation and intracellular trafficking within macrophages. Infect. Immun. 76, 1059–1067.
Saldias, M. S., and Valvano, M. A. (2009). Interactions of Burkholderia cenocepacia and other Burkholderia cepacia complex bacteria with epithelial and phagocytic cells. Microbiology 155, 2809–2817.
Scholte, B. J., Davidson, D. J., Wilke, M., and De Jonge, H. R. (2004). Animal models of cystic fibrosis. J. Cyst. Fibros. 3, 183–190.
Segal, B. H., Leto, T. L., Gallin, J. I., Malech, H. L., and Holland, S. M. (2000). Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore) 79, 170–200.
Seger, R. A. (2008). Modern management of chronic granulomatous disease. Br. J. Haematol. 140, 255–266.
Sheppard, M. N., and Nicholson, A. G. (2002). The pathology of cystic fibrosis. Curr. Diagn. Pathol. 8, 50–59.
Shinkai, M., and Rubin, B. K. (2005). Macrolides and airway inflammation in children. Paediatr. Respir. Rev. 6, 227–235.
Silipo, A., Molinaro, A., Cescutti, P., Bedini, E., Rizzo, R., Parrilli, M., and Lanzetta, R. (2005). Complete structural characterization of the lipid A fraction of a clinical strain of B. cepacia genomovar I lipopolysaccharide. Glycobiology 15, 561–570.
Silipo, A., Molinaro, A., Ierano, T., De Soyza, A., Sturiale, L., Garozzo, D., Aldridge, C., Corris, P. A., Khan, C. M., Lanzetta, R., and Parrilli, M. (2007). The complete structure and pro-inflammatory activity of the lipooligosaccharide of the highly epidemic and virulent gram-negative bacterium Burkholderia cenocepacia ET-12 (strain J2315). Chemistry (Easton) 13, 3501–3511.
Smith, J. J., Travis, S. M., Greenberg, E. P., and Welsh, M. J. (1996). Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 85, 229–236.
Sousa, S. A., Ulrich, M., Bragonzi, A., Burke, M., Worlitzsch, D., Leitao, J. H., Meisner, C., Eberl, L., Sa-Correia, I., and Doring, G. (2007). Virulence of Burkholderia cepacia complex strains in gp91phox-/- mice. Cell. Microbiol. 9, 2817–2825.
Taggart, C., Coakley, R. J., Greally, P., Canny, G., O’Neill, S. J., and McElvaney, N. G. (2000). Increased elastase release by CF neutrophils is mediated by tumor necrosis factor-alpha and interleukin-8. Am. J. Physiol. Lung Cell. Mol. Physiol. 278, L33–41.
Tirouvanziam, R., Gernez, Y., Conrad, C. K., Moss, R. B., Schrijver, I., Dunn, C. E., Davies, Z. A., Herzenberg, L. A., and Herzenberg, L. A. (2008). Profound functional and signaling changes in viable inflammatory neutrophils homing to cystic fibrosis airways. Proc. Natl. Acad. Sci. U.S.A. 105, 4335–4339.
Tosi, M. F., Zakem, H., and Berger, M. (1990). Neutrophil elastase cleaves C3bi on opsonized Pseudomonas as well as CR1 on neutrophils to create a functionally important opsonin receptor mismatch. J. Clin. Invest. 86, 300–308.
Vanlaere, E., Baldwin, A., Gevers, D., Henry, D., De Brandt, E., LiPuma, J. J., Mahenthiralingam, E., Speert, D. P., Dowson, C., and Vandamme, P. (2009). Taxon K, a complex within the Burkholderia cepacia complex, comprises at least two novel species, Burkholderia contaminans sp. nov. and Burkholderia lata sp. nov. Int. J. Syst. Evol. Microbiol. 59, 102–111.
Vejrazka, M., Micek, R., and Stipek, S. (2005). Apocynin inhibits NADPH oxidase in phagocytes but stimulates ROS production in non-phagocytic cells. Biochim. Biophys. Acta 1722, 143–147.
Vinion-Dubiel, A. D., and Goldberg, J. B. (2003). Review: lipopolysaccharide on Burkholderia cepacia complex. J. Endotoxin Res. 9, 201–213.
Walker, T. S., Tomlin, K. L., Worthen, G. S., Poch, K. R., Lieber, J. G., Saavedra, M. T., Fessler, M. B., Malcolm, K. C., Vasil, M. L., and Nick, J. A. (2005). Enhanced Pseudomonas aeruginosa biofilm development mediated by human neutrophils. Infect. Immun. 73, 3693–3701.
Wiedow, O., and Meyer-Hoffert, U. (2005). Neutrophil serine proteases: potential key regulators of cell-signalling during inflammation. J. Int. Med. 257, 319–328.
Winkelstein, J. A., Marino, M. C., Johnston, R. B. Jr., Boyle, J., Curnutte, J., Gallin, J. I., Malech, H. L., Holland, S. M., Ochs, H., Quie, P., Buckley, R. H., Foster, C. B., Chanock, S. J., and Dickler, H. (2000). Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore) 79, 155–169.
Young, R. L., Malcolm, K. C., Kret, J. E., Caceres, S. M., Poch, K. R., Nichols, D. P., Taylor-Cousar, J. L., Saavedra, M. T., Randell, S. H., Vasil, M. L., Burns, J. L., Moskowitz, S. M., and Nick, J. A. (2011). Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: evidence of acquired resistance within the CF airway, independent of CFTR. PLoS ONE 6, e23637. doi:10.1371/journal.pone.0023637.
Zarember, K. A., and Malech, H.L. (2011). “The roles of neutrophils in innate immunity,” in Regulation of Innate Immune Function, Chapt. 5, eds. C. B. Marsh, S. Tridandapani, and M. G. Piper (Kerala: Research Signpost Press), 99–140.
Zelazny, A. M., Ding, L., Elloumi, H. Z., Brinster, L. R., Benedetti, F., Czapiga, M., Ulrich, R. L., Ballentine, S. J., Goldberg, J. B., Sampaio, E. P., and Holland, S. M. (2009). Virulence and cellular interactions of Burkholderia multivorans in chronic granulomatous disease. Infect. Immun. 77, 4337–4344.
Zhen, L., King, A. A., Xiao, Y., Chanock, S. J., Orkin, S. H., and Dinauer, M. C. (1993). Gene targeting of X chromosome-linked chronic granulomatous disease locus in a human myeloid leukemia cell line and rescue by expression of recombinant gp91phox. Proc. Natl. Acad. Sci. U.S.A. 90, 9832–9836.
Keywords: Burkholderia cepacia complex, Burkholderia, chronic granulomatous disease, cystic fibrosis, neutrophil, immunocompromised
Citation: Porter LA and Goldberg JB (2011) Influence of neutrophil defects on Burkholderia cepacia complex pathogenesis. Front. Cell. Inf. Microbio. 1:9. doi: 10.3389/fcimb.2011.00009
Received: 30 September 2011;
Accepted: 01 November 2011;
Published online: 18 November 2011.
Edited by:
D. Scott Merrell, Uniformed Services University, USAReviewed by:
Umadevi S. Sajjan, University of Michigan, USAKol A. Zarember, National Institutes of Health, USA
Copyright: © 2011 Porter and Goldberg. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence: Joanna B. Goldberg, Department of Microbiology, Immunology, and Cancer Biology, University of Virginia Health System, 7230 Jordan Hall, 1300 Jefferson Park Avenue, Box 800-734, Charlottesville, VA 22908-0734, USA. e-mail:amJnMmJAdmlyZ2luaWEuZWR1