

- Department of Pediatrics and Communicable Diseases, University of Michigan, Ann Arbor, MI, USA
Burkholderia cenocepacia is an opportunistic respiratory pathogen of individuals with cystic fibrosis (CF). Some strains of B. cenocepacia are highly transmissible and resistant to almost all antibiotics. Approximately one-third of B. cenocepacia infected CF patients go on to develop fatal “cepacia syndrome.” During the last two decades, substantial progress has been made with regards to evasion of host innate defense mechanisms by B. cenocepacia. Almost all strains of B. cenocepacia have the capacity to survive and replicate intracellularly in both airway epithelial cells and macrophages, which are primary sentinels of the lung and play a pivotal role in clearance of infecting bacteria. Those strains of B. cenocepacia, which express both cable pili and the associated 22 kDa adhesin are also capable of transmigrating across airway epithelium and persist in mouse models of infection. In this review, we will discuss how this type of interaction between B. cenocepacia and host may lead to persistence of bacteria as well as lung inflammation in CF patients.
The two powerful host defense mechanisms against infecting bacteria are innate and adaptive immune systems. The innate immune system provides immediate protection against intruding bacteria by triggering appropriate host responses that control bacterial proliferation, dissemination, and elimination. On the other hand, the adaptive immune system is responsible for elimination of bacteria at later phases of infection and in the generation of an immunological memory. A variety of cell types including mucosal epithelial cells, macrophages, neutrophils, dendritic cells, natural killer cells, and eosinophils contribute to the innate immune system. Since airway epithelium lining the respiratory tract is the first point of contact for inhaled bacterial pathogens, it has evolved to eliminate pathogens efficiently by more than one mechanism (Diamond et al., 2000; Bals and Hiemstra, 2004; Mayer and Dalpke, 2007; Vareille et al., 2011). These include mucociliary apparatus which traps and sweeps the intruding bacteria away from the lungs (Knowles and Boucher, 2002), potent antimicrobial agents that kill invading bacterial pathogens (Bals et al., 1998; Hiemstra, 2001), junctional complexes between the airway epithelial cells which prevent bacterial pathogens from gaining access to the submucosal compartment (Pohl et al., 2009); and pattern recognition molecules that stimulate pro-inflammatory cytokines upon recognition of bacteria or bacterial products leading to recruitment and activation of innate immune cells and to the induction of expression of co-stimulatory molecules on the antigen presenting cells (Parker and Prince, 2011). The latter response activates the adaptive immune system (Medzhitov and Janeway, 1997). Bacterial pathogens which escape from epithelial defenses are taken up by the recruited professional phagocytes and killed. Despite these multiple layers of host defense, some bacterial pathogens cause persistent infection suggesting that they have evolved to evade or subvert host defenses. Opportunistic pathogens, such as, Pseudomonas aeruginosa and the members of Burkholderia cepacia complex can cause both acute and chronic infections preferentially in cystic fibrosis (CF) patients because, they are thought to have a defective innate immune system (Chmiel and Davis, 2003; Jacquot et al., 2008). In this review, we will discuss how Burkholderia cenocepacia, a member of B. cepacia complex evades the innate immune system and cause persistent respiratory infections in CF patients.
Burkholderia cenocepacia Infection in Individuals with Cystic Fibrosis
Although most members of B. cepacia complex have been isolated from the lungs of CF patients, B. cenocepacia is one of the most commonly isolated species in North America and Europe (Mahenthiralingam et al., 2001; Lipuma, 2005; Drevinek and Mahenthiralingam, 2010). In addition, certain strains of B. cenocepacia are associated with heightened transmissibility (Govan et al., 1993; Sun et al., 1995; Pitt et al., 1996; Clode et al., 2000; Woods et al., 2004), poor clinical outcome, and increased risk of developing fatal “cepacia syndrome” (Frangolias et al., 1999; Ledson et al., 2002; Courtney et al., 2004; Jones et al., 2004). B. cenocepacia is also associated with bacteremia in non-CF adult critically ill-patients (Siddiqui et al., 2001; Woods et al., 2004; Bressler et al., 2007). These observations suggest a tropism of this organism to chronically injured lungs.
By using genetic analysis based on recA gene sequencing, B. cenocepacia was further divided into four phylogenetic lineages IIIA to IIID, but most of the CF isolates belong to IIIA and IIIB (Mahenthiralingam et al., 2000; Vandamme et al., 2003). In the United States, PHDC and Midwest clones are dominant epidemic lineages and belong to B. cenocepacia IIIB (Chen et al., 2001; Coenye and Lipuma, 2003). Strains from the PHDC lineage are also found in Europe (Coenye et al., 2004). In contrast, ET12 (one of the clonal lineages of B. cenocepacia IIIA) is dominant in Canada, UK, and other European countries and was responsible for most of the “cepacia syndrome”-related deaths documented in Canada and UK during the early 1980s (Isles et al., 1984; Govan et al., 1996; Speert et al., 2002). Based on epidemiological studies, isolates belonging to the ET12 lineage are considered to be highly transmissible and virulent. As a result, most of the studies are focused on understanding the mechanisms by which these organisms evade host innate defense mechanisms.
Invasion of Airway Epithelium by B. cenocepacia
The main components of mucus that overlays airway epithelium are high molecular weight glycoproteins called mucins. These mucin molecules cross link with each other to provide a structural framework for the mucus barrier (Rose et al., 2001). Under normal conditions, binding of bacteria to airway mucins is generally considered beneficial for the host, as the bacteria bound to mucins are subsequently cleared by mucociliary action. In addition, surface mucus also contains antimicrobial products which kill trapped bacteria (Singh et al., 1998; Moskwa et al., 2007). In contrast, under the conditions of impaired mucociliary clearance and reduced activity or absence of antimicrobial products as observed in CF patients (Singh et al., 1998; Boucher, 2004, 2007; Conner et al., 2007; Moskwa et al., 2007), bacteria bound to mucin may persist in the airway lumen and gain access to airway epithelial cells. Previously, we have demonstrated that the majority of clinical isolates collected during the B. cepacia epidemic in Toronto CF center bind to mucin and this binding was mediated by a 22-kDa adhesin protein associated with cable pili (Sajjan and Forstner, 1992; Sajjan et al., 1992). All the mucin binding isolates belong to the ET12 lineage and were found to express both cable pili and the 22-kDa adhesin (Sajjan and Forstner, Unpublished observations). The isolates which showed the highest binding to mucin were recovered from patients who ultimately died due to fatal “cepacia syndrome,” suggesting that cable pili and the associated 22 kDa adhesin may be necessary for persistent bacterial infections leading to “cepacia syndrome” (Sajjan et al., 1992). Consistent with this notion, we found that cable and adhesin positive B. cenocepacia isolates preferentially persist in the apical mucus layer as microcolonies (biofilm) in CF airway epithelial cells differentiated into a mucociliary phenotype (Sajjan et al., 2004; Figure 1). We also found individual bacteria deeper in the epithelial cell layer 24 h after infection, indicating that some bacteria escape from the mucus layer and invade the underlying epithelial cells. In contrast, normal airway epithelial cells differentiated into a mucociliary phenotype trapped the added B. cenocepacia in the apical mucus and prevented bacteria from invading the cells. Compared to normal, CF cell cultures also showed 100-fold more bacteria possibly due to the inefficient killing of bacteria by airway epithelial cell-derived antimicrobials. CF airway epithelial cells have been demonstrated to be defective in producing antimicrobial products (Singh et al., 1998; Conner et al., 2007; Moskwa et al., 2007). However, B. cenocepacia which is inherently resistant to many antibiotics is also resistant to killing by a cationic peptide, β-defensin (Baird et al., 1999). β-defensin is expressed by both airway epithelial cells and inflammatory cells and therefore present abundantly in airway lumen (Singh et al., 1998). This may partly explain the persistence of bacteria (although fewer in numbers) in the apical mucus layer of normal airway epithelial cell cultures. However, Schwab et al. (2002) demonstrated that cable and adhesin positive B. cenocepacia also persist in the apical mucus in the form of microcolonies and also invade mucociliary-differentiated normal airway epithelial cells leading to extensive cell damage. This may be due to higher infection doses used in this study, which could overwhelm the innate immune defenses of airway epithelial cells in culture.
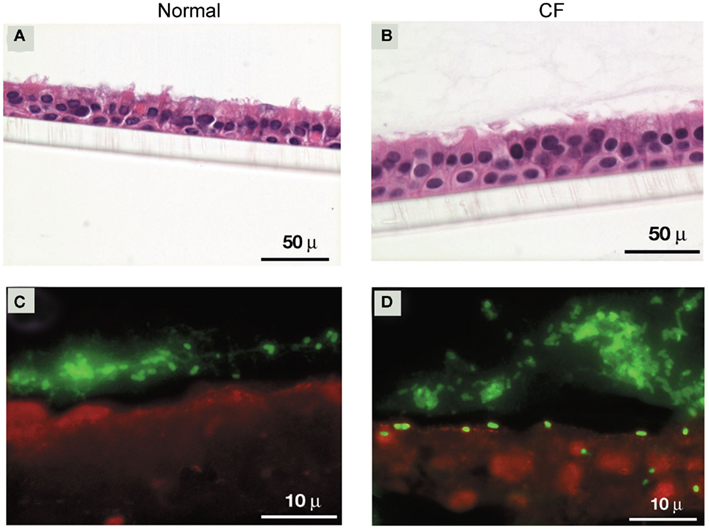
Figure 1. Cable and adhesin positive B. cenocepacia invade well-differentiated CF, but not normal airway epithelial cells. (A,B) Cross sections of B. cenocepacia-infected normal and CF airway epithelial cell cultures stained with hemotoxylin and eosin. (C,D) Localization of bacteria in normal and CF airway epithelial cells infected with B. cenocepacia.
Removal of apical mucus prior to infection with B. cenocepacia increased bacterial invasion of airway epithelium in both normal and CF airway epithelial cell cultures (Sajjan et al., 2004). However, while invaded bacteria in normal airway epithelial cells were contained in endocytic vesicles and killed, bacteria in CF epithelial cells appeared to be intact and also caused extensive damage. Consistent with these observations, cable and adhesin positive bacteria were also found to be less destructive than other strains of B. cenocepacia and Burkholderia multivorans in normal polarized cells (Duff et al., 2006). B. cenocepacia strains other than ET12 also transmigrate across normal polarized airway epithelial cell cultures by causing dissociation of occludin from the tight junction complex (Kim et al., 2005). Similarly, B. multivorans and non-CF isolates belonging to the genera Burkholderia stabilis and B. cepacia also invaded polarized normal airway epithelial cells by paracytosis and lead to loss of zona occludens and occludin from the tight junction complex (Duff et al., 2006).
Cable and adhesin positive B. cenocepacia also invade squamous-differentiated normal airway epithelial cells efficiently (Sajjan et al., 2002; Urban et al., 2005). Squamous epithelium represents respiratory epithelium undergoing squamous metaplasia and/or repair following epithelial injury, and express cytokeratin 13 (Leube and Rustad, 1991; Roger et al., 1999). Mutants defective in expression of cable pili and the 22-kDa adhesin do not bind to isolated cytokeratin 13. These mutants are also attenuated in their capacity to transmigrate across squamous epithelium (Urban et al., 2005), suggesting a role for cable pili and the 22-kDa adhesin in this process. It is not uncommon for pathogenic bacteria to utilize cytokeratins as receptors. For instance, undefined type of cytokeratins on injured corneal epithelial cells were identified as receptors for pili isolated from P. aeruginosa PAK strain (Wu et al., 1996). Interaction with cytokeratin 10 was proposed to contribute to colonization of nasal mucosa by Staphylococcus aureus (O’brien et al., 2002). Since CF airway epithelium undergoes repeated injury and repair, it may express increased cytokeratin 13. Therefore, it is possible that cable and the 22-kDa adhesin positive B. cenocepacia which has the capacity to bind to cytokeratin 13 may readily bind and invade CF airway epithelium expressing cytokeratin 13.
Neither CF nor normal airway epithelial cells which are differentiated into the mucociliary phenotype express cytokeratin 13. Yet, cable and adhesin positive B. cenocepacia invade CF airway epithelial cells and to some extent normal epithelial cells (Sajjan et al., 2004). Other strains of B. cenocepacia and B. multivorans also invade mucociliary airway epithelial cells (Schwab et al., 2002). B. cenocepacia other than ET12 strain and B. multivorans were also shown to traverse polarized airway epithelial cell lines by dissociating zona occludens-1 and/or occludin from the tight junction complex (Kim et al., 2005; Duff et al., 2006). Such observations suggest interaction of these organisms with cellular receptors other than cytokeratin 13 that facilitate entry of bacteria into cells. Consistent with this notion, both cable piliated and non-cable piliated B. cenocepacia were shown to interact with glycolipid receptors, primarily with galactose-containing globotriaosylceramides Gb2 and Gb3 and moderately with asialo GM1 and asialoGM2 (Sylvester et al., 1996). Similar to these findings, uncharacterized members of the B. cepacia complex were shown to bind to purified asialo GM1 and asialo GM2 (Krivan et al., 1988). However, the contribution of bacterial interaction with glycolipid receptors in invasion of epithelial cells or other host responses was not investigated in these earlier studies. Interestingly, adherence to unspecified glycosphingolipids was demonstrated to promote invasion of epithelial cells by B. multivorans and cable piliated B. cenocepacia (Mullen et al., 2010). Several investigators have reported that P. aeruginosa interact with asialo GM1 (Imundo et al., 1995; De Bentzmann et al., 1996; Davies et al., 1999). Such interaction was shown to activate cytotoxicity and internalization of bacteria by epithelial cells (Feldman et al., 1998; Comolli et al., 1999).
In addition, flagella also appear to be required for optimal invasion of A549 (alveolar type II epithelial) cells by B. cenocepacia as non-motile mutants of B. cenocepacia strain (belonging to the ET12 lineage) with a disruption in genes encoding flagellar genes (fliG or fliI) were attenuated in invading A549 cells (Tomich et al., 2002). Flagella of P. aeruginosa has been shown to interact with asialo GM1 (Feldman et al., 1998; Adamo et al., 2004) and this interaction increased invasion of epithelial cells by this organism (Feldman et al., 1998). Expression of asialo GM1 is increased on the apical surface of CF airway epithelial cells (Muir et al., 2004). Therefore, it is likely that binding of B. cenocepacia with asialo GM1 mediated by flagella may promote invasion of airway epithelial cells by this organism.
Burkholderia cenocepacia Stimulates Disproportionate Pro-Inflammatory Cytokine Response by Subverting Innate Immune Receptors
Interaction of bacteria or bacterial products with innate immune receptors such as toll-like receptors (TLR) is expected to be beneficial for the host, because it elicits pro-inflammatory cytokine responses that are required for recruitment of neutrophils. However, abnormal stimulation of TLR may lead to excessive chemokine response and increased recruitment of neutrophils. Similarly, interaction of bacteria with cytokine receptors may activate autocrine pathway resulting in disproportionately increased production of chemokines and recruitment of neutrophils. This combined with impaired clearance of recruited neutrophils by efferocytosis as observed in CF lungs may lead to lung injury (Vandivier et al., 2006, 2009).
Bacterial products such as lipopolysaccharides (LPS), flagella, pili, and lipopeptides are recognized by different TLRs in both lung epithelial cells and inflammatory cells and stimulate production of pro-inflammatory cytokines. LPS isolated from B. cepacia complex was found to elicit ninefold higher pro-inflammatory cytokines than LPS from P. aeruginosa in leukocytes (Shaw et al., 1995; Zughaier et al., 1999). Following this, De Soyza et al. (2004) examined whole bacterial cell lysates of representative isolates belonging to all the members of B. cepacia complex to stimulate pro-inflammatory cytokines in a human monocytic cell line, U937. They found that in general, lysates of B. cenocepacia and B. multivorans species are significantly more potent stimulators of pro-inflammatory cytokines than the other species. Based on the structural analysis of lipid A moiety from a limited number of strains, the difference between pro-inflammatory activity of LPS was proposed to be due to differences in acylation of lipid A (De Soyza et al., 2004). The strains that lack primary acylation chain in lipid A stimulated the least pro-inflammatory responses in macrophages. Later, highly purified LPS from different isolates belonging to B. cenocepacia ET12 lineage or B. multivorans was demonstrated to elicit varying levels of TNF-α in a Mono Mac 6 (human monocytic cell line; Bamford et al., 2007). This was dependent on the interaction of purified LPS with TLR4 and the activation of MyD88-dependent signaling pathway.
Recently, it was demonstrated that O antigen of LPS from B. cenocepacia also stimulate IL-1β in murine macrophages and this was dependent on both TLR4 and caspase-1 (Kotrange et al., 2011). Knockdown of TLR4 or caspase-1 abrogated B. cenocepacia-induced IL-1β. Knockdown of MyD88, an adaptor protein downstream of TLR4 also completely inhibited B. cenocepacia-induced IL-1β, indicating the requirement of MyD88-dependent TLR4 signaling in this process. Caspase-1 is normally activated by NOD-like receptors, which are a family of intracellular pattern recognition receptors and sense microbial signatures in the cytosol. So far, 22 NLR’s have been identified in humans. NOD1 and NOD2 are the most widely studied NLRs, which detect components from both gram positive and Gram negative bacteria and activate inflammasome leading to caspase-1 expression (Chamaillard et al., 2003; Girardin et al., 2003a,b). However activation of inflammasome also requires a second signal that is usually elicited by one of the TLRs and based on the observations from Kotrange et al. (2011) TLR4 appears to be involved in B. cenocepacia O antigen-stimulated inflammasome activation and caspase-1 expression. The identity of NLR that sense B. cenocepacia O antigen is yet to be determined.
Interestingly, LPS from B. cenocepacia isolates K56-2 or BC7 was found to be less efficient in stimulating TNF-α in monocytes than other B. cenocepacia isolates (De Soyza et al., 2004). Yet, CFTR knockout mice infected with either K56-2 or BC7 show more severe lung inflammation than mice infected with isolates from other species (Chattoraj et al., 2010). These two B. cenocepacia isolates also elicited more IL-8 expression in primary CF airway epithelial cells than the other B. cenocepacia isolates examined (Sajjan et al., 2004) suggesting that in addition to LPS, other pro-inflammatory factors may also contribute to B. cenocepacia-induced excessive stimulation of pro-inflammatory cytokines, particularly in CF lungs. Consistent with this notion, binding of P. aeruginosa flagella to asialo GM1 was shown to cooperate with TLR5 in stimulating IL-8 production and to induce nucleotide signaling leading ERK1/2 activation in airway epithelial cells (Adamo et al., 2004; Mcnamara et al., 2006). Since asialo GM1 expression is increased in CF airway epithelial cells it is possible that such cooperative interactions between asialo GM1 and TLR5 are operative in these cells. Accordingly, B. cenocepacia-induced pro-inflammatory responses in airway epithelial cells was shown to involve TLR5, but not TLR2 or TLR4 (De et al., 2008).
Flagella from B. cenocepacia K56-2 was demonstrated to elicit chemokine and cytokine responses both in vitro and in vivo by interacting with TLR5 (Urban et al., 2004). However, it was not demonstrated whether this interaction stimulates higher chemokine responses in CF than in normal cells. Interestingly, we found that some strains of B. cenocepacia including BC7 binds to TNF receptor 1 (TNFR1) and activates TNFR signaling leading to IL-8 production in CF airway epithelial cells (Sajjan et al., 2008b). Further, we observed co-localization of B. cenocepacia with TNFR1 in the airway epithelium of a CF patient infected with cable and the 22-kDa adhesin positive B. cenocepacia. Although activation of TNFR1 by TNF-α plays a critical role in host defense, pathogenic bacteria that have the capacity to bind to this receptor can subvert the pathway to heighten the host inflammatory responses. As observed for B. cenocepacia, S. aureus has also been shown to bind to TNFR1 in airway epithelial cells and induce a TNFR1-related signaling pathway leading to IL-8 expression (Gomez et al., 2004). Protein A, a major surface protein present in almost all strains of S. aureus has been identified as a bacterial ligand. In a murine model of lung infection, the interaction of protein A with TNFR1 results in recruitment of neutrophils to airways, leading to severe inflammation. However whether interaction of B. cenocepacia with TNFR1 increases lung inflammation in vivo is yet to be examined. B. cenocepacia ligand that interacts with TNFRI is also not known.
Burkholderia cenocepacia Subverts the Endocytic Pathway
Various studies have shown that B. cenocepacia has the capacity to survive intracellularly. This trait may contribute to the organism’s ability to persist and evade host defenses in vivo during chronic infection. Burns et al. (1996) were the first to demonstrate that a strain of B. cepacia complex isolated from a CF patient invade airway epithelial cells in vitro and the bacteria-positive vesicles were found not to fuse with lysosomes. Immunolocalization studies using lung sections from CF patients who were chronically infected with strains of the B. cepacia complex indicated presence of B. cepacia frequently in the intercellular junctions of airway epithelium, inside the airway epithelial cells, and macrophages, but rarely in the airway lumen (Sajjan et al., 2001a). These findings confirmed that the strains of the B. cepacia complex are capable of surviving intracellularly in both airway epithelial cells and macrophages during chronic infection. B. cenocepacia elaborated factors such as flagella and lipase appear to promote bacterial invasion of lung epithelial cells, but not survival or replication (Tomich et al., 2002; Mullen et al., 2010). Interestingly, exogenously added IL-8 also increased invasion of A549 cells by B. cenocepacia complex in a dose dependent manner. Based on this observation, it was postulated that strains that elicit more IL-8 may invade cells with higher propensity (Kaza et al., 2011).
Intracellular survival and replication of B. cenocepacia appears to require type IV secretion system (T4SS; Sajjan et al., 2006, 2008a), similar to that observed for L. pneumophila and Brucella (reviewed in De Jong et al., 2010; Hubber and Roy, 2010). Analysis of the completely sequenced genome of B. cenocepacia belonging to the ET12 lineage Engledow et al. (2004) identified two T4SSs. One of these is encoded on chromosome 2 and bears homology to the VirB/D4 T4SS of Agrobacterium tumefaciens (Christie et al., 2005). The second, Ptw T4SS, is encoded by a 92-kb resident megaplasmid and is a chimeric system composed ofVirB/D4 and F-specific subunits (Engledow et al., 2004; Christie et al., 2005). Insertional inactivation of ptwD4 of the Ptw system attenuated intracellular survival and replication of B. cenocepacia strain K56-2 (Sajjan et al., 2008a). PtwD4 is homologous to coupling proteins of other T4SSs and involved in effector translocation (Engledow et al., 2004). Both wild-type and ptwD4 mutant invade CF airway epithelial cells and monocyte derived macrophages at a similar level. However, 24 h post infection, higher number of wild-type than theptwD4 mutant was recovered from both CF airway epithelial cells and monocyte derived macrophages. The observed difference between the wild-type and ptwD4 mutant was found to be due to altered intracellular trafficking. Immediately after entry into airway epithelial cells or macrophages, both wild-type and the ptwD4 mutant co-localized to early endosomes. However, while most of the wild-type bacteria containing vacuoles failed to acquire cathepsin D (a lysosomal marker), twice as many ptwD4 mutant containing vacuoles acquired cathepsin D, suggesting that unlike wild-type bacteria containing vacuoles, a proportion of ptwD4 mutant containing vacuoles fuse normally with lysosomes and the bacteria are subsequently killed by a typical endosomal pathway. Mutant bacteria that escape the typical endosomal pathway were targeted to autophagosomes which eventually fused with lysosome and bacteria in the autophagolysosome were killed. In contrast, a majority of vacuoles containing wild-type bacteria excluded early endosomal marker and acquired markers of autophagosomes suggesting presence of bacteria in the autophagosomes. However, unlike ptwD4 mutant containing autophagosomes, wild-type bacteria containing autophagosomes did not fuse with the lysosomes. As a result, wild-type bacteria that persist in the autophagosomes appear to escape from the autophagosomes and replicate in endoplasmic reticulum (Figure 2). These observations demonstrate how B. cenocepacia can subvert the normal endosomal pathway to its benefit.
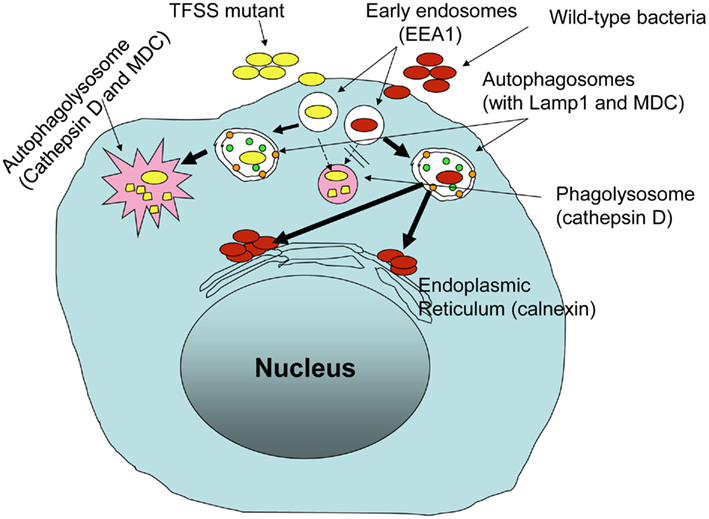
Figure 2. Burkholderia cenocepacia Ptw T4SS is required for subversion of endosomal pathway in host innate immune cells.
To understand the mechanism by which B. cenocepacia prevent fusion of endosomes with lysosome, Huynh et al. (2010) performed elegant studies using a synchronized phagocytosis assay and fluorescence recovery after photo-bleaching techniques. Similar to above observations, these authors also found defective fusion of B. cenocepacia containing vacuoles with the lysosomes in macrophages. Rab7 plays an important role in directing the fusion of phagosomes not only with late endosomes, but also with the lysosomes. Therefore using GFP–Rab7 transfected macrophages, a more sensitive method for detecting Rab7 recruitment to endosomes than the conventional immunolocalization, the authors showed that Rab7 was recruited to majority of B. cenocepacia containing vacuoles (Huynh et al., 2010). Despite this, B. cenocepacia containing vacuoles failed to fuse with the lysosomes. This prompted the authors to examine the cycling of recruited Rab7 between the guanosine triphosphate (GTP) to guanosine diphosphate (GDP)-bound forms of Rab7, as this is required for efficient fusion of bacteria containing vacuoles with lysosomes (Vieira et al., 2003). Fluorescence recovery after photo-bleaching of GFP–Rab7 was used to examine the cycling of GTP to GDP-bound form of Rab7 in B. cenocepacia infected cells. In the steady state, the rate of fluorescence recovery after bleaching of the entire bacteria containing vacuole is dictated by exchange of the photo-bleached membrane bound Rab7 with fluorescent cytosolic (unbleached) Rab7, a process that is determined in part by the rate of conversion of Rab7–GDP to the GTP-bound form (Vieira et al., 2003). The rate of fluorescence recovery in vacuoles containing live B. cenocepacia was remarkably slower and less compared to those containing dead bacteria suggesting that these organisms may prevent the fusion of endosomes by retaining inactive GDP-bound form of Rab7.
In addition to preventing fusion of endosomes with lysosomes, B. cenocepacia also delays acidification of vacuoles in macrophages (Lamothe et al., 2007; Saldias and Valvano, 2009). The bacterial factor which plays a role in this process is thought to be regulated by alternative sigma factor RpoE, which controls the expression of genes required for extracellular stress response (Flannagan and Valvano, 2008). Inactivation of rpoE reduced the ability of intracellular B. cenocepacia to prevent acidification of bacteria containing vacuoles. As a result bacteria were targeted to the classical endocytic pathway. However the precise RpoE controlled gene product that is responsible for preventing acidification of bacteria containing vacuole is yet to be identified.
Burkholderia cenocepacia is Resistant to Killing by Host-Derived Antimicrobials
Another mechanism by which B. cenocepacia evade host defenses is via inactivating and/or preventing expression of host elaborated antimicrobials. The LPS of B. cenocepacia and other members of Burkholderia species is unique in that it contains 4-amino-4-deoxy-L-arabinose (Ara4N) residues in the lipid A and inner core oligosaccharide (Vinion-Dubiel and Goldberg, 2003). Ara4N moieties are positively charged, and therefore they reduce the net charge on the external membrane. This renders bacteria resistant to cationic antimicrobial peptides, including defensins which are enriched in the respiratory tract (Albrecht et al., 2002; Vinion-Dubiel and Goldberg, 2003; Loutet et al., 2006).
B. cenocepacia secretes zinc metalloproteases (Zmp)A and ZmpB (Kooi et al., 2005, 2006) in addition to other proteases. ZmpA is positively regulated by the CepR quorum-sensing system, whereas ZmpB is positively regulated by both CepIR and CcilR quorum-sensing systems (Gingues et al., 2005; Kooi et al., 2006). Both ZmpA and ZmpB have broad-spectrum proteolytic activity and degrade matrix proteins, antiproteinases, and α2-macroglobulin. ZmpA also degrades interferon-γ, and ZmpB degrades transferrin and immunoglobulins (Kooi et al., 2005, 2006). These proteases were also found to degrade antimicrobial peptides. Both proteases cleave elafin and secretory leukocyte inhibitors. While ZmpA degrades cathelicidin LL37, ZmpB cleaves β-defensin (Kooi and Sokol, 2009). Mutants of CepIR quorum-sensing system are less invasive and cause less lung inflammation than their wild-type counterparts in CF mice (Sokol et al., 2003), suggesting that ZmpA and ZmpB may contribute to bacterial evasion of antimicrobial defenses in vivo (Sokol et al., 2003).
B. cenocepacia is also resistant to killing by phagocytes. Some strains of B. cenocepacia produce large amounts of exopolysaccharides (EPS), which makes bacteria mucoid (Cerantola et al., 2000). Some of the CF clinical isolates were found to express EPS when cultured on solid media (Herasimenka et al., 2007) and also in a mouse model of infection (Conway et al., 2004). B. cenocepacia EPS differ chemically from P. aeruginosa exopolysaccharide alginate (Cerantola et al., 1996; Sist et al., 2003; Conway et al., 2004), but can still inactivate cathelicidin peptides similar to alginate (Foschiatti et al., 2009). The EPS from B. cenocepacia also inhibits neutrophil chemotaxis and scavenges reactive oxygen species in vitro (Bylund et al., 2006). Subsequently B. cenocepacia was also demonstrated to delay the assembly of NADPH oxidase enzyme complex in the phagosomes (Keith et al., 2009). NADPH oxidase is the major source of superoxide, a precursor of potent microbicidal reactive oxygen species such as hydrogen peroxide, hydroxyl radical, and hypochlorous acid (Fang, 2004).
Summary
The picture emerging from these studies is that B. cenocepacia, an important opportunistic pathogen in CF patients can evade the innate immune system by multiple mechanisms. This results in either chronic bacterial colonization, or acute infection, which is associated with severe lung inflammation and sometimes development of necrotizing pneumonia and death. The majority of B. cenocepacia strains express factors that protect bacteria from host-derived antimicrobial factors, thus providing a mechanism for evading antimicrobial host defenses. Some strains of B. cenocepacia, particularly those isolated from patients who succumbed to fatal “cepacia syndrome” bind to multiple host receptors including TNFR1 and elicit heightened IL-8 response (Sajjan et al., 2008b). This may result in excessive recruitment of neutrophils ultimately leading to extensive lung damage in CF patients, as mechanisms for removal of apoptotic neutrophils is defective in these patients (Vandivier et al., 2009). Consistent with this idea, strains that are capable of binding to TNFR1 caused severe inflammation in lungs of CF mice (Sajjan et al., 2001b; Chattoraj et al., 2010). However, further studies using TNFR1 knockout mice are required to establish direct contribution of B. cenocepacia binding to TNFR1 in development of severe lung disease. In addition, these isolates also express the cable pilus and the associated 22 kDa adhesin and each of which were found to be required for eliciting maximal IL-8 in vitro and lung inflammation in vivo (Urban et al., 2005; Goldberg et al., 2011).
Some B. cenocepacia strains also have the capacity to invade and reside inside airway epithelial cells or macrophages by subverting the normal endocytic pathway (Sajjan et al., 2006; Lamothe et al., 2007; Huynh et al., 2010). This may provide a suitable niche for the bacteria to persist chronically in the lungs of CF patients without being recognized by innate immune cells. Consistent with this notion, lung sections from patients who were chronically colonized with B. cenocepacia showed bacteria inside the airway epithelial cells and macrophages (Sajjan et al., 2001a). Although not much is known about the bacterial factors that subvert the endocytic pathway, Ptw T4SS appears to contribute to this process (Sajjan et al., 2008a). Investigations on identification of Ptw T4SS effectors and how they interact with the components of endocytic pathway will substantially enhance our understanding of the mechanisms by which these organisms subvert the endocytic pathway.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Benjamin Unger for proof reading the manuscript. This work was supported by grant from Cystic Fibrosis Foundation (SAJJAN06I0) and NIH grants AT4793 and HL897720 (to US).
References
Adamo, R., Sokol, S., Soong, G., Gomez, M. I., and Prince, A. (2004). Pseudomonas aeruginosa flagella activate airway epithelial cells through asialoGM1 and toll-like receptor 2 as well as toll-like receptor 5. Am. J. Respir. Cell Mol. Biol. 30, 627–634.
Albrecht, M. T., Wang, W., Shamova, O., Lehrer, R. I., and Schiller, N. L. (2002). Binding of protegrin-1 to Pseudomonas aeruginosa and Burkholderia cepacia. Respir. Res. 3, 18.
Baird, R. M., Brown, H., Smith, A. W., and Watson, M. L. (1999). Burkholderia cepacia is resistant to the antimicrobial activity of airway epithelial cells. Immunopharmacology 44, 267–272.
Bals, R., and Hiemstra, P. S. (2004). Innate immunity in the lung: how epithelial cells fight against respiratory pathogens. Eur. Respir. J. 23, 327–333.
Bals, R., Wang, X., Zasloff, M., and Wilson, J. M. (1998). The peptide antibiotic LL-37/hCAP-18 is expressed in epithelia of the human lung where it has broad antimicrobial activity at the airway surface. Proc. Natl. Acad. Sci. U.S.A. 95, 9541–9546.
Bamford, S., Ryley, H., and Jackson, S. K. (2007). Highly purified lipopolysaccharides from Burkholderia cepacia complex clinical isolates induce inflammatory cytokine responses via TLR4-mediated MAPK signalling pathways and activation of NFkappaB. Cell. Microbiol. 9, 532–543.
Boucher, R. C. (2004). New concepts of the pathogenesis of cystic fibrosis lung disease. Eur. Respir. J. 23, 146–158.
Boucher, R. C. (2007). Evidence for airway surface dehydration as the initiating event in CF airway disease. J. Intern. Med. 261, 5–16.
Bressler, A. M., Kaye, K. S., Lipuma, J. J., Alexander, B. D., Moore, C. M., Reller, L. B., and Woods, C. W. (2007). Risk factors for Burkholderia cepacia complex bacteremia among intensive care unit patients without cystic fibrosis: a case-control study. Infect. Control Hosp. Epidemiol. 28, 951–958.
Burns, J. L., Jonas, M., Chi, E. Y., Clark, D. K., Berger, A., and Griffith, A. (1996). Invasion of respiratory epithelial cells by Burkholderia (Pseudomonas) cepacia. Infect. Immun. 64, 4054–4059.
Bylund, J., Burgess, L. A., Cescutti, P., Ernst, R. K., and Speert, D. P. (2006). Exopolysaccharides from Burkholderia cenocepacia inhibit neutrophil chemotaxis and scavenge reactive oxygen species. J. Biol. Chem. 281, 2526–2532.
Cerantola, S., Bounery, J., Segonds, C., Marty, N., and Montrozier, H. (2000). Exopolysaccharide production by mucoid and non-mucoid strains of Burkholderia cepacia. FEMS Microbiol. Lett. 185, 243–246.
Cerantola, S., Marty, N., and Montrozier, H. (1996). Structural studies of the acidic exopolysaccharide produced by a mucoid strain of Burkholderia cepacia, isolated from cystic fibrosis. Carbohydr. Res. 285, 59–67.
Chamaillard, M., Hashimoto, M., Horie, Y., Masumoto, J., Qiu, S., Saab, L., Ogura, Y., Kawasaki, A., Fukase, K., Kusumoto, S., Valvano, M. A., Foster, S. J., Mak, T. W., Nunez, G., and Inohara, N. (2003). An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 4, 702–707.
Chattoraj, S. S., Murthy, R., Ganesan, S., Goldberg, J. B., Zhao, Y., Hershenson, M. B., and Sajjan, U. S. (2010). Pseudomonas aeruginosa alginate promotes Burkholderia cenocepacia persistence in cystic fibrosis transmembrane conductance regulator knockout mice. Infect. Immun. 78, 984–993.
Chen, J. S., Witzmann, K. A., Spilker, T., Fink, R. J., and Lipuma, J. J. (2001). Endemicity and inter-city spread of Burkholderia cepacia genomovar III in cystic fibrosis. J. Pediatr. 139, 643–649.
Chmiel, J. F., and Davis, P. B. (2003). State of the art: why do the lungs of patients with cystic fibrosis become infected and why can’t they clear the infection? Respir. Res. 4, 8.
Christie, P. J., Atmakuri, K., Krishnamoorthy, V., Jakubowski, S., and Cascales, E. (2005). Biogenesis, architecture, and function of bacterial type IV secretion systems. Annu. Rev. Microbiol. 59, 451–485.
Clode, F. E., Kaufmann, M. E., Malnick, H., and Pitt, T. L. (2000). Distribution of genes encoding putative transmissibility factors among epidemic and nonepidemic strains of Burkholderia cepacia from cystic fibrosis patients in the United Kingdom. J. Clin. Microbiol. 38, 1763–1766.
Coenye, T., and Lipuma, J. J. (2003). Population structure analysis of Burkholderia cepacia genomovar III: varying degrees of genetic recombination characterize major clonal complexes. Microbiology 149, 77–88.
Coenye, T., Spilker, T., Van Schoor, A., Lipuma, J. J., and Vandamme, P. (2004). Recovery of Burkholderia cenocepacia strain PHDC from cystic fibrosis patients in Europe. Thorax 59, 952–954.
Comolli, J. C., Waite, L. L., Mostov, K. E., and Engel, J. N. (1999). Pili binding to asialo-GM1 on epithelial cells can mediate cytotoxicity or bacterial internalization by Pseudomonas aeruginosa. Infect. Immun. 67, 3207–3214.
Conner, G. E., Wijkstrom-Frei, C., Randell, S. H., Fernandez, V. E., and Salathe, M. (2007). The lactoperoxidase system links anion transport to host defense in cystic fibrosis. FEBS Lett. 581, 271–278.
Conway, B. A., Chu, K. K., Bylund, J., Altman, E., and Speert, D. P. (2004). Production of exopolysaccharide by Burkholderia cenocepacia results in altered cell-surface interactions and altered bacterial clearance in mice. J. Infect. Dis. 190, 957–966.
Courtney, J. M., Dunbar, K. E., Mcdowell, A., Moore, J. E., Warke, T. J., Stevenson, M., and Elborn, J. S. (2004). Clinical outcome of Burkholderia cepacia complex infection in cystic fibrosis adults. J. Cyst. Fibros. 3, 93–98.
Davies, J., Dewar, A., Bush, A., Pitt, T., Gruenert, D., Geddes, D. M., and Alton, E. W. (1999). Reduction in the adherence of Pseudomonas aeruginosa to native cystic fibrosis epithelium with anti-asialoGM1 antibody and neuraminidase inhibition. Eur. Respir. J. 13, 565–570.
De, C. V. G. M., Le Goffic, R., Balloy, V., Plotkowski, M. C., Chignard, M., and Si-Tahar, M. (2008). TLR 5, but neither TLR2 nor TLR4, is involved in lung epithelial cell response to Burkholderia cenocepacia. FEMS Immunol. Med. Microbiol. 54, 37–44.
De Bentzmann, S., Roger, P., Dupuit, F., Bajolet-Laudinat, O., Fuchey, C., Plotkowski, M. C., and Puchelle, E. (1996). Asialo GM1 is a receptor for Pseudomonas aeruginosa adherence to regenerating respiratory epithelial cells. Infect. Immun. 64, 1582–1588.
De Jong, M. F., Rolan, H. G., and Tsolis, R. M. (2010). Innate immune encounters of the (type) 4th kind: Brucella. Cell. Microbiol. 12, 1195–1202.
De Soyza, A., Ellis, C. D., Khan, C. M., Corris, P. A., and Demarco De Hormaeche, R. (2004). Burkholderia cenocepacia lipopolysaccharide, lipid A, and proinflammatory activity. Am. J. Respir. Crit. Care Med. 170, 70–77.
Diamond, G., Legarda, D., and Ryan, L. K. (2000). The innate immune response of the respiratory epithelium. Immunol. Rev. 173, 27–38.
Drevinek, P., and Mahenthiralingam, E. (2010). Burkholderia cenocepacia in cystic fibrosis: epidemiology and molecular mechanisms of virulence. Clin. Microbiol. Infect. 16, 821–830.
Duff, C., Murphy, P. G., Callaghan, M., and Mcclean, S. (2006). Differences in invasion and translocation of Burkholderia cepacia complex species in polarised lung epithelial cells in vitro. Microb. Pathog. 41, 183–192.
Engledow, A. S., Medrano, E. G., Mahenthiralingam, E., Lipuma, J. J., and Gonzalez, C. F. (2004). Involvement of a plasmid-encoded type IV secretion system in the plant tissue water soaking phenotype of Burkholderia cenocepacia. J. Bacteriol. 186, 6015–6024.
Fang, F. C. (2004). Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2, 820–832.
Feldman, M., Bryan, R., Rajan, S., Scheffler, L., Brunnert, S., Tang, H., and Prince, A. (1998). Role of flagella in pathogenesis of Pseudomonas aeruginosa pulmonary infection. Infect. Immun. 66, 43–51.
Flannagan, R. S., and Valvano, M. A. (2008). Burkholderia cenocepacia requires RpoE for growth under stress conditions and delay of phagolysosomal fusion in macrophages. Microbiology 154, 643–653.
Foschiatti, M., Cescutti, P., Tossi, A., and Rizzo, R. (2009). Inhibition of cathelicidin activity by bacterial exopolysaccharides. Mol. Microbiol. 72, 1137–1146.
Frangolias, D. D., Mahenthiralingam, E., Rae, S., Raboud, J. M., Davidson, A. G., Wittmann, R., and Wilcox, P. G. (1999). Burkholderia cepacia in cystic fibrosis. Variable disease course. Am. J. Respir. Crit. Care Med. 160, 1572–1577.
Gingues, S., Kooi, C., Visser, M. B., Subsin, B., and Sokol, P. A. (2005). Distribution and expression of the ZmpA metalloprotease in the Burkholderia cepacia complex. J. Bacteriol. 187, 8247–8255.
Girardin, S. E., Boneca, I. G., Carneiro, L. A., Antignac, A., Jehanno, M., Viala, J., Tedin, K., Taha, M. K., Labigne, A., Zahringer, U., Coyle, A. J., Distefano, P. S., Bertin, J., Sansonetti, P. J., and Philpott, D. J. (2003a). Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300, 1584–1587.
Girardin, S. E., Boneca, I. G., Viala, J., Chamaillard, M., Labigne, A., Thomas, G., Philpott, D. J., and Sansonetti, P. J. (2003b). Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 278, 8869–8872.
Goldberg, J. B., Ganesan, S., Comstock, A. T., Zhao, Y., and Sajjan, U. S. (2011). Cable pili and the associated 22 kda adhesin contribute to Burkholderia cenocepacia persistence in vivo. PLoS ONE 6, e22435. doi: 10.1371/journal.pone.0022435
Gomez, M. I., Lee, A., Reddy, B., Muir, A., Soong, G., Pitt, A., Cheung, A., and Prince, A. (2004). Staphylococcus aureus protein A induces airway epithelial inflammatory responses by activating TNFR1. Nat. Med. 10, 842–848.
Govan, J. R., Brown, P. H., Maddison, J., Doherty, C. J., Nelson, J. W., Dodd, M., Greening, A. P., and Webb, A. K. (1993). Evidence for transmission of Pseudomonas cepacia by social contact in cystic fibrosis. Lancet 342, 15–19.
Govan, J. R., Hughes, J. E., and Vandamme, P. (1996). Burkholderia cepacia: medical, taxonomic and ecological issues. J. Med. Microbiol. 45, 395–407.
Herasimenka, Y., Cescutti, P., Impallomeni, G., Campana, S., Taccetti, G., Ravenni, N., Zanetti, F., and Rizzo, R. (2007). Exopolysaccharides produced by clinical strains belonging to the Burkholderia cepacia complex. J. Cyst. Fibros. 6, 145–152.
Hiemstra, P. S. (2001). Epithelial antimicrobial peptides and proteins: their role in host defence and inflammation. Paediatr. Respir. Rev. 2, 306–310.
Hubber, A., and Roy, C. R. (2010). Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26, 261–283.
Huynh, K. K., Plumb, J. D., Downey, G. P., Valvano, M. A., and Grinstein, S. (2010). Inactivation of macrophage Rab7 by Burkholderia cenocepacia. J. Innate Immun. 2, 522–533.
Imundo, L., Barasch, J., Prince, A., and Al-Awqati, Q. (1995). Cystic fibrosis epithelial cells have a receptor for pathogenic bacteria on their apical surface. Proc. Natl. Acad. Sci. U.S.A. 92, 3019–3023.
Isles, A., Maclusky, I., Corey, M., Gold, R., Prober, C., Fleming, P., and Levison, H. (1984). Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. J. Pediatr. 104, 206–210.
Jacquot, J., Tabary, O., Le Rouzic, P., and Clement, A. (2008). Airway epithelial cell inflammatory signalling in cystic fibrosis. Int. J. Biochem. Cell Biol. 40, 1703–1715.
Jones, A. M., Dodd, M. E., Govan, J. R., Barcus, V., Doherty, C. J., Morris, J., and Webb, A. K. (2004). Burkholderia cenocepacia and Burkholderia multivorans: influence on survival in cystic fibrosis. Thorax 59, 948–951.
Kaza, S. K., Mcclean, S., and Callaghan, M. (2011). IL-8 released from human lung epithelial cells induced by cystic fibrosis pathogens Burkholderia cepacia complex affects the growth and intracellular survival of bacteria. Int. J. Med. Microbiol. 301, 26–33.
Keith, K. E., Hynes, D. W., Sholdice, J. E., and Valvano, M. A. (2009). Delayed association of the NADPH oxidase complex with macrophage vacuoles containing the opportunistic pathogen Burkholderia cenocepacia. Microbiology 155, 1004–1015.
Kim, J. Y., Sajjan, U. S., Krasan, G. P., and Lipuma, J. J. (2005). Disruption of tight junctions during traversal of the respiratory epithelium by Burkholderia cenocepacia. Infect. Immun. 73, 7107–7112.
Knowles, M. R., and Boucher, R. C. (2002). Mucus clearance as a primary innate defense mechanism for mammalian airways. J. Clin. Invest. 109, 571–577.
Kooi, C., Corbett, C. R., and Sokol, P. A. (2005). Functional analysis of the Burkholderia cenocepacia ZmpA metalloprotease. J. Bacteriol. 187, 4421–4429.
Kooi, C., and Sokol, P. A. (2009). Burkholderia cenocepacia zinc metalloproteases influence resistance to antimicrobial peptides. Microbiology 155, 2818–2825.
Kooi, C., Subsin, B., Chen, R., Pohorelic, B., and Sokol, P. A. (2006). Burkholderia cenocepacia ZmpB is a broad-specificity zinc metalloprotease involved in virulence. Infect. Immun. 74, 4083–4093.
Kotrange, S., Kopp, B., Akhter, A., Abdelaziz, D., Abu Khweek, A., Caution, K., Abdulrahman, B., Wewers, M. D., Mccoy, K., Marsh, C., Loutet, S. A., Ortega, X., Valvano, M. A., and Amer, A. O. (2011). Burkholderia cenocepacia O polysaccharide chain contributes to caspase-1-dependent IL-1beta production in macrophages. J. Leukoc. Biol. 89, 481–488.
Krivan, H. C., Roberts, D. D., and Ginsburg, V. (1988). Many pulmonary pathogenic bacteria bind specifically to the carbohydrate sequence GalNAc beta 1-4Gal found in some glycolipids. Proc. Natl. Acad. Sci. U.S.A. 85, 6157–6161.
Lamothe, J., Huynh, K. K., Grinstein, S., and Valvano, M. A. (2007). Intracellular survival of Burkholderia cenocepacia in macrophages is associated with a delay in the maturation of bacteria-containing vacuoles. Cell. Microbiol. 9, 40–53.
Ledson, M. J., Gallagher, M. J., Jackson, M., Hart, C. A., and Walshaw, M. J. (2002). Outcome of Burkholderia cepacia colonisation in an adult cystic fibrosis centre. Thorax 57, 142–145.
Leube, R. E., and Rustad, T. J. (1991). Squamous cell metaplasia in the human lung: molecular characteristics of epithelial stratification. Virchows Arch. B Cell Pathol. 61, 227–253.
Lipuma, J. J. (2005). Update on the Burkholderia cepacia complex. Curr. Opin. Pulm. Med. 11, 528–533.
Loutet, S. A., Flannagan, R. S., Kooi, C., Sokol, P. A., and Valvano, M. A. (2006). A complete lipopolysaccharide inner core oligosaccharide is required for resistance of Burkholderia cenocepacia to antimicrobial peptides and bacterial survival in vivo. J. Bacteriol. 188, 2073–2080.
Mahenthiralingam, E., Bischof, J., Byrne, S. K., Radomski, C., Davies, J. E., Av-Gay, Y., and Vandamme, P. (2000). DNA-based diagnostic approaches for identification of Burkholderia cepacia complex, Burkholderia vietnamiensis, Burkholderia multivorans, Burkholderia stabilis, and Burkholderia cepacia genomovars I and III. J. Clin. Microbiol. 38, 3165–3173.
Mahenthiralingam, E., Vandamme, P., Campbell, M. E., Henry, D. A., Gravelle, A. M., Wong, L. T., Davidson, A. G., Wilcox, P. G., Nakielna, B., and Speert, D. P. (2001). Infection with Burkholderia cepacia complex genomovars in patients with cystic fibrosis: virulent transmissible strains of genomovar III can replace Burkholderia multivorans. Clin. Infect. Dis. 33, 1469–1475.
Mayer, A. K., and Dalpke, A. H. (2007). Regulation of local immunity by airway epithelial cells. Arch. Immunol. Ther. Exp. (Warsz.) 55, 353–362.
Mcnamara, N., Gallup, M., Sucher, A., Maltseva, I., Mckemy, D., and Basbaum, C. (2006). AsialoGM1 and TLR5 cooperate in flagellin-induced nucleotide signaling to activate Erk1/2. Am. J. Respir. Cell Mol. Biol. 34, 653–660.
Medzhitov, R., and Janeway, C. A. Jr. (1997). Innate immunity: impact on the adaptive immune response. Curr. Opin. Immunol. 9, 4–9.
Moskwa, P., Lorentzen, D., Excoffon, K. J., Zabner, J., Mccray, P. B. Jr., Nauseef, W. M., Dupuy, C., and Banfi, B. (2007). A novel host defense system of airways is defective in cystic fibrosis. Am. J. Respir. Crit. Care Med. 175, 174–183.
Muir, A., Soong, G., Sokol, S., Reddy, B., Gomez, M. I., Van Heeckeren, A., and Prince, A. (2004). Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 30, 777–783.
Mullen, T., Callaghan, M., and Mcclean, S. (2010). Invasion of Burkholderia cepacia complex isolates into lung epithelial cells involves glycolipid receptors. Microb. Pathog. 49, 381–387.
O’brien, L. M., Walsh, E. J., Massey, R. C., Peacock, S. J., and Foster, T. J. (2002). Staphylococcus aureus clumping factor B (ClfB) promotes adherence to human type I cytokeratin 10: implications for nasal colonization. Cell. Microbiol. 4, 759–770.
Parker, D., and Prince, A. (2011). Innate immunity in the respiratory epithelium. Am. J. Respir. Cell Mol. Biol. 45, 189–201.
Pitt, T. L., Kaufmann, M. E., Patel, P. S., Benge, L. C., Gaskin, S., and Livermore, D. M. (1996). Type characterisation and antibiotic susceptibility of Burkholderia (Pseudomonas) cepacia isolates from patients with cystic fibrosis in the United Kingdom and the Republic of Ireland. J. Med. Microbiol. 44, 203–210.
Pohl, C., Hermanns, M. I., Uboldi, C., Bock, M., Fuchs, S., Dei-Anang, J., Mayer, E., Kehe, K., Kummer, W., and Kirkpatrick, C. J. (2009). Barrier functions and paracellular integrity in human cell culture models of the proximal respiratory unit. Eur. J. Pharm. Biopharm. 72, 339–349.
Roger, P., Puchelle, E., Bajolet-Laudinat, O., Tournier, J. M., Debordeaux, C., Plotkowski, M. C., Cohen, J. H., Sheppard, D., and De Bentzmann, S. (1999). Fibronectin and alpha5beta1 integrin mediate binding of Pseudomonas aeruginosa to repairing airway epithelium. Eur. Respir. J. 13, 1301–1309.
Rose, M. C., Nickola, T. J., and Voynow, J. A. (2001). Airway mucus obstruction: mucin glycoproteins, MUC gene regulation and goblet cell hyperplasia. Am. J. Respir. Cell Mol. Biol. 25, 533–537.
Sajjan, S. U., Carmody, L. A., Gonzalez, C. F., and Lipuma, J. J. (2008a). A type IV secretion system contributes to intracellular survival and replication of Burkholderia cenocepacia. Infect. Immun. 76, 5447–5455.
Sajjan, U. S., Hershenson, M. B., Forstner, J. F., and Lipuma, J. J. (2008b). Burkholderia cenocepacia ET12 strain activates TNFR1 signalling in cystic fibrosis airway epithelial cells. Cell. Microbiol. 10, 188–201.
Sajjan, S. U., and Forstner, J. F. (1992). Identification of the mucin-binding adhesin of Pseudomonas cepacia isolated from patients with cystic fibrosis. Infect. Immun. 60, 1434–1440.
Sajjan, U., Ackerley, C., and Forstner, J. (2002). Interaction of cblA/adhesin-positive Burkholderia cepacia with squamous epithelium. Cell. Microbiol. 4, 73–86.
Sajjan, U., Corey, M., Humar, A., Tullis, E., Cutz, E., Ackerley, C., and Forstner, J. (2001a). Immunolocalisation of Burkholderia cepacia in the lungs of cystic fibrosis patients. J. Med. Microbiol. 50, 535–546.
Sajjan, U., Thanassoulis, G., Cherapanov, V., Lu, A., Sjolin, C., Steer, B., Wu, Y. J., Rotstein, O. D., Kent, G., Mckerlie, C., Forstner, J., and Downey, G. P. (2001b). Enhanced susceptibility to pulmonary infection with Burkholderia cepacia in Cftr(−/−) mice. Infect. Immun. 69, 5138–5150.
Sajjan, U., Keshavjee, S., and Forstner, J. (2004). Responses of well-differentiated airway epithelial cell cultures from healthy donors and patients with cystic fibrosis to Burkholderia cenocepacia infection. Infect. Immun. 72, 4188–4199.
Sajjan, U. S., Corey, M., Karmali, M. A., and Forstner, J. F. (1992). Binding of Pseudomonas cepacia to normal human intestinal mucin and respiratory mucin from patients with cystic fibrosis. J. Clin. Invest. 89, 648–656.
Sajjan, U. S., Yang, J. H., Hershenson, M. B., and Lipuma, J. J. (2006). Intracellular trafficking and replication of Burkholderia cenocepacia in human cystic fibrosis airway epithelial cells. Cell. Microbiol. 8, 1456–1466.
Saldias, M. S., and Valvano, M. A. (2009). Interactions of Burkholderia cenocepacia and other Burkholderia cepacia complex bacteria with epithelial and phagocytic cells. Microbiology 155, 2809–2817.
Schwab, U., Leigh, M., Ribeiro, C., Yankaskas, J., Burns, K., Gilligan, P., Sokol, P., and Boucher, R. (2002). Patterns of epithelial cell invasion by different species of the Burkholderia cepacia complex in well-differentiated human airway epithelia. Infect. Immun. 70, 4547–4555.
Shaw, D., Poxton, I. R., and Govan, J. R. (1995). Biological activity of Burkholderia (Pseudomonas) cepacia lipopolysaccharide. FEMS Immunol. Med. Microbiol. 11, 99–106.
Siddiqui, A. H., Mulligan, M. E., Mahenthiralingam, E., Hebden, J., Brewrink, J., Qaiyumi, S., Johnson, J. A., and Lipuma, J. J. (2001). An episodic outbreak of genetically related Burkholderia cepacia among non-cystic fibrosis patients at a university hospital. Infect. Control Hosp. Epidemiol. 22, 419–422.
Singh, P. K., Jia, H. P., Wiles, K., Hesselberth, J., Liu, L., Conway, B. A., Greenberg, E. P., Valore, E. V., Welsh, M. J., Ganz, T., Tack, B. F., and Mccray, P. B. Jr. (1998). Production of beta-defensins by human airway epithelia. Proc. Natl. Acad. Sci. U.S.A. 95, 14961–14966.
Sist, P., Cescutti, P., Skerlavaj, S., Urbani, R., Leitao, J. H., Sa-Correia, I., and Rizzo, R. (2003). Macromolecular and solution properties of cepacian: the exopolysaccharide produced by a strain of Burkholderia cepacia isolated from a cystic fibrosis patient. Carbohydr. Res. 338, 1861–1867.
Sokol, P. A., Sajjan, U., Visser, M. B., Gingues, S., Forstner, J., and Kooi, C. (2003). The CepIR quorum-sensing system contributes to the virulence of Burkholderia cenocepacia respiratory infections. Microbiology 149, 3649–3658.
Speert, D. P., Henry, D., Vandamme, P., Corey, M., and Mahenthiralingam, E. (2002). Epidemiology of Burkholderia cepacia complex in patients with cystic fibrosis, Canada. Emerging Infect. Dis. 8, 181–187.
Sun, L., Jiang, R. Z., Steinbach, S., Holmes, A., Campanelli, C., Forstner, J., Sajjan, U., Tan, Y., Riley, M., and Goldstein, R. (1995). The emergence of a highly transmissible lineage of cbl+ Pseudomonas (Burkholderia) cepacia causing CF centre epidemics in North America and Britain. Nat. Med. 1, 661–666.
Sylvester, F. A., Sajjan, U. S., and Forstner, J. F. (1996). Burkholderia (basonym Pseudomonas) cepacia binding to lipid receptors. Infect. Immun. 64, 1420–1425.
Tomich, M., Herfst, C. A., Golden, J. W., and Mohr, C. D. (2002). Role of flagella in host cell invasion by Burkholderia cepacia. Infect. Immun. 70, 1799–1806.
Urban, T. A., Goldberg, J. B., Forstner, J. F., and Sajjan, U. S. (2005). Cable pili and the 22-kilodalton adhesin are required for Burkholderia cenocepacia binding to and transmigration across the squamous epithelium. Infect. Immun. 73, 5426–5437.
Urban, T. A., Griffith, A., Torok, A. M., Smolkin, M. E., Burns, J. L., and Goldberg, J. B. (2004). Contribution of Burkholderia cenocepacia flagella to infectivity and inflammation. Infect. Immun. 72, 5126–5134.
Vandamme, P., Holmes, B., Coenye, T., Goris, J., Mahenthiralingam, E., LiPuma, J. J., and Govan, J. R. (2003). Burkholderia cenocepacia sp. nov. – a new twist to an old story. Res. Microbiol. 154, 91–96.
Vandivier, R. W., Henson, P. M., and Douglas, I. S. (2006). Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest 129, 1673–1682.
Vandivier, R. W., Richens, T. R., Horstmann, S. A., Decathelineau, A. M., Ghosh, M., Reynolds, S. D., Xiao, Y. Q., Riches, D. W., Plumb, J., Vachon, E., Downey, G. P., and Henson, P. M. (2009). Dysfunctional cystic fibrosis transmembrane conductance regulator inhibits phagocytosis of apoptotic cells with proinflammatory consequences. Am. J. Physiol. Lung Cell Mol. Physiol. 297, L677–L686.
Vareille, M., Kieninger, E., Edwards, M. R., and Regamey, N. (2011). The airway epithelium: soldier in the fight against respiratory viruses. Clin. Microbiol. Rev. 24, 210–229.
Vieira, O. V., Bucci, C., Harrison, R. E., Trimble, W. S., Lanzetti, L., Gruenberg, J., Schreiber, A. D., Stahl, P. D., and Grinstein, S. (2003). Modulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3-kinase. Mol. Cell Biol. 23, 2501–2514.
Vinion-Dubiel, A. D., and Goldberg, J. B. (2003). Lipopolysaccharide of Burkholderia cepacia complex. J. Endotoxin Res. 9, 201–213.
Woods, C. W., Bressler, A. M., Lipuma, J. J., Alexander, B. D., Clements, D. A., Weber, D. J., Moore, C. M., Reller, L. B., and Kaye, K. S. (2004). Virulence associated with outbreak-related strains of Burkholderia cepacia complex among a cohort of patients with bacteremia. Clin. Infect. Dis. 38, 1243–1250.
Wu, X., Kurpakus, M., and Hazlett, L. D. (1996). Some P. aeruginosa pilus-binding proteins of human corneal epithelium are cytokeratins. Curr. Eye Res. 15, 782–791.
Keywords: cystic fibrosis, innate immunity, inflammation, bacterial pathogenesis, lung infection, cable pili, airway epithelium, macrophages
Citation: Ganesan S and Sajjan US (2012) Host evasion by Burkholderia cenocepacia. Front. Cell. Inf. Microbio. 1:25. doi: 10.3389/fcimb.2011.00025
Received: 16 September 2011; Accepted: 22 December 2011;
Published online: 11 January 2012.
Edited by:
Joanna Goldberg, University of Virginia Health System, USAReviewed by:
Mikhail A. Gavrilin, Ohio State University, USALeo Eberl, University of Zurich, Switzerland
Copyright: @ 2012 Ganesan and Sajjan. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Umadevi S. Sajjan, University of Michigan, 1150 West Medical Center Drive, Room 3570, MSRBII, Box 5688, Ann Arbor, MI 48109-0688, USA. e-mail:dXNhamphbkB1bWljaC5lZHU=