

- 1Division of Pulmonary, Allergy and Critical Care Medicine, Department of Medicine, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
- 2Department of Immunology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
Ligand-mediated activation of toll-like receptors (TLRs) not only induces inflammation but also immune suppression, which is an emerging area of investigation. Multiple negative feedback intracellular mechanisms have been described that are brought into play to prevent uncontrolled TLR activation. However, the identification of TLR-induced regulatory myeloid cells is a relatively recent development that has ramifications in pathogen-induced disease state as well as in cancer. Our efforts to understand how a high dose of lipopolysaccharide (LPS), a ligand of TLR4, suppresses allergic airway inflammation led to the identification of myeloid cells that are CD11b+Griint(Ly6Gint)F4/80+ and are phenotypically and morphologically similar to myeloid-derived suppressor cells (MDSCs) which are best studied in the context of cancer. MDSCs have been also detected during infection by various bacteria, parasites and viruses, which can engage different TLRs. These TLR-induced myeloid cells produce different types of mediators to influence immune response and inflammation that can be either beneficial or detrimental to the host. One beneficial function of TLR4/MyD88-triggered MDSCs in the lung is to efferocytose apoptotic neutrophils to help resolve inflammation elicited during bacterial pneumonia. A better understanding of the generation and function of these regulatory cells would be helpful to harness their potential or suppress their function for disease-specific immune regulation.
Microorganism-associated molecular patterns are recognized by pattern-recognition receptors such as Toll-like receptors (TLRs), NOD-like receptors and RIG-I-like receptors in different cells of infected hosts to initiate a rapid innate immune response as the first step in defense against the invading pathogen. The innate immune response, in turn, induces an appropriate adaptive immune response to effectively protect the host with the establishment of a memory response for a rapid recall response during subsequent exposure to the pathogen. After the identification of the toll protein in Drosophila as an essential host-defense mechanism in an organism that lacks the adaptive immune system (Lemaitre et al., 1996), TLR4 was the first TLR to be identified in mammals (Medzhitov et al., 1997; Poltorak et al., 1998; Qureshi et al., 1999). The desire to identify a counterpart to the Drosophila Toll in the mammalian system was largely driven by the need to understand the pathogenesis of bacterial infection-induced sepsis syndrome (Rittirsch et al., 2008). Thus, lipopolysaccharide (LPS), a component of the outer membrane of Gram-negative bacteria, was identified as a ligand of TLR4. Since then, 10 and 12 functional TLRs have been characterized in humans and mice, respectively (Kawai and Akira, 2010). In addition to bacterial LPS, the TLR4 complex is triggered by multiple additional ligands. This process ultimately results in downstream activation of NF-κB or IRF-1 via the adaptors MyD88 and TRIF to induce transcription of cytokine and chemokine genes and a host of other genes that collectively ramp up the host's immune defense mechanisms.
Regulation of TLR-Induced Intracellular Responses
Although TLR induced pro-inflammatory response is crucial for the elimination of the invading pathogen, uncontrolled immune activation leads to collateral tissue damage. Therefore, stringent negative feedback is necessary to temper TLR-induced signaling in target cells. These measures include a range of extracellular and intracellular decoy receptors, membrane-bound suppressors, intracellular negative regulators, degradation of TLRs, and TLR-induced apoptosis (Liew et al., 2005; Kondo et al., 2012). A distinct TLR-induced immunosuppressive mechanism in different organs, as discovered in some laboratories including our own, is the development of myeloid-derived suppressor cells (MDSCs). In this review, we will discuss the characteristics of MDSCs induced by TLR agonists such as LPS and by pathogens and the emerging field of MDSC-mediated suppression of immune responses.
Development of a Regulatory Myeloid Cell, MDSC, by LPS-TLR4 Signaling to Control Immune Responses
LPS exerts differential effects on immune responses in the lung depending on the dose. In the context of exposure to the model allergen ovalbumin (OVA), a very low dose (< 1 ng) of LPS does not induce adaptive immunity and instead tolerance to OVA develops (Oriss et al., 2005). An intermediate dose (100 ng) of LPS induces a Th2-biased response to OVA and promotes allergic airway inflammation (Eisenbarth et al., 2002). Higher doses of LPS (1–100 μg), however, inhibit allergic inflammation (Gerhold et al., 2002, 2008; Rodriguez et al., 2003; Delayre-Orthez et al., 2005). Our efforts to understand the mechanism(s) by which a high dose of LPS inhibits allergic airway inflammation led to the discovery of a new role for the MDSC as a TLR4/MyD88- but not TRIF-induced cell type that can influence T cell responses in the lung (Arora et al., 2010). Figure 1 illustrates the induction of lung MDSCs in response to LPS or bacterial infection and the function of these cells in the lung tissue.
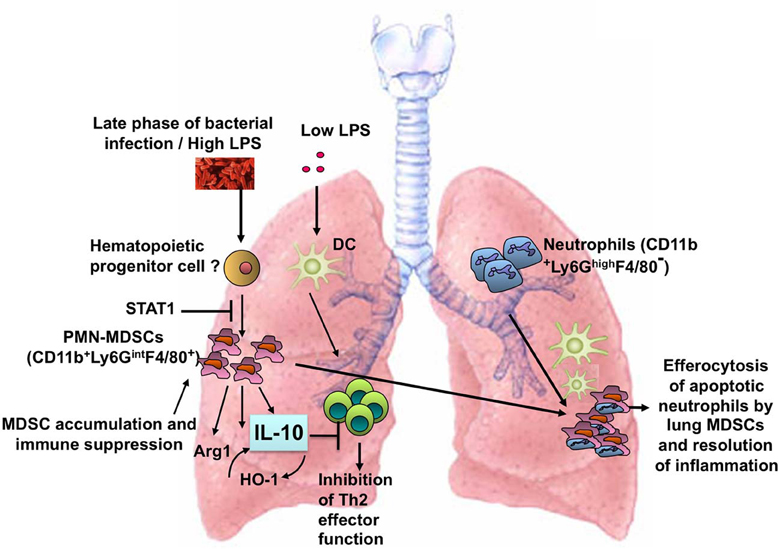
Figure 1. Induction of MDSCs in the lung in response to a high dose of LPS or bacterial infection. The lung MDSCs express Arg1 and produce IL-10 and suppress Th2 effector function in the context of allergic airway inflammation and phagocytose apoptotic neutrophils during bacterial pneumonia.
In mice, MDSCs are uniformly characterized as CD11b+Gr1+ cells. Since anti-Gr1 recognizes both the Ly6G and Ly6C epitopes, use of antibodies with monospecificity toward one or the other has shown differential expression of these molecules on different types of MDSCs. Typically, CD11b+Ly6Chigh MDSCs are mononuclear and considered to be monocytic (Mo-MDSC) while CD11b+Ly6G+Ly6Clow/− MDSCs consist of multilobed nuclei and represent granulocytic or neutrophilic MDSCs (PMN-MDSC). Additional cell surface molecules including the α-chain of the receptor for IL-4 and IL-13 (CD124), the mouse macrophage-expressed molecule, F4/80, M-CSF-1R or c-fms (CD115) and the costimulatory molecule CD80 have also been identified in different combinations on some subsets of MDSCs (Gabrilovich and Nagaraj, 2009; Ostrand-Rosenberg and Sinha, 2009; Peranzoni et al., 2010). LPS-induced MDSCs in the lung have the phenotype CD11b+Ly6GintLy6Clow/−F4/80+CD80+.
Much before LPS was shown to be a stimulus for MDSC accumulation, multiple factors, associated with chronic inflammation and cancer such as VEGF, GM-CSF, G-CSF, IL-1β, IL6, and PGE2 were described as inducers of MDSCs (Gabrilovich and Nagaraj, 2009). However, the cell surface phenotype and mediator employed by the MDSCs induced by the various types of agents to suppress immune responses are not the same as discussed below.
LPS-Induced Lung MDSCs are Tissue Resident and can Suppress Effector T Cell Function
A previous study described proliferation of migratory hematopoietic stem and progenitor cells (HSPCs) within extramedullary tissues in response to LPS (Massberg et al., 2007). Although ~70% of the myeloid cells generated in the presence of LPS expressed the dendritic cell (DC) marker CD11c, a subset of lower frequency (10%) expressed Gr1 at intermediate to high levels and may have been related to MDSCs. In our study, after infusion of GFP+ lineageneg (lin−) bone marrow progenitor cells into naïve mice, LPS instillation into the lungs promoted the accumulation of GFP+CD11b+Gr1int(Ly6Gint) cells in the lung tissue (Arora et al., 2010).
LPS-induced Gr1int cells lack CCR7, which is essential for the migration to lymph nodes (Debes et al., 2005). LPS-induced Gr1int cells were not detectable in the lung-draining lymph nodes (LNs) and have been largely identified as tissue-dwelling cells (Arora et al., 2010; Poe et al., 2013). The earlier study also showed that LPS stimulation not only enhances the local proliferation and differentiation of HSPCs but also reduces the migratory capacity of HSPCs within extramedullary tissues by interfering with S1P-S1P1-dependent signaling (Massberg et al., 2007). It was similarly shown in vitro that incubation of HSPCs with TLR ligands triggers HSPC proliferation and rapid myeloid differentiation (Nagai et al., 2006). Intradermal injection of Salmonella typhimurium or LPS was shown to induce a potent local innate inflammatory response that blocked DC differentiation and migration to the draining LNs (Rotta et al., 2003).
MDSCs are well-known for their ability to inhibit T-cell proliferation and immune responses (Nagaraj et al., 2013). MDSCs utilize multiple mechanisms to exert their suppressive functions and recent studies suggest communication between MDSCs and T cells which is not just limited to cancer but to different types of inflammation-associated conditions which has been comprehensively reviewed recently (Nagaraj et al., 2013). The best-described mechanisms/mediators related to immunosuppression by MDSCs include expression of Arginase 1, nitric oxide, peroxynitrite and reactive oxygen species. MDSCs can also block T-cell activation by deprivation of L-cysteine, an essential amino acid required for T-cell activation and function (Srivastava et al., 2010). Various other mechanisms used by MDSCs to suppress immune responses have also been suggested which include upregulation of cyclooxygenase 2 and prostaglandin E2 (Rodriguez et al., 2005), secretion of TGF-β (Yang et al., 2008; Filipazzi et al., 2012) and induction of Tregs (Huang et al., 2006; Serafini et al., 2008; Nagaraj et al., 2013). Our study showed that the IL-10/Arg1 axis is involved in the suppressive activity of lung MDSCs on Th2 cells (Arora et al., 2010). In another study, HO-1 was implicated in the suppression of alloreactive responses by LPS-induced MDSCs (De Wilde et al., 2009). HO-1 was previously shown to promote IL-10 expression and conversely IL-10 can induce HO-1 expression (Ricchetti et al., 2004; Chauveau et al., 2005). In view of the diversity of immune signals to which MDSCs are exposed in different biological contexts, it is expected that the mechanism of suppression by MDSCs induced by various agents would also vary.
Our study has shown that after repeated exposure of mice to LPS, the MDSCs accumulate with a delayed kinetics in the lung tissue and their numbers increase significantly over baseline only 3 days after LPS instillation once every day (Poe et al., 2013). Since the MDSCs in the lung remain in the tissue after each LPS instillation unlike dendritic cells (DCs) which traffic readily to the LNs (Arora et al., 2010), this differential response causes selective enrichment of the MDSCs over DCs in the tissue (Arora et al., 2010). The LPS/TLR4-induced lung MDSCs suppress the ability of lung DCs to promote Th2 cytokine production, upregulate GATA-3 or induce STAT5 activation in primed Th2 cells, both transcription factors being critical in Th2 effector function (Zhang et al., 1997; Zheng and Flavell, 1997; Zhu et al., 2003). Since STAT5 activation promotes T cell viability (Wofford et al., 2008; Hand et al., 2010), it is possible that lung MDSCs compromise Th2 cell survival thereby reducing the size of the memory T cell pool (Hu et al., 2001; De Wilde et al., 2009). Thus, collectively, it appears that an important effector function of TLR-induced MDSCs is not directed to the LN to influence the development of adaptive immune functions but to control immune responses in the tissue. Of note, flagellin (TLR5)-induced MDSCs have been identified in the lung in the context of cystic fibrosis and associated infection by Pseudomonas aeruginosa (Rieber et al., 2012). Although it is proposed that these MDSCs may inhibit T cell responses against the bacteria based on the ability of the cells to inhibit T cell proliferation ex vivo, it is unknown whether these TLR5-induced MDSCs indeed traffic to the lung-draining LNs where effects on T cell proliferation would be most relevant. Alternatively, these MDSCs may also function to dampen effector T cell function in the tissue.
Bacterial Pneumonia and Resolution of Inflammation: a Novel Role for TLR-Induced MDSCs
Given that a high dose of a bacterial component, LPS, was found to be a potent stimulus for MDSC development, it seemed logical to us that bacterial infection of the lung would also trigger MDSC expansion. Toward this end, the response to the Gram-negative bacterium, Klebsiella pneumoniae, was studied. K. pneumoniae is a common pathogen in hospital-acquired pneumonia, particularly in chronically ill subjects (Neuhauser et al., 2003). Bacterial pneumonia remains a leading cause of morbidity and mortality (Ware and Matthay, 2000; Lee and Downey, 2001; Abraham, 2003; Mizgerd, 2006, 2008; Tsai and Grayson, 2008; Craig et al., 2009; Balamayooran et al., 2010). Three million cases of pneumonia are reported annually in the United States alone, which causes 40,000–70,000 deaths each year (Mizgerd, 2008). K. pneumoniae induces massive neutrophil influx followed by extensive lung destruction and TLR4/MyD88 signaling has an important role in host defense against K. pneumoniae (Chan et al., 2009; Wieland et al., 2011). A number of studies have documented the role of both alveolar macrophages and neutrophils in innate host defense against bacterial pathogens including K. pneumoniae (Broug-Holub et al., 1997; Craig et al., 2009). However, much less is known about the relationship between these two cell types with regard to bacterial clearance and resolution of lung inflammation. It is critical that neutrophil-associated (immune-mediated) pathology be controlled to prevent collateral damage during defense against the invading pathogen (Lee and Downey, 2001; Abraham, 2003). The problem is, therefore, 2-fold. Bacteria must be cleared and the ensuing host inflammatory mechanisms used for bacterial clearance must also be attenuated to prevent lung tissue damage and high mortality. The significance of this question can be best appreciated in the context of pneumonia precipitating ALI/ARDS (acute lung injury/acute respiratory distress syndrome) in which excessive lung inflammation is an overriding factor of injury suggesting inadequate control by anti-inflammatory mechanisms.
Defense against pathogenic bacteria involves a set of well-orchestrated events in which the first step is recognition of the invading pathogen by the host in which TLRs play an important role. A key purpose of sensing pathogens via TLRs is to rapidly engage an innate immune response to clear the pathogen (Barton, 2008; Soehnlein and Lindbom, 2010). The cells of the innate immune system that are well-characterized with respect to phagocytosis and killing of internalized bacteria are alveolar macrophages and neutrophils (Barton, 2008; Soehnlein and Lindbom, 2010). Typically, alveolar macrophages participate initially but are taken over by neutrophils that are rapidly recruited to the site of infection aided by chemokines, whose major sources are lung epithelial cells and macrophages (Bergeron et al., 1999). Neutrophils generate various noxious products including reactive oxygen species and proteases that are not only harmful for the pathogen but also for the host's own cells (Lee and Downey, 2001; Abraham, 2003; Mizgerd, 2006, 2008; Balamayooran et al., 2010). Therefore, once the pathogen is cleared, the immediate next goal of the host is to mount an appropriate anti-inflammatory response to limit further neutrophil recruitment. Neutrophils also have a relatively short life span and rapidly begin to undergo apoptosis at the site of inflammation. To prevent lung injury, these apoptotic cells need to be rapidly cleared by phagocytes, a process termed efferocytosis. It is at this last phase of the host's response to infection that the MDSCs step in. As observed with LPS exposure, after infection, MDSCs do not accumulate rapidly in the lung but rather develop late after infection (Poe et al., 2013). This makes perfect sense since the lung MDSCs produce IL-10, which if produced too early is detrimental to bacterial clearance since it impedes neutrophil recruitment (Poe et al., 2013). We showed that the lung MDSCs efficiently efferocytose apoptotic neutrophils, which is aided by IL-10 produced by the MDSCs. The successful clearance of pathogens, dampening of neutrophil infiltration and clearance of dead neutrophils ultimately restores tissue homeostasis.
In the context of cancer, MDSCs have been shown to promote the development of regulatory T cells (Tregs) (Nagaraj et al., 2013). Whether this is also true for lung MDSCs during infection remains to be determined. However, Tregs were implicated in suppression of lung inflammation in a model of LPS-induced lung injury (D'Alessio et al., 2009).
TLR-Induced Lung MDSC Development and the JAK/STAT Pathway
GM-CSF has been implicated in innate immune responses in the lung in response to LPS (Bozinovski et al., 2002). In vitro, the combination of LPS and GM-CSF induces differentiation of lin-progenitor cells into MDSCs, which suggests co-operation between TLR/MyD88 and Jak2/STAT5 pathways in MDSC development (Arora et al., 2010). LPS-induced MDSCs in the lung produce GM-CSF and IL-6, which have the ability to activate STAT5 and STAT3, respectively, in cells in an autocrine or paracrine fashion. STAT5 activation promotes MDSC survival (Condamine and Gabrilovich, 2011). Jak2/STAT3 signaling has been also shown to play a crucial role in tumor-associated MDSC generation (Nefedova et al., 2004; Condamine and Gabrilovich, 2011). Autocrine IL-6-induced STAT3 signaling downstream of Hsp72/TLR2 was implicated in MDSC-suppressive function (Chalmin et al., 2010). In our studies, neutralization of IL-6 blunted MDSC numbers in ex vivo cultures of the cells (Poe et al., 2013). Collectively, these studies suggest that TLR4 activation by a high dose of LPS in precursor cells induces GM-CSF and IL-6 production causing STAT5 and STAT3 activation respectively, which can support the development and function of MDSCs. In keeping with the noted antagonism between STAT1 and STAT3, STAT1-deficient mice were examined in our study for lung MDSC numbers as compared to wild-type mice after bacterial infection. Indeed, lung MDSC numbers almost doubled in STAT1−/− mice in response to bacterial infection with fewer neutrophils as compared to that in wild-type mice (Poe et al., 2013). These observations suggest that increasing MDSC numbers via STAT1 inhibition in combination with antibiotics may be beneficial in the context of non-resolving pneumonia.
MDSC Induction by Pathogens VIS-À-VIS Use of TLR Agonists in Cancer
Not only in the lung, in other tissues as well, infection by bacteria, viruses and parasites can promote MDSC accumulation. Table 1 outlines a comparison of MDSCs that have been detected during infection by various pathogens and those that are most commonly associated with cancer, albeit much less information is available about the former. The common inducer in both contexts is inflammation. Upon reviewing currently available information it is evident that an important role of MDSCs is to exercise balance between host-defense-associated inflammation mounted during an immune response and inflammation-mediated tissue pathology (Ortega-Gomez et al., 2013). In the case of sepsis, MDSC development induced by acute phase proteins was shown to be crucial for control of systemic inflammation and sepsis-induced mortality (Sander et al., 2010). While our study shows a beneficial effect of MDSCs in mediating resolution of inflammation during bacterial pneumonia, in the case of infection by influenza virus, MDSC effector function needs to be regulated by iNKT cells in the absence of which immune suppression exercised by MDSCs causes increased viral titer and mortality (De Santo et al., 2008). A recent study has proposed that host susceptibility to co-infection by viruses and bacteria results not due to exaggerated activation of some well-described immune pathways but due to failure to tolerate excessive tissue damage after infection (Jamieson et al., 2013). However, since lung MDSCs have been shown to regulate susceptibility to single infection by either viruses (De Santo et al., 2008) or bacteria (Poe et al., 2013), it is possible that MDSCs regulate both inflammation and tissue damage during co-infection via one or more of its mediators. In this regard, the kinetics of MDSC development post infection (De Santo et al., 2008; Poe et al., 2013) may dictate the ultimate outcome.
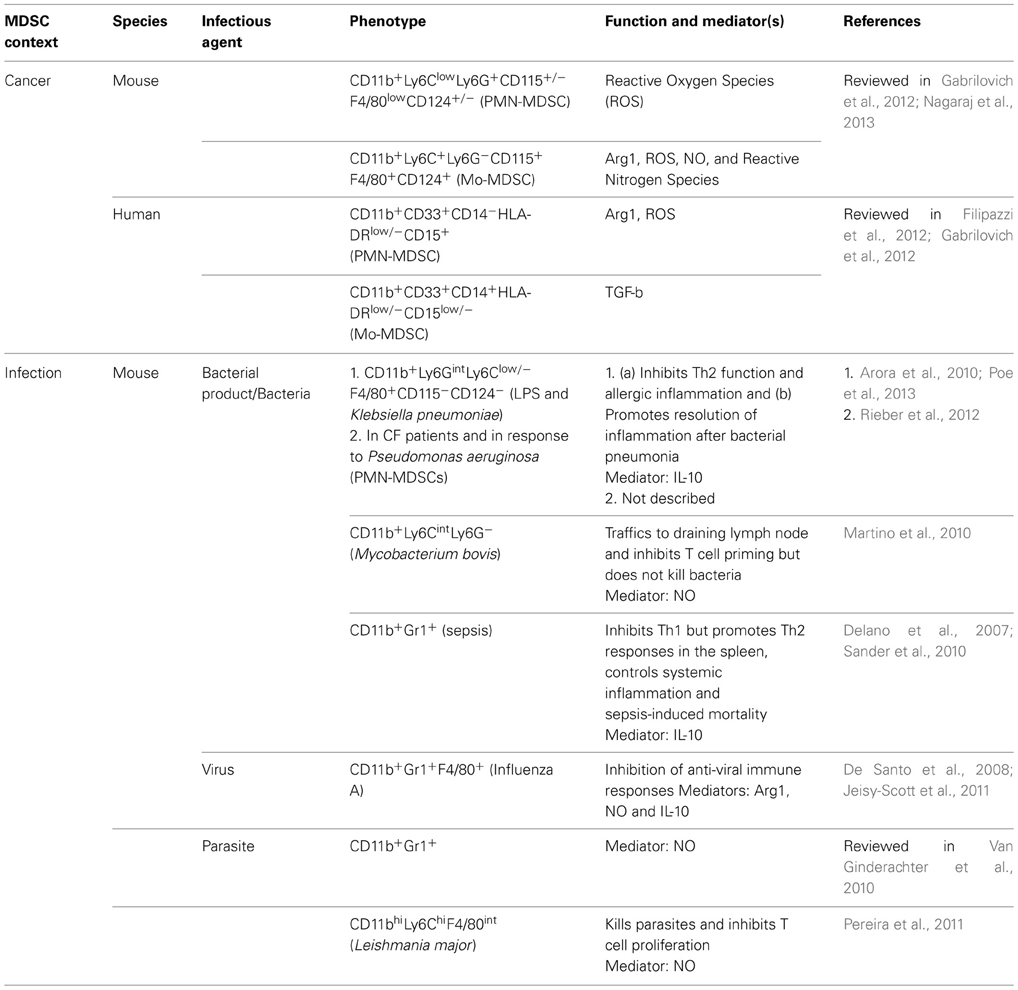
Table 1. MDSCs in cancer and infection.
Similar to our observation of the ability of a combination of GM-CSF and LPS to induce MDSCs in vitro (Arora et al., 2010), a combination of IFN-γ and LPS was found to enhance the development and activation of bone marrow-derived MDSCs while simultaneously inhibiting the differentiation of bone marrow cells to DCs (Greifenberg et al., 2009). Not only LPS, but various other danger-associated molecular patterns (DAMPs) such as S100A, and heat shock proteins released from dying cells that also engage TLR4 or other TLRs have the potential to promote accumulation of MDSCs or enhance their function (Cheng et al., 2008; Bunt et al., 2009). Hsp72 from tumor-derived exosomes was shown to promote STAT3-dependent immunosuppressive function of MDSCs in a TLR2/MyD88 dependent manner (Chalmin et al., 2010). Also, the fact that various pathogens that can activate diverse TLRs can also promote MDSCs suggests that the ability to cause MDSC-mediated immunosuppression is not just restricted to TLR4.
Immunosuppression resulting from TLR engagement is of particular importance in cancer therapy. In fact, most anti-cancer strategies involving TLR agonist monotherapy have proven unsuccessful in clinical trials (Guha, 2012). For example, in a pre-clinical study, failure of imiquimod, a TLR7 agonist, in antitumor therapy was associated with IL-10-mediated immunosuppression. IL-10 neutralization significantly enhanced the efficacy of imiquimod (Lu et al., 2010). In contrast, use of TLR agonists in combination with a cancer vaccination approach or chemotherapy has yielded more promising results toward the induction of anti-tumor CTL responses (Ridnour et al., 2013). Most importantly, while TLR signaling and MDSCs portend poor prognosis for cancers (Ridnour et al., 2013), they appear to be beneficial in regulating tissue inflammation in the lung (Arora et al., 2010; Poe et al., 2013) or systemic inflammation during polymicrobial sepsis (Sander et al., 2010).
Concluding Remarks
TLR signaling elicited by pathogens or by components of pathogens in concert with signals imparted by growth factors such as GM-CSF and IFN-γ has the potential to induce MDSCs in different organs including the lung. MDSCs can be either beneficial or detrimental to the host depending on the infectious agent, involved organ and mediator(s) released. High LPS dose or bacterial infection elicits MDSCs in the lung that play an important role in regulating immune response in the context of diverse pulmonary inflammatory conditions such as allergic airway inflammation and bacterial pneumonia. These TLR4-induced MDSCs accumulate with delayed kinetics after high dose LPS exposure or bacterial infection and have features of PMN-MDSCs. The MDSCs can suppress Th2 effector function and efferocytose apoptotic neutrophils and the common mediator involved is IL-10. The net outcome in each case is resolution of inflammation. Thus, a possible therapeutic approach for non-resolving pneumonia is boosting MDSC numbers via STAT1 inhibition in combination with antibiotics where suppression of unremitting inflammation along with complete elimination of the infectious agent is the desired goal. Unquestionably, a fertile area of future research is to understand context-specific function of MDSCs to suppress or stimulate them for therapeutic benefits.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by US National Institutes of Health grants HL 077430 and AI 048927 (to Anuradha Ray), AI 100012 (to Prabir Ray), and HL 113956 (to Anuradha Ray and Prabir Ray).
References
Abraham, E. (2003). Neutrophils and acute lung injury. Crit. Care Med. 31, S195–S199. doi: 10.1097/01.CCM.0000057843.47705.E8
Arora, M., Poe, S. L., Oriss, T. B., Krishnamoorthy, N., Yarlagadda, M., Wenzel, et al. (2010). TLR4/MyD88-induced CD11b+Gr-1 int F4/80+ non-migratory myeloid cells suppress Th2 effector function in the lung. Mucosal Immunol. 3, 578–593. doi: 10.1038/mi.2010.41
Balamayooran, G., Batra, S., Fessler, M. B., Happel, K. I., and Jeyaseelan, S. (2010). Mechanisms of neutrophil accumulation in the lungs against bacteria. Am. J. Respir. Cell Mol. Biol. 43, 5–16. doi: 10.1165/rcmb.2009-0047TR
Barton, G. M. (2008). A calculated response: control of inflammation by the innate immune system. J. Clin. Invest. 118, 413–420. doi: 10.1172/JCI34431
Bergeron, Y., Deslauriers, A. M., Ouellet, N., Gauthier, M. C., and Bergeron, M. G. (1999). Influence of cefodizime on pulmonary inflammatory response to heat-killed Klebsiella pneumoniae in mice. Antimicrob. Agents Chemother. 43, 2291–2294.
Bozinovski, S., Jones, J. E., Vlahos, R., Hamilton, J. A., and Anderson, G. P. (2002). Granulocyte/macrophage-colony-stimulating factor (GM-CSF) regulates lung innate immunity to lipopolysaccharide through Akt/Erk activation of NFkappa B and AP-1 in vivo. J. Biol. Chem. 277, 42808–42814. doi: 10.1074/jbc.M207840200
Broug-Holub, E., Toews, G. B., van Iwaarden, J. F., Strieter, R. M., Kunkel, S. L., Paine, R. 3rd., et al. (1997). Alveolar macrophages are required for protective pulmonary defenses in murine Klebsiella pneumonia: elimination of alveolar macrophages increases neutrophil recruitment but decreases bacterial clearance and survival. Infect. Immun. 65, 1139–1146.
Bunt, S. K., Clements, V. K., Hanson, E. M., Sinha, P., and Ostrand-Rosenberg, S. (2009). Inflammation enhances myeloid-derived suppressor cell cross-talk by signaling through Toll-like receptor 4. J. Leukoc. Biol. 85, 996–1004. doi: 10.1189/jlb.0708446
Chalmin, F., Ladoire, S., Mignot, G., Vincent, J., Bruchard, M., Remy-Martin, J. P., et al. (2010). Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Invest. 120, 457–471. doi: 10.1172/JCI40483
Chan, Y. R., Liu, J. S., Pociask, D. A., Zheng, M., Mietzner, T. A., Berger, T., et al. (2009). Lipocalin 2 is required for pulmonary host defense against Klebsiella infection. J. Immunol. 182, 4947–4956. doi: 10.4049/jimmunol.0803282
Chauveau, C., Remy, S., Royer, P. J., Hill, M., Tanguy-Royer, S., Hubert, F. X., et al. (2005). Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood 106, 1694–1702. doi: 10.1182/blood-2005-02-0494
Cheng, P., Corzo, C. A., Luetteke, N., Yu, B., Nagaraj, S., Bui, M. M., et al. (2008). Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 205, 2235–2249. doi: 10.1084/jem.20080132
Condamine, T., and Gabrilovich, D. I. (2011). Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 32, 19–25. doi: 10.1016/j.it.2010.10.002
Craig, A., Mai, J., Cai, S., and Jeyaseelan, S. (2009). Neutrophil recruitment to the lungs during bacterial pneumonia. Infect. Immun. 77, 568–575. doi: 10.1128/IAI.00832-08
D'Alessio, F. R., Tsushima, K., Aggarwal, N. R., West, E. E., Willett, M. H., Britos, M. F., et al. (2009). CD4+CD25+Foxp3+ Tregs resolve experimental lung injury in mice and are present in humans with acute lung injury. J. Clin. Invest. 119, 2898–2913. doi: 10.1172/JCI36498
Debes, G. F., Arnold, C. N., Young, A. J., Krautwald, S., Lipp, M., Hay, J. B., et al. (2005). Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat. Immunol. 6, 889–894. doi: 10.1038/ni1238
Delano, M. J., Scumpia, P. O., Weinstein, J. S., Coco, D., Nagaraj, S., Kelly-Scumpia, K. M., et al. (2007). MyD88-dependent expansion of an immature GR-1(+)CD11b(+) population induces T cell suppression and Th2 polarization in sepsis. J. Exp. Med. 204, 1463–1474. doi: 10.1084/jem.20062602
Delayre-Orthez, C., Becker, J., de Blay, F., Frossard, N., and Pons, F. (2005). Exposure to endotoxins during sensitization prevents further endotoxin-induced exacerbation of airway inflammation in a mouse model of allergic asthma. Int. Arch. Allergy Immunol. 138, 298–304. doi: 10.1159/000088867
De Santo, C., Salio, M., Masri, S. H., Lee, L. Y., Dong, T., Speak, A. O., et al. (2008). Invariant NKT cells reduce the immunosuppressive activity of influenza A virus-induced myeloid-derived suppressor cells in mice and humans. J. Clin. Invest. 118, 4036–4048. doi: 10.1172/JCI36264
De Wilde, V., Van Rompaey, N., Hill, M., Lebrun, J. F., Lemaitre, P., Lhomme, F., et al. (2009). Endotoxin-induced myeloid-derived suppressor cells inhibit alloimmune responses via heme oxygenase-1. Am. J. Transplant. 9, 2034–2047. doi: 10.1111/j.1600-6143.2009.02757.x
Eisenbarth, S. C., Piggott, D. A., Huleatt, J. W., Visintin, I., Herrick, C. A., and Bottomly, K. (2002). Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J. Exp. Med. 196, 1645–1651. doi: 10.1084/jem.20021340
Filipazzi, P., Huber, V., and Rivoltini, L. (2012). Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol. Immunother. 61, 255–263. doi: 10.1007/s00262-011-1161-9
Gabrilovich, D. I., and Nagaraj, S. (2009). Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9, 162–174. doi: 10.1038/nri2506
Gabrilovich, D. I., Ostrand-Rosenberg, S., and Bronte, V. (2012). Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 12, 253–268. doi: 10.1038/nri3175
Gerhold, K., Avagyan, A., Reichert, E., Blumchen, K., Wahn, U., and Hamelmann, E. (2008). Lipopolysaccharides modulate allergen-specific immune regulation in a murine model of mucosal tolerance induction. Int. Arch. Allergy Immunol. 147, 25–34. doi: 10.1159/000128583
Gerhold, K., Blumchen, K., Bock, A., Seib, C., Stock, P., Kallinich, T., et al. (2002). Endotoxins prevent murine IgE production, T(H)2 immune responses, and development of airway eosinophilia but not airway hyperreactivity. J. Allergy Clin. Immunol. 110, 110–116. doi: 10.1067/mai.2002.125831
Greifenberg, V., Ribechini, E., Rossner, S., and Lutz, M. B. (2009). Myeloid-derived suppressor cell activation by combined LPS and IFN-gamma treatment impairs DC development. Eur. J. Immunol. 39, 2865–2876. doi: 10.1002/eji.200939486
Guha, M. (2012). Anticancer TLR agonists on the ropes. Nat. Rev. Drug Discov. 11, 503–505. doi: 10.1038/nrd3775
Hand, T. W., Cui, W., Jung, Y. W., Sefik, E., Joshi, N. S., Chandele, A., et al. (2010). Differential effects of STAT5 and PI3K/AKT signaling on effector and memory CD8 T-cell survival. Proc. Natl. Acad. Sci. U.S.A. 107, 16601–16606. doi: 10.1073/pnas.1003457107
Hu, H., Huston, G., Duso, D., Lepak, N., Roman, E., and Swain, S. L. (2001). CD4(+) T cell effectors can become memory cells with high efficiency and without further division. Nat. Immunol. 2, 705–710. doi: 10.1038/90643
Huang, B., Pan, P. Y., Li, Q., Sato, A. I., Levy, D. E., Bromberg, J., et al. (2006). Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 66, 1123–1131. doi: 10.1158/0008-5472.CAN-05-1299
Jamieson, A. M., Pasman, L., Yu, S., Gamradt, P., Homer, R. J., Decker, T., et al. (2013). Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science 34, 1230–1234. doi: 10.1126/science.1233632
Jeisy-Scott, V., Davis, W. G., Patel, J. R., Bowzard, J. B., Shieh, W. J., and Zaki, S. R. (2011). Increased MDSC accumulation and Th2 biased response to influenza A virus infection in the absence of TLR7 in mice. PLoS ONE 6:e25242. doi: 10.1371/journal.pone.0025242
Kawai, T., and Akira, S. (2010). The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 11, 373–384. doi: 10.1038/ni.1863
Kondo, T., Kawai, T., and Akira, S. (2012). Dissecting negative regulation of Toll-like receptor signaling. Trends Immunol. 33, 449–458. doi: 10.1016/j.it.2012.05.002
Lee, W. L., and Downey, G. P. (2001). Neutrophil activation and acute lung injury. Curr. Opin. Crit. Care 7, 1–7. doi: 10.1097/00075198-200102000-00001
Lemaitre, B., Nicolas, E., Michaut, L., Reichhart, J. M., and Hoffmann, J. A. (1996). The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86, 973–983. doi: 10.1016/S0092-8674(00)80172-5
Liew, F. Y., Xu, D., Brint, E. K., and O'Neill, L. A. (2005). Negative regulation of toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 5, 446–458. doi: 10.1038/nri1630
Lu, H., Wagner, W. M., Gad, E., Yang, Y., Duan, H., Amon, L. M., et al. (2010). Treatment failure of a TLR-7 agonist occurs due to self-regulation of acute inflammation and can be overcome by IL-10 blockade. J. Immunol. 184, 5360–5367. doi: 10.4049/jimmunol.0902997
Martino, A., Badell, E., Abadie, V., Balloy, V., Chignard, M., Mistou, M. Y., et al. (2010). Mycobacterium bovis bacillus Calmette-Guerin vaccination mobilizes innate myeloid-derived suppressor cells restraining in vivo T cell priming via IL-1R-dependent nitric oxide production. J. Immunol. 184, 2038–2047. doi: 10.4049/jimmunol.0903348
Massberg, S., Schaerli, P., Knezevic-Maramica, I., Kollnberger, M., Tubo, N., Moseman, E. A., et al. (2007). Immunosurveillance by hematopoietic progenitor cells trafficking through blood, lymph, and peripheral tissues. Cell 131, 994–1008. doi: 10.1016/j.cell.2007.09.047
Medzhitov, R., Preston-Hurlburt, P., and Janeway, C. A. Jr. (1997). A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388, 394–397. doi: 10.1038/41131
Mizgerd, J. P. (2006). Lung infection–a public health priority. PLoS Med. 3:e76. doi: 10.1371/journal.pmed.0030076
Mizgerd, J. P. (2008). Acute lower respiratory tract infection. N. Engl. J. Med. 358, 716–727. doi: 10.1056/NEJMra074111
Nagai, Y., Garrett, K. P., Ohta, S., Bahrun, U., Kouro, T., Akira, S., et al. (2006). Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 24, 801–812. doi: 10.1016/j.immuni.2006.04.008
Nagaraj, S., Youn, J. I., and Gabrilovich, D. I. (2013). Reciprocal Relationship between Myeloid-Derived Suppressor Cells and T Cells. J. Immunol. 191, 17–23. doi: 10.4049/jimmunol.1300654
Nefedova, Y., Huang, M., Kusmartsev, S., Bhattacharya, R., Cheng, P., Salup, R., et al. (2004). Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J. Immunol. 172, 464–474.
Neuhauser, M. M., Weinstein, R. A., Rydman, R., Danziger, L. H., Karam, G., and Quinn, J. P. (2003). Antibiotic resistance among gram-negative bacilli in US intensive care units: implications for fluoroquinolone use. JAMA 289, 885–888. doi: 10.1001/jama.289.7.885
Oriss, T. B., Ostroukhova, M., Seguin-Devaux, C., Dixon-McCarthy, B., Stolz, D. B., Watkins, S. C., et al. (2005). Dynamics of dendritic cell phenotype and interactions with CD4+ T cells in airway inflammation and tolerance. J. Immunol. 174, 854–863.
Ortega-Gomez, A., Perretti, M., and Soehnlein, O. (2013). Resolution of infllammation:an integrated view. EMBO Mol. Med. 5, 661–674. doi: 10.1002/emmm.201202382
Ostrand-Rosenberg, S., and Sinha, P. (2009). Myeloid-derived suppressor cells: linking inflammation and cancer. J. Immunol. 182, 4499–4506. doi: 10.4049/jimmunol.0802740
Peranzoni, E., Zilio, S., Marigo, I., Dolcetti, L., Zanovello, P., Mandruzzato, S., et al. (2010). Myeloid-derived suppressor cell heterogeneity and subset definition. Curr. Opin. Immunol. 22, 238–244. doi: 10.1016/j.coi.2010.01.021
Pereira, W. F., Ribeiro-Gomes, F. L., Guillermo, L. V., Vellozo, N. S., Montalvao, F., and Dosreis, G. A. (2011). Myeloid-derived suppressor cells help protective immunity to Leishmania major infection despite suppressed T cell responses. J. Leukoc. Biol. 90, 1191–1197. doi: 10.1189/jlb.1110608
Poe, S. L., Arora, M., Oriss, T. B., Yarlagadda, M., Isse, K., Khare, A., et al. (2013). STAT1-regulated lung MDSC-like cells produce IL-10 and efferocytose apoptotic neutrophils with relevance in resolution of bacterial pneumonia. Mucosal Immunol. 6, 189–199. doi: 10.1038/mi.2012.62
Poltorak, A., He, X., Smirnova, I., Liu, M. Y., Van Huffel, C., Du, X., et al. (1998). Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088. doi: 10.1126/science.282.5396.2085
Qureshi, S. T., Lariviere, L., Leveque, G., Clermont, S., Moore, K. J., Gros, P., et al. (1999). Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). J. Exp. Med. 189, 615–625. doi: 10.1084/jem.189.4.615
Ricchetti, G. A., Williams, L. M., and Foxwell, B. M. (2004). Heme oxygenase 1 expression induced by IL-10 requires STAT-3 and phosphoinositol-3 kinase and is inhibited by lipopolysaccharide. J. Leukoc. Biol. 76, 719–726. doi: 10.1189/jlb.0104046
Ridnour, L. A., Cheng, R. Y., Switzer, C. H., Heinecke, J. L., Ambs, S., Glynn, S., et al. (2013). Molecular pathways: toll-like receptors in the tumor microenvironment–poor prognosis or new therapeutic opportunity. Clin. Cancer Res. 19, 1340–1346. doi: 10.1158/1078-0432.CCR-12-0408
Rieber, N., Brand, A., Hector, A., Graepler-Mainka, U., Ost, M., Schafer, I., et al. (2012). Flagellin induces myeloid-derived suppressor cells:implications for cystic fibrosis infection in cystic fibrosis lung disease. J. Immunol. 190, 1276–1284. doi: 10.4049/jimmunol.1202144
Rittirsch, D., Flierl, M. A., and Ward, P. A. (2008). Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 8, 776–787. doi: 10.1038/nri2402
Rodriguez, D., Keller, A. C., Faquim-Mauro, E. L., de Macedo, M. S., Cunha, F. Q., Lefort, J., et al. (2003). Bacterial lipopolysaccharide signaling through Toll-like receptor 4 suppresses asthma-like responses via nitric oxide synthase 2 activity. J. Immunol. 171, 1001–1008.
Rodriguez, P. C., Hernandez, C. P., Quiceno, D., Dubinett, S. M., Zabaleta, J., Ochoa, J. B., et al. (2005). Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J. Exp. Med. 202, 931–939. doi: 10.1084/jem.20050715
Rotta, G., Edwards, E. W., Sangaletti, S., Bennett, C., Ronzoni, S., Colombo, M. P., et al. (2003). Lipopolysaccharide or whole bacteria block the conversion of inflammatory monocytes into dendritic cells in vivo. J. Exp. Med. 198, 1253–1263. doi: 10.1084/jem.20030335
Sander, L. E., Sackett, S. D., Dierssen, U., Beraza, N., Linke, R. P., and Muller, M. (2010). Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J. Exp. Med. 207, 1453–1464. doi: 10.1084/jem.20091474
Serafini, P., Mgebroff, S., Noonan, K., and Borrello, I. (2008). Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 68, 5439–5449. doi: 10.1158/0008-5472.CAN-07-6621
Soehnlein, O., and Lindbom, L. (2010). Phagocyte partnership during the onset and resolution of inflammation. Nat. Rev. Immunol. 10, 427–439. doi: 10.1038/nri2779
Srivastava, M. K., Sinha, P., Clements, V. K., Rodriguez, P., and Ostrand-Rosenberg, S. (2010). Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 70, 68–77. doi: 10.1158/0008-5472.CAN-09-2587
Tsai, K. S., and Grayson, M. H. (2008). Pulmonary defense mechanisms against pneumonia and sepsis. Curr. Opin. Pulm. Med. 14, 260–265. doi: 10.1097/MCP.0b013e3282f76457
Van Ginderachter, J. A., Beschin, A., De Baetselier, P., and Raes, G. (2010). Myeloid-derived suppressor cells in parasitic infections. Eur. J. Immunol. 40, 2976–2985. doi: 10.1002/eji.201040911
Ware, L. B., and Matthay, M. A. (2000). The acute respiratory distress syndrome. N. Engl. J. Med. 342, 1334–1349. doi: 10.1056/NEJM200005043421806
Wieland, C. W., van Lieshout, M. H., Hoogendijk, A. J., and van der Poll, T. (2011). Host defense during Klebsiella pneumonia relies on hematopoietic expressed TLR4 and TLR2. Eur. Respir. J. 37, 848–857. doi: 10.1183/09031936.00076510
Wofford, J. A., Wieman, H. L., Jacobs, S. R., Zhao, Y., and Rathmell, J. C. (2008). IL-7 promotes Glut1 trafficking and glucose uptake via STAT5-mediated activation of Akt to support T-cell survival. Blood 111, 2101–2111. doi: 10.1182/blood-2007-06-096297
Yang, L., Huang, J., Ren, X., Gorska, A. E., Chytil, A., Aakre, M., et al. (2008). Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 13, 23–35. doi: 10.1016/j.ccr.2007.12.004
Zhang, D. H., Cohn, L., Ray, P., Bottomly, K., and Ray, A. (1997). Transcription factor GATA-3 is differentially expressed in murine Th1 and Th2 cells and controls Th2-specific expression of the interleukin-5 gene. J. Biol. Chem. 272, 21597–21603. doi: 10.1074/jbc.272.34.21597
Zheng, W., and Flavell, R. A. (1997). The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89, 587–596. doi: 10.1016/S0092-8674(00)80240-8
Keywords: TLR, LPS, bacteria, MDSC, pneumonia, lung, resolution, inflammation
Citation: Ray A, Chakraborty K and Ray P (2013) Immunosuppressive MDSCs induced by TLR signaling during infection and role in resolution of inflammation. Front. Cell. Infect. Microbiol. 3:52. doi: 10.3389/fcimb.2013.00052
Received: 31 July 2013; Accepted: 27 August 2013;
Published online: 18 September 2013.
Edited by:
Nelson Gekara, Umea University, SwedenReviewed by:
Syed R. Ali, University of California, San Diego, USAAndy Wullaert, Ghent University, Belgium
Copyright © 2013 Ray, Chakraborty and Ray. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anuradha Ray and Prabir Ray, Division of Pulmonary, Allergy and Critical Care Medicine, Department of Medicine, University of Pittsburgh School of Medicine, 3459 Fifth Avenue, MUH A628 NW, Pittsburgh, PA 15213, USA e-mail:cmF5YUBwaXR0LmVkdQ==;cmF5cEBwaXR0LmVkdQ==