 Erdem Bangi
Erdem Bangi- Department of Developmental and Regenerative Biology, Icahn School of Medicine at Mount Sinai, New York, NY, USA
Recent studies show that both cellular and humoral aspects of innate immunity play important roles during tumor progression. These interactions have traditionally been explored in vertebrate model systems. In recent years, Drosophila has emerged as a genetically tractable model system for studying key aspects of tumorigenesis including proliferation, invasion, and metastasis. The absence of adaptive immunity in Drosophila provides a unique opportunity to study the interactions between innate immune system and cancer in different genetic contexts. In this review, I discuss recent advances made by using Drosophila models of cancer to study the role of innate immune pathways Toll/Imd, JNK, and JAK-STAT, microbial infection and inflammation during tumor progression.
The interaction between the tumor and the immune system is a complex, multi-step process in which both innate and adaptive branches of the immune system participate (Finn, 2012). The outcome of this antitumor response is variable and unpredictable; it can be tumor suppressive or tumor promoting depending on the immunogenicity and genetic composition of the tumor and the strength of patient's immune response (Finn, 2012). Several recent studies report that at least some aspects of the relationship between the immune system and cancer are also conserved in flies (Pastor-Pareja et al., 2008; Apidianakis et al., 2009; Cordero et al., 2010; Bangi et al., 2012): Drosophila immune system also recognizes and responds to tumors and this response can be tumor promoting or tumor suppressive depending on the genetic composition of the tumor. Here, I briefly summarize studies that use Drosophila to explore the role of the innate immune system during tumor progression.
Inflammation, Tumor Associated Hemocytes (TAHs), and Invasion
The first potential link between cancer and inflammation was proposed 2000 years ago, when the Roman physician Galenos suggested that cancers evolved from inflammatory lesions (Trinchieri, 2012). The first experimental evidence supporting this remarkable observation would not emerge until 1863, when German scientist and physician Rudolf Virchow observed that leukocytes were associated with neoplastic tissues, re-establishing this forgotten link between cancer and inflammation (Balkwill and Mantovani, 2001). Now, it is a well-established fact that inflammation impacts every aspect of of tumor development and progression (Trinchieri, 2012).
An early step in the anti-tumor response is the recruitment of macrophages and other blood cells mediating the innate immune response to the tumor site (Finn, 2012). These cells phagocytose tumor cells and secrete inflammatory cytokines to both maintain the innate immune response and promote and support activation of the adaptive immune response (Finn, 2012). While the Drosophila immune system shows evidence of some primed responses (Kvell et al., 2007), flies lack adaptive immunity as we know it in mammals. However, both the cellular and humoral aspects of the innate immune response and the pathways that mediate them are highly conserved (Hoffmann et al., 1999).
The cellular arm of the Drosophila immune response consists of circulating blood cells called hemocytes. There are three morphologically distinct types of hemocytes in Drosophila that share a common developmental and evolutionary origin with mammalian blood cells (Hartenstein, 2006). Plasmatocytes are the most common hemocyte type in Drosophila, comprising more than 95% of all hemocytes. Plasmatocytes resemble mammalian phagocytes and like macrophages, they are recruited to sites of infections or wounds to phagocytose apoptotic cells, invading microbes, and other foreign bodies (Tepass et al., 1994; Franc et al., 1999; Elrod-Erickson et al., 2000). Like their mammalian counterparts, Drosophila hemocytes are also recruited to epithelial tumors (Pastor-Pareja et al., 2008). Epithelial tumors are often established in Drosophila by generating patches of epithelial cells (clones) mutant for apical/basal polarity genes such as scrib (scr), lethal giant larvae (lgl), or discs large (dlg) while also expressing the oncogenic form of Drosophila dRas1 (e.g., scrib−/− dRas1V12) (Pastor-Pareja et al., 2008; Gonzalez, 2013). Cells mutant for apical/basal polarity genes alone are quickly eliminated from the epithelium by apoptosis in a JNK dependent manner (Rudrapatna et al., 2012). However, co-expressing dRas1V12 in these polarity-defective cells leads to invasive tumors as JNK pathway activation in these tumors promotes MMP expression, basement membrane degradation and invasion instead of apoptosis (Brumby and Richardson, 2003; Pagliarini and Xu, 2003).
Using a scrib−/− dRas1V12 tumor model, Pastor-Pareja and collegues showed that hemocytes infiltrate epithelial tumors in Drosophila (Pastor-Pareja et al., 2008). Tumor bearing animals also show increased numbers of circulating hemocytes and enlarged lymph glands as a result of increased hemocyte proliferation. Interestingly, this anti-tumor response is remarkably similar to the immune response to experimentally induced aseptic wounds, consistent with the idea that tumors are like wounds that never heal (Dvorak, 1986).
The mechanism by which hemocytes are recruited to tumors is not clear. However, Tumor Associated Hemocytes (TAHs) are preferentially found in the regions of the tumor where the basement membrane is disrupted (Pastor-Pareja et al., 2008). Basement membrane disruption in the absence of tumors by overexpression of MMP2 is sufficient to induce hemocyte recruitment but not proliferation, indicating that basement membrane break-down is only one of the signals mediating this immune response.
Local activation of JNK signaling in the tumor cells is critical for the maintenance of the anti-tumor response (Pastor-Pareja et al., 2008). JNK signaling promotes the secretion of JAK-STAT activating cytokines (Upd ligands) from the tumor; this initiates a positive feedback loop that activates upd expression in hemocytes and the fat body (also the site of antimicrobial peptide expression and release in response to infection). The increased JAK-STAT pathway activity in the hemocytes is required to induce hemocyte proliferation in response to tumors.
Tumor Necrosis Factor (TNF) signaling is another critical component of the inflammatory response activated in response to microbial infection, tissue damage and malignant cells (Waters et al., 2013a). While both tumor suppressive and tumor promoting roles for this pathway have been well established, the molecular mechanisms mediating these different responses are less clear (Waters et al., 2013b). Drosophila has a highly conserved but simplified TNF pathway with a single TNF ligand called Eiger (Egr) (Igaki et al., 2002; Moreno et al., 2002). Removal of scrib−/− or lgl−/− cells from the epithelium also requires TNF/Eiger indicating a conserved role for TNF signaling as a tumor suppressor pathway in Drosophila within these genetic contexts (Igaki et al., 2009; Cordero et al., 2010).
Cordero and collegues showed that hemocyte attachment and infiltration of tumors provoke tumor cells to induce high levels of egr expression in TAHs (Cordero et al., 2010). By transfusing hemocytes into tumor bearing larvae with egr mutant or wildtype hemocytes, Cordero and collegues show that egr expression in TAHs is required to induce JNK signaling and MMP expression in tumor cells and that these defects can be partially rescued by transfusing animals with egr+/+ hemocytes. Most importantly, removing egr from TAH's has drastically different consequences on tumors with different genotypes: scrib−/− tumors cannot be eliminated from the tissue without egr+/+ hemocytes, indicating a tumor suppressive role for TAHs and TNF signaling in this genetic context (Figures 1A,C). In contrast, Egr signaling from the TAHs is essential for scrib−/− dRas1V12 cells to become invasive tumors (Figures 1B,C) indicating a tumor promoting role for this pathway in this genetic context.
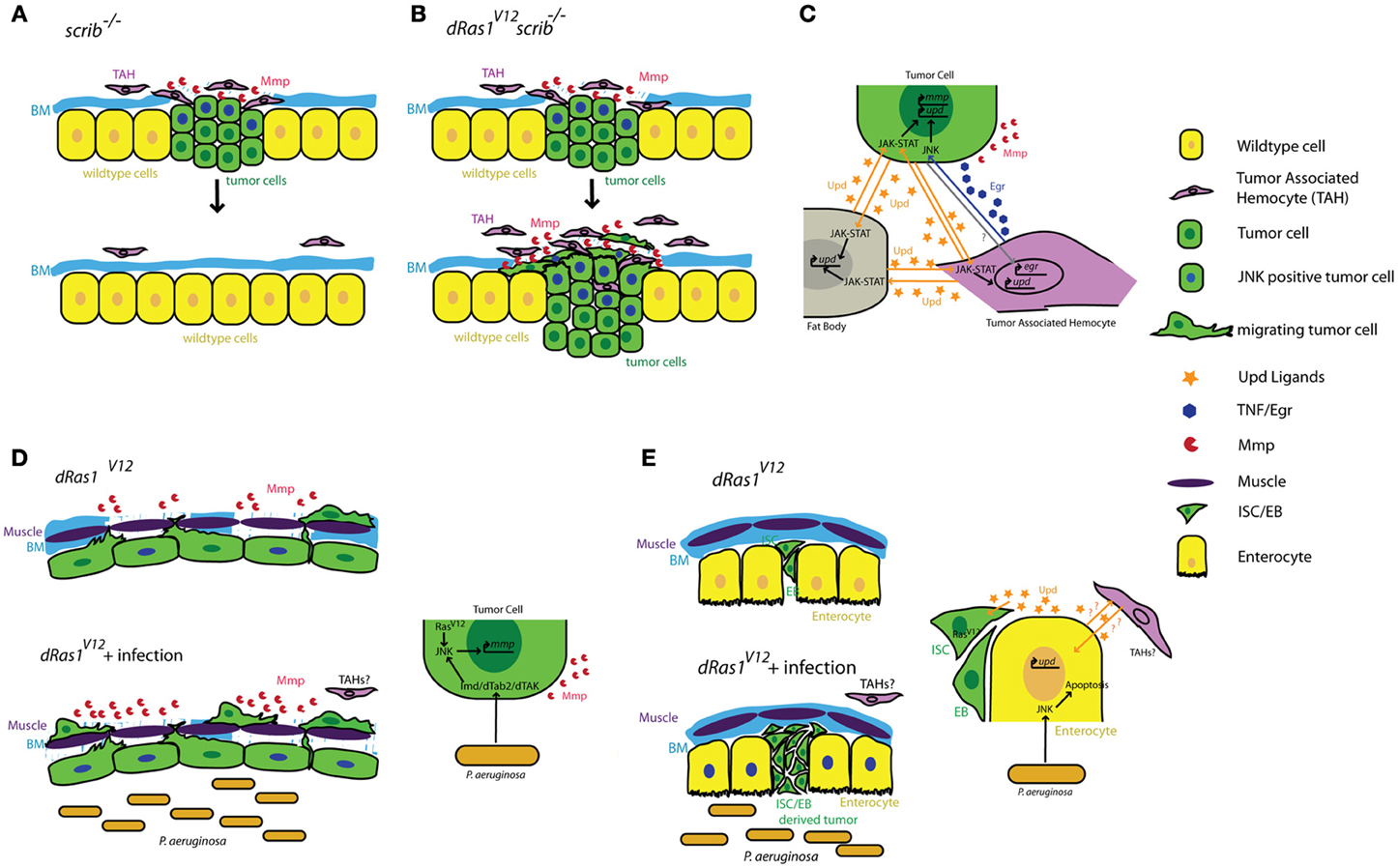
Figure 1. Summary of tumor cell-immune system interactions in Drosophila. (A) Clones of cells defective for cell polarity genes such as scrib (green) generated in the context of a wild type tissue (yellow) are rapidly eliminated from the epithelium with the help of tumor associated hemocytes (TAHs, purple). (B) This antitumor immune response is subverted in scrib−/− dRas1V12 cells, leading to the establishment of an invasive tumor. (C) Reciprocal interactions between tumor cells (green) TAHs (purple), and fat body (gray) result in both local and systemic release of inflammatory cytokines in a positive feedback loop. (D) Microbial infection synergizes with dRas1V12 to induce activation of JNK signaling (blue nuclei) and Mmp expression (red) in transformed hindgut epithelial cells, leading to migration and dissemination. (E) Microbial infection induces apoptosis of differentiated enterocytes (yellow), and synergizes with with dRas1V12 to induce hyperplastic ISC/EB-like tumors. BM, Basement Membrane; Mmp, Matrix Metalloprotease; ISC, Intestinal Stem Cell; EB, Enteroblast.
Activation of JNK signaling and induction of MMP1 expression are a part of the normal immune response to facilitate delamination of abnormal cells from the epithelium and promote further infiltration of the wound or infection by hemocytes. As both JNK and TNF pathways are strong inducers of cell death, these MMP expressing cells are normally quickly eliminated by apoptosis to ensure tissue integrity. However, these studies suggest that if these JNK/MMP1 positive tumor cells persist long enough in the tissue, for instance as a result of additional mutations that prevent apoptosis, they can further promote degradation of the basement membrane and infiltration by additional TAHs. This in turn leads to a positive feedback loop that increases the number of JNK/MMP positive cells within the tumor and thereby its metastatic potential (Figures 1A–C).
Aspects of bacterial infection can also be studied by directly expressing pathogen-derived proteins in host tissues. For instance, Drosophila models of H. pylori infection have been generated by expressing the H. pylori virulence factor CagA in Drosophila tissues (Botham et al., 2008; Wandler and Guillemin, 2012). Certain virulent strains of H. pylori possess a secretion system that allows them to directly inject the CagA protein into gastric epithelial cells and can promote the development of gastric carcinoma in a small percentage of infected individuals (Peek and Blaser, 2002; Hatakeyama, 2008; Wroblewski et al., 2010). Wandler and Guillemin showed that CagA expression in discrete domains in the Drosophila wing disc epithelium leads to the activation of apoptosis in a subset of CagA expressing cells in a JNK signaling dependent fashion (Wandler and Guillemin, 2012). Interestingly, loss of egr function in the whole animal increased the number of apoptotic CagA expressing cells, but not when egr was only reduced in CagA expressing cells. This suggests a non-cell-autonomous role for Egr in apoptotic cell clearance. The authors propose a model whereby loss of Egr from the neighboring wildtype epithelial cells mediate elimination of apoptotic CagA expressing cells from the epithelium. CagA expression also synergized with oncogenic Ras to facilitate JNK mediated tumor progression and invasion, however, the role of Egr in this context has not been investigated. Furthermore, potential roles for the core immune signaling pathways and the cellular immune response in this process remain unexplored possibilities. It will be interesting to see if hemocytes also associate with tumors in this paradigm and whether similar pro-tumor and anti-tumor roles for Egr/TNF signaling can be elucidated.
Toll/Imd Signaling, Microbial Infection, and Cancer
Recognition or pathogen and damage associated molecular patterns by the immune system is a key component of mounting an effective host defense. In Drosophila, this innate immune response is mediated by two pathways: Recognition of Gram positive bacteria and fungi depends on secreted factors that regulate the processing and activation of the Toll receptor ligand Spatzle (Spz) (Lemaitre et al., 1996). Subsequent activation of the Toll pathway leads to the expression and secretion of antimicrobial peptides (AMPs) mediated by NFκB related molecules Dorsal and Dif (Valanne et al., 2011). On the other hand, Gram negative bacteria are recognized by pattern recognition receptors called PGRPs, ultimately leading to activation of another NFκB related molecule called Relish as well as JNK pathway in an Imd dependent fashion (Choe et al., 2002; Ramet et al., 2002; Kallio et al., 2005). In mammals, Toll Related Receptor (TLR) signaling is activated by direct binding of pathogen associated molecules, leading to NFκB-mediated induction of AMP expression (Takeuchi and Akira, 2010). In addition, pathogen associated peptidoglycan fragments are recognized by NOD-like Receptors (NLRs), which leads to activation of NFκB and JNK pathways (Lavelle et al., 2010). Even though there are some differences in the activation mechanisms of these pathways, most of the downstream pathway components and their roles are highly conserved between mammals and Drosophila (see reference 30 for an in-depth comparative analysis).
Stimulation of innate immune responses by microbial components can also modulate migratory potential of epithelial cells (Wang et al., 2003; Merrell et al., 2006) and recent identification of functionally active TLRs in several tumor cell lines point to important roles for TLR signaling in epithelial tumor progression and metastasis (Huang et al., 2005, 2008; Kelly et al., 2006; Rakoff-Nahoum and Medzhitov, 2009). In recent years several groups took advantage of the high degree of conservation of core immune signaling pathways in Drosophila to explore the relationship between innate immune responses and tumor progression.
The gastrointestinal tract is a prominent component of both mammalian and Drosophila immune systems. The intestinal epithelium expresses several TLRs and studies both in murine models and in Drosophila reveal that intestinal epithelial cells respond to microbial infection by secreting AMPs, a Toll/Imd/TLR signaling mediated process (O'Neil et al., 1999; Apidianakis et al., 2005). Interestingly, chronic activation of the immune response is thought to facilitate intestinal tumorigenesis in genetically predisposed individuals (Pasparakis, 2008; Secher et al., 2010), again suggesting a pro-tumorigenic role for Toll/Imd/TLR signaling in the intestine. We found that acute activation of the Imd pathway in response to microbial infection interacts with pre-existing oncogenic mutations to promote tumorigenesis in a dRas1V12 induced model of colon cancer in Drosophila (Bangi et al., 2012). When targeted to the hindgut epithelium—the functional equivalent of the mammalian colon—dRas1V12 activates JNK signaling and MMP expression in a subset of the hindgut epithelial cells. These transformed cells eventually migrate out of the epithelium to colonize distant sites within the animal. While JNK/MMP positive cells do not migrate themselves, both JNK signaling and MMP expression is necessary for the dissemination phenotype. Microbial infection of these animals using a previously established infection paradigm by oral feeding of the Gram negative bacterium Pseudomonas aeruginosa (Apidianakis and Rahme, 2009, 2011) leads to a significant enhancement of dRas1V12 induced dissemination in an Imd dependent fashion. Microbial infection in this case increases the metastatic potential of the tumor by increasing the number of JNK/MMP1 positive cells, thereby further compromising the integrity of the tissue and facilitating the migration of dRas1V12 transformed cells (Figure 1D).
By contrast in the midgut, microbial infection synergizes with dRas1V12 to induce intestinal hyperplasia but not invasion or dissemination; in this model, dRas1V12 was targeted to intestinal stem cells (ISCs) and undifferentiated enteroblasts (EBs), the immediate progeny of ISCs (Apidianakis and Rahme, 2009; Pitsouli et al., 2009) (Figure 1E). Hyperplasia is driven by bacteria-induced death of differentiated midgut cells. Curiously, JNK induced secretion of JAK-STAT inducing cytokines (Upd-1, -2, -3) by the dying midgut cells is known to be a key mediator of tissue regeneration (Jiang et al., 2009), reminiscent of the positive feedback loop created between TAHs and tumor cells in the imaginal disc tumor models discussed above (Pastor-Pareja et al., 2008). Adult hemocytes have been reported to respond to microbial infection by phagocytosing invading pathogens in multiple infection paradigms (Elrod-Erickson et al., 2000; Kocks et al., 2005; Nehme et al., 2007). However, there is no evidence that they infiltrate the adult gut as part of the immune response and whether they contribute to hyperplasia and dissemination phenotypes in these intestinal cancer models have not been investigated.
Antiviral Immunity and Cancer
In addition to bacterial and fungal infection paradigms, several Drosophila models of viral infection also exist; these include models that use natural viruses that infect Drosophila as well as several viruses that cause disease in humans and those that directly express various viral proteins in Drosophila tissues (Bier and Guichard, 2012; Merkling and van Rij, 2013). The major immune defense against viral infection in insects is the RNA interference pathway, however, several recent reports indicate possible roles for the evolutionarily conserved core immune signaling pathways Toll, Imd, and JAK-STAT in antiviral immunity (Dostert et al., 2005; Zambon et al., 2005; Costa et al., 2009). It would be interesting to combine these viral infection models with available Drosophila cancer models to explore interactions between viral infection, antiviral immunity and cancer.
Drosophila Offers New Tools to Explore Links between Immunology and Cancer
The presence of an antitumor immune response in Drosophila opens up new avenues of research in the field of tumor immunology. The absence of an adaptive immune response precludes modeling certain aspects of immune response. However, signaling pathways that mediate the interactions between tumor cells and the innate immune system (JNK, JAK-STAT, TNF, Toll/Imd/TLR) as well as the way these pathways interact with each other are highly conserved in flies.
The sophisticated genetic tools available in Drosophila can be used for genetic dissection of conserved aspects of the anti-tumor immune response. For instance, multiple independent targeted and inducible expression systems are available in Drosophila (del Valle Rodriguez et al., 2012), making it possible to separately label and genetically manipulate tumor cells and cells of the immune system. An increasing number of genetically complex tumor models are being reported in Drosophila (Gonzalez, 2013). For instance, 30 multigenic models of colon cancer in the adult Drosophila gut have recently been generated and characterized in our laboratory (Bangi et. al., in review). These models allow us to explore the mechanisms by which the innate immune system reacts to tumors with different genetic compositions.
Lastly, Drosophila is emerging as a useful platform for cancer drug discovery: flies provide a high degree of conservation of cancer relevant pathways as well as appropriate sensitivity to compounds targeting these pathways (Bangi et al., 2011; Gonzalez, 2013). Compound screens in Drosophila using organismal lethality or other complex phenotypic read outs of cancer are revealing new anti-cancer agents with promising activity in mammalian models (Dar et al., 2012). With these tools, Drosophila can be useful both as a genetic model system for tumor immunology but also as a drug discovery platform to screen for compounds that target the immune system and its interactions with tumor cells.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I would like to thank Ross Cagan for his feedback on the manuscript. Erdem Bangi is supported by grants R01-CA109730 and R01-EY011495.
References
Apidianakis, Y., Mindrinos, M. N., Xiao, W., Lau, G. W., Baldini, R. L., Davis, R. W., et al. (2005). Profiling early infection responses: pseudomonas aeruginosa eludes host defenses by suppressing antimicrobial peptide gene expression. Proc. Natl. Acad. Sci. U.S.A. 102, 2573–2578. doi: 10.1073/pnas.0409588102
Apidianakis, Y., Pitsouli, C., Perrimon, N., and Rahme, L. (2009). Synergy between bacterial infection and genetic predisposition in intestinal dysplasia. Proc. Natl. Acad. Sci. U.S.A. 106, 20883–20888. doi: 10.1073/pnas.0911797106
Apidianakis, Y., and Rahme, L. G. (2009). Drosophila melanogaster as a model host for studying Pseudomonas aeruginosa infection. Nat. Protoc. 4, 1285–1294. doi: 10.1038/nprot.2009.124
Apidianakis, Y., and Rahme, L. G. (2011). Drosophila melanogaster as a model for human intestinal infection and pathology. Dis. Model. Mech. 4, 21–30. doi: 10.1242/dmm.003970
Balkwill, F., and Mantovani, A. (2001). Inflammation and cancer: back to Virchow? Lancet 357, 539–545. doi: 10.1016/S0140-6736(00)04046-0
Bangi, E., Garza, D., and Hild, M. (2011). In vivo analysis of compound activity and mechanism of action using epistasis in Drosophila. J. Chem. Biol. 4, 55–68. doi: 10.1007/s12154-010-0051-5
Bangi, E., Pitsouli, C., Rahme, L. G., Cagan, R., and Apidianakis, Y. (2012). Immune response to bacteria induces dissemination of Ras-activated Drosophila hindgut cells. EMBO Rep. 13, 569–576. doi: 10.1038/embor.2012.44
Bier, E., and Guichard, A. (2012). Deconstructing host-pathogen interactions in Drosophila. Dis. Model. Mech. 5, 48–61. doi: 10.1242/dmm.000406
Botham, C. M., Wandler, A. M., and Guillemin, K. (2008). A transgenic Drosophila model demonstrates that the Helicobacter pylori CagA protein functions as a eukaryotic Gab adaptor. PLoS Pathog. 4:e1000064. doi: 10.1371/journal.ppat.1000064
Brumby, A. M., and Richardson, H. E. (2003). Scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 22, 5769–5779. doi: 10.1093/emboj/cdg548
Choe, K. M., Werner, T., Stoven, S., Hultmark, D., and Anderson, K. V. (2002). Requirement for a peptidoglycan recognition protein (PGRP) in Relish activation and antibacterial immune responses in Drosophila. Science 296, 359–362. doi: 10.1126/science.1070216
Cordero, J. B., Macagno, J. P., Stefanatos, R. K., Strathdee, K. E., Cagan, R. L., and Vidal, M. (2010). Oncogenic ras diverts a host TNF Tumor suppressor activity into Tumor promoter. Dev. Cell 18, 999–1011. doi: 10.1016/j.devcel.2010.05.014
Costa, A., Jan, E., Sarnow, P., and Schneider, D. (2009). The Imd pathway is involved in antiviral immune responses in Drosophila. PLoS ONE 4:e7436. doi: 10.1371/journal.pone.0007436
Dar, A. C., Das, T. K., Shokat, K. M., and Cagan, R. L. (2012). Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature 486, 80–84. doi: 10.1038/nature11127
del Valle Rodriguez, A., Didiano, D., and Desplan, C. (2012). Power tools for gene expression and clonal analysis in Drosophila. Nat. Methods 9, 47–55. doi: 10.1038/nmeth.1800
Dostert, C., Jouanguy, E., Irving, P., Troxler, L., Galiana-Arnoux, D., Hetru, C., et al. (2005). The Jak-STAT signaling pathway is required but not sufficient for the antiviral response of drosophila. Nat. Immunol. 6, 946–953. doi: 10.1038/ni1237
Dvorak, H. F. (1986). Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N.Engl. J. Med. 315, 1650–1659. doi: 10.1056/NEJM198612253152606
Elrod-Erickson, M., Mishra, S., and Schneider, D. (2000). Interactions between the cellular and humoral immune responses in Drosophila. Curr. Biol. 10, 781–784. doi: 10.1016/S0960-9822(00)00569-8
Finn, O. J. (2012). Immuno-oncology: understanding the function and dysfunction of the immune system in cancer. Ann. Oncol. 23 (Suppl. 8), viii6–viii 9. doi: 10.1093/annonc/mds256
Franc, N. C., Heitzler, P., Ezekowitz, R. A., and White, K. (1999). Requirement for croquemort in phagocytosis of apoptotic cells in Drosophila. Science 284, 1991–1994. doi: 10.1126/science.284.5422.1991
Gonzalez, C. (2013). Drosophila melanogaster: a model and a tool to investigate malignancy and identify new therapeutics. Nat. Rev. Cancer 13, 172–183. doi: 10.1038/nrc3461
Hartenstein, V. (2006). Blood cells and blood cell development in the animal kingdom. Annu. Rev. Cell Dev. Biol. 22, 677–712. doi: 10.1146/annurev.cellbio.22.010605.093317
Hatakeyama, M. (2008). SagA of CagA in Helicobacter pylori pathogenesis. Curr. Opin. Microbiol. 11, 30–37. doi: 10.1016/j.mib.2007.12.003
Hoffmann, J. A., Kafatos, F. C., Janeway, C. A., and Ezekowitz, R. A. (1999). Phylogenetic perspectives in innate immunity. Science 284, 1313–1318. doi: 10.1126/science.284.5418.1313
Huang, B., Zhao, J., Li, H., He, K. L., Chen, Y., Chen, S. H., et al. (2005). Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res. 65, 5009–5014. doi: 10.1158/0008-5472.CAN-05-0784
Huang, B., Zhao, J., Unkeless, J. C., Feng, Z. H., and Xiong, H. (2008) TLR signaling by tumor and immune cells: a double-edged sword. Oncogene 27, 218–224. doi: 10.1038/sj.onc.1210904
Igaki, T., Kanda, H., Yamamoto-Goto, Y., Kanuka, H., Kuranaga, E., Aigaki, T., et al. (2002). Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 21, 3009–3018. doi: 10.1093/emboj/cdf306
Igaki, T., Pastor-Pareja, J. C., Aonuma, H., Miura, M., and Xu, T. (2009). Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev. Cell 16, 458–465. doi: 10.1016/j.devcel.2009.01.002
Jiang, H., Patel, P. H., Kohlmaier, A., Grenley, M. O., McEwen, D. G., and Edgar, B. A. (2009). Cytokine/Jak/Stat signaling mediates regeneration and homeostasis in the Drosophila Midgut. Cell 137, 1343–1355. doi: 10.1016/j.cell.2009.05.014
Kallio, J., Leinonen, A., Ulvila, J., Valanne, S., Ezekowitz, R. A., and Ramet, M. (2005). Functional analysis of immune response genes in Drosophila identifies JNK pathway as a regulator of antimicrobial peptide gene expression in S2 cells. Microbes Infect. 7, 811–819. doi: 10.1016/j.micinf.2005.03.014
Kelly, M. G., Alvero, A. B., Chen, R., Silasi, D. A., Abrahams, V. M., Chan, S., et al. (2006). TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 66, 3859–3868. doi: 10.1158/0008-5472.CAN-05-3948
Kocks, C., Cho, J. H., Nehme, N., Ulvila, J., Pearson, A. M., Meister, M., et al. (2005). Eater, a transmembrane protein mediating phagocytosis of bacterial pathogens in Drosophila. Cell 123, 335–346. doi: 10.1016/j.cell.2005.08.034
Kvell, K., Cooper, E. L., Engelmann, P., Bovari, J., and Nemeth, P. (2007). Blurring borders: innate immunity with adaptive features. Clin. Dev. Immunol. 2007, 83671. doi: 10.1155/2007/83671
Lavelle, E. C., Murphy, C., O'Neill, L. A., and Creagh, E. M. (2010). The role of TLRs, NLRs, and RLRs in mucosal innate immunity and homeostasis. Mucosal Immunol. 3, 17–28. doi: 10.1038/mi.2009.124
Lemaitre, B., Nicolas, E., Michaut, L., Reichhart, J. M., and Hoffmann, J. A. (1996). The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86, 973–983. doi: 10.1016/S0092-8674(00)80172-5
Merkling, S. H., and van Rij, R. P. (2013). Beyond RNAi: antiviral defense strategies in Drosophila and mosquito. J. Insect. Physiol. 59, 159–170. doi: 10.1016/j.jinsphys.2012.07.004
Merrell, M. A., Ilvesaro, J. M., Lehtonen, N., Sorsa, T., Gehrs, B., Rosenthal, E., et al. (2006). Toll-like receptor 9 agonists promote cellular invasion by increasing matrix metalloproteinase activity. Mol. Cancer Res. 4, 437–447. doi: 10.1158/1541-7786.MCR-06-0007
Moreno, E., Yan, M., and Basler, K. (2002). Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr. Biol. 12, 1263–1268. doi: 10.1016/S0960-9822(02)00954-5
Nehme, N. T., Liegeois, S., Kele, B., Giammarinaro, P., Pradel, E., Hoffmann, J. A., et al. (2007). A model of bacterial intestinal infections in Drosophila melanogaster. PLoS Pathog. 3:e173. doi: 10.1371/journal.ppat.0030173
O'Neil, D. A., Porter, E. M., Elewaut, D., Anderson, G. M., Eckmann, L., Ganz, T., et al. (1999). Expression and regulation of the human beta-defensins hBD-1 and hBD-2 in intestinal epithelium. J. Immunol. 163, 6718–6724.
Pagliarini, R. A., and Xu, T. (2003). A genetic screen in Drosophila for metastatic behavior. Science 302, 1227–1231. doi: 10.1126/science.1088474
Pasparakis, M. (2008). IKK/NF-kappaB signaling in intestinal epithelial cells controls immune homeostasis in the gut. Mucosal Immunol. 1 (Suppl. 1), S54–S57. doi: 10.1038/mi.2008.53
Pastor-Pareja, J. C., Wu, M., and Xu, T. (2008). An innate immune response of blood cells to tumors and tissue damage in Drosophila. Dis. Model. Mech. 1, 144–154. discussion: 53. doi: 10.1242/dmm.000950
Peek, R. M. Jr., and Blaser, M. J. (2002). Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2, 28–37. doi: 10.1038/nrc703
Pitsouli, C., Apidianakis, Y., and Perrimon, N. (2009). Homeostasis in infected epithelia: stem cells take the lead. Cell Host Microbe 6, 301–307. doi: 10.1016/j.chom.2009.10.001
Rakoff-Nahoum, S., and Medzhitov, R. (2009). Toll-like receptors and cancer. Nat. Rev. Cancer 9, 57–63. doi: 10.1038/nrc2541
Ramet, M., Manfruelli, P., Pearson, A., Mathey-Prevot, B., and Ezekowitz, R. A. (2002). Functional genomic analysis of phagocytosis and identification of a Drosophila receptor for E. coli. Nature 416, 644–648. doi: 10.1038/nature735
Rudrapatna, V. A., Cagan, R. L., and Das, T. K. (2012). Drosophila cancer models. Dev. Dyn. 241, 107–118. doi: 10.1002/dvdy.22771
Secher, T., Gaillot, O., Ryffel, B., and Chamaillard, M. (2010). Remote control of intestinal tumorigenesis by innate immunity. Cancer Res. 70, 1749–1752. doi: 10.1158/0008-5472.CAN-09-3401
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820. doi: 10.1016/j.cell.2010.01.022
Tepass, U., Fessler, L. I., Aziz, A., and Hartenstein, V. (1994). Embryonic origin of hemocytes and their relationship to cell death in Drosophila. Development 120, 1829–1837.
Trinchieri, G. (2012). Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu. Rev. Immunol. 30, 677–706. doi: 10.1146/annurev-immunol-020711-075008
Valanne, S., Wang, J. H., and Ramet, M. (2011). The Drosophila Toll signaling pathway. J. Immunol. 186, 649–656. doi: 10.4049/jimmunol.1002302
Wandler, A. M., and Guillemin, K. (2012). Transgenic expression of the Helicobacter pylori virulence factor CagA promotes apoptosis or tumorigenesis through JNK activation in Drosophila. PLoS Pathog. 8:e1002939. doi: 10.1371/journal.ppat.1002939
Wang, J. H., Manning, B. J., Wu, Q. D., Blankson, S., Bouchier-Hayes, D., Redmond, H. P., et al. (2003). Endotoxin/lipopolysaccharide activates NF-kappa B and enhances tumor cell adhesion and invasion through a beta 1 integrin-dependent mechanism. J. Immunol. 170, 795–804.
Waters, J. P., Pober, J. S., and Bradley, J. R. (2013a). Tumour necrosis factor in infectious disease. J. Pathol. 230, 132–147. doi: 10.1002/path.4187
Waters, J. P., Pober, J. S., and Bradley, J. R. (2013b). Tumour necrosis factor and cancer. J. Pathol. 230, 241–248. doi: 10.1002/path.4188
Wroblewski, L. E., Peek, R. M. Jr., and Wilson, K. T. (2010). Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin. Microbiol. Rev. 23, 713–739. doi: 10.1128/CMR.00011-10
Keywords: Drosophila, innate immune response, inflammation, infection, cancer
Citation: Bangi E (2013) Drosophila at the intersection of infection, inflammation, and cancer. Front. Cell. Infect. Microbiol. 3:103. doi: 10.3389/fcimb.2013.00103
Received: 05 September 2013; Accepted: 06 December 2013;
Published online: 19 December 2013.
Edited by:
Yiorgos Apidianakis, University of Cyprus, CyprusReviewed by:
Terry Kwok, Monash University, AustraliaSung Ouk Kim, University of Western Ontario, Canada
Copyright © 2013 Bangi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erdem Bangi, Department of Developmental and Regenerative Biology, Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Annenberg 25-40 New York, NY 10029, USA e-mail:ZXJkZW0uYmFuZ2lAbXNzbS5lZHU=