 Lee-Ann H. Allen
Lee-Ann H. Allen- Inflammation Program and the Departments of Internal Medicine and Microbiology, University of Iowa and the VA Medical Center, Iowa City, IA, USA
The central role of neutrophils in innate immunity and host defense has long been recognized, and the ability of these cells to efficiently engulf and kill invading bacteria has been extensively studied, as has the role of neutrophil apoptosis in resolution of the inflammatory response. In the past few years additional immunoregulatory properties of neutrophils were discovered, and it is now clear that these cells play a much greater role in control of the immune response than was previously appreciated. In this regard, it is noteworthy that Francisella tularensis is one of relatively few pathogens that can successfully parasitize neutrophils as well as macrophages, DC and epithelial cells. Herein we will review the mechanisms used by F. tularensis to evade elimination by neutrophils. We will also reprise effects of this pathogen on neutrophil migration and lifespan as compared with other infectious and inflammatory disease states. In addition, we will discuss the evidence which suggests that neutrophils contribute to disease progression rather than effective defense during tularemia, and consider whether manipulation of neutrophil migration or turnover may be suitable adjunctive therapeutic strategies.
Neutrophils in Innate Host Defense
Polymorphonuclear leukocytes (PMNs) are the most abundant leukocyte population in human blood and are rapidly mobilized to sites of infection (Kennedy and Deleo, 2009). In this locale they phagocytose microbes and utilize a combination of NADPH oxidase-derived reactive oxygen species (ROS), cytotoxic granule components and antimicrobial peptides to generate a highly lethal intraphagosomal environment (Nauseef, 2007; Kennedy and Deleo, 2009). In contrast to other leukocytes, neutrophils are short-lived and are preprogrammed to undergo constitutive (spontaneous) apoptosis 18–24 h after release into circulation, and under normal circumstances, PMN apoptosis is further accelerated by phagocytosis and oxidant production (Watson et al., 1996; Kobayashi et al., 2003c; Kobayashi and Deleo, 2009). Ground-breaking studies of DeLeo and colleagues revealed that both constitutive and infection-induced PMN death are controlled not only at the level of intracellular signaling, but also by global changes in gene expression (Kobayashi et al., 2002, 2003a,c), discoveries which necessitated revision of the long-standing notion that mature neutrophils were nearly transcriptionally inert (Jack and Fearon, 1988; Kobayashi and Deleo, 2009). Tight spatial and temporal control of PMN apoptosis is critical for elimination of infection and resolution of the inflammatory response, and during this process phagocytic and proinflammatory capacity are down-regulated, release of toxic cell components is prevented, and tissue damage is minimized (Kobayashi et al., 2003b,c; Fox et al., 2010). If this process is perturbed PMNs can develop a proinflammatory phenotype that promotes necrosis and granuloma formation and sustains infection (Kobayashi et al., 2003b, 2004). For this reason, defects in PMN turnover are indicative of an ineffective and dysregulated inflammatory response (Nathan, 2002). In keeping with this, recent studies revealed that neutrophils have immunoregulatory properties that directly influence the function of NK cells, DCs, macrophages and lymphocytes (Mantovani et al., 2011).
Neutrophils and Tularemia Pathogenesis
Francisella tularensis is a facultative intracellular pathogen that is distributed throughout the Northern hemisphere and two subspecies of this bacterium, F. tularensis subspecies tularensis (type A) and F. tularensis subspecies holarctica (type B) account for nearly all cases of human tularemia. Most studies of this organism have focused on macrophages as major vehicles for intracellular growth and bacterial dissemination from sites of infection to the liver and spleen (Chong and Celli, 2010). Nevertheless, F. tularensis is unusual in its ability to infect neutrophils and epithelial cells as well as mononuclear phagocytes, but relatively little is known about the shared and distinct contributions of these other cell types to disease (McLendon et al., 2006).
Aerosol infection of rhesus monkeys with virulent type B F. tularensis strains defined prominent features of pneumonic tularemia, and these studies were among the first to suggest a key role for neutrophils in tissue destruction and disease progression (Tulis et al., 1970; Schricker et al., 1972; Hall et al., 1973). These data demonstrate that large numbers of PMNs are present the lungs, and from day 2 onward alveoli and bronchioles become progressively clogged with neutrophils, bacteria and necrotic debris. Granulomas also begin to organize wherein live PMNs, bacteria and necrotic debris become enveloped by epithelial syncytia. A similar disease course has been described using rats, rabbits and mice (Dunaeva and Shlygina, 1975), and ex vivo analyses indicate that PMNs contain viable bacteria. Rhesus monkeys, rabbits, mice (and many humans) do not survive acute infection with type A F. tularensis (Eigelsbach et al., 1962; Schricker et al., 1972). These organisms replicate much faster than type B isolates, and progression to moribund status is characterized by an accumulation of neutrophils and bacteria in the lung and extensive necrotic tissue damage (Conlan et al., 2003; Lamps et al., 2004; Bosio et al., 2007). In keeping with the histopathology data, flow cytometry analysis of mouse lung cells indicates that whereas alveolar macrophages account for ~70% of F. tularensis-infected lung cells on the first day of infection, neutrophils are the major infected cell population by day 3, and the fraction of infected DCs and alveolar type II (ATII) cells is relatively low (Hall et al., 2008).
It has long been known that neutropenia or inherited defects in PMN function markedly increase susceptibility to infection. For this reason, neutrophil-depleting antibodies are often used to determine the role of this cell type in different diseases. Results of studies that used the first antibody developed for this purpose, RB6-8C5, suggested that PMNs are critical for host defense against F. tularensis (Sjostedt et al., 1994). However, subsequent studies revealed that RB6-8C5 causes depletion of both inflammatory monocytes and PMNs, necessitating a reinterpretation of prior results (Daley et al., 2008; Dunay et al., 2010). A subsequent study that used a lower dose of RB6-8C5 that is somewhat more selective for PMNs did not identify a role for these cells in control of F. tularensis (Kuolee et al., 2011). Using a different approach, Malik et al. discovered that inhibition of PMN migration into the lung allowed mice to survive what would otherwise be a lethal dose of type A F. tularensis or the live vaccine strain (LVS) (Malik et al., 2007). At the same time, studies of Elkins and colleagues suggest that tularemia severity is significantly increased under conditions that induce neutrophilia, and this correlates directly with enhanced hepatotoxicity (Bosio and Elkins, 2001; Mellilo et al., 2013). On balance, the data indicate that PMNs do not contribute to effective host defense during tularemia, and instead contribute to disease progression.
Neutrophil Chemotaxis
Mechanisms of neutrophil migration to sites of infection have been extensively studied (Craig et al., 2009; Balamayooran et al., 2010; Sadik et al., 2012; Fullerton et al., 2013). In the lung, inhaled bacteria typically interact with alveolar macrophages and ATII cells, and signaling downstream of pattern recognition receptors, including TLRs, triggers secretion of major neutrophil chemotactic agents, particularly IL-8 (CXCL8) in humans and KC in mice, as well as GRO-α, MIP-2 and MCP-1. Additional players include IL-1β, LIX/CXCL5, G-CSF, complement factor C5a and the eicosanoids LTB4 and PGE2, as well as Proline-Glycine-Proline (PGP), a peptide generated from collagen in the extracellular matrix by matrix metalloproteinases MMP-8 and MMP-9. As neutrophil function and phenotype are affected by the mechanism of recruitment, it is noteworthy that IL-8, KC, GRO-α, MIP-2, LIX, and PGP all bind CXCR1 and/or CXCR2 on PMNs. Neutrophils directly contribute to this process as well via secretion IL-8, IL-1β, LTB4, and MMP-9.
During tularemia a select subset of signals trigger PMN migration into the lung as the amount of IL-8 released by infected ATII cells, macrophages and endothelial cells is relatively low, and neither IL-8 nor MCP-1 appears to be essential (Gentry et al., 2007; Moreland et al., 2009). In marked contrast, F. tularensis specifically stimulates expression of MMP-9, and PGP is a major PMN chemoattractant in this system, as lung PMNs are markedly diminished in MMP-9 null mice and these animals are able to survive what would otherwise be a lethal dose of type A F. tularensis or the LVS (Malik et al., 2007), as noted above. In this regard, it is of interest that PGP is sufficient to recruit and maintain tissue neutrophils in in absence of other chemotactic agents and its stability and activity are enhanced by N-acetylation (Snelgrove et al., 2010), but whether this modification is induced during tularemia is unknown. PGE2 is produced by F. tularensis-infected macrophages and ATII cells (Woolard et al., 2007) and infected PMNs upregulate IL-1β (Schwartz et al., 2013), but the extent to which these agents contribute to PMN chemotaxis and phenotypic modulation during tularemia remains to be determined. A model that integrates these data is shown in Figure 1.
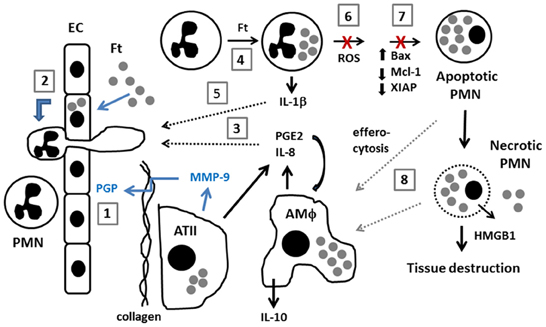
Figure 1. Model of neutrophil dynamics in the F. tularensis-infected lung. Inhaled F. tularensis rapidly infects alveolar type II (ATII) cells and macrophages (AMΦ) (Gentry et al., 2007). (1) MMP-9, likely secreted by ATII cells, cleaves collagen, generating PGP which directly stimulates PMN recruitment from the bloodstream (Malik et al., 2007). (2) PMN migration is also stimulated by direct infection of pulmonary endothelial cells (EC) by an IL-8 and MCP-1-independent mechanism (Moreland et al., 2009). (3) F. tularensis stimulates release of PGE2 and IL-8 from AMΦ and ATII cells (Gentry et al., 2007; Woolard et al., 2007). PGE2 stimulates macrophage production of IL-10 (Hunt et al., 2012). (4,5) F. tularensis infects PMNs and upregulates IL-1β (Schwartz et al., 2013). The extent to which PGE2, IL-8 and IL-1β contribute to PMN chemotaxis and phenotypic modulation during tularemia remains to be determined (dotted black arrows). (6,7) F. tularensis inhibits PMN NADPH oxidase activity and prevents changes in gene expression that are critical for constitutive and phagocytosis-induced apoptosis (McCaffrey and Allen, 2006; McCaffrey et al., 2010; Schwartz et al., 2012a, 2013). (8) Efferocytosis of apoptotic PMNs is critical for control of infection and resolution of inflammation. Defects in apoptosis favor PMN necrosis, and subsequent release of cytotoxic cell components, and danger molecules such as HMGB1 exacerbate tissue destruction. Recent data suggest that efferocytosis and/or clearance of necrotic cell debris may be impaired (Mares et al., 2011) (dotted gray arrows).
F. tularensis Disrupts Neutrophil Defense Mechanisms
Francisella tularensis lipopolysaccharide (LPS) has an atypical structure that does not signal through toll like receptors 4 or 2. Nevertheless, LPS O-antigen and capsular polysaccharides act in concert to protect this organism from the lytic effects of serum complement (McLendon et al., 2006). This is significant as neutrophil uptake of F. tularensis is markedly enhanced by complement factors in normal human serum, and under these conditions phagocytosis is mediated by complement receptor 1 and complement receptor 3 (Schwartz et al., 2012b). Neutrophil receptors that confer inefficient phagocytosis of unopsonized F. tularensis remain obscure.
Under normal circumstances, phagocytosis is coupled to rapid activation of the Nox2 NADPH oxidase complex, such that toxic oxidants kill most bacteria within 60 min of infection (Deleo et al., 1999; Kobayashi et al., 2003a; Nauseef, 2007). In marked contrast, we and others demonstrated using several strains of type A and type B F. tularensis as well as LVS that this organism does not trigger a respiratory burst in human or monkey PMNs (Proctor et al., 1975; McCaffrey and Allen, 2006; McCaffrey et al., 2010). At the molecular level, forming F. tularensis phagosomes exclude flavocytochrome b558, which contains the catalytic core of the NADPH oxidase and also acts as a docking site in the membrane for the cytosolic subunits that are also essential for ROS production (McCaffrey and Allen, 2006; McCaffrey et al., 2010). Within minutes, the ability of infected PMNs to be activated by heterologous particulate and soluble stimuli is also profoundly impaired, and in this case F. tularensis acts at a later stage to inhibit the activity of enzyme complexes at the membrane (McCaffrey and Allen, 2006; McCaffrey et al., 2010). A similar, if not identical, mechanism of post-assembly NADPH oxidase inhibition also undermines the ability of specific IgG in anti-F. tularensis immune serum to enhance bacterial killing via neutrophil activation (McCaffrey and Allen, 2006; McCaffrey et al., 2010).
Evasion of toxic oxidants at early stages of infection is followed by phagosome egress, and release of bacteria into neutrophil cytosol, and F. tularensis replicates to some extent in this locale, though but not to the same extent as in macrophages (McCaffrey and Allen, 2006; Schulert et al., 2009; Schwartz et al., 2012a; Long et al., 2013). FevR and MigR, major virulence regulators of F. tularensis, are essential for inhibition of neutrophil activation (Buchan et al., 2009; McCaffrey et al., 2010). The genes in the MigR/FevR regulon required for NADPH oxidase inhibition are as yet unknown, but are distinct from pathogenicity island genes required for phagosome escape and intracellular growth such as iglI and iglJ (McCaffrey et al., 2010; Long et al., 2013). Other genes such as carA, carB, and pyrB act indirectly via effects on pyrimidine biosynthesis (Schulert et al., 2009), and the role of the acid phosphatase AcpA in NADPH oxidase inhibition and virulence is controversial (Reilly et al., 1996; Child et al., 2010; McCaffrey et al., 2010; Mohapatra et al., 2010).
F. tularensis Prolongs Neutrophil Lifespan Via Effects on Signaling and Gene Expression
Neutrophils are short-lived and undergo constitutive apoptosis at a rate of 1011 cell per day in humans (Kennedy and Deleo, 2009). Although the specific events that initiate constitutive PMN apoptosis are unknown, it is clear that cell death is tightly regulated and that both intrinsic and extrinsic pathway caspases contribute to this process along with complex changes in gene expression that comprise and “apoptosis differentiation program,” as noted above. Since the seminal studies of Watson (Watson et al., 1996) it has also been clear that phagocytosis and NADPH oxidase-derived ROS profoundly accelerate PMN apoptosis as compared with unstimulated controls (Kobayashi et al., 2003a, 2004). This “phagocytosis-induced cell death” (PICD) respone is also regulated at the level of gene expression, and is critical for effective host defense. Blockade of NADPH oxidase activity by F. tularensis suggested that this organism may not accelerate PMN death, and as such may not induce PICD. Indeed, our recent biochemical studies and analysis of PMN gene expression provide definitive evidence that F. tularensis not only fails to induce PICD, but also inhibits constitutive neutrophil apoptosis via effects on the intrinsic and extrinsic pathways, and in this manner profoundly prolongs cell lifespan (Schwartz et al., 2012a, 2013)
Specifically, the biochemical data indicate that the vast majority of infected PMNs do not progress to an apoptotic morphology within 48 h of infection with live F. tularensis, and in keeping with this PS externalization, processing and activation of caspases-8, −9, and −3, and DNA fragmentation are markedly impaired (Schwartz et al., 2012a). At the same time, F. tularensis significantly alters the expression of over 3400 human neutrophil genes between 3 and 24 h of infection, including 365 unique genes linked to apoptosis and cell survival (Schwartz et al., 2013). Of particular note are effects of F. tularensis on BAX, a proapoptotic member of the Bcl-2 family of proteins that plays a pivotal role in the intrinsic apoptotic pathway via disruption of the outer mitochondrial membrane. Upregulation of BAX is a hallmark of the PICD response (Kobayashi et al., 2002, 2003a, 2004). However, BAX mRNA and protein are progressively downregulated by F. tularensis (Schwartz et al., 2013). It is therefore likely that blockade of the respiratory burst and downregulation of BAX synergize to prevent PICD during F. tularensis infection. At the same time, upregulation of several prosurvival factors and anti-apoptosis genes collaborate to diminish and delay the constitutive apoptosis program in PMN. Although not all of these data can be discussed here, sustained expression BIRC4, which encodes X-linked inhibitor of apoptosis protein (XIAP), and CAST which encodes calpastatin, likely account in large part for defective processing and activation of intrinsic pathway caspases (Schwartz et al., 2013). At the same time not all genes associated with enhanced PMN survival are modulated by this pathogen, as expression of IL-8 (CXCL8) is not induced and this cytokine is not secreted by PMNs infected with live F. tularensis (Schwartz et al., 2012a, 2013). In contrast, IL-8 appears critical for sustained survival of neutrophils infected with the obligate intracellular pathogen Chlamydia pneumoniae (van Zandbergen et al., 2004), and as such the data indicate that these two pathogens use distinct mechanisms to modulate PMN lifespan.
It is also important to note that apoptosis normally downregulates PMN functional capacity, and if apoptosis is inhibited these cells exhibit a sustained proinflammatory phenotype (Kobayashi et al., 2003b; Kennedy and Deleo, 2009). Consistent with this, F. tularensis-infected PMNs show enhanced expression of VEGF, IL6, IL1B, CXCL1, OSM, and IL1RN (Schwartz et al., 2013). Together, enhanced lifespan and proinflammatory capacity increase the probably of cell progression to secondary necrosis, which is characterized by spilling of DAMPs, alarmins, and other cytotoxic molecules that further amplify inflammation and cause extensive host tissue destruction (Kobayashi et al., 2003c; Fox et al., 2010; Silva, 2010).
Targeting PMNs as a Candidate Adjunctive Theraputic Strategy
Neutrophil accumulation and enhanced longevity, granuloma formation, and extensive tissue necrosis are benchmarks that define a defective inflammatory response (Nathan, 2002), and are also characteristic features of tissues infected with F. tularensis (Tulis et al., 1970; Schricker et al., 1972; Hall et al., 1973). The fact that blockade of PMN influx into the lung favors host survival without markedly altering tissue bacterial load (Malik et al., 2007) is consistent with an immunoregulatory role for PMNs in tularemia pathogenesis (Mantovani et al., 2011), as are the effects of F. tularensis on neutrophil activation state, lifespan and proinflammatory capacity that are noted above and summarized in Figure 1. Considered together, the data support a model in which neutrophils play a unique role in tularemia pathogenesis via dysregulation of the inflammatory response that is distinct from the role of macrophages as major vehicles for bacterial growth and dissemination. Thus, suitable points of therapeutic intervention may include PMN chemotaxis, apoptosis or activation state.
Inhibition of PMN apoptosis and aggressive neutrophilic inflammation also exacerbate the severity of pneumococcal meningitis (Koedel et al., 2009) and contribute significantly to lung destruction in human patients with chronic obstructive pulmonary disease (COPD) (Weathington et al., 2006). As in tularemia, PMN accumulation in the lungs of humans with COPD is driven by MMP-9-dependent production of PGP, and published data suggest that targeting this pathway with anti-PGP antibodies suppresses neutrophil responses and appears to have some therapeutic benefit in mouse COPD models (Weathington et al., 2006). Similarly, Arginine-Threonine-Arginine peptides block IL-8 and PGP signaling at the level of CXCR1 and CXCR2 to inhibit PMN migration and activation, and in this manner ameliorate lung destruction during emphysema (van Houwelingen et al., 2008). Other interventions that directly target this mechanism of PMN chemotaxis include small molecule inhibitors of MMPs such as GM6001, CP-471,474, and RS113,456 and enhancement of endogenous mechanisms of MMPs inhibition via intratracheal delivery of recombinant TIMPs (tissue inhibitors of metalloproteinases) (Djekic et al., 2009). As protein-based therapeutics are very expensive they are not optimal for treatment of chronic illnesses, yet could be of considerable benefit in the context of acute infectious diseases, including tularemia.
Other potential therapeutic targets include lipid mediators of the eicosanoid family, which are dysregulated in many critical illnesses. In particular, resolvin E1 is of interest as it is effective at concentrations as low as 1 nM, is beneficial for treatment of aspiration pneumonia, and is known to decrease lung PMNs via effects on ROS production, PICD and efferocytosis (Fullerton et al., 2013). Aspirin-triggered resolvins also appear to reduce mortality associated with systemic inflammatory response syndrome. On the other hand, excess or sustained production of PGE2 by macrophages and epithelial cells induces a state of “injurious resolution” that compromises Fc receptor function and NADPH oxidase activity and alters macrophage phenotype. This condition can occur in burn patients or infection with Aspergillus, and is reversed in by aspirin or cyclooxygenase inhibitors which reduce PGE2 levels by 95% (Fullerton et al., 2013). Although further analysis of eicosanoid profiles during tularemia is needed, these lipid mediators are of interest as PGE2 is induced by F. tularensis and dampens at least some aspects of the immune response (Woolard et al., 2007).
Other studies have begun to examine the potential therapeutic utility of PMN apoptosis induction. Cyclin-dependent kinases (CDKs) regulate growth of most cell types, yet are critical regulators of PMN viability and lifespan (Witko-Sarsat et al., 2011). Inhibition of CDKs with R-roscovitine and other related compounds can induce apoptosis and resolution of PMN-dominant inflammatory responses (Leitch et al., 2010), and in this manner accelerate recovery of mice with pneumococcal meningitis (Koedel et al., 2009). Moreover, our recent data indicate that CDK7 and CDK2 are induced in PMNs by F. tularensis (Schwartz et al., 2013). In summary, given the potentially short widow between the onset of symptoms, severe disease and death, future studies should consider whether agents that modulate PMN chemotaxis, directly target PMN apoptosis, or modulate the inflammatory response may be useful as adjunctive therapeutic agents when combined with antibiotics for treatment of tularemia.
Relevance to Other Infectious Diseases
As in tularemia, PMNs are the most commonly infected cell type in the airway of persons infected with Mycobacterium tuberculosis (Lowe et al., 2012). PMN lifespan is prolonged, and protective CD4+ T cell-driven adaptive immune responses are curtailed at the level of DCs, as downstream responses are less efficient when these cells are directly infected than when antigens are acquired by efferocytosis of apoptotic, infected PMNs (Blomgran and Ernst, 2011). Thus, neutrophilia is associated with impaired control of infection and correlates directly with the severity of cavitary disease and tissue damage. Neutrophils may kill some M. tuberculosis early in infection, but PMN depletion at later stages appears to be beneficial (Lowe et al., 2012). Many factors drive PMN accumulation during tuberculosis including macrophage and DC-derived IL-8, G-CSF, and LTB4, as well as IL-1β, IL-8, and LTB4 from PMNs themselves (Lowe et al., 2012). A role for PGE2 has also been described, and MMP-9 may play a specific role in recruitment of PMNs to granulomas (Lowe et al., 2012; Hawn et al., 2013).
Brucella abortus replicates in an ER-derived vacuole in macrophages. Similar to F. tularensis, B. abortus LPS has low bioactivity, yet in contrast to F. tularensis, there are relatively few PMNs in the circulation or at sites of infection (Barquero-Calvo et al., 2007). At the same time, B. abortus is not efficiently killed by human PMNs in vitro (Kreutzer et al., 1979), and neutrophils do not appear to contribute directly to bacterial killing. Rather, control of infection is favored by PMN depletion during late stages of infection, which enhances lymphocyte activation and proinflammatory cytokine production ((Barquero-Calvo et al., 2007, 2013). Arthritis is a complication of B. abortus infection, and local secretion of MMP-2 and MMP-9 plays an important role in neutrophil recruitment and joint damage (Scian et al., 2011). In contrast, C5a, IL-1β, and LTB4 play dominant roles in neutrophil-mediated joint damage in rheumatoid arthritis (Sadik et al., 2012).
Finally, MMP-8 and MMP-9 also drive PMN recruitment to the lungs during cystic fibrosis, and this is perpetuated and amplified by MMP-9 released by PMN degranulation. PGP, particularly in its highly active N-acetylated form, is elevated in patient sputum (Xu et al., 2011). Thus, strategies that target MMPs may also be useful in this disease that is characterized by chronic neutrophilic inflammation. Considered together, the data summarized here demonstrate that PMNs play complex and important roles in infection, and therapeutic strategies that target this cell type are being developed and may have wide utility alone or in combination with antibiotics.
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Lee-Ann H. Allen is supported in part by funds from the National Institutes of Health, NIAID R01, AI0738-35 and P01 AI044642, and by VA Merit Review Grant 1I01BX002108-01.
Abbreviations
ATII, alveolar type II cells; CDK, cyclin-dependent kinase; COPD, chronic obstructive pulmonary disease; DAMPs, danger-associated molecular patterns; LVS, live vaccine strain; MMP-9, matrix metalloproteinase-9; PICD, phagocytosis-induced cell death; PMN, Polymorphonuclear leukocyte; ROS, reactive oxygen species.
References
Balamayooran, G., Batra, S., Fessler, M. B., Happel, K. I., and Jeyaseelan, S. (2010). Mechanisms of neutrophil accumulation in the lungs against bacteria. Am. J. Respir. Cell Mol. Biol. 43, 5–16. doi: 10.1165/rcmb.2009-0047TR
Barquero-Calvo, E., Chaves-Olarte, E., Weiss, D. S., Guzman-Veri, C., Chacon-Diaz, C., Rucavado, A., et al. (2007). Brucella abortus uses a stealthy strategy to avoid activation of the innate immune system during the onset of infection. PLoS ONE 2:e631. doi: 10.1371/journal.pone.0000631
Barquero-Calvo, E., Martirosyan, A., Ordoñez-Rueda, D., Arce-Gorvel, V., Alfaro-Alarcón, A., Lepidi, H., et al. (2013). Neutrophils exert a suppressive effect on Th1 responses to intracellular pathogen Brucella abortus. PLoS Pathog. 9:e1003167. doi: 10.1371/journal.ppat.1003167
Blomgran, R., and Ernst, J. D. (2011). Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J. Immunol. 186, 7110–7119. doi: 10.4049/jimmunol.1100001
Bosio, C. M., Bielefeldt-Ohmann, H., and Belisle, J. T. (2007). Active suppression of the pulmonary immune response by francisella tularensis Schu4. J. Immunol. 178, 4538–4547.
Bosio, C. M., and Elkins, K. L. (2001). Susceptibility to secondary francisella tularensis live vaccine strain infection in B-Cell-Deficient Mice is associated with neutrophilia but not with defects in specific T-Cell-Mediated Immunity. Infect. Immun. 69, 194–203. doi: 10.1128/IAI.69.1.194-203.2001
Buchan, B. W., McCaffrey, R. L., Lindemann, S. R., Allen, L.-A. H., and Jones, B. D. (2009). Identification of migR, a regulatory element of the Francisella tularensis live vaccine strain iglABCD virulence operon required for normal replication and trafficking in macrophages. Infect. Immun. 77, 2517–2529. doi: 10.1128/IAI.00229-09
Child, R., Wehrly, T. D., Rockx-Brouwer, D., Dorward, D. W., and Celli, J. (2010). Acid Phosphatases do not contribute to the pathogenesis of Type A Francisella tularensis. Infect. Immun. 78, 59–67. doi: 10.1128/IAI.00965-09
Chong, A., and Celli, J. (2010). The Francisella intracellular life cycle: towards molecular mechanisms of intracellular survival and proliferation. Front. Microbiol. 1:138. doi: 10.3389/fmicb.2010.00138
Conlan, J. W., Chen, W., Shen, H., Webb, A., and Kuolee, R. (2003). Experimental tularemia in mice challenged by aerosol or intradermally with virulent strains of Francisella tularensis: bacteriologic and histopathologic studies. Microb. Pathog. 34, 239. doi: 10.1016/S0882-4010(03)00046-9
Craig, A., Mai, J., Cai, S., and Jeyaseelan, S. (2009). Neutrophil recruitment to the lungs during bacterial pneumonia. Infect. Immun. 77, 568–575. doi: 10.1128/IAI.00832-08
Daley, J. M., Thomay, A. A., Connolly, M. D., Reichner, J. S., and Albina, J. E. (2008). Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 83, 64–70. doi: 10.1189/jlb.0407247
Deleo, F. R., Allen, L.-A. H., Apicella, M., and Nauseef, W. M. (1999). NADPH oxidase activation and assembly during phagocytosis. J. Immunol. 163, 6732–6740.
Djekic, U. V., Gaggar, A., and Weathington, N. M. (2009). Attacking the multi-tiered proteolytic pathology of COPD: new insights from basic and translational studies. Pharmacol. Ther. 121, 132–146. doi: 10.1016/j.pharmthera.2008.09.008
Dunaeva, T. N., and Shlygina, K. N. (1975). Phagocytic activity of the neutrophils in tularemia in animals with varying infective sensitivity. Zh. Mikrobiol. Epidomiol. Immunobiol. 10, 22–26.
Dunay, I. R., Fuchs, A., and Sibley, L. D. (2010). Inflammatory monocytes but not neutrophils are necessary to control infection with Toxoplasma gondii in mice. Infect. Immun. 78, 1564–1570. doi: 10.1128/IAI.00472-09
Eigelsbach, H. T., Tulis, J. J., McGavran, M. H., and White, J. D. (1962). Live tularemia vaccine I. Host parastie relationship in monkeys vaccinated intracutaneously or aerogenically. J. Bacteriol. 84, 1020–1027.
Fox, S., Leitch, A. E., Duffin, R., Haslett, C., and Rossi, A. G. (2010). Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J. Innate Immun. 2, 216–227. doi: 10.1159/000284367
Fullerton, J. N., O'Brien, A. J., and Gilroy, D. W. (2013). Lipid mediators in immune dysfunction after severe inflammation. Trends Immunol. doi: 10.1016/j.it.2013.10.008. (in press).
Gentry, M., Taormina, J., Pyles, R. B., Yeager, L., Kirtley, M., Popov, V. L., et al. (2007). Role of primary human alveolar epithelial cells in host defense against Francisella tularensis Infection. Infect. Immun. 75, 3969–3978. doi: 10.1128/IAI.00157-07
Hall, J. D., Woolard, M. D., Gunn, B. M., Craven, R. R., Taft-Benz, S., Frelinger, J. A., et al. (2008). Infected-Host-Cell repertoire and cellular response in the lung following inhalation of francisella tularensis Schu S4, LVS, or U112. Infect. Immun. 76, 5843–5852. doi: 10.1128/IAI.01176-08
Hall, W. C., Kovatch, R. M., and Schricker, R. L. (1973). Tularemic pneumonia: pathogenesis of the aerosol-induced disease in monkeys. J. Pathol. 110, 193–201. doi: 10.1002/path.1711100302
Hawn, T. R., Matheson, A. I., Maley, S. N., and Vandal, O. (2013). Host-directed theraputics for tuberculosis: can we harness the host? Microbiol. Mol. Biol. Rev. 77, 608–627. doi: 10.1128/MMBR.00032-13
Hunt, D., Wilson, J. E., Weih, K. A., Ishido, S., Harton, J. A., Roche, P. A., et al. (2012). Francisella tularensis Elicits IL-10 via a PGE2-Inducible Factor, to drive macrophage MARCH1 expression and Class II down-regulation. PLoS ONE 7:e37330. doi: 10.1371/journal.pone.0037330
Jack, R. M., and Fearon, D. T. (1988). Selective synthesis of mRNA and proteins by human peripheral blood neutrophils. J. Immunol. 140, 4286–4293.
Kennedy, A., and Deleo, F. (2009). Neutrophil apoptosis and the resolution of infection. Immunol. Res. 43, 25–61. doi: 10.1007/s12026-008-8049-6
Kobayashi, S. D., Braughton, K. R., Whitney, A. R., Voyich, J. M., Schwan, T. G., Musser, J. M., et al. (2003a). Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc. Natl. Acad. Sci. U.S.A. 100, 10948–10953. doi: 10.1073/pnas.1833375100
Kobayashi, S. D., Voyich, J. M., Braughton, K. R., and Deleo, F. R. (2003b). Down-regulation of proinflammatory capacity during apoptosis in human polymorphonuclear leukocytes. J. Immunol. 170, 3357–3368.
Kobayashi, S. D., Voyich, J. M., Somerville, G. A., Braughton, K. R., Malech, H. L., Musser, J. M., et al. (2003c). An apoptosis-differentiation program in human polymorphonuclear leukocytes facilitates resolution of inflammation. J. Leukoc. Biol. 73, 315–322. doi: 10.1189/jlb.1002481
Kobayashi, S. D., and Deleo, F. R. (2009). Role of neutrophils in innate immunity: a systems biology-level approach. Wiley Interdiscip. Rev. Syst. Biol. Med. 1, 309–333. doi: 10.1002/wsbm.32
Kobayashi, S. D., Voyich, J. M., Braughton, K. R., Whitney, A. R., Nauseef, W. M., Malech, H. L., et al. (2004). Gene expression profiling provides insight into the pathophysiology of chronic granulomatous disease. J. Immunol. 172, 636–643.
Kobayashi, S. D., Voyich, J. M., Buhl, C. L., Stahl, R. M., and Deleo, F. R. (2002). Global changes in gene expression by human polymorphonuclear leukocytes during receptor-mediated phagocytosis: cell fate is regulated at the level of gene expression. Proc. Nat. Acad. Sci. U.S.A. 99, 6901–6906. doi: 10.1073/pnas.092148299
Koedel, U., Frankenberg, T., Kirschnek, S., Obermaier, B., Häcker, H., Paul, R., et al. (2009). Apoptosis is essential for neutrophil functional shutdown and determines tissue damage in experimental Pneumococcal meningitis. PLoS Pathog. 5:e1000461. doi: 10.1371/journal.ppat.1000461
Kreutzer, D. L., Dreyfus, L. A., and Robertson, D. C. (1979). Interaction of polymorphonuclear leukocytes with smooth and rough strains of Brucella abortus. Infect. Immun. 23, 737–742.
Kuolee, R., Harris, G., Conlan, J. W., and Chen, W. (2011). Role of neutrophils and NADPH phagocyte oxidase in host defense against respiratory infection with virulent Francisella tularensis in mice. Microbes Infect. 13, 447–456. doi: 10.1016/j.micinf.2011.01.010
Lamps, L. W., Havens, J. M., Sjostedt, A., Page, D. L., and Scott, M. A. (2004). Histologic and molecular diagnosis of tularemia: a potential bioterrorism agent endemic to North America. Mod. Pathol. 17, 489–495. doi: 10.1038/modpathol.3800087
Leitch, A. E., Riley, N. A. A., Sheldrake, T., Festa, M., Fox, S., Duffin, R., et al. (2010). The cyclin-dependent kinase inhibitor R-roscovitine down-regulates Mcl-1 to override pro-inflammatory signalling and drive neutrophil apoptosis. Eur. J. Immunol. 40, 1127–1138. doi: 10.1002/eji.200939664
Long, M. E., Lindemann, S. R., Rasmussen, J. A., Jones, B. D., and Allen, L.-A. H. (2013). Disruption of Francisella tularensis Schu S4 iglI, iglJ, and pdpC genes results in attenuation for growth in human macrophages and in vivo virulence in mice and reveals a unique phenotype for pdpC. Infect. Immun. 81, 850–861. doi: 10.1128/IAI.00822-12
Lowe, D. M., Redford, P. S., Wilkinson, R. J., O'Garra, A., and Martineau, A. R. (2012). Neutrophils in tuberculosis: friend or foe? Trends Immunol. 33, 14–25. doi: 10.1016/j.it.2011.10.003
Malik, M., Bakshi, C. S., McCabe, K., Catlett, S. V., Shah, A., Singh, R., et al. (2007). Matrix metalloproteinase 9 activity enhances host susceptibility to pulmonary infection with type A and B strains of Francisella tularensis. J. Immunol. 178, 1013–1020.
Mantovani, A., Cassatella, M. A., Costantini, C., and Jaillon, S. (2011). Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 11, 519–531. doi: 10.1038/nri3024
Mares, C. A., Sharma, J., Li, Q., Rangel, E. L., Morris, E. G., Enriquez, M. I., et al. (2011). Defect in efferocytosis leads to alternative activation of macrophages in Francisella infections. Immunol. Cell Biol. 89, 167–172. doi: 10.1038/icb.2010.81
McCaffrey, R. L., and Allen, L.-A. H. (2006). Pivotal advance: Francisella tularensis evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J. Leukoc. Biol. 80, 1224–1230. doi: 10.1189/jlb.0406287
McCaffrey, R. L., Schwartz, J. T., Lindemann, S. R., Moreland, J. G., Buchan, B. W., Jones, B. D., et al. (2010). Multiple mechanisms of NADPH oxidase inhibition by type A and type B Francisella tularensis. J. Leukoc. Biol. 88, 791–805. doi: 10.1189/jlb.1209811
McLendon, M. K., Apicella, M., and Allen, L.-A. H. (2006). Francisella tularensis: taxomony, genetics and immunopathogenesis of a potenial agent of biowarfare. Annu. Rev. Microbiol. 60, 167–185. doi: 10.1146/annurev.micro.60.080805.142126
Mellilo, A. A., Foreman, O., and Elkins, K. L. (2013). IL-12Rb2 is critical for survival of primary Francisella tularensis LVS infection J. Leukoc. Biol. 93, 657–667. doi: 10.1189/jlb.1012485
Mohapatra, N. P., Soni, S., Rajaram, M. V. S., Dang, P. M.-C., Reilly, T. J., El-Benna, J., et al. (2010). Francisella acid phosphatases inactivate the NADPH oxidase in human phagocytes. J. Immmunol. 184, 5141–5150. doi: 10.4049/jimmunol.0903413
Moreland, J. G., Hook, J. S., Bailey, G., Ulland, T., and Nauseef, W. M. (2009). Francisella tularensis directly interacts with the endothelium and recruits neutrophils with a blunted inflammatory phenotype. Am. J. Physiol. Lung Cell Mol. Physiol. 296, L1076–L1084. doi: 10.1152/ajplung.90332.2008
Nauseef, W. M. (2007). How human neutrophils kill and degrade microbes: an integrated view. Immunol. Rev. 219, 88–102. doi: 10.1111/j.1600-065X.2007.00550.x
Proctor, R. A., White, J. D., Ayala, E., and Canonico, P. G. (1975). Phagocytosis of Francisella tularensis by rhesus monkey peripheral leukocytes. Infect. Immun. 11, 146–152.
Reilly, T. J., Baron, G. S., Nano, F. E., and Kuhlenschmidt, M. S. (1996). Characterization and sequencing of a respiratory burst-inhibiting acid phosphatase from Francisella tularensis. J. Biol. Chem. 271, 10973–10983. doi: 10.1074/jbc.271.18.10973
Sadik, C. D., Kim, N. D., Iwakura, Y., and Luster, A. D. (2012). Neutrophils orchestrate their own recruitment in murine arthritis through C5aR and FcγR signaling. Proc. Natl. Acad. Sci. U.S.A. 109, E3177–E3185. doi: 10.1073/pnas.1213797109
Schricker, R. L., Eigelsbach, H. T., Mitten, J. O., and Hall, W. C. (1972). Pathogenesis of tularemia in monkeys aerogenically exposed to Francisella tularensis 425. Infect. Immun. 5, 734–744.
Schulert, G. S., McCaffrey, R. L., Buchan, B. W., Lindemann, S. R., Hollenback, C., Jones, B. D., et al. (2009). Francisella tularensis genes required for inhibition of the neutrophil respiratory burst and intramacrophage growth identified by random transposon mutagenesis of strain LVS. Infect. Immun. 77, 1324–1336. doi: 10.1128/IAI.01318-08
Schwartz, J. T., Bandyopadhyay, S., Kobayashi, S. D., McCracken, J., Whitney, A. R., Deleo, F. R., et al. (2013). Francisella tualrensis alters human neutrophil gene expression: insights into the molecular basis ob delayed neutrophil apoptosis. J. Innate Immun. 5, 5124–5136. doi: 10.1159/000342430
Schwartz, J. T., Barker, J. H., Kaufman, J., Fayram, D. C., McCracken, J. M., and Allen, L.-A. H. (2012a). Francisella tularensis inhibits the intrinsic and extrinsic pathways to delay constitutive apoptosis and prolong human neutrophil lifespan. J. Immmunol. 188, 3351–3363. doi: 10.4049/jimmunol.1102863
Schwartz, J. T., Barker, J. H., Long, M. E., Kaufman, J., McCracken, J., and Allen, L.-A. H. (2012b). Natural IgM mediates complement-dependent uptake of Francisella tularensis by human neutrophils via complement receptors 1 and 3 in nonimmune serum. J. Immunol. 189, 3064–3077. doi: 10.4049/jimmunol.1200816
Scian, R., Barrionuevo, P., Giambartolomei, G. H., de Simone, E. A., Vanzuilli, S. I., Fossati, C. A., et al. (2011). Potential role of fibroblast-like synoviocytes in joint damage induced by Brucella abortus infection through production and induction of matrix metalloproteinases. Infect. Immun. 79, 3619–3632. doi: 10.1128/IAI.05408-11
Silva, M. T. (2010). Bacteria-induced phagocyte secondary necrosis as a pathogenicity mechanism. J. Leukoc. Biol. 88, 885–896. doi: 10.1189/jlb.0410205
Sjostedt, A., Conlan, J. W., and North, R. J. (1994). Neutrophils are critical for host defense against primary infection with the facultative intracellular bacterium Francisella tularensis in mice and participate in defense against reinfection. Infect. Immun. 62, 2779–2783.
Snelgrove, R. J., Jackson, P. L., Hardison, M. T., Noerager, B. D., Kinloch, A., Gaggar, A., et al. (2010). A critical role for LTA4H in limiting chronic pulmonary neutrophilic inflammation. Science 330, 90–94. doi: 10.1126/science.1190594
Tulis, J. J., Eigelsbach, H. T., and Kerpsack, R. W. (1970). Host-parasite relationship in Monkeys administered live tularemia vaccine. Am J. Pathol. 58, 329–336.
van Houwelingen, A. H., Weathington, N. M., Verweij, V., Blalock, J. E., Nijkamp, F. P., and Folkerts, G. (2008). Induction of lung emphysema is prevented by l-arginine-threonine-arginine. FASEB J. 22, 3403–3408. doi: 10.1096/fj.07-096230
van Zandbergen, G., Gieffers, J., Kothe, H., Rupp, J., Bollinger, A., Aga, E., et al. (2004). Chlamydia pneumoniae multiply in neutrophil granulocytes and delay their spontaneous apoptosis. J. Immunol. 172, 1768–1776.
Watson, R. W., Redmond, H. P., Wang, J. H., Condron, C., and Bouchier-Hayes, D. (1996). Neutrophils undergo apoptosis following ingestion of Escherichia coli. J. Immunol. 156, 3986–3992.
Weathington, N. M., van Houwelingen, A. H., Noerager, B. D., Jackson, P. L., Kraneveld, A. D., Galin, F. S., et al. (2006). A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat. Med. 12, 317–323. doi: 10.1038/nm1361
Witko-Sarsat, V., Pederzoli-Ribeil, M., Hirsh, E., Sozzani, S., and Cassatella, M. A. (2011). Regulating neutrophil apoptosis: new players enter the game. Trends Immunol. 32, 117–124. doi: 10.1016/j.it.2011.01.001
Woolard, M. D., Wilson, J. E., Hensley, L. L., Jania, L. A., Kawula, T. H., Drake, J. R., et al. (2007). Francisella tularensis-infected macrophages release prostaglandin E2 that blocks T Cell proliferation and promotes a Th2-like response. J. Immunol. 178, 2065–2074.
Keywords: neutrophils, apoptosis, inflammation, Francisella tularensis, innate immunity
Citation: Allen L-AH (2013) Neutrophils: potential therapeutic targets in tularemia? Front. Cell. Infect. Microbiol. 3:109. doi: 10.3389/fcimb.2013.00109
Received: 06 November 2013; Paper pending published: 26 November 2013;
Accepted: 14 December 2013; Published online: 27 December 2013.
Edited by:
Max Maurin, Université Aix-Marseille II, FranceCopyright © 2013 Allen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lee-Ann H. Allen, Inflammation Program and the Departments of Internal Medicine and Microbiology, University of Iowa and the VA Medical Center, 2501 Crosspark Rd., MTF D-154, Coralville, IA 52241, USA e-mail:bGVlLWFubi1hbGxlbkB1aW93YS5lZHU=