 Jianwu Pei
Jianwu Pei Melissa Kahl-McDonagh
Melissa Kahl-McDonagh Thomas A. Ficht
Thomas A. Ficht- Department of Veterinary Pathobiology, Texas A&M University and Texas Agricultural Experiment Station, College Station, TX, USA
It has long been observed that smooth Brucella can dissociate into rough mutants that are cytotoxic to macrophages. However, the in vivo biological significance and/or mechanistic details of Brucella dissociation and cytotoxicity remain incomplete. In the current report, a plaque assay was developed using Brucella strains exhibiting varying degrees of cytotoxicity. Infected monolayers were observed daily using phase contrast microscopy for plaque formation while Brucella uptake and replication were monitored using an immunofluorescence assay (IFA). Visible plaques were detected at 4–5 days post infection (p.i.) with cytotoxic Brucella 16MΔmanBA at an MOI of 0.1. IFA staining demonstrated that the plaques consisted of macrophages with replicating Brucella. Visible plaques were not detected in monolayers infected with non-cytotoxic 16MΔmanBAΔvirB2 at an MOI of 0.1. However, IFA staining did reveal small groups of macrophages (foci) with replicating Brucella in the monolayers infected with 16MΔmanBAΔvirB2. The size of the foci observed in macrophage monolayers infected with rough Brucella correlated directly with cytotoxicity measured in liquid culture, suggesting that cytotoxicity was essential for Brucella egress and dissemination. In monolayers infected with 16M, small and large foci were observed. Double antibody staining revealed spontaneous rough mutants within the large, but not the small foci in 16M infected monolayers. Furthermore, plaque formation was observed in the large foci derived from 16M infections. Finally, the addition of gentamicin to the culture medium inhibited plaque formation, suggesting that cell-to-cell spread occurred only following release of the organisms from the cells. Taken together, these results demonstrate that Brucella-induced cytotoxicity is critical for Brucella egress and dissemination.
Introduction
Brucella is a genus of Gram-negative, facultative intracellular bacteria that cause brucellosis in a variety of animals and undulant fever in humans. Ten species have been described to date (Whatmore, 2009), and three species, B. melitensis, B. abortus, and B. suis provide the major threat to agriculture and public health worldwide (Boschiroli et al., 2001).
Macrophages/monocytes are the primary target cells in which Brucella replicate and cause persistent infection and as such are essential dissemination within the host (Baldwin and Winter, 1994; Liautard et al., 1996). Recent studies have shown that Brucella modulates the fate of infected macrophages and monocytes. Smooth Brucella infection inhibits macrophage and monocyte apoptosis by targeting the intrinsic (mitochondrial) and extrinsic (death receptor) pathways (Galdiero et al., 2000; Gross et al., 2000; Eskra et al., 2003; Fernandez-Prada et al., 2003; Tolomeo et al., 2003; He et al., 2006; Covert et al., 2009). In contrast, Brucella rough mutant infection results in type four-secretion system (T4SS) dependent macrophage cell death (Pei and Ficht, 2004; Pei et al., 2006, 2008b; De Jong et al., 2008; Zhong et al., 2009). It has been shown in a number of intracellular bacterial species that the regulation of host cell apoptosis is important to pathogenesis. Prevention of host cell apoptosis provides a hospitable intracellular niche for multiplication (Hacker and Fischer, 2002; Faherty and Maurelli, 2008) while induction of host cell death promotes bacterial release (Weinrauch and Zychlinsky, 1999; Gao and Kwaik, 2000b).
Although initial observations documenting rough Brucella-induced cell death in guinea pig macrophages occurred 50 years ago (Freeman et al., 1961), the mechanisms responsible have only been recently investigated (Pei and Ficht, 2004; Pei et al., 2006, 2008b; De Jong et al., 2008; Chen and He, 2009; Zhong et al., 2009; Chen et al., 2011), and the biological significance of the Brucella cytotoxicity remains undefined. In the current study, a plaque formation assay was developed to better evaluate Brucella-induced cytotoxicity. Comparison using Brucella strains with different levels of cytotoxicity provide direct evidence that cytotoxicity plays an important role in Brucella egress and dissemination in culture.
Materials and Methods
Bacteria Strains and Media
Bacterial strains used in these experiments include B. melitensis 16M, rough mutants 16MΔmanBA and 16MΔmanBAΔvirB2 (Pei et al., 2008b), and the rough B. abortus vaccine strain RB51. Brucella strains were routinely grown in tryptic soy agar (TSA) or tryptic soy broth (TSB) as described previously (Pei and Ficht, 2004).
Cell Culture and Infection
Murine macrophage-like cells J774.A1 (ATCC, TIB-67) were grown in DMEM with 10% (v/v) fetal bovine serum, 1 mM L-glutamine, and 1 mM non-essential amino acid as described previously (Pei and Ficht, 2004). For plaque assays, 1.25 × 105 cells were seeded into each well of a 24-well plate and incubated overnight at 37°C in atmosphere containing 5% CO2 prior to inoculation with Brucella at various multiplicities of infection (MOI). Infections were synchronized by centrifugation at 200× g for 5 min at room temperature and the plates were incubated at 37°C for 20 min. Cell monolayers were washed with PBS (pH 7.4) three times, and complete DMEM containing 50 μg/ml of gentamicin was added to kill extracellular bacteria with incubation at 37°C for 1 h (Pei and Ficht, 2004). Brucella uptake was determined following a 1-h incubation by washing the monolayers with PBS and lysing the cells with 0.5% (w/v) Tween 20 in distilled water. CFUs present in the lysates were determined as described previously (Pei and Ficht, 2004).
Plaque Formation Assay (Oaks et al., 1985)
J774.A1 macrophages cultured in 24-well plates (1.25 × 105 cells/well) were infected with Brucella as described above. Medium was replaced with 1 ml of warm (45°C) complete DMEM without gentamicin containing 1% (w/v) ultra-pure agarose (Gibco. Gaithersburg, MD). One milliliter of complete DMEM (with or without gentamicin) was added to each well following agarose solidification. Liquid media were changed every 2 days, and cell monolayers were observed daily using phase contrast light microscopy for plaque formation. Following incubation, liquid medium was removed and replaced with sufficient formalin to fix the cells and kill Brucella [3.7% (v/v) formaldehyde final] with incubation overnight at room temperature.
Immunofluorescence Assay (IFA)
Following fixation, the agarose was carefully removed and the cell monolayer washed with PBS. Infected cells were stained with goat anti Brucella serum and rabbit anti-rough Brucella monospecific serum (1:1000) in PBS-TT (PBS with 0.05% (v/v) Tween-20 and 0.05% (v/v) Triton X-100) for smooth and rough Brucella, respectively. Following three washes with PBS-T (PBS with 0.05% Tween-20), the cells were incubated with secondary antibodies including donkey anti goat IgG Alexa Fluor 488, chicken anti rabbit IgG Alexa Fluor 488, or chicken anti rabbit IgG Alexa Fluor 594 (Molecular Probes) (1:1000 in PBS-TT). Bacteria were revealed using IX70 fluorescence microscopy (Olympus).
Double Antibody Staining
To differentiate rough mutants from smooth Brucella, J774.A1 macrophages were inoculated with 16M, 16MΔmanBA, or a mixture of 16M and 16MΔmanBA (10:1). The cells were fixed at 1 h after infection and incubated with rabbit anti Brucella mono-specific M serum (1:500 in PBS-TT) and goat anti B. ovis serum (1:500 in PBS-TT). Following three washes with PBS-T, the cells were incubated with chicken anti rabbit IgG Alexa Fluor 488 and donkey anti goat IgG Alexa Fluor 594 (1:1000 in PBS-TT). Bacteria were revealed using IX70 fluorescence microscopy.
Plaque-Forming Unit
To enumerate plaque-forming units (PFUs) in each well, PFUs were averaged over five randomly selected fields in 16MΔmanBA infected monolayers using phase contrast and IX70 fluorescence microscopy (10× objective). Since, the area covered by each field under 10× objective lens is 1.798 mm2 and the total surface area is 200 mm2 the number of PFU/well is equal to the average PFU/field × (200/1.798).
Results
Plaque Formation Associated with Brucella Cytotoxicity
Previous studies have shown that Brucella rough mutants proliferate in murine macrophage J774.A1 causing oncotic and necrotic cell death (Pei and Ficht, 2004; Pei et al., 2006). Therefore, infection with cytotoxic Brucella in macrophage monolayers with low MOI was predicted to produce plaques, resulting from replication and release of bacteria that infect neighboring cells to cause localized lysis. To test this hypothesis, a plaque assay was developed using the cytotoxic mutant 16MΔmanBA while the non-cytotoxic Brucella mutant 16MΔmanBAΔvirB2 was employed as control (Pei et al., 2008b). In order to detect individual plaques, J774.A1 macrophages were infected with Brucella at low MOI (1.0 or 0.1). All the macrophages in the wells infected with 16MΔmanBA at 1 MOI were lysed within 4 days, and no individual plaques were observed. Plaques were visible between 4 and 5 days in monolayers infected with 16MΔmanBA at an MOI of 0.1 (Figure 1A). Neither cell death nor plaques were observed in cell monolayers infected with 16MΔmanBAΔvirB2 at MOIs of 1 or 0.1 (data not shown) (Figure 1B). Since the MOI used was 0.1, at most only one in 10 macrophage are expected to be infected, and this was confirmed by IFA staining of infected cells at 1 h p.i. (shown in the following section). The results suggested that rough, cytotoxic Brucella replicated in macrophages causing cell death, and the bacteria released re-infected neighboring cells to cause a localized cell death. The process repeated itself until an area of dead cells (plaque) was observed (Figure 1A). MOI of 0.1 was used in all plaque assays described in this report unless otherwise specified.

Figure 1. Plaque formation in Brucella-infected macrophage monolayers associated with cytotoxicity. J774.A1 macrophages were infected at an MOI of 0.1 with B. melitensis mutant 16MΔmanBA (A), 16MΔmanBAΔvirB2 0.1 (B), or left uninfected, as control (C). The agarose (1% w/v) overlay and DMEM were added at 1 h p.i. as described in Materials and Methods, and incubated an additional 5 days. The cells were fixed overnight in 3.7% (w/v) formaldehyde and observed using IX70 microscopy. Scale bar, 100 μm.
To further determine whether the plaques observed in 16MΔmanBA infected cells (Figure 2A) were caused by Brucella replication, the infected cell monolayer was stained using the Immunofluorescence assay (IFA) for Brucella at 4 days p.i. The results revealed that significant replication of 16MΔmanBA occurred in cells comprising the plaques (Figure 2B). These results indicated that plaque formation was a direct result of the intracellular replication of the cytotoxic Brucella mutants.

Figure 2. Plaque formation corresponds with increased intracellular Brucella replication. J774.A1 macrophages were infected with 16MΔmanBA at an MOI of 0.1. An agarose over lay and DMEM were added at 1 h p.i. and incubated an additional 4 days. Following fixation in 3.7% (w/v) formaldehyde, rough Brucella were visualized via IFA and IX70 microscopy under phase contrast field (A) and fluorescent field (B) (showing Brucella replication in green) microscopy. Scale bar, 100 μm.
Brucella Cytotoxicity was Essential for Bacteria Egress and Subsequent Reinfection
The results described in the previous section indicated that plaques were composed of dead, lysed or shedding cells containing replicating Brucella. However, differences in plaque size were apparent depending on the strain employed and it was hypothesized that plaque size was directly related to Brucella cytotoxicity, as measured by LDH release (Pei and Ficht, 2004). To test this hypothesis, J774.A1 macrophages were infected with Brucella strains exhibiting different levels of cytotoxicity: 16MΔmanBA (high cytotoxicity), 16MΔmanBAΔvirB2 (no cytotoxicity), and B. abortus rough vaccine strain RB51 (low cytotoxicity) (Pei and Ficht, 2004; Pei et al., 2008b). Brucella replication and spread (designated foci) were detected by 4 days p.i. using IFA. As predicted, foci of infected macrophage monolayers containing RB51 were smaller than those in 16MΔmanBA-infected cells, but larger than the foci in monolayers infected with non-cytotoxic mutant 16MΔmanBAΔvirB2 (Figure 3). These results suggested that cytotoxicity was directly related to Brucella dissemination and subsequent re-infection presumably as a result of enhanced egress, suggesting a possible role for cytotoxicity in the spread of infection.
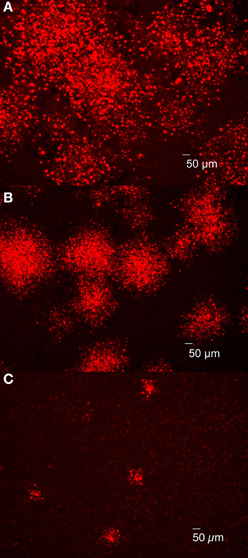
Figure 3. Brucella cytotoxicity was essential for bacterial egress and subsequent re-infection. J774.A1 macrophages were infected with 16MΔmanBA (A), RB51 (B) or 16MΔmanBAΔvirB2 (C) at an MOI of 0.1. Agarose overlay and DMEM were added at 1 h p.i. and incubated for 4 days. Following formaldehyde fixation, the cells were stained via IFA to detect rough Brucella observed using IX70 microscopy. Scale bar, 50 μm.
To further determine whether plaques can develop from an individual bacterium, uptake and replication of cytotoxic and non-cytotoxic Brucella were examined in J774.A1 monolayers infected with 16MΔmanBA or 16MΔmanBAΔvirB2. The infected monolayers were fixed at 1 h p.i. and visualized via rough-specific Brucella IFA staining to evaluate bacterial uptake. The cell monolayers were fixed at 2, 3, and 4 days p.i. and stained to monitor plaque (cytotoxicity) and focus (replication) formation. Staining of the infected cells following 1 h incubation confirmed a low-level of bacterial uptake, per infected cell (Figures 4A,E) and no demonstrable bacterial aggregation. By day 2, individual cells containing replicating Brucella were detected in the monolayers (Figures 4B,F), indicating that the bacteria replicated within the macrophages. By day 3, groups of cells containing Brucella were observed in the infected monolayers (Figures 4C,G). By day 4, large plaques consisting of dead cells were observed in the 16MΔmanBA infected cells using light microscopy (as shown in Figure 2A), and the cells within the foci were full of Brucella as revealed by IFA (Figure 4D). Although no plaques were observed in 16MΔmanBAΔvirB2 infected monolayers at day 4 using light microscopy, foci containing large numbers of 16MΔmanBAΔvirB2 were observed following IFA staining (Figure 4H). However, the foci detected by IFA were much smaller than those in the monolayers infected with 16MΔmanBA (Figure 4D). These results suggest that individual plaques or foci are formed by replication of a single bacterium, and further confirmed that the cytotoxicity was important for re-infection via extracellular spread to neighboring cells via egress from the host macrophage.
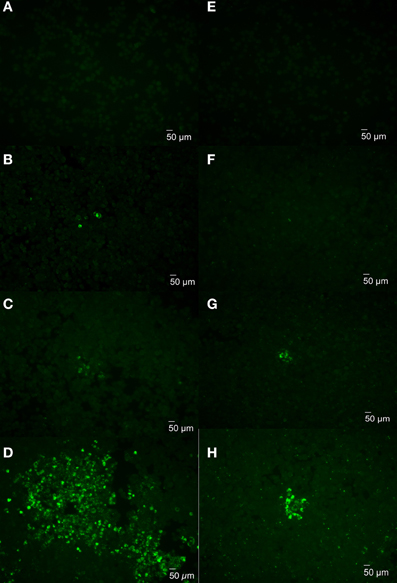
Figure 4. Monitoring plaque formation during Brucella infection. J774.A1 macrophages were infected with 16MΔmanBA (A–D) or 16MΔmanBAΔvirB2 (E–H) at an MOI of 0.1. Agarose overlay and DMEM were added at 1 h p.i. and incubated an additional 4 days. The cells were fixed at 1 h (A,E), 2 days (B,F), 3 days (C,G), and 4 days (D,H) p.i., and stained to visualize intracellular Brucella via IFA. Scale bar, 50 μm.
Brucella Dissociation Assists Organism Dissemination
Previous studies in our lab revealed that Brucella dissociation occurs in vitro and in vivo at elevated frequency and can result in an accumulation of rough variants in infected macrophages (Turse et al., 2011). However, previous studies have also revealed low levels of cytotoxicity associated with infection by wild type smooth Brucella at elevated MOI (Pei et al., 2008b). In order to determine whether ongoing dissociation, rather than low-level toxicity, is necessary for bacterial egress and dissemination, infection foci were evaluated for dissociation using a double antibody staining method described in the Materials and Methods. The results confirm the binding specificities of the rabbit anti-Brucella mono-specific M serum for wild type 16M (Figures 5A,C), and the goat anti-B. ovis serum for rough variants (Figures 5B,D). Antibody specificity and the capacity to distinguish smooth vs. rough Brucella during a mixed infection were confirmed using cells infected with a 10:1 mixture (smooth to rough) of Brucella, in which smooth Brucella were revealed in green (Figure 5E) and rough Brucella were revealed in red (Figure 5F).

Figure 5. Double antibody staining differentiates rough variants from smooth Brucella. J774.A1 macrophages were inoculated at an MOI of 50 with 16M (A,B), 16MΔmanBA (C,D) or a 10:1 mixture of 16M and 16MΔmanBA (E,F). The cells were fixed at 1 h p.i. and incubated with rabbit mono-specific anti-M serum (1:500 in PBS-TT) and goat anti-B. ovis serum (1:500 in PBS-TT). Intracellular smooth (A,C,E) and rough (B,D,F) Brucella were revealed following incubation with conjugated secondary antibodies chicken anti rabbit IgG Alexa Fluor 488 and donkey anti goat IgG Alexa Fluor 594 (1:1000 in PBS-TT). Panels A+B, C+D, E+F are derived by merging the appropriate channels. Scale bar, 5 μm.
To detect rough dissociation in infected cells, J774.A1 macrophage monolayers were infected with 16M and fixed at 1 h and 4 days p.i. Because no antibiotic was added during experimentation, overgrowth of 16M in the media prevented further analysis beyond 4 days p.i. Double antibody staining of the cells fixed at 1 h p.i. confirmed a low-level bacterial uptake without bacterial aggregation or detectable dissociation (data not shown). Staining of the cells fixed at 4 days p.i. revealed two types of foci: small foci consisting of a few cells with replicating Brucella (Figure 6A) and large foci in which the cells that had not sloughed-off the surface of the plate were retained (Figures 6B,C). Double antibody staining confirmed the presence of significant numbers of rough Brucella (Figures 6D,F) mixed with smooth Brucella (Figures 6E,F) in the large foci that were not detected in the small foci (data not shown). These results are consistent with the hypothesis that rough dissociation enhances Brucella dissemination.

Figure 6. Brucella dissociation detected in large foci following 16M infection. J774.A1 macrophages were infected with 16M at an MOI of 0.1. Agarose overlay and fresh DMEM were added at 1 h p.i. and incubation was continued for an additional 4 days. Following fixation in 3.7 % (w/v) formaldehyde, double antibody staining (rabbit mono-specific anti-M serum and goat anti B. ovis serum) was performed as described in the legend to Figure 5. Small (A) and large (B) foci were observed. Cell death was observed in the large foci (panel C, a bright field image of panel B). Rough variants (red, D) arising during growth of the parental organism (green, E) were detected in the large foci. Panel (F) is the merged image of panels (D,E), an enlarged square shown in (B). Scale bars, 50 μm in (A–C); 10 μm in (D–F).
To determine whether each Brucella-infected macrophage formed a plaque or focus of infection, PFUs in each well were evaluated via IFA staining at 4 days p.i. and compared with the uptake colony forming unit (CFUs) determined by gentamicin protection assay at 1 h p.i. The results indicated that 26.5% of the invading 16MΔmanBA formed plaques; only 4.6% of the invading 16M and 0.25% of the invading 16MΔmanBAΔviB2 formed foci.
Inhibition of Brucella Dissemination with the Addition of Gentamicin to Growth Medium
Bacterial spread from cell to cell can be accomplished by invasion of neighboring cells without release into the medium, such as Listeria monocytogenes (Mounier et al., 1990), or following release and re-infection of neighboring cells. To determine whether Brucella spread from cell to cell directly or via medium, experiments were performed in which gentamicin (50 μg/ml) was added to the overlay and DMEM so as to inactivate bacteria on the cell surface or released into the media and, as such, are not protected by intracellular uptake. Comparison of rough B. melitensis 16MΔmanBA and smooth 16M via IFA staining revealed small foci consisting of cells with replicating Brucella formed in monolayers infected with 16MΔmanBA (Figure 7A) and 16M (Figure 7B). But, neither plaques nor cell death were detected via phase contrast microscopy by 4 days p.i. (data not shown). In addition, the size of the foci was much smaller compared with those present in the infected monolayers without gentamicin treatment (Figures 7C,D). These results are consistent with the hypothesis that Brucella dissemination occurs via release into the medium with subsequent cell reinfection, and does not occur via cell-to-cell contact.
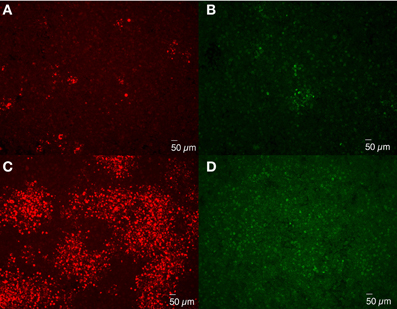
Figure 7. Extracellular cell-to-cell dissemination of Brucella. J774.A1 macrophages were infected with 16MΔmanBA (A,C) or 16M (B,D) at an MOI of 0.1. An agarose overlay was added at 1 h p.i. along with fresh DMEM supplemented with (A,B) or without (C,D) 50 μg/ml of gentamicin. Following an additional 4 days of incubation, the cells were fixed and stained to visualize intracellular Brucella via IFA. Scale bar, 50 μm.
Discussion
Pathogens have developed various strategies to evade host innate and adaptive immune systems (Finlay and McFadden, 2006), one of which is to manipulate host cell viability (Guiney, 2005). Similarly, both necrotic/apoptotic and anti-apoptotic cell death have been reported in macrophage during Brucella infection (Freeman et al., 1961; Gross et al., 2000; Fernandez-Prada et al., 2003; Pei and Ficht, 2004). Although several mechanisms have been thoroughly studied (Eskra et al., 2003; He et al., 2006; Pei et al., 2006, 2008b; De Jong et al., 2008; Chen and He, 2009; Zhong et al., 2009; Chen et al., 2011), their biological significance has not been established. The current study demonstrates that Brucella cytotoxicity enhances bacterial egress and dissemination.
An extensive amount of work has been performed regarding Brucella invasion and subversion of host cell functions thought to be critical to establishment of a hospitable replicative niche. However, another critical aspect of Brucella pathogenesis is organism egress from the host cell and continued dissemination. Seven potential exiting strategies or mechanisms used by different pathogens have been described in the literature to date (Hybiske and Stephens, 2008). Legionella generates pores in phagosomal or cellular membranes during different stages of infection inducing host cell cytotoxicity that is required for bacterial egress (Kirby et al., 1998; Alli et al., 2000; Gao and Kwaik, 2000a; Molmeret et al., 2002; Zink et al., 2002; Chen et al., 2004). Salmonella replication induces macrophage oncosis resulting in bacterial release from the cell (Sano et al., 2007). Similarly, our previous studies have revealed that rough Brucella infection induced pore formation on macrophage cell membranes to cause cell necrosis/oncosis (Pei et al., 2006). We provide support for this idea in the current study by demonstrating that Brucella cytotoxicity determines focus size, consistent with the idea that Brucella cytotoxicity is important for bacterial egress and dissemination.
Plaque formation was not apparent in the presence of gentamicin indicating that Brucella infection of neighboring cells occurs via extracellular dissemination of the organism. The reduced size of the foci formed under these conditions confirms the importance of extracellular spread of the organism to infection. Although the results obtained do not rule out the capacity of Brucella to disseminate using actin-based protrusion (used by Listeria monocytogenes, Shigella flexneri, Rickettsia rickettsi, Rickettsia conorii, Burkholderia pseudomallei, and Mycobacterium marinum), budding (by Orientia tsutsugamushi), or extrusion (Chlamydia spp.) (Hybiske and Stephens, 2008); the difference in the size of the foci observed suggests that Brucella dissemination is primarily the result of cell lysis and extracellular dissemination through the medium.
It has been proposed that two steps are required for Legionella release from infected cells. First, replicating Legionella form pores in the phagosomal membranes causing phagosome disruption. Second, bacteria released into the cytoplasm form pores in cell membrane resulting in cell lysis (Molmeret and Abu Kwaik, 2002). Our previous study has shown that rough Brucella are retained within intact vacuoles identified within dead macrophages (Pei et al., 2006), suggesting that phagosomal membranes are not disrupted, and suggesting that only a single step is involved in the release of Brucella from macrophages. A complete or well-defined description of Brucella-containing vacuoles (BCV) is not available, but the result described suggests that a putative Brucella “cytotoxin” might only form pores on the cell membrane, not on the BCV membrane. Inhibition of plaque formation by gentamicin treatment suggested that these BCVs do not protect the organisms from gentamicin. Since rough Brucella are sensitive to complement- and cationic peptide-mediated lysis, organisms released in intact BCV derived from lysed macrophages may be protected by the vacuoles and phagocytosed by surviving macrophages to start a new round of infection. Therefore, the one-step release may facilitate Brucella dissemination.
At the MOI used in the plaque assays, i.e., ≤0.1, bacterial uptake by infected cells is not expected to exceed more than one bacterium per infected cell. Yet, massive bacterial replication can be detected as soon as 4 days p.i. (Figures 2, 4). This is consistent with previous reports documenting rough Brucella mutant replication in HeLa cells and macrophages (Godfroid et al., 1998; Ugalde et al., 2000; Porte et al., 2003; Pei and Ficht, 2004; Pei et al., 2008b). These results demonstrate that Brucella cytotoxicity is not simply due to an uptake of excessively high numbers of Brucella, but derive from the replication of cytotoxic Brucella within the macrophages. The results also reveal that individual plaques are formed from cells infected with a single bacterium. The small foci associated with non-cytotoxic 16MΔmanBAΔvirB2 result from their poor release from infected cells, despite significant levels of intracellular replication. It is not clear why the foci in non-cytotoxic strain infected monolayers was lower than that in cytotoxic strain infected monolayers. However, one possibility is that the foci formed by the non-cytotoxic strain are so small that they are easily missed during enumeration.
The T4SS is the predominant virulence factor identified in Brucella to date. It is widely accepted that the T4SS is essential for Brucella survival within host cells, and is consistent with the attenuated survival of Brucella ΔvirB mutants in the mouse model (Hong et al., 2000). Yet, despite the well-defined role of the T4SS in Brucella intracellular trafficking (Comerci et al., 2001; Delrue et al., 2001; Celli et al., 2003), recent reports show that virB mutants survive in the host as well as wild type organisms for the first 3 days in vivo (Roux et al., 2007). Since the T4SS is essential for Brucella cytotoxicity (De Jong et al., 2008; Pei et al., 2008b; Zhong et al., 2009) and subsequent bacterial dissemination, in vivo attenuation of virB mutants could be explained by a failure to disseminate within the host just as well as failing to obtain a replication niche. This hypothesis is supported by our recent studies showing that the 16MΔmanBAΔvirB2 mutant was cleared from infected mice within 1 week, while ΔmanBA and ΔvirB2 single mutants persist in mice beyond 4 weeks (Pei and Ficht, unpublished data).
To evade the immune system for survival in the host, many pathogenic Gram-negative bacteria have the ability to alter their LPS structure, including smooth-rough variation (Lukacova et al., 2008). The current results suggest that the spontaneous appearance of rough variants from smooth Brucella may function in the dissemination of infection. Brucella dissociation, shown to be enhanced in acidic environments (Braun, 1946a), may induce dissociation and subsequently assist Brucella dissemination from within acidic phagosomes (Porte et al., 1999; Boschiroli et al., 2002). It should be pointed out that dissociated Brucella can still revert to smooth phenotype (Braun, 1947). Therefore, it is possible that some smooth Brucella dissociated into a rough phenotype when needed to disseminate, and can still revert to smooth phenotype following egress to resist intracellular killing.
It has been demonstrated that Brucella dissociation is genetically based (Braun, 1946a; Mancilla et al., 2010, 2012, 2013; Turse et al., 2011). Each individual bacterium may have a different dissociation rate. In the current study, the dissociation rate was not determined, but a study conducted by Braun showed that the percentage of rough organisms in individual cultures was different (Braun, 1946a). This could be the reason why different size foci were observed following smooth Brucella infection. Mancilla et al. demonstrated that phage integrase-mediated excision of genomic island 2 (GI-2) and ISBm1-mediated excision of wbkA glycosyltransferase gene were partially responsible for Brucella rough dissociation (Mancilla et al., 2010). Although knockout of these genes could not eliminate the dissociation, it would be interesting to determine the disseminating abilities of these mutants in comparison with the wild type. Starr et al. reported recently that forming autophagic Brucella-containing vacuoles (aBCV) promoted Brucella egress in HeLa cells. However, the phenomenon was not tested in macrophages in this report (Starr et al., 2012). Since autophagy is involved in Brucella survival and replication in macrophages (Qin et al., 2008; Guo et al., 2012; Starr et al., 2012), its role in Brucella egress from macrophage needs to be investigated.
Brucella dissociation was observed more than 60 years ago (Stearns and Roepke, 1941; Braun, 1945, 1946a,b). However, the biological significance was not identified. Our current study revealed that Brucella dissociation enhanced bacterial dissemination, which may enhance Brucella virulence. A working model was proposed based on the results from this report and published studies (Figure 8). During smooth Brucella infection, the organism traffics to ER-like compartments and replicates (Celli et al., 2003). During replication, some of the organisms dissociate into a rough phenotype and accumulate in the host cells (Turse et al., 2011). The rough mutants with enhanced T4SS produce more cytotoxic factors (Pei et al., 2008b). Once rough mutant accumulation reaches a threshold level, the host cell will die from necrosis and apoptosis (Pei and Ficht, 2004; Pei et al., 2006; De Jong et al., 2008; Chen and He, 2009). The cell contents including the organisms will be released. Smooth Brucella will subsequently infect more macrophages and start a new round of replication and dissociation. Rough mutants may be killed by complement or other cationic peptide-mediated lysis (Allen et al., 1998). Rough Brucella induced cytokine and chemokine release (Rittig et al., 2003; Pei et al., 2008a) and macrophage necrotic cell death result in inflammatory responses, which in turn recruits more macrophages to the infection sites to help Brucella dissemination (Figure 8). This working model is strongly supported by the undulant fever presentation of human brucellosis.
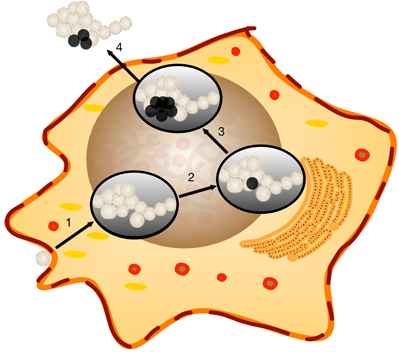
Figure 8. Working model of Brucella host cell egress. Brucella dissociation has been previously reported to continue unabated within the host cell. The properties of rough derivatives would lend themselves to bacterial dissemination and the induction of an inflammatory response sufficient to attract new target cells. Step 1, Smooth Brucella, that are resistant to extracellular killing mechanisms, successfully invades target macrophages where replication occurs within a Brucella-containing vacuole (BCV). Step 2, dissociation occurs in association with replication within the BCV as observed in vitro. Step 3, cytotoxic activity, enhanced in rough mutants, begins to break down the the cellular membrane. Step 4, Brucella are released from the cell and successive rounds of replication and dissociation continue. Rough Brucella infection and macrophage necrotic cell death induce inflammatory responses, which recruit more macrophages to the infection sites.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by grants to Thomas A. Ficht from NIH (R01-AI48496), the Western Regional Center for Excellence-WRCE (1U54-AI057156), and Hatch Project TEX09219.
References
Allen, C. A., Adams, L. G., and Ficht, T. A. (1998). Transposon-derived Brucella abortus rough mutants are attenuated and exhibit reduced intracellular survival. Infect. Immun. 66, 1008–1016.
Alli, O. A., Gao, L. Y., Pedersen, L. L., Zink, S., Radulic, M., Doric, M., et al. (2000). Temporal pore formation-mediated egress from macrophages and alveolar epithelial cells by Legionella pneumophila. Infect. Immun. 68, 6431–6440. doi: 10.1128/IAI.68.11.6431-6440.2000
Boschiroli, M. L., Foulongne, V., and O'Callaghan, D. (2001). Brucellosis: a worldwide zoonosis. Curr. Opin. Microbiol. 4, 58–64. doi: 10.1016/S1369-5274(00)00165-X
Boschiroli, M. L., Ouahrani-Bettache, S., Foulongne, V., Michaux-Charachon, S., Bourg, G., Allardet-Servent, A., et al. (2002). The Brucella suis virB operon is induced intracellularly in macrophages. Proc. Natl. Acad. Sci. U.S.A. 99, 1544–1549. doi: 10.1073/pnas.032514299
Braun, W. (1945). Factors controlling bacterial dissociation. Science 101, 182–183. doi: 10.1126/science.101.2616.182
Braun, W. (1946a). Dissociation in Brucella abortus: a demonstration of the role of inherent and environmental factors in bacterial variation. J. Bacteriol. 51, 327–349.
Braun, W. (1946b). Dissociation in Brucella abortus: a demonstration of the role of inherent and environmental factors in bacterial variation. J. Bacteriol. 52, 243–249.
Braun, W. (1947). Bacterial dissociation: a critical review of a phenomenon of bacterial variation. Bacteriol. Rev. 11, 75–114.
Celli, J., De Chastellier, C., Franchini, D. M., Pizarro-Cerda, J., Moreno, E., and Gorvel, J. P. (2003). Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J. Exp. Med. 198, 545–556. doi: 10.1084/jem.20030088
Chen, F., Ding, X., Ding, Y., Xiang, Z., Li, X., Ghosh, D., et al. (2011). Proinflammatory caspase-2-mediated macrophage cell death induced by a rough attenuated Brucella suis strain. Infect. Immun. 79, 2460–2469. doi: 10.1128/IAI.00050-11
Chen, F., and He, Y. (2009). Caspase-2 mediated apoptotic and necrotic murine macrophage cell death induced by rough Brucella abortus. PLoS ONE 4:e6830. doi: 10.1371/journal.pone.0006830
Chen, J., De Felipe, K. S., Clarke, M., Lu, H., Anderson, O. R., Segal, G., et al. (2004). Legionella effectors that promote nonlytic release from protozoa. Science 303, 1358–1361. doi: 10.1126/science.1094226
Comerci, D. J., Martinez-Lorenzo, M. J., Sieira, R., Gorvel, J. P., and Ugalde, R. A. (2001). Essential role of the VirB machinery in the maturation of the Brucella abortus-containing vacuole. Cell. Microbiol. 3, 159–168. doi: 10.1046/j.1462-5822.2001.00102.x
Covert, J., Mathison, A. J., Eskra, L., Banai, M., and Splitter, G. (2009). Brucella melitensis, B. neotomae and B. ovis elicit common and distinctive macrophage defense transcriptional responses. Exp. Biol. Med. (Maywood) 234, 1450–1467. doi: 10.3181/0904-RM-124
De Jong, M. F., Sun, Y. H., Den Hartigh, A. B., Van Dijl, J. M., and Tsolis, R. M. (2008). Identification of VceA and VceC, two members of the VjbR regulon that are translocated into macrophages by the Brucella type IV secretion system. Mol. Microbiol. 70, 1378–1396. doi: 10.1111/j.1365-2958.2008.06487.x
Delrue, R. M., Martinez-Lorenzo, M., Lestrate, P., Danese, I., Bielarz, V., Mertens, P., et al. (2001). Identification of Brucella spp. genes involved in intracellular trafficking. Cell. Microbiol. 3, 487–497. doi: 10.1046/j.1462-5822.2001.00131.x
Eskra, L., Mathison, A., and Splitter, G. (2003). Microarray analysis of mRNA levels from RAW264.7 macrophages infected with Brucella abortus. Infect. Immun. 71, 1125–1133. doi: 10.1128/IAI.71.3.1125-1133.2003
Faherty, C. S., and Maurelli, A. T. (2008). Staying alive: bacterial inhibition of apoptosis during infection. Trends Microbiol. 16, 173–180. doi: 10.1016/j.tim.2008.02.001
Fernandez-Prada, C. M., Zelazowska, E. B., Nikolich, M., Hadfield, T. L., Roop, R. M. 2nd., Robertson, G. L., et al. (2003). Interactions between Brucella melitensis and human phagocytes: bacterial surface O-Polysaccharide inhibits phagocytosis, bacterial killing, and subsequent host cell apoptosis. Infect. Immun. 71, 2110–2119. doi: 10.1128/IAI.71.4.2110-2119.2003
Finlay, B. B., and McFadden, G. (2006). Anti-immunology: evasion of the host immune system by bacterial and viral pathogens. Cell 124, 767–782. doi: 10.1016/j.cell.2006.01.034
Freeman, B. A., Kross, D. J., and Circo, R. (1961). Host-parasite relationships in brucellosis. II. destruction of macrophage cultures by Brucella of different virulence. J. Infect. Dis. 108, 333–338. doi: 10.1093/infdis/108.3.333
Galdiero, E., Romano Carratelli, C., Vitiello, M., Nuzzo, I., Del Vecchio, E., Bentivoglio, C., et al. (2000). HSP and apoptosis in leukocytes from infected or vaccinated animals by Brucella abortus. New Microbiol. 23, 271.
Gao, L. Y., and Kwaik, Y. A. (2000a). The mechanism of killing and exiting the protozoan host Acanthamoeba polyphaga by Legionella pneumophila. Environ. Microbiol. 2, 79–90. doi: 10.1046/j.1462-2920.2000.00076.x
Gao, L. Y., and Kwaik, Y. A. (2000b). The modulation of host cell apoptosis by intracellular bacterial pathogens. Trends Microbiol. 8, 306–313. doi: 10.1016/S0966-842X(00)01784-4
Godfroid, F., Taminiau, B., Danese, I., Denoel, P., Tibor, A., Weynants, V., et al. (1998). Identification of the perosamine synthetase gene of Brucella melitensis 16M and involvement of lipopolysaccharide O side chain in Brucella survival in mice and in macrophages. Infect. Immun. 66, 5485–5493.
Gross, A., Terraza, A., Ouahrani-Bettache, S., Liautard, J. P., and Dornand, J. (2000). In vitro Brucella suis infection prevents the programmed cell death of human monocytic cells. Infect. Immun. 68, 342–351. doi: 10.1128/IAI.68.1.342-351.2000
Guiney, D. G. (2005). The role of host cell death in Salmonella infections. Curr. Top. Microbiol. Immunol. 289, 131–150. doi: 10.1007/3-540-27320-4_6
Guo, F., Zhang, H., Chen, C., Hu, S., Wang, Y., Qiao, J., et al. (2012). Autophagy favors Brucella melitensis survival in infected macrophages. Cell. Mol. Biol. Lett. 17, 249–257. doi: 10.2478/s11658-012-0009-4
Hacker, G., and Fischer, S. F. (2002). Bacterial anti-apoptotic activities. FEMS Microbiol. Lett. 211, 1–6. doi: 10.1016/S0378-1097(02)00654-7
He, Y., Reichow, S., Ramamoorthy, S., Ding, X., Lathigra, R., Craig, J. C., et al. (2006). Brucella melitensis triggers time-dependent modulation of apoptosis and down-regulation of mitochondrion-associated gene expression in mouse macrophages. Infect. Immun. 74, 5035–5046. doi: 10.1128/IAI.01998-05
Hong, P. C., Tsolis, R. M., and Ficht, T. A. (2000). Identification of genes required for chronic persistence of Brucella abortus in mice. Infect. Immun. 68, 4102–4107. doi: 10.1128/IAI.68.7.4102-4107.2000
Hybiske, K., and Stephens, R. S. (2008). Exit strategies of intracellular pathogens. Nat. Rev. Microbiol. 6, 99–110. doi: 10.1038/nrmicro1821
Kirby, J. E., Vogel, J. P., Andrews, H. L., and Isberg, R. R. (1998). Evidence for pore-forming ability by Legionella pneumophila. Mol. Microbiol. 27, 323–336. doi: 10.1046/j.1365-2958.1998.00680.x
Liautard, J. P., Gross, A., Dornand, J., and Kohler, S. (1996). Interactions between professional phagocytes and Brucella spp. Microbiologia 12, 197–206.
Lukacova, M., Barak, I., and Kazar, J. (2008). Role of structural variations of polysaccharide antigens in the pathogenicity of Gram-negative bacteria. Clin. Microbiol. Infect. 14, 200–206. doi: 10.1111/j.1469-0691.2007.01876.x
Mancilla, M., Grillo, M. J., De Miguel, M. J., Lopez-Goni, I., San-Roman, B., Zabalza-Barangua, A., et al. (2013). Deletion of the GI-2 integrase and the wbkA flanking transposase improves the stability of Brucella melitensis Rev 1 vaccine. Vet. Res. 44:105. doi: 10.1186/1297-9716-44-105
Mancilla, M., Lopez-Goni, I., Moriyon, I., and Zarraga, A. M. (2010). Genomic island-2 is an unstable genetic element contributing to Brucella lipopolysaccharide spontaneous smooth to rough dissociation. J. Bacteriol. 192, 6346–6351. doi: 10.1128/JB.00838-10
Mancilla, M., Marin, C. M., Blasco, J. M., Zarraga, A. M., Lopez-Goni, I., and Moriyon, I. (2012). Spontaneous excision of the O-polysaccharide wbkA glycosyltranferase gene is a cause of dissociation of smooth to rough Brucella colonies. J. Bacteriol. 194, 1860–1867. doi: 10.1128/JB.06561-11
Molmeret, M., and Abu Kwaik, Y. (2002). How does Legionella pneumophila exit the host cell? Trends Microbiol. 10, 258–260. doi: 10.1016/S0966-842X(02)02359-4
Molmeret, M., Alli, O. A., Radulic, M., Susa, M., Doric, M., and Kwaik, Y. A. (2002). The C-terminus of IcmT is essential for pore formation and for intracellular trafficking of Legionella pneumophila within Acanthamoeba polyphaga. Mol. Microbiol. 43, 1139–1150. doi: 10.1046/j.1365-2958.2002.02842.x
Mounier, J., Ryter, A., Coquis-Rondon, M., and Sansonetti, P. J. (1990). Intracellular and cell-to-cell spread of Listeria monocytogenes involves interaction with F-actin in the enterocyte like cell line Caco-2. Infect. Immun. 58, 1048–1058.
Oaks, E. V., Wingfield, M. E., and Formal, S. B. (1985). Plaque formation by virulent Shigella flexneri. Infect. Immun. 48, 124–129.
Pei, J., and Ficht, T. A. (2004). Brucella abortus rough muntants are cytopathic for macrophages in culture. Infect. Immun. 72, 440–450. doi: 10.1128/IAI.72.1.440-450.2004
Pei, J., Turse, J. E., and Ficht, T. A. (2008a). Evidence that Brucella abortus OPS dictating uptake and restricts NF-κB activation in murine macrophages. Microbes Infect. 10, 582–590. doi: 10.1016/j.micinf.2008.01.005
Pei, J., Turse, J. E., Wu, Q., and Ficht, T. A. (2006). Brucella abortus rough mutants induce macrophage oncosis that requires bacterial protein synthesis and direct interaction with the macrophage. Infect. Immun. 74, 2667–2675. doi: 10.1128/IAI.74.5.2667-2675.2006
Pei, J., Wu, Q., Kahl-McDonagh, M., and Ficht, T. A. (2008b). Cytotoxicity in macrophage infected with rough Brucella is type IV secretion system-dependent. Infect. Immun. 76, 30–37. doi: 10.1128/IAI.00379-07
Porte, F., Liautard, J. P., and Kohler, S. (1999). Early acidification of phagosomes containing Brucella suis is essential for intracellular survival in murine macrophages. Infect. Immun. 67, 4041–4047.
Porte, F., Naroeni, A., Ouahrani-Bettache, S., and Liautard, J. P. (2003). Role of the Brucella suis lipopolysaccharide O antigen in phagosomal genesis and in inhibition of phagosome-lysosome fusion in murine macrophages. Infect. Immun. 71, 1481–1490. doi: 10.1128/IAI.71.3.1481-1490.2003
Qin, Q. M., Pei, J., Ancona, V., Shaw, B. D., Ficht, T. A., and De Figueiredo, P. (2008). RNAi screen of endoplasmic reticulum-associated host factors reveals a role for IRE1alpha in supporting Brucella replication. PLoS Pathog. 4:e1000110. doi: 10.1371/journal.ppat.1000110
Rittig, M. G., Kaufmann, A., Robins, A., Shaw, B., Sprenger, H., Gemsa, D., et al. (2003). Smooth and rough lipopolysaccharide phenotypes of Brucella induce different intracellular trafficking and cytokine/chemokine release in human monocytes. J. Leukoc. Biol. 74, 1045–1055. doi: 10.1189/jlb.0103015
Roux, C. M., Rolan, H. G., Santos, R. L., Beremand, P. D., Thomas, T. L., Adams, L. G., et al. (2007). Brucella requires a functional Type IV secretion system to elicit innate immune responses in mice. Cell. Microbiol. 9, 1851–1869. doi: 10.1111/j.1462-5822.2007.00922.x
Sano, G., Takada, Y., Goto, S., Maruyama, K., Shindo, Y., Oka, K., et al. (2007). Flagella facilitate escape of Salmonella from oncotic macrophages. J. Bacteriol. 189, 8224–8232. doi: 10.1128/JB.00898-07
Starr, T., Child, R., Wehrly, T. D., Hansen, B., Hwang, S., López-Otin, C., et al. (2012). Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe 11, 33–45. doi: 10.1016/j.chom.2011.12.002
Stearns, T. W., and Roepke, M. H. (1941). The effect of dissociation on the electrophoretic mobility of Brucella. J. Bacteriol. 42, 745–755.
Tolomeo, M., Carlo, P. D., Abbadessa, V., Titone, L., Miceli, S., Barbusca, E., et al. (2003). Monocyte and lymphocyte apoptosis resistance in acute and chronic brucellosis and its possible implications in clinical management. Clin. Infect. Dis. 36, 1533–1538. doi: 10.1086/375223
Turse, J. E., Pei, J., and Ficht, T. (2011). Lipopolysaccharide-deficient Brucella variants arise spontaneously during infection. Front. Microbiol. 2:54. doi: 10.3389/fmicb.2011.00054
Ugalde, J. E., Czibener, C., Feldman, M. F., and Ugalde, R. A. (2000). Identification and characterization of the Brucella abortus phosphoglucomutase gene: role of lipopolysaccharide in virulence and intracellular multiplication. Infect. Immun. 68, 5716–5723. doi: 10.1128/IAI.68.10.5716-5723.2000
Weinrauch, Y., and Zychlinsky, A. (1999). The induction of apoptosis by bacterial pathogens. Annu. Rev. Microbiol. 53, 155–187. doi: 10.1146/annurev.micro.53.1.155
Whatmore, A. M. (2009). Current understanding of the genetic diversity of Brucella, an expanding genus of zoonotic pathogens. Infect. Genet. Evol. 9, 1168–1184. doi: 10.1016/j.meegid.2009.07.001
Zhong, Z., Wang, Y., Qiao, F., Wang, Z., Du, X., Xu, J., et al. (2009). Cytotoxicity of Brucella smooth strains for macrophages is mediated by increased secretion of the type IV secretion system. Microbiology 155, 3392–3402. doi: 10.1099/mic.0.030619-0
Keywords: Brucella dissociation, macrophage cytotoxicity, egress and dissemination, plaque assay, infection foci
Citation: Pei J, Kahl-McDonagh M and Ficht TA (2014) Brucella dissociation is essential for macrophage egress and bacterial dissemination. Front. Cell. Infect. Microbiol. 4:23. doi: 10.3389/fcimb.2014.00023
Received: 11 August 2013; Accepted: 10 February 2014;
Published online: 05 March 2014.
Edited by:
Yongqun “Oliver” He, University of Michigan Medical School, USAReviewed by:
Srinand Sreevastan, University of Minnesota, USAMartin R. Roop II, East Carolina University, USA
Copyright © 2014 Pei, Kahl-McDonagh and Ficht. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas A. Ficht, Department of Veterinary Pathobiology, Texas A&M University, Veterinary Medicine Research Building, 634-676 University Drive, College Station, TX77843-4467, USA e-mail:dGZpY2h0QGN2bS50YW11LmVkdQ==