 Kalliopi K. Gkouskou
Kalliopi K. Gkouskou Chrysoula Deligianni
Chrysoula Deligianni Christos Tsatsanis
Christos Tsatsanis Aristides G. Eliopoulos
Aristides G. Eliopoulos- 1Molecular and Cellular Biology Laboratory, Division of Basic Sciences, University of Crete Medical School, Heraklion, Greece
- 2Laboratory of Translational Medicine and Experimental Therapeutics, University of Crete Medical School, Heraklion, Greece
- 3Department of Clinical Chemistry, University of Crete Medical School, Heraklion, Greece
- 4Laboratory of Cancer Biology, Institute of Molecular Biology and Biotechnology–FORTH, Heraklion, Greece
The intestine and the intestinal immune system have evolved through a symbiotic homeostasis under which a highly diverse microbial flora is maintained in the gastrointestinal tract while pathogenic bacteria are recognized and eliminated. Disruption of the balance between the immune system and the gut microbiota results in the development of multiple pathologies in humans. Inflammatory bowel diseases (IBD) have been associated with alterations in the composition of intestinal flora but whether these changes are causal or result of inflammation is still under dispute. Various chemical and genetic models of IBD have been developed and utilized to elucidate the complex relationship between intestinal epithelium, immune system and the gut microbiota. In this review we describe some of the most commonly used mouse models of colitis and Crohn's disease (CD) and summarize the current knowledge of how changes in microbiota composition may affect intestinal disease pathogenesis. The pursuit of gut-microbiota interactions will no doubt continue to provide invaluable insight into the complex biology of IBD.
Introduction
The lower gastrointestinal tract of healthy adult humans contains more than 100 trillion bacteria (Ley et al., 2008), termed the gut “microbiota,” which are involved in complex interactions with host mucosal epithelial and immune cells and shape fundamental physiological processes such as digestion, energy homeostasis, and development of gut-associated lymphoid tissues (Bakhtiar et al., 2013). Surface antigens and metabolic end-products of gut microbiota modulate the activation of resident immune cells and the production of cytokines which protect against potential pathogens (Cario, 2013). However, this homeostatic relationship is perturbed in inflammatory bowel diseases (IBD), a group of chronic relapsing and remitting disorders of the gastrointestinal tract manifesting as Crohn's disease (CD) or ulcerative colitis (UC). UC usually affects only the rectum and shows continuous inflammation, whereas CD may occur anywhere along the gastrointestinal tract and is characterized by discontinuous lesions in the intestinal wall.
One of the most important and devastating complications of the long-standing inflammation in IBD is colorectal cancer development. The first case of UC-associated carcinoma of the intestine was reported by Crohn and Rosenberg (1925), and CD was connected to cancer in 1945 (Warren and Sommers, 1948). Subsequent studies confirmed that patients with IBD, especially UC, have increased risk for developing colorectal cancer and this risk increases further with the severity of inflammation (reviewed in Danese and Mantovani, 2010; Rubin et al., 2012).
The realization of the intimate relationship between the microbiota and intestinal homeostasis has spurred large collaborative efforts aiming to identify and characterize the microorganisms which associate with health and disease in humans. The European MetaHIT [Metagenomics of the Human Intestinal Tract, (Qin et al., 2010)] project and the Human Microbiome Project [HMP, (Peterson et al., 2009)] explore multi-“omic” data to define the role of human microbiome in health and disease along with the development of a reference set of microbial genome sequences. However, experimental model systems such as the mouse and Drosophila continue to provide critical insight into how host-microbiota homeostasis is established, maintained or perturbed (Kostic et al., 2013).
Herein, we review the phenotypic, cellular, and molecular characteristics of some of the most widely-used mouse models of experimental IBD and colitis-associated cancer (CAC) and the impact of microbiota on these pathologies (Figure 1).
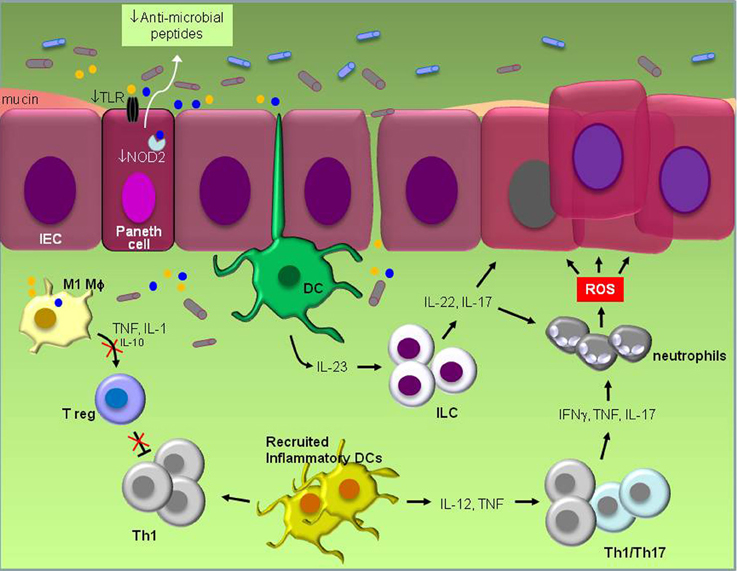
Figure 1. Schematic representation of known pathogenic events in experimental IBD. Defective TLR and NOD signaling in Paneth epithelial cells leads to reduced “sensing” of bacterial products (yellow and blue circles) and reduced production of anti-microbial peptides. The ensuing disruption of microbiota balance which may also be influenced by the frequent use of antibiotics and/or diet stimulates inflammation that is largely orchestrated by resident dendritic cells (DCs). Their activation by products of pathogenic bacteria induces IL-23 which in turn engages innate lymphoid cells (ILC) to produce IL-22 and IL-17. Inflammation also results in the recruitment of inflammatory DCs which secrete IL-12 and TNF and increase IFNγ, TNF and IL-17-producing Th1/Th17 cells. Cytokines secreted by ILCs and Th1/Th17 cells promote both the recruitment of neutrophils that produce DNA-damaging reactive oxygen species (ROS) and the survival of intestinal epithelial cells (IEC) by the engagement of STAT3 signal transduction, eventually leading to malignant transformation. Suppression of regulatory T cell (Treg) activity by pro-inflammatory M1 macrophages which secrete high TNF and IL-1 but low IL-10 levels unleashes inflammation and allows macrophages to produce oxidative products and mutagens which are believed to contribute to carcinogenesis. Reduced production of mucus by Goblet cells impacts on microbial composition and gastrointestinal barrier function.
Chemical and Genetic Mouse Models of Inflammatory Bowel Disease and Colitis-Associated Colon Cancer
Dextran Sodium Sulfate-Induced Colitis
An established model of IBD employs the chemical Dextran Sodium Sulfate (DSS). DSS administered to the drinking water in repeated cycles triggers a state of chronic intestinal inflammation by binding to medium-chain-length fatty acids present in the mouse colon, inducing disruption of colonic epithelial barrier (Laroui et al., 2012). The ensuing tissue damage allows exposure of innate immune cells to commensal bacteria accompanied by a robust Th1-type immune response to eliminate infiltrating pathogens and promote tissue healing. Multiple cell types participate in the pathogenesis of DSS-induced colitis including gut epithelial cells, CD4+ and CD8+ T lymphocytes, regulatory T cells, neutrophils and macrophages, resembling the pathogenic events in human colitis. Mucosal macrophages may prime the local inflammatory response through both phagocytosis of DSS and activation by bacteria products. The contribution of macrophage polarization phenotype to the development of CAC has been described using this model including the demonstration that Akt2 deficient mice are partly protected from DSS-induced colitis because of a macrophage phenotype shift from M1 to M2 in the colonic mucosa (Arranz et al., 2012).
Chronic inflammation induced by prolonged administration of DSS results in malignancy only in a small proportion of animals (Okayasu et al., 1990, 1996) but adenocarcinoma development readily occurs upon intraperitoneal injection of the mutagen azoxymethane (AOM) followed by repeated DSS cycles (reviewed in Wirtz et al., 2007; Chen and Huang, 2009). AOM is metabolized in vivo to methylazoxymethanol (MAM) by cytochrome P450 (Sohn et al., 2001). MAM and its derivatives are direct DNA mutagens although tumor formation requires additional cellular and molecular events associated with chronic inflammatory imbalance. Indeed, the degree of inflammation correlates with the development of dysplasia in minor lesion aberrant crypt foci and is linked to the nuclear translocation of β-catenin (Cooper et al., 2000). Impairment of indoleamine 2,3 dioxygenase-1 (IDO-1) activity, a molecule which catabolizes tryptophan in the kynurenine pathway and is expressed in inflamed and neoplastic intestinal epithelial cells, reduces nuclear β-catenin and cell proliferation (Thaker et al., 2013). Inflammatory cytokines such as TNF, IL-6, and IL-1α which have been implicated in human IBD and IBD-associated colorectal carcinogenesis, also largely dictate the outcome of AOM/DSS-induced pathology (Becker et al., 2004; Van Hauwermeiren et al., 2013; Bersudsky et al., in press). Interestingly, mice deficient in myeloid translocation gene related-1 (MTGR1) are resistant to AOM/DSS-induced CAC despite the preservation of an active inflammatory infiltrate. Tumor resistance in these animals arises from increased malignant cell death and impaired wound-healing (Barrett et al., 2011), suggesting that in addition to the severity of inflammation, AOM/DSS-induced carcinogenesis depends on apoptosis and wound-healing regulatory pathways.
Mutations in p53 are abundant in both sporadic and IBD-associated colorectal cancer in humans, suggesting a pivotal role for this tumor suppressor in intestinal disease pathogenesis. However, whereas p53 mutations are late genetic events in sporadic CRC, they are observed in inflamed colonic tissue well before neoplastic lesions become detectable (Hussain et al., 2000). Thus, p53 mutations probably have an initiating role in human IBD-associated cancer. In the mouse colon, AOM/DSS-induced pathology is largely amplified by either mutations or loss of WT p53. Knock-in mice carrying a germline mutated p53 allele encoding p53R172H, the mouse equivalent of the human hot spot mutant p53R175H (Lang et al., 2004), develop adenocarcinomas even in the absence of AOM treatment (Cooks et al., 2013). The accelerated tumorigenesis in these animals results from a combination of amplified and prolonged inflammation and augmented capacity of mutated p53-containing epithelial cells to evade apoptosis. P53-deficient or p53+/− mice also develop multiple tumors upon exposure to DSS without the requirement of AOM administration (Fujii et al., 2004; Chang et al., 2007). Therefore, AOM/DSS induces a state of chronic intestinal inflammation which progresses to cancer with molecular, histopathological and phenotypic characteristics that resemble the human disease.
Another carcinogen used in combination with DSS is 1, 2-dimethylhydrazine (DMH). DMH is metabolized in liver and its derivatives induce the production of diazonium by gut epithelial cells. The aforementioned metabolite exerts mutagenic effects through oxidative stress and methylation events (Hamiza et al., 2012).
TNBS-Induced Inflammatory Bowel Disease
Intrarectal administration of the contact sensitizing allergen 2,4,6-trinitrobenzenesulfonic acid (TNBS) initiates acute T cell-mediated, IL-12 driven intestinal inflammation (Scheiffele and Fuss, 2002; Neurath and Finotto, 2009). Ethanol is required to disrupt the mucosal barrier, whereas TNBS is proposed to haptenize microbiota or colonic autologous proteins rendering them immunogenic. The overall phenotypic and histopathological features of TNBS-induced colitis mostly resemble those characterizing CD. Recently, the TNBS model was used for the identification of rVEGF164b, a VEGF-A isoform, as an inhibitory molecule of angiogenesis in IBD (Cromer et al., 2013). Thus, TNBS is considered as a suitable model to study both gut inflammation and the mechanism involved in colonic healing in IBD. Using this model we have recently described the efficacy of antisense oligonucleotides targeting CD40, a TNF family receptor that triggers Th1 and innate immune responses upon stimulation by its ligand, in treating early stage and established colitis (Arranz et al., 2013).
Adenomatous Polyposis Coli Mutation-Induced Adenoma Model
Mutations in the Adenomatous polyposis coli (APC) gene in humans are critically involved in familial adenomatous polyposis (FAP) and represent an early genetic aberration in sporadic colorectal cancer (Liang et al., 2013). The multiple intestinal neoplasia (Min) mouse, one of the first genetic models used to study intestinal cancer in rodents, bears a point mutation in the Apc gene (Apcmin/+) and develops numerous adenomas. Exposure of Apcmin/+ mice to DSS alone mimics CAC and results in accelerated tumorigenesis (Tanaka et al., 2006). In addition to inflammation, Apcmin/+-induced carcinogenesis can be influenced by oxidative stress. Thus, Cheung et al. (2012) showed that ablation of nuclear factor-erythroid 2 related factor 2 (Nrf2) attenuates anti-oxidative stress pathways and increases proliferation in the intestinal crypts leading to enhanced intestinal carcinogenesis in Apcmin/+ mice. This observation is pertinent to the role of gut microbiome in disease pathogenesis, identifying microbial metabolites as modulators of carcinogenesis in part through induction of chronic oxidative stress (Arthur et al., 2012).
IKK-γ (NEMO) Deficiency in Intestinal Epithelial Cells
Intestinal epithelial-cell-specific inhibition of NF-κB through conditional ablation of NEMO/IKKγ, the regulatory subunit of the IKK signaling complex essential for NF-κB activation, spontaneously causes severe chronic intestinal inflammation in mice (Nenci et al., 2007). Histological examination of colon sections from these animals revealed extensive apoptosis of colonic epithelial cells leading to disruption of epithelial integrity and translocation of bacteria from the lumen into the mucosa. Notably, these mice exhibit reduced expression of defensin-3, an antimicrobial peptide primarily produced by specialized intestinal epithelial cells, called Paneth. Low defensin copy number has been reported to correlate with predisposition to IBD in humans (Wehkamp et al., 2006) and unpublished data from our laboratory suggest that defensin expression is higher in the proximal compared to distal colon reflecting their differential susceptibility to DSS-induced pathology (Gkouskou and Eliopoulos, in preparation).
Interleukin-10 (IL-10)-Dependent Inflammatory Bowel Disease
Genome-wide association studies have identified SNPs flanking the IL-10 gene to be linked to UC (Franke et al., 2008). IL-10-deficient mice exhibit intolerance to their intestinal microbiota, have altered responses to inflammatory or autoimmune stimuli and develop spontaneous enterocolitis and adenocarcinoma (Sturlan et al., 2001). A similar intestinal phenotype was observed in mice with a T cell specific IL-10 deficiency, underscoring the importance of T cell derived IL-10 and IL-10-dependent regulatory T-cells in the regulation of mucosal T cell responses and disease pathogenesis (Erdman et al., 2003).
T Cell Adoptive Transfer Model
Initially developed by the group of Fiona Powrie (Powrie et al., 1994), mouse CD4+ T lymphocytes which express high CD45RB (CD4+CD45RBHi) can be adoptively transferred into immunodeficient SCID or RAG1/2−/− mice, where they traffic to the intestine and induce gut inflammation. Recipient mice repopulated with CD4+CD45RBLo T cells or total CD4+ T lymphocytes do not develop colitis, despite their ability to colonize the host gut. This phenomenon is attributed to the presence of CD25+FoxP3+ regulatory T cells within the CD4+CD45RBLo population (Read et al., 2000) and adoptive transfer of CD4+CD25− T cells has thus been proposed as the most suitable T cell transfer model of enterocolitis (Kjellev et al., 2006). IL-10 appears to have an important role in the pathogenesis of the disease in this model as SCID mice administered both CD4+CD45RBHi and regulatory T cells together with anti-IL-10 receptor antibodies develop colitis (Kjellev et al., 2006).
The Gut Microbiota in Mouse Models of IBD
Several lines of evidence support a role for the microbiota in experimental colitis. Early studies reported a significant increase in members of Bacteroidaceae and Clostridium spp. families, in particular Bacteroides distasonis and Clostridium ramosum, in the intestines of mice exposed to DSS (Okayasu et al., 1990) (Table 1). Subsequent reports showed elevated 16S rRNA gene copy numbers of the mucin-degrading Gram-negative bacterium Akkermansia muciniphila and of Enterobacteriaceae to correlate with disease activity in mice administered DSS (Hakansson et al., 2014). A breakthrough in appreciating the major impact of gut microbiota on disease pathogenesis came by the observations that treatment with antibiotics or germ-free breeding of various mouse models of IBD is associated with significantly less severe bowel inflammation and carcinogenesis. Thus, Dove and colleagues showed that ApcMin/+ mice housed in germ-free environment display more than 50% reduction in tumor development compared to the same animals housed in standard specific pathogen-free (SPF) conditions (Dove et al., 1997). IL-10 deficient mice were also found to be resistant to spontaneous colitis when kept in germ-free environment (Sellon et al., 1998).
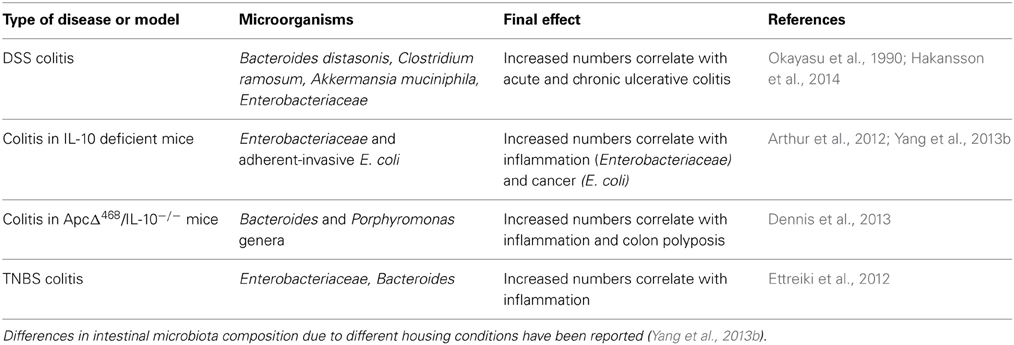
Table 1. Microorganisms reported to associate with IBD in the mouse.
Analysis of different classes of antibiotics indicated differential and localized roles of bacteria species in the establishment and perpetuation of colitis in IL-10−/− mice after colonization with SPF bacteria. Ciprofloxacin was found to be most effective in caecal inflammation by controlling aerobic organisms, including E. coli and E. faecalis, whereas metronidazole was preferentially active in the colon and selectively decreased anaerobic bacteria and Bacteroides spp. (Hoentjen et al., 2003). Interestingly, whereas induction of colitis in IL-10−/− mice born under SPF conditions and in mice exposed to DSS is prevented by ciprofloxacin and metronidazole respectively, these antibiotics have minimal effect after the onset of colitis (Hans et al., 2000; Madsen et al., 2000). In contrast, vancomycin-imipenem reduces total luminal bacteria, eliminates specific aerobic and anaerobic organisms and effectively treats established disease (Hoentjen et al., 2003). These results suggest that some intestinal bacteria species may orchestrate the initiation of inflammation whereas other subsets may have a role in perpetuating colitis (Rath et al., 2001). In line with this notion, colonic polyps developed in ApcΔ468/IL-10−/− mice are significantly enriched in two genera of the phylum Bacteroidetes, namely Bacteroides and Porphyromonas as compared with uninvolved tissue (Dennis et al., 2013) (Table 1). The interplay between oncogenes and microbiota species in the development of gut pathologies is also highlighted by studies in Drosophila which have demonstrated that the human pathogen Pseudomonas aeruginosa synergizes with the RasV12 oncogene to induce intestinal dysplasia and metastasis-like phenotype (Apidianakis et al., 2009; Bangi et al., 2012).
Further evidence supporting the significance of microbes in colitis development has been provided by studies describing a communicable form of colitis induced by deficiency of T-bet in cells of the innate immune system. T-bet is a transcription factor with a pivotal role in the development of Th1 cells and in the regulation of adaptive and innate immune responses. Loss of T-bet in mice lacking B and T cells (T-bet−/−/RAG-1−/−) results in spontaneous colitis which is transmissible to wild-type animals (which express T-bet) upon cross-fostering or co-housing (Garrett et al., 2007).
Nutrition plays an important role in the establishment of microbial flora which in turn affects metabolism of several macro- and micronutrients. For example, a high Firmicutes to Bacteroidetes ratio and low microbial diversity is indicative of a high-calorie diet and obesity in humans (Ley et al., 2006). A telling example of how genetics, microbiota and the immune system may interact to promote chronic gut inflammation is highlighted by a recent study by Devkota et al. (2012) which demonstrated that the ingestion of saturated fat by IL-10−/− mice induces a more severe form of chronic colitis compared to the disease that normally develops in these animals. This diet was shown to stimulate the formation of taurocholine-conjugated bile acids leading to intestinal dysbiosis characterized by the overgrowth of the rare sulfate-reducing pathogenic bacteria Bilophilia wadsworthia (Devkota et al., 2012). The modulation of microbiota species and density has also highlighted the important role of bacteria in gut homeostasis and disease. Thus, administration of VSL#3 probiotics, a mixture of Lactobacillus, Bifidobacterium and Streptococcus salivarious strains, has shown to confer beneficial effects on various mouse models of colitis and in humans suffering from IBD (Isaacs and Herfarth, 2008). Intriguingly, VSL#3 does not reduce colitis-associated colon cancer in the mouse (Arthur et al., 2013).
Direct evidence for the role of pathogenic bacteria in IBD has been provided by studies using the aerobic bacterium Helicobacter hepaticus. Immunodeficient RAG−/− mice infected with H. hepaticus and treated with AOM develop invasive colon carcinoma after 3–5 months, at the sites of highest inflammation in the colon and cecum (Fox et al., 2011). This model has also assisted in the identification of a genetic interval on the telomeric part of mouse chromosome 3 designated Hiccs (Helicobacter hepaticus-induced colitis and associated cancer susceptibility), which harbors 8 genes and 5 micro RNAs and confers protection against H. hepaticus-induced chronic colitis and inflammation-driven colon cancer (Boulard et al., 2012).
What are the mechanisms by which bacteria dysbiosis triggers inflammatory bowel disease? Several studies have highlighted a prominent role for TLR and NOD family members as key sensors of and responders to microbe-associated molecular patterns. The effects of Nod2 mutations are of particular interest because they have been implicated in human IBD and Nod2 knockout mice have diminished ability to prevent intestinal colonization of pathogenic bacteria (Petnicki-Ocwieja et al., 2009; Couturier-Maillard et al., 2013). Impaired TLR and NOD function in Paneth epithelial cells affects their capacity to produce antimicrobial factors which kill pathogenic bacteria, resulting in a shift in the composition of gut microbiota (Figure 1). Frequent use of antibiotics or personal habits, including diet may also influence this shift. The concomitant release of ATP, other metabolic products and DNA by microbia (Atarashi et al., 2008; Hall et al., 2008) may lead to increased production of IL-23 by resident monocytes, such as dendritic cells, which in turn stimulates innate lymphoid cells to secrete IL-17, IL-22, and IFNγ (Buonocore et al., 2010). IL-17 is of particular relevance to colitis as it is linked to reduced regulatory T cell (Treg) activity. Resident Treg produce IL-10 which inhibits Th1 cells and monocyte effector functions associated with inflammation. Suppression of Treg activity thereby unleashes inflammation, leading to a switch in the differentiation program of Ly6Chi monocytes from anti-inflammatory M2 macrophages to inflammatory dendritic cells and M1 macrophages in the colon (Rivollier et al., 2012) which produce a plethora of pro-inflammatory cytokines, oxidative products and mutagens such as trans-4-hydroxy-2-nonenal (4-HNE) (Yang et al., 2013a). Reactive oxygen species (ROS) generated by recruited neutrophils may also cause DNA damage in epithelial cells.
The production by pathogenic bacteria of secondary bile acids that have carcinogenic effects is believed to contribute to the dysbiosis-inflammation-tumorigenesis axis (Sommer and Backhed, 2013). Additional host genetic factors may influence the cross-talk between microbiota and IBD. For example, production of mucus by Goblet cells, especially mucin 2 (MUC2), has a significant impact on microbial composition and gastrointestinal barrier function. Altered MUC2 expression and/or glycosylation leads to accompanying intestinal pathologies, including IBD and colon cancer (Yang et al., 2008).
Conclusions and Future Directions
In the intestine, the symbiotic relationship between the host and the microbiota influences nutrition, metabolism, immune system functions, development and normal physiology, as well as susceptibility to IBD and CAC. Accumulating experimental, epidemiological, and clinical evidence highlights the potential of targeting the dysbiosis-inflammation-tumorigenesis axis for the development of new therapeutic strategies for IBD and colorectal cancer. Much of the current knowledge of the regulation of this axis comes from studies exploring the effects of particular pathogenic bacteria using chemical or genetic models of CAC. High-throughput human microbiome studies confirm that the genetic make-up, environmental factors and personal habits influence the bacteria communities among individuals; however, further studies are warranted to fully appreciate how a particular microbiota is established and orchestrates the immune responses toward the development of colitis and CAC. The establishment of “humanized” gnotobiotic mice, animals that carry only human-derived gut microbes (Turnbaugh et al., 2009) is expected to improve human disease modeling and provide further insight into how environmental factors, including diet, may influence the microbiota and shape gut physiology and disease pathogenesis. Similarly, it would be important to assess changes in the gut flora during aging and evaluate their impact on IBD susceptibility. In line with this notion, recent studies in Drosophila show that immunosenescence associated with aging results in dysbiosis and triggers an inflammatory response which promotes intestinal stem cell over-proliferation and dysplasia (Guo et al., 2014). Further studies are also needed to determine whether changes in particular microbiota species induced by inflammation may impact on progression to cancer.
Future research could also lead to the development of beneficial (probiotic) microbes or inhibitors of specific microbes and/or their products which “normalize” the intestinal flora and can improve human health. As the current repertoire of probiotics is limited, further studies to explore the potential of fecal microbiota transplantation (FMT) therapy, the infusion of fecal bacteria from a healthy individual into a recipient patient, for the treatment of intestinal disorders are warranted. FMT has demonstrated tremendous efficacy in treating refractory Clostridium difficile infection, and there are case reports of successful management of UC using FMT in humans (Lemon et al., 2012). A more focused approach requires the identification of species or bacterial products and metabolites which normalize the inflamed gut mucosa. In this regard, the isolation of 17 human clostridia species and the discovery of microbial-derived short-chain fatty acids that can stimulate the expansion of Treg cells in mice (Atarashi et al., 2013; Smith et al., 2013) opens up new therapeutic options for the treatment of IBD.
The microbiome plays an important role in immunity and energy metabolism and will thus be important to determine if the microbial gut ecology may also impact on non-gastrointestinal diseases, including obesity, cancer and neurological disorders.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the European Commission FP7 programmes INFLA-CARE (EC contract number 223151) to Aristides G. Eliopoulos and “Translational Potential” (TransPOT; EC contract number 285948) to Aristides G. Eliopoulos and Christos Tsatsanis. Aristides G. Eliopoulos also acknowledges co-funding of this review by the General Secretariat of Research and Technology of Greece through the Operational Program Competitiveness and Entrepreneurship (OPC II), NSRF 2007-2013, action “SYNERGASIA 2011,” Project THERA-CAN (contract number 11ΣYN_1_485).
References
Apidianakis, Y., Pitsouli, C., Perrimon, N., and Rahme, L. (2009). Synergy between bacterial infection and genetic predisposition in intestinal dysplasia. Proc. Natl. Acad. Sci. U.S.A. 106, 20883–20888. doi: 10.1073/pnas.0911797106
Arranz, A., Doxaki, C., Vergadi, E., Martinez de la Torre, Y., Vaporidi, K., Lagoudaki, E. D., et al. (2012). Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc. Natl. Acad. Sci. U.S.A. 109, 9517–9522. doi: 10.1073/pnas.1119038109
Arranz, A., Reinsch, C., Papadakis, K. A., Dieckmann, A., Rauchhaus, U., Androulidaki, A., et al. (2013). Treatment of experimental murine colitis with CD40 antisense oligonucleotides delivered in amphoteric liposomes. J. Control. Release 165, 163–172. doi: 10.1016/j.jconrel.2012.11.008
Arthur, J. C., Gharaibeh, R. Z., Uronis, J. M., Perez-Chanona, E., Sha, W., Tomkovich, S., et al. (2013). VSL#3 probiotic modifies mucosal microbial composition but does not reduce colitis-associated colorectal cancer. Sci. Rep. 3, 2868. doi: 10.1038/srep02868
Arthur, J. C., Perez-Chanona, E., Muhlbauer, M., Tomkovich, S., Uronis, J. M., Fan, T. J., et al. (2012). Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123. doi: 10.1126/science.1224820
Atarashi, K., Nishimura, J., Shima, T., Umesaki, Y., Yamamoto, M., Onoue, M., et al. (2008). ATP drives lamina propria T(H)17 cell differentiation. Nature 455, 808–812. doi: 10.1038/nature07240
Atarashi, K., Tanoue, T., Oshima, K., Suda, W., Nagano, Y., Nishikawa, H., et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236. doi: 10.1038/nature12331
Bakhtiar, S. M., LeBlanc, J. G., Salvucci, E., Ali, A., Martin, R., Langella, P., et al. (2013). Implications of the human microbiome in inflammatory bowel diseases. FEMS Microbiol. Lett. 342, 10–17. doi: 10.1111/1574-6968.12111
Bangi, E., Pitsouli, C., Rahme, L. G., Cagan, R., and Apidianakis, Y. (2012). Immune response to bacteria induces dissemination of Ras-activated Drosophila hindgut cells. EMBO Rep. 13, 569–576. doi: 10.1038/embor.2012.44
Barrett, C. W., Fingleton, B., Williams, A., Ning, W., Fischer, M. A., Washington, M. K., et al. (2011). MTGR1 is required for tumorigenesis in the murine AOM/DSS colitis-associated carcinoma model. Cancer Res. 71, 1302–1312. doi: 10.1158/0008-5472.CAN-10-3317
Becker, C., Fantini, M. C., Schramm, C., Lehr, H. A., Wirtz, S., Nikolaev, A., et al. (2004). TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 21, 491–501. doi: 10.1016/j.immuni.2004.07.020
Bersudsky, M., Luski, L., Fishman, D., White, R. M., Ziv-Sokolovskaya, N., Dotan, S., et al. (in press). Non-redundant properties of IL-1alpha and IL-1beta during acute colon inflammation in mice. Gut doi: 10.1136/gutjnl-2012-303329
Boulard, O., Kirchberger, S., Royston, D. J., Maloy, K. J., and Powrie, F. M. (2012). Identification of a genetic locus controlling bacteria-driven colitis and associated cancer through effects on innate inflammation. J. Exp. Med. 209, 1309–1324. doi: 10.1084/jem.20120239
Buonocore, S., Ahern, P. P., Uhlig, H. H., Ivanov, I. I., Littman, D. R., Maloy, K. J., et al. (2010). Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464, 1371–1375. doi: 10.1038/nature08949
Cario, E. (2013). Microbiota and innate immunity in intestinal inflammation and neoplasia. Curr. Opin. Gastroenterol. 29, 85–91. doi: 10.1097/MOG.0b013e32835a670e
Chang, W. C., Coudry, R. A., Clapper, M. L., Zhang, X., Williams, K. L., Spittle, C. S., et al. (2007). Loss of p53 enhances the induction of colitis-associated neoplasia by dextran sulfate sodium. Carcinogenesis 28, 2375–2381. doi: 10.1093/carcin/bgm134
Chen, J., and Huang, X. F. (2009). The signal pathways in azoxymethane-induced colon cancer and preventive implications. Cancer Biol. Ther. 8, 1313–1317. doi: 10.4161/cbt.8.14.8983
Cheung, K. L., Lee, J. H., Khor, T. O., Wu, T. Y., Li, G. X., Chan, J., et al. (2012). Nrf2 knockout enhances intestinal tumorigenesis in Apc(min/+) mice due to attenuation of anti-oxidative stress pathway while potentiates inflammation. Mol. Carcinog. 53, 77–84. doi: 10.1002/mc.21950
Cooks, T., Pateras, I. S., Tarcic, O., Solomon, H., Schetter, A. J., Wilder, S., et al. (2013). Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 23, 634–646. doi: 10.1016/j.ccr.2013.03.022
Cooper, H. S., Murthy, S., Kido, K., Yoshitake, H., and Flanigan, A. (2000). Dysplasia and cancer in the dextran sulfate sodium mouse colitis model. Relevance to colitis-associated neoplasia in the human: a study of histopathology, B-catenin and p53 expression and the role of inflammation. Carcinogenesis 21, 757–768. doi: 10.1093/carcin/21.4.757
Couturier-Maillard, A., Secher, T., Rehman, A., Normand, S., De Arcangelis, A., Haesler, R., et al. (2013). NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J. Clin. Invest. 123, 700–711. doi: 10.1172/JCI62236
Crohn, B., and Rosenberg, H. (1925). The sigmoidoscopic picture of chronic ulcerative colitis (non-specific). Am. J. Med. Sci. 170, 220–228. doi: 10.1097/00000441-192508010-00006
Cromer, W. E., Ganta, C. V., Patel, M., Traylor, J., Kevil, C. G., Alexander, J. S., et al. (2013). VEGF-A isoform modulation in an preclinical TNBS model of ulcerative colitis: protective effects of a VEGF164b therapy. J. Transl. Med. 11, 207. doi: 10.1186/1479-5876-11-207
Danese, S., and Mantovani, A. (2010). Inflammatory bowel disease and intestinal cancer: a paradigm of the Yin-Yang interplay between inflammation and cancer. Oncogene 29, 3313–3323. doi: 10.1038/onc.2010.109
Dennis, K. L., Wang, Y., Blatner, N. R., Wang, S., Saadalla, A., Trudeau, E., et al. (2013). Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10-producing T cells. Cancer Res. 73, 5905–5913. doi: 10.1158/0008-5472.CAN-13-1511
Devkota, S., Wang, Y., Musch, M. W., Leone, V., Fehlner-Peach, H., Nadimpalli, A., et al. (2012). Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 487, 104–108. doi: 10.1038/nature11225
Dove, W. F., Clipson, L., Gould, K. A., Luongo, C., Marshall, D. J., Moser, A. R., et al. (1997). Intestinal neoplasia in the ApcMin mouse: independence from the microbial and natural killer (beige locus) status. Cancer Res. 57, 812–884.
Erdman, S. E., Rao, V. P., Poutahidis, T., Ihrig, M. M., Ge, Z., Feng, Y., et al. (2003). CD4(+)CD25(+) regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res. 63, 6042–6050.
Ettreiki, C., Gadonna-Widehem, P., Mangin, I., Coeffier, M., Delayre-Orthez, C., and Anton, P. M. (2012). Juvenile ferric iron prevents microbiota dysbiosis and colitis in adult rodents. World J. Gastroenterol. 18, 2619–2629. doi: 10.3748/wjg.v18.i21.2619
Fox, J. G., Ge, Z., Whary, M. T., Erdman, S. E., and Horwitz, B. H. (2011). Helicobacter hepaticus infection in mice: models for understanding lower bowel inflammation and cancer. Mucosal Immunol. 4, 22–30. doi: 10.1038/mi.2010.61
Franke, A., Balschun, T., Karlsen, T. H., Sventoraityte, J., Nikolaus, S., Mayr, G., et al. (2008). Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat. Genet. 40, 1319–1323. doi: 10.1038/ng.221
Fujii, S., Fujimori, T., Kawamata, H., Takeda, J., Kitajima, K., Omotehara, F., et al. (2004). Development of colonic neoplasia in p53 deficient mice with experimental colitis induced by dextran sulphate sodium. Gut 53, 710–716. doi: 10.1136/gut.2003.028779
Garrett, W. S., Lord, G. M., Punit, S., Lugo-Villarino, G., Mazmanian, S. K., Ito, S., et al. (2007). Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell 131, 33–45. doi: 10.1016/j.cell.2007.08.017
Guo, L., Karpac, J., Tran, S. L., and Jasper, H. (2014). PGRP-SC2 promotes gut immune homeostasis to limit commensal dysbiosis and extend lifespan. Cell 156, 109–122. doi: 10.1016/j.cell.2013.12.018
Hakansson, A., Tormo-Badia, N., Baridi, A., Xu, J., Molin, G., Hagslatt, M. L., et al. (2014). Immunological alteration and changes of gut microbiota after dextran sulfate sodium (DSS) administration in mice. Clin. Exp. Med. doi: 10.1007/s10238-013-0270-5. [Epub ahead of print].
Hall, J. A., Bouladoux, N., Sun, C. M., Wohlfert, E. A., Blank, R. B., Zhu, Q., et al. (2008). Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity 29, 637–649. doi: 10.1016/j.immuni.2008.08.009
Hamiza, O. O., Rehman, M. U., Tahir, M., Khan, R., Khan, A. Q., Lateef, A., et al. (2012). Amelioration of 1,2 Dimethylhydrazine (DMH) induced colon oxidative stress, inflammation and tumor promotion response by tannic acid in Wistar rats. Asian Pac. J. Cancer Prev. 13, 4393–4402. doi: 10.7314/APJCP.2012.13.9.4393
Hans, W., Scholmerich, J., Gross, V., and Falk, W. (2000). The role of the resident intestinal flora in acute and chronic dextran sulfate sodium-induced colitis in mice. Eur. J. Gastroenterol. Hepatol. 12, 267–273. doi: 10.1097/00042737-200012030-00002
Hoentjen, F., Harmsen, H. J., Braat, H., Torrice, C. D., Mann, B. A., Sartor, R. B., et al. (2003). Antibiotics with a selective aerobic or anaerobic spectrum have different therapeutic activities in various regions of the colon in interleukin 10 gene deficient mice. Gut 52, 1721–1727. doi: 10.1136/gut.52.12.1721
Hussain, S. P., Amstad, P., Raja, K., Ambs, S., Nagashima, M., Bennett, W. P., et al. (2000). Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: a cancer-prone chronic inflammatory disease. Cancer Res. 60, 3333–3337.
Isaacs, K., and Herfarth, H. (2008). Role of probiotic therapy in IBD. Inflamm. Bowel Dis. 14, 1597–1605. doi: 10.1002/ibd.20465
Kjellev, S., Lundsgaard, D., Poulsen, S. S., and Markholst, H. (2006). Reconstitution of Scid mice with CD4+CD25- T cells leads to rapid colitis: an improved model for pharmacologic testing. Int. Immunopharmacol. 6, 1341–1354. doi: 10.1016/j.intimp.2006.04.017
Kostic, A. D., Howitt, M. R., and Garrett, W. S. (2013). Exploring host-microbiota interactions in animal models and humans. Genes Dev. 27, 701–718. doi: 10.1101/gad.212522.112
Lang, G. A., Iwakuma, T., Suh, Y. A., Liu, G., Rao, V. A., Parant, J. M., et al. (2004). Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119, 861–872. doi: 10.1016/j.cell.2004.11.006
Laroui, H., Ingersoll, S. A., Liu, H. C., Baker, M. T., Ayyadurai, S., Charania, M. A., et al. (2012). Dextran sodium sulfate (DSS) induces colitis in mice by forming nano-lipocomplexes with medium-chain-length fatty acids in the colon. PLoS ONE 7:e32084. doi: 10.1371/journal.pone.0032084
Lemon, K. P., Armitage, G. C., Relman, D. A., and Fischbach, M. A. (2012). Microbiota-targeted therapies: an ecological perspective. Sci. Transl. Med. 4, 137rv5. doi: 10.1126/scitranslmed.3004183
Ley, R. E., Hamady, M., Lozupone, C., Turnbaugh, P. J., Ramey, R. R., Bircher, J. S., et al. (2008). Evolution of mammals and their gut microbes. Science 320, 1647–1651. doi: 10.1126/science.1155725
Ley, R. E., Turnbaugh, P. J., Klein, S., and Gordon, J. I. (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023. doi: 10.1038/4441022a
Liang, J., Lin, C., Hu, F., Wang, F., Zhu, L., Yao, X., et al. (2013). APC polymorphisms and the risk of colorectal neoplasia: a HuGE review and meta-analysis. Am. J. Epidemiol. 177, 1169–1179. doi: 10.1093/aje/kws382
Madsen, K. L., Doyle, J. S., Tavernini, M. M., Jewell, L. D., Rennie, R. P., and Fedorak, R. N. (2000). Antibiotic therapy attenuates colitis in interleukin 10 gene-deficient mice. Gastroenterology 118, 1094–1105. doi: 10.1016/S0016-5085(00)70362-3
Nenci, A., Becker, C., Wullaert, A., Gareus, R., van Loo, G., Danese, S., et al. (2007). Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 446, 557–561. doi: 10.1038/nature05698
Neurath, M. F., and Finotto, S. (2009). Translating inflammatory bowel disease research into clinical medicine. Immunity 31, 357–361. doi: 10.1016/j.immuni.2009.08.016
Okayasu, I., Hatakeyama, S., Yamada, M., Ohkusa, T., Inagaki, Y., and Nakaya, R. (1990). A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 98, 694–702.
Okayasu, I., Ohkusa, T., Kajiura, K., Kanno, J., and Sakamoto, S. (1996). Promotion of colorectal neoplasia in experimental murine ulcerative colitis. Gut 39, 87–92. doi: 10.1136/gut.39.1.87
Peterson, J., Garges, S., Giovanni, M., McInnes, P., Wang, L., Schloss, J. A., et al. (2009). The NIH Human Microbiome Project. Genome Res. 19, 2317–2323. doi: 10.1101/gr.096651.109
Petnicki-Ocwieja, T., Hrncir, T., Liu, Y. J., Biswas, A., Hudcovic, T., Tlaskalova-Hogenova, H., et al. (2009). Nod2 is required for the regulation of commensal microbiota in the intestine. Proc. Natl. Acad. Sci. U.S.A. 106, 15813–15818. doi: 10.1073/pnas.0907722106
Powrie, F., Leach, M. W., Mauze, S., Menon, S., Caddle, L. B., and Coffman, R. L. (1994). Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1, 553–562. doi: 10.1016/1074-7613(94)90045-0
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. doi: 10.1038/nature08821
Rath, H. C., Schultz, M., Freitag, R., Dieleman, L. A., Li, F., Linde, H. J., et al. (2001). Different subsets of enteric bacteria induce and perpetuate experimental colitis in rats and mice. Infect. Immun. 69, 2277–2285. doi: 10.1128/IAI.69.4.2277-2285.2001
Read, S., Malmstrom, V., and Powrie, F. (2000). Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25(+)CD4(+) regulatory cells that control intestinal inflammation. J. Exp. Med. 192, 295–302. doi: 10.1084/jem.192.2.295
Rivollier, A., He, J., Kole, A., Valatas, V., and Kelsall, B. L. (2012). Inflammation switches the differentiation program of Ly6Chi monocytes from antiinflammatory macrophages to inflammatory dendritic cells in the colon. J. Exp. Med. 209, 139–155. doi: 10.1084/jem.20101387
Rubin, D. C., Shaker, A., and Levin, M. S. (2012). Chronic intestinal inflammation: inflammatory bowel disease and colitis-associated colon cancer. Front. Immunol. 3:107. doi: 10.3389/fimmu.2012.00107
Scheiffele, F., and Fuss, I. J. (2002). Induction of TNBS colitis in mice. Curr. Protoco. Immunol. Chapter 15, Unit 15.19. doi: 10.1002/0471142735.im1519s49
Sellon, R. K., Tonkonogy, S., Schultz, M., Dieleman, L. A., Grenther, W., Balish, E., et al. (1998). Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect. Immun. 66, 5224–5231.
Smith, P. M., Howitt, M. R., Panikov, N., Michaud, M., Gallini, C. A., Bohlooly, Y. M., et al. (2013). The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573. doi: 10.1126/science.1241165
Sohn, O. S., Fiala, E. S., Requeijo, S. P., Weisburger, J. H., and Gonzalez, F. J. (2001). Differential effects of CYP2E1 status on the metabolic activation of the colon carcinogens azoxymethane and methylazoxymethanol. Cancer Res. 61, 8435–8440.
Sommer, F., and Backhed, F. (2013). The gut microbiota–masters of host development and physiology. Nat. Rev. Microbiol. 11, 227–238. doi: 10.1038/nrmicro2974
Sturlan, S., Oberhuber, G., Beinhauer, B. G., Tichy, B., Kappel, S., Wang, J., et al. (2001). Interleukin-10-deficient mice and inflammatory bowel disease associated cancer development. Carcinogenesis 22, 665–671. doi: 10.1093/carcin/22.4.665
Tanaka, T., Kohno, H., Suzuki, R., Hata, K., Sugie, S., Niho, N., et al. (2006). Dextran sodium sulfate strongly promotes colorectal carcinogenesis in Apc(Min/+) mice: inflammatory stimuli by dextran sodium sulfate results in development of multiple colonic neoplasms. Int. J. Cancer 118, 25–34. doi: 10.1002/ijc.21282
Thaker, A. I., Rao, M. S., Bishnupuri, K. S., Kerr, T. A., Foster, L., Marinshaw, J. M., et al. (2013). IDO1 metabolites activate beta-catenin signaling to promote cancer cell proliferation and colon tumorigenesis in mice. Gastroenterology 145, 416–425.e1–e4. doi: 10.1053/j.gastro.2013.05.002
Turnbaugh, P. J., Ridaura, V. K., Faith, J. J., Rey, F. E., Knight, R., and Gordon, J. I. (2009). The effect of diet on the human gut microbiome: a metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 1, 6ra14. doi: 10.1126/scitranslmed.3000322
Van Hauwermeiren, F., Armaka, M., Karagianni, N., Kranidioti, K., Vandenbroucke, R. E., Loges, S., et al. (2013). Safe TNF-based antitumor therapy following p55TNFR reduction in intestinal epithelium. J. Clin. Invest. 123, 2590–2603. doi: 10.1172/JCI65624
Warren, S., and Sommers, S. C. (1948). Cicatrizing enteritis as a pathologic entity; analysis of 120 cases. Am. J. Pathol. 24, 475–501.
Wehkamp, J., Chu, H., Shen, B., Feathers, R. W., Kays, R. J., Lee, S. K., et al. (2006). Paneth cell antimicrobial peptides: topographical distribution and quantification in human gastrointestinal tissues. FEBS Lett. 580, 5344–5350. doi: 10.1016/j.febslet.2006.08.083
Wirtz, S., Neufert, C., Weigmann, B., and Neurath, M. F. (2007). Chemically induced mouse models of intestinal inflammation. Nat. Protoc. 2, 541–546. doi: 10.1038/nprot.2007.41
Yang, I., Eibach, D., Kops, F., Brenneke, B., Woltemate, S., Schulze, J., et al. (2013b). Intestinal microbiota composition of interleukin-10 deficient C57BL/6J mice and susceptibility to Helicobacter hepaticus-induced colitis. PLoS ONE 8:e70783. doi: 10.1371/journal.pone.0070783
Keywords: microbiota, colitis, mouse models, IBD, Crohn's disease
Citation: Gkouskou KK, Deligianni C, Tsatsanis C and Eliopoulos AG (2014) The gut microbiota in mouse models of inflammatory bowel disease. Front. Cell. Infect. Microbiol. 4:28. doi: 10.3389/fcimb.2014.00028
Received: 09 January 2014; Accepted: 14 February 2014;
Published online: 28 February 2014.
Edited by:
Yiorgos Apidianakis, University of Cyprus, CyprusReviewed by:
Triantafyllos Chavakis, Technische Univerasität Dresden, GermanyChristos Polytarchou, University of California Los Angeles, USA
Copyright © 2014 Gkouskou, Deligianni, Tsatsanis and Eliopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aristides G. Eliopoulos, University of Crete Medical School, GR-71003 Heraklion, Crete, Greece e-mail:ZWxpb3BhZ0BtZWQudW9jLmdy
†These authors have contributed equally to this work.