 Dinesh Kumar Parandhaman1,2
Dinesh Kumar Parandhaman1,2 Sujatha Narayanan1*
Sujatha Narayanan1*- 1Department of Immunology, National Institute for Research in Tuberculosis, Chennai, India
- 2Department of Immunology, International Centre for Genetic Engineering and Biotechnology, New Delhi, India
Cell death or senescence is a fundamental event that helps maintain cellular homeostasis, shapes the growth of organism, and provides protective immunity against invading pathogens. Decreased or increased cell death is detrimental both in infectious and non-infectious diseases. Cell death is executed both by regulated enzymic reactions and non-enzymic sudden collapse. In this brief review we have tried to summarize various cell death modalities and their impact on the pathogenesis of Mycobacterium tuberculosis.
Introduction
Cell death is a primordial event in embryogenesis, metamorphosis, and in innate immune response against the invading pathogens. Cell death as a defense mechanism is also documented in the plant kingdom (Kabbage et al., 2013). Cell death is executed in a series of ordered biochemical cascades and is referred as programmed cell death or PCD.
Till early 2000, cell death was discussed as dichotomy in terms of either apoptosis or necrosis. However, with the growth of science many distinct modes of cell death with well-organized signaling cascades were unraveled. Currently, there exists nine different forms of cell death namely apoptosis (Fink and Cookson, 2005), autophagy (Fink and Cookson, 2005), mitoptosis (Chaabane et al., 2012), necrosis (Fink and Cookson, 2005), necroptosis (Galluzzi and Kroemer, 2008), netosis (Remijsen et al., 2011), oncosis (Fink and Cookson, 2005), pyroptosis (Fink and Cookson, 2005), and pyronecrosis (Willingham et al., 2007). It is still a puzzle whether these pathways are different features of the same response or physiologically distinct responses. Apoptosis as an defense mechanism initiates both innate and adaptive immunity (Behar et al., 2010). However, pathogenic organisms have developed mechanisms to modulate apoptosis for their survival. Apoptosis of the infected cells have been reported to be a favorable outcome for the dissemination of infections like Yersinia, Francisella, etc. (Ruckdeschel et al., 1997; Wickstrum et al., 2009). On the contrary, impairment of apoptosis provides a survival niche to many intracellular pathogens including Mycobacterium tuberculosis (Behar et al., 2010), leads to auto immunity, cancer and degenerative disorders (Elmore, 2007). Studies in M. tuberculosis have identified a causal relationship between virulence of the strain and induction of apoptosis. Inhibition of apoptosis favors M. tuberculosis survival in many ways like preventing bactericidal effects, T-cell priming, etc. (Velmurugan et al., 2007). In contrast, a recent report states that apoptosis inducing strains could disseminate M. tuberculosis infection (Aguilo et al., 2013). Necrotic cell death of burdened M. tuberculosis infected cells was shown to pave way for re-infection (Butler et al., 2012). In here, we summarize various apoptotic modalities and their role in the pathogenesis of M. tuberculosis. Furthermore, we share our experience in analyzing these responses in M. tuberculosis infection.
Models of Cell Death
Apoptosis
First represented in the article by Kerr, Wyllie, and Currie in 1972 (Elmore, 2007). Apoptosis is an energy dependent regulatory process that disintegrates the dying cell by enclosing the cytoplasmic contents inside membrane bound vesicles called apoptotic bodies. These apoptotic bodies are engulfed by the phagocytic cells by a process called efferocytosis thereby efficiently clearing the dying cell without any inflammatory responses (Lee et al., 2009). Three pathways namely extrinsic/ligand-mediated pathway, intrinsic/mitochondrial pathway, and the granzyme B-mediated pathway regulate the process of apoptosis upon activation by physiological or pathological conditions (Elmore, 2007). The major players in apoptosis are caspases, adaptor proteins, tumor necrosis factor (TNF) receptor (TNF-R) super family, and Bcl-2 family of proteins (Strasser et al., 2000). There are three categories of caspases; initiators (caspase-2,-8,-9,-10), effectors or executioners (caspase-3,-6,-7), and inflammatory caspases (caspase-1,-4,-5) (Elmore, 2007). Caspase-activated DNases activate endonuclease that produce the typical internucleosomal DNA cleavage during apoptosis (Strasser et al., 2000). Adapter proteins play a major role in apoptosis as a link between caspases and the TNF-R by mediating homotypic interactions between the domains death domain, the death effector domain, and the caspase recruitment domains (Strasser et al., 2000).
Bcl-2 family of proteins are classified into three types that fall into pro-survival and pro-apoptotic categories based on the amino acid sequence homology to Bcl-2 homology regions BH1–BH4. Pro -survival Bcl-xL, Bcl-w, A1/Bfl-1, Mcl-1, and Boo/Diva have three or four bcl-2 homology regions while the pro-apoptotic members called Bax-like death factors Bax, Bcl-xS, Bak, and Bok/Mtd contain two or three homology regions (Pecina-Slaus, 2010). The third group of proteins Bad, Bik/Nbk, Bid, Hrk/DP5, Bim/Bod, and Blk, etc. that possess only a BH3 region are potent inducers of apoptosis (Strasser et al., 2000).
Apoptotic pathways
• Extrinsic pathway is initiated by binding of the ligands like TNF-α, FasL, CD95L, TRAIL, etc. to their respective receptors TNFR, Fas/CD95, and DR3 on the cell surface. This activates the initiator caspases such as caspases 8 and 10 that results in the formation and activation of death inducing signaling complex (DISC) that activates caspase 3 (Pecina-Slaus, 2010; Kalimuthu and Se-Kwon, 2013). Caspase 3 activation leads to cleavage of various death substrates that results in the characteristic hallmarks of apoptosis like DNA fragmentation, membrane blebbing, etc. (Kalimuthu and Se-Kwon, 2013).
• Intrinsic pathway of apoptosis is trigged due to the intracellular death signals. Mitochondrial enzyme endonuclease G, Bcl-2 family of proteins like Bax, Bid, and other mitochondrial proteins AIF, DIABLO [SMAC (second mitochondria-derived activator of caspases)], and cytochrome C plays a major role in this response (Kalimuthu and Se-Kwon, 2013). Upon the stimulus, the BH3-only protein Bid activates Bax and Bak that results in conformational change and oligomerization, forming an oligomeric pore in the outer mitochondrial membrane called permeability transition pores (Ferri and Kroemer, 2001; Kalimuthu and Se-Kwon, 2013). This results in the release of cytochrome C and other pro-apoptotic factors from the mitochondria into the cytosol. Cytochrome C interacts with Apaf and activates caspase-9 forming a multi-protein subunit complex called casposome (apoptosome) comprising cytochrome C, Apaf-1, procaspase-9, and ATP. In the absence of death stimulus, inhibitor of apoptosis family proteins (IAP) inactivates the caspase activity by direct binding. However, upon apoptotic stimuli IAPs are negatively regulated by SMAC and that leads to the activation of caspase-3 (Pecina-Slaus, 2010; Kalimuthu and Se-Kwon, 2013). Furthermore, extrinsic pathway was found to influence the intrinsic pathway of apoptosis by truncation of Bid (Cillessen et al., 2007).
• Granzyme B-mediated pathway utilizing the extrinsic mode of apoptosis is used by cytotoxic T lymphocytes as a mechanism to kill its target. Besides this, the secretion of pore forming granules containing serine proteases granzyme A and granzyme B also execute apoptosis that is both dependent and independent of caspase activation (Elmore, 2007).
Autophagy
It is a regulated homeostatic response conserved in all living cells degrading their own cytoplasm. Autophagy is a predominant cell survival response that is involved either in nutrient turnover or energy production during stress or removal of long lived cells or to protect against invading intracellular pathogens (Chaabane et al., 2012). Three forms of autophagy namely macroautophagy, microautophagy, and chaperone-mediated autophagy exist. During the autophagy, damaged organelle is lined with an isolation membrane called the phagophore that enlarges forming the double membrane structure called autophagosome. The autophagosome fuses either with late endosomes or lysosomes causing cell death (Levine and Deretic, 2007; Remijsen et al., 2011). Autophagy is regulated by autophagy-related proteins, serine/threonine kinase, mammalian target of rapamycin (mTOR), class I and class III phosphoinositide 3-kinases (PI3Ks) (Levine and Deretic, 2007; Su et al., 2013).
Mitoptosis
Apoptotic changes inside the mitochondria are called mitoptosis. Mitoptosis is still in infancy and no specific factors have been identified. The identification is based on morphological changes like disintegrating cristae, swollen mitochondria, etc. (Chaabane et al., 2012).
Necrosis
Accidental cell death induced due to pathological or physiological conditions are called necrosis. During necrosis, swelling of organelles like endoplasmic reticulum, mitochondria occurs thereby rupturing the plasma membrane. This leaks the intracellular contents of the necrotic cell into the intercellular space causing inflammatory responses (Fink and Cookson, 2005; Chaabane et al., 2012).
Necroptosis
In the year 2008, Hitomi et al. reported that necrosis could be a regulated process of cell death. The activation of serine/threonine kinase RIP1, BH3 only protein Bmf, and mitochondrial dysfunction executes necroptosis (Galluzzi and Kroemer, 2008).
NETosis
In 2004, the findings of Brinkman group unveiled another cell death program named by Steinberg in 2007 called NETosis (Mesa and Vasquez, 2013). One among the defense mechanisms used by neutrophils is the extrusion of intracellular material in the form of extracellular traps (ETs) to the surrounding extracellular medium. This concentrates the microbicidal substances to trap and kill pathogens (Mesa and Vasquez, 2013). Release of ETs by neutrophils is called NETs and mast cells as MCETs. NETs are composed of DNA and histones, and they are resistant to degradation by proteases, insensitive to caspase inhibition and necrostatins (cytoprotective agents) (Mesa and Vasquez, 2013). During NETosis both the nuclear and granular membranes disintegrate leaving the plasma membrane intact (Remijsen et al., 2011). NETosis is activated by pathogens, platelets activated with LPS and in eosinophils (Remijsen et al., 2011). Formation of NET is both nuclear and mitochondrial in origin.
Oncosis
It is the swelling of cells that involves rapid plasma membrane breakdown, and swollen nuclei without internucleosomal DNA fragmentation. Oncosis depletes cellular energy and leads to failure of the ionic pumps in the plasma membrane. It is elicited by agents that disrupt the ATP production of the cell (Fink and Cookson, 2005).
Pyroptosis
Apoptosis in general does not induce an inflammatory response. However, apoptosis in Shigella, Salmonella, Francisella, and Legionella infections produce inflammatory responses that are called as pyroptosis (Carneiro et al., 2009; Lee et al., 2011). Pyroptosis is executed by the formation of inflammasomes by bacterial products involving NLRC 4 (Nod-like receptor—NLR), that activates caspase-1 and the processing of IL-1β and IL-18 cytokines promoting cell death (Fink and Cookson, 2005; Carneiro et al., 2009).
Pyronecrosis
Cathepsin B-dependent apoptosis that is independent of caspase-1 activation and inflammasome formation is called pyronecrosis. This mode of apoptosis is observed in shigellosis (Willingham et al., 2007; Carneiro et al., 2009).
Other Apoptotic Models
• Tumor suppressor protein 53 (TP53) induced apoptosis involves the transcriptional induction of redox proteins, generation of reactive oxygen species, and oxidative degradation of mitochondrial components that results in cell death. TP53 was shown to transcriptionally regulate proapoptotic proteins like Bax and NOXA (Yamada et al., 2002).
• NF-kB expression is implicated in the survival of living cells. NF-kB family contains five proteins namely c-Rel, RelA, RelB, p50/p105, and p52/p100. NF-kB as a homo or hetero dimers bind to the kB sites on their target DNA and regulate their expression (Barkett and Gilmore, 1999). NF-kB is activated by various stimuli like pathogens, mitogens, proinflammatory cytokines, etc. It plays a major role in immune responses and affects the expression of genes c-IAP-1 and c-IAP-2, Fas ligand, c-myc, p53, etc. involved in apoptosis (Zhang and Ghosh, 2001). Two TNF receptors TNFRSF8 and TNFRSF9 were shown to promote apoptosis, former activating, and latter inactivating NF-kB expression (Wang et al., 2008).
Apoptosis and Mycobacterium Tuberculosis
M. tuberculosis infections with virulent strains have been reported to inhibit macrophage apoptosis (Behar et al., 2010). Varied mechanisms of apoptotic suppression have been reported in M. tuberculosis infections (Table 1) unraveling the tactics of this pathogen to generate a protective niche inside the host. Among the various cell death modalities described above, only three apoptotic responses were documented in M. tuberculosis infection namely apoptosis (nuoG, SecA2, pknE, lpqH, esxA (ESAT-6), PE_PGRS33, pstS-1, Rv3654c, and Rv3655c), pyroptosis (zmp1, Rv3364c), and autophagy (eis) (Hinchey et al., 2007; Velmurugan et al., 2007; Jayakumar et al., 2008; Master et al., 2008; Sanchez et al., 2009, 2012; Danelishvili et al., 2010, 2012; Shin et al., 2010).
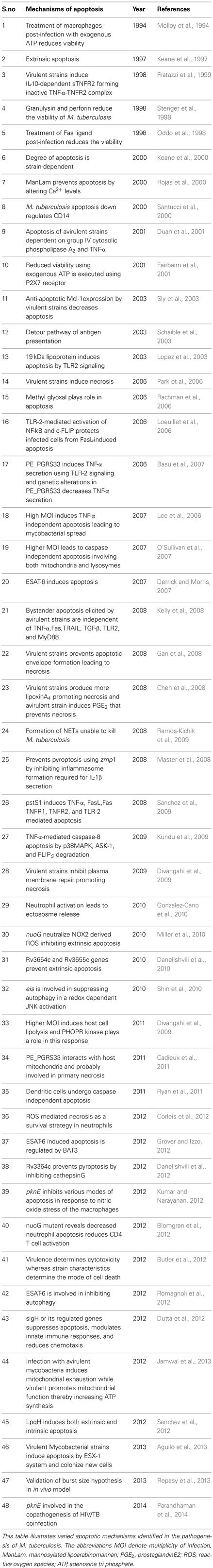
Table 1. Apoptotic mechanisms in the pathogenesis of M. tuberculosis.
Serine/Threonine Protein Kinases (STPK)
Two component signaling systems were considered as the standalone mechanism of signaling in prokaryotes in response to environmental cues. However with the availability of various molecular techniques serine, threonine, and tyrosine mediated phosphorylation events unique to eukaryotes were documented in pathogenic prokaryotes like M. tuberculosis, Streptococcus species, Staphylococcus spp, Pseudomonas spp, etc. (Chao et al., 2009; Chakraborti et al., 2011). Among the 11 STPKs that M. tuberculosis encodes, only five of them pknE, pknG, pknH, pknI, and pknK were reported to support intracellular survival (Walburger et al., 2004; Papavinasasundaram et al., 2005; Jayakumar et al., 2008; Gopalaswamy et al., 2009; Malhotra et al., 2010). Our data for the first time proved that PknE was the only STPK to inhibit apoptosis (Jayakumar et al., 2008).
pknE in Innate Immunity
The function of pknE was established from our studies using the deletion mutant ΔpknE generated using specialized transduction. Deletion of pknE had reduced intracellular survival, increased apoptosis, and reduced proinflammatory responses (Jayakumar et al., 2008). Subsequent molecular pathogenesis studies revealed that the deletion of pknE promotes macrophage cell death dependent on intrinsic pathway of apoptosis, TP53, and Arg2. This apoptosis was independent of TNF-α, iNOS, Akt, Arg1, and pro-inflammatory cytokines (Kumar and Narayanan, 2012). M. tuberculosis encounters reactive nitrogen and oxygen intermediates inside the macrophages as one among the host defenses. Characterization of the promoter of the pknE gene showed its elevated expression during nitric oxide (NO) stress (Jayakumar et al., 2008). Macrophage experiments performed using NO donor sodium nitroprusside to mimic the host microbicidal activity confirmed that, pknE in response to NO stress suppresses innate immune responses (Kumar and Narayanan, 2012). In vitro studies carried with the deletion mutant showed defective growth in pH 7.0 and lysozyme (a cell wall-damaging agent) with better survival in pH 5.5, SDS (surfactant stress), and kanamycin (a second-line anti-tuberculosis drug). ΔpknE was reduced in cell size during growth in liquid media and exhibited hypervirulence in a guinea pig model of infection (Kumar et al., 2012). The data from the in vitro studies highlighted the role of pknE in adaptive responses of M. tuberculosis. Recently we reported that, deletion of pknE results in defective phosphorylation kinetics of MAPKs (p38MAPK, Erk½, and SAPK/JNK) and their transcription factors ATF-2 and c-JUN. Deletion of pknE also revealed crosstalks in the host macrophages where Erk½ signaling was found to be influenced by SAPK/JNK and p38 pathways independently. Modulations in intra cellular signaling altered the expression of coreceptors CCR5 and CXCR4 in macrophages infected with the deletion mutant of pknE that were authenticated using HIV tropic strains (Parandhaman et al., 2014). For the first time, our data showed that difference in apoptosis and intracellular signaling events, and the virulence capacity of the M. tuberculosis strain could influence the copathogenesis of HIV infection (Parandhaman et al., 2014). Collectively the reports show that pknE has a role suppression of innate immunity and help M. tuberculosis to adapt to the different environmental condition that it encounters.
Conclusion
Molecular techniques have revolutionized our understanding of pathogenic organisms and their interactions with the immune system. Pathogenic organisms have evolved host mimicking properties and utilize the host responses for their own survival and propagation. This review has addressed the various mechanisms of cell death that is vital for initiating an innate and adaptive immunity against the invading pathogen. As novel cell death paradigms evolve, it adds to the complexity of how temporally and spatially the immune system coordinates these responses. Most of the cell death models described here disrupt the energy source of the cell, mitochondria indicating whether these paradigms are interconnected response of a single biochemical event and this still remains a puzzle. Adding complexity to this conundrum is that, pathogenic organisms like M. tuberculosis is able to inhibit the various apoptotic models that were discovered so far. This arise the question whether M. tuberculosis by educating itself avoids cell death or has antigens that are poor inducers of cell death and that await further studies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aguilo, J. I., Alonso, H., Uranga, S., Marinova, D., Arbues, A., De Martino, A., et al. (2013). ESX-1-induced apoptosis is involved in cell-to-cell spread of Mycobacterium tuberculosis. Cell. Microbiol. 15, 1994–2005. doi: 10.1111/cmi.12169
Barkett, M., and Gilmore, T. D. (1999). Control of apoptosis by Rel/NF-kappaB transcription factors. Oncogene 18, 6910–6924. doi: 10.1038/sj.onc.1203238
Basu, S., Pathak, S. K., Banerjee, A., Pathak, S., Bhattacharyya, A., Yang, Z., et al. (2007). Execution of macrophage apoptosis by PE_PGRS33 of Mycobacterium tuberculosis is mediated by Toll-like receptor 2-dependent release of tumor necrosis factor-alpha. J. Biol. Chem. 282, 1039–1050. doi: 10.1074/jbc.M604379200
Behar, S. M., Divangahi, M., and Remold, H. G. (2010). Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat. Rev. Microbiol. 8, 668–674. doi: 10.1038/nrmicro2387
Blomgran, R., Desvignes, L., Briken, V., and Ernst, J. D. (2012). Mycobacterium tuberculosis inhibits neutrophil apoptosis, leading to delayed activation of naive CD4 T cells. Cell Host Microbe 11, 81–90. doi: 10.1016/j.chom.2011.11.012
Butler, R. E., Brodin, P., Jang, J., Jang, M. S., Robertson, B. D., Gicquel, B., et al. (2012). The balance of apoptotic and necrotic cell death in Mycobacterium tuberculosis infected macrophages is not dependent on bacterial virulence. PLoS ONE 7:e47573. doi: 10.1371/journal.pone.0047573
Cadieux, N., Parra, M., Cohen, H., Maric, D., Morris, S. L., and Brennan, M. J. (2011). Induction of cell death after localization to the host cell mitochondria by the Mycobacterium tuberculosis PE_PGRS33 protein. Microbiology 157, 793–804. doi: 10.1099/mic.0.041996-0
Carneiro, L. A., Travassos, L. H., Soares, F., Tattoli, I., Magalhaes, J. G., Bozza, M. T., et al. (2009). Shigella induces mitochondrial dysfunction and cell death in nonmyleoid cells. Cell Host Microbe 5, 123–136. doi: 10.1016/j.chom.2008.12.011
Chaabane, W., User, S. D., El-Gazzah, M., Jaksik, R., Sajjadi, E., Rzeszowska-Wolny, J., et al. (2012). Autophagy, apoptosis, mitoptosis and necrosis: interdependence between those pathways and effects on cancer. Arch. Immunol. Ther. Exp. (Warsz.) 61, 43–58. doi: 10.1007/s00005-012-0205-y
Chakraborti, P. K., Matange, N., Nandicoori, V. K., Singh, Y., Tyagi, J. S., and Visweswariah, S. S. (2011). Signalling mechanisms in Mycobacteria. Tuberculosis (Edinb.) 91, 432–440. doi: 10.1016/j.tube.2011.04.005
Chao, J., Wong, D., Zheng, X., Poirier, V., Bach, H., Hmama, Z., et al. (2009). Protein kinase and phosphatase signaling in Mycobacterium tuberculosis physiology and pathogenesis. Biochim. Biophys. Acta 1804, 620–627. doi: 10.1016/j.bbapap.2009.09.008
Chen, M., Divangahi, M., Gan, H., Shin, D. S., Hong, S., Lee, D. M., et al. (2008). Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J. Exp. Med. 205, 2791–2801. doi: 10.1084/jem.20080767
Cillessen, S. A., Hess, C. J., Hooijberg, E., Castricum, K. C., Kortman, P., Denkers, F., et al. (2007). Inhibition of the intrinsic apoptosis pathway downstream of caspase-9 activation causes chemotherapy resistance in diffuse large B-cell lymphoma. Clin. Cancer Res. 13, 7012–7021. doi: 10.1158/1078-0432.CCR-06-2891
Corleis, B., Korbel, D., Wilson, R., Bylund, J., Chee, R., and Schaible, U. E. (2012). Escape of Mycobacterium tuberculosis from oxidative killing by neutrophils. Cell. Microbiol. 14, 1109–1121. doi: 10.1111/j.1462-5822.2012.01783.x
Danelishvili, L., Everman, J. L., McNamara, M. J., and Bermudez, L. E. (2012). Inhibition of the plasma-membrane-associated serine protease cathepsin G by Mycobacterium tuberculosis Rv3364c suppresses caspase-1 and pyroptosis in macrophages. Front. Microbiol. 2:281. doi: 10.3389/fmicb.2011.00281
Danelishvili, L., Yamazaki, Y., Selker, J., and Bermudez, L. E. (2010). Secreted Mycobacterium tuberculosis Rv3654c and Rv3655c proteins participate in the suppression of macrophage apoptosis. PLoS ONE 5:e10474. doi: 10.1371/journal.pone.0010474
Derrick, S. C., and Morris, S. L. (2007). The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell. Microbiol. 9, 1547–1555. doi: 10.1111/j.1462-5822.2007.00892.x
Divangahi, M., Chen, M., Gan, H., Desjardins, D., Hickman, T. T., Lee, D. M., et al. (2009). Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 10, 899–906. doi: 10.1038/ni.1758
Duan, L., Gan, H., Arm, J., and Remold, H. G. (2001). Cytosolic phospholipase A2 participates with TNF-alpha in the induction of apoptosis of human macrophages infected with Mycobacterium tuberculosis H37Ra. J. Immunol. 166, 7469–7476.
Dutta, N. K., Mehra, S., Martinez, A. N., Alvarez, X., Renner, N. A., Morici, L. A., et al. (2012). The stress-response factor SigH modulates the interaction between Mycobacterium tuberculosis and host phagocytes. PLoS ONE 7:e28958. doi: 10.1371/journal.pone.0028958
Elmore, S. (2007). Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516. doi: 10.1080/01926230701320337
Fairbairn, I. P., Stober, C. B., Kumararatne, D. S., and Lammas, D. A. (2001). ATP-mediated killing of intracellular mycobacteria by macrophages is a P2X(7)-dependent process inducing bacterial death by phagosome-lysosome fusion. J. Immunol. 167, 3300–3307.
Ferri, K. F., and Kroemer, G. (2001). Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 3, E255–E263. doi: 10.1038/ncb1101-e255
Fink, S. L., and Cookson, B. T. (2005). Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 73, 1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005
Fratazzi, C., Arbeit, R. D., Carini, C., Balcewicz-Sablinska, M. K., Keane, J., Kornfeld, H., et al. (1999). Macrophage apoptosis in mycobacterial infections. J. Leukoc. Biol. 66, 763–764.
Galluzzi, L., and Kroemer, G. (2008). Necroptosis: a specialized pathway of programmed necrosis. Cell 135, 1161–1163. doi: 10.1016/j.cell.2008.12.004
Gan, H., Lee, J., Ren, F., Chen, M., Kornfeld, H., and Remold, H. G. (2008). Mycobacterium tuberculosis blocks crosslinking of annexin-1 and apoptotic envelope formation on infected macrophages to maintain virulence. Nat. Immunol. 9, 1189–1197. doi: 10.1038/ni.1654
Gonzalez-Cano, P., Mondragon-Flores, R., Sanchez-Torres, L. E., Gonzalez-Pozos, S., Silva-Miranda, M., Monroy-Ostria, A., et al. (2010). Mycobacterium tuberculosis H37Rv induces ectosome release in human polymorphonuclear neutrophils. Tuberculosis (Edinb.) 90, 125–134. doi: 10.1016/j.tube.2010.01.002
Gopalaswamy, R., Narayanan, S., Chen, B., Jacobs, W. R., and Av-Gay, Y. (2009). The serine/threonine protein kinase PknI controls the growth of Mycobacterium tuberculosis upon infection. FEMS Microbiol. Lett. 295, 23–29. doi: 10.1111/j.1574-6968.2009.01570.x
Grover, A., and Izzo, A. A. (2012). BAT3 regulates Mycobacterium tuberculosis protein ESAT-6-mediated apoptosis of macrophages. PLoS ONE 7:e40836. doi: 10.1371/journal.pone.0040836
Hinchey, J., Lee, S., Jeon, B. Y., Basaraba, R. J., Venkataswamy, M. M., Chen, B., et al. (2007). Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. J. Clin. Invest. 117, 2279–2288. doi: 10.1172/JCI31947
Jamwal, S., Midha, M. K., Verma, H. N., Basu, A., Rao, K. V., and Manivel, V. (2013). Characterizing virulence-specific perturbations in the mitochondrial function of macrophages infected with Mycobacterium tuberculosis. Sci. Rep. 3:1328. doi: 10.1038/srep01328
Jayakumar, D., Jacobs, W. R. Jr., and Narayanan, S. (2008). Protein kinase E of Mycobacterium tuberculosis has a role in the nitric oxide stress response and apoptosis in a human macrophage model of infection. Cell. Microbiol. 10, 365–374. doi: 10.1111/j.1462-5822.2007.01049.x
Kabbage, M., Williams, B., and Dickman, M. B. (2013). Cell death control: the interplay of apoptosis and autophagy in the pathogenicity of Sclerotinia sclerotiorum. PLoS Pathog. 9:e1003287. doi: 10.1371/journal.ppat.1003287
Kalimuthu, S., and Se-Kwon, K. (2013). Cell survival and apoptosis signaling as therapeutic target for cancer: marine bioactive compounds. Int. J. Mol. Sci. 14, 2334–2354. doi: 10.3390/ijms14022334
Keane, J., Balcewicz-Sablinska, M. K., Remold, H. G., Chupp, G. L., Meek, B. B., Fenton, M. J., et al. (1997). Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect. Immun. 65, 298–304.
Keane, J., Remold, H. G., and Kornfeld, H. (2000). Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J. Immunol. 164, 2016–2020.
Kelly, D. M., Ten Bokum, A. M., O'Leary, S. M., O'Sullivan, M. P., and Keane, J. (2008). Bystander macrophage apoptosis after Mycobacterium tuberculosis H37Ra infection. Infect. Immun. 76, 351–360. doi: 10.1128/IAI.00614-07
Kumar, D., and Narayanan, S. (2012). pknE, a serine/threonine kinase of Mycobacterium tuberculosis modulates multiple apoptotic paradigms. Infect. Genet. Evol. 12, 737–747. doi: 10.1016/j.meegid.2011.09.008
Kumar, D., Palaniyandi, K., Challu, V. K., Kumar, P., and Narayanan, S. (2012). PknE, a serine/threonine protein kinase from Mycobacterium tuberculosis has a role in adaptive responses. Arch. Microbiol. 195, 75–80. doi: 10.1007/s00203-012-0848-4
Kundu, M., Pathak, S. K., Kumawat, K., Basu, S., Chatterjee, G., Pathak, S., et al. (2009). A TNF- and c-Cbl-dependent FLIP(S)-degradation pathway and its function in Mycobacterium tuberculosis-induced macrophage apoptosis. Nat. Immunol. 10, 918–926. doi: 10.1038/ni.1754
Lee, J., Hartman, M., and Kornfeld, H. (2009). Macrophage apoptosis in tuberculosis. Yonsei Med. J. 50, 1–11. doi: 10.3349/ymj.2009.50.1.1
Lee, J., Remold, H. G., Ieong, M. H., and Kornfeld, H. (2006). Macrophage apoptosis in response to high intracellular burden of Mycobacterium tuberculosis is mediated by a novel caspase-independent pathway. J. Immunol. 176, 4267–4274.
Lee, J., Repasy, T., Papavinasasundaram, K., Sassetti, C., and Kornfeld, H. (2011). Mycobacterium tuberculosis induces an atypical cell death mode to escape from infected macrophages. PLoS ONE 6:e18367. doi: 10.1371/journal.pone.0018367
Levine, B., and Deretic, V. (2007). Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 7, 767–777. doi: 10.1038/nri2161
Loeuillet, C., Martinon, F., Perez, C., Munoz, M., Thome, M., and Meylan, P. R. (2006). Mycobacterium tuberculosis subverts innate immunity to evade specific effectors. J. Immunol. 177, 6245–6255.
Lopez, M., Sly, L. M., Luu, Y., Young, D., Cooper, H., and Reiner, N. E. (2003). The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J. Immunol. 170, 2409–2416.
Malhotra, V., Arteaga-Cortes, L. T., Clay, G., and Clark-Curtiss, J. E. (2010). Mycobacterium tuberculosis protein kinase K confers survival advantage during early infection in mice and regulates growth in culture and during persistent infection: implications for immune modulation. Microbiology 156, 2829–2841. doi: 10.1099/mic.0.040675-0
Master, S. S., Rampini, S. K., Davis, A. S., Keller, C., Ehlers, S., Springer, B., et al. (2008). Mycobacterium tuberculosis prevents inflammasome activation. Cell Host Microbe 3, 224–232. doi: 10.1016/j.chom.2008.03.003
Miller, J. L., Velmurugan, K., Cowan, M. J., and Briken, V. (2010). The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis. PLoS Pathog. 6:e1000864. doi: 10.1371/journal.ppat.1000864
Molloy, A., Laochumroonvorapong, P., and Kaplan, G. (1994). Apoptosis, but not necrosis, of infected monocytes is coupled with killing of intracellular bacillus Calmette-Guerin. J. Exp. Med. 180, 1499–1509. doi: 10.1084/jem.180.4.1499
Oddo, M., Renno, T., Attinger, A., Bakker, T., Macdonald, H. R., and Meylan, P. R. (1998). Fas ligand-induced apoptosis of infected human macrophages reduces the viability of intracellular Mycobacterium tuberculosis. J. Immunol. 160, 5448–5454.
O'Sullivan, M. P., O'Leary, S., Kelly, D. M., and Keane, J. (2007). A caspase-independent pathway mediates macrophage cell death in response to Mycobacterium tuberculosis infection. Infect. Immun. 75, 1984–1993. doi: 10.1128/IAI.01107-06
Papavinasasundaram, K. G., Chan, B., Chung, J. H., Colston, M. J., Davis, E. O., and Av-Gay, Y. (2005). Deletion of the Mycobacterium tuberculosis pknH gene confers a higher bacillary load during the chronic phase of infection in BALB/c mice. J. Bacteriol. 187, 5751–5760. doi: 10.1128/JB.187.16.5751-5760.2005
Parandhaman, D. K., Hanna, L. E., and Narayanan, S. (2014). PknE, a serine/threonine protein kinase of Mycobacterium tuberculosis initiates survival crosstalk that also impacts HIV coinfection. PLoS ONE 9:e83541. doi: 10.1371/journal.pone.0083541
Park, J. S., Tamayo, M. H., Gonzalez-Juarrero, M., Orme, I. M., and Ordway, D. J. (2006). Virulent clinical isolates of Mycobacterium tuberculosis grow rapidly and induce cellular necrosis but minimal apoptosis in murine macrophages. J. Leukoc. Biol. 79, 80–86. doi: 10.1189/jlb.0505250
Pecina-Slaus, N. (2010). Wnt signal transduction pathway and apoptosis: a review. Cancer Cell Int. 10, 22. doi: 10.1186/1475-2867-10-22
Rachman, H., Kim, N., Ulrichs, T., Baumann, S., Pradl, L., Nasser Eddine, A., et al. (2006). Critical role of methylglyoxal and AGE in mycobacteria-induced macrophage apoptosis and activation. PLoS ONE 1:e29. doi: 10.1371/journal.pone.0000029
Ramos-Kichik, V., Mondragon-Flores, R., Mondragon-Castelan, M., Gonzalez-Pozos, S., Muniz-Hernandez, S., Rojas-Espinosa, O., et al. (2009). Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (Edinb.) 89, 29–37. doi: 10.1016/j.tube.2008.09.009
Remijsen, Q., Kuijpers, T. W., Wirawan, E., Lippens, S., Vandenabeele, P., and Vanden Berghe, T. (2011). Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 18, 581–588. doi: 10.1038/cdd.2011.1
Repasy, T., Lee, J., Marino, S., Martinez, N., Kirschner, D. E., Hendricks, G., et al. (2013). Intracellular bacillary burden reflects a burst size for Mycobacterium tuberculosis in vivo. PLoS Pathog. 9:e1003190. doi: 10.1371/journal.ppat.1003190
Rojas, M., Garcia, L. F., Nigou, J., Puzo, G., and Olivier, M. (2000). Mannosylated lipoarabinomannan antagonizes Mycobacterium tuberculosis-induced macrophage apoptosis by altering Ca+2-dependent cell signaling. J. Infect. Dis. 182, 240–251. doi: 10.1086/315676
Romagnoli, A., Etna, M. P., Giacomini, E., Pardini, M., Remoli, M. E., Corazzari, M., et al. (2012). ESX-1 dependent impairment of autophagic flux by Mycobacterium tuberculosis in human dendritic cells. Autophagy 8, 1357–1370. doi: 10.4161/auto.20881
Ruckdeschel, K., Roggenkamp, A., Lafont, V., Mangeat, P., Heesemann, J., and Rouot, B. (1997). Interaction of Yersinia enterocolitica with macrophages leads to macrophage cell death through apoptosis. Infect. Immun. 65, 4813–4821.
Ryan, R. C., O'Sullivan, M. P., and Keane, J. (2011). Mycobacterium tuberculosis infection induces non-apoptotic cell death of human dendritic cells. BMC Microbiol. 11:237. doi: 10.1186/1471-2180-11-237
Sanchez, A., Espinosa, P., Esparza, M. A., Colon, M., Bernal, G., and Mancilla, R. (2009). Mycobacterium tuberculosis 38-kDa lipoprotein is apoptogenic for human monocyte-derived macrophages. Scand. J. Immunol. 69, 20–28. doi: 10.1111/j.1365-3083.2008.02193.x
Sanchez, A., Espinosa, P., Garcia, T., and Mancilla, R. (2012). The 19 kDa Mycobacterium tuberculosis lipoprotein (LpqH) induces macrophage apoptosis through extrinsic and intrinsic pathways: a role for the mitochondrial apoptosis-inducing factor. Clin. Dev. Immunol. 2012:950503. doi: 10.1155/2012/950503
Santucci, M. B., Amicosante, M., Cicconi, R., Montesano, C., Casarini, M., Giosue, S., et al. (2000). Mycobacterium tuberculosis-induced apoptosis in monocytes/macrophages: early membrane modifications and intracellular mycobacterial viability. J. Infect. Dis. 181, 1506–1509. doi: 10.1086/315371
Schaible, U. E., Winau, F., Sieling, P. A., Fischer, K., Collins, H. L., Hagens, K., et al. (2003). Apoptosis facilitates antigen presentation to T lymphocytes through MHC-I and CD1 in tuberculosis. Nat. Med. 9, 1039–1046. doi: 10.1038/nm906
Shin, D. M., Jeon, B. Y., Lee, H. M., Jin, H. S., Yuk, J. M., Song, C. H., et al. (2010). Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog. 6:e1001230. doi: 10.1371/journal.ppat.1001230
Sly, L. M., Hingley-Wilson, S. M., Reiner, N. E., and McMaster, W. R. (2003). Survival of Mycobacterium tuberculosis in host macrophages involves resistance to apoptosis dependent upon induction of antiapoptotic Bcl-2 family member Mcl-1. J. Immunol. 170, 430–437.
Stenger, S., Hanson, D. A., Teitelbaum, R., Dewan, P., Niazi, K. R., Froelich, C. J., et al. (1998). An antimicrobial activity of cytolytic T cells mediated by granulysin. Science 282, 121–125. doi: 10.1126/science.282.5386.121
Strasser, A., O'Connor, L., and Dixit, V. M. (2000). Apoptosis signaling. Annu. Rev. Biochem. 69, 217–245. doi: 10.1146/annurev.biochem.69.1.217
Su, M., Mei, Y., and Sinha, S. (2013). Role of the crosstalk between autophagy and apoptosis in cancer. J. Oncol. 2013:102735. doi: 10.1155/2013/102735
Velmurugan, K., Chen, B., Miller, J. L., Azogue, S., Gurses, S., Hsu, T., et al. (2007). Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLoS Pathog. 3:e110. doi: 10.1371/journal.ppat.0030110
Walburger, A., Koul, A., Ferrari, G., Nguyen, L., Prescianotto-Baschong, C., Huygen, K., et al. (2004). Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 304, 1800–1804. doi: 10.1126/science.1099384
Wang, M., Windgassen, D., and Papoutsakis, E. T. (2008). A global transcriptional view of apoptosis in human T-cell activation. BMC Med. Genomics 1:53. doi: 10.1186/1755-8794-1-53
Wickstrum, J. R., Bokhari, S. M., Fischer, J. L., Pinson, D. M., Yeh, H. W., Horvat, R. T., et al. (2009). Francisella tularensis induces extensive caspase-3 activation and apoptotic cell death in the tissues of infected mice. Infect. Immun. 77, 4827–4836. doi: 10.1128/IAI.00246-09
Willingham, S. B., Bergstralh, D. T., O'Connor, W., Morrison, A. C., Taxman, D. J., Duncan, J. A., et al. (2007). Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe 2, 147–159. doi: 10.1016/j.chom.2007.07.009
Yamada, T., Goto, M., Punj, V., Zaborina, O., Kimbara, K., Das Gupta, T. K., et al. (2002). The bacterial redox protein azurin induces apoptosis in J774 macrophages through complex formation and stabilization of the tumor suppressor protein p53. Infect. Immun. 70, 7054–7062. doi: 10.1128/IAI.70.12.7054-7062.2002
Keywords: apoptosis, pyroptosis, intrinsic pathway, extrinsic pathway, autophagy, Mycobacterium tuberculosis
Citation: Parandhaman DK and Narayanan S (2014) Cell death paradigms in the pathogenesis of Mycobacterium tuberculosis infection. Front. Cell. Infect. Microbiol. 4:31. doi: 10.3389/fcimb.2014.00031
Received: 20 December 2013; Accepted: 17 February 2014;
Published online: 05 March 2014.
Edited by:
Yongqun “Oliver” He, University of Michigan Medical School, USAReviewed by:
Dario S. Zamboni, Universidade de São Paulo, BrazilSubramanian Dhandayuthapani, Texas Tech Health Sciences Center, USA
Copyright © 2014 Parandhaman and Narayanan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sujatha Narayanan, Department of Immunology, National Institute for Research in Tuberculosis (Formerly Tuberculosis Research Centre), Indian Council of Medical Research, Mayor Sathayamoorthy Road, Chetpet, Chennai 600031, Tamilnadu, India e-mail:c3VqYXRoYS5zdWphdGhhMzZAZ21haWwuY29t