Abstract
The Asian tiger mosquito Aedes (Stegomya) albopictus is an invasive species that has spread across the world in the last two decades, showing a great capacity to adapt to contrasting climates and environments. While demonstrated in many insects, the contribution of bacterial symbionts in Aedes ecology is a challenging aspect that needs to be investigated. Also some bacterial species have already been identified in Ae. albopictus using classical methods, but a more accurate survey of mosquito-associated bacterial diversity is needed to decipher the potential biological functions of bacterial symbionts in mediating or constraining insect adaptation. We surveyed the bacteria associated with field populations of Ae. albopictus from Madagascar by pyrosequencing 16S rRNA gene amplicons. Different aspects of amplicon preparation and sequencing depth were tested to optimize the breadth of bacterial diversity identified. The results revealed that all mosquitoes collected from different sites have a bacterial microbiota dominated by a single taxon, Wolbachia pipientis, which accounted for about 99% of all 92,615 sequences obtained. As Ae. albopictus is known to harbor two Wolbachia strains (wAlbA and wAlbB), a quantitative PCR was used to estimate the relative densities, (i.e., the bacteria-to-host gene ratios) of each strains in individual mosquitoes. Relative densities were between 6.25 × 100.01 and 5.47 × 100.1 for wAlbA and between 2.03 × 100.1 and 1.4 × 101 for wAlbB. Apart from Wolbachia, a total of 31 bacterial taxa were identified at the genus level using different method variations. Diversity index values were low and probably underestimated the true diversity due to the high abundance of Wolbachia sequences vastly outnumbering sequences from other taxa. Further studies should implement alternative strategies to specifically discard from analysis any sequences from Wolbachia, the dominant endosymbiotic bacterium in Ae. albopictus from this area.
Introduction
Invasive mosquitoes give cause for worldwide concern not only because of their potential ecological impact, but also they are vectors of a wide range of pathogens affecting both humans and animals (Medlock et al., 2012). In particular, the tiger mosquito Aedes albopictus has rapidly spread across the world from its native South-East Asia and is considered as one of the world's 100 most dangerous invasive species (Benedict et al., 2007). Ae. albopictus was found to be competent for more than 20 arboviruses (Gratz, 2004). Ae. albopictus was involved in the transmission of the Chikungunya and Dengue viruses worldwide, including to Europe (Bonilauri et al., 2008; Grandadam et al., 2011; Caron et al., 2012), and was also implicated as a vector of the parasitic nematode Dirofilaria (Cancrini et al., 2007).
In light of the recent theory of the holobiont unit, it is now necessary to enlarge our view of how commensal host-bacterial relationships function in insects (Feldhaar, 2011; Minard et al., 2013a). Insects establish numerous types of symbiotic associations ranging from parasitism to mutualism with their microbial communities (Toft and Andersson, 2010). Recent studies have highlighted how bacterial endosymbioses influence evolutionary and ecological processes through effects on insect host biology (Douglas, 2011). However, most examples demonstrate how symbiotic microbiota are essential to phytophagous insect functions such as host plant specialization (Tsuchida et al., 2004), reproduction (Simon et al., 2011), protection against natural enemies (Oliver et al., 2003), and tolerance to environmental stress (Brumin et al., 2011; Feldhaar, 2011). It is therefore necessary to assess how the bacterial microbiota contributes to the biology of hematophagous insects, particularly invasive mosquitoes.
Until now the composition of bacterial communities associated with Ae. albopictus has mainly been investigated using classical microbiological and molecular techniques. Various culture-dependent isolation methods were used to identify cultivable bacteria (Zouache et al., 2009; Chouaia et al., 2010; Minard et al., 2013a,b; Valiente Moro et al., 2013). In parallel, non-culture-based methods such as DGGE fingerprinting (Chouaia et al., 2010; Zouache et al., 2011) or taxonomic microarray hybridization (Zouache et al., 2012) were employed. Together these approaches have unraveled some of the bacterial diversity present in both laboratory-reared and field-caught Ae. albopictus mosquitoes. However, these relatively low-throughput techniques cannot provide comprehensive coverage of the microbial composition and diversity in mosquito hosts. The recent rapid development of high-throughput sequencing methods has made it possible to detect a deeper level of microbial diversity in animal hosts, it has been demonstrated for symbiotic microbiotas of insect guts (Shi et al., 2010; Fukatsu, 2012). To date very few studies have used Next-Generation Sequencing technologies to explore bacterial microbiota in mosquitoes. Osei-Poku et al. (2012) successfully used pyrosequencing of 16S rRNA genes to investigate the diversity of gut bacterial communities in field-caught mosquitoes and showed that bacterial composition varies greatly between populations. When pyrosequencing was used to determine the composition of the midgut microbiota of the malaria mosquito vector Anopheles gambiae, much richer information was obtained than from low-throughout methods (Wang et al., 2011; Boissière et al., 2012).
The goal of the present study was to survey the bacterial diversity associated with Ae. albopictus mosquitoes by pyrosequencing 16S rDNA genes. The effect of varying different methodological parameters like those for amplicon preparation and sequencing depth on the bacterial diversity results was assessed.
Materials and methods
Mosquito collection
The sampling area and mosquito capture procedure were approved by Madagascar National Parks. Mosquito specimens were collected from various natural breeding sites on the east coast of Madagascar (Toamasina) in the Antsinanana region (18° 8′ 59.64″ S) in December 2010. Butterfly nets were used to catch adult mosquitoes flying near the grass or around the capturers. Aedes albopictus specimens were identified using morphological characteristics and pictorial keys (Rueda, 2004) and then female and male mosquitoes were separated. Only non-blooded female mosquitoes were used for the analysis, as previous studies have demonstrated that blood meals have a dynamic effect on bacterial diversity (Wang et al., 2011; Boissière et al., 2012). Mosquitoes were stored in 70% ethanol at −80°C until use.
Genomic DNA extraction
All DNA extractions were performed in a sterile environment under a laminar hood to avoid contamination. Mosquitoes were surface-disinfected with 70% ethanol and rinsed with sterile water, as previously described (Minard et al., 2013b). Each mosquito was individually crushed using a Bioblock Scientific MM 2000 mill (Retsch, France) in an Eppendorf tube with 5-mm diameter inox beads and 200 μl of extraction buffer (2% hexadecyltrimethyl ammonium bromide, 1.4 M NaCl, 0.02 M EDTA, 0.1 M Tris pH 8.0, 0.2% 2-β-mercaptoethanol) and heated at 60°C for 2 h. Lipids and proteins were extracted with phenol-chloroform-isoamyl alcohol (25:24:1; v/v/v) and then chloroform:isoamyl alcohol (24:1; v/v). DNA was precipitated with isopropyl alcohol and centrifugation at 16,100 g for 30 min at 4°C. DNA pellets were washed with cold ethanol, air-dried, and resuspended in 20 μl TE buffer (2 mM Tris, 1 mM EDTA).
Primers and PCR amplification
Hypervariable rrs gene V5-V6 regions of about 280 bp were amplified with 784F 5′-AGGATTAGATACCCTGGTA-3′ and 1061R 5′-CRRCACGAGCTGACGAC-3′ primers (Andersson et al., 2008). These primers were selected in silico with the RDP Probe Match tool according to the following criteria (Cole et al., 2009): hybridization with 94% of sequences belonging to the RDP Bacteria domain database with good quality and ≥1200 bp long with 2 mismatches allowed in primer sequences. In addition, Basic Local Alignment Search Tool (BLAST) was used to check that the chosen primers did not match Ae. albopictus 18S and mitochondrial 16S rDNA gene sequences. PCR amplification was performed using 1.75 U of Expand High Fidelity Enzyme Mix (Roche, Switzerland) with 1× Expand High Fidelity Buffer (Roche, Switzerland), 0.06 mg mL−1 of T4 gene 32 protein (Roche, France), 0.06 mg mL−1 of bovine serum albumin (New England Biolabs, France), 40 μM of dNTP mix, 200 nM of each primer (Invitrogen, France) and 30 ng of template DNA. PCR products were purified with QiaAmp purification kit (Qiagen, France) following the manufacturer's recommendations.
Preparation of samples for 454 pyrosequencing and bioinformatic processing
Amplicon tagging and pyrosequencing were performed by a commercial laboratory (DNAvision, Belgium) using the Roche 454 FLX Titanium platform (Roche, Switzerland). Various modalities were tested for in-depth exploration of bacterial diversity (Figure 1) including: (i) constituting a pool of DNA then performing PCR amplification vs. pooling PCR of several amplicons generated from different individual samples; (ii) sequencing a single amplicon vs. a pool of amplicons; and (iii) evaluating the depth of sequencing (number of reads) necessary for the most complete examination of the bacterial diversity. A total of eight rrs V5-V6 amplicon libraries were sequenced at two different ranges of about 3200 reads per sample (S1, S2, S3, S4, S5) and 16,000 reads per sample (S3′, P1, P2) (Figure 1). Data were analyzed by first trimming sequence quality with different cutoffs using standard filtering tools in the Mothur package (Schloss et al., 2009). Two errors on primer sequence and one error on barcode sequence were allowed. Sequences shorter than 250 bp were discarded and chimeric sequences were removed using UCHIME and manual analysis (Edgar et al., 2011). Sequences were grouped into operational taxonomic units (OTUs) by clustering at 97% similarity. Singleton reads assigned to OTUs were discarded from the analysis if they were not found in at least two different samples as singletons are likely to be sequencing errors which can lead to an overestimation of diversity. Rarefaction curves were built to estimate sample coverage. Richness, α diversity and β diversity were calculated to compare samples with respectively richness estimators (Chao, Jackknife, Abundance Coverage Estimator), diversity indices (Simpson, Shannon) and Bray–Curtis dissimilarity. Bray–Curtis dissimilarity values were used to construct a neighbor joining tree with the BIONJ algorithm (Gascuel, 1997) using R software (R Development Core Team, 2009). OTU consensus sequences were screened against the SILVA database (release 111) with a 5% dissimilarity cut-off and an e-value of 10−12. Taxonomic assignment of sequence clusters was performed with local BLASTALL searches. A new sample, named ind, was added to the analysis. This sample was generated by pooling all the reads from each individual sample (S1 to S5) (Figure 1). All sff files were deposited at EMBL European Nucleotide Archive (www.ebi.ac.uk/ena/) under project accession number PRJEB4976.
Figure 1
Quantification of Wolbachia
The relative density of Wolbachia pipientis was measured by real-time quantitative PCR specifically targeting the wsp genes for each of the Wolbachia strains wAlbA and wAlbB (Zouache et al., 2011). Amplification using the LightCycler LC480 apparatus (Roche) was performed using 5 ng of DNA from a single female mosquito (10 females in total), as previously described (Zouache et al., 2011). Standard curves were constructed using a dilution series (101–108 molecules) of the pQuantAlb plasmid containing wsp and actin fragments (Tortosa et al., 2008). For each gene, three technical replicates were processed per DNA sample.
Results
Bacterial species richness and diversity in mosquitoes
The V5-V6 regions of bacterial rrs genes amplified from female Ae. albopictus adults were sequenced by high-throughput pyrosequencing. From the total of 92,615 reads obtained, approximately 79,895 were judged to be of good quality. For samples from individual mosquitoes (S1, S2, S3, S4, S5), read numbers varied from 1583 (S2) to 5048 (S5). For P1, P2, Ind, and S3′, read numbers were 17,584, 20,447, 19,132, and 22,199, respectively. Sequences clustered at 97% similarity into 417 OTUs. The coverage of the sequencing was determined by using rarefaction analysis. The rarefaction curves of samples from single mosquitoes did not reach saturation (Figure 2). On the contrary, saturation was almost attained for the ind pool of individual sample sequences (S1–S5), P1 sequences obtained from amplicons generated from a pool of DNA and P2 sequences obtained from a pool of amplicons (i.e., after amplification of a single sample). The comparison of rarefaction curves between S3 (4276 reads) and S3′ (22,296 reads) clearly indicated that for a single mosquito individual a minimal sequence number close to 16,000 reads is sufficient for a good estimation of sample richness. Indeed, diversity indices Chao1, ACE1, Jackknife for S3 are about half of the respective S3′ values (Table 1). However, the Simpson and Shannon diversity indices of S3 and S3′ were similar. In addition, analyses of Bray–Curtis distances showed a low level of dissimilarity (BC = 0.03939) between S3 and S3′. Other dissimilarity scores revealed that overall the P2 and ind pools (BC = 0.059085) are more similar to each other than to P1 (Figure 3).
Figure 2
Table 1
| Richness | Chao1 | ACE1 | Jackknife | λ | H′ | |
|---|---|---|---|---|---|---|
| S1 | 32 | 44.0000 | 94.3809 | 48.0000 | 0.5537 | 0.8149 |
| S2 | 23 | 36.7500 | 48.4876 | 34.8822 | 0.4548 | 0.9647 |
| S3 | 34 | 43.7500 | 60.0596 | 47.0000 | 0.5092 | 0.8708 |
| S4 | 44 | 69.3000 | 133.3393 | 68.9360 | 0.6461 | 0.7645 |
| S5 | 35 | 42.8000 | 48.1620 | 48.0000 | 0.8159 | 0.4642 |
| S3′ | 88 | 99.3226 | 112.2252 | 115.0000 | 0.5159 | 0.8063 |
| P1 | 66 | 75.5000 | 85.8432 | 86.0000 | 0.6802 | 0.6256 |
| P2 | 82 | 94.0370 | 107.1057 | 108.0000 | 0.6039 | 0.7106 |
| ind | 91 | 99.5556 | 106.7494 | 113.0000 | 0.5967 | 0.8214 |
Richness and diversity index.
Indexes were calculated for each sample with 3% distant OTUs.
Figure 3
Wolbachia sequences are the predominant 16S rDNA amplicons
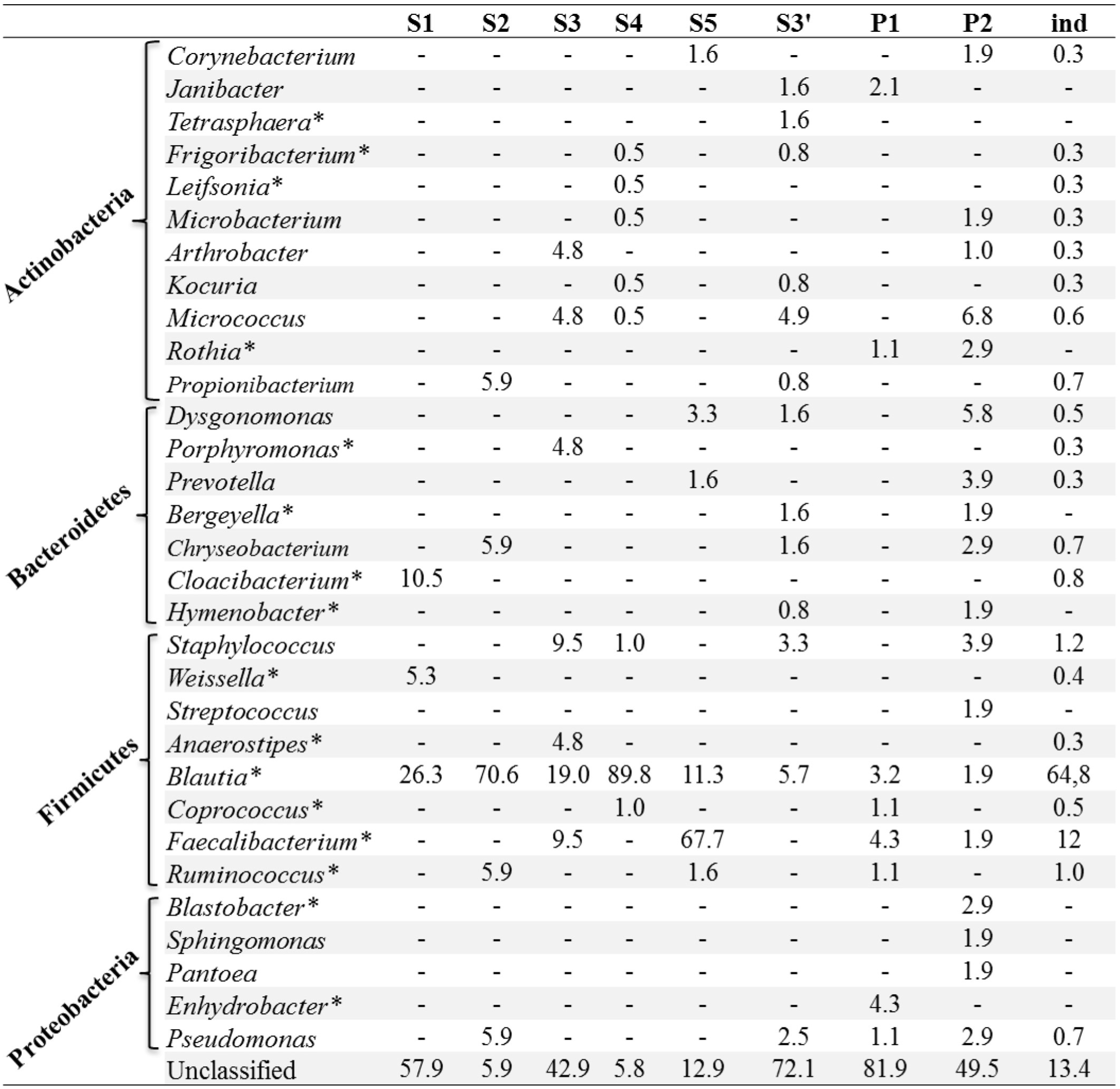
A total of 417 OTUs were assigned to 4 phyla and 32 genera (Figure 4). Sequences from Proteobacteria are the predominant sequences found in all samples with all the experimental modalities tested (>99% of reads). About 96 to 99% of reads belonged to the Wolbachia genus and this largely explains the low diversity values obtained in each condition (Table 1). To confirm sequences assignment to Wolbachia, a local alignment of the two most abundant OTUs (Wolb G1 and Wolb G2) was performed against the non-redundant NCBI database to allow the selection of sequences from the most closely related taxa. The latter sequences were then aligned with Wolb G1 and Wolb G2 sequences and with 15 Wolbachia sequences retrieved from GenBank. This alignment highlighted the presence of a specific 25-bp sequence in the Wolbachia rrs gene (Figure S1). The alignment of this region with public databases was also performed using the SeqMatch tool (http://rdp.cme.msu.edu). The sequence showed 100% similarity and query coverage with other Wolbachia sequences but only 57% query coverage and 100% similarity with the nearest neighbor sequence from different taxa including Candidatus mikurensis, Ehrlichia ruminantium, and Anaplasma platys. This confirmed that the 25-bp sequence was specific to Wolbachia.
Figure 4
The numbers of Wolbachia wAlbA and wAlbB strains present in 10 mosquito individuals were quantified by clade-specific quantitative PCR of wsp, a gene encoding for Wolbachia surface protein, normalized to actin, a mosquito housekeeping gene. For each strain a relatively high gene copy number was found in all individuals tested (Figure 5). The wsp/actin ratios were between 6.25 × 100.01 and 5.47 × 100.1 for wAlbA and between 2.03 × 100.1 and 1.4 × 101 for wAlbB. Significant bacterial density differences were found between individual mosquitoes for both wAlbA (p-value = 8.499 × 10−15, Kruskall–Wallis test) and wAlbB (p-value = 3.616 × 10−14, Kruskall–Wallis).
Figure 5
Taxonomic analysis and relative abundance of other bacterial taxa
Apart from Wolbachia, a total of 31 bacterial taxa were identified at the genus level from less than 1% of all the sequences (Figure 4). Firmicutes bacteria were found to be the dominant group (67%), followed by Actinobacteria (4%), Bacteroidetes (3%), and Proteobacteria (2%). In terms of OTU richness and genus content, ind and P2 were the most diverse with 22 and 19 genera identified, respectively (Tables 1, 2). Sequences corresponding to Blautia, a Firmicutes genus, were found in all the datasets from individual and pooled samples, ranging from 3.2% (for P1) to 89.2% (for S4) of sequences (Table 2). Moreover, 88 sequences from a single OTU were assigned to Lachnospiaceae family with no similarity at genus level. Overall, the proportion of sequences affiliated to known or unclassified bacterial genera varied among samples.
Discussion
The application of next-generation sequencing techniques to insect microbiota research makes it possible to gain a deeper knowledge of its microbial diversity. The goal of the present study was to assess the diversity of bacteria associated with the Asian tiger mosquito using high-throughput sequencing. Previous studies of bacterial diversity in various arthropods using a similar approach gave rise to different methodological parameters (Khöler et al., 2012; Moran et al., 2012; Osei-Poku et al., 2012; Ridley et al., 2012), so we chose to carry out a pilot study using pyrosequencing on a hypervariable region of 16S rRNA genes. Various experimental modalities were tested by varying the sample preparation (processing DNA samples from individuals or mixing several 16S amplicons either before or after PCR amplification) and depth of sequencing (number of reads). The primers chosen targeted the V5-V6 hypervariable region of 16S rRNA which allows a large number of bacterial taxa to be detected from sequences available in databases (94% of RDP database) and takes into account other limiting factors such as amplicon size and a lack of hybridization with host DNA.
The results obtained show first that a depth of at least 16,000 reads was necessary to accurately reach the saturation of richness discovery curves. Indeed, the observed richness was made to approach the estimated richness by increasing the depth of sequencing as demonstrated by comparing S3 (4276 reads) and S3′ (22,296 reads). In addition, as these two datasets originated from the same amplicon sample sequenced independently, the low Bray–Curtis dissimilarity levels between them reflects experimental reproducibility. Another factor evaluated was whether the pooling of samples would prove a cost-effective way to compare a huge number of samples. The drawback is that pooling may hide variability in the bacterial composition among samples. Here, we showed that it is preferable to pool amplicons rather than DNA before amplification as the latter method clearly failed to detect rare sequences.
In this study, pyrosequencing amplicons from Ae. albopictus bacterial microbiota revealed a lower Simpson diversity index than previously obtained with fingerprint methods (Zouache et al., 2011). However, using 454 pyrosequencing of 16S rRNA fragments, Osei-Poku et al. (2012) found that 23 genera occurred at a frequency of more than 1% in the guts of at least one individual of different mosquito species. They also reported very low bacterial diversity within individuals, with a single OTU typically making up two-thirds of all the bacteria in all field-caught individuals from seven species studied, which is similar to our results. Interestingly, the results presented here reveal the highest genus richness that we have yet found in Ae. albopictus using various methods (Zouache et al., 2011, 2012; Valiente Moro et al., 2013). The results show that high throughput sequencing is an efficient way to deeply investigate microbiota composition in mosquitoes.
We conclude that Wolbachia was the principle component of Ae. albopictus microbiota in different individuals as surprisingly about 99% of reads were Wolbachia sequences independent of the experimental method used. The relative densities of Wolbachia wAlbA and wAlbB in both field-caught and lab-reared Ae. albopictus have been measured by quantitative PCR and the values we report here are similar to those previously found in field-caught populations (Tortosa et al., 2010; Zouache et al., 2011, 2012). All mosquito samples analyzed here were positive for both strains. Possibly the abundance of Wolbachia and strain bi-infection confers a fitness advantage like that described in Wolbachia-superinfected mosquitoes (Dobson et al., 2002, 2004). This selective pressure that acts on hatching rates and lifespan in laboratory conditions could be a reason for the predominance of Wolbachia in field populations mitigating the metabolic cost of symbiosis. Cytoplasmic incompatibility induced by Wolbachia is also responsible for a high prevalence of superinfected females (Kittayapong et al., 2002; Dobson et al., 2004; Zouache et al., 2011).
Sequences affiliated to Blautia were the second most abundant after those from Wolbachia. This is the first report of the presence of this genus in Ae. albopictus and even in mosquitoes. Blautia are anaerobic acetogenic members of Firmicutes and some have been isolated from mammal guts (Bernalier et al., 1996; Gagen et al., 2010). In addition to Blautia, we discovered other anaerobic bacteria in Ae. albopictus, including Faecalibacterium, Ruminococcus, and Coprococcus. Some of these are thought to be involved in the metabolism of organic compounds such as acetic acid in the human gut (Tsai et al., 1976; Wrzosek et al., 2013). Of the 32 bacterial genera identified overall, 9 had been previously found in Ae. albopictus and 8 others had already been described in other mosquito species using low throughput molecular techniques (reviewed in Minard et al., 2013a). Among these bacteria, the genus Pantoea is one of the most prevalent bacterial cultivable groups identified in field-caught Ae. albopictus from Madagascar (Valiente Moro et al., 2013). The confirmation of the presence of these bacterial taxa in Ae. albopictus highlights the urgency of characterizing the metabolic aspects of these host-microbe interactions in relation to habitat specialization. Stinkingly, some human opportunistic pathogens belong to genera that were identified in mosquitoes such as Pseudomonas, Staphylococcus, Chryseobacterium, and Dysgonomonas (Hironaga et al., 2008; Asaad et al., 2013; Dettman et al., 2013; Ozcan et al., 2013; Coates et al., 2014). Linked to the contact between mosquito proboscis and human circulation during females' blood feeding, these observations argue the possibility of a mosquito-mediated bacterial transmission. Finally, a number of sequences were not assigned to any bacterial family but sequencing longer amplicons might improve their identification rate.
Conclusion
Here we developed a high-throughput sequencing protocol to phylotype bacterial communities associated with Ae. albopictus mosquitoes. To our knowledge, this is the first time that such an approach has been used to explore Ae. albopictus microbiota. Although the results reveal the most complete picture of the bacterial microbiota in any given insect, they also show the limits of the high-throughput sequencing approach when there is a dominant endosymbiotic species. Alternative strategies such as the use of blocking primers (Vestheim et al., 2011) will be implemented to specifically discard sequences of the particularly abundant Wolbachia endosymbiont. The NGS approach developed in this study will be helpful to perform a large scale analysis of mosquito-associated microbiota from different geographic locations and would allow exploring the role of bacteria in invasive capacities of mosquitoes.
Statements
Author contributions
Guillaume Minard, Claire Valiente Moro, and Patrick Mavingui conceived and designed the work. Guillaume Minard, Florence-Hélène Tran, and Van Tran-Van performed the experiments. Guillaume Minard and Audrey Dubost managed the data mining. Guillaume Minard, Claire Valiente Moro, and Patrick Mavingui analyzed and interpreted the data, and drafted the manuscript and the final version was approved by all the authors.
Acknowledgments
We thank Maxime Bruto for his help in phylogeny analysis. Funding for this project was provided by grants from EC2CO CNRS and CMIRA Région Rhône-Alpes. This research was also partially funded by the ERA-NET BiodivERsA, with the national funders FRB France, FWF Austria, and DFG Germany, part of the 2012-2013 BiodivERsA call for research proposals, and was carried out within the frameworks of GDRIs “Biodiversité et Développement Durable à Madagascar” and “Biodiversity and Infectious Diseases in Southeast Asia.”
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fcimb.2014.00059/abstract
Figure S1Alignment of Wolbachia reads. Wolbachia reads Wolb G1 and Wolb G2 were aligned with sequences from most closely related taxa (based on V5-V6 rrs sequences) and other Wolbachia sequences retrieved from GenBank, using Jalview software. A 25-bp Wolbachia specific region is highlighted.
References
1
AnderssonA. F.LindbergM.JakobssonH.NyrénP.EngstrandL. (2008). Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE3:e2836. 10.1371/journal.pone.0002836
2
AsaadM. A.Al-AyedM. S.QureshM. A. (2013). Emergence of unusual nonfermenting gram-negative nosocomial pathogens in a Saudi hospital. Jpn. J. Infect. Dis. 66, 507–511. 10.7883/yoken.66.507
3
BenedictM. Q.LevineR. S.HawleyW. A.LounibosL. P. (2007). Spread of the tiger: global risk of invasion by the mosquito Aedes albopictus. Vector Borne Zoonotic Dis. 7, 76–85. 10.1089/vbz.2006.0562
4
BernalierA.WillemsA.LeclercM.RochetV.CollinsM. D. (1996). Ruminococcus hydrogenotrophicus sp. nov., a new H2/CO2-utilizing acetogenic bacterium isolated from human feces. Arch. Microbiol. 166, 176–183. 10.1007/s002030050373
5
BoissièreA.TchioffoM. T.BacharD.AbateL.MarieA.NsangoS. E.et al. (2012). Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLOS Pathog. 8:e1002742. 10.1371/journal.ppat.1002742
6
BonilauriP.BelliniR.CalzolariM.AngeliniR.VenturiL.FallacaraF.et al. (2008). Chikungunya virus in Aedes albopictus, Italy. Emerg. Infect. Dis. 14, 852–854. 10.3201/eid1405.071144
7
BruminM.KontsedalovS.GhanimM. (2011). Rickettsia influences thermotolerance in the whitefly Bemisia tabaci B biotype. Insect Sci. 18, 57–56. 10.1111/j.1744-7917.2010.01396.x
8
CancriniG.ScaramozzinoP.GabrielliS.Di PaoloM.TomaL.RomiR. (2007). Aedes albopictus and Culex pipiens implicated as natural vectors of Dirofilaria repens in central Italy. J. Med. Entomol. 44, 1064–1066. 10.1603/0022-2585(2007)44[1064:AAACPI]2.0.CO;2
9
CaronM.PaupyC.GrardG.BecquartP.MomboI.NsoB. B.et al. (2012). Recent introduction and rapid dissemination of Chikungunya virus and Dengue virus serotype 2 associated with human and mosquito coinfections in Gabon, central Africa. Clin. Infect. Dis. 55, e45–e53. 10.1093/cid/cis530
10
ChouaiaB.RossiP.MontagnaM.RicciI.CrottiE.DamianiC.et al. (2010). Molecular evidence for multiple infections as revealed by typing of Asaia bacterial symbionts of four mosquito species. Appl. Environ. Microbiol. 76, 7444–7450. 10.1128/AEM.01747-10
11
CoatesR.MoranJ.HorsbughM. J. (2014). Staphylococci: colonizers and pathogens of human skin. Future Microbiol. 9, 75–91. 10.2217/fmb.13.145
12
ColeJ. R.WangQ.CardenasE.FishJ.ChaiB.FarrisR. J.et al. (2009). The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37, D141–D145. 10.1093/nar/gkn879
13
DettmanJ. R.RodrigueN.AaronS. D.KassenR. (2013). Evolutionary genomics of epidemic strains of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 110, 21065–21070. 10.1073/pnas.1307862110
14
DobsonS. L.MarslandE. J.RattanadechakulW. (2002). Mutualistic Wolbachia infection in Aedes albopictus: accelerating cytoplasmic drive. Genetics160, 1087–1094.
15
DobsonS. L.RattanadechakulW.MarslandE. J. (2004). Fitness advantage and cytoplasmic incompatibility in Wolbachia single- and superinfected Aedes albopictus. Heredity93, 135–142. 10.1038/sj.hdy.6800458
16
DouglasA. E. (2011). Lessons from studying insect symbioses. Cell Host Microbe10, 359–367. 10.1016/j.chom.2011.09.001
17
EdgarR. C.HaasB. J.ClementeJ. C.QuinceC.KnightR. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics27, 2194–2200. 10.1093/bioinformatics/btr381
18
FeldhaarH. (2011). Bacterial symbionts as mediators of ecologically important traits of insect hosts. Ecol. Entomol. 36, 533–543. 10.1111/j.1365-2311.2011.01318.x
19
FukatsuT. (2012). Next-generation sequencing sheds light on intricate regulation of insect gut microbiota. Mol. Ecol. 21, 5908–5910. 10.1111/mec.12090
20
GagenE. J.DenmanS. E.PadmanabhaJ.ZadbukeS.Al JassimR.MorrisonM.et al. (2010). Functional gene analysis suggests different acetogen populations in the bovine rumen and tammar wallaby forestomach. Appl. Environ. Microbiol. 76, 7785–7795. 10.1128/AEM.01679-10
21
GascuelO. (1997). BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695. 10.1093/oxfordjournals.molbev.a025808
22
GrandadamM.CaroV.PlumetS.ThibergeJ. M.SouarèsY.FaillouxA. B.et al. (2011). Chikungunya virus, southeastern France. Emerg. Infect. Dis. 17, 910–913. 10.3201/eid1705.101873
23
GratzN. G. (2004). Critical review of the vector status of Aedes albopictus. Med. Vet. Entomol. 18, 215–227. 10.1111/j.0269-283X.2004.00513.x
24
HironagaM.YamaneK.InabaM.HagaY.ArakawaY. (2008). Characterization and antimicrobial succeptibility of Dysgonomonas capnocytophagoides isolated from human blood sampe. Jpn. J. Infect. Dis. 61, 212–213.
25
KittayapongP.BaisleyK. J.SharpeR. G.BaimaiV.O'NeillS. L. (2002). Maternal transmission efficiency of Wolbachia superinfections in Aedes albopictus populations in Thailand. Am. J. Trop. Med. Hyg. 66, 103–107.
26
KhölerT.DietrichC.ScheffrahnR. H.BruneA. (2012). High-resolution analysis of gut environment and bacterial microbiota reveals functional compartmentation of the gut in wood-feeding higher termites (Nasutitermes spp.). Appl. Environ. Microbiol. 78, 4691–4701. 10.1128/AEM.00683-12
27
MedlockJ. M.HansfordK. M.SchaffnerF.VersteirtV.HendrickxG.ZellerH.et al. (2012). A review of the invasive mosquitoes in Europe: ecology, public health risks, and control options. Vector Borne Zoonotic Dis. 12, 435–447. 10.1089/vbz.2011.0814
28
MinardG.MavinguiP.MoroC. V. (2013a). Diversity and function of bacterial microbiota in the mosquito holobiont. Parasit. Vectors6, 146. 10.1186/1756-3305-6-146
29
MinardG.TranF. H.RaharimalalaF. N.HellardE.RavelonandroP.MavinguiP.et al. (2013b). Prevalence, genomic and metabolic profiles of Acinetobacter and Asaia associated with field-caught Aedes albopictus from Madagascar. FEMS Microbiol. Ecol. 83, 63–73. 10.1111/j.1574-6941.2012.01455.x
30
MoranN. A.HansenA. K.PowellJ. E.SabreeZ. L. (2012). Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees. PLoS ONE7:e36393. 10.1371/journal.pone.0036393
31
OliverK. M.RussellJ. A.MoranN. A.HunterM. S. (2003). Facultative bacterial symbionts in aphids confer resistance to parasitic wasps. Proc. Natl. Acad. Sci. U.S.A. 100, 1803–1807. 10.1073/pnas.0335320100
32
Osei-PokuJ.MbogoC. M.PalmerW. J.JigginsF. M. (2012). Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Mol. Ecol. 21, 5138–5150. 10.1111/j.1365-294X.2012.05759.x
33
OzcanN.TubaD.TekinA.KelekciS.CanS.EzinO.et al. (2013). Is Chryseobacterium indologenes a shunt-lover bacterium? A case report and review of the literature. Infez. Med. 4, 312–316.
34
R Development Core Team. (2009). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: http://www.R-project.org
35
RidleyE. V.WongA. C.WestmillerS.DouglasA. E. (2012). Impact of the resident microbiota on the nutritional phenotype of Drosophila melanogaster. PLoS ONE7:e36765. 10.1371/journal.pone.0036765
36
RuedaL. M. (2004). Pictorial keys for the identification of mosquitoes (Diptera: Culicidae) associated with Dengue Virus Transmission. Zootaxa589, 33–41.
37
SchlossP. D.WestcottS. L.RyabinT.HallJ. R.HartmannM.HollisterE. B.et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. 10.1128/AEM.01541-09
38
ShiW.SyrenneR.SunJ. Z.YuanJ. S. (2010). Molecular approaches to study the insect gut symbiotic microbiota at the “omics” age. Insect Sci. 17, 199–219. 10.1111/j.1744-7917.2010.01340.x
39
SimonJ. C.BoutinS.TsuchidaT.KogaR.Le GallicJ. F.FrantzA.et al. (2011). Facultative symbiont infections affect aphid reproduction. PLoS ONE6:e21831. 10.1371/journal.pone.0021831
40
ToftC.AnderssonS. G. (2010). Evolutionary microbial genomics: insights into bacterial host adaptation. Nat. Rev. Genetics. 11, 465–475. 10.1038/nrg2798
41
TortosaP.CharlatS.LabbéP.DehecqJ. S.BarréH.WeillM. (2010). Wolbachia age-sex-specific density in Aedes albopictus: a host evolutionary response to cytoplasmic incompatibility?PLoS ONE5:e9700. 10.1371/journal.pone.0009700
42
TortosaP.CourtiolA.MoutaillerS.FaillouxA. B.WeillM. (2008). Chikungunya-Wolbachia interplay in Aedes albopictus. Insect. Mol. Biol. 17, 677–684. 10.1111/j.1365-2583.2008.00842.x
43
TsaiC. G.GatesD. M.IndledewW. M.JonesG. A. (1976). Products of anaerobic phloroglucinol degradation by Coprococcus sp. Pe15. Can. J. Microbiol. 22, 159–164. 10.1139/m76-022
44
TsuchidaT.KogaR.FukatsuT. (2004). Host plant specialization governed by facultative symbiont. Science303, 1989. 10.1126/science.1094611
45
Valiente MoroC.TranF. H.RaharimalalaF. N.RavelonandroP.MavinguiP. (2013). Diversity of culturable bacteria including Pantoea in wild mosquito Aedes albopictus. BMC Microbiol. 13:70. 10.1186/1471-2180-13-70
46
VestheimH.DeagleB. E.JarmanS. N. (2011). Application of blocking oligonucleotides to improve signal-to-noise ratio in a PCR. Methods Mol. Biol. 687, 265–274. 10.1007/978-1-60761-944-4_19
47
WangY.GilbreathT. M.III.KukutlaP.YanG.XuJ. (2011). Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS ONE6:e24767. 10.1371/journal.pone.0024767
48
WrzosekL.MiquelS.NoordineM. L.BouetS.Joncquel Chevalier-CurtM.RobertV.et al. (2013). Bacteroides thetaiotaomicron and Faecalibacterium prausnitzii influence the production of mucus glycans and the development of goblet cells in the colonic epithelium of a gnotobiotic model rodent. BMC Biol. 11:61. 10.1186/1741-7007-11-61
49
ZouacheK.MichellandR. J.FaillouxA. B.GrundmannG. L.MavinguiP. (2012). Chikungunya virus impacts the diversity of symbiotic bacteria in mosquito vector. Mol. Ecol. 21, 2297–2309. 10.1111/j.1365-294X.2012.05526.x
50
ZouacheK.RaharimalalaF. N.RaquinV.Tran-VanV.RavelosonL. H.RavelonandroP.et al. (2011). Bacterial diversity of field-caught mosquitoes, Aedes albopictus and Ae. aegypti, from different geographic regions of Madagascar. FEMS Microbiol. Ecol. 75, 377–389. 10.1111/j.1574-6941.2010.01012.x
51
ZouacheK.VoroninD.Tran-VanV.MoussonL.FaillouxA. B.MavinguiP. (2009). Persistent Wolbachia and cultivable bacteria infection in the reproductive and somatic tissues of the mosquito vector Aedes albopictus. PLoS ONE4:e6388. 10.1371/journal.pone.0006388
Summary
Keywords
high-throughput sequencing, 16S rDNA, bacterial diversity, Wolbachia, field-caught mosquitoes
Citation
Minard G, Tran F-H, Dubost A, Tran-Van V, Mavingui P and Valiente Moro C (2014) Pyrosequencing 16S rRNA genes of bacteria associated with wild tiger mosquito Aedes albopictus: a pilot study. Front. Cell. Infect. Microbiol. 4:59. doi: 10.3389/fcimb.2014.00059
Received
27 January 2014
Accepted
21 April 2014
Published
14 May 2014
Volume
4 - 2014
Edited by
Muriel Vayssier-Taussat, INRA, France
Reviewed by
Fadi Bittar, Aix-Marseille Université, URMITE, UM63, CNRS 7278, IRD 198, Inserm 1095, France; Damien F. Meyer, CIRAD, France
Copyright
© 2014 Minard, Tran, Dubost, Tran-Van, Mavingui and Valiente Moro.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patrick Mavingui and Claire Valiente Moro, Ecologie Microbienne, UMR CNRS 5557, Université de Lyon 1, Bâtiment Lwoff, 10, rue Raphaël Dubois, 69100 Villeurbanne, France e-mail: patrick.mavingui@univ-lyon1.fr; claire.valiente-moro@univ-lyon1.fr
This article was submitted to the journal Frontiers in Cellular and Infection Microbiology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.