 Katie Haning
Katie Haning Seung Hee Cho
Seung Hee Cho Lydia M. Contreras
Lydia M. Contreras- 1McKetta Department of Chemical Engineering, Cockrell School of Engineering, The University of Texas at Austin, Austin, TX, USA
- 2Department of Molecular Biosciences, Institute for Cellular and Molecular Biology, The University of Texas at Austin, Austin, TX, USA
Mycobacteria represent a class of powerful pathogens, including those causing tuberculosis and leprosy, which continue to be worldwide health challenges. In the last 20 years, an abundance of non-coding, small RNAs (sRNAs) have been discovered in model bacteria and gained significant attention as regulators of cellular responses, including pathogenesis. Naturally, a search in mycobacteria followed, revealing over 200 sRNAs thus far. Characterization of these sRNAs is only beginning, but differential expression under environmental stresses suggests relevance to mycobacterial pathogenesis. This review provides a comprehensive overview of the current knowledge of sRNAs in mycobacteria, including historical perspective and techniques used for identification and characterization.
Introduction
Mycobacterial species threaten human health worldwide, causing infectious diseases such as tuberculosis and leprosy. More than 140 species have been reported in the gram-positive genus Mycobacterium, which is divided into three major categories: Mycobaterium tuberculosis complex, Mycobacterium leprae, and non-tuberculosis mycobacteria (Jagielski et al., 2014). The majority of species are non-tuberculosis mycobacteria related to non-pathogenic organisms that live in water or soil. Diagnosis of non-tuberculosis mycobacteria is complex since they are diverse in growth temperatures, growth rates, and drug susceptibility, as well as in clinical relevance (Cosma et al., 2003). M. leprae and Mycobacterium ulcerans are considered to be highly successful pathogens for causing leprosy and Buruli ulcers, respectively. However, M. tuberculosis is one of the most common pathogens, causing tuberculosis in humans and animals. M. bovis and other five closely related species are also classified within the M. tuberculosis complex (shown in Figure 1). Although genome sequences are highly related among species in the M. tuberculosis complex, phenotypic properties and hosts vary by species (Cole et al., 1998; Garnier et al., 2003).
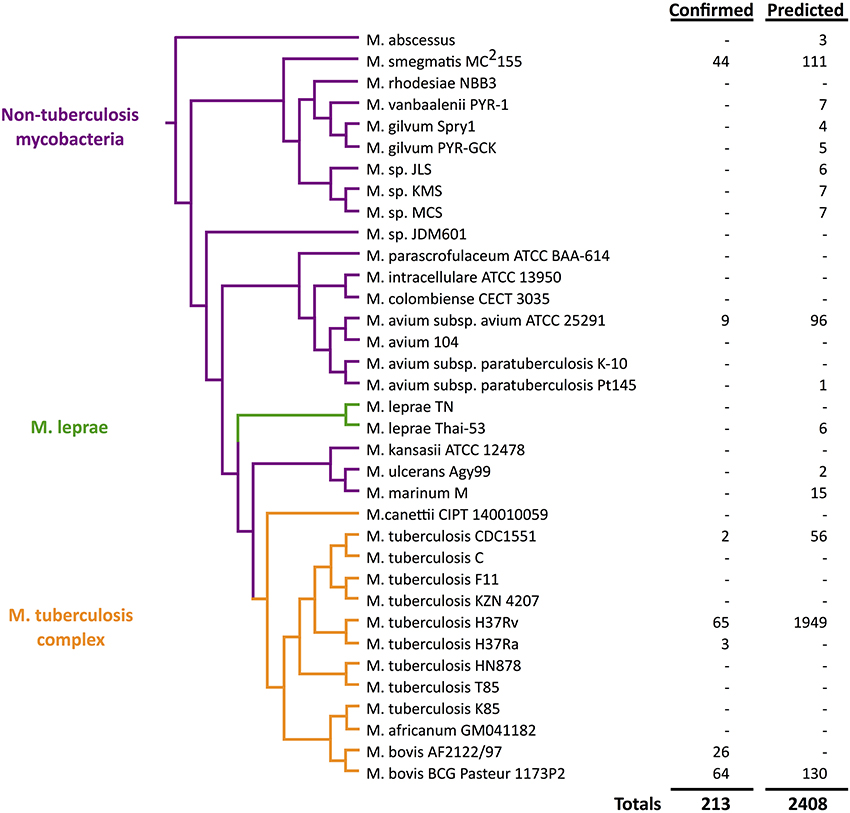
Figure 1. Quantitative view of sRNA discovery in mycobacteria by phylogeny. Confirmed sRNAs have been experimentally validated while unconfirmed sRNAs include unverified computational predictions (Wattam et al., 2014).
M. tuberculosis is transmitted by aerosol and has evolutionarily developed various strategies to evade host immune systems. Upon invasion, M. tuberculosis infects host macrophages. Infection with attenuated strains of M. tuberculosis causes apoptosis to macrophages and, consequently, induces adaptive immunity by recruiting T cells (Behar et al., 2010). However, virulent M. tuberculosis might inhibit apoptosis, favoring macrophage necrosis, and ultimately translocate into the cytosol. Once it can survive against macrophages, M. tuberculosis starts to quickly replicate its genome and to infect adjacent cells (Keane et al., 2000; Wilkinson et al., 2005). It is worth noting that within granuloma, formed with macrophages, neutrophils, and other immune cells, M. tuberculosis persists for a long time through various evasion strategies that can lead to reactivation and, eventually, disease (Huynh et al., 2011).
The life cycle of M. tuberculosis is unique in that it has two distinct metabolic states: one is an active replicative state and the other is a persistent state (Wayne and Sohaskey, 2001). M. tuberculosis can hide from the host defense mechanism by non-proliferative persistent states. This is the most contributing feature of M. tuberculosis for successful survival in host cells and is referred to as latent infection, but can be reactivated. It has been known that continuous activation of macrophage is important for preventing reactivation of the infection (Flynn et al., 1998). As such, control of latency in mycobacteria depends on host responses such as maintenance of granuloma, cytokines, and chemokines (Flynn and Chan, 2001).
Development of effective drugs against mycobacteria is still an active area of research. The keys to these efforts lie in the mechanisms of how mycobacteria switch from latent states to aggressive, disease-causing forms. Recently, sequence-based approaches for mycobacteria revealed extensive numbers of non-coding RNAs that include intergenic small RNAs (sRNAs) (Arnvig et al., 2011). Regulatory RNAs participate in adaptive responses of bacteria against environmental change such as transitions during pathogenesis. As interest escalates in sRNAs across mycobacterial species, there is a need to compile current knowledge in the field to unify and focus future work to gain highly sought medical advantages. The focus of this review is to unfold the history of sRNA investigation in mycobacterial species. We begin with a brief narration of sRNA discovery and characterization in pathogens and then extensively document progress in the search and characterization of mycobacterial sRNAs in particular.
Small RNAs in Pathogenesis: from Discovery to Targeting
Mycobacteria and other pathogens must be highly adaptive to endure and exploit environmental changes presented by hosts and their immune responses. Traditionally, various transcription factors were credited for such cellular reprogramming by turning on and off expression of relevant genes in response to environmental stimuli. However, over the last 15 years, understanding of bacterial gene expression has expanded beyond the DNA level, now including multiple layers of regulation. Particularly interesting are sRNAs, typically 50–500 nucleotides in length, known to act as global regulators of cellular phenotypes (Gottesman and Storz, 2011). Although previously thought to be strictly untranslated regions, some sRNAs have now been classified as bifunctional, encoding small peptides (Bobrovskyy and Vanderpool, 2013). Although our focus is on sRNAs, we have also included limited discussions of recently identified riboswitches in mycobacteria.
Efforts of sRNA Discovery in Model Organisms Highlight Biological Relevance
The first bacterial sRNA was discovered almost 50 years ago in Escherichia coli MRE600 (ATCC 29417), but its function remained unknown for three decades, surfacing shortly after the complete genome of E. coli K-12 was published (Hindley, 1967; Blattner, 1997; Wassarman and Storz, 2000). At this time, 10 sRNAs were known in E. coli, mostly discovered by chance during studies of individual genetic systems (Wassarman et al., 1999). However, availability of the fully sequenced E. coli K-12 genome led to the booming of computational approaches, enabling large-scale systematic searches of intergenic regions for sRNAs. In fact, 14 novel sRNAs were reported in a single study of E. coli in 2001, more than doubling the previously known list (Argaman et al., 2001). Recent developments in deep sequencing and high-density microarray technologies have continued to allow extensive genome-wide studies of sRNAs and their functions. Now, ~100 sRNAs are known to exist in E. coli and it has become clear that many are involved in regulating stress responses (Gottesman et al., 2006; Park et al., 2013). These sRNAs show significant conservation among other organisms, including pathogenic bacteria (Hershberg et al., 2003).
Investigations in E. coli have increasingly shifted toward mechanistic studies and revealed that regulatory sRNAs typically function by base-pairing with target mRNAs, thereby altering transcription, mRNA stability, or translation (Gottesman, 2004). Regulatory sRNAs are generally classified by their genomic locations with respect to their targets. Cis-encoded sRNAs are encoded on the same gene as a target mRNA, but in the opposite, complementary direction, while trans-encoded sRNAs are located apart from their targets in the genome. Many trans-encoded sRNAs have been shown to depend on Hfq, an RNA-binding protein providing stability, in order to function (Zhang et al., 2003); this has been particularly prevalent in gram-negative bacteria. Some sRNAs have also been discovered to regulate proteins directly (Romeo et al., 2013). Lastly, riboswitches represent another class of powerful RNA regulators, typically found in the 5′ untranslated regions (5′ UTRs) of their mRNA targets and directly responsive to environmental changes. In this category, RNA thermometers have been found to respond structurally to changes in temperature to regulate translation (Kortmann and Narberhaus, 2012). It is worth noting that sRNA classification continues to present a major challenge with increasing diversity.
With improved mechanistic understanding, there is enormous interest in inhibiting and/or mimicking expression of natural sRNA actions to achieve desired changes in targeted genes (Na et al., 2013; Vazquez-Anderson and Contreras, 2013). Figure 2 summarizes the large-scale sRNA searches, validation experiments, mechanistic and functional studies, and ultimately, targeting efforts. In parallel with such progress in model organisms, investigations emerged to follow this path in more unique classes of bacteria.
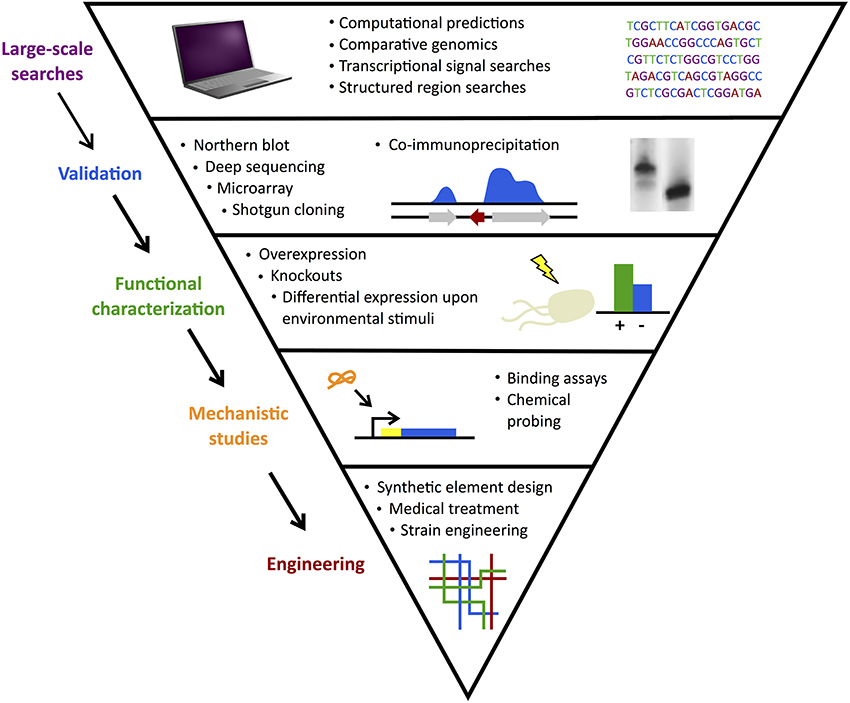
Figure 2. sRNA search and characterization. Discovery of sRNAs often begins with large-scale computational searches followed by experimental validation. Functional characterization of confirmed candidates identifies their gene or protein targets and mechanistic studies elucidate their methods of action. Finally, sRNAs can be used in engineering efforts to develop useful applications from synthetic elements to medical treatments.
sRNAs in Pathogens Pursued for Medical Advantages
The search of sRNAs has sparked special interest in the context of microbial pathogens. The emergence of studies of sRNAs in pathogenesis has been supported by the increasing availability of non-model microbial genomes as well as by the uncovered roles of sRNAs in environmental stress responses. For instance, the increasingly well-annotated collection of sRNAs in E. coli has facilitated discovery of sRNAs in other organisms by computational homology searches and other bioinformatic tools (Lu et al., 2011). Combining deep sequencing and transposon mutagenesis, a recent study identified 89 sRNAs in Streptococcus pneumonia and presented evidence of their importance in pathogenesis (Mann et al., 2012). Several investigations in non-model organisms have capitalized on the presence of Hfq in some pathogens to co-immunoprecipite bound RNA targets, followed by deep sequencing. In a model pathogen, Salmonella enterica serovar Typhimurium, this approach has revealed 100–200 sRNAs associated with Hfq and suggested that Hfq may regulate expression of more than one-fifth of all mRNAs (Sittka et al., 2008; Chao et al., 2012; Kröger et al., 2012). Although Hfq-dependent mechanisms have not yet been as prevalently detected in gram-positive bacteria (Romby and Charpentier, 2009), combined computational and experimental approaches have led to successful sRNA searches and validation in a variety of gram-positive pathogens that include Staphylococcus aureus, Listeria monocytogenes, and S. pneumonia (Papenfort and Vogel, 2010).
The sRNAs discovered in pathogens thus far exert diverse functions that include regulation of transcription factors, virulence genes, quorum sensing, and outer membrane dynamics in response to a variety of environmental inputs like temperature, pH, metabolite, oxidative, and anaerobic stresses. A few examples include the following: in Helicobacter pylori, a bacterial pathogen that colonizes the human stomach, a cis-encoded 5'ureB-sRNA enhances truncation of gastric acid acclimation operon ureAB at neutral pH, but releases its control in acidic environments to allow survival (Wen et al., 2012). Multiple trans-acting Qrr sRNAs in Vibrio cholerae collaborate with Hfq to regulate quorum sensing and biofilm formation, important for host transmission (Zhao et al., 2013). In Shigella flexneri, sRNAs CsrB and CsrC bind to protein CsrA to regulate carbon metabolism, attachment, and invasion in pathogenesis (Gore and Payne, 2010). Riboswitches may regulate 2% of genes in gram-positive pathogens such as S. aureus and L. monocytogenes in response to metabolites and pH (Caldelari et al., 2013). An RNA thermometer in L. monocytogenes inhibits translation of prfA, which encodes a key transcription factor for virulence (Johansson et al., 2002). Recent reviews provide more extensive catalogs of sRNA function and mechanisms in microbial pathogens (Gripenland et al., 2010; Papenfort and Vogel, 2010; Caldelari et al., 2013) as well as species-specific discussions for L. monocytogenes (Mellin and Cossart, 2012), Pseudomonas aeruginosa (Sonnleitner et al., 2012), S. aureus (Romilly et al., 2012; Tomasini et al., 2014), S. Typhimurium (Hébrard et al., 2012), and V. cholerae (Bardill and Hammer, 2012).
With increasing resistance of bacteria to traditional antimicrobials, sRNAs represent new medical targets to disarm pathogens. Riboswitches in particular are being pursued as antibiotic targets due to remarkable specificity and high affinity for their ligands (Lünse et al., 2014). At this point, one compound targeting the guanine riboswitch of S. aureus has been shown to successfully reduce bacterial concentrations in animals after infection (Ster et al., 2013). Also in S. aureus, sRNA SprX has been shown to regulate glycopeptide antibiotic resistance through antisense repression of sporulation protein SpoVG (Eyraud et al., 2014). This recent literature confirms the promise of sRNAs as new territory for controlling gene expression in pathogenesis.
sRNAs in Mycobacteria
Between 2003 and 2006, 30 new mycobacterial species were discovered, bringing the known total to 120 (Tortoli, 2006). These discoveries have resulted in significant investment toward understanding sigma factors and other regulatory proteins as potential keys to crippling virulence exerted by many of the mycobacterial pathogens (Rodrigue et al., 2006). Some common species that have been particularly studied are M. tuberculosis, M. leprae, and M. ulcerans due to their common pathogenesis in humans (Remus et al., 2003). M. bovis BCG is widely used as a vaccine against M. tuberculosis and is genetically similar, making it also a major focus of study (Skeiky and Sadoff, 2006). M. smegmatis serves as a model mycobacterial species due to ease of genetic manipulation, fast growth, and non-pathogenic nature (Shiloh and DiGiuseppe Champion, 2010).
Early Computational Searches for sRNAs in Mycobacteria
The growing interest in mycobacteria gene regulation has paralleled the launching of bioinformatics databases such as Rfam, a collection of non-coding RNA families that allows classification of new regulatory RNAs based on sequence and structure homology in over 200 complete genomes (Griffiths-Jones, 2005). One of the earliest mycobacterial genomes included in the Rfam database was M. tuberculosis CDC1551. This led to the prediction of three initial sRNAs in mycobacteria.
Although important for setting the stage to identify (and validate) the expression of more sRNAs in mycobacteria, the pure homology-based approach of Rfam limited early studies by having to rely on previously identified pools of sRNAs. A second type of bioinformatics search, sRNAPredict2, which predicted sRNAs by co-localization of genetic features common to sRNA-encoded regions, was also applied to the prediction of sRNAs in mycobacteria. This algorithm identified 56 new sRNAs in M. tuberculosis CDC1551, a much higher number that did not correspond to the three previously annotated by Rfam (Livny et al., 2006). Although these early computational efforts lacked consistency, most importantly, they provided evidence that multiple genomic regions could encode sRNAs in mycobacteria.
As we unfold major themes of the sRNA story in mycobacteria, refer to Figure 3 for the chronological alignment of these events. Also, note that Supplementary Table S1 lists all confirmed sRNAs in mycobacterial species to date along with characteristic data such as genomic location, differential expression, and known homology between species. Amazingly, more than half of the M. tuberculosis H37Rv sRNAs have been confirmed since the most recent sRNA review of this species (Arnvig and Young, 2012). Unconfirmed sRNA predictions are not included in Supplementary Table S1, but are quantified in Figure 1, a view of sRNA discovery by phylogeny within mycobacterial species, and listed in Supplementary Table S2. We use a universal sRNA nomenclature reported in Lamichhane et al. (2013) throughout this review to allow convenient cross-reference among the sRNAs being discussed.
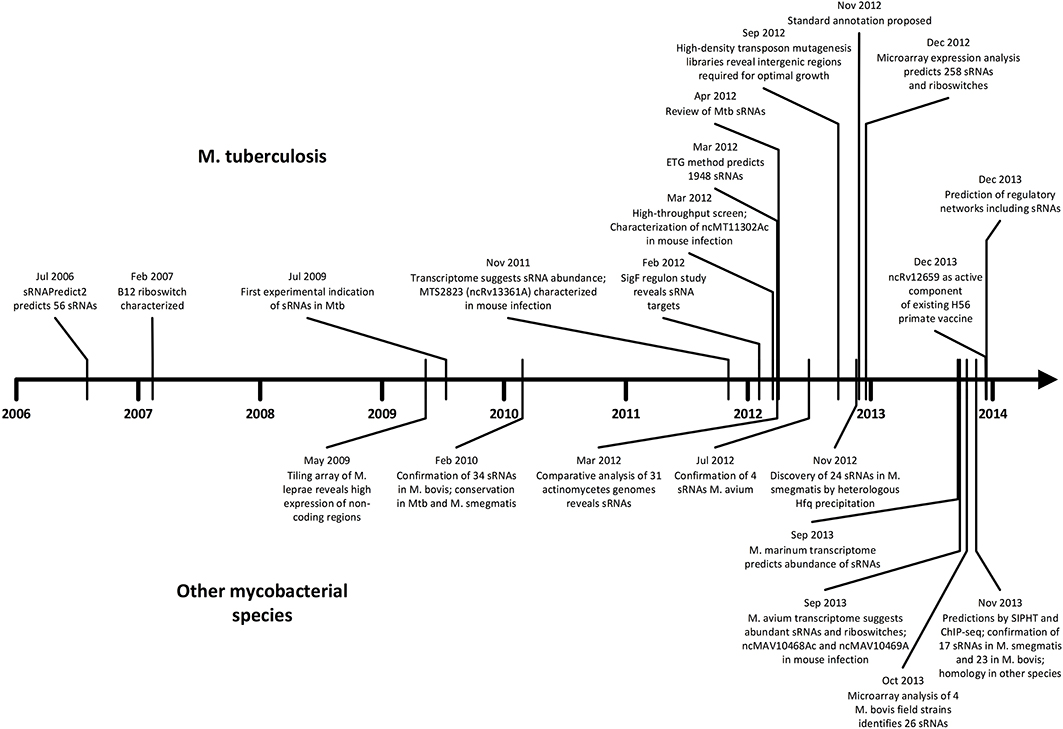
Figure 3. Timeline of sRNA developments in mycobacteria. Progress in M. tuberculosis (upper) and other mycobacterial species (lower) are shown in parallel in chronological order. The surge of studies in the last few years suggests momentum toward further discovery, mechanistic studies, and medical applications.
Early Experimental Indication of Non-Coding RNA Regulators
One of the first experimental indications of the presence of responsive regulatory RNA elements in mycobacteria was obtained fortuitously by studies that aimed to understand the role of vitamin B12 in the growth inhibition of M. tuberculosis CD1551. In the context of this work, a B12 riboswitch (ncMT2183Ac) was found to regulate transcription of metE, a gene encoding methionine synthase (Warner et al., 2007). As previously suggested for other bacteria, this exposed potential for using antibiotics and/or small molecules to target riboswitch gatekeepers of essential genes expressed during pathogenesis. Only a couple of years after this study, tiling array analysis of M. leprae Thai-53 infected rats revealed high expression of 68 non-coding regions throughout the genome, representing 32.5% of the total relative expression profile (Akama et al., 2009). Although this data could have been interpreted as simply transcriptional degradation under these host conditions, earlier data in other mycobacterial species encouraged the alternative: that an abundance of sRNAs was expressed in this strain of M. leprae.
To date, riboswitch motifs remain largely uncharacterized in M. tuberculosis and further analysis of existing datasets could lead to confirmation of these important regulators. For example, two Mbox riboswitches (also called Ykok leaders) are predicted in M. tuberculosis: one located in the 5′ UTR of a hypothetical protein induced during Mg-starvation and the other in the 5′ UTR of a predicted magnesium transporter (Arnvig and Young, 2012). It has been hypothesized that these regulatory elements act similarly to their Bacillus subtilis homologs by inhibiting transcription of the downstream gene when bound to Mg2+.
Confirmation of the First sRNAs in Mycobacteria
The first complete experimental confirmation of sRNAs in mycobacteria was published in 2009, revealing five trans-encoded and four cis-encoded sRNAs in M. tuberculosis H37Rv (Arnvig and Young, 2009). These sRNAs were discovered by screening cDNA libraries of low molecular weight RNAs (20–75 nucleotides) in exponential and stationary growth phases. All reported sRNAs (see Supplementary Table S1) were confirmed by northern blotting analysis and their transcriptional ends were mapped by 5′ and 3′ RACE (Arnvig and Young, 2009). The functional significance of these sRNAs was reported in the context of oxidative stress, where M. tuberculosis H37Rv cells were cultured to exponential phase and then H2O2 was added to 10 mM for 1 h. Under these specific stress conditions, sRNAs B55 (ncRv10609AA), F6 (ncRv10243A), and ASpks (ncRv2048A) showed differential expression of 2-fold or greater (relative to their unstressed state). F6 was similarly expressed under pH stress, in which exponentially growing cells were suspended in acidified medium of pH 5.0 and allowed to grow for another 24 h. These conditions mimic the changing macrophage environment during active infection from pH ~6.2 to ~5.0 and during the accumulation of free fatty acids from the host (Vandal et al., 2009).
Evidence of the functional role of F6 (later reported as Mcr14) indirectly resulted from studies of the SigF regulon in M. tuberculosis H37Rv, where this sigma factor was shown to regulate its transcription (Hartkoorn et al., 2012). Furthermore, slow cell growth of H37Rv had been shown both upon F6/Mcr14 overexpression (under strong rrnB promoter of M. smegmatis) and upon induction of SigF (presumably due to its ability to up-regulate F6). It has also been suggested that some SigF binding sites are not associated with mRNA transcripts and are likely to bind to sRNAs not yet annotated.
Interestingly, none of the first experimentally confirmed sRNAs were predicted computationally. This discrepancy can be rationalized by the computational challenge presented by the uniqueness of the promoters and terminators in M. tuberculosis. Furthermore, the genomes of mycobacterial species are much higher in GC content (~65%) relative to other model bacteria (e.g., E. coli, GC content of ~50%) where earlier computational sRNA searches had been successful (DiChiara et al., 2010). For these reasons, one question of even more importance in mycobacterial species has been: How many computationally predicted sRNAs represent false positives? We have reported mixed results with predictions made by SIPHT and WU-BLAST (DiChiara et al., 2010; Tsai et al., 2013; Cho et al., 2014).
A Rising Number of Newly Identified sRNAs
The year following the first report of sRNAs in M. tuberculosis, a new approach coupled low-molecular cloning and computational methods in M. bovis BCG, M. tuberculosis H37Rv, and M. smegmatis MC2155 (DiChiara et al., 2010). The cloning in these model strains began with construction and screening of cDNA libraries from both log and stationary phases, yielding 116 sRNA candidates. After elimination of rRNA, tRNA, and other annotated elements, 60 candidates were selected and tested in M. bovis by northern blotting analysis, leading to 19 confirmed sRNAs (listed in Supplementary Table S1). In parallel, the computational approach began with predictions by the SIPHT program, a high-throughput progeny of sRNAPredict2, which identified 144 sRNA candidates (Livny et al., 2008). The 67 that showed partial conservation in other mycobacterial species were tested by northern blotting in M. bovis and 21 additional sRNAs were confirmed (listed in Supplementary Table S1). Three sRNAs were confirmed by both cloning and computation methods, bringing the total to 37. Mcr6 (ncBCG3782Ac), Mcr14/Mpr13 (ncBCG10281A), and Mpr19 (ncBCG0526A) had been identified by previous studies in M. tuberculosis H37Rv as F6 (ncRv10243A), Mpr19 (ncRv13660Ac), and B11 (ncRv13660Ac), respectively. A unique approach taken by this study was to check for homologs of confirmed M. bovis sRNAs by using the same probes in northern blotting analysis in additional species. With this method, 20 new sRNAs were confirmed in M. tuberculosis H37Rv and 15 in M. smegmatis MC2155. The higher number identified and verified in M. tuberculosis (relative to M. smegmatis) is not surprising given the shorter evolutionary distance between M. bovis and M. tuberculosis relative to the distance between M. bovis and M. smegmatis, as shown in Figure 1 (Devulder, 2005). Only three M. bovis sRNAs were identified by both cloning and computational methods and only three M. tuberculosis H37Rv sRNAs had been previously discovered, conveying the importance of combining various approaches.
A follow-up study (Tsai et al., 2013) used the same SIPHT predictions in M. bovis BCG and M. smegmatis to test the remaining candidates. As part of that work, 23 additional novel sRNAs were confirmed by northern blotting analysis in M. bovis BCG and 17 in M. smegmatis MC2155 (listed in Supplementary Table S1). With M. smegmatis as the primary species of interest, this approach revealed nine homologs in M. bovis and four in M. tuberculosis (listed in Supplementary Table S1). By analysis of existing ChIP-seq data from M. tuberculosis (Galagan et al., 2012), at least four sRNA 5′ ends were matched with otherwise uncharacterized transcription factors. From this series of studies, a stronger connection between pathogenesis and differential expression of sRNAs in mycobacteria emerged. A powerful example was the differential expression of Mcr11, an sRNA between two cAMP metabolism genes, in M. bovis (ncBCG1323Ac) and M. tuberculosis (ncRv11264Ac) under host-like conditions, such as low pH (5.5) and hypoxia (1.3% O2 + 5% CO2). Perhaps most importantly, this study proposed the use of conservation analysis across multiple mycobacterial species to begin posing questions related to the potential pathogenic vs. housekeeping functions of the newly uncovered sRNAs. New questions were raised for future studies that included: Which sRNAs are uniquely conserved among the pathogenic species? Do conserved sRNAs by sequence perform the same function across phylogeny? This study also was one of the first to adhere to the recommended annotation (also being used in this review).
Implications of a Missing Hfq Homolog to the Search and Characterization of sRNAs
A challenge in mycobacterial sRNA studies has been the lack of identification of an Hfq homolog, preventing Hfq co-immunoprecipitation approaches for sRNA discovery. The lack of Hfq has also raised questions about alternative chaperones or inherent stability granted by C-rich stretches observed in the rising number of confirmed sRNAs. It is important to note that in 2012, a review of non-coding RNAs specific to M. tuberculosis (Arnvig and Young, 2012) posed the notion that hypothetical protein Rv2367 of unknown function could serve as a potential RNA chaperone alternative to Hfq based on its homology to YbeY, a protein in Sinorhizobium meliloti shown to perform certain Hfq-like functions (Pandey et al., 2011). Rv2367, however, has significantly lower expression than Hfq in E. coli, strengthening the argument that C-rich stretches of mycobacterial sRNAs may provide enough stability to the sequences without the need of a functionally-equivalent chaperone.
It is also worth noting that a novel method has exploited the conserved sRNA-binding ability of E. coli Hfq by expressing it in M. smegmatis and using co-immunoprecipitation and deep sequencing to identify sRNAs (Li et al., 2012). The approach found 12 trans-encoded and 12 cis-encoded sRNAs confirmed by northern blotting analysis and mapped by 5' RACE (listed in Supplementary Table S1). Interestingly, five of the cis-encoded sRNAs are immediately upstream of known transposases. Some of the identified M. smegmatis sRNAs also show homology to regions in other mycobacteria, especially in non-pathogenic species known for rapid growth, suggesting that these sRNAs could be involved in enhancing growth efficiency. All of them showed differential expression between exponential and stationary phases. Homologs to previously identified sRNAs C8 (ncRv13722Ac) and B11 (ncRv13660Ac) in M. tuberculosis H37Rv (Arnvig and Young, 2009) were also identified in M. smegmatis (ncMSMEG16286A and ncMSMEG6172Ac, respectively). Consistent with previous studies, multiple sizes of individual sRNAs were also observed here, raising questions about how sRNAs are processed and the potential role of sigma factors in mycobacteria. Although, sRNA regulation of transposition has been studied in E. coli, mechanisms in mycobacteria remain largely obscure.
An Explosion in sRNA Identification Guided by High-Throughput Sequencing
Transcriptome profiling of M. tuberculosis H37Rv revealed high-density reads in intergenic regions (other than from those encoding rRNAs and tRNAs), representing a potential abundance of sRNAs (Arnvig et al., 2011). In fact, 28% of the total transcriptome represented intergenic reads, consistent with other organisms. The most highly expressed sRNA detected in exponential phase was MTS2823 (ncRv13661A), which also increased 10-fold in stationary phase. Overexpression of MTS2823 up-regulated two genes (Rv2035, a potential activator of HspG, and Rv3229c, a fatty acyl desaturase) and down-regulated a large set of about 300 genes, including a methyl citrate synthase that is reduced by 15-fold. It was suggested that MTS2823 may have functional homology with 6S RNA, but the mechanism remains unclear. MTS1338 (ncRv1734A) was also induced in stationary phase, depending at least partially on the DosRS hypoxia-responsive regulator system. In the same study, significant accumulation of sRNAs was observed in infected lungs of mice, especially MTS2823, MTS0997 (ncRv11264Ac), and MTS1338, suggesting potential involvement in pathogenesis. Later work showed that M. tuberculosis H37Rv sRNAs MTS0479 (ncRv10609AA) and MTS1338 are not present in M. avium (Ignatov et al., 2013). Since M. avium is conditionally pathogenic and M. tuberculosis H37Rv is highly pathogenic, these two sRNAs could be key players in pathogenesis. These studies marked an important step in the successful application of high-throughput genome-wide sequencing technology for sRNA identification as well as in establishing direct medical relevance to pathogenesis by use of mouse models.
The next genome-wide search in M. tuberculosis examined strain CDC1551 (Pelly et al., 2012). Twelve sRNA candidates were identified; four of the intergenic candidates were novel sRNAs and the other eight all contained sequences within the region between genes MT1302 and MT1303, which showed homology to a single sRNA identified in M. bovis BCG. This abundant sRNA was labeled ncrMT1302 (ncMT1130Ac) and examined further under nitric oxide (250 μM diethylenetriamine nitric oxide adduct), low pH (5.5), and limited nutrition (PBS + 0.05% Tween 80) stresses for 6 h during exponential phase. Expression levels of ncrMT1302 were reduced in all cases, most dramatically at low pH with a 7-fold reduction. To detect antibiotic responses, total RNA was exposed to the minimal inhibitory concentrations (MIC) of isoniazid or rifampicin for 3 h before ncrMT1302 expression was observed by northern blot analysis. Isoniazid showed no significant effect, but rifampicin caused reduction in ncrMT1302 expression. MT1302 encodes an adenylyl cyclase that converts ATP to cAMP and MT1303 encodes a hypothetical protein that is transcribed in the presence of cAMP in low-oxygen conditions. In a strain lacking the cAMP-producing gene MT1302, ncrMT1302 was less abundant and no longer differentially expressed under stress. This sRNA also contains a potential binding site for transcription factor Cmr (cAMP and macrophage regulator) and was confirmed in the lungs of mice during infection.
A new method for sRNA identification involved building an effective target genome (ETG) and then combining transcriptome data with comparative genomics (Pellin et al., 2012). The target genome in this case included only intergenic regions, thus coding regions were removed before the dataset was filtered further by RNA-seq reads and conservation between genomes. Applied to M. tuberculosis H37Rv, this approach generated a list of 1948 candidate sRNAs, which included six of the nine identified by Arnvig and Young (Arnvig and Young, 2009). This method also predicted 17 of the 37 found in M. bovis BCG by DiChiara et al. (2010). All candidates identified were compared with the Rfam database and many showed homology with known families. Most notably, Rfam matches included SAM-IV, TPP family, and Ykok leader riboswitches.
In addition to large scale transcriptomic studies, a comparative analysis of 31 genomes of mycobacteria and related actinomycetes by sequence and gene expression allowed identification of 50 predicted non-coding RNAs (McGuire et al., 2012). This set of genomes included 8 strains of the M. tuberculosis complex (M. tuberculosis and M. bovis) and 11 other mycobacteria including M. leprae, M. ulcerans, M. marinum, M. avium, and M. smegmatis. Four more novel sRNAs were confirmed by northern blotting analysis in M. tuberculosis H37Rv (listed in Supplementary Table S1) with homology in M. smegmatis MC2155 (listed in Supplementary Table S2). This massive dataset highlighted the evolutionary importance of lipid metabolism and its regulation among these species in regards to pathogenesis. Figure 1 illustrates the presence of sRNAs across mycobacterial phylogeny.
Increased Studies Beyond M. Tuberculosis, M. Bovis, and M. Smegmatis
By cloning and homology search, four intergenic sRNAs have been directly detected in M. avium subsp. avium TMC724 (listed in Supplementary Table S1) (Ignatov et al., 2012). Because the genome of this strain is not available, the sRNAs were mapped by RACE to the M. avium hominissuis 104 genome. A follow-up study of M. avium TMC724 predicted 86 cis-encoded sRNAs, 10 trans-encoded sRNAs, and five riboswitches, which are listed in Supplementary Table S2 (Ignatov et al., 2013). The predicted riboswitches included three Ykok leaders (Mg2+ sensing) and one SAM-IV (S-adenosylmethionine sensing); both types had been previously spotted in M. tuberculosis H37Rv. The trans-encoded sRNAs showed conservation with M. tuberculosis and M. ulcerans. Of six intergenic sRNAs, only igMAV_1034-1035 (ncMAV11034Bc) had no homolog in M. tuberculosis. The two mostly highly expressed sRNAs in this study, igMAV_0468-0469 (ncMAV10468Ac) and igMAV_0469-0470 (ncMAV10469A), were tested in mice genetically susceptible and genetically resistant to M. avium infection. The resistant mice showed much lower expression of these sRNAs in lung tissue relative to non-resistant mice. However, the M. tuberculosis homolog to igMAV 0468-0469, sRNA MTS2823 (ncRv13661A), was shown to accumulate to high levels in the lungs of mice resistant to tuberculosis, potentially indicating a difference between M. avium and M. tuberculosis in the mouse infection model.
In addition to increased studies in M. avium, transcriptome analysis has been recently applied to the search of regulatory sRNAs in M. marinum during exponential and stationary phases. In these studies, it was observed that read-rich antisense and intergenic regions accounted for 23% of the total transcriptome in exponential phase and 40% in early stationary phase (Wang et al., 2013). A very highly expressed sRNA candidate, MMAR_5556, is homologous to MTS2823 (ncRv13661A), previously shown as the most abundant sRNA in M. tuberculosis H37Rv. Three sRNAs predicted were matched with Rfam as a TPP family riboswitch, an ALIL pseudoknot, and a 6C RNA.
Efforts to Unify Rapidly Increasing Knowledge in the Field
As focus begins to shift to the functional characterization of mycobacterial sRNAs, the need to unify all research efforts in cross-species discovery and characterization has become more prevalent. A challenge highlighted by increased sRNA discovery studies across mycobacterial species was the lack of universal sRNA nomenclature across the emerging literature. For instance, F6, Mcr14, Mcr13, and MTS0194 represent the same sRNA (ncRv10243A). To address this issue, an official recommendation for systematic annotation of non-coding RNA in mycobacteria was recently published (Lamichhane et al., 2013). The suggested format includes species designation and genomic location. For example, an sRNA identified within the open reading frame (ORF) of gene Rv1234 in M. tuberculosis H37Rv would be labeled “ncRv1234” if on the plus strand or “ncRv1234c” if on the minus strand. If the sRNA were located completely between ORFs, it would carry the number of the upstream ORF and a “1” would be added in front to indicate an intergenic region, such as “ncRv11234” or “ncRv11234c.” Finally, the direction of the sRNA should be indicated by “A” if in the plus strand orientation or “B” if in the minus strand orientation, such as “ncRv1234A” or “ncRv11234B.” In this way, the sRNA names would give useful information and be consistent between studies. We have adhered to this nomenclature in this review, where we have taken the challenge to begin consolidating all mycobacterial sRNAs.
From Discovery to Functional Characterization
As seen above, cis-regulatory element functions are suggested by their locations relative to known ORFs. Although not greatly exploited in mycobacterial species, bioinformatics tools such as TargetRNA have been used in Salmonella and Listeria pathogens to predict sRNA interactions in silico (Tjaden, 2008; Wurtzel et al., 2012; Yu and Schneiders, 2012). Still, experimental methods are necessary to confirm these suggested interactions. Efforts to elucidate sRNA functions have traditionally relied on microarray for transcriptome analysis under various stress conditions or under sRNA overexpression or deletion. Even with the development of high-density tiling arrays, this technology is becoming obsolete in favor of increasingly affordable high-throughput methods.
The sRNA candidates identified with the ETG transcriptome and comparative genome approach in M. tuberculosis H37Rv (Pellin et al., 2012) have been examined by microarray to confirm expression (Miotto et al., 2012). Of the 1373 predicted in exponential phase, 258 were confirmed by microarray including 22 intergenic, 84 in 5′ or 3′ UTRs (including potential riboswitches), and 152 antisense sRNAs. Twenty of 23 candidates tested were confirmed by northern blot analysis and mapped by 5′ RACE (listed in Supplementary Table S1). A computational analysis of pathway regulation showed that membrane-bound proteins were especially likely subjects of antisense regulation. Additionally, about 100 sRNAs < 50 nucleotides were detected, but the functions of these micro-sized RNAs are unknown.
In an effort to define genomic regions required for growth in M. tuberculosis H37Rv, two 100,000-clone libraries were generated by high-density transposon mutagenesis (Zhang et al., 2012). Transposon-mapping probes were developed and amplified to allow deep sequencing of their neighboring genomic regions, mapping insertion sites. Sliding window analysis was used to scan the genome for insertion site counts in order to find underrepresented regions, likely to be essential in function. The search identified 25 intergenic regions as essential for growth including tmRNA, the RNA component of RNaseP, and 19 regions with unknown function. One limitation of this study was that the smallest search window for intergenic regions was 250 basepairs, preventing detection of shorter sRNAs. This study showed consistency with a previous microarray approach (Sassetti et al., 2003), but provided much higher resolution.
Perhaps the most currently favored high-throughput alternative to microarray is ChIP-seq, which combines traditional chromatin immunoprecipitation with deep sequencing to map in vivo DNA-protein interactions across the genome. Advantages of ChIP-seq over microarray include single-nucleotide resolution (compared to 30–100 bp resolution for microarray), low cost, and small amount of required DNA (10–50 ng for ChIP-seq compared to >1 μg for microarray) (Park, 2009).
By combining ChIP-seq and microarray data, sRNA regulation networks were predicted for M. tuberculosis H37Rv and are consistent with previously known sRNA functions (Peterson, 2013). The context likelihood of readiness (CLR) algorithm was used to infer the networks and false positives were filtered out by comparison to overexpression microarray data. As a proof-of-concept, M. tuberculosis was grown under hypoxia stress over 14 days and examined by microarray, revealing 58 trans-acting sRNAs significantly induced or repressed, and the sRNA hypoxia regulatory network was successfully generated. These networks can be visualized in a spider web fashion to clearly show direct and indirect connections. Sigma factor SigH was predicted to regulate sRNA ncRv13596A, linking hypoxia and cholesterol metabolism, and DosR was predicted to regulate ncRv1102A and ncRv1734A, linking hypoxia also to changing phthiocerol dimycocerosates. SigF and ncRv10243c were associated with the cell wall and plasma membrane, consistent with E. coli homologs with a known mechanism.
In other organisms, ultraviolet crosslinking and immunoprecipitation (CLIP) has been used to detect RNA-protein binding sites in vivo with deep sequencing. Photoactivatable ribonucleoside-enhanced CLIP (PAR-CLIP) allows single cross-linked nucleotide resolution, but requires cells to incorporate photoactivatable nucleoside analogs like 4-thiouridine or 6-thioguanosine (König et al., 2012). CLIP-seq provides strand-specific data unlike ChIP-seq (Wang et al., 2014). At this point, CLIP has been most used to determine alternative splicing mechanisms in mammalian systems (König et al., 2014). Mycobacterial sRNA studies may pursue PAR-CLIP or other CLIP variations to obtain strand-specific binding insight in the future. Other technologies are available for characterization of discovered sRNA interactions including reporter gene fusions (gfp, lacZ, luc), in vitro RNA-RNA footprinting, and in vitro toeprinting (Podkaminski et al., 2014). In particular, toeprinting has been successful in characterizing sRNAs that negatively regulate their targets. With these techniques, mycobacterial studies will benefit from precedents set in widely studied organisms.
sRNA Characterization for Medical Applications
Recent studies have employed high-throughput sRNA characterization methods to analyze medical problems. Remarkably, in response to an epidemic of bovine tuberculosis in Great Britain, four field strains of M. bovis were processed by sequencing, microarray, transcriptome analysis, RACE, and recombinant DNA technology to observe effects of synonymous point mutations (sSNPs) (Golby et al., 2013). Unexpectedly, some of these sSNPs of coding strands resulted in expression of antisense transcripts on the opposite strand, challenging the definition of “silent mutation” and suggesting the potential of sRNAs in these loci. A high-density tiled microarray was designed to examine changes in expression of non-coding RNAs across the genomes. Transcripts were considered confirmed sRNAs only if detected by two or more overlapping probes. In this way, 26 sRNAs were found (listed in Supplementary Table S1). Seven intergenic transcripts were located within the direct repeat (DR) locus, a suggested CRISPR locus. One antisense sRNA as_Mb1618c (ncMb1618Ac) overlaps with a predicted secretory lipase gene was only present in strain 1121/01. Similarly, as_Mb1914c (ncMb1914Ac) and as_echA21 (ncMb3803Ac) were only expressed in strain 2451/01, encoded antisense to a short chain dehydrogenase and an enoyl-CoA hydratase, respectively. These sRNAs unique to individual strains may be evolutionary adaptations to different environments.
The H56 vaccine for primate M. tuberculosis includes Rv2660c as an antigen, which overlaps with sRNA ncRv12659 (Houghton et al., 2013). The Rv2660c locus was originally selected as an antigen due to its enhanced transcription during M. tuberculosis H37Rv starvation, but by RNA-seq and northern blotting, the sRNA was shown to be the true cause of this increased expression. Because ncRv12659 accumulates to high levels during infection, it can serve as a potential biomarker. Still, the mechanism by which ncRv12659 leads to this increased transcription is unknown. Interestingly, only the 5′ portion of ncRv12659 was detected during infection, raising questions about sRNA processing. Truncation could be the result of premature termination or degradation of an unstable 3′ portion.
The Path Forward
The surge of sRNA studies in mycobacteria in the last 3 years favors continued momentum toward medical applications. Still, many species have yet to be studied and a large number of sRNAs remain uncharacterized. High-throughput experimental methods would aid in bridging the gap between the ever-growing pool of predicted sRNA candidates and the set of confirmed molecules with known functions. Work remains to be done computationally and experimentally to map the complex sRNA control networks of pathogens. Future work could employ ChIP-seq to map gene regulatory networks and truly begin addressing the need to understand the functional role of these sRNAs in mycobacterial species. Likewise, increased phenotypic data for individual sRNA knockout or overexpression strains could enhance efforts to map sRNA regulatory networks.
Despite the limited number of functionally characterized sRNAs in mycobacteria, the door is already open for exploiting known sRNA functions in pathogens to gain medical advantages. The development of antibiotics to cripple sRNA-enabled virulence could be a powerful, new approach, especially as microbes show resistance to conventional compounds. In particular, abundant and uniquely expressed sRNAs during infection could serve as useful biomarkers or as therapeutic targets.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
This work was supported by the National Science Foundation (NSF) Graduate Research Fellowship Program to Katie Haning as well as the Welch Foundation (F-1756) and NSF (CBET-1254754 and MCB-1330862) to Lydia M. Contreras.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fcimb.2014.00096/abstract
References
Akama, T., Suzuki, K., Tanigawa, K., Kawashima, A., Wu, H., Nakata, N., et al. (2009). Whole-genome tiling array analysis of Mycobacterium leprae RNA reveals high expression of pseudogenes and noncoding regions. J. Bacteriol. 191, 3321–3327. doi: 10.1128/JB.00120-09
Argaman, L., Hershberg, R., Vogel, J., Bejerano, G., Wagner, E. G. H., Margalit, H., et al. (2001). Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr. Biol. 11, 941–950. doi: 10.1016/S0960-9822(01)00270-6
Arnvig, K. B., Comas, I., Thomson, N. R., Houghton, J., Boshoff, H. I., Croucher, N. J., et al. (2011). Sequence-based analysis uncovers an abundance of non-coding rna in the total transcriptome of Mycobacterium tuberculosis. PLoS Pathog. 7:e1002342. doi: 10.1371/journal.ppat.1002342
Arnvig, K. B., and Young, D. B. (2009). Identification of small RNAs in Mycobacterium tuberculosis. Mol. Microbiol. 73, 397–408. doi: 10.1111/j.1365-2958.2009.06777.x
Arnvig, K. B., and Young, D. B. (2012). Non-coding RNA and its potential role in Mycobacterium tuberculosis pathogenesis. RNA Biol. 9, 427–436. doi: 10.4161/rna.20105
Bardill, J. P., and Hammer, B. K. (2012). Non-coding sRNAs regulate virulence in the bacterial pathogen Vibrio cholerae. RNA Biol. 9, 392–401. doi: 10.4161/rna.19975
Behar, S. M., Divangahi, M., and Remold, H. G. (2010). Evasion of innate immunity by Mycobacterium tuberculosis: is deaht an exit strategy? Nat. Rev. Microbiol. 8, 668–674. doi: 10.1038/nrmicro2387
Blattner, F. R. (1997). The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1462. doi: 10.1126/science.277.5331.1453
Bobrovskyy, M., and Vanderpool, C. K. (2013). Regulation of bacterial metabolism by small rnas using diverse mechanisms. Annu. Rev. Genet. 47, 209–232. doi: 10.1146/annurev-genet-111212-133445
Caldelari, I., Chao, Y., Romby, P., and Vogel, J. (2013). RNA-mediated regulation in pathogenic bacteria. Cold Spring Harb. Perspect. Med. 3:a010298. doi: 10.1101/cshperspect.a010298
Chao, Y., Papenfort, K., Reinhardt, R., Sharma, C. M., and Vogel, J. (2012). An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J. 31, 4005–4019. doi: 10.1038/emboj.2012.229
Cho, S. H., Lei, R., Henninger, T. D., and Contreras, L. M. (2014). Discovery of ethanol responsive small RNAs in Zymomonas mobilis. Appl. Environ. Microbiol. 80, 4189–4198. doi: 10.1128/AEM.00429-14
Cole, S. T., Brosch, R., Parkhill, J., Garnier, T., Churcher, C., Harris, D., et al. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. doi: 10.1038/31159
Cosma, C. L., Sherman, D. R., and Ramakrishnan, L. (2003). The secret lives of the pathogenic mycobacteria. Annu. Rev. Microbiol. 57, 641–676. doi: 10.1146/annurev.micro.57.030502.091033
Devulder, G. (2005). A multigene approach to phylogenetic analysis using the genus Mycobacterium as a model. Int. J. Syst. Evol. Microbiol. 55, 293–302. doi: 10.1099/ijs.0.63222-0
DiChiara, J. M., Contreras-Martinez, L. M., Livny, J., Smith, D., McDonough, K. A., and Belfort, M. (2010). Multiple small RNAs identified in Mycobacterium bovis BCG are also expressed in Mycobacterium tuberculosis and Mycobacterium smegmatis. Nucleic Acids Res. 38, 4067–4078. doi: 10.1093/nar/gkq101
Eyraud, A., Tattevin, P., Chabelskaya, S., and Felden, B. (2014). A small RNA controls a protein regulator involved in antibiotic resistance in Staphylococcus aureus. Nucleic Acids Res. 42, 4892–4905. doi: 10.1093/nar/gku149
Flynn, J. L., and Chan, J. (2001). Tuberculosis: latency and reactivation. Infect. Immun. 69, 4195–4201. doi: 10.1128/IAI.69.7.4195-4201.2001
Flynn, J. L., Scanga, C. A., Tanaka, K. E., and Chan, J. (1998). Effects of aminoguanidine on latent murine tuberculosis. J. Immunol. 160, 1796–1803.
Galagan, J., Lyubetskaya, A., and Gomes, A. (2012). ChIP-Seq and the complexity of bacterial transcriptional regulation. Curr. Top. Microbiol. Immunol. 363, 43–68. doi: 10.1007/82_2012_257
Garnier, T., Eiglmeier, K., Camus, J.-C., Medina, N., Mansoor, H., Pryor, M., et al. (2003). The complete genome sequence of Mycobacterium bovis. Proc. Natl. Acad. Sci. U.S.A. 100, 7877–7882. doi: 10.1073/pnas.1130426100
Golby, P., Nunez, J., Witney, A., Hinds, J., Quail, M. A., Bentley, S., et al. (2013). Genome-level analyses of Mycobacterium bovis lineages reveal the role of SNPs and antisense transcription in differential gene expression. BMC Genomics 14:710. doi: 10.1186/1471-2164-14-710
Gore, A. L., and Payne, S. M. (2010). CsrA and Cra influence Shigella flexneri Pathogenesis. Infect. Immun. 78, 4674–4682. doi: 10.1128/IAI.00589-10
Gottesman, S. (2004). The small RNA regulators of Escherichia coli: roles and mechanisms. Annu. Rev. Microbiol. 58, 303–328. doi: 10.1146/annurev.micro.58.030603.123841
Gottesman, S., McCullen, C., Guillier, M., Vanderpool, C., Majdalani, N., Benhammou, J., et al. (2006). Small RNA regulators and the bacterial response to stress. Cold Spring Harb. Symp. Quant. Biol. 71, 1–11. doi: 10.1101/sqb.2006.71.016
Gottesman, S., and Storz, G. (2011). Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb. Perspect. Biol. 3:a003798. doi: 10.1101/cshperspect.a003798
Griffiths-Jones, S. (2005). Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124. doi: 10.1093/nar/gki081
Gripenland, J., Netterling, S., Loh, E., Tiensuu, T., Toledo-Arana, A., and Johansson, J. (2010). RNAs: regulators of bacterial virulence. Nat. Rev. Microbiol. 8, 857–866. doi: 10.1038/nrmicro2457
Hartkoorn, R. C., Sala, C., Uplekar, S., Busso, P., Rougemont, J., and Cole, S. T. (2012). Genome-wide definition of the SigF regulon in Mycobacterium tuberculosis. J. Bacteriol. 194, 2001–2009. doi: 10.1128/JB.06692-11
Hébrard, M., Kröger, C., Srikumar, S., Colgan, A., Händler, K., and Hinton, J. C. D. (2012). sRNAs and the virulence of Salmonella enterica serovar Typhimurium. RNA Biol. 9, 437–445. doi: 10.4161/rna.20480
Hershberg, R., Altuvia, S., and Margalit, H. (2003). A survey of small RNA-encoding genes in Escherichia coli. Nucleic Acids Res. 31, 1813–1820. doi: 10.1093/nar/gkg297
Hindley, J. (1967). Fractionation of P32-labelled ribonucleic acids on polyacrylamide gels and their characterization by fingerprinting. J. Mol. Biol. 30, 125–136. doi: 10.1016/0022-2836(67)90248-3
Houghton, J., Cortes, T., Schubert, O., Rose, G., Rodgers, A., De Ste Croix, M., et al. (2013). A small RNA encoded in the Rv2660c locus of Mycobacterium tuberculosis is induced during starvation and infection. PLoS ONE 8:e80047. doi: 10.1371/journal.pone.0080047
Huynh, K. K., Joshi, S. A., and Brown, E. J. (2011). A delicate dance: host response to mycobacteria. Curr. Opin. Immunol. 23, 464–472. doi: 10.1016/j.coi.2011.06.002
Ignatov, D., Malakho, S., Majorov, K., Skvortsov, T., Apt, A., and Azhikina, T. (2013). RNA-Seq analysis of Mycobacterium avium non-coding transcriptome. PLoS ONE 8:e74209. doi: 10.1371/journal.pone.0074209
Ignatov, D. V., Mefodieva, L. G., Majorov, K. B., Skvortsov, T. A., and Azhikina, T. L. (2012). Novel small RNAs from Mycobacterium avium. Russ. J. Bioorg. Chem. 38, 447–449. doi: 10.1134/S1068162012040085
Jagielski, T., van Ingen, J., Rastogi, N., Dziadek, J., Mazur, P. K., and Bielecki, J. (2014). Current methods in the molecular typing of Mycobacterium tuberculosis and other mycobacteria. Biomed Res. Int. 2014:645802. doi: 10.1155/2014/645802
Johansson, J., Mandin, P., Renzoni, A., Chiaruttini, C., Springer, M., and Cossart, P. (2002). An RNA thermosensor controls expression of virulence genes in Listeria monocytogenes. Cell 110, 551–561. doi: 10.1016/S0092-8674(02)00905-4
Keane, J., Remold, H. G., and Kornfeld, H. (2000). Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J. Immunol. 164, 2016–2020. doi: 10.4049/jimmunol.164.4.2016
König, J., McGlincy, N. J., and Ule, J. (2014). “Global analysis of protein-RNA interactions with single-nucleotide resolution using iCLIP,” in Handbook of RNA Biochemistry, eds R. K. Hartmann, A. Bindereif, A. Schon, and E. Westhof (Weinheim: Wiley-VCH), 899–918.
König, J., Zarnack, K., Luscombe, N. M., and Ule, J. (2012). Protein–RNA interactions: new genomic technologies and perspectives. Nat. Rev. Genet. 13, 77–83. doi: 10.1038/nrg3141
Kortmann, J., and Narberhaus, F. (2012). Bacterial RNA thermometers: molecular zippers and switches. Nat. Rev. Microbiol. 10, 255–265. doi: 10.1038/nrmicro2730
Kröger, C., Dillon, S. C., Cameron, A. D., Papenfort, K., Sivasankaran, S. K., Hokamp, K., et al. (2012). The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc. Natl. Acad. Sci. U.S.A. 109, E1277–E1286. doi: 10.1073/pnas.1201061109
Lamichhane, G., Arnvig, K. B., and McDonough, K. A. (2013). Definition and annotation of (myco)bacterial non-coding RNA. Tuberculosis 93, 26–29. doi: 10.1016/j.tube.2012.11.010
Li, S.-K., Ng, P. K.-S., Qin, H., Lau, J. K.-Y., Lau, J. P.-Y., Tsui, S. K.-W., et al. (2012). Identification of small RNAs in Mycobacterium smegmatis using heterologous Hfq. RNA 19, 74–84. doi: 10.1261/rna.034116.112
Livny, J., Brencic, A., Lory, S., and Waldor, M. K. (2006). Identification of 17 Pseudomonas aeruginosa sRNAs and prediction of sRNA-encoding genes in 10 diverse pathogens using the bioinformatic tool sRNAPredict2. Nucleic Acids Res. 34, 3484–3493. doi: 10.1093/nar/gkl453
Livny, J., Teonadi, H., Livny, M., and Waldor, M. K. (2008). High-throughput, kingdom-wide prediction and annotation of bacterial non-coding RNAs. PLoS ONE 3:e3197. doi: 10.1371/journal.pone.0003197
Lünse, C. E., Schüller, A., and Mayer, G. (2014). The promise of riboswitches as potential antibacterial drug targets. Int. J. Med. Microbiol. 304, 79–92. doi: 10.1016/j.ijmm.2013.09.002
Lu, X., Goodrich-Blair, H., and Tjaden, B. (2011). Assessing computational tools for the discovery of small RNA genes in bacteria. RNA 17, 1635–1647. doi: 10.1261/rna.2689811
Mann, B., van Opijnen, T., Wang, J., Obert, C., Wang, Y.-D., Carter, R., et al. (2012). Control of virulence by small RNAs in Streptococcus pneumoniae. PLoS Pathog. 8:e1002788. doi: 10.1371/journal.ppat.1002788
McGuire, A. M., Weiner, B., Park, S. T., Wapinski, I., Raman, S., Dolganov, G., et al. (2012). Comparative analysis of Mycobacterium and related Actinomycetes yields insight into the evolution of Mycobacterium tuberculosis pathogenesis. BMC Genomics 13:120. doi: 10.1186/1471-2164-13-120
Mellin, J. R., and Cossart, P. (2012). The non-coding RNA world of the bacterial pathogen Listeria monocytogenes. RNA Biol. 9, 372–378. doi: 10.4161/rna.19235
Miotto, P., Forti, F., Ambrosi, A., Pellin, D., Veiga, D. F., Balazsi, G., et al. (2012). Genome-wide discovery of small RNAs in Mycobacterium tuberculosis. PLoS ONE 7:e51950. doi: 10.1371/journal.pone.0051950
Na, D., Yoo, S. M., Chung, H., Park, H., Park, J. H., and Lee, S. Y. (2013). Metabolic engineering of Escherichia coli using synthetic small regulatory RNAs. Nat. Biotechnol. 31, 170–174. doi: 10.1038/nbt.2461
Pandey, S. P., Minesinger, B. K., Kumar, J., and Walker, G. C. (2011). A highly conserved protein of unknown function in Sinorhizobium meliloti affects sRNA regulation similar to Hfq. Nucleic Acids Res. 39, 4691–4708. doi: 10.1093/nar/gkr060
Papenfort, K., and Vogel, J. (2010). Regulatory RNA in bacterial pathogens. Cell Host Microbe 8, 116–127. doi: 10.1016/j.chom.2010.06.008
Park, H., Bak, G., Kim, S. C., and Lee, Y. (2013). Exploring sRNA-mediated gene silencing mechanisms using artificial small RNAs derived from a natural RNA scaffold in Escherichia coli. Nucleic Acids Res. 41, 3787–3804. doi: 10.1093/nar/gkt061
Park, P. J. (2009). ChIP–seq: advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669–680. doi: 10.1038/nrg2641
Pellin, D., Miotto, P., Ambrosi, A., Cirillo, D. M., and Di Serio, C. (2012). A Genome-wide identification analysis of small regulatory RNAs in Mycobacterium tuberculosis by RNA-seq and conservation analysis. PLoS ONE 7:e32723. doi: 10.1371/journal.pone.0032723
Pelly, S., Bishai, W. R., and Lamichhane, G. (2012). A screen for non-coding RNA in Mycobacterium tuberculosis reveals a cAMP-responsive RNA that is expressed during infection. Gene 500, 85–92. doi: 10.1016/j.gene.2012.03.044
Peterson, M. W. (2013). Network Reconstruction by ChIP-Seq in Mycobacterium Tuberculosis and Neurospora Crassa (Doctoral dissertation). Ann Arbor, MI: ProQuest (Publication No. 3575360).
Podkaminski, D., Bouvier, M., and Vogel, J. (2014). “Identification and characterization of small non-coding RNAs in bacteria,” in Handbook of RNA Biochemistry: Second, Completely Revised and Enlarged Edition (Weinheim: Wiley-VCH), 719–786. Available online at: http://onlinelibrary.wiley.com/doi/10.1002/9783527647064.ch33/summary (Accessed April 15, 2014).
Remus, N., Alcais, A., and Abel, L. (2003). Human genetics of common mycobacterial infections. Immunol. Res. 28, 109–129. doi: 10.1385/IR:28:2:109
Rodrigue, S., Provvedi, R., Jacques, P. -É, Gaudreau, L., and Manganelli, R. (2006). The σ factors of Mycobacterium tuberculosis. FEMS Microbiol. Rev. 30, 926–941. doi: 10.1111/j.1574-6976.2006.00040.x
Romby, P., and Charpentier, E. (2009). An overview of RNAs with regulatory functions in gram-positive bacteria. Cell. Mol. Life Sci. 67, 217–237. doi: 10.1007/s00018-009-0162-8
Romeo, T., Vakulskas, C. A., and Babitzke, P. (2013). Post-transcriptional regulation on a global scale: form and function of Csr/Rsm systems: the Csr global regulatory system. Environ. Microbiol. 15, 313–324. doi: 10.1111/j.1462-2920.2012.02794.x
Romilly, C., Caldelari, I., Parmentier, D., Lioliou, E., Romby, P., and Fechter, P. (2012). Current knowledge on regulatory RNAs and their machineries in Staphylococcus aureus. RNA Biol. 9, 402–413. doi: 10.4161/rna.20103
Sassetti, C. M., Boyd, D. H., and Rubin, E. J. (2003). Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48, 77–84. doi: 10.1046/j.1365-2958.2003.03425.x
Shiloh, M. U., and DiGiuseppe Champion, P. A. (2010). To catch a killer. What can mycobacterial models teach us about Mycobacterium tuberculosis pathogenesis? Curr. Opin. Microbiol. 13, 86–92. doi: 10.1016/j.mib.2009.11.006
Sittka, A., Lucchini, S., Papenfort, K., Sharma, C. M., Rolle, K., Binnewies, T. T., et al. (2008). Deep sequencing analysis of small noncoding rna and mrna targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 4:e1000163. doi: 10.1371/journal.pgen.1000163
Skeiky, Y. A., and Sadoff, J. C. (2006). Advances in tuberculosis vaccine strategies. Nat. Rev. Microbiol. 4, 469–476. doi: 10.1038/nrmicro1419
Sonnleitner, E., Romeo, A., and Bläsi, U. (2012). Small regulatory RNAs in Pseudomonas aeruginosa. RNA Biol. 9, 364–371. doi: 10.4161/rna.19231
Ster, C., Allard, M., Boulanger, S., Lamontagne Boulet, M., Mulhbacher, J., Lafontaine, D. A., et al. (2013). Experimental treatment of Staphylococcus aureus bovine intramammary infection using a guanine riboswitch ligand analog. J. Dairy Sci. 96, 1000–1008. doi: 10.3168/jds.2012-5890
Tjaden, B. (2008). TargetRNA: a tool for predicting targets of small RNA action in bacteria. Nucleic Acids Res. 36, W109–W113. doi: 10.1093/nar/gkn264
Tomasini, A., François, P., Howden, B. P., Fechter, P., Romby, P., and Caldelari, I. (2014). The importance of regulatory RNAs in Staphylococcus aureus. Infect. Genet. Evol. 21, 616–626. doi: 10.1016/j.meegid.2013.11.016
Tortoli, E. (2006). The new mycobacteria: an update. FEMS Immunol. Med. Microbiol. 48, 159–178. doi: 10.1111/j.1574-695X.2006.00123.x
Tsai, C.-H., Baranowski, C., Livny, J., McDonough, K. A., Wade, J. T., and Contreras, L. M. (2013). Identification of novel sRNAs in mycobacterial species. PLoS ONE 8:e79411. doi: 10.1371/journal.pone.0079411
Vandal, O. H., Nathan, C. F., and Ehrt, S. (2009). Acid resistance in Mycobacterium tuberculosis. J. Bacteriol. 191, 4714–4721. doi: 10.1128/JB.00305-09
Vazquez-Anderson, J., and Contreras, L. M. (2013). Regulatory RNAs: charming gene management styles for synthetic biology applications. RNA Biol. 10, 1778–1797. doi: 10.4161/rna.27102
Wang, S., Dong, X., Zhu, Y., Wang, C., Sun, G., Luo, T., et al. (2013). Revealing of Mycobacterium marinum Transcriptome by RNA-seq. PLoS ONE 8:e75828. doi: 10.1371/journal.pone.0075828
Wang, T., Xie, Y., and Xiao, G. (2014). dCLIP: a computational approach for comparative CLIP-seq analyses. Genome Biol. 15:R11. doi: 10.1186/gb-2014-15-1-r11
Warner, D. F., Savvi, S., Mizrahi, V., and Dawes, S. S. (2007). A riboswitch regulates expression of the coenzyme B12-independent methionine synthase in Mycobacterium tuberculosis: implications for differential methionine synthase function in strains H37Rv and CDC1551. J. Bacteriol. 189, 3655–3659. doi: 10.1128/JB.00040-07
Wassarman, K. M., and Storz, G. (2000). 6S RNA Regulates E. coli RNA polymerase activity. Cell 101, 613–623. doi: 10.1016/S0092-8674(00)80873-9
Wassarman, K. M., Zhang, A., and Storz, G. (1999). Small RNAs in E. coli. Trends Microbiol. 7, 37–45. doi: 10.1016/S0966-842X(98)01379-1
Wattam, A. R., Abraham, D., Dalay, O., Disz, T. L., Driscoll, T., Gabbard, J. L., et al. (2014). PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 42, D581–D591. doi: 10.1093/nar/gkt1099
Wayne, L. G., and Sohaskey, C. D. (2001). Nonreplicating persistence of Mycobacterium tuberculosis. Annu. Rev. Microbiol. 55, 139–163. doi: 10.1146/annurev.micro.55.1.139
Wen, Y., Feng, J., and Sachs, G. (2012). Helicobacter pylori 5'ureB-sRNA, a cis-encoded antisense small RNA, negatively regulates ureAB expression by transcription termination. J. Bacteriol. 195, 444–452. doi: 10.1128/JB.01022-12
Wilkinson, K. A., Stewart, G. R., Newton, S. M., Vordermeier, H. M., Wain, J. R., Murphy, H. N., et al. (2005). Infection biology of a novel -crystallin of Mycobacterium tuberculosis: Acr2. J. Immunol. 174, 4237–4243. doi: 10.4049/jimmunol.174.7.4237
Wurtzel, O., Sesto, N., Mellin, J. R., Karunker, I., Edelheit, S., Bécavin, C., et al. (2012). Comparative transcriptomics of pathogenic and non-pathogenic Listeria species. Mol. Syst. Biol. 8:583. doi: 10.1038/msb.2012.11
Yu, J., and Schneiders, T. (2012). Tigecycline challenge triggers sRNA production in Salmonella enterica serovar Typhimurium. BMC Microbiol. 12:195. doi: 10.1186/1471-2180-12-195
Zhang, A., Wassarman, K. M., Rosenow, C., Tjaden, B. C., Storz, G., and Gottesman, S. (2003). Global analysis of small RNA and mRNA targets of Hfq: global analysis of Hfq targets. Mol. Microbiol. 50, 1111–1124. doi: 10.1046/j.1365-2958.2003.03734.x
Zhang, Y. J., Ioerger, T. R., Huttenhower, C., Long, J. E., Sassetti, C. M., Sacchettini, J. C., et al. (2012). Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS Pathog. 8:e1002946. doi: 10.1371/journal.ppat.1002946
Zhao, X., Koestler, B. J., Waters, C. M., and Hammer, B. K. (2013). Post-transcriptional activation of a diguanylate cyclase by quorum sensing small RNAs promotes biofilm formation in Vibrio cholerae: post-transcriptional activation of a V. cholerae DGC. Mol. Microbiol. 89, 989–1002. doi: 10.1111/mmi.12325
Keywords: small RNA, mycobacteria, regulation, pathogenesis, non-coding RNAs, Mycobacterium tuberculosis
Citation: Haning K, Cho SH and Contreras LM (2014) Small RNAs in mycobacteria: an unfolding story. Front. Cell. Infect. Microbiol. 4:96. doi: 10.3389/fcimb.2014.00096
Received: 28 May 2014; Accepted: 03 July 2014;
Published online: 24 July 2014.
Edited by:
Wyndham W. Lathem, Northwestern University Feinberg School of Medicine, USAReviewed by:
Guangchun Bai, Albany Medical College, USAErin R. Murphy, Ohio University Heritage College of Osteopathic Medicine, USA
Copyright © 2014 Haning, Cho and Contreras. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lydia M. Contreras, McKetta Department of Chemical Engineering, Cockrell School of Engineering, The University of Texas at Austin, 200 E. Dean Keeton St. Stop C0400, Austin, TX 78712, USA e-mail:bGNvbnRyZXJAY2hlLnV0ZXhhcy5lZHU=