 Thuy Do
Thuy Do Evelyn C. Sheehy2
Evelyn C. Sheehy2 Tonnie Mulli
Tonnie Mulli Francis Hughes
Francis Hughes David Beighton
David Beighton- 1Department of Oral Biology, School of Dentistry, University of Leeds, Leeds, UK
- 2Department of Periodontology, Dental Institute, King's College London, London, UK
- 3Department of Periodontology, School of Dental Sciences, University of Nairobi, Nairobi, Kenya
Veillonella spp. are predominant bacteria found in all oral biofilms. In this study, a metatranscriptomic approach was used to investigate the gene expression levels of three oral Veillonella spp. (V. parvula, V. dispar and V. atypica) in whole stimulated saliva from caries-free volunteers and in carious lesions (n = 11 for each group). In the lesions the greatest proportion of reads were assigned to V. parvula and genes with the highest level of expression in carious samples were those coding for membrane transport systems. All three Veillonella spp. increased expression of genes involved in the catabolism of lactate and succinate, notably the alpha- and beta-subunits of L(+)-tartrate dehydratase (EC 4.2.1.32). There was also significantly increased expression of histidine biosynthesis pathway in V. parvula, suggesting higher intra-cellular levels of histidine that could provide intra-cellular buffering capacity and, therefore, assist survival in the acidic environment. Various other systems such as potassium uptake systems were also up regulated that may aid in the survival and proliferation of V. parvula in carious lesions.
Introduction
Veillonella are obligate anaerobic Gram-negative small cocci isolated from the oral cavity and intestinal tract of humans and animals that gain energy from the utilization of short-chain organic acids, particularly lactate and succinate (Delwiche et al., 1985). The human Veillonella are Veillonella parvula, V. atypica, V. dispar, V. montpellierensis, V. denticariosi, and V. rogosae (Mays et al., 1982; Rogosa, 1984; Jumas-Bilak et al., 2004; Byun et al., 2007; Arif et al., 2008). The predominant Veillonella species on the tongue were V. rogosae, V. atypical, and V. dispar (Beighton et al., 2008; Mashima et al., 2011). V. parvula has often been detected as the predominant Veillonella species isolated from active occlusal carious lesions (Arif et al., 2008; Beighton et al., 2008). Based on these studies, each Veillonella species seems to occupy different intra-oral habitats with limited degree of overlap between species. With the pH of carious lesions reported to be below 5 (Hojo et al., 1994), the bacteria's ability to colonize and proliferate in such an environment necessitates them to exhibit a phenotype characterized by acid resistance. The objective of this study was to determine and compare the transcriptome of three of the predominant human oral Veillonella (V. parvula, V. dispar, and V. atypica) present in caries lesions and in the saliva of caries-free individuals.
Many bacterial genome sequence data are now publicly available, making it possible to exploit the opportunities offered by next generation sequencing (NGS) approaches to determine the in vivo expression of specific bacterial genes of individual species present in mixed-population biofilms. The short reads obtained from NGS can be aligned to bacterial genomes, enabling transcriptomic analysis of species without the need for species-specific protocols, as is necessary with the micro-array approach. The functional potential of the oral microbiome has been investigated using metagenomic approaches in which genomic DNA is extracted, sequenced and the resulting sequences annotated by comparison to extant complete and partial genome sequences (Belda-Ferre et al., 2012; Luo et al., 2012). To investigate gene expression, the metatranscriptome of an individual species within a natural biofilm may be determined using RNA sequencing (RNA-seq). The application of RNA-seq to the study of bacterial transcriptomes has been reviewed by Pinto et al. (2011) and McLean (2014). Several studies have also recently described the use of RNA-Seq as a tool to investigate the oral microbiome in health and disease (Duran-Pinedo et al., 2014; Jorth et al., 2014) as well as interrogate specific metabolic pathways in oral bacterial species in vitro (Zeng et al., 2013).
In this study, we adopted a metatranscriptomic approach to investigate the level of genes expressed by the three Veillonella in both active carious lesions and saliva of caries-free subjects, in order to observe metabolic activities occurring in their natural environment, which may give an insight into their intra-oral distribution.
Materials and Methods
Samples Collection and RNA Isolation
Ethical approval was obtained for the collection of carious lesions (n = 11) and saliva (n = 11) samples. All subjects (n = 22) gave informed consent prior to collection of the clinical material. Extracted teeth with large occlusal soft, active carious lesions were obtained from patients attending dental clinics at Guy's Hospital dental surgery. The teeth were immediately placed in 5 ml RNAprotect® Bacteria Reagent (Qiagen) and transferred to the laboratory. The superficial biofilm was carefully removed and discarded. The infected soft dentine was collected using sterile excavators, and placed in 1 ml RNAprotect reagent, disaggregated, centrifuged (4°C at 10,000 × g) and the pellets stored at −80°C. Whole mouth wax-stimulated saliva samples, collected for 5 min, were obtained from caries-free volunteers who refrained from eating for at least 2 h prior to sampling. Immediately after collection, RNAprotect reagent was added to the saliva (1:1 v/v), the samples were centrifuged and the pellets stored at −80°C until further processing. Total RNA was extracted using the UltraClean® Microbial RNA isolation kit (MOBIO Laboratories, Inc.), including a DNase treatment step using the RNase-Free DNase Set (Qiagen) prior RNA elution.
cDNA Synthesis and Library Preparation for High-Throughput Sequencing
A minimum of 100 ng of total RNA was extracted from each clinical sample. The total RNA was processed using reagents provided in the Illumina® TruSeq™ RNA Sample Preparation Kit. Briefly, the RNA extracts were further purified, and fragmented. First and second strands cDNA were synthesized with Superscript II Reverse Transcriptase (Invitrogen). End repair was performed on the nucleic acid fragments, 3′ ends were adenylated and adapter indexes ligated. The processed cDNA were amplified and further purified, prior library validation with the Agilent DNA 1000 Bioanalyzer (Agilent Technologies) and dsDNA BR Qubit assays (Invitrogen). The resulting libraries were processed for cluster generation using the TruSeq paired end cluster kit v.2, Illumina Inc., and an equimolar amount of each library was run in a separate flowcell lane. Paired end sequencing was then carried out using a Genome Analyzer IIx Illumina platform to produce 76 bp reads.
Data Handling and Gene Expression Analyses
A FASTQ file was obtained for each of the 22 cDNA libraries. Initial checks of the sequencing read base qualities were done via the local server provided by the Genomics facilities at Guy's Hospital Biomedical Research Centre, the data were then imported into the CLC Genomics Workbench software (CLC Bio, Qiagen). Within the CLC environment, adapter sequences were removed and for each sample file a short read mapping was performed simultaneously against 144 annotated oral bacterial genomes which were previously imported from various databases (the DNA Data Bank of Japan, NCBI, the Broad Institute and HOMD databases) (Supplementary File 5). The read mapping was carried out using the RNA-Seq analysis package default settings (mismatch cost: 2, insertion cost: 3, deletion cost: 3, length fraction: 0.8, and similarity fraction: 0.8; with the maximum number of hits for a read set to 1) within the CLC software, which employs the CLC Assembly Cell (CLC3) read mapper (http://www.clcbio.com/products/clc-assembly-cell/).
In this study, we are concerned with reads that mapped to 3 Veillonella strains: V. parvula DSM2008, V. dispar ATCC 17748, and V. atypica ACS 0049 V Sch6 only (Table 1). In order to facilitate comparison between these Veillonella strains, a RAST annotation (Rapid Annotation using Subsystem Technology) (Aziz et al., 2008) was carried out on their genomes and used with the other oral strains in the read mapping. All of the 22 sequence data files were processed for read mapping against the 144 oral genomes. Results were exported as excel files containing raw read counts determined for each of the genes from the 144 oral strains (total of 351,456 genes) (Supplementary File 1). In order to compare expression levels between the 22 biological samples, the raw count data were gathered into a single excel spreadsheet for normalization (Supplementary File 1). The read counts were scaled by determining the effective library size of each sample, using the estimateSizeFactors and counts accessor functions within the Bioconductor R package DESeq (Anders and Huber, 2010), which provided an output table displaying normalized expression values for each gene and for each of the 22 samples. Data corresponding to the 3 Veillonella strains were manually extracted from the spreadsheet and used separately for further analysis to infer on their gene expression levels in caries lesions and caries-free saliva samples. Median values were calculated for both caries and saliva sample groups (n = 11 each) (Supplementary File 2), which we called relative median expression (RME) values. The RME values of identical genes found in the 3 Veillonella strains were summed and ranked from highest to lowest values, to observe the most highly expressed Veillonella transcripts in caries and caries-free saliva samples (Supplementary File 2). The gene identities were obtained from the RAST annotations and was supplemented by BLAST searching within Uniprot (http://www.uniprot.org/), InterProScan (http://www.ebi.ac.uk/Tools/pfa/iprscan/) and PATRIC (http://patricbrc.org/) when necessary.

Table 1. Characteristics of the 3 Veillonella strains selected in this study, with their relative proportions in caries and saliva metatranscriptomes (CDS refers to coding genes).
The raw read count data from all 144 oral strains were also used to carry out differential gene expression analysis between both sample groups using the statistical software R package DESeq2 (Love et al., 2014) based on the negative binomial model. Differential expression analysis results for the 3 Veillonella strains were manually extracted from the total R result outputs into excel spreadsheets, and the largest negative and positive Log2 Fold Change values, with adjusted p-values (padj) < 10−3 were considered as significant.
The Supplementary File 1 contains the raw count input information used for the DESeq and DESeq2 analyses.
Sequence Data Accession Numbers
RNA-Seq sequencing data are available from the National Center for Biotechnology Information (NCBI) Sequence Read Archive; biosamples accession numbers for this study are SRS741215 and SRS752041.
Results and Discussion
Analysis of Read Count and Ecological Considerations
Here we have determined gene expression levels by mapping reads to bacterial species which form part of the oral microbial populations. The total number of mapped reads ranged between 25,593,022 and 88,238,546 for the caries-free saliva samples and between 20,088,245 and 32,910,299 for the caries samples (Supplementary File 1). In the carious lesions, 16.62 ± 11.17 per cent, 2.18 ± 1.13 per cent and 0.91 ± 0.43 percent of the mapped reads were assigned to V. parvula, V. dispar, and V. atypica, respectively, compared with 4.76 ± 7.21, 7.08 ± 5.07, and 4.09 ± 3.47 in the saliva samples (all p < 0.05) (Table 1). The pattern of the distribution of reads mirrored the reported distribution of these three species based on cultivable bacterial studies (Arif et al., 2008; Beighton et al., 2008). Belda-Ferre et al. (2012) also reported V. parvula to be the most predominant species in biofilm infecting dentine, with 166 contigs (>500 bp) assigned to V. parvula from their metagenomic data.
The major environmental factors affecting the Veillonella strains in the carious lesions and in saliva are suspected to be the low pH and availability of organic acids (lactate and succinate) required for the generation of ATP. The acidic environment within carious lesions is unlikely to be homogenous despite lactic acid being the major organic acid present (Palmer et al., 2006), resulting in areas that might be more alkaline (i.e., pH > 6). Nevertheless, it should be expected that the concentration of organic acids in saliva is less than that of carious lesions, since subjects had refrained from eating for 2 h prior sample collection, hence organic acids and dietary components should have cleared from the mouth. Moreover, we should emphasize that the microbiota present in wax-stimulated saliva is likely to derive from the intra-oral mucosal surfaces and from the supra-gingival plaque, providing an average composition of intra-oral surfaces, but mostly of the tongue surface (Simon-Soro et al., 2013). These ecological aspects have been taken into account and explain the differences in metabolic activities occurring within the Veillonella species in both sample groups.
Gene Expression Analysis
The combined expression level of the 3 Veillonella species was determined for each condition. The relative mean expression (RME) values for each identical gene product were added and the top 30 most highly expressed gene products in saliva were ranked. The corresponding values for the caries samples are also displayed together in Figure 1.
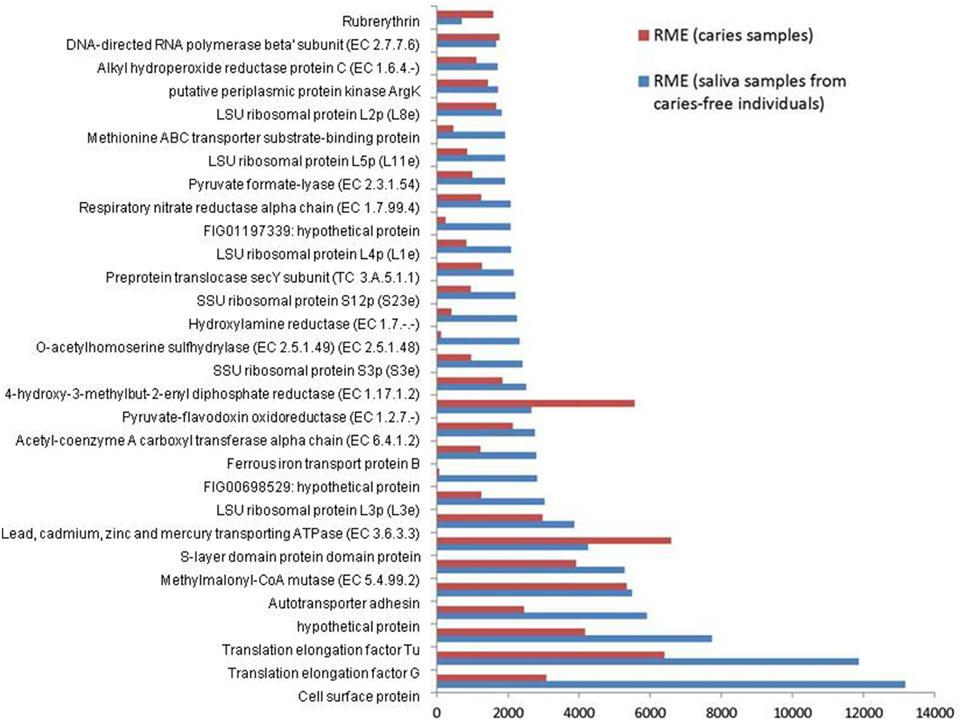
Figure 1. Relative median expression (RME) levels in 3 Veillonella strains (V. parvula DSM2008, V. dispar ATCC 17748 and V. atypica ACS 0049 V Sch6). RME were calculated from the median values of normalized read counts in the caries (n = 11) and saliva (n = 11) samples. The 30 highest RME values were sorted in ascending order for the genes in saliva samples and are displayed with the RME values of corresponding genes in caries samples.
Overall, the 3 Veillonella species present in the caries and saliva samples display a similar profile of transcripts. V. parvula expressed more genes in the caries samples, whereas V. dispar expressed more genes in the saliva samples (Table 1, Supplementary File 2).
The most abundant transcripts were related to the production of cell surface proteins (RME = 13175), outer membrane synthesis (S-layer proteins, RME = 6592), translation elongation factors (G and Tu, RME = 11860 and 7731 respectively), transport systems (RME = 5481), ribosomal subunit proteins (protein biosynthesis, RME = 3028), and carbohydrate metabolism (particularly the glyoxylate and dicarboxylate metabolism, EC 4.2.1.32, EC 6.4.1.2, EC 1.1.1.37; RME = 2147, 2126, and 1461 respectively). These results are consistent with those described by Peterson et al. (2014) in plaque biofilm. Similarly Benítez-Páez et al. (2014) found evidence of overrepresentation of translation functions, together with high expressions of elongation factors Tu and G, emphasizing their importance and involvement in oral biofilm formation especially in early biofilms.
We also report high levels of transcripts encoding membrane transport proteins (cadmium-exporting ATPase, RME = 3861; autotransporter adhesin, RME = 5481; ABC tranporters, RME = 1998), as well as transcripts involved in oxidative stress protection (rubrerythrin, RME = 1577; and alkyl hydroperoxide reductase protein C, EC 1.6.4.-, RME = 1700), in both caries and saliva groups (Supplementary File 2). The overall similarity in transcription profiles in both sample groups suggest that the selected Veillonella species are actively expressing genes that are involved in cellular maintenance and survival within diverse environments.
Differential Expression Analysis
Differential gene expression between the caries and saliva groups was investigated using the R package DESeq2 (Love et al., 2014).
Sample to sample distances were calculated within the DESeq2 package. The principal components analysis and the heatmap of Euclidian distance between samples were based on the metatranscriptomic data mapped to 144 oral strains, and show caries and saliva samples to form distinct clusters. The PCA plot displays larger differences between saliva samples than between caries samples (Figure 2), suggesting that metabolic functions in the caries lesions are more conserved that in the caries-free samples. Likewise, the heatmap indicates the overall similarity between samples of the same group, with the exception of saliva sample number 9 (H9 in Figure 3), which seems to cluster with the caries samples, indicating that it shares similar functions found in caries.
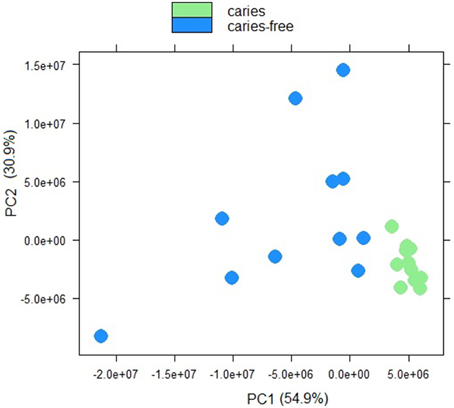
Figure 2. Principal component analysis plot displaying sample-to-sample distances for caries and saliva samples. The PCA plot is based on the differential expression analysis of 144 oral bacterial strains, carried out using the R package DEseq2 (Love et al., 2014).
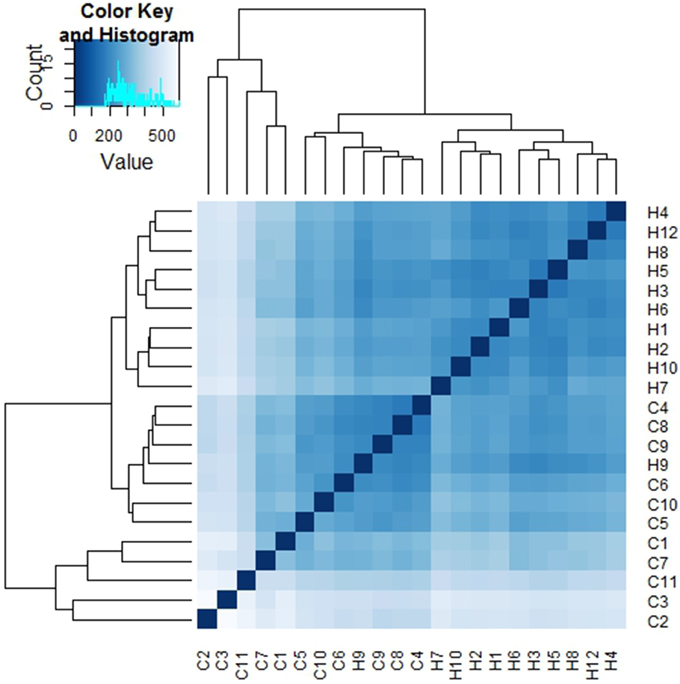
Figure 3. Heatmap of Euclidian distances between samples (n = 22). The heatmap was constructed using the R package DESeq2 (Love et al., 2014), and is based on the differential expression analysis of 144 oral bacterial strains.
Jorth et al. (2014) found more similar functional features in microbiota associated with disease compared to health-associated microbiota, even though great variations in the oral microbial composition were observed between and within patients. Other papers have described inter-patients variations in terms of bacterial profiles, and these seem to reduce in diversity when changing from healthy to a disease status (Munson et al., 2004; Preza et al., 2008). Our data suggest that in the caries lesions, metabolic functions in the 3 Veillonella species are more similar, than in caries-free saliva samples.
In order to identify the main functional differences between the caries lesions and saliva samples, output data from the DESeq2 analysis were sorted according to the log2 fold change values (Supplementary File 3). Since the transcriptomic data (n = 22) were analyzed with the caries-free vs. caries condition (used as the default DESeq2 condition setting), negative log2 fold change values, with corresponding Benjamini-Hochberg (BH) adjusted p-values (padj) < 10−3 considered as significant (Benjamini and Hochberg, 1995), indicate genes with the strongest down-regulation in saliva (or strongest up-regulation in caries). Conversely, the largest log2 fold change values, with corresponding significant BH padj < 10−3, indicate genes which are the most differentially expressed in saliva. Only the top 15 genes in both conditions are displayed in Table 2, and ranked according to the log2FoldChange values. A heatmap was also constructed within the DESeq2 package, and displays the top 30 differentially expressed genes across all 22 samples for the 3 Veillonella species (Supplementary File 4).
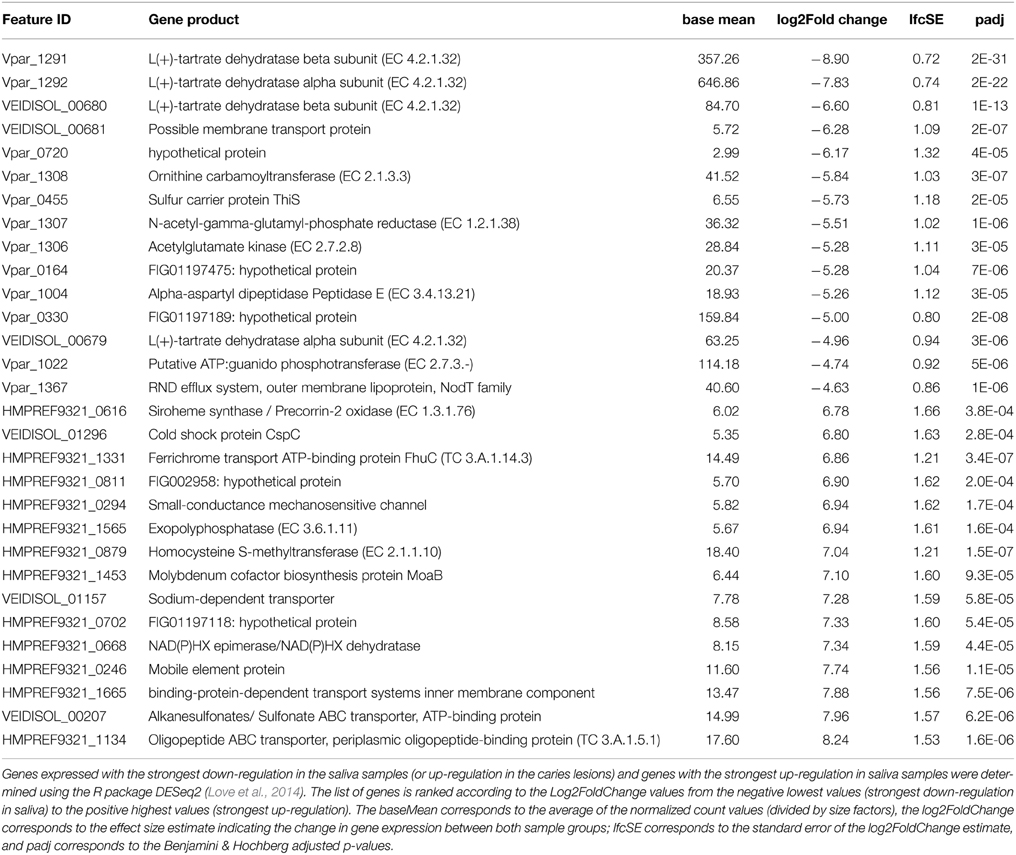
Table 2. Up-regulated genes in the caries samples (top of table) and in the saliva samples (bottom of table).
Genes that were differentially expressed in caries lesions (padj < 10−3) were those expressed by V. parvula, and were mainly involved in pyruvate metabolism, transferases and membrane transport systems (including the biosynthesis of efflux pump components, ABC transporter and sulfur carrier proteins) (Table 2), inferring a role of these functions in disease. Similar findings were reported by Benítez-Páez et al. (2014) who found that ABC transporters were significantly up-regulated in mature biofilms, with cell motility function associated with bacterial chemotaxis, whereas Duran-Pinedo et al. (2014) reported significant levels of ABC transporters in periodontitis samples that seem associated with high levels of expression of virulence factors. Other specific pathways associated with disease have also been reported. In the case of periodontitis, a significant enrichment in butyrate production was detected (Jorth et al., 2014), iron acquisition and membrane synthesis have also been described as important metabolic activities defining disease (Duran-Pinedo et al., 2014).
However, in our data all 3 Veillonella species (especially V. parvula) expressed genes involved in glyoxylate and dicarboxylate metabolism, and alanine aspartate and glutamate metabolism, in particular genes encoding the alpha- and beta-subunits of L(+)-tartrate dehydratase (EC 4.2.1.32). These are involved in the production of ATP through catabolism of lactate and succinate. Overall, the data suggest that all species responded to growth in the carious lesions by increasing the expression of many genes associated with the utilization of lactate and succinate with the consequent generation of ATP via the sodium ion-translocating methylmalonyl-CoA decarboxylase (Buckel, 2001). We also found significant up-regulation of genes encoding aspartate aminotransferases (Vpar_1105, Vpar_0075, HMPREF9321_0571, HMPREF9321_1684) in both caries and saliva samples (Supplementary File 3); these enzymes catalyze the reaction L-aspartate + 2-oxoglutarate into oxaloacetate + L-glutamate and may be an alternative method of entering intermediates into the lactate metabolic pathway, for producing ATP.
Genes involved in histidine metabolism were also up-regulated in caries by V. parvula, but not in the other 2 species (Supplementary File 3). Of particular importance is the up-regulation of ATP phosphoribosyltransferase (EC 2.4.2.17) which has a central role in histidine biosynthesis. Similar up-regulation was observed in Corynebacterium glutamicum and Salmonella typhimurium, as well as in Lactobacillus casei, in response to acid adaptation (20 min at pH 4.5) (Foster, 1995; Brockmann-Gretza and Kalinowski, 2006; Broadbent et al., 2010). It was suggested that the up-regulation of the histidine operon resulted in increased intra-cellular levels of His which may contribute to intracellular buffering capacity as the pKa value of the imidazole groups of histidine and histidine-containing peptides is near 6.0 and these have been shown to contribute to intracellular buffering in vertebrate cells (Abe, 2000).
Additionally, a potassium uptake system in V. parvula (Vpar_1334 and Vpar_1335; KtrA and KtrB) was also significantly up-regulated in caries, but not by the other 2 species. K+ uptake in prokaryotes is essential for maintenance of cytoplasmic pH (Csonka and Epstein, 1996; Stumpe et al., 1996), this system may also assist in the survival of V. parvula in the acidic environment of the carious dentine. In V. dispar and V. dispar, these genes were significantly up-regulated in saliva, which may explain their lower ability to control their intracellular pH in the caries lesions. Clearly, V. parvula exhibits several distinct systems for intracellular pH control which do not appear to function as well in either V. atypica or V. dispar, and this may explain the ability of V. parvula to be better fitted to growth and proliferation in the acidic environment of carious lesions compared to the other two species.
Most of the differentially expressed genes in the saliva samples are those expressed by V. atypica and V. dispar, and encode for the oligopeptide, sulfonate transporter systems, and cysteine and methionine metabolism (EC 2.1.1.10). Others include genes involved in purine metabolism (EC 3.6.1.11), ferrichrome and other transport systems, molybdenum cofactor biosynthesis, as well as oxidoreductases which involve the use of NAD+ or NADP+ as acceptor in the chemical reaction leading to the formation of siroheme from uroporphyrinogen III (EC 1.3.1.76) (Table 2).
General stress response genes have also been identified in the carious lesions and saliva samples in all 3 Veillonella species (Supplementary File 3). Several genes encoding heat shock and chaperonin proteins were found up-regulated in caries (Vpar_1034, Vpar_1035, Vpar_0881), and others up-regulated in saliva (VEIDISOL_01212, HMPREF9321_0106, VEIDISOL_01142). Stress proteins such as chaperonin heat shock protein 33 (HMPREF9321_0536) and putative peroxide-responsive repressor PerR (HMPREF9321_0995) were up-regulated by V. atypica in the saliva samples (Supplementary File 3). In V. parvula and V. dispar, several genes associated with extracellular S-layer formation, were significantly up-regulated, which is a well characterized stress-associated response (Xiao et al., 2012).
Conclusion
Recent reports of metagenomic and metatranscriptomic analyses of oral samples are providing extensive information on the microbial populations and functions characterizing health and disease. These studies have also confirmed previous culturable observations regarding the intra-oral distribution of particular species but also found novel taxa which have not previously been identified amongst cultured bacteria and phyla for which only limited cultivated isolates are extant.
Here we have applied a metatranscriptomic approach to study 3 predominant oral Veillonella spp. in their natural habitat and have shown that their gene expression profiles are overall similar in both caries lesions and saliva (caries-free) samples. However, through differential expression analysis, V. parvula seems to exhibit a distinct method of intra-cellular pH control not evident in the other two species investigated, which might explain the preponderance of V. parvula in carious lesions and the reduced ability of V. atypica and V. dispar to proliferate in this acid environment. Other important functions related to membrane transport systems are reported to be over-expressed in the caries lesions inferring a role in disease.
We have shown here that RNA-Seq is a powerful technique that can be used to observe the transcriptome of selected species or strain, provided their genome sequence data are available. The obvious drawbacks from such technique relate to the limited number of reference genomes available for reads mapping, and also to the fact that non-core genome sequences are not captured using the current methodology. Further analyses including larger samples and samples from similar biofilms such as plaque instead of saliva would be beneficial to add to our understanding of the oral microbial functions during initiation and development of disease.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research was supported by the Dental Institute, King's College London and the Department of Health via the National Institute for Health Research (NIHR) comprehensive Biomedical Research Centre award to Guy's and St Thomas' NHS Foundation Trust in partnership with King's College London and King's College Hospital NHS Foundation Trust. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fcimb.2015.00025/abstract
Supplementary File 1. Read count data for the 144 oral strains (including V. parvula DSM2008, V. dispar ATCC 17748 and V. atypica ACS 0049 V Sch6), used as input file for analyses using DESeq and DESeq2 in R.
Supplementary File 2. Relative median expression (RME) values based on the median and 75th percentile (Q3) values of normalized read counts in the caries (n = 11) and saliva samples (n = 11), for the 3 Veillonella species.
Supplementary File 3. Data output from the differential expression analysis for the 3 Veillonella species, obtained from DESeq2 analysis.
Supplementary File 4. Heatmap constructed using the R package DESeq2, displaying the highest most variable genes across all samples (n = 22), the analysis was based on the 3 Veillonella strains only.
Supplementary File 5. List of reference strains used in the CLC Genomics Workbench (CLC Bio, Qiagen) for RNA-Seq analysis.
References
Abe, H. (2000). Role of histidine-related compounds as intracellular proton buffering constituents in vertebrate muscle. Biochemistry 65, 757–765. Available online at: http://www.protein.bio.msu.ru/biokhimiya/contents/v65/pdf/bcm_0757.pdf
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Arif, N., Do, T., Byun, R., Sheehy, E., Clark, D., and Gilbert, S. C. (2008). Veillonella rogosae sp. nov., an anaerobic, Gram-negative coccus isolated from dental plaque. Int. J. Syst. Evol. Microbiol. 58, 581–584. doi: 10.1099/ijs.0.65093-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Aziz, R. K., Bartels, D., Best, A. A., Dejongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Beighton, D., Clark, D., Hanakuka, B., Gilbert, S., and Do, T. (2008). The predominant cultivable Veillonella spp. of the tongue of healthy adults identified using rpoB sequencing. Oral Microbiol. Immunol. 23, 344–347. doi: 10.1111/j.1399-302X.2007.00424.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Belda-Ferre, P., Alcaraz, L. D., Cabrera-Rubio, R., Romero, H., Simón-Soro, A., Pignatelli, M., et al. (2012). The oral metagenome in health and disease. ISME J. 6, 46–56. doi: 10.1038/ismej.2011.85
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Benítez-Páez, A., Belda-Ferre, P., Simón-Soro, A., and Mira, A. (2014). Microbiota diversity and gene expression dynamics in human oral biofilms. BMC Genomics 15:311. doi: 10.1186/1471-2164-15-311
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289–300.
Broadbent, J. R., Larsen, R. L., Deibel, V., and Steele, J. L. (2010). Physiological and transcriptional response of Lactobacillus casei ATCC 334 to acid stress. J. Bacteriol. 192, 2445–2458. doi: 10.1128/JB.01618-09
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Brockmann-Gretza, O., and Kalinowski, J. (2006). Global gene expression during stringent response in Corynebacterium glutamicum in presence and absence of the rel gene encoding (p)ppGpp synthase. BMC Genomics 7:230. doi: 10.1186/1471-2164-7-230
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Buckel, W. (2001). Sodium ion-translocating decarboxylases. Biochim. Biophys. Acta 1505, 15–27. doi: 10.1016/S0005-2728(00)00273-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Byun, R., Carlier, J. P., Jacques, N. A., Marchandin, H., and Hunter, N. (2007). Veillonella denticariosi sp. nov., isolated from human carious dentine. Int. J. Syst. Evol. Microbiol. 57, 2844–2848. doi: 10.1099/ijs.0.65096-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Csonka, L. N., and Epstein, W. (1996). “Osmoregulation,” in Escherichia coli and Salmonella: Cellular and Molecular Biology, eds F. Neidhardt, R. Curtis III, J. Ingraham, E. Lin, K. Low, and S. E. A. Magasanik (Washington DC: American Society for Microbiology Press), 1210–1223.
Delwiche, E. A., Pestka, J. J., and Tortorello, M. L. (1985). The veillonellae: gram-negative cocci with a unique physiology. Annu. Rev. Microbiol. 39, 175–193. doi: 10.1146/annurev.mi.39.100185.001135
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Duran-Pinedo, A. E., Chen, T., Teles, R., Starr, J. R., Wang, X., Krishnan, K., et al. (2014). Community-wide transcriptome of the oral microbiome in subjects with and without periodontitis. ISME J. 8, 1659–1672. doi: 10.1038/ismej.2014.23
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Foster, J. W. (1995). Low pH adaptation and the acid tolerance response of Salmonella typhimurium. Crit. Rev. Microbiol. 21, 215–237. doi: 10.3109/10408419509113541
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Hojo, S., Komatsu, M., Okuda, R., Takahashi, N., and Yamada, T. (1994). Acid profiles and pH of carious dentin in active and arrested lesions. J. Dent. Res. 73, 1853–1857.
Jorth, P., Turner, K. H., Gumus, P., Nizam, N., Buduneli, N., and Whiteley, M. (2014). Metatranscriptomics of the human oral microbiome during health and disease. MBio 5, e01012–e01014. doi: 10.1128/mBio.01012-14
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Jumas-Bilak, E., Carlier, J. P., Jean-Pierre, H., Teyssier, C., Gay, B., Campos, J., et al. (2004). Veillonella montpellierensis sp. nov., a novel, anaerobic, Gram-negative coccus isolated from human clinical samples. Int. J. Syst. Evol. Microbiol. 54, 1311–1316. doi: 10.1099/ijs.0.02952-0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi: 10.1186/s13059-014-0550-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Luo, C., Tsementzi, D., Kyrpides, N., Read, T., and Konstantinidis, K. T. (2012). Direct comparisons of Illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS ONE 7:e30087. doi: 10.1371/journal.pone.0030087
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mashima, I., Kamaguchi, A., and Nakazawa, F. (2011). The distribution and frequency of oral veillonella spp. in the tongue biofilm of healthy young adults. Curr. Microbiol. 63, 403–407. doi: 10.1007/s00284-011-9993-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Mays, T. D., Holdeman, L. V., Moore, W. E. C., Rogosa, M., and Johnson, J. L. (1982). Taxonomy of the genus Veillonella. Int. J. Syst. Bacteriol. 32, 28–36.
McLean, J. S. (2014). Advancements toward a systems level understanding of the human oral microbiome. Front. Cell. Infect. Microbiol. 4:98. doi: 10.3389/fcimb.2014.00098
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Munson, M. A., Banerjee, A., Watson, T. F., and Wade, W. G. (2004). Molecular analysis of the microflora associated with dental caries. J. Clin. Microbiol. 42, 3023–3029. doi: 10.1128/JCM.42.7.3023-3029.2004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Palmer, R. J. J., Diaz, P. I., and Kolenbrander, P. E. (2006). Rapid succession within the Veillonella population of a developing human oral biofilm in situ. J. Bacteriol. 188, 4117–4124. doi: 10.1128/JB.01958-05
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Peterson, S. N., Meissner, T., Su, A. I., Snesrud, E., Ong, A. C., Schork, N. J., et al. (2014). Functional expression of dental plaque microbiota. Front. Cell. Infect. Microbiol. 4:108. doi: 10.3389/fcimb.2014.00108
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Pinto, A. C., Melo-Barbosa, H. P., Miyoshi, A., Silva, A., and Azevedo, V. (2011). Application of RNA-seq to reveal the transcript profile in bacteria. Genet. Mol. Res. 10, 1707–1718. doi: 10.4238/vol10-3gmr1554
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Preza, D., Olsen, I., Aas, J. A., Willumsen, T., Grinde, B., and Paster, B. J. (2008). Bacterial profiles of root caries in elderly patients. J. Clin. Microbiol. 46, 2015–2021. doi: 10.1128/JCM.02411-07
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Rogosa, M. (1984). “Anaerobic Gram-negative cocci,” in Bergey's Manual of Systematic Bacteriology, Vol. 1, eds N. R. Krieg and J. G. Holt (Baltimore, MD: Williams & Wilkins), 680–685.
Simon-Soro, A., Tomas, I., Cabrera-Rubio, R., Catalan, M. D., Nyvad, B., and Mira, A. (2013). Microbial geography of the oral cavity. J. Dent. Res. 92, 616–621. doi: 10.1177/0022034513488119
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Stumpe, S., Schlosser, A., Schleyer, M., and Bakker, E. (1996). “K+ circulation across the prokaryotic cell membrane: K+-uptake systems. Transport processes in eukaryotic and prokaryotic organisms,” in Handbook of Biological Physics, eds K. Konings, H. Kaback, and J. Lolkema (Amsterdam: Elsevier), 473–499.
Xiao, J., Klein, M. I., Falsetta, M. L., Lu, B., Delahunty, C. M., Yates, J. R. III, et al. (2012). The Exopolysaccharide Matrix Modulates the Interaction between 3D Architecture and Virulence of a Mixed-Species Oral Biofilm. PLoS Pathog. 8:e1002623. doi: 10.1371/journal.ppat.1002623
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Zeng, L., Choi, S. C., Danko, C. G., Siepel, A., Stanhope, M. J., and Burne, R. A. (2013). Gene regulation by CcpA and catabolite repression explored by RNA-Seq in Streptococcus mutans. PLoS ONE 8:e60465. doi: 10.1371/journal.pone.0060465
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: Veillonella, caries, stress proteins, pH regulation, RNA-sequencing
Citation: Do T, Sheehy EC, Mulli T, Hughes F and Beighton D (2015) Transcriptomic analysis of three Veillonella spp. present in carious dentine and in the saliva of caries-free individuals Front. Cell. Infect. Microbiol. 5:25. doi: 10.3389/fcimb.2015.00025
Received: 27 June 2014; Accepted: 04 March 2015;
Published: 26 March 2015.
Edited by:
Alex Mira, Center for Advanced Research in Public Health, SpainReviewed by:
Alex Mira, Center for Advanced Research in Public Health, SpainAlfonso Benitez-Paez, Bioinformatics Analysis Group - GABi. Centro de Investigación y Desarrollo en Biotecnología, Colombia
Luis David Alcaraz, Universidad Nacional Autónoma de México, Mexico
Copyright © 2015 Do, Sheehy, Mulli, Hughes and Beighton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thuy Do, Department of Oral Biology, School of Dentistry, University of Leeds, Wellcome Trust Brenner Building, Level 7, St James' Hospital, Beckett Street, Leeds LS9 7TF, UKbi50LmRvQGxlZWRzLmFjLnVr