 Shaun Steele
Shaun Steele Jason Brunton
Jason Brunton Thomas Kawula
Thomas Kawula- Department of Microbiology and Immunology, School of Medicine, University of North Carolina, Chapel Hill, NC, USA
Following entry into host cells intracellular pathogens must simultaneously evade innate host defense mechanisms and acquire energy and anabolic substrates from the nutrient-limited intracellular environment. Most of the potential intracellular nutrient sources are stored within complex macromolecules that are not immediately accessible by intracellular pathogens. To obtain nutrients for proliferation, intracellular pathogens must compete with the host cell for newly-imported simple nutrients or degrade host nutrient storage structures into their constituent components (fatty acids, carbohydrates, and amino acids). It is becoming increasingly evident that intracellular pathogens have evolved a wide variety of strategies to accomplish this task. One recurrent microbial strategy is to exploit host degradative processes that break down host macromolecules into simple nutrients that the microbe can use. Herein we focus on how a subset of bacterial, viral, and eukaryotic pathogens leverage the host process of autophagy to acquire nutrients that support their growth within infected cells.
Introduction
Food and reproduction are basic necessities for life. Intracellular pathogens infect host cells and are dependent on them for nutrients to propagate. While there is an abundance of food inside host cells, molecules are mostly sequestered in complex compounds or structures such as glycogen, lipid droplets, and proteins; forms that are not readily usable by microbial intruders. Therefore, simply gaining access to the interior of a host cell and avoiding potent innate antimicrobial host defenses is not sufficient to guarantee successful occupation and growth. Once inside, pathogens must either stimulate host cell import of metabolites or degrade intracellular storage molecules into compounds that can be transported and metabolized. There are multiple mechanisms by which intracellular pathogens accomplish this goal. For example, Mycobacterium tuberculosis encodes proteins to degrade host-derived lipids, such as cholesterol, for a carbon source (Griffin et al., 2012). Pathogens can also take advantage of host signaling pathways to acquire nutrients. Both Brucella abortus and Salmonella enterica thrive on the increased glucose that is imported upon activation of various peroxisome proliferation-activated receptors (PPARs) in alternatively activated monocytes (Eisele et al., 2013; Xavier et al., 2013). Recently, several pathogens have been demonstrated to exploit host cell macroautophagy for nutrients. Autophagy is a critical mechanism that host cells use to increase nutrient availability when stressed. Since infection should exert a wide range of stresses on cells, it is not surprising that a diverse range of microbes have evolved strategies to extract the products of autophagy.
Autophagy is a highly conserved, multi-faceted eukaryotic process that maintains cellular homeostasis by degrading cytosolic material. Autophagy was noted as early as 1957 during the characterization of kidney cells by transmission electron microscopy (Clark, 1957, Deter and de Duve, 1967). In 1964, autophagy was identified as a mechanism to degrade cytosolic components and mitochondria under starvation conditions (Malkoff and Buetow, 1964). Since then, autophagy has been linked to a wide range of functions including antigen presentation through major histocompatibility complex II (MHC-II), unconventional secretion of inflammatory mediators, and cell viability (Ogata et al., 2006; Munz, 2009; Dupont et al., 2011).
Autophagy is divided into several subsets based on the components being degraded. Bulk autophagy refers to non-specific cytoplasmic turnover while selective autophagy refers to autophagic degradation of specific structures. There are several distinct types of selective autophagy, which target specific cellular components such as mitochondria (mitophagy) or lipids (lipophagy). During infections, intracellular microbes are recognized, targeted, and degraded through a form of selective autophagy termed xenophagy. Although xenophagy is efficient at destroying microbes that enter the cytosol, intracellular pathogens have developed numerous evasion strategies to avoid destruction by xenophagy, including the degradation or inhibition of autophagy components, camouflaging itself in host proteins, or blocking autophagosome maturation (Table 1).
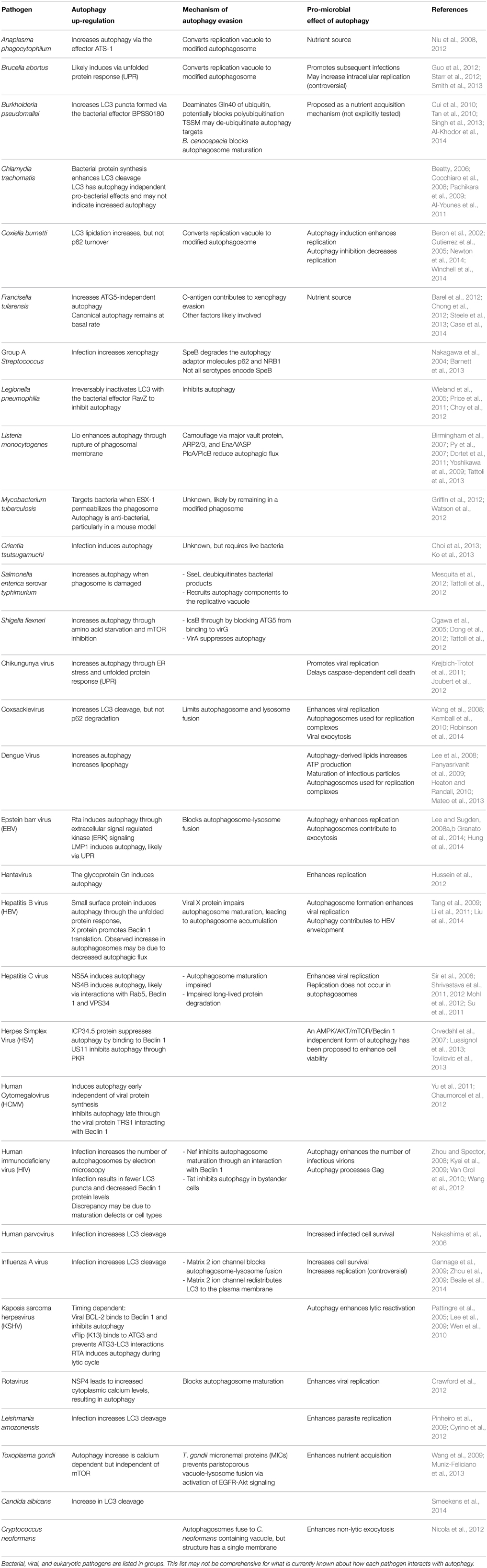
Table 1. A summary of the mechanisms employed by select pathogens to induce autophagy, evade destruction through xenophagy, and pro-microbial benefits of autophagy induction.
Several pathogens that evade xenophagic killing have incorporated autophagy into their intracellular life cycle. These microbes exploit autophagy to sustain host cell viability, increase nutrient production, and/or for non-lytic exocytosis (Table 1). Viruses also use autophagy or autophagy components for viral assembly and maturation (Table 1). In this review, we will focus on how pathogens avoid destruction by xenophagy while harvesting nutrients from autophagic degradation of host components.
What is Autophagy?
Autophagy is a constitutive process that degrades long lived-proteins, organelles, and aggregates. In mouse embryonic fibroblasts (MEFs), a common cell line used for autophagy research, the basal rate of autophagy is approximately 1–2% of the cytosolic volume of the cell (Nishida et al., 2009). A wide range of stimuli increase autophagy over the basal rate. Two major autophagy signaling nodes are the activation of the energy sensing protein AMP-activated protein kinase (AMPK) and inhibition of the mammalian target of rapamycin (mTOR). AMPK is activated in response to a low ATP to AMP ratio, such as during glucose deprivation (Carling et al., 1987; Sato et al., 1993). AMPK induces autophagy directly by phosphorylating ULK1 or indirectly through mTOR inhibition (Kim et al., 2011). mTOR is inhibited by several other stress factors besides AMPK, such as amino acid starvation or hypoxia (Jung et al., 2010).
For an in-depth review of canonical autophagy signaling, see the following reviews (Jewell et al., 2013; Galluzzi et al., 2014; Noda and Inagaki, 2015). Briefly, AMPK activation or mTOR inhibition result in ULK1 activation (Kim et al., 2011). ULK1 phosphorylates Beclin 1 and activates the kinase VPS34. ULK1 and Beclin 1–VPS34 associated complexes localize to an open, double membrane structure termed the phagophore. The phagophore is elongated by the ATG5-ATG12-ATG16L complex (Walczak and Martens, 2013). The phagophore expands to engulf cytoplasmic material while forming a double membrane vacuole termed the autophagosome. Unprocessed LC3 is cytosolic (LC3-I), but LC3 is cleaved, lipidated with phosphatidylethanolamine (LC3-II), and embedded into the autophagic membrane upon autophagy initiation (Kabeya et al., 2000). Molecules targeted for autophagic degradation are polyubiquitinated and adaptor proteins including p62, OPTINEURIN, or NDP52 bind to both LC3-II and ubiquitinated molecules (Thurston et al., 2009; Zheng et al., 2009; Wild et al., 2011). The autophagosome then fuses with a lysosome to become an autolysosome. The adaptor molecule NDP52 was recently shown to also regulate the fusion of a subset of bacteria containing autophagosomes to lysosomes by mediating binding between LC3 (which is embedded in the autophagosome), Myosin VI (a myosin motor protein that moves toward the minus end of actin) and Tom-1 (which associates with lysosomes) (Verlhac et al., 2015). The contents within the autolysosome are degraded into their components and exported to the cytosol.
Canonical autophagy is the best characterized form of autophagy, but there are several forms of non-canonical autophagy. These non-canonical forms also generate double membrane, degradative vacuoles with the same basic maturation process (phagophore to autophagosome to autolysosome). However, these non-canonical autophagosomes are initiated through different mechanisms and do not use all of the proteins or protein complexes required for canonical autophagy. One recurrent form of non-canonical autophagy associated with pathogenesis is ATG5-independent autophagy. ATG5-independent autophagy uses some of the same machinery as canonical autophagy, such as ULK1 and Beclin 1, but does not require ATG5, ATG7, or LC3 (Nishida et al., 2009). LC3 cleavage and ATG5 knockouts are commonly used to assay for xenophagy; pathogens may preferentially induce ATG5-independent autophagy to avoid xenophagy.
ATG5-independent autophagy is induced by starvation and correlates with mTOR inhibition, but mTOR inhibition alone is not sufficient to induce this form of autophagy (Nishida et al., 2009; Steele et al., 2013). ATG5-independent autophagy is critical for Francisella tularensis replication and the ability of B. abortus to infect neighboring cells (Starr et al., 2012; Steele et al., 2013). Mycobacterium marinum enters autophagosome-like vacuoles in an ATG5-independent manner although the function of this vacuole is unknown (Collins et al., 2009). It is unclear how ATG5-independent autophagy is preferentially induced over canonical autophagy during these infections.
Pathogens Induce Xenophagy
During infections, intracellular microbes are recognized, targeted, and degraded through a form of selective autophagy termed xenophagy. Inhibition of mTOR induces xenophagy in response to extracellular or phagocytosed microbes through Toll-like recepetors (TLRs). TLRs recognize conserved microbial factors and initiate several anti-microbial processes, including xenophagy via Myd88 and Trif interacting with Beclin 1 (Delgado et al., 2008; Shi and Kehrl, 2008). Cell to cell signaling can also induce autophagy. Interferon gamma (IFN-γ) activates autophagy through IRGM1 in human cells while CD40 ligation stimulates autophagy through PI3K and Rab7; priming cells to resist microbes (Andrade et al., 2006; Singh et al., 2006).
After phagocytosis, many pathogens escape the phagosome to replicate within the cytosol. The host cell mounts a xenophagic response to the membrane damage that occurs during phagosomal escape (Tattoli et al., 2012). Once microbes reach the cytosol, they can be targeted for xenophagy through immune surveillance or by causing cell stress. Several molecules, such as Nod-1 and Nod-2, identify microbial components within the cytosol to target microbes for xenophagy. Nod-1 and Nod-2 induce xenophagy and microbial antigen processing in response to bacterial peptidoglycan (Cooney et al., 2010; Travassos et al., 2010). Microbes also induce xenophagy through a number of cell stress mechanisms. B. abortus secreted TcpB induces endoplasmic reticulum stress via the unfolded protein response (UPR) pathway while Toxoplasma gondii increases intracellular calcium levels to induce autophagy (Qin et al., 2008; Wang et al., 2009; Smith et al., 2013).
Lastly, xenophagy can also be directly induced by microbial proteins (Table 1). For example, Shigella flexneri exported VirG polymerizes actin to propel the bacteria through the cytosol (Makino et al., 1986). ATG5 binds to VirG and initiates autophagosome formation without upstream autophagy signaling (Ogawa et al., 2005). However, S. flexneri also produces IcsB which blocks ATG5 from binding to VirG, thus inhibiting xenophagy (Ogawa et al., 2005). Likewise, the viral protein NS4B in Hepatitis C virus (HCV) induces autophagy by interacting with a Rab5/Beclin 1/VPS34 complex (Su et al., 2011).
Pathogens have Evolved Complex Xenophagy Evasion Mechanisms
Xenophagy is typically extremely effective at destroying microbes that enter the cytosol. For example, some serotypes of Group A Streptococcus (GAS) invade host cells, escape into the cytosol, and are then destroyed by xenophagy (Nakagawa et al., 2004; Joubert et al., 2009). Xenophagy effectively blocks these serotypes from using the cytosol as a replicative niche. To defend themselves, most intracellular pathogens have evolved mechanisms to either inhibit or evade xenophagy (Table 1). Some GAS serotypes encode SpeB, which degrades the xenophagy adaptor proteins p62 and NRB1 (Barnett et al., 2013). GAS serotypes that are normally destroyed by xenophagy can be functionally complemented for xenophagy evasion and intracellular replication by expressing SpeB (Barnett et al., 2013).
To inhibit autophagy, pathogens frequently impair the function of xenophagy machinery. The RavZ protein secreted by Legionella pneumophilia inactivates LC3, effectively blocking autophagy in infected cells (Choy et al., 2012). Human Cytomegalovirus (HCMV), Herpes Simplex virus, and Kaposis sarcoma herpesvirus inactivate Beclin 1 to inhibit autophagy at specific points in their life cycle (Table 1). Many viruses, such as Coxsackievirus, Hepatitis B virus, and HIV, inhibit autophagosome-lysosome fusion, functionally inhibiting xenophagy (Table 1). The exact mechanism by which these viruses block autophagosome maturation is unknown, but many different RNA viruses encode proteins that interact with LC3, p62, NDP52, or NRB1 (Gregoire et al., 2011; Judith et al., 2013). These proteins have several roles in xenophagy, but microbes may alter autophagosome maturation by manipulating these proteins (Verlhac et al., 2015).
A few pathogens evade xenophagy without inhibiting autophagy. Listeria monocytogenes camouflages itself by binding to the host proteins ARP2/3, major vault protein (MVP), and ena/VASP (Yoshikawa et al., 2009; Dortet et al., 2011). Many bacterial and eukaryotic pathogens modify phagosomes and are likely hidden from xenophagy targeting by remaining within a modified vacuole. M. tuberculosis and S. enterica typically reside in modified phagosomes but bacteria that disrupt the phagosomal membrane are rapidly destroyed by xenophagy (Tattoli et al., 2012; Watson et al., 2012). Vacuolar M. tuberculosis and T. gondii are degraded via autophagy when autophagy is stimulated by external sources, such as CD40 ligation or IFN-γ (Andrade et al., 2006; Ling et al., 2006; Singh et al., 2006). Certain pathogens evade xenophagy by altering or destroying the components that target the microbes for degradation. S. enterica de-ubiquitinates aggregates with the effector protein SseL to prevent the aggregates from being degraded via autophagy (Mesquita et al., 2012). Likewise, B. pseudomallei encodes the de-ubiquitinase TssM which blocks several innate immune signals including the NF-kB and type 1 IFN pathways and has been proposed as a potential autophagy evasion mechanism (Tan et al., 2010; Gong et al., 2011). A few other cytosolic pathogens, such as Orientia tsutsugamuchi and F. tularensis, induce autophagy but the mechanisms of xenophagy evasion are not clear (Choi et al., 2013; Ko et al., 2013; Steele et al., 2013).
Pathogens Harvest Autophagy Derived Nutrients for Replication
Intracellular microbes acquire nutrients from a range of sources, but generally rely on macromolecule degradation or nutrient import. Most basic nutrients within cells (amino acids, fatty acids, and carbohydrates) are incorporated into macromolecules (proteins, lipid droplets, and glycogen, respectively). In uninfected cells, these macromolecules are primarily degraded by autophagy to increase the amount of basic nutrients so the cell can build new structures. Thus, autophagy can increase the intracellular pool of nutrients that pathogens can access. Microbes can divert the nutrient by-products of autophagy toward microbial replication rather than for use by the cell. Dengue virus, F. tularensis, Anaplasma phagocytophilum, and T. gondii all induce autophagy, evade autophagic degradation, and harvest the autophagy derived nutrients for replication through different mechanisms (Wang et al., 2009; Heaton and Randall, 2010; Niu et al., 2012; Steele et al., 2013). Additionally, B. pseudomallei, Coxiella burnetii, and Leishmania amozonensis have impaired replication when autophagy is inhibited and nutrient acquisition has been implicated as a potential explanation for this phenotype.
Dengue virus requires the degradation of lipid droplets via autophagy for optimal replication (Heaton and Randall, 2010). Dengue virus infections increase cellular levels of autophagy and the resulting autophagosomes form around and degrade lipid droplets. The triglycerides derived from the lipid droplets are catabolized via mitochondrial β-oxidation, generating ATP. Thus, autophagy produces energy for the cell to indirectly enhance viral replication (Heaton and Randall, 2010). In addition to energy production, Dengue virus modifies autophagosomes or amphisomes to form a replicative niche (Lee et al., 2008; Panyasrivanit et al., 2009; Mateo et al., 2013). Rather than being degraded through xenophagy, autophagy contributes to the maturation of infectious particles (Mateo et al., 2013).
F. tularensis replicates in the cytosol of infected cells and induces an ATG5-independent, non-canonical form of autophagy. F. tularensis harvests amino acids from ATG5-independent autophagy for optimal intracellular replication. The amino acids are used for protein synthesis and are also metabolized as a major carbon source. F. tularensis bacteria are frequently adjacent to autophagosomes (Steele et al., 2013), indicating that F. tularensis is in the optimal physical location to compete with the host for autophagy derived nutrients. Although F. tularensis bacteria are frequently adjacent to autophagosomes, live bacteria are rarely degraded by xenophagy (Chong et al., 2012; Steele et al., 2013). O-antigen contributes to F. tularensis xenophagy evasion, but other effectors are also likely to be involved (Case et al., 2014).
A. phagocytophilum replicates in a vacuolar compartment and recruits autophagosomes directly to its replicative inclusions. A. phagocytophilum induces autophagy with the type IV secretion system (T4SS) effector Ats-1. Ats-1 binds to Beclin 1 and induces autophagosome nucleation directly rather than signaling through mTOR. Ats-1 induced autophagosomes localize with the inclusion membrane, suggesting that autophagosomes fuse with the inclusion body so that the bacteria can acquire the by-products of autophagic degradation. Inhibition of autophagy decreases A. phagocytophilum replication due to amino acid deficiency (Niu et al., 2008, 2012). Likewise, C. burnetti induces autophagy to enhance replication (Gutierrez et al., 2005; Newton et al., 2014; Winchell et al., 2014). C burnetti enters cells upon phagocytosis and modifies the phagosome to form a C. burnetti containing vacuoles (CCV). CCVs promiscuously fuse with other CCVs, endosomes and autophagosomes using the T4SS effector Cig2 (Newton et al., 2014; Winchell et al., 2014). When autophagy is impaired, CCVs do not fuse with one another and there is a severe replication defect (Gutierrez et al., 2005; Newton et al., 2014). The autophagosomes recruited to the CCV contain LC3, p62, and LAMP-1, suggesting that the autophagosomes that are recruited to CCVs have already fused with lyosomes (Klionsky et al., 2012; Winchell et al., 2014). Since artificially enhancing autophagy further increases C. burnetti replication, the fusion of autophagosomes with the CCV has been postulated as a nutrient and membrane acquisition mechanism (Gutierrez et al., 2005; Winchell et al., 2014).
T. gondii induces autophagy in infected host cells in a calcium dependent, mTOR independent manner (Wang et al., 2009). Inhibiting autophagy decreases T. gondii replication and parasite replication is rescued by supplementing with additional amino acids (Wang et al., 2009). Unlike its bacterial counterparts, fusion of T. gondii containing parasitophorous vacuoles (PVs) with autophagosomes leads to parasite destruction (Muniz-Feliciano et al., 2013). T. gondii activates EGFR and AKT to inhibit PV-autophagosome fusion with EGF-MICs, primarily MIC3 and MIC6 (Muniz-Feliciano et al., 2013).
Exploiting autophagy for nutrients is a recurrent theme in the pathogenesis of a diverse range of microbes. Several other microbes have enhanced replication when autophagy is induced and impaired intracellular replication when autophagy is inhibited, such as Chikungunya virus, B. pseudomallei and L. amazonensis (Krejbich-Trotot et al., 2011; Cyrino et al., 2012; Singh et al., 2013). B. pseudomallei encodes the protein BPSS0180, which induces autophagy and is required for optimal intracellular replication (Singh et al., 2013). Similarly, L. amazonensis induces autophagy and has a replication defect when cells are deficient for autophagy (Cyrino et al., 2012). The role of autophagy in enhancing replication of these pathogens is unknown, but nutrient acquisition is a likely explanation for these phenotypes.
Conclusions and Perspectives
Autophagy has been linked to both nutrient acquisition and pathogen destruction for decades and it has recently become clear that a diverse range of pathogens harvest autophagy derived products to enhance replication (Wang et al., 2009; Heaton and Randall, 2010; Niu et al., 2012; Steele et al., 2013). Autophagy derived nutrient acquisition is relatively straightforward in bacterial pathogens. F. tularensis is adjacent to autophagosomes while C. burnetii and A. phagocytophilum recruit autophagosomes to their replicative vacuoles (Gutierrez et al., 2005; Niu et al., 2012; Steele et al., 2013; Newton et al., 2014). These pathogens likely harvest the autophagy derived nutrients immediately after macromolecules are degraded. T. gondii acquires nutrients via host cell autophagy, but it is unclear how the parasites out-compete the host for autophagy by-products (Wang et al., 2009).
In contrast, the role of autophagy in viral nutrient acquisition is less straightforward. For example, Dengue virus does not directly incorporate the autophagy by-products into structural components. Instead, autophagy increases the amount of intracellular ATP, providing energy to the cell to support viral replication (Heaton and Randall, 2010). Additionally, many viruses benefit from other facets of autophagy, such as enhanced cell viability or the maturation of infectious particles, further confounding the role of autophagy in viral nutrient acquisition (Table 1). Viruses that induce autophagy are likely to benefit from the increase in intracellular nutrients, although this benefit may be indirect, as with Dengue virus. It is important to note that autophagy still degrades host macromolecules in cells infected with microbes that block autophagy, indicating that autophagy derived nutrients are available to microbes that can outcompete the host (Sir et al., 2008). Further investigation is needed to determine if autophagy derived nutrients are incorporated into viral macromolecules.
Autophagy is one of many mechanisms that intracellular pathogens usurp to acquire nutrients. Intracellular pathogens must acquire nutrients from the host cell to survive and propagate. These pathogens can acquire nutrients either by altering eukaryotic cell metabolism to increase nutrient import or by degrading macromolecules within the host through processes such as autophagy. For example, HCMV increases the expression of GLUT4 to increase glucose import for replication while L. pneumophila co-opts proteosomal degradation for amino acids (Price et al., 2011; Yu et al., 2011). It is likely that intracellular pathogens exploit multiple, additive mechanisms for nutrient acquisition. F. tularensis requires autophagy and L. pneumophila requires proteosomal degradation for optimal replication, but both of these pathogens also up-regulate the expression of the host amino acid transporter SLC1A5 in macrophages which further enhances replication by increasing amino acid transport into the infected cell (Wieland et al., 2005; Barel et al., 2012). Inhibiting macromolecule degradation or import for these bacteria impairs, but does not block, replication, suggesting that these different nutrient acquisition strategies are additive. This is likely a common theme for microbial nutrient acquisition.
Research into how pathogens acquire nutrients is still in its infancy, particularly in how microbes acquire nutrients via autophagy. ATG22, Avt3, and Avt4 contribute to amino acid efflux from autophagosomes in yeast, but the mechanisms for nutrient efflux from mammalian cell autophagosomes is not well-defined (Yang et al., 2006). Other critical questions that remain largely unanswered are which nutrients microbes acquire from autophagy and the degree to which microbes rely on autophagy for nutrients. Either glucose or amino acids rescues T. gondii replication in autophagy deficient cells whereas F. tularensis and A. phagocytophilum primarily acquire amino acids from autophagy, but may also harvest other autophagy derived nutrients such as lipids (Wang et al., 2009; Niu et al., 2012; Steele et al., 2013). Even microbes that do not induce autophagy or actively inhibit autophagy can compete with the host for autophagy derived nutrients. Defining host cell metabolic processes that specific pathogens depend on to acquire useable nutrients could reveal new infection treatment strategies based on interfering with host processes to augment or supplant more traditional antimicrobial therapies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Al-Khodor, S., Marshall-Batty, K., Nair, V., Ding, L., Greenberg, D. E., and Fraser, I. D. (2014). Burkholderia cenocepacia J2315 escapes to the cytosol and actively subverts autophagy in human macrophages. Cell Microbiol. 16, 378–395. doi: 10.1111/cmi.12223
Al-Younes, H. M., Al-Zeer, M. A., Khalil, H., Gussmann, J., Karlas, A., Machuy, N., et al. (2011). Autophagy-independent function of MAP-LC3 during intracellular propagation of Chlamydia trachomatis. Autophagy 7, 814–828. doi: 10.4161/auto.7.8.15597
Andrade, R. M., Wessendarp, M., Gubbels, M. J., Striepen, B., and Subauste, C. S. (2006). CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J. Clin. Invest. 116, 2366–2377. doi: 10.1172/JCI28796
Barel, M., Meibom, K., Dubail, I., Botella, J., and Charbit, A. (2012). Francisella tularensis regulates the expression of the amino acid transporter SLC1A5 in infected THP-1 human monocytes. Cell Microbiol. 14, 1769–1783. doi: 10.1111/j.1462-5822.2012.01837.x
Barnett, T. C., Liebl, D., Seymour, L. M., Gillen, C. M., Lim, J. Y., Larock, C. N., et al. (2013). The globally disseminated M1T1 clone of group A Streptococcus evades autophagy for intracellular replication. Cell Host Microbe 14, 675–682. doi: 10.1016/j.chom.2013.11.003
Beale, R., Wise, H., Stuart, A., Ravenhill, B. J., Digard, P., and Randow, F. (2014). A LC3-interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe 15, 239–247. doi: 10.1016/j.chom.2014.01.006
Beatty, W. L. (2006). Trafficking from CD63-positive late endocytic multivesicular bodies is essential for intracellular development of Chlamydia trachomatis. J. Cell Sci. 119, 350–359. doi: 10.1242/jcs.02733
Beron, W., Gutierrez, M. G., Rabinovitch, M., and Colombo, M. I. (2002). Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect Immun. 70, 5816–5821. doi: 10.1128/IAI.70.10.5816-5821.2002
Birmingham, C. L., Canadien, V., Gouin, E., Troy, E. B., Yoshimori, T., Cossart, P., et al. (2007). Listeria monocytogenes evades killing by autophagy during colonization of host cells. Autophagy 3, 442–451. doi: 10.4161/auto.4450
Carling, D., Zammit, V. A., and Hardie, D. G. (1987). A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 223, 217–222. doi: 10.1016/0014-5793(87)80292-2
Case, E. D., Chong, A., Wehrly, T. D., Hansen, B., Child, R., Hwang, S., et al. (2014). The Francisella O-antigen mediates survival in the macrophage cytosol via autophagy avoidance. Cell Microbiol. 16, 862–877. doi: 10.1111/cmi.12246
Chaumorcel, M., Lussignol, M., Mouna, L., Cavignac, Y., Fahie, K., Cotte-Laffitte, J., et al. (2012). The human cytomegalovirus protein TRS1 inhibits autophagy via its interaction with Beclin 1. J. Virol. 86, 2571–2584. doi: 10.1128/JVI.05746-11
Choi, J. H., Cheong, T. C., Ha, N. Y., Ko, Y., Cho, C. H., Jeon, J. H., et al. (2013). Orientia tsutsugamushi subverts dendritic cell functions by escaping from autophagy and impairing their migration. PLoS Negl. Trop. Dis. 7:e1981. doi: 10.1371/journal.pntd.0001981
Chong, A., Wehrly, T. D., Child, R., Hansen, B., Hwang, S., Virgin, H. W., et al. (2012). Cytosolic clearance of replication-deficient mutants reveals Francisella tularensis interactions with the autophagic pathway. Autophagy 8, 1342–1356. doi: 10.4161/auto.20808
Choy, A., Dancourt, J., Mugo, B., O'Connor, T. J., Isberg, R. R., Melia, T. J., et al. (2012). The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 338, 1072–1076. doi: 10.1126/science.1227026
Clark, S. L. Jr. (1957). Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J. Biophys. Biochem. Cytol. 3, 349–362. doi: 10.1083/jcb.3.3.349
Cocchiaro, J. L., Kumar, Y., Fischer, E. R., Hackstadt, T., and Valdivia, R. H. (2008). Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. Proc. Natl. Acad. Sci. U.S.A. 105, 9379–9384. doi: 10.1073/pnas.0712241105
Collins, C. A., De Maziere, A., van Dijk, S., Carlsson, F., Klumperman, J., and Brown, E. J. (2009). Atg5-independent sequestration of ubiquitinated mycobacteria. PLoS Pathog. 5:e1000430. doi: 10.1371/journal.ppat.1000430
Cooney, R., Baker, J., Brain, O., Danis, B., Pichulik, T., Allan, P., et al. (2010). NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 16, 90–97. doi: 10.1038/nm.2069
Crawford, S. E., Hyser, J. M., Utama, B., and Estes, M. K. (2012). Autophagy hijacked through viroporin-activated calcium/calmodulin-dependent kinase kinase-beta signaling is required for rotavirus replication. Proc. Natl. Acad. Sci. U.S.A. 109, E3405–E3413. doi: 10.1073/pnas.1216539109
Cui, J., Yao, Q., Li, S., Ding, X., Lu, Q., Mao, H., et al. (2010). Glutamine deamidation and dysfunction of ubiquitin/NEDD8 induced by a bacterial effector family. Science 329, 1215–1218. doi: 10.1126/science.1193844
Cyrino, L. T., Araujo, A. P., Joazeiro, P. P., Vicente, C. P., and Giorgio, S. (2012). In vivo and in vitro Leishmania amazonensis infection induces autophagy in macrophages. Tissue Cell 44, 401–408. doi: 10.1016/j.tice.2012.08.003
Delgado, M. A., Elmaoued, R. A., Davis, A. S., Kyei, G., and Deretic, V. (2008). Toll-like receptors control autophagy. EMBO J. 27, 1110–1121. doi: 10.1038/emboj.2008.31
Deter, R. L., and de Duve, C. (1967). Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J. Cell Biol. 33, 437–449 doi: 10.1083/jcb.33.2.437
Dong, N., Zhu, Y., Lu, Q., Hu, L., Zheng, Y., and Shao, F. (2012). Structurally distinct bacterial TBC-like GAPs link Arf GTPase to Rab1 inactivation to counteract host defenses. Cell 150, 1029–1041. doi: 10.1016/j.cell.2012.06.050
Dortet, L., Mostowy, S., Samba-Louaka, A., Gouin, E., Nahori, M. A., Wiemer, E. A., et al. (2011). Recruitment of the major vault protein by InlK: a Listeria monocytogenes strategy to avoid autophagy. PLoS Pathog. 7:e1002168. doi: 10.1371/journal.ppat.1002168
Dupont, N., Jiang, S., Pilli, M., Ornatowski, W., Bhattacharya, D., and Deretic, V. (2011). Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. EMBO J. 30, 4701–4711. doi: 10.1038/emboj.2011.398
Eisele, N. A., Ruby, T., Jacobson, A., Manzanillo, P. S., Cox, J. S., Lam, L., et al. (2013). Salmonella require the fatty acid regulator PPARÎ' for the establishment of a metabolic environment essential for long term persistence. Cell Host Microbe 14, 171–182. doi: 10.1016/j.chom.2013.07.010
Galluzzi, L., Pietrocola, F., Levine, B., and Kroemer, G. (2014). Metabolic control of autophagy. Cell 159, 1263–1276. doi: 10.1016/j.cell.2014.11.006
Gannage, M., Dormann, D., Albrecht, R., Dengjel, J., Torossi, T., Ramer, P. C., et al. (2009). Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6, 367–380. doi: 10.1016/j.chom.2009.09.005
Gong, L., Cullinane, M., Treerat, P., Ramm, G., Prescott, M., Adler, B., et al. (2011). The Burkholderia pseudomallei Type III secretion system and BopA are required for evasion of LC3-associated phagocytosis. PLoS ONE 6:e17852. doi: 10.1371/journal.pone.0017852
Granato, M., Santarelli, R., Farina, A., Gonnella, R., Lotti, L. V., Faggioni, A., et al. (2014). Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. J. Virol. 88, 12715–12726. doi: 10.1128/JVI.02199-14
Gregoire, I. P., Richetta, C., Meyniel-Schicklin, L., Borel, S., Pradezynski, F., Diaz, O., et al. (2011). IRGM is a common target of RNA viruses that subvert the autophagy network. PLoS Pathog. 7:e1002422. doi: 10.1371/journal.ppat.1002422
Griffin, J. E., Pandey, A. K., Gilmore, S. A., Mizrahi, V., McKinney, J. D., Bertozzi, C. R., et al. (2012). Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem. Biol. 19, 218–227. doi: 10.1016/j.chembiol.2011.12.016
Guo, F., Zhang, H., Chen, C., Hu, S., Wang, Y., Qiao, J., et al. (2012). Autophagy favors Brucella melitensis survival in infected macrophages. Cell Mol. Biol. Lett. 17, 249–257. doi: 10.2478/s11658-012-0009-4
Gutierrez, M. G., Vazquez, C. L., Munafo, D. B., Zoppino, F. C., Beron, W., Rabinovitch, M., et al. (2005). Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell Microbiol. 7, 981–993. doi: 10.1111/j.1462-5822.2005.00527.x
Heaton, N. S., and Randall, G. (2010). Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe 8, 422–432. doi: 10.1016/j.chom.2010.10.006
Hung, C. H., Chen, L. W., Wang, W. H., Chang, P. J., Chiu, Y. F., Hung, C. C., et al. (2014). Regulation of autophagic activation by Rta of Epstein-Barr virus via the extracellular signal-regulated kinase pathway. J. Virol. 88, 12133–12145. doi: 10.1128/JVI.02033-14
Hussein, I. T., Cheng, E., Ganaie, S. S., Werle, M. J., Sheema, S., Haque, A., et al. (2012). Autophagic clearance of Sin Nombre hantavirus glycoprotein Gn promotes virus replication in cells. J. Virol. 86, 7520–7529. doi: 10.1128/JVI.07204-11
Jewell, J. L., Russell, R. C., and Guan, K. L. (2013). Amino acid signalling upstream of mTOR, Nat. Rev. Mol. Cell Biol. 14, 133–139. doi: 10.1038/nrm3522
Joubert, P. E., Meiffren, G., Gregoire, I. P., Pontini, G., Richetta, C., Flacher, M., et al. (2009). Autophagy induction by the pathogen receptor CD46. Cell Host Microbe 6, 354–366. doi: 10.1016/j.chom.2009.09.006
Joubert, P. E., Werneke, S. W., de la Calle, C., Guivel-Benhassine, F., Giodini, A., Peduto, L., et al. (2012). Chikungunya virus-induced autophagy delays caspase-dependent cell death. J. Exp. Med. 209, 1029–1047. doi: 10.1084/jem.20110996
Judith, D., Mostowy, S., Bourai, M., Gangneux, N., Lelek, M., Lucas-Hourani, M., et al. (2013). Species-specific impact of the autophagy machinery on Chikungunya virus infection. EMBO Rep. 14, 534–544. doi: 10.1038/embor.2013.51
Jung, C. H., Ro, S. H., Cao, J., Otto, N. M., and Kim, D. H. (2010). mTOR regulation of autophagy. FEBS Lett. 584, 1287–1295. doi: 10.1016/j.febslet.2010.01.017
Kabeya, Y., Mizushima, N., Ueno, T., Yamamoto, A., Kirisako, T., Noda, T., et al. (2000). LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728. doi: 10.1093/emboj/19.21.5720
Kemball, C. C., Alirezaei, M., Flynn, C. T., Wood, M. R., Harkins, S., Kiosses, W. B., et al. (2010). Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J. Virol. 84, 12110–12124. doi: 10.1128/JVI.01417-10
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Klionsky, D. J., Abdalla, F. C., Abeliovich, H., Abraham, R. T., Acevedo-Arozena, A., Adeli, K., et al. (2012). Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544 doi: 10.4161/auto.19496
Ko, Y., Choi, J. H., Ha, N. Y., Kim, I. S., Cho, N. H., and Choi, M. S. (2013). Active escape of Orientia tsutsugamushi from cellular autophagy. Infect. Immun. 81, 552–559. doi: 10.1128/IAI.00861-12
Krejbich-Trotot, P., Gay, B., Li-Pat-Yuen, G., Hoarau, J. J., Jaffar-Bandjee, M. C., Briant, L., et al. (2011). Chikungunya triggers an autophagic process which promotes viral replication. Virol. J. 8:432 doi: 10.1186/1743-422X-8-432
Kyei, G. B., Dinkins, C., Davis, A. S., Roberts, E., Singh, S. B., Dong, C., et al. (2009). Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J. Cell Biol. 186, 255–268. doi: 10.1083/jcb.200903070
Lee, D. Y., and Sugden, B. (2008a). The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood 111, 2280–2289. doi: 10.1182/blood-2007-07-100032
Lee, D. Y., and Sugden, B. (2008b). The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 27, 2833–2842. doi: 10.1038/sj.onc.1210946
Lee, J. S., Li, Q., Lee, J. Y., Lee, S. H., Jeong, J. H., Lee, H. R., et al. (2009). FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 11, 1355–1362. doi: 10.1038/ncb1980
Lee, Y. R., Lei, H. Y., Liu, M. T., Wang, J. R., Chen, S. H., Jiang-Shieh, Y. F., et al. (2008). Autophagic machinery activated by dengue virus enhances virus replication. Virology 374, 240–248. doi: 10.1016/j.virol.2008.02.016
Li, J., Liu, Y., Wang, Z., Liu, K., Wang, Y., Liu, J., et al. (2011). Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. J. Virol. 85, 6319–6333. doi: 10.1128/JVI.02627-10
Ling, Y. M., Shaw, M. H., Ayala, C., Coppens, I., Taylor, G. A., Ferguson, D. J., et al. (2006). Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J. Exp. Med. 203, 2063–2071. doi: 10.1084/jem.20061318
Liu, B., Fang, M., Hu, Y., Huang, B., Li, N., Chang, C., et al. (2014). Hepatitis B virus X protein inhibits autophagic degradation by impairing lysosomal maturation. Autophagy 10, 416–430. doi: 10.4161/auto.27286
Lussignol, M., Queval, C., Bernet-Camard, M. F., Cotte-Laffitte, J., Beau, I., Codogno, P., et al. (2013). The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J. Virol. 87, 859–871. doi: 10.1128/JVI.01158-12
Makino, S., Sasakawa, C., Kamata, K., Kurata, T., and Yoshikawa, M. (1986). A genetic determinant required for continuous reinfection of adjacent cells on large plasmid in S. flexneri 2a. Cell 46, 551–555.
Malkoff, D. B., and Buetow, D. E. (1964). Ultrastructural changes during carbon starvation in euglena gracilis. Exp. Cell Res. 35, 58–68. doi: 10.1016/0014-4827(64)90071-0
Mateo, R., Nagamine, C. M., Spagnolo, J., Mendez, E., Rahe, M., Gale, M., et al. (2013). Inhibition of cellular autophagy deranges dengue virion maturation. J. Virol. 87, 1312–1321. doi: 10.1128/JVI.02177-12
Mesquita, F. S., Thomas, M., Sachse, M., Santos, A. J., Figueira, R., and Holden, D. W. (2012). The Salmonella deubiquitinase SseL inhibits selective autophagy of cytosolic aggregates. PLoS Pathog. 8:e1002743. doi: 10.1371/journal.ppat.1002743
Mohl, B. P., Tedbury, P. R., Griffin, S., and Harris, M. (2012). Hepatitis C virus-induced autophagy is independent of the unfolded protein response. J. Virol. 86, 10724–10732. doi: 10.1128/JVI.01667-12
Muniz-Feliciano, L., van Grol, J., Portillo, J. A., Liew, L., Liu, B., Carlin, C. R., et al. (2013). Toxoplasma gondii-induced activation of EGFR prevents autophagy protein-mediated killing of the parasite. PLoS Pathog. 9:e1003809. doi: 10.1371/journal.ppat.1003809
Munz, C. (2009). Enhancing immunity through autophagy. Annu. Rev. Immunol. 27, 423–449. doi: 10.1146/annurev.immunol.021908.132537
Nakagawa, I., Amano, A., Mizushima, N., Yamamoto, A., Yamaguchi, H., Kamimoto, T., et al. (2004). Autophagy defends cells against invading group A Streptococcus. Science 306, 1037–1040. doi: 10.1126/science.1103966
Nakashima, A., Tanaka, N., Tamai, K., Kyuuma, M., Ishikawa, Y., Sato, H., et al. (2006). Survival of parvovirus B19-infected cells by cellular autophagy. Virology 349, 254–263. doi: 10.1016/j.virol.2006.03.029
Newton, H. J., Kohler, L. J., McDonough, J. A., Temoche-Diaz, M., Crabill, E., Hartland, E. L., et al. (2014). A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog. 10:e1004286. doi: 10.1371/journal.ppat.1004286
Nicola, A. M., Albuquerque, P., Martinez, L. R., Dal-Rosso, R. A., Saylor, C., De Jesus, M., et al. (2012). Macrophage autophagy in immunity to Cryptococcus neoformans and Candida albicans. Infect. Immun. 80, 3065–3076. doi: 10.1128/IAI.00358-12
Nishida, Y., Arakawa, S., Fujitani, K., Yamaguchi, H., Mizuta, T., Kanaseki, T., et al. (2009). Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461, 654–658. doi: 10.1038/nature08455
Niu, H., Xiong, Q., Yamamoto, A., Hayashi-Nishino, M., and Rikihisa, Y. (2012). Autophagosomes induced by a bacterial Beclin 1 binding protein facilitate obligatory intracellular infection. Proc. Natl. Acad. Sci. U.S.A. 109, 20800–20807. doi: 10.1073/pnas.1218674109
Niu, H., Yamaguchi, M., and Rikihisa, Y. (2008). Subversion of cellular autophagy by Anaplasma phagocytophilum. Cell Microbiol. 10, 593–605. doi: 10.1111/j.1462-5822.2007.01068.x
Noda, N. N., and Inagaki, F. (2015). Mechanisms of autophagy. Annu. Rev. Biophys. doi: 10.1146/annurev-biophys-060414-034248. [Epub ahead of print].
Ogata, M., Hino, S., Saito, A., Morikawa, K., Kondo, S., Kanemoto, S., et al. (2006). Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 26, 9220–9231. doi: 10.1128/MCB.01453-06
Ogawa, M., Yoshimori, T., Suzuki, T., Sagara, H., Mizushima, N., and Sasakawa, C. (2005). Escape of intracellular Shigella from autophagy. Science 307, 727–731. doi: 10.1126/science.1106036
Orvedahl, A., Alexander, D., Talloczy, Z., Sun, Q., Wei, Y., Zhang, W., et al. (2007). HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1, 23–35. doi: 10.1016/j.chom.2006.12.001
Pachikara, N., Zhang, H., Pan, Z., Jin, S., and Fan, H. (2009). Productive Chlamydia trachomatis lymphogranuloma venereum 434 infection in cells with augmented or inactivated autophagic activities. FEMS Microbiol. Lett. 292, 240–249. doi: 10.1111/j.1574-6968.2009.01494.x
Panyasrivanit, M., Khakpoor, A., Wikan, N., and Smith, D. R. (2009). Co-localization of constituents of the dengue virus translation and replication machinery with amphisomes. J. Gen. Virol. 90, 448–456. doi: 10.1099/vir.0.005355-0
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., et al. (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927-939. doi: 10.1016/j.cell.2005.07.002
Pinheiro, R. O., Nunes, M. P., Pinheiro, C. S., D'Avila, H., Bozza, P. T., Takiya, C. M., et al. (2009). Induction of autophagy correlates with increased parasite load of Leishmania amazonensis in BALB/c but not C57BL/6 macrophages. Microbes Infect. 11, 181–190. doi: 10.1016/j.micinf.2008.11.006
Price, C. T., Al-Quadan, T., Santic, M., Rosenshine, I., and Abu Kwaik, Y. (2011). Host proteasomal degradation generates amino acids essential for intracellular bacterial growth. Science 334, 1553–1557. doi: 10.1126/science.1212868
Py, B. F., Lipinski, M. M., and Yuan, J. (2007). Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy 3, 117–125. doi: 10.4161/auto.3618
Qin, Q. M., Pei, J., Ancona, V., Shaw, B. D., Ficht, T. A., and de Figueiredo, P. (2008). RNAi screen of endoplasmic reticulum-associated host factors reveals a role for IRE1alpha in supporting Brucella replication. PLoS Pathog 4:e1000110. doi: 10.1371/journal.ppat.1000110
Robinson, S. M., Tsueng, G., Sin, J., Mangale, V., Rahawi, S., McIntyre, L. L., et al. (2014). Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 10:e1004045. doi: 10.1371/journal.ppat.1004045
Sato, R., Goldstein, J. L., and Brown, M. S. (1993). Replacement of serine-871 of hamster 3-hydroxy-3-methylglutaryl-CoA reductase prevents phosphorylation by AMP-activated kinase and blocks inhibition of sterol synthesis induced by ATP depletion. Proc. Natl. Acad. Sci. U.S.A. 90, 9261–9265. doi: 10.1073/pnas.90.20.9261
Shi, C. S., and Kehrl, J. H. (2008). MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 283, 33175–33182. doi: 10.1074/jbc.M804478200
Shrivastava, S., Bhanja Chowdhury, J., Steele, R., Ray, R., and Ray, R. B. (2012). Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J. Virol. 86, 8705–8712. doi: 10.1128/JVI.00616-12
Shrivastava, S., Raychoudhuri, A., Steele, R., Ray, R., and Ray, R. B. (2011). Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 53, 406–414. doi: 10.1002/hep.24073
Singh, A. P., Lai, S. C., Nandi, T., Chua, H. H., Ooi, W. F., Ong, C., et al. (2013). Evolutionary analysis of Burkholderia pseudomallei identifies putative novel virulence genes, including a microbial regulator of host cell autophagy. J. Bacteriol. 195, 5487–5498. doi: 10.1128/JB.00718-13
Singh, S. B., Davis, A. S., Taylor, G. A., and Deretic, V. (2006). Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313, 1438–1441. doi: 10.1126/science.1129577
Sir, D., Chen, W., Choi, J., Wakita, T., Yen, T. S. B., and Ou, J. J. (2008). Induction of Incomplete autophagic response by Hepatitis C Virus via the unfolded protein response. Hepatology 48, 1054–1061. doi: 10.1002/hep.22464
Smeekens, S. P., Malireddi, R. K., Plantinga, T. S., Buffen, K., Oosting, M., Joosten, L. A., et al. (2014). Autophagy is redundant for the host defense against systemic Candida albicans infections. Eur. J. Clin. Microbiol. Infect. Dis. 33, 711–722. doi: 10.1007/s10096-013-2002-x
Smith, J. A., Khan, M., Magnani, D. D., Harms, J. S., Durward, M., Radhakrishnan, G. K., et al. (2013). Brucella induces an unfolded protein response via TcpB that supports intracellular replication in macrophages. PLoS Pathog. 9:e1003785. doi: 10.1371/journal.ppat.1003785
Starr, T., Child, R., Wehrly, T. D., Hansen, B., Hwang, S., Lopez-Otin, C., et al. (2012). Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe 11, 33–45. doi: 10.1016/j.chom.2011.12.002
Steele, S., Brunton, J., Ziehr, B., Taft-Benz, S., Moorman, N., and Kawula, T. (2013). Francisella tularensis harvests nutrients derived via ATG5-independent autophagy to support intracellular growth. PLoS Pathog. 9:e1003562. doi: 10.1371/journal.ppat.1003562
Su, W. C., Chao, T. C., Huang, Y. L., Weng, S. C., Jeng, K. S., and Lai, M. M. (2011). Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 85, 10561–10571. doi: 10.1128/JVI.00173-11
Tan, K. S., Chen, Y., Lim, Y. C., Tan, G. Y., Liu, Y., Lim, Y. T., et al. (2010). Suppression of host innate immune response by Burkholderia pseudomallei through the virulence factor TssM. J. Immunol. 184, 5160–5171. doi: 10.4049/jimmunol.0902663
Tang, H., Da, L., Mao, Y., Li, Y., Li, D., Xu, Z., et al. (2009). Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology 49, 60–71. doi: 10.1002/hep.22581
Tattoli, I., Sorbara, M. T., Vuckovic, D., Ling, A., Soares, F., Carneiro, L. A., et al. (2012). Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11, 563–575. doi: 10.1016/j.chom.2012.04.012
Tattoli, I., Sorbara, M. T., Yang, C., Tooze, S. A., Philpott, D. J., and Girardin, S. E. (2013). Listeria phospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures. EMBO J. 32, 3066–3078. doi: 10.1038/emboj.2013.234
Thurston, T. L., Ryzhakov, G., Bloor, S., von Muhlinen, N., and Randow, F. (2009). The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 10, 1215–1221. doi: 10.1038/ni.1800
Tovilovic, G., Ristic, B., Siljic, M., Nikolic, V., Kravic-Stevovic, T., Dulovic, M., et al. (2013). mTOR-independent autophagy counteracts apoptosis in herpes simplex virus type 1-infected U251 glioma cells. Microbes Infect. 15, 615–624. doi: 10.1016/j.micinf.2013.04.012
Travassos, L. H., Carneiro, L. A., Ramjeet, M., Hussey, S., Kim, Y. G., Magalhaes, J. G., et al. (2010). Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 11, 55–62. doi: 10.1038/ni.1823
Van Grol, J., Subauste, C., Andrade, R. M., Fujinaga, K., Nelson, J., and Subauste, C. S. (2010). HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLoS ONE 5:e11733. doi: 10.1371/journal.pone.0011733
Verlhac, P., Gregoire, I. P., Azocar, O., Petkova, D. S., Baguet, J., Viret, C., et al. (2015). Autophagy Receptor NDP52 regulates pathogen-containing autophagosome maturation. Cell Host Microbe. 17, 515–525. doi: 10.1016/j.chom.2015.02.008
Walczak, M., and Martens, S. (2013). Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy 9, 424–425. doi: 10.4161/auto.22931
Wang, X., Gao, Y., Tan, J., Devadas, K., Ragupathy, V., Takeda, K., et al. (2012). HIV-1 and HIV-2 infections induce autophagy in Jurkat and CD4+ T cells. Cell Signal. 24, 1414–1419. doi: 10.1016/j.cellsig.2012.02.016
Wang, Y., Weiss, L. M., and Orlofsky, A. (2009). Host cell autophagy is induced by Toxoplasma gondii and contributes to parasite growth. J. Biol. Chem. 284, 1694–1701. doi: 10.1074/jbc.M807890200
Watson, R. O., Manzanillo, P. S., and Cox, J. S. (2012). Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150, 803–815. doi: 10.1016/j.cell.2012.06.040
Wen, H. J., Yang, Z., Zhou, Y., and Wood, C. (2010). Enhancement of autophagy during lytic replication by the kaposi's sarcoma-associated herpesvirus replication and transcription activator. J. Virol. 84, 7448–7458. doi: 10.1128/JVI.00024-10
Wieland, H., Ullrich, S., Lang, F., and Neumeister, B. (2005). Intracellular multiplication of Legionella pneumophila depends on host cell amino acid transporter SLC1A5. Mol. Microbiol. 55, 1528–1537. doi: 10.1111/j.1365-2958.2005.04490.x
Wild, P., Farhan, H., McEwan, D. G., Wagner, S., Rogov, V. V., Brady, N. R., et al. (2011). Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333, 228–233. doi: 10.1126/science.1205405
Winchell, C. G., Graham, J. G., Kurten, R. C., and Voth, D. E. (2014). Coxiella burnetii Type IV secretion-dependent recruitment of macrophage autophagosomes. Infect. Immun. 82, 2229–2238. doi: 10.1128/IAI.01236-13
Wong, J., Zhang, J., Si, X., Gao, G., Mao, I., McManus, B. M., et al. (2008). Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 82, 9143–9153. doi: 10.1128/JVI.00641-08
Xavier, M. N., Winter, M. G., Spees, A. M., den Hartigh, A. B., Nguyen, K., Roux, C. M., et al. (2013). PPARgamma-mediated increase in glucose availability sustains chronic Brucella abortus infection in alternatively activated macrophages. Cell Host Microbe 14, 159–170. doi: 10.1016/j.chom.2013.07.009
Yang, Z., Huang, J., Geng, J., Nair, U., and Klionsky, D. J. (2006). Atg22 recycles amino acids to link the degradative and recycling functions of autophagy. Mol. Biol. Cell 17, 5094–5104. doi: 10.1091/mbc.E06-06-0479
Yoshikawa, Y., Ogawa, M., Hain, T., Yoshida, M., Fukumatsu, M., Kim, M., et al. (2009). Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 11, 1233–1240. doi: 10.1038/ncb1967
Yu, Y., Maguire, T. G., and Alwine, J. C. (2011). Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J. Virol. 85, 1573–1580. doi: 10.1128/JVI.01967-10
Zheng, Y. T., Shahnazari, S., Brech, A., Lamark, T., Johansen, T., and Brumell, J. H. (2009). The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J. Immunol. 183, 5909–5916. doi: 10.4049/jimmunol.0900441
Zhou, D., and Spector, S. A. (2008). Human immunodeficiency virus type-1 infection inhibits autophagy. AIDS 22, 695–699. doi: 10.1097/QAD.0b013e3282f4a836
Keywords: autophagy, xenophagy, nutrient acquisition, intracellular pathogens, immune evasion
Citation: Steele S, Brunton J and Kawula T (2015) The role of autophagy in intracellular pathogen nutrient acquisition. Front. Cell. Infect. Microbiol. 5:51. doi: 10.3389/fcimb.2015.00051
Received: 30 April 2015; Accepted: 26 May 2015;
Published: 09 June 2015.
Edited by:
Alain Charbit, Paris Descartes University, FranceReviewed by:
Maria Isabel Colombo, Universidad Nacional de Cuyo-Consejo Nacional de Investigaciones Científicas y Técnicas, ArgentinaEric Ghigo, Centre National de la Recherche Scientifique, France
Copyright © 2015 Steele, Brunton and Kawula. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Thomas Kawula, Department of Microbiology and Immunology, School of Medicine, University of North Carolina, CB#7290, Chapel Hill, NC 27599, USA,a2F3dWxhQG1lZC51bmMuZWR1