 Hiroshi Ashida
Hiroshi Ashida Chihiro Sasakawa
Chihiro Sasakawa- 1Division of Bacterial Infection Biology, Institute of Medical Science, University of Tokyo, Tokyo, Japan
- 2Nippon Institute for Biological Science, Tokyo, Japan
- 3Medical Mycology Research Center, Chiba University, Chiba, Japan
Shigella spp. are highly adapted human pathogens that cause bacillary dysentery (shigellosis). Via the type III secretion system (T3SS), Shigella deliver a subset of virulence proteins (effectors) that are responsible for pathogenesis, with functions including pyroptosis, invasion of the epithelial cells, intracellular survival, and evasion of host immune responses. Intriguingly, T3SS effector activity and strategies are not unique to Shigella, but are shared by many other bacterial pathogens, including Salmonella, Yersinia, and enteropathogenic Escherichia coli (EPEC). Therefore, studying Shigella T3SS effectors will not only improve our understanding of bacterial infection systems, but also provide a molecular basis for developing live bacterial vaccines and antibacterial drugs. One of Shigella T3SS effectors, IpaH family proteins, which have E3 ubiquitin ligase activity and are widely conserved among other bacterial pathogens, are very relevant because they promote bacterial survival by triggering cell death and modulating the host immune responses. Here, we describe selected examples of Shigella pathogenesis, with particular emphasis on the roles of IpaH family effectors, which shed new light on bacterial survival strategies and provide clues about how to overcome bacterial infections.
Introduction
The interplay between enteric pathogens and the gastrointestinal (GI) tract is a critical determinant of the fate of bacterial infection and disease progression. Shigella spp. are highly adapted human pathogens that cause shigellosis, a disease that provokes severe bloody and mucous diarrhea, which still remains a major public health concern in developing countries. No safe and effective Shigella vaccine currently exists, and antibiotic-resistant bacteria have recently been observed, increasing the threat of outbreaks due to lack of effective treatments (Barry et al., 2013). Therefore, there is an urgent need to understand the interplay between bacterial infection and host cellular and immune responses in order to develop safe and effective Shigella vaccines or novel treatment.
After ingestion via the fecal–oral route, Shigella preferentially invade the intestinal epithelium via M cells overlying the follicle-associated epithelium. Once Shigella are endocytosed by M cells, they enter resident macrophages and induce inflammatory cell death (Wassef et al., 1989; Perdomo et al., 1994). After being released from dying macrophages, the bacteria invade surrounding epithelial cells, escape from the vacuole, and multiply within the cytoplasm (Zychlinsky et al., 1992). Subsequently, Shigella move by inducing actin polymerization at one pole of the bacterium, thereby spreading to neighboring cells (Suzuki et al., 1998, 2000; Egile et al., 1999). Thus, shigellosis is the consequence of multiple pathogenic events, including macrophage cell death, invasion of and multiplication within epithelial cells, cell-cell spread, and severe intestinal inflammation.
The infection strategies of Shigella depend on the delivery of bacterial virulence proteins, called “effectors,” into host cells via the type III secretion system (T3SS) (Carayol and Tran Van Nhieu, 2013; Ashida et al., 2015). The effectors, which are delivered into both myeloid cells (macrophages, dendritic cells, T, and B cells) and nonmyeloid cells (epithelial cells), mimic or subvert host cellular functions, allowing Shigella to colonize the intestinal epithelium. The diverse roles of effectors include bacterial invasion into cells, intracellular survival and multiplication, maintenance of an infectious foothold, and modulation of the host immune response. Recent studies showed that many T3SS effectors have novel and specific enzymatic activities distinct from those of mammalian enzymes, e.g., phosphothreonine lyase, deamidase, and E3 ubiquitin ligase (Li et al., 2007; Rohde et al., 2007; Sanada et al., 2012). Moreover, T3SS effector activity and function is not specific to Shigella but is shared by other enteric pathogens, including Salmonella, Yersinia, and EPEC. Therefore, the study of effector functions will improve our understanding of bacterial infection mechanisms and facilitate development of novel vaccines or drugs to overcome bacterial infection. Shigella is a good model for such studies because it has many effectors, some of which are common to other pathogens, and many of its strategies for infection are widely conserved among other enteric bacterial pathogens. In this review, we will highlight the role of one class of Shigella T3SS effector, the IpaH family.
IpaH Family Effectors
Shigella possesses 12 ipaH genes, which reside on both the large plasmid and the chromosome. The encoded IpaH proteins are injected into host cells via the T3SS (Ashida et al., 2007). IpaH family proteins contain N-terminal leucine-rich repeats (LRRs) and have E3 ubiquitin ligase activity in their conserved C-terminal regions (Rohde et al., 2007; Ashida et al., 2014a). Ubiquitination is an important post-translational modification that regulates several cellular functions, including cell signaling, protein degradation, transcription, and endocytosis (Pickart, 2001). Ubiquitination is accomplished via a series of reactions catalyzed by a multienzymatic cascade: E1 (ubiquitin-activating enzyme), E2 (ubiquitin-conjugating enzyme), and E3 (ubiquitin ligase). The ubiquitination cascade starts with ATP-dependent activation of ubiquitin via formation of a thioester linkage between the carboxyl-terminal Gly of ubiquitin and a Cys of E1. Activated ubiquitin is transferred to the active-site Cys of E2, and finally E3 ligase mediates the transfer of ubiquitin from the E2 to specific substrate proteins (mainly via substrate Lys residues). E3 ligases can be categorized into two groups based on their structures and functions: HECT (Homologous to the E6-AP Carboxyl Terminus)-type and RING (Really Interesting New Gene)/U-box-type. HECT-type E3 ligases catalyze ubiquitin transfer by accepting ubiquitin from E2 via formation of a thioester bond with their catalytic cysteine residue, and then transfer ubiquitin to their target substrates. On the other hand, RING/U-box-type E3 ligases catalyze direct ubiquitin transfer by acting as scaffold molecules to bind and recruit the E2-ubiquitin complex, and then directly transfer ubiquitin from E2 to E3-bound substrates. Recent reports showed that some bacterial pathogens deliver several types of T3SS effectors with E3 ligase activity (Ashida et al., 2014a). Because the ubiquitination pathway is absent in bacteria, bacteria might deliver E3 ligase effectors and hijack the mammalian ubiquitination pathway in order to counteract host responses. IpaH family proteins are widely conserved among animal and plant pathogens, including Shigella (IpaH), Salmonella (SspH1, SspH2, and SlrP), Edwardsiella, Bradyrhizobium, Rhizobium, and some Pseudomonas species, illustrating the importance of these effectors in bacterial infection. Although IpaH family proteins have E3 ubiquitin ligase activity and their C-terminal domains contain a single conserved Cys that form a Cys-ubiquitin intermediate similar to that of HECT-type ligases, the catalytic domains of IpaH family members differ at the sequence and structural levels from eukaryotic E3 ubiquitin ligases. Consequently, IpaH family proteins are now considered to constitute a new class of E3 ubiquitin ligases, NEL (Novel E3 ligase), distinct from typical RING-, and HECT-types of E3 ubiquitin ligases (Singer et al., 2008; Zhu et al., 2008; Quezada et al., 2009). Intriguingly, IpaH family proteins exhibit auto-inhibition to prevent ubiquitination of unintended proteins. Specifically, the LRR domain sequesters and masks the catalytic Cys residue in the C-terminal region prior to substrate binding. Once the LRR binds to substrate proteins, a conformational change occurs, releasing the auto-inhibition of IpaH E3 ligase activity (Quezada et al., 2009; Chou et al., 2012; Keszei et al., 2014).
Although IpaH family proteins are highly similar to one another, the sequences of their LRR regions, regarded as substrate recognition sites, and subcellular localizations (e.g., nucleus, cytoplasm, or plasma membrane) are different. These observations imply that each IpaH family protein has a specific host target protein due to its unique subcellular localization and substrate recognition domain, and thus makes a distinct contribution to promotion of bacterial pathogenesis. Consistent with this, recent studies have revealed the role of IpaH family members in disabling distinct target proteins in host cells.
Shigella IpaH Effectors Target NF-κB Signaling
The host innate immune system can detect markers of Shigella infection as pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs), leading to severe inflammatory colitis aimed at preventing and clearing bacterial infection. However, many enteric bacterial pathogens, including Shigella, are able to optimize the host inflammatory responses to allow pathogen survival in such inflamed environments by delivering a subset of T3SS effector proteins (Rahman and McFadden, 2011; Ashida et al., 2015). Because the nuclear factor κB (NF-κB) plays a pivotal role in the bacteria-induced inflammatory response, Shigella deliver effectors that target and inhibit NF-κB signaling to modulate the host inflammatory response (Ashida et al., 2015; Rahman and McFadden, 2011). Recognition of PAMPs or DAMPs by various sensors, including Toll-like receptors (TLRs) and Nod-like receptors (NLRs), and subsequent signal transduction through Myd88, TRAF, RIP, TAK1, and TAB2/3 trigger the activation of IKK kinase complex. Once the IKK complex is activated, it phosphorylates IκB (inhibitor of NF-κB), leading to its ubiquitination and proteasome degradation. NF-κB then translocates to the nucleus and activates the transcription of proinflammatory cytokines (Hayden and Ghosh, 2012). Because ubiquitination is an essential aspect of this regulatory mechanism, Shigella deliver three IpaH effectors (IpaH9.8, IpaH0722, and IpaH4.5) that subvert NF-κB activation by manipulating the host ubiquitin system (Figure 1). After Shigella invasion of epithelial cells, the host sensor Nod1 recognizes peptidoglycans released from Shigella as PAMPs, triggering NF-κB activation. IpaH9.8 preferentially prevents Nod1-dependent NF-κB activation. Specifically, IpaH9.8 interacts with NEMO/IKKγ, which is critical for downstream activation of NF-κB, and targets NEMO (lysines 309 and 321 residues) for ubiquitination. IpaH9.8 also interacts with ABIN-1, a ubiquitin-binding adaptor protein and bridge IpaH9.8-NEMO, to further promote polyubiquitination of NEMO. Ubiquitination of NEMO by IpaH9.8 leads to proteasomal degradation, thereby reducing NF-κB activation (Ashida et al., 2010). Consistent with this model, the level of NEMO protein and Shigella-induced NF-κB activation were decreased by IpaH9.8 E3 ligase activity in NEMO-WT cells, but not in cells stably expressing NEMO-K309R/K321R, a mutant lacking the IpaH9.8-mediated ubiquitination site. The reduction in NF-κB activation due to the activity of this E3 ligase effector during Shigella infection results in downregulation of host inflammatory responses, such as cytokine production and neutrophil recruitment, and contributes to bacterial colonization in mouse models of lung infection (Ashida et al., 2010). In addition to NEMO, IpaH9.8 also binds to the host splicing factor U2AF35 and inhibits the U2AF35-dependent mRNA splicing reaction (Okuda et al., 2005). Because IpaH9.8 ubiquitinates U2AF35 in vitro, it has been proposed that IpaH9.8 diminishes the amount of U2AF35 and downregulates U2AF35-dependent pro-inflammatory gene expression (Seyedarabi et al., 2010).
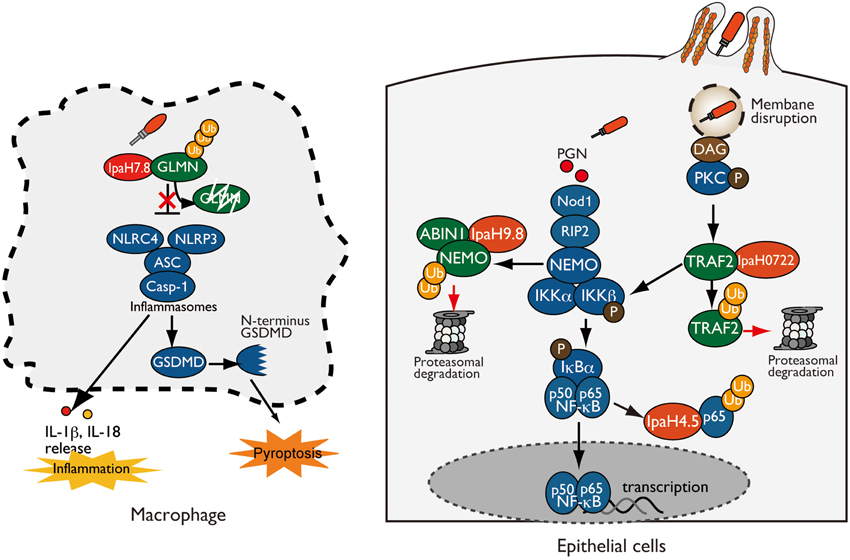
Figure 1. Shigella IpaH effectors target host substrates for ubiquitination. (left: Macrophage) IpaH7.8 interacts with and ubiquitinates GLMN, leading to its proteasome-dependent degradation. Degradation of GLMN abolishes its ability to inhibit inflammasomes, resulting in NLRP3/NLRC4 inflammasome activation and pyroptosis. (right: Epithelial cell) IpaH0722, IpaH9.8, and IpaH4.5 target and ubiquitinate TRAF2, NEMO, and p65, respectively, which undergo proteasome degradation, resulting in inhibition of NF-κB.
Another IpaH family protein, IpaH0722 localizes to host cell plasma membrane via the modulation of S-palmitoylation at N-terminal Cys14 and Cys18 residues. IpaH0722 E3 ligase preferentially inhibits phorbol myristate acetate (PMA)-mediated NF-κB activation in NF-κB-luciferase reporter assays. Since PMA mimics the role of diacylglycerol (DAG) in the activation of the protein kinase C (PKC)-NF-κB pathway, IpaH0722 also inhibits PKC-mediated NF-κB activation (Ashida et al., 2013). Intriguingly, DAG is recognized as the danger signals that are generated by vacuolar membrane ruptures in Shigella infection. When Shigella invades epithelial cells, the bacteria are surrounded by a vacuolar membrane, but they rapidly disrupt this membrane and disseminate into the cytoplasm. At this step, Shigella-mediated vacuolar membrane ruptures are recognized as DAMPs, triggering NF-κB activation through the recruitment and activation of PKC. To counteract PKC–NF-κB activation, Shigella delivers IpaH0722. IpaH0722 interacts with TRAF2, a molecule downstream of PKC, and targets it for ubiquitination. TRAF2 ubiquitinated by IpaH0722 is degraded by the proteasome, thereby inhibiting PKC-mediated NF-κB activation (Ashida et al., 2013). In addition to IpaH9.8 and IpaH0722, Shigella also prevent NF-κB activation by delivering IpaH4.5, which targets the p65 subunit of the NF-κB complex targets it for ubiquitination in vitro (Wang et al., 2013).
In addition to the IpaH family, other Shigella effectors, such as OspG, OspI, and OspZ, also manipulate the ubiquitination system to inhibit NF-κB activation. OspG, which is homologous to EPEC NleH, is a serine/threonine kinase that binds to ubiquitin and ubiquitinated E2s to prevent phospho-IκBα ubiquitination, which is required for NF-κB activation (Kim et al., 2005; Zhou et al., 2013; Grishin et al., 2014; Pruneda et al., 2014). Another Shigella effector, OspI, has deamidase activity, which is shared by other effectors such as Cif from EPEC and CHBP from Burkholderia pseudomallei. OspI targets Ubc13, an E2 protein required for TRAF6 E3 ligase, and converts Gln-100 of Ubc13 to Glu-100, thereby inactivating its E2 activity. Inactivation of Ubc13 by OspI blocks TRAF6-dependent NF-κB signaling, thereby downregulating host inflammation (Sanada et al., 2012). OspZ from S. flexneri 6 and S. boydii blocks the nuclear translocation of NF-κB subunit p65, thereby inhibiting NF-κB activation (Newton et al., 2010). Although it remains unknown whether OspZ shares the same enzymatic activity, EPEC T3SS effector NleE has methyltransferase activity that specifically modifies zinc-finger cysteines of TAB2 and TAB3, ubiquitin-chain binding proteins involved in the NF-κB activation. Following cysteine methylation by NleE, TAB2, and TAB3 lose their zinc ions and ubiquitin-chain binding activities, making them unable to activate NF-κB (Zhang et al., 2011).
Furthermore, Shigella hijack epigenetic modification to modulate host innate immune responses. Shigella T3SS effector OspF, which is homologous to Salmonella SpvC and Pseudomonas syringae HopAl1, has a unique phosphothreonine lyase activity (Arbibe et al., 2007; Kramer et al., 2007; Li et al., 2007). OspF irreversibly dephosphorylates and inactivates MAPKs. This inactivation of MAPK further inhibits downstream phosphorylation of histone H3 at Ser10 at the promoters of a subset of innate immune genes, and promotes chromatin condensation, resulting in inhibition of transcriptional activation by masking NF-κB binding sites (Arbibe et al., 2007). Recent study further showed a novel function of OspF, which alters the activity of the chromatin reader Heterochromatin Protein 1γ (HP1γ) and reprograms host gene expression during Shigella infection via its phosphothreonine lyase activity (Harouz et al., 2014).
Shigella IpaH Effector Targets Inflammasomes
In addition to NF-κB signaling, Shigella use IpaH proteins to target inflammasome activation. As mentioned above, Shigella invade resident macrophages, disrupt the phagosome vacuole, disseminate into and multiply within the cytosol, and induce a form of inflammatory cell death called pyroptosis, which is accompanied by NLRP3 or NLRC4 inflammasome activation and leads to IL-1β and IL-18 secretion (Suzuki et al., 2007; Willingham et al., 2007; Ashida et al., 2011, 2014b; Davis et al., 2011; Suzuki et al., 2014a). Pyroptosis is a specialized form of cell death that is regulated by activated inflammatory caspases (caspase-1 or caspase-11). Caspase-1 is activated by canonical inflammasome (multiprotein complexes, such as NLRP3 or NLRC4 and ASC) via recognition of T3SS components, whereas caspase-11 directly binds to cytosolic LPS and forms non-canonical inflammasomes; both complexes trigger the release of proinflammatory cytokines (IL-1β and IL-18) and pyroptosis (Kayagaki et al., 2011, 2013; Shi et al., 2014). Recent studies show that activated caspase-1 and caspase-11 cleave GSDMD, and the cleaved N-terminal domain of GSDMD induces pyroptosis accompanied by cell lysis and release of cell contents (Kayagaki et al., 2015; Shi et al., 2015).
Although pyroptosis seems like a host defense system aimed at clearing bacterial infection, Shigella deliver IpaH7.8 to induce inflammasome activation and pyroptosis via its E3 ligase activity in order to escape from macrophages; otherwise, the bacteria will be killed (Figure 1) (Suzuki et al., 2014b). In a mouse model of lung infection, mice infected with the Shigella WT or ΔipaH7.8/WT (IpaH7.8 WT complemented) strain exhibited severe macrophage cell death and IL-1β secretion, whereas those infected with the ΔipaH7.8 or ΔipaH7.8/CA (E3 ligase-deficient mutant) strain did not. Notably, colonizing bacterial number was significantly higher in mice infected with the Shigella WT or ΔipaH7.8/WT strain than in mice infected with ΔipaH7.8 or ΔipaH7.8/CA mutants, illustrating the importance of IpaH7.8-mediated cell death in Shigella infection. IpaH7.8 interacts with GLMN, which acts either directly or indirectly as a negative regulator of the NLRP3/NLRC4 inflammasome, and targets it for ubiquitination. GLMN ubiquitinated by IpaH7.8 is subsequently degraded by the proteasome. GLMN degradation abolishes its inhibitory activity and thus triggers NLRP3/NLRC4 inflammasome activation and pyroptosis. To further support this idea, Shigella-induced cytotoxicity and IL-1β secretion levels were remarkably higher in macrophages derived from GLMN+∕− mice than in those from GLMN+∕+ mice. Furthermore, the number of internalized bacterial number was also higher in the lungs of GLMN+∕− mice (Suzuki et al., 2014b). Therefore, induction of pyroptosis by IpaH7.8 is more beneficial to Shigella than the host: specifically, it results in further invasion of surrounding epithelial cells and spread to neighboring cells, thereby promoting bacterial survival.
Conclusion
The lack of a licensed vaccine and the emergence of antibiotic-resistant Shigella highlight the difficulty of controlling this pathogen and the need for new antimicrobial strategies. Studying the course of Shigella infection, especially the interaction between bacterial effectors and host cells, might provide a means to resolve this problem. In this report, we highlighted Shigella infection strategies mediated by IpaH family effectors, including NF-κB inhibition and macrophage killing. As mentioned above, IpaH family effectors are novel E3 ubiquitin ligases that are widely conserved among Gram-negative bacterial pathogens. The conservation of IpaH effectors among Shigella and bacterial pathogens, and their critical roles in pathogenesis, make them good targets for antibacterial drug or vaccine candidates. Recent studies show that each IpaH protein interacts with a specific host target protein via its LRR region, and then modulates the ubiquitination of that target. In addition to the Shigella IpaH family effectors, SspH1, SspH2, and SlrP from Salmonella target and ubiquitinate PKN1, SGT1, and Nod1, and thioredoxin, respectively (Haraga and Miller, 2006; Bernal-Bayard and Ramos-Morales, 2009; Bhavsar et al., 2013; Keszei et al., 2014). Although, the LRR regions of IpaH family effectors are slightly different, each protein contains a catalytic Cys residue that is required for transferring ubiquitin to substrate proteins. Therefore, an inhibitor that could block or mask the catalytic Cys residue of IpaH, or a conserved IpaH C-terminal-based vaccine, would provide broad coverage across against many bacterial pathogens. Therefore, the study of IpaH family effectors may lead to the development of a single drug that is effective against many infectious bacterial diseases.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by a Grant-in-Aid for Specially Promoted Research (23000012, to CS) and a Grant-in-Aid for Scientific Research (C) (25460527 to HA). Part of this work was supported by grants from the Naito Foundation (HA), the Mochida Memorial Foundation for Medical and Pharmaceutical Research (HA), research grant of Astellas Foundation for Research on Metabolic Disorders (HA). This work was jointly supported by a Yakult Honsha Co, Ltd.
References
Arbibe, L., Kim, D. W., Batsche, E., Pedron, T., Mateescu, B., Muchardt, C., et al. (2007). An injected bacterial effector targets chromatin access for transcription factor NF-κB to alter transcription of host genes involved in immune responses. Nat. Immunol. 8, 47–56. doi: 10.1038/ni1423
Ashida, H., Kim, M., and Sasakawa, C. (2014a). Exploitation of the host ubiquitin system by human bacterial pathogens. Nat. Rev. Microbiol. 12, 399–413. doi: 10.1038/nrmicro3259
Ashida, H., Kim, M., and Sasakawa, C. (2014b). Manipulation of the host cell death pathway by Shigella. Cell. Microbiol. 16, 1757–1766. doi: 10.1111/cmi.12367
Ashida, H., Kim, M., Schmidt-Supprian, M., Ma, A., Ogawa, M., and Sasakawa, C. (2010). A bacterial E3 ubiquitin ligase IpaH9.8 targets NEMO/IKKγ to dampen the host NF-κB-mediated inflammatory response. Nat. Cell Biol. 12, 66–73. doi: 10.1038/ncb2006
Ashida, H., Mimuro, H., Ogawa, M., Kobayashi, T., Sanada, T., Kim, M., et al. (2011). Cell death and infection: a double-edged sword for host and pathogen survival. J. Cell Biol. 195, 931–942. doi: 10.1083/jcb.201108081
Ashida, H., Mimuro, H., and Sasakawa, C. (2015). Shigella manipulates host immune responses by delivering effector proteins with specific roles. Front. Immunol. 6:219. doi: 10.3389/fimmu.2015.00219
Ashida, H., Nakano, H., and Sasakawa, C. (2013). Shigella IpaH0722 E3 ubiquitin ligase effector targets TRAF2 to inhibit PKC-NF-κB activity in invaded epithelial cells. PLoS Pathog. 9:e1003409. doi: 10.1371/journal.ppat.1003409
Ashida, H., Toyotome, T., Nagai, T., and Sasakawa, C. (2007). Shigella chromosomal IpaH proteins are secreted via the type III secretion system and act as effectors. Mol. Microbiol. 63, 680–693. doi: 10.1111/j.1365-2958.2006.05547.x
Barry, E. M., Pasetti, M. F., Sztein, M. B., Fasano, A., Kotloff, K. L., and Levine, M. M. (2013). Progress and pitfalls in Shigella vaccine research. Nat. Rev. Gastroenterol. Hepatol. 10, 245–255. doi: 10.1038/nrgastro.2013.12
Bernal-Bayard, J., and Ramos-Morales, F. (2009). Salmonella type III secretion effector SlrP is an E3 ubiquitin ligase for mammalian thioredoxin. J. Biol. Chem. 284, 27587–27595. doi: 10.1074/jbc.M109.010363
Bhavsar, A. P., Brown, N. F., Stoepel, J., Wiermer, M., Martin, D. D., Hsu, K. J., et al. (2013). The Salmonella Type III Effector SspH2 Specifically Exploits the NLR Co-chaperone Activity of SGT1 to Subvert Immunity. PLoS Pathog. 9:e1003518. doi: 10.1371/journal.ppat.1003518
Carayol, N., and Tran Van Nhieu, G. (2013). Tips and tricks about Shigella invasion of epithelial cells. Curr. Opin Microbiol. 16, 1–6. doi: 10.1016/j.mib.2012.11.010
Chou, Y. C., Keszei, A. F., Rohde, J. R., Tyers, M., and Sicheri, F. (2012). Conserved structural mechanisms for autoinhibition in IpaH ubiquitin ligases. J. Biol. Chem. 287, 268–275. doi: 10.1074/jbc.M111.316265
Davis, B. K., Roberts, R. A., Huang, M. T., Willingham, S. B., Conti, B. J., Brickey, W. J., et al. (2011). Cutting edge: NLRC5-dependent activation of the inflammasome. J. Immunol. 186, 1333–1337. doi: 10.4049/jimmunol.1003111
Egile, C., Loisel, T. P., Laurent, V., Li, R., Pantaloni, D., Sansonetti, P. J., et al. (1999). Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J. Cell Biol. 146, 1319–1332. doi: 10.1083/jcb.146.6.1319
Grishin, A. M., Condos, T. E., Barber, K. R., Campbell-Valois, F. X., Parsot, C., Shaw, G. S., et al. (2014). Structural basis for the inhibition of host protein ubiquitination by Shigella effector kinase OspG. Structure 22, 878–888. doi: 10.1016/j.str.2014.04.010
Haraga, A., and Miller, S. I. (2006). A Salmonella type III secretion effector interacts with the mammalian serine/threonine protein kinase PKN1. Cell. Microbiol. 8, 837–846. doi: 10.1111/j.1462-5822.2005.00670.x
Harouz, H., Rachez, C., Meijer, B. M., Marteyn, B., Donnadieu, F., Cammas, F., et al. (2014). Shigella flexneri targets the HP1γ subcode through the phosphothreonine lyase OspF. EMBO J. 33, 2606–2622. doi: 10.15252/embj.201489244
Hayden, M. S., and Ghosh, S. (2012). NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 26, 203–234. doi: 10.1101/gad.183434.111
Kayagaki, N., Stowe, I. B., Lee, B. L., O'Rourke, K., Anderson, K., Warming, S., et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature 526, 666–671. doi: 10.1038/nature15541
Kayagaki, N., Warming, S., Lamkanfi, M., Vande Walle, L., Louie, S., Dong, J., et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. doi: 10.1038/nature10558
Kayagaki, N., Wong, M. T., Stowe, I. B., Ramani, S. R., Gonzalez, L. C., Akashi-Takamura, S., et al. (2013). Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249. doi: 10.1126/science.1240248
Keszei, A. F., Tang, X., McCormick, C., Zeqiraj, E., Rohde, J. R., Tyers, M., et al. (2014). Structure of an SspH1-PKN1 complex reveals the basis for host substrate recognition and mechanism of activation for a bacterial E3 ubiquitin ligase. Mol. Cell. Biol. 34, 362–373. doi: 10.1128/MCB.01360-13
Kim, D. W., Lenzen, G., Page, A. L., Legrain, P., Sansonetti, P. J., and Parsot, C. (2005). The Shigella flexneri effector OspG interferes with innate immune responses by targeting ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. U.S.A. 102, 14046–14051. doi: 10.1073/pnas.0504466102
Kramer, R. W., Slagowski, N. L., Eze, N. A., Giddings, K. S., Morrison, M. F., Siggers, K. A., et al. (2007). Yeast functional genomic screens lead to identification of a role for a bacterial effector in innate immunity regulation. PLoS Pathog. 3:e21. doi: 10.1371/journal.ppat.0030021
Li, H., Xu, H., Zhou, Y., Zhang, J., Long, C., Li, S., et al. (2007). The phosphothreonine lyase activity of a bacterial type III effector family. Science 315, 1000–1003. doi: 10.1126/science.1138960
Newton, H. J., Pearson, J. S., Badea, L., Kelly, M., Lucas, M., Holloway, G., et al. (2010). The type III effectors NleE and NleB from enteropathogenic E. coli and OspZ from Shigella block nuclear translocation of NF-kB p65. PLoS Pathog. 6:e1000898. doi: 10.1371/journal.ppat.1000898
Okuda, J., Toyotome, T., Kataoka, N., Ohno, M., Abe, H., Shimura, Y., et al. (2005). Shigella effector IpaH9.8 binds to a splicing factor U2AF(35) to modulate host immune responses. Biochem. Biophys. Res. Commun. 333, 531–539. doi: 10.1016/j.bbrc.2005.05.145
Perdomo, O. J., Cavaillon, J. M., Huerre, M., Ohayon, H., Gounon, P., and Sansonetti, P. J. (1994). Acute inflammation causes epithelial invasion and mucosal destruction in experimental shigellosis. J. Exp. Med. 180, 1307–1319. doi: 10.1084/jem.180.4.1307
Pickart, C. M. (2001). Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 70, 503–533. doi: 10.1146/annurev.biochem.70.1.503
Pruneda, J. N., Smith, F. D., Daurie, A., Swaney, D. L., Villén, J., Scott, J. D., et al. (2014). E2~Ub conjugates regulate the kinase activity of Shigella effector OspG during pathogenesis. EMBO J. 33, 437–449. doi: 10.1002/embj.201386386
Quezada, C. M., Hicks, S. W., Galán, J. E., and Stebbins, C. E. (2009). A family of Salmonella virulence factors functions as a distinct class of autoregulated E3 ubiquitin ligases. Proc. Natl. Acad. Sci. U.S.A. 106, 4864–4869. doi: 10.1073/pnas.0811058106
Rahman, M. M., and McFadden, G. (2011). Modulation of NF-κB signalling by microbial pathogens. Nat. Rev. Microbiol. 9, 291–306. doi: 10.1038/nrmicro2539
Rohde, J. R., Breitkreutz, A., Chenal, A., Sansonetti, P. J., and Parsot, C. (2007). Type III secretion effectors of the IpaH family are E3 ubiquitin ligase. Cell Host Microbe. 1, 77–83. doi: 10.1016/j.chom.2007.02.002
Sanada, T., Kim, M., Mimuro, H., Suzuki, M., Ogawa, M., Oyama, A., et al. (2012). The Shigella flexneri effector OspI deamidates UBC13 to dampen the inflammatory response. Nature 483, 623–626. doi: 10.1038/nature10894
Seyedarabi, A., Sullivan, J. A., Sasakawa, C., and Pickersgill, R. W. (2010). A disulfide driven domain swap switches off the activity of Shigella IpaH9.8 E3 ligase. FEBS Lett. 584, 4163–4168. doi: 10.1016/j.febslet.2010.09.006
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Shi, J., Zhao, Y., Wang, Y., Gao, W., Ding, J., Li, P., et al. (2014). Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192. doi: 10.1038/nature13683
Singer, A. U., Rohde, J. R., Lam, R., Skarina, T., Kagan, O., DiLeo, R., et al. (2008). Structure of the Shigella T3SS effector IpaH defines a new class of E3 ubiquitin ligases. Nat. Struct. Mol. Biol. 15, 1293–1301. doi: 10.1038/nsmb.1511
Suzuki, S., Franchi, L., He, Y., Muñoz-Planillo, R., Mimuro, H., Suzuki, T., et al. (2014a). Shigella type III secretion protein MxiI is recognized by Naip2 to induce Nlrc4 inflammasome activation independently of Pkcδ. PLoS Pathog. 10:e1003926. doi: 10.1371/journal.ppat.1003926
Suzuki, S., Mimuro, H., Kim, M., Ogawa, M., Ashida, H., Toyotome, T., et al. (2014b). Shigella IpaH7.8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to demolish macrophages. Proc. Natl. Acad. Sci. U.S.A. 111, E4254–4263. doi: 10.1073/pnas.1324021111
Suzuki, T., Franchi, L., Toma, C., Ashida, H., Ogawa, M., Yoshikawa, Y., et al. (2007). Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 3:e111. doi: 10.1371/journal.ppat.0030111
Suzuki, T., Miki, H., Takenawa, T., and Sasakawa, C. (1998). Neural Wiskott-Aldrich syndrome protein is implicated in the actin-based motility of Shigella flexneri. EMBO J. 17, 2767–2776. doi: 10.1093/emboj/17.10.2767
Suzuki, T., Mimuro, H., Miki, H., Takenawa, T., Sasaki, T., Nakanishi, H., et al. (2000). Rho family GTPase Cdc42 is essential for the actin-based motility of Shigella in mammalian cells. J. Exp. Med. 191, 1905–1920. doi: 10.1084/jem.191.11.1905
Wang, F., Jiang, Z., Li, Y., He, X., Zhao, J., Yang, X., et al. (2013). Shigella flexneri T3SS effector IpaH4.5 modulates the host inflammatory response via interaction with NF-κB p65 protein. Cell. Microbiol. 15, 474–485. doi: 10.1111/cmi.12052
Wassef, J. S., Keren, D. F., and Mailloux, J. L. (1989). Role of M cells in initial antigen uptake and in ulcer formation in the rabbit intestinal loop model of shigellosis. Infect. Immun. 57, 858–863.
Willingham, S. B., Bergstralh, D. T., O'Connor, W., Morrison, A. C., Taxman, D. J., Duncan, J. A., et al. (2007). Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe. 2, 147–159. doi: 10.1016/j.chom.2007.07.009
Zhang, L., Ding, X., Cui, J., Xu, H., Chen, J., Gong, Y. N., et al. (2011). Cysteine methylation disrupts ubiquitin-chain sensing in NF-κB activation. Nature 481, 204–208. doi: 10.1038/nature10690
Zhou, Y., Dong, N., Hu, L., and Shao, F. (2013). The Shigella type three secretion system effector OspG directly and specifically binds to host ubiquitin for activation. PLoS ONE 8:e57558. doi: 10.1371/journal.pone.0057558
Zhu, Y., Li, H., Hu, L., Wang, J., Zhou, Y., Pang, Z., et al. (2008). Structure of a Shigella effector reveals a new class of ubiquitin ligases. Nat. Struct. Mol. Biol. 15, 1302–1308. doi: 10.1038/nsmb.1517
Keywords: NF-kB, Shigella, effector, ubiquitin, E3 ligase
Citation: Ashida H and Sasakawa C (2016) Shigella IpaH Family Effectors as a Versatile Model for Studying Pathogenic Bacteria. Front. Cell. Infect. Microbiol. 5:100. doi: 10.3389/fcimb.2015.00100
Received: 14 October 2015; Accepted: 14 December 2015;
Published: 06 January 2016.
Edited by:
William D. Picking, University of Kansas, USAReviewed by:
Jost Enninga, Pasteur Institute, FranceTeresa Estrada-Garcia, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, Mexico
Copyright © 2016 Ashida and Sasakawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroshi Ashida, aGFzaGlkYUBpbXMudS10b2t5by5hYy5qcA==