 Mariette Bonnet1,2,3,4
Mariette Bonnet1,2,3,4 Guy Tran Van Nhieu1,2,3,4*
Guy Tran Van Nhieu1,2,3,4*- 1Equipe Communication Intercellulaire et Infections Microbiennes, Centre de Recherche Interdisciplinaire en Biologie, Collège de France, Paris, France
- 2Institut National de la Santé et de la Recherche Médicale U1050, Paris, France
- 3Centre National de la Recherche Scientifique, UMR7241, Paris, France
- 4MEMOLIFE Laboratory of Excellence and Paris Science Lettre, Paris, France
Shigella, the causative agent of bacillary dysentery invades intestinal epithelial cells using a type III secretion system (T3SS). Through the injection of type III effectors, Shigella manipulates the actin cytoskeleton to induce its internalization in epithelial cells. At early invasion stages, Shigella induces atypical Ca2+ responses confined at entry sites allowing local cytoskeletal remodeling for bacteria engulfment. Global Ca2+ increase in the cell triggers the opening of connexin hemichannels at the plasma membrane that releases ATP in the extracellular milieu, favoring Shigella invasion and spreading through purinergic receptor signaling. During intracellular replication, Shigella regulates inflammatory and death pathways to disseminate within the epithelium. At later stages of infection, Shigella downregulates hemichannel opening and the release of extracellular ATP to dampen inflammatory signals. To avoid premature cell death, Shigella activates cell survival by upregulating the PI3K/Akt pathway and downregulating the levels of p53. Furthermore, Shigella interferes with pro-apoptotic caspases, and orients infected cells toward a slow necrotic cell death linked to mitochondrial Ca2+ overload. In this review, we will focus on the role of Ca2+ responses and their regulation by Shigella during the different stages of bacterial infection.
Introduction
Shigella, the causative agent of bacillary dysentery, invades the colonic mucosa, where it induces a strong inflammatory response responsible for massive destruction of the epithelium (Ashida et al., 2015). Shigella crosses the intestinal barrier by transcytosis through M cells to reach the subepithelial tissue and invades colonocytes through the basolateral side (Sansonetti et al., 1996). Recent evidence suggests that Shigella can also invade colonocytes at the level of mouth crypts (Arena et al., 2015). Although less efficient, colonocyte invasion via the apical site may correspond to a discrete route enabling bacterial intracellular replication while dampening inflammatory responses.
Shigella invasion is a tightly regulated process involving the type III secretion system (T3SS) (Carayol and Tran Van Nhieu, 2013). By subverting cytoskeleton components, Shigella type III effectors trigger actin polymerization and membrane ruffling to induce its internalization by epithelial cell in a macropinocytic-like process (Valencia-Gallardo et al., 2015). Following invasion, Shigella escapes rapidly from the newly formed vacuole to reach the host cell cytosol, its replicative niche (Ray et al., 2010). The formation of an actin comet tail at one pole of the bacterium propels it in the cytoplasm and allows spread from cell to cell within the epithelium (Schroeder and Hilbi, 2008). In recent years, it has become clear that bacterial dissemination within the epithelium is critically dependent on the timely control of cell processes, such as autophagy, inflammatory signals and cell death pathways. While, as illustrated in this issue, various studies have described the involvement of type III effectors in these molecular processes, the role of second messengers has been relatively overlooked. Specifically, the role of Ca2+ signaling during pathogenesis is still poorly characterized, despite its importance and versatility. Here, we will review how Shigella hijacks Ca2+ signaling to promote invasiveness while tuning its deleterious effect to avoid premature cell death and inflammation.
Ca2+ signaling is involved in virtually every cell biological processes. At basal state, the cytosolic Ca2+ concentration is low, in the hundreds of nanomolar range. In response to the activation of cell surface receptors, such as the G-protein coupled receptor (GPCR) at the plasma membrane (PM), the cytosolic Ca2+ concentration increases to reach a micromolar range (Figure 1A). Under physiological conditions, Ca2+ increases are transient and often oscillatory. The base for Ca2+ oscillations relies on an interplay between Ca2+ channels and pumps at the plasma and internal membranes. For example, Ca2+ increases can result from Ca2+ influx i.e., the uptake of extracellular Ca2+ by Ca2+ channels at the PM, or the release of Ca2+ from intracellular stores. In non-excitable cells, Ca2+ release is predominantly mediated by inositol-1,4,5-trisphophate receptors (InsP3Rs) on the endoplasmic reticulum (ER), which are InsP3-gated Ca2+ release channels. Following GPCR stimulation, InsP3 is generated by the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) by phospholipase C (PLC). Increase in InsP3 levels triggers the opening of InsP3Rs and Ca2+ release. The consecutive Ca2+ depletion from the ER activates Ca2+ entry across the PM, a process called store-operated Ca2+ entry (SOCE) carried out by the interaction of the Ca2+ depletion sensor stromal interaction molecule (STIMs) on the ER and the Ca2+ release-activated Ca2+ channel protein ORAI on the PM. Restoration of basal cytosolic Ca2+ concentration is rapidly achieved by extrusion across the PM by the Na+/Ca2+ exchanger or the Ca2+ ATPase PMCA and Ca2+ refilling into the ER by the Ca2+ ATPase SERCA (Bootman, 2012, Figure 1A).
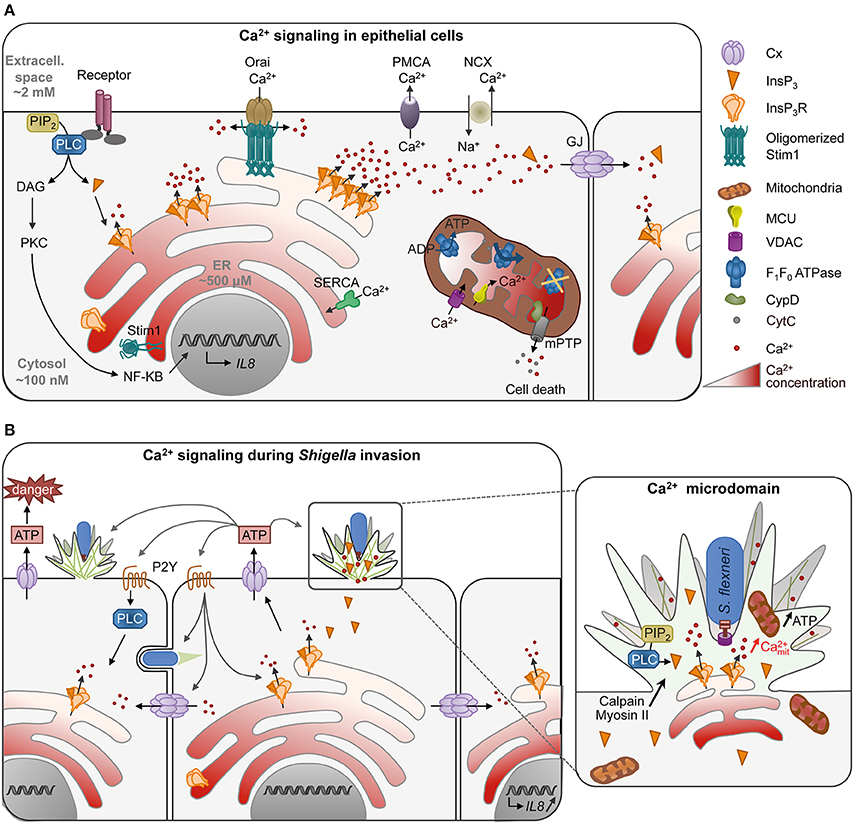
Figure 1. Ca2+ signaling in epithelial cells in normal conditions (A) or during Shigella invasion (B). (A) Activation of cell receptor such as the G-protein coupled receptor at the plasma membrane (PM) stimulates the production of InsP3 by PLC hydrolysis of PIP2. Binding of InsP3 to its receptor InsP3R on the ER releases Ca2+ in the cytosol, translated into cellular processes such as transcription of cytokines by NF-κB. Low InsP3 production induces the opening of a single (blips) or a few InsP3R (puffs) resulting in Ca2+ microdomains, while high InsP3 levels generate global Ca2+ responses. Ca2+ waves can further propagate to the neighboring cells through gap junctions (GJ, 2 hexameric connexins apposed to each other forming a channel) to set up an intercellular wave. Ca2+ depletion in the ER induces the oligomerization of the Ca2+ level sensor protein STIM (stromal interaction molecule), which then interacts with the Ca2+ channel protein ORAI on the PM allowing Ca2+ influx. Restoration of basal cytosolic Ca2+ concentration is rapidly achieved by extrusion of Ca2+ across the PM by the Na+/Ca2+ exchanger or the Ca2+ ATPase PMCA or Ca2+ refilling into the ER by the Ca2+ ATPase SERCA. Mitochondria, often in vicinity to the ER, can buffer cytosolic Ca2+ by taking up Ca2+ through the mitochondrial outer membrane channel VDAC (Voltage-Dependent Anion Channel) and the Ca2+ uniporter MCU (Mitochondrial Ca2+ Uniporter) on the inner membrane. Ca2+ elevation in mitochondria activates mitochondrial functions such as the F1/F0 ATPsynthase. Ca2+ overload, however, inhibits ATP production and induces the opening of the mitochondrial permeability transition pore (mPTP) through CypD activation. mPTP releases small solutes such as Ca2+ and cytochrome c leading to cell death. (B) During invasion of epithelial cells, Shigella induces Ca2+ signaling by recruiting PLC at entry site. Due to the dense actin meshwork and recruitment of InsP3R in the foci, Ca2+ microdomains are confined in the entry site. Mitochondria trapped or in close vicinity to the entry site are activated with an increase of mitochondrial Ca2+ and produce more ATP necessary for actin foci formation. Ca2+ increase is required for efficient bacteria invasion through activation of Ca2+-dependent processes such as calpain and myosin II. Shigella invasion also induces global Ca2+ responses, which induce connexin (Cx) hemichannel opening at the PM, releasing ATP in the extracellular milieu. ATP signaling in turn increases the number of bacteria captured and entering the cell at a given invasion site, as well as bacterial invasion in the neighboring cells. It also increases global Ca2+ responses through purinergic reeptor signaling (P2Y). Global Ca2+ responses can further propagate to neighboring cells through gap junctions, leading to the transcription of IL-8 by bystander cells. S. flexneri is represented in blue with a Type III secretion system.
Sustained increases in high Ca2+ concentrations, however, lead to cell death due to mitochondrial Ca2+ overload (Calí et al., 2012). Upon Ca2+ release, mitochondria—associated or in close vicinity to ER membranes—take up Ca2+ via the mitochondrial outer membrane channel VDAC (Voltage-Dependent Anion Channel) and the Ca2+ uniporter MCU (Mitochondrial Ca2+ Uniporter). Increase in mitochondrial Ca2+ activates mitochondrial functions including ATP synthesis from oxidative phosphorylation. Because of their slow Ca2+ uptake rate, mitochondria also buffer cytosolic Ca2+ variations and play an important role in shaping physiological Ca2+ signals (Olson et al., 2012). Sustained mitochondrial Ca2+ accumulation, however, triggers the irreversible opening of the mitochondrial permeability transition pore (mPTP) mediated by cyclophilin D (CypD), influx of solutes into the mitochondrial matrix leading to the swelling of mitochondria and permeabilization of mitochondrial membranes. The release of the pro-apoptotic factor cytochrome C as well as ROS eventually leads to cell death by apoptosis or necrosis (Orrenius et al., 2015, Figure 1A).
Ca2+ oscillations can be explained by the bi-phasic regulation of InsP3Rs by cytosolic Ca2+, and vary in amplitude, frequency and duration of responses (Dupont et al., 2008). The cell's ability to trigger different types of oscillatory responses leading to the activation of various processes is the base of signal encoding during Ca2+ signaling (Smedler and Uhlén, 2014). Changes in the Ca2+ basal concentration are translated into distinct cellular processes by Ca2+-modulated proteins, in a process termed Ca2+ decoding (Carafoli, 2003). Global Ca2+ responses have been implicated in activation of the transcription factor NF-κB that regulates pro-inflammatory cytokines expression in response to bacterial invasion (Dolmetsch et al., 1997; Gewirtz et al., 2000). Global Ca2+ responses can further propagate to neighboring cells through gap junction channels composed of two hexameric connexins (Cx) (Leybaert and Sanderson, 2012, Figure 1A).
Besides these features associated with specific profiles of global Ca2+ responses, Ca2+ signals can also locally display spatial organization. Ca2+ microdomains have been described in response to weak InsP3 levels, leading to opening of a single (i.e., blips) or a small cluster of InsP3Rs (i.e., puffs) (LaFerla, 2002, Figure 1A). They are transient, short-lived Ca2+ responses that remain confined to a small cytoplasmic region at Ca2+ releasing sites. Ca2+ microdomains have been implicated in various processes involving cytoskeletal reorganization, such as phagocytosis (Nunes et al., 2012), chemotaxis (Tsai et al., 2015), or in filopodial dynamics through activation of calpain, a Ca2+-dependent cysteine protease (Kerstein et al., 2015).
Shigella Utilizes Ca2+ Signaling During Invasion of Epithelial Cells
Upon host cell invasion, Shigella induces local and global Ca2+ responses dependent on InsP3-mediated signaling with a pattern that differs significantly from classical agonist-induced Ca2+ response (Tran Van Nhieu et al., 2003, 2013, Figure 1B). Local Ca2+ responses are induced as early as 5 min after bacterial contact with epithelial cells, with a peak of responses at 15 min. Some of these local Ca2+ responses, called RATP for “Responses Associated with Trespassing Pathogens,” are highly atypical because they can last tens of seconds and are confined to the bacterial invasion site. They involve PLC-β1 and δ1 activated at Shigella entry sites, leading to the local accumulation of InsP3. The bacterial stimuli inducing PLCs' activation have not been identified yet, but depend on a functional T3SS. While injected effectors are dispensable, mutants defective for the translocon components IpaB or IpaC fail to recruit PLCs (Tran Van Nhieu et al., 2013). IpaB and IpaC are both predicted to contain coiled-coil domains and structural studies have shown that IpaB coiled-coil domain shared similarities with pore-forming toxins (Barta et al., 2012). Insertion of type III translocon in the PM may trigger Ca2+ signaling during Shigella invasion, since destabilization of the PM by pore-formin toxins is sufficient to activate PLCs, Ca2+ release and cytoskeletal reorganization (García-Sáez et al., 2011; Schwan et al., 2014). The particular duration of the local Ca2+ responses is due to the recruitment of InsP3Rs and the dense actin meshwork that restricts the diffusion of InsP3 at the entry site (Tran Van Nhieu et al., 2013, Figure 1B).
Shortly after the peak of long-lasting local Ca2+ responses, Shigella elicits isolated global Ca2+ responses with slow dynamics. These responses are not essential for Shigella entry but amplify it, through the release of ATP in the extracellular milieu by Cx-hemichannel opening at the PM (Tran Van Nhieu et al., 2003; Clair et al., 2008). Secreted ATP stimulates cellular functions in an autocrine or paracrine manner, such as Ca2+ signaling, through pathways involving P2Y purinergic receptors (Sáez and Leybaert, 2014; Figure 1B).
Local Ca2+ Signals are Involved in Shigella Entry
Inhibition of the local Ca2+ responses by the Ca2+ chelator BAPTA, as well as transfection of InsP3-5 phosphatase impaired Shigella induced actin foci formation, indicating that they participate in the invasion process. These local Ca2+ responses are likely to have multiple other implications during Shigella invasion. For example, calpain activation by cytosolic Ca2+ increase participates in the cytoskeleton reorganization necessary for bacterial entry (Bergounioux et al., 2012). Calpain targets several components regulating cytoskeleton reorganization. Some of these, such as the Src tyrosine kinase and cortactin, have been implicated in Shigella invasion, and local sustained Ca2+ increases could mediate their calpain-dependent regulation at invasions sites (Franco and Huttenlocher, 2005; Jeong et al., 2013). Myosin II, a Ca2+ dependent motor protein, has been implicated in Salmonella invasion into host cells, a process sharing similarities with Shigella invasion (Hänisch et al., 2011). Myosin II is recruited in Shigella entry foci (Clerc and Sansonetti, 1987). Ca2+ elevation in the foci could thus participate in actomyosin contraction during invasion (Valencia-Gallardo et al., 2015). Mitochondria are detected in the dense actin meshwork at bacterial invasion sites and display mitochondrial Ca2+ increase (Tran Van Nhieu et al., 2013). Mitochondrial activation through InsP3-mediated Ca2+ increase may lead to increased ATP production necessary for high-energy demanding processes such as actin filament elongation and myosin activation required for Shigella engulfment in the cell (Kuiper et al., 2008; Griffiths and Rutter, 2009).
The confinement of RATP at the entry site may also impact on other aspects linked to mitochondrial signals during the early stages of infection. The uptake of Ca2+ by mitochondria might also restrict Ca2+ increase to the entry site and block its diffusion to the remainder of the cell, as proposed in other systems (Rizzuto et al., 2012). Mitochondrial Ca2+ buffering has been implicated in various processes including vesicle secretion (Celsi et al., 2009). Since exocytic processes triggered during Shigella invasion participate in the subsequent lysis of the bacteria-containing vacuole (Mellouk et al., 2014), it is possible that mitochondrial Ca2+ also regulates later stages following Shigella invasion.
Global Ca2+ Responses Enhance Shigella Invasion and Dissemination Mediated by Cx Signaling
Cx-mediated signaling favors both Shigella invasion and dissemination in epithelial cells (Tran Van Nhieu et al., 2013). The basis for enhancement of bacterial dissemination is not understood, but may involve processes similar to the ones described for the stimulation of invasion promoted by ATP released by Cx-hemichannels. Extracellular ATP stimulates bacterial capture by filopodia, a process that precedes Shigella invasion. The Erk MAP-kinase is stimulated by extracellular ATP and was found to control the elongation and retraction of filopodia, by regulating the rate of actin retrograde flow and the actin dynamics at the base of the filopodia in the cell cortex. ATP signaling thus increases the number of bacteria captured and entering the cell at a given site, as well as bacterial invasion in neighboring cells at early stage of infection (Romero et al., 2011, Figure 1B). While there is a lack of in vivo evidence for the relevance of increased bacterial invasion linked to extracellular ATP released from Cx-hemichanels, it is expected to play at initial local sites of bacterial breaching of the intestinal epithelium at the onset of the infectious process. Global Ca2+ increases could favor dissemination, either directly through the activation of myosin required for protrusion engulfment (Rathman et al., 2000), or by affecting junctional integrity (Lum and Morona, 2014). Cxs enable the propagation of Ca2+ responses among neighboring cells. Classically, these intercellular waves are described as a result of InsP3 diffusion through gap junctions, but can also result from signaling through Cx-hemichannels (Leybaert and Sanderson, 2012). In addition, such Cx-mediated intercellular Ca2+ waves stimulate IL-8 expression in bystander cells through cell-cell propagation of the activation signal of the NF-κB transcription factor and MAP-kinases, a process that may occur at later infectious stages. Thus, through Cx-mediated signaling, epithelial cells may amplify inflammatory signals and innate immunity against bacterial infection (Kasper et al., 2010). Cxs have been described to favor adherence and invasion of other pathogenic bacteria in epithelial cells, therefore facilitating bacterial infection in vivo (Velasquez Almonacid et al., 2009; Guttman et al., 2010; Puhar et al., 2013; Simpson et al., 2013).
Ca2+-Induced Slow Necrotic Cell Death
Shigella invasion is perceived as a genotoxic stress by epithelial cells, triggering the activation of the p53 pro-apoptotic signaling pathway at early stages of infection (Bergounioux et al., 2012, Figure 2). In addition, the pro-apoptotic growth arrest and DNA damage 45α (Gadd45α) factor is strongly induced, resulting in the induction of the intrinsic apoptosis pathway via loss of mitochondrial potential and caspase-9 activation (Lembo-Fazio et al., 2011). However, Shigella deploys different strategies to prevent rapid apoptotic death, including the degradation of p53 through Ca2+- and VirA dependent activation of calpain (Bergounioux et al., 2012), the activation of the PI3K/Akt pro-survival pathway by the type III effector IpgD and transcription of anti-apoptotic processes (Pendaries et al., 2006; Clark and Maurelli, 2007; Carneiro et al., 2009). Shigella also interferes with apoptotic processes by inhibiting the mitochondria-mediated release of cytochrome c through the pilus protein FimA (Sukumaran et al., 2010), and inhibiting caspases through the type III effectors Spa15 and OspC3 (Faherty and Maurelli, 2009; Kobayashi et al., 2013, Figure 2).
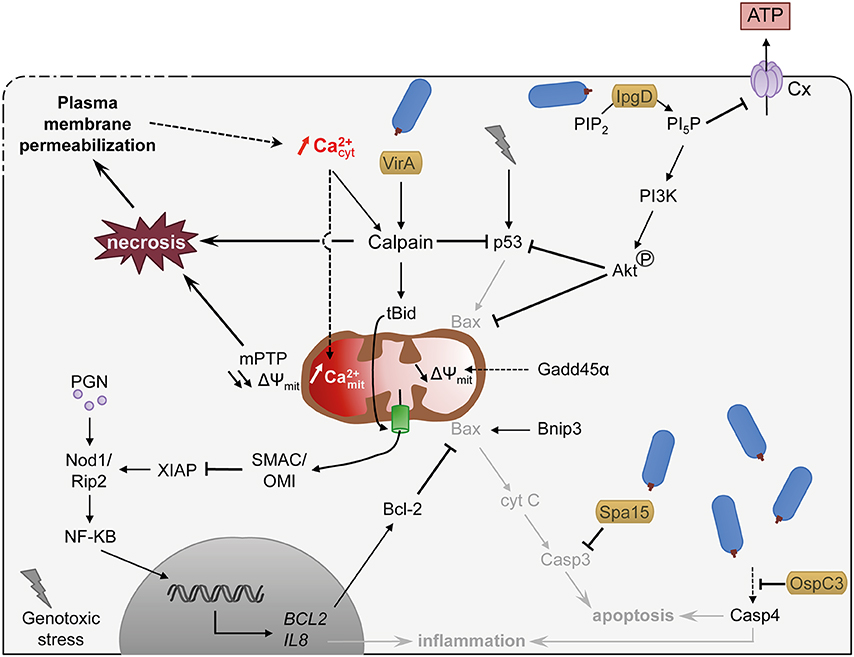
Figure 2. Shigella exploits Ca2+ signaling to delay cell death and to dampen inflammation. Shigella invasion into host cell is perceived as genotoxic stress and induces apoptotic signaling mediated in part by the pro-apoptotic factor p53. Shigella, however, delays apoptotic cell death by different means: (i) promoting p53 degradation through Ca2+ increase and VirA dependent activation of calpain and IpgD-dependent activation of the PI3K/Akt survival pathway, (ii) maintaining mitochondria integrity by inhibiting Bax through Akt phosphorylation and NF-κB-dependent transcription activation of Bcl-2, (iii) inhibiting caspases by type III effectors. Shigella also dampens inflammation through IpgD-mediated closure of connexin hemichannel (Cx, in light violet) and calpain-mediated cleavage of Bid, which releases mitochondrial SMAC/OMI and antagonizes the inflammatory action of XIAP. Sustained cytosolic Ca2+ increase leads to mitochondrial Ca2+ overload, which induces a prolonged opening of the mPTP and ultimately necrosis. Necrosis is also promoted by pathologic activation of calpain. Altogether, the epithelial cell dies from a slow necrotic death. Plasma membrane permeabilization observed at later stages of infection might induce cytosolic Ca2+ increase, favoring necrotic cell death through mitochondrial overload. Dashed arrows indicate activation by an unknown mechanism. S. flexneri is drawn in blue. Type III effectors are labeled in yellow. Pathways hampered by Shigella are indicated in light gray. Lightning bolt represents genotoxic stress.
As a consequence, Shigella-infected epithelial cells do not die from apoptosis but from a slow necrotic death associated with PM permeabilization and increased cytosolic Ca2+ (Carneiro et al., 2009; Dupont et al., 2009, Figure 2). The cause for the increased cytosolic Ca2+ during cell infection by Shigella is not clearly established. It could be a direct result from Ca2+ influx linked to PM damage (Carneiro et al., 2009). Alternatively, it could result from the Shigella-induced activation of Bax/Bak or ER stress, shown in other studies to induce Ca2+ leakage from the ER (Distelhorst and Bootman, 2011; Tattoli et al., 2012; Tsalikis et al., 2015). The anti-apoptotic factor Bcl-2 is upregulated during Shigella invasion and delays Shigella-induced necrotic death (Carneiro et al., 2009). Bcl-2 is well characterized for its antagonistic function against MOMP (mitochondrial outer membrane permeability) and was proposed to counteract the pro-apoptotic role of Bnip3 during Shigella infection. Bcl-2, however, can also inhibit InsP3-mediated Ca2+ release and the mitochondrial Ca2+ overload (Distelhorst and Bootman, 2011), suggesting that it could also delay cell death through the modulation of Ca2+ responses.
Sustained increase in cytosolic Ca2+ favors the necrotic death of infected epithelial cells through diverse pathways. It leads to the pathological activation of calpain, committing the cells toward necrotic death (Łopatniuk and Witkowski, 2011; Bergounioux et al., 2012, Figure 2). It eventually also leads to increase in mitochondrial Ca2+, triggering mPTP opening mediated by CypD, resulting in loss of mitochondrial membrane potential, and eventually causes mitochondrial collapse, i.e., mitochondria swelling, loss of ATP production and ROS generation (Carneiro et al., 2009). The collapse of mitochondria depresses the levels of intracellular ATP below what is required for the execution of apoptosis, thus leading to necrotic death, for lack of ATP (Bernardi et al., 2015; Karch and Molkentin, 2015).
Shigella Manipulates Ca2+ Signals to Limit Innate Immunity and Inflammation
As described elsewhere in this issue, Shigella utilizes different pathways to dampen innate immunity and inflammation. For example, the type III effector OspC3 inhibits caspase 4 activation, which would otherwise induce inflammatory cell death. In addition to a variety of type III effectors preventing the activation of NF-κB or MAP-kinases (Ashida et al., 2013), intracellular Shigella remodels its LPS into a poorly immunogenic form to better disseminate within the gut microenvironment (Paciello et al., 2013).
Ca2+-dependent calpain activation also dampens inflammatory signals during Shigella invasion. Calpain cleaves the pro-apoptotic BH3-only member of the Bcl-2 protein family BID, to trigger its translocation in mitochondria and the release of SMAC (second mitochondria-derived activator of caspases) and the mitochondrial serine protease Omi/HtrA2 (High-Temperature Requirement). SMAC and Omi/HtrA2 then antagonize the inflammatory action of XIAP (X-linked inhibitor of apoptosis protein; Andree et al., 2014, Figure 2).
Extracellular ATP acts as a local endogenous danger signal, triggering a protective inflammatory host response to eradicate pathogens (Bours et al., 2006). For example, extracellular ATP activates the differentiation of Th17 lymphocytes (Atarashi et al., 2008) and induces intracellular Ca2+ signaling leading to the activation of the NLRP3 (nucleotide-binding domain, leucine-rich-repeat-containing family, pyrin domain-containing 3) inflammasome (Murakami et al., 2012). ATP released by Ca2+-mediated opening of Cx-hemichannel during Shigella infection acts as a potent danger signal, triggering the release of cytokines oriented toward a proinflammatory Th17 cell response (Tran Van Nhieu et al., 2003; Puhar et al., 2013). At later stages following bacterial invasion, IpgD induces the closure of Cx-hemichannel thereby limiting ATP release in the extracellular medium and dampening inflammation (Puhar et al., 2013, Figure 2). The mechanism involved is not entirely clear but involves the IpgD-mediated production of phosphatidylinositol 5-phosphate (PI5P).
Concluding Remarks
We have discussed how Shigella uses Ca2+ signals from the onset of cell invasion to the death of infected cells. Local Ca2+ signals affect the dynamics of cytoskeletal reorganization to promote bacterial invasion. Global Ca2+ signals, usually associated with the induction of inflammatory signals, are not required for invasion and are involved at later stages of infection by Shigella to promote slow death. Between these two extremes of the cell infectious cycle and because of the versatility of Ca2+ signaling, processes key to Shigella virulence are likely to be regulated by Ca2+ signals, spatially and temporally regulated during the course of infection. The dual role of Ca2+-dependent calpain, involved in Shigella invasion and the orchestration of the death of infected cells illustrates this notion. How Ca2+ signals are generated and how they regulate the various processes associated with bacterial infection are key issues that cannot be ignored. Many pathogenic bacteria have been described to trigger Ca2+ responses during cell infection, involved in various key processes of infection such as bacterial adhesion, invasion, intracellular replication, or stimulating inflammation (Ruschkowski et al., 1992; Pace et al., 1993; Gewirtz et al., 2000; Asmat et al., 2014; Czyz et al., 2014; Jolly et al., 2014). The role of virulence factors has been studied through their catalytic activity or cellular compounds that they target. The role of bacterial factors on the regulation of second messenger such as InsP3, or ions such as Ca2+, has been seldom addressed, despite of their key role in fundamental cellular processes. The in-depth understanding of Shigella-induced signaling will implicate the integration of the diversion of the function of such second messengers during infection.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the Institut National de la Santé et de la Recherche Médicale, the Collège de France, and Agence Nationale pour la Recherche (ANR_12-BSV3-0017 MITOPATHO) grants. MB is a fellow from DIM Malinf (Domaines d'Interets Majeurs, Infections digestives, hépatiques et urogénitales), Conseil Régional Ile de France and from Fondation pour la Recherche Médicale (FRM), grant n°SPF20130526673.
References
Andree, M., Seeger, J. M., Schüll, S., Coutelle, O., Wagner-Stippich, D., Wiegmann, K., et al. (2014). BID-dependent release of mitochondrial SMAC dampens XIAP-mediated immunity against Shigella. EMBO J. 33, 2171–2187. doi: 10.15252/embj.201387244
Arena, E. T., Campbell-Valois, F. X., Tinevez, J. Y., Nigro, G., Sachse, M., Moya-Nilges, M., et al. (2015). Bioimage analysis of Shigella infection reveals targeting of colonic crypts. Proc. Natl. Acad. Sci. U.S.A. 112, E3282–E3290. doi: 10.1073/pnas.1509091112
Ashida, H., Mimuro, H., and Sasakawa, C. (2015). Shigella manipulates host immune responses by delivering effector proteins with specific roles. Front. Immunol. 6:219. doi: 10.3389/fimmu.2015.00219
Ashida, H., Nakano, H., and Sasakawa, C. (2013). Shigella IpaH0722 E3 ubiquitin ligase effector targets TRAF2 to inhibit PKC-NF-κB activity in invaded epithelial cells. PLoS Pathog. 9:e1003409. doi: 10.1371/journal.ppat.1003409
Asmat, T. M., Tenenbaum, T., Jonsson, A. B., Schwerk, C., and Schroten, H. (2014). Impact of calcium signaling during infection of Neisseria meningitidis to human brain microvascular endothelial cells. PLoS ONE 9:e114474. doi: 10.1371/journal.pone.0114474
Atarashi, K., Nishimura, J., Shima, T., Umesaki, Y., Yamamoto, M., Onoue, M., et al. (2008). ATP drives lamina propria TH17 cell differentiation. Nature 455, 808–812. doi: 10.1038/nature07240
Barta, M. L., Dickenson, N. E., Patil, M., Keightley, A., Wyckoff, G. J., Picking, W. D., et al. (2012). The structures of coiled-coil domains from type III secretion system translocators reveal homology to pore-forming toxins. J. Mol. Biol. 417, 395–405. doi: 10.1016/j.jmb.2012.01.026
Bergounioux, J., Elisee, R., Prunier, A. L., Donnadieu, F., Sperandio, B., Sansonetti, P., et al. (2012). Calpain activation by the Shigella flexneri effector VirA regulates key steps in the formation and life of the bacterium's epithelial niche. Cell Host Microbe 11, 240–252. doi: 10.1016/j.chom.2012.01.013
Bernardi, P., Rasola, A., Forte, M., and Lippe, G. (2015). The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol. Rev. 95, 1111–1155. doi: 10.1152/physrev.00001.2015
Bootman, M. D. (2012). Calcium signaling. Cold. Spring. Harb. Perspect. Biol. 4:a011171. doi: 10.1101/cshperspect.a011171
Bours, M. J., Swennen, E. L., Di Virgilio, F., Cronstein, B. N., and Dagnelie, P. C. (2006). Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol. Ther. 112, 358–404. doi: 10.1016/j.pharmthera.2005.04.013
Calí, T., Ottolini, D., and Brini, M. (2012). Mitochondrial Ca2+ as a key regulator of mitochondrial activities. Adv. Exp. Med. Biol. 942, 53–73. doi: 10.1007/978-94-007-2869-1_3
Carafoli, E. (2003). The calcium-signalling saga: tap water and protein crystals. Nat. Rev. Mol. Cell. Biol. 4, 326–332. doi: 10.1038/nrm1073
Carayol, N., and Tran Van Nhieu, G. (2013). Tips and tricks about Shigella invasion of epithelial cells. Curr. Opin. Microbiol. 16, 32–37. doi: 10.1016/j.mib.2012.11.010
Carneiro, L. A., Travassos, L. H., Soares, F., Tattoli, I., Magalhaes, J. G., Bozza, M. T., et al. (2009). Shigella induces mitochondrial dysfunction and cell death in nonmyleoid cells. Cell Host Microbe 5, 123–136. doi: 10.1016/j.chom.2008.12.011
Celsi, F., Pizzo, P., Brini, M., Leo, S., Fotino, C., Pinton, P., et al. (2009). Mitochondria, calcium and cell death: a deadly triad in neurodegeneration. Biochim. Biophys. Acta 1787, 335–344. doi: 10.1016/j.bbabio.2009.02.021
Clair, C., Combettes, L., Pierre, F., Sansonetti, P., and Tran Van Nhieu, G. (2008). Extracellular-loop peptide antibodies reveal a predominant hemichannel organization of connexins in polarized intestinal cells. Exp. Cell. Res. 314, 1250–1265. doi: 10.1016/j.yexcr.2007.12.021
Clark, C. S., and Maurelli, A. T. (2007). Shigella flexneri inhibits staurosporine-induced apoptosis in epithelial cells. Infect. Immun. 75, 2531–2539. doi: 10.1128/IAI.01866-06
Clerc, P., and Sansonetti, P. J. (1987). Entry of Shigella flexneri into HeLa cells: evidence for directed phagocytosis involving actin polymerization and myosin accumulation. Infect. Immun. 55, 2681–2688.
Czyz, D. M., Potluri, L. P., Jain-Gupta, N., Riley, S. P., Martinez, J. J., Steck, T. L., et al. (2014). Host-directed antimicrobial drugs with broad-spectrum efficacy against intracellular bacterial pathogens. mBio 5, e01534–14. doi: 10.1128/mBio.01534-14
Distelhorst, C. W., and Bootman, M. D. (2011). Bcl-2 interaction with the inositol 1,4,5-trisphosphate receptor: role in Ca2+ signaling and disease. Cell Calcium 50, 234–241. doi: 10.1016/j.ceca.2011.05.011
Dolmetsch, R. E., Lewis, R. S., Goodnow, C. C., and Healy, J. I. (1997). Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 386, 855–858. doi: 10.1038/386855a0
Dupont, G., Abou-Lovergne, A., and Combettes, L. (2008). Stochastic aspects of oscillatory Ca2+ dynamics in hepatocytes. Biophys. J. 95, 2193–2202. doi: 10.1529/biophysj.108.133777
Dupont, N., Lacas-Gervais, S., Bertout, J., Paz, I., Freche, B., Tran Van Nhieu, G., et al. (2009). Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe 6, 137–149. doi: 10.1016/j.chom.2009.07.005
Faherty, C. S., and Maurelli, A. T. (2009). Spa15 of Shigella flexneri is secreted through the type III secretion system and prevents staurosporine-induced apoptosis. Infect. Immun. 77, 5281–5290. doi: 10.1128/IAI.00800-09
Franco, S. J., and Huttenlocher, A. (2005). Regulating cell migration: calpains make the cut. J. Cell Sci. 118, 3829–3838. doi: 10.1242/jcs.02562
García-Sáez, A. J., Buschhorn, S. B., Keller, H., Anderluh, G., Simons, K., and Schwille, P. (2011). Oligomerization and pore formation by equinatoxin II inhibit endocytosis and lead to plasma membrane reorganization. J. Biol. Chem. 286, 37768–37777. doi: 10.1074/jbc.M111.281592
Gewirtz, A. T., Rao, A. S., Simon, P. O. Jr., Merlin, D., Carnes, D., Madara, J. L., et al. (2000). Salmonella typhimurium induces epithelial IL-8 expression via Ca2+-mediated activation of the NF-κB pathway. J. Clin. Invest. 105, 79–92. doi: 10.1172/JCI8066
Griffiths, E. J., and Rutter, G. A. (2009). Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim. Biophys. Acta. 1787, 1324–1333. doi: 10.1016/j.bbabio.2009.01.019
Guttman, J. A., Lin, A. E., Li, Y., Bechberger, J., Naus, C. C., Vogl, A. W., et al. (2010). Gap junction hemichannels contribute to the generation of diarrhoea during infectious enteric disease. Gut 59, 218–226. doi: 10.1136/gut.2008.170464
Hänisch, J., Kölm, R., Wozniczka, M., Bumann, D., Rottner, K., and Stradal, T. E. (2011). Activation of a RhoA/myosin II-dependent but Arp2/3 complex-independent pathway facilitates Salmonella invasion. Cell Host Microbe 9, 273–285. doi: 10.1016/j.chom.2011.03.009
Jeong, S. Y., Martchenko, M., and Cohen, S. N. (2013). Calpain-dependent cytoskeletal rearrangement exploited for anthrax toxin endocytosis. Proc. Natl. Acad. Sci. U.S.A. 110, E4007–E4015. doi: 10.1073/pnas.1316852110
Jolly, C., Winfree, S., Hansen, B., and Steele-Mortimer, O. (2014). The Annexin A2/p11 complex is required for efficient invasion of Salmonella Typhimurium in epithelial cells. Cell. Microbiol. 16, 64–77. doi: 10.1111/cmi.12180
Karch, J., and Molkentin, J. D. (2015). Regulated necrotic cell death: the passive aggressive side of Bax and Bak. Circ. Res. 116, 1800–1809. doi: 10.1161/CIRCRESAHA.116.305421
Kasper, C. A., Sorg, I., Schmutz, C., Tschon, T., Wischnewski, H., Kim, M. L., et al. (2010). Cell-cell propagation of NF-κB transcription factor and MAP kinase activation amplifies innate immunity against bacterial infection. Immunity 33, 804–816. doi: 10.1016/j.immuni.2010.10.015
Kerstein, P. C., Nichol, R. H., and Gomez, T. M. (2015). Mechanochemical regulation of growth cone motility. Front. Cell. Neurosci. 9:244. doi: 10.3389/fncel.2015.00244
Kobayashi, T., Ogawa, M., Sanada, T., Mimuro, H., Kim, M., Ashida, H., et al. (2013). The Shigella OspC3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe 13, 570–583. doi: 10.1016/j.chom.2013.04.012
Kuiper, J. W., Pluk, H., Oerlemans, F., van Leeuwen, F. N., de Lange, F., Fransen, J., et al. (2008). Creatine kinase-mediated ATP supply fuels actin-based events in phagocytosis. PLoS Biol. 6:e51. doi: 10.1371/journal.pbio.0060051
LaFerla, F. M. (2002). Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat. Rev. Neurosci. 3, 862–872. doi: 10.1038/nrn960
Lembo-Fazio, L., Nigro, G., Noël, G., Rossi, G., Chiara, F., Tsilingiri, K., et al. (2011). Gadd45α activity is the principal effector of Shigella mitochondria-dependent epithelial cell death in vitro and ex vivo. Cell Death Dis. 2:e122. doi: 10.1038/cddis.2011.4
Leybaert, L., and Sanderson, M. J. (2012). Intercellular Ca2+ waves: mechanisms and function. Physiol. Rev. 92, 1359–1392. doi: 10.1152/physrev.00029.2011
Łopatniuk, P., and Witkowski, J. M. (2011). Conventional calpains and programmed cell death. Acta Biochim. Pol. 58, 287–296.
Lum, M., and Morona, R. (2014). Myosin IIA is essential for Shigella flexneri cell-to-cell spread. Pathog. Dis. 72, 174–187. doi: 10.1111/2049-632X.12202
Mellouk, N., Weiner, A., Aulner, N., Schmitt, C., Elbaum, M., Shorte, S. L., et al. (2014). Shigella subverts the host recycling compartment to rupture its vacuole. Cell Host Microbe 16, 517–530. doi: 10.1016/j.chom.2014.09.005
Murakami, T., Ockinger, J., Yu, J., Byles, V., McColl, A., Hofer, A. M., et al. (2012). Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. U.S.A. 109, 11282–11287. doi: 10.1073/pnas.1117765109
Nunes, P., Cornut, D., Bochet, V., Hasler, U., Oh-Hora, M., Waldburger, J. M., et al. (2012). STIM1 juxtaposes ER to phagosomes, generating Ca2+ hotspots that boost phagocytosis. Curr. Biol. 22, 1990–1997. doi: 10.1016/j.cub.2012.08.049
Olson, M. L., Chalmers, S., and McCarron, J. G. (2012). Mitochondrial organization and Ca2+ uptake. Biochem. Soc. Trans. 40, 158–167. doi: 10.1042/BST20110705
Orrenius, S., Gogvadze, V., and Zhivotovsky, B. (2015). Calcium and mitochondria in the regulation of cell death. Biochem. Biophys. Res. Commun. 460, 72–81. doi: 10.1016/j.bbrc.2015.01.137
Pace, J., Hayman, M. J., and Galán, J. E. (1993). Signal transduction and invasion of epithelial cells by S. typhimurium.Cell 72, 505–514.
Paciello, I., Silipo, A., Lembo-Fazio, L., Curcurù, L., Zumsteg, A., Noël, G., et al. (2013). Intracellular Shigella remodels its LPS to dampen the innate immune recognition and evade inflammasome activation. Proc. Natl. Acad. Sci. U.S.A. 110, E4345–4354. doi: 10.1073/pnas.1303641110
Pendaries, C., Tronchère, H., Arbibe, L., Mounier, J., Gozani, O., Cantley, L., et al. (2006). PtdIns5P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. EMBO J. 25, 1024–1034. doi: 10.1038/sj.emboj.7601001
Puhar, A., Tronchère, H., Payrastre, B., Tran Van Nhieu, G., and Sansonetti, P. J. (2013). A Shigella effector dampens inflammation by regulating epithelial release of danger signal ATP through production of the lipid mediator PtdIns5P. Immunity 39, 1121–1131. doi: 10.1016/j.immuni.2013.11.013
Rathman, M., de Lanerolle, P., Ohayon, H., Gounon, P., and Sansonetti, P. (2000). Myosin light chain kinase plays an essential role in S. flexneri dissemination. J. Cell Sci. 113(Pt 19), 3375–3386.
Ray, K., Bobard, A., Danckaert, A., Paz-Haftel, I., Clair, C., Ehsani, S., et al. (2010). Tracking the dynamic interplay between bacterial and host factors during pathogen-induced vacuole rupture in real time. Cell. Microbiol. 12, 545–556. doi: 10.1111/j.1462-5822.2010.01428.x
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell. Biol. 13, 566–578. doi: 10.1038/nrm3412
Romero, S., Grompone, G., Carayol, N., Mounier, J., Guadagnini, S., Prévost, M. C., et al. (2011). ATP-mediated Erk1/2 activation stimulates bacterial capture by filopodia, which precedes Shigella invasion of epithelial cells. Cell Host Microbe 9, 508–519. doi: 10.1016/j.chom.2011.05.005
Ruschkowski, S., Rosenshine, I., and Finlay, B. B. (1992). Salmonella typhimurium induces an inositol phosphate flux in infected epithelial cells. FEMS Microbiol. Lett. 74, 121–126.
Sáez, J. C., and Leybaert, L. (2014). Hunting for connexin hemichannels. FEBS Lett. 588, 1205–1211. doi: 10.1016/j.febslet.2014.03.004
Sansonetti, P. J., Arondel, J., Cantey, J. R., Prévost, M. C., and Huerre, M. (1996). Infection of rabbit Peyer's patches by Shigella flexneri: effect of adhesive or invasive bacterial phenotypes on follicle-associated epithelium. Infect. Immun. 64, 2752–2764.
Schroeder, G. N., and Hilbi, H. (2008). Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin. Microbiol. Rev. 21, 134–156. doi: 10.1128/CMR.00032-07
Schwan, C., Kruppke, A. S., Nölke, T., Schumacher, L., Koch-Nolte, F., Kudryashev, M., et al. (2014). Clostridium difficile toxin CDT hijacks microtubule organization and reroutes vesicle traffic to increase pathogen adherence. Proc. Natl. Acad. Sci. U.S.A. 111, 2313–2318. doi: 10.1073/pnas.1311589111
Simpson, C., Kelsell, D. P., and Marchès, O. (2013). Connexin 26 facilitates gastrointestinal bacterial infection in vitro. Cell Tissue Res. 351, 107–116. doi: 10.1007/s00441-012-1502-9
Smedler, E., and Uhlén, P. (2014). Frequency decoding of calcium oscillations. Biochim. Biophys. Acta. 1840, 964–969. doi: 10.1016/j.bbagen.2013.11.015
Sukumaran, S. K., Fu, N. Y., Tin, C. B., Wan, K. F., Lee, S. S., and Yu, V. C. (2010). A soluble form of the pilus protein FimA targets the VDAC-hexokinase complex at mitochondria to suppress host cell apoptosis. Mol. Cell 37, 768–783. doi: 10.1016/j.molcel.2010.02.015
Tattoli, I., Sorbara, M. T., Vuckovic, D., Ling, A., Soares, F., Carneiro, L. A., et al. (2012). Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11, 563–575. doi: 10.1016/j.chom.2012.04.012
Tran Van Nhieu, G., Clair, C., Bruzzone, R., Mesnil, M., Sansonetti, P., and Combettes, L. (2003). Connexin-dependent inter-cellular communication increases invasion and dissemination of Shigella in epithelial cells. Nat. Cell. Biol. 5, 720–726. doi: 10.1038/ncb1021
Tran Van Nhieu, G., Kai Liu, B., Zhang, J., Pierre, F., Prigent, S., Sansonetti, P., et al. (2013). Actin-based confinement of calcium responses during Shigella invasion. Nat. Commun. 4, 1567. doi: 10.1038/ncomms2561
Tsai, F. C., Kuo, G. H., Chang, S. W., and Tsai, P. J. (2015). Ca2+ signaling in cytoskeletal reorganization, cell migration, and cancer metastasis. Biomed. Res. Int. 2015:409245. doi: 10.1155/2015/409245
Tsalikis, J., Tattoli, I., Ling, A., Sorbara, M. T., Croitoru, D. O., Philpott, D. J., et al. (2015). Intracellular bacterial pathogens trigger the formation of U small nuclear RNA bodies (U Bodies) through metabolic stress induction. J. Biol. Chem. 290, 20904–20918. doi: 10.1074/jbc.M115.659466
Valencia-Gallardo, C. M., Carayol, N., and Tran Van Nhieu, G. (2015). Cytoskeletal mechanics during Shigella invasion and dissemination in epithelial cells. Cell. Microbiol. 17, 174–182. doi: 10.1111/cmi.12400
Keywords: Shigella invasion, Ca2+ signaling, mitochondria-induced cell death, host cell survival, inflammation
Citation: Bonnet M and Tran Van Nhieu G (2016) How Shigella Utilizes Ca2+ Jagged Edge Signals during Invasion of Epithelial Cells. Front. Cell. Infect. Microbiol. 6:16. doi: 10.3389/fcimb.2016.00016
Received: 10 December 2015; Accepted: 25 January 2016;
Published: 10 February 2016.
Edited by:
Alfredo G. Torres, University of Texas Medical Branch, USAReviewed by:
William D. Picking, University of Kansas, USAAmanda G. Oglesby-Sherrouse, University of Maryland School of Pharmacy, USA
Copyright © 2016 Bonnet and Tran Van Nhieu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guy Tran Van Nhieu, Z3V5LnRyYW4tdmFuLW5oaWV1QGNvbGxlZ2UtZGUtZnJhbmNlLmZy