 Nora Mellouk
Nora Mellouk Jost Enninga*
Jost Enninga*- Dynamics of Host-Pathogen Interactions Unit, Institut Pasteur, Paris, France
Shigella is a Gram-negative bacterial pathogen, which causes bacillary dysentery in humans. A crucial step of Shigella infection is its invasion of epithelial cells. Using a type III secretion system, Shigella injects several bacterial effectors ultimately leading to bacterial internalization within a vacuole. Then, Shigella escapes rapidly from the vacuole, it replicates within the cytosol and spreads from cell-to-cell. The molecular mechanism of vacuolar rupture used by Shigella has been studied in some detail during the recent years and new paradigms are emerging about the underlying molecular events. For decades, bacterial effector proteins were portrayed as main actors inducing vacuolar rupture. This includes the effector/translocators IpaB and IpaC. More recently, this has been challenged and an implication of the host cell in the process of vacuolar rupture has been put forward. This includes the bacterial subversion of host trafficking regulators, such as the Rab GTPase Rab11. The involvement of the host in determining bacterial vacuolar integrity has also been found for other bacterial pathogens, particularly for Salmonella. Here, we will discuss our current view of host factor and pathogen effector implications during Shigella vacuolar rupture and the steps leading to it.
Introduction
Upon type 3 secretion system (T3SS)-triggered internalization into epithelial cells, Shigella rapidly ruptures its vacuolar membrane to reach the host cytosol for proliferation and cell-to-cell spread. Despite the importance of vacuolar rupture for intracellular bacterial proliferation and propagation, the underlying molecular mechanism has only recently been studied in more detail, and we still lack a precise understanding of the overall processes leading to and determining it (Ray et al., 2009, 2010; Carayol and Tran Van Nhieu, 2013a). Here, we will give an overview on the formation of the Shigella-containing vacuole, we will discuss the involvment of bacterial and host factors in the destablization of the vacuolar membrane, and we will compare it with vacuolar rupture by other bacterial pathogens.
The Steps Leading to the Formation of the Shigella-Containing Vacuole
Shigella internalization relies on elaborate plasma membrane and actin rearrangements, which are spatiotemporally controlled by the interplay between bacterial and cellular factors. Several Shigella T3SS effectors, namely IpaB/IpaC, IpgB1/2, and IpaD directly or indirectly modulate GTPase activation of the Rho family, including Rac1, Cdc42, and RhoA as well as host kinases to promote actin polymerization at the bacterial entry site. This leads to plasma membrane reorganization required for efficient formation of the Shigella-containing vacuole (Mounier et al., 1999; Carayol and Tran Van Nhieu, 2013a). Concomitantly, Shigella also alters the cellular levels of phosphoinositides (PIs) within targeted cells through the T3SS effector IpgD, thereby subverting several host pathways. IpgD is a phosphatidylinositol-phosphatase that specifically depletes PI(4,5)P2 resulting in the formation of PI(5)P (Niebuhr et al., 2002). IpgD is not required for bacterial entry, however it impacts on the way how the bacterium is internalized: Unlike the Shigella wildtype (WT) invasion site characterized by massive filopodia-like extensions and membrane ruffles, ipgD only induce a small albeit dense actin cup at the vicinity of the bacteria. Strikingly, ectopic expression of IpgD in epithelial cells results in a decrease in membrane-cytoskeleton tethering force and eventually causes membrane blebbing (Allaoui et al., 1993; Niebuhr et al., 2000, 2002; Mellouk et al., 2014). Therefore, it is likely that the depletion of PI(4,5)P2 by IpgD weakens plasma membrane-cortical actin interactions thereby facilitating membrane extensions (Saarikangas et al., 2010). In addition, IpgD-dependent production of PI(5)P during the internalization process of Shigella in epithelial cells recruits and activates the epidermal growth factor receptor (EGFR), independently of its bona fide ligand. In turn, EGFR activation stimulates the PI3K/Akt pathway activation. Importantly, PI(5)P production mediates a sustained Akt activation by promoting accumulation of active EGFR in early endosomes (EEs), protecting it from lysosomal degradation (Pendaries et al., 2006; Ramel et al., 2011). During these processes, PI(5)P allows the recruitment of the adaptor protein TOM1, which delays EGFR degradation and bulk endocytosis (Boal et al., 2015). Together, these findings suggest a primordial role of IpgD in the remodeling of the plasma membrane and the underlying actin cortex, as well as a regulator of lipid signaling during the communication of the Shigella-containing vacuole with its surrounding.
Direct Destabilization of Shigella-Containing Vacuoles by Bacterial Effectors
The step of vacuolar rupture takes place rapidly after bacterial internalization within epithelial cells. Studies that allow its tracking in real time revealed that the Shigella-containing vacuole gets damaged in <10 min (Paz et al., 2010; Ray et al., 2010). Early work implicated the T3SS effectors/translocators IpaB and IpaC in vacuolar rupture due to their ability to insert into the host cell membrane for the delivery of bacterial effectors. Collectively, these studies demonstrated that IpaB and IpaC can disrupt lipid vesicles (liposomes) in vitro and are required for contact-mediated hemolysis by Shigella as well as bacterial phagosomal escape in macrophages (High et al., 1992; Ménard et al., 1993; De Geyter et al., 1997; Blocker et al., 1999; De Geyter et al., 2000). In addition, IpaB was shown to be required for Shigella-induced macrophage death via direct binding and activation of caspase-1 (High et al., 1992; Hilbi et al., 1998). However, because IpaB and IpaC act both, as translocators and effectors, discriminating between these two functions represents a major experimental difficulty, both in macrophages and in epithelial cells. Furthermore, since bacterial uptake into epithelial cells also depends on a functional T3SS, it has been challenging to assess the direct contribution of IpaB and IpaC in the subsequent step of vacuolar escape in this cell type. Two recent studies provided new insight on IpaB function(s) using meticulous purification protocols allowing the preservation of its active conformation. Senerovic et al. showed that purified IpaB could oligomerize and form large channels, which would promote potassium influx at acidic pH. They further showed that IpaB-induced ions fluxes could activate caspase-1 via the inflammasome, thereby promoting macrophage pyroptosis. Intriguingly, the reported caspase-1/inflammasome-mediated death of macrophages required endocytosis of purified IpaB in a dynamin-dependent manner, however IpaB remained at the surface of epithelial cells (showing a defect of internalization) and thus did not affect their viability (Senerovic et al., 2012). In addition, Dickenson et al. showed that besides forming large channels, purified IpaB could also assemble into tetramers, and form small pore-like structures that presumably serve as scaffold for translocon insertion into membrane (Dickenson et al., 2013). Together, these works highlight distinct functions of IpaB, either serving as a structural translocator component for bacterial effector delivery into host cells or as a potent effector to induce endolysosomal leakage and promote macrophages pyroptosis. Noteworthy, T3SS translocators with high homology to IpaB and IpaC are present in a number of vacuolar-bound Gram-negative bacterial pathogens such as Salmonella and Yersinia. Moreover, contact-mediated hemolysis also occurs with Yersinia and Salmonella and also requires their respective translocator YopB and SipB (Håkansson et al., 1996; Hume et al., 2003; Coburn et al., 2007). Collectively, this suggests additional or other mechanisms controlling vacuolar membrane integrity during Shigella infection. For instance, the T3SS effector IpaH7.8 may promote Shigella vacuolar escape in macrophages, although its function is not yet established (Fernandez-Prada et al., 2000). Once Shigella gains access to the cytosol, vacuolar membrane remnants are polyubiquitinated, they recruit autophagic markers and adaptors such as LC3 and p62 as well as inflammasome components, and they are targeted to autophagic degradation, thereby dampening inflammatory response and promoting host cell survival (Dupont et al., 2009). LC3 has also been found surrounding the double-membranous vacuoles that Shigella forms after cell-to-cell spread, and two bacterial factors, namely IcsB and VirA have been implicated in the process of bacterial escape interfering with the LC3 recruitment (Campbell-Valois et al., 2015). Along these lines, constituents of the autophagy machinery have been shown to repair damaged vacuoles during Salmonella enterica infection (Kreibich et al., 2015). This indicates a link between the autophagy machinery, membrane repair, and their subversion through bacterial effectors, nevertheless an understanding of the underlying mechanism has remained illusive and requires further studies.
Identification of Host Factors Involved in Shigella Vacuolar Rupture
Compared to intravacuolar pathogens, considerably less is known about the interplay between cytosolic pathogens with the endocytic/exocytic pathways prior to their vacuolar escape, very likely because they rapidly escape into the cytosol and therefore were mostly assumed to not interact selectively with the host vesicular trafficking (Cossart and Roy, 2010; Fredlund and Enninga, 2014). Nonetheless a better understanding of the early events of vacuolar progression preceding cytosolic escape could provide novel insights into the pre-requisite for vacuolar rupture and potentially unravel new host factors subverted by bacterial pathogens to promote intracellular survival and proliferation. Noteworthy, Listeria has been shown to delay Rab5-GDP exchange and expression of constitutively active Rab5 induced bacterial degradation whereas dominant-negative Rab5 promoted bacterial survival and proliferation (Prada-Delgado et al., 2005). Albeit the mechanism of Rab5 modulation by Listeria is not yet established, it suggests that Listeria has evolved a strategy to avoid vacuolar maturation along the endolysosomal pathway prior to its escape into the cytosol.
To explore the involvement of host trafficking pathways in Shigella vacuolar rupture, an imaging-based vacuolar reporter assay was implemented in conjunction with siRNA screening (Ray et al., 2010; Keller et al., 2013). High-content/medium-throughput screens using an siRNA library targeting membrane traffic identified multiple host factors likely involved in Shigella uptake and vacuolar membrane rupture (Mellouk et al., 2014). In agreement with a subversion of the host cytoskeleton machinery to trigger bacterial entry, several host factors were found that promote actin polymerization such as the nucleator ARP2/3 complex and the Rho-GTPase Cdc42 (Tran Van Nhieu et al., 1999; Ehsani et al., 2012; Carayol and Tran Van Nhieu, 2013b). More surprisingly, a subset of endosomal factors was identified, particularly involved in sorting and/or recycling pathways notably including the EE markers Rab5 and EEA1, the sorting nexins (SNX1 and 2), and the recycling endosome markers Rab4 and Rab11. Accordingly, these findings indicate a complex mechanism underlying Shigella vacuolar escape that implicates the hijacking of several host pathways by the pathogen to efficiently gain access to the host cytosol, pinpointing a key role of endocytic and recycling regulators.
Host Membrane Trafficking Regulators and Vacuolar Rupture
The intravacuolar lifestyle requires the subversion of distinct endosomal and/or secretory pathways to diverge from the degradative lysosomal pathway and to build a unique niche for bacterial survival and proliferation. Accordingly, intravacuolar pathogens are well-described to selectively interact with host vesicles notably by hijacking key regulators such as Rab GTPases, SNAREs and PIPs to allow nutrient acquisition and vacuole expansion (Brumell and Scidmore, 2007; Weber et al., 2009; Cossart and Roy, 2010). Strikingly, altering the interplay of pathogen-containing vacuoles with the host membrane trafficking machinery has been shown to have dramatic consequences on their intracellular fate (Kumar and Valdivia, 2009; Creasey and Isberg, 2014). For instance, expression of either constitutively active form of Rab5 or dominant-negative form of Rab7 results in Salmonella-containing vacuole (SCV) disruption, implying that the Rab5-to-Rab7 conversion that promotes subsequent fusion with late endosomes (LEs) is crucial to maintain the SCV integrity (Brumell et al., 2002). Accumulating evidences suggest that Legionella manipulates multiple host cellular pathways using a wide range of type 4 secretion system effectors to promote intracellular growth in a partially redundant manner (Isberg et al., 2009; Hilbi and Haas, 2012). Two studies particularly support this emerging theme. Hoffmann et al. undertook a proteomic analysis of purified Legionella-containing vacuoles (LCVs) from macrophages and identified 9 Rabs (Rab1, 2, 4, 8, 10, 11, 14, 21, and 32) specifically localized on WT but not icmT mutant LCVs. Strikingly, subsequent knockdown of Rabs by siRNA showed that a subset of endocytic Rabs such as Rab5, Rab14, and Rab21 restrict intracellular replication whereas several secretory Rabs including Rab8, Rab10, and Rab32 had the opposite effect by promoting intracellular growth. Surprisingly, although numerous effectors target Rab1 function, knocking down Rab1 did not impair bacterial growth (Hoffmann et al., 2014). In this regard, O'Connor et al. implemented an innovative genetic screening approach that combines bacterial mutants and siRNA targeting of host factors to uncover the importance of functional redundancy of Legionella virulence. In particular, they revealed that the Legionella double mutant LidA/WipB exhibited intracellular growth defects concomitant with an increase in LCV disruption, ultimately promoting both bacterial degradation and macrophages apoptosis (O'Connor et al., 2012).
As mentioned above, the siRNA screen on host factors involved in Shigella vacuolar escape suggested that endocytic and recycling pathways could be involved in the early stage of Shigella invasion (Mellouk et al., 2014). Live-imaging and quantification of the obtained data revealed the massive accumulation of Rab11-positive vesicles at the invasion site of Shigella prior to vacuolar rupture (Figure 1, left side). It also indicated the transitory presence of Rab5-positive vesicles whereas Rab4-positive vesicles were not enriched. Because Shigella rapidly escapes into the cytosol in less than 10 min, such accumulation of Rab11 at the invasion site was unforeseen (Paz et al., 2010; Ray et al., 2010). Indeed, although a number of intravacuolar pathogens including Salmonella, Legionella, and Chlamydia also recruit Rab11-positive vesicles, it commonly appears to be at an “intermediate or late” stage of vacuolar maturation presumably promoting bacterial proliferation by delivering nutrients and membrane to the growing bacterial vacuoles (Smith, 2005; Rejman Lipinski et al., 2009; Hoffmann et al., 2014).
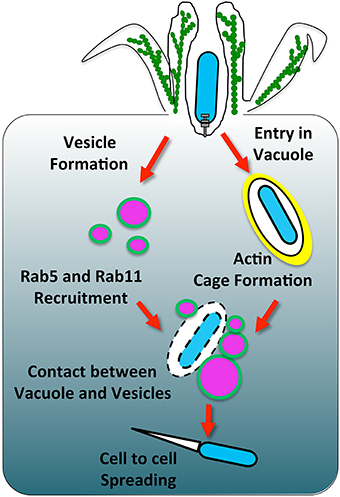
Figure 1. The involvement of host membrane trafficking in Shigella vacuolar rupture. Shigella induces its uptake into a vacuole that gets surrounded by an actin cage. Concomitantly, vesicles around the bacteria are modulated by IpgD to deplete PI(4,5)P2 and form PI(5)P (highlighted in green). This change of lipid content leads to the recruitment of the Rab GTPases Rab5 and Rab11. Vesicles around the Shigella-containing vacuole make contact with the bacterial compartment. During these contacts, the vacuole ruptures through a mechanism that requires further characterization.
Rab11 knockdown significantly delayed the vacuolar rupture of Shigella without disturbing its entry into epithelial cells (Mellouk et al., 2014). Interestingly, a genome-wide siRNA screen for Chlamydia infection in Drosophila cells where Listeria infection was also assessed in a secondary screen indicated that Rab11 knockdown inhibits both Chlamydia and Listeria infection (Derré et al., 2007). Later on, it was shown that both Rab6 and Rab11 knockdown prevent Golgi fragmentation induced by Chlamydia, thereby causing a defect in bacterial growth by impairing lipid transport to the inclusion (Rejman Lipinski et al., 2009). The preliminary results concerning Listeria require further investigations in a more physiological model, but it is tempting to speculate that the findings on Rab11 involvement in Shigella vacuolar rupture may be extended to other cytosolic pathogens.
A Link between Rab11 and PI Subversion
Several pathogens hijack Rab11 cellular function, however we only start to understand the underlying mechanism for controlling Rab11 recruitment to the bacteria-containing vacuole (Guichard et al., 2014). Nevertheless, a widespread strategy used by intravacuolar pathogens to diverge from the degradative pathway and establish their replicative niche is the subversion of phosphoinositides (Weber et al., 2009). For instance, a key feature of Legionella-containing vacuole maturation is the accumulation of PI(4)P, most likely involving bacterial and host cellular phosphatases and kinases. In turn, PI(4)P anchors the effector DrrA (also called SidM), a Rab1-GEF required for Rab1 recruitment to the LCV (Machner and Isberg, 2006; Brombacher et al., 2009; Weber et al., 2013). Mycobacterium depletes PI(3)P by inhibiting the host PI3K hVPS34 and by the bacterial PI(3)P phosphatase, SapM thereby preventing further phagolysosomal maturation (Fratti et al., 2003; Vergne et al., 2005). In contrast, Salmonella generates PI(3)P on the SCV via the T3SS effector SopB to subsequently promote fusion with LEs. Although SopB may directly hydrolyse PI(3,4,5)P3 at the plasma membrane or PI(3,5) P2 on endosomes, it seems that SopB indirectly mediates PI(3)P accumulation on the SCV presumably by depleting PI(4,5)P2 to promote the recruitment of Rab5 and its effector hVPS34 (Marcus et al., 2001; Terebiznik et al., 2002; Hernandez et al., 2004; Mallo et al., 2008). Recently, Bakowski et al. showed that SopB depletion of PI(4,5)P2 increases the surface charge of the SCV, thereby preventing the accumulation of Rab8B, 13, 23, and 35 commonly targeted to the plasma membrane through their polycationic prenyl motif, and this potentially plays a role in the avoidance of the SCV-lysosome fusion (Bakowski et al., 2010). Collectively, bacterial pathogens thoroughly subvert PIs, which alter membrane identity subsequently affecting trafficking. Likewise, Shigella also modulates PIs through its effector IpgD, which acts as a phosphatidylinositol-phosphatase that specifically dephosphorylates PI(4,5)P2 into PI(5)P (Niebuhr et al., 2002). Results by us showed that the IpgD inositol phosphatase activity is absolutely required for the recruitment of Rab11-positive vesicles to the invasion site of Shigella. Using large volume correlative light and electron microscopy (CLEM), the ultrastructural details of the Shigella WT and ipgD invasion site were further characterized within epithelial cells and revealed that indeed Rab11-positive vesicles accumulate at the vicinity of the Shigella WT-containing vacuole but not around the ipgD-containing vacuole. More broadly barely any vesicles were observed at the ipgD invasion site, indicating that IpgD is not only required for Rab11 recruitment but also for the abundant presence of miscellaneous vesicles at the bacterial invasion site. The origin of these vesicles requires further investigations but based on their heterogeneous morphology we suggest that they could undergo successive fusion and fission events.
At the Shigella invasion site (characterized by membrane and actin rearrangements) it was found that: (i) PI(4,5)P2 was dramatically depleted in an IpgD-dependent manner, (ii) its precursor PI(4)P was enriched in an IpgD-independent manner presumably together with membrane ruffling rather than true enrichment, (iii) PI(5)P was enriched in an IpgD-dependent manner, and (iv) PI(3)P was not enriched in both cases (Mellouk et al., 2014). Secondly, at the Shigella-containing vacuole neither PI(4,5)P2, PI(5)P, PI(4)P, nor PI(3)P were found enriched, independently of IpgD. However, PI(3)P dynamically localized to a number of large vesicles present at the invasion site of Shigella WT but not ipgD, resembling PI(3)P-positive macropinosomes. These recent results corroborate the study from Niebuhr et al. where biochemical analysis revealed a global drop of PI(4,5)P2 cellular levels with a concomitant increase in PI(5)P controlled by the IpgD effector (Niebuhr et al., 2002). Furthermore, in our own studies we observed a certain enrichment of PI(5)P in the surrounding of the Shigella-containing vacuole but not particularly on its surface similarly to the previously reported data (Pendaries et al., 2006).
Yet in the case of Salmonella, SopB (homolog of IpgD) not only depletes PI(4,5)P2 at the plasma membrane but is also required for PI(4,5)P2 clearance at the surface of the SCV, thereby pinpointing a divergence between Salmonella and Shigella remodeling of PIs (Terebiznik et al., 2002; Mallo et al., 2008). In addition, despite the presence of large PI(3)P-positive vesicles at the invasion site, the Shigella-containing vacuole remains PI(3)P-negative. This corroborates the findings on Rab5 which is also transiently recruited at the invasion site without decorating the Shigella-containing vacuole (Mellouk et al., 2014). By contrast, as mentioned above Salmonella promotes the recruitment of Rab5 and the accumulation of PI(3)P on the SCV via SopB. Collectively, this suggested that Shigella diverges very rapidly from the classical endosomal (and intravacuolar pathogen) maturation by altering the PIP signature on the Shigella-containing vacuole and its surrounding trafficking notably by recruiting Rab11 and avoiding Rab5, ultimately leading to its vacuolar escape. Further studies are required to investigate the molecular mechanism of Rab11 recruitment via IpgD-mediated modulation and to decipher how Shigella avoids PI(3)P/Rab5, thereby shedding light on the intricate interplay between Rabs and PIPs.
Actin Surrounding the Shigella-Containing Vacuole and Its Rupture
In agreement with the fact that Rab11 promotes efficient vacuolar rupture and that ipgD completely abolishes the Rab11 recruitment to the invasion site, an ipgD mutant delays vacuolar rupture significantly. The ipgD mutant-containing vacuoles are predominantly surrounded by an F-actin meshwork we would like to coin an “actin cage,” which appears to confine the bacteria for extended periods of time. In contrast, Shigella WT-containing vacuoles are rarely observed confined within actin cages, suggesting that Shigella either prevents or rapidly disassembles the actin cage in an IpgD-dependent manner prior to vacuolar escape (Figure 1, right side). Previous observations of actin cages around Shigella WT-containing vacuoles have been reported, however the underlying mechanism controlling their dynamics remain uncovered (Ehsani et al., 2012). Given that PI(4,5)P2 dynamically controls numerous actin-binding proteins, the actin cage dynamics may be directly coordinated by IpgD modulation of the PI(4,5)P2 level at the invasion site. In particular, IpgD-mediated PI(4,5)P2 depletion is believed to be important for disrupting the linkage between the plasma membrane and cortical actin, thereby promoting membrane protrusions. In a similar manner, IpgD could prevent F-actin linkage to the vacuolar membrane, potentially by acting on cortical actin regulators such as the ERM-family proteins (Niebuhr et al., 2002; Fehon et al., 2010; Saarikangas et al., 2010). Remarkably, chemical depolymerization of the actin cage structure around the ipgD mutant-containing vacuole induces rapid rupture, further supporting that the actin cage confinement impairs efficient vacuolar escape (Mellouk et al., 2014). Noteworthy, Salmonella and Chlamydia promote the assembly of an F-actin meshwork resembling the actin cage structures observed in our study, which are crucial to maintain their vacuole integrity (Méresse et al., 2001; Kumar and Valdivia, 2008). Similarly, the actin cage could stabilize the ipgD mutant-containing vacuole by direct structural support.
Vacuolar Rupture By other Bacterial Pathogens
Interestingly, intravacuolar pathogens have evolved intricate strategies to maintain vacuolar integrity concomitantly with vacuolar expansion to accommodate bacterial replication. Similarly to the process of vacuolar rupture induced by cytosolic pathogens, the process of vacuolar integrity maintenance is not well-understood. However, it appears that the recruitment of potentially “destabilizing” host factors needs to be counterbalance to avoid vacuolar rupture (Kumar and Valdivia, 2009; Creasey and Isberg, 2014). For instance, numerous Salmonella effectors regulate the interaction of the SCV with cytoskeletal motors. In particular, PipB2 and SopD2 recruit kinesin-1 whereas SifA binds to the host SKIP to promote Sif formation. Importantly, in the absence of SifA, kinesin-1 accumulates on the SCV leading to vacuolar rupture (Beuzón et al., 2000; Boucrot et al., 2005; Dumont et al., 2010; Schroeder et al., 2010). In contrast, the Salmonella double mutant sifA/sseJ maintains vacuolar integrity, indicating that SseJ rather promotes vacuolar destabilization. SseJ is a phospholipase that promotes the depletion of cholesterol from the SCV, thus increasing membrane fluidity that may facilitate tubulation from the SCV but may also increase sensitivity to cytoskeleton motor-dependent forces leading to a loss of vacuolar integrity (Ohlson et al., 2005; Lossi et al., 2008). Likewise, Legionella secretes PlaA (homolog to SseJ), which also lead to vacuolar rupture in the absence of another bacterial effector, SdhA (Creasey and Isberg, 2012).
Vacuolar rupture induced by Listeria monocytogenes or Rickettsia was commonly believed to be directly and solely driven by bacterial effectors, notably through the pore-forming toxin listeriolysin O (LLO) and phospholipases (such as Listeria phospholipases C and Rickettsia phospholipases A and D) (Whitworth et al., 2005; Pizarro-Cerdá et al., 2012; Rahman et al., 2013). This has been changing due to an increasing number of reports revealing the implication of host factors in the process of vacuolar rupture for a number of bacterial pathogens. For instance, LLO-mediated Listeria vacuolar escape requires the host factors gamma-interferon-inducible lysosomal thiol reductase (GILT) and cystic fibrosis transmembrane conductance regulator (CFTR) to potentiate its activity (Singh et al., 2008; Radtke et al., 2011).
In contrast, the molecular mechanisms underlying Rickettsia and Francisella access to the host cytosol are poorly understood. Rickettsia species produce hemolysin C and phospholipases that seem to play a role in vacuolar escape (Renesto et al., 2003; Whitworth et al., 2005; Rahman et al., 2013). On the other hand, the Francisella-containing vacuole (FCV) subverts the endolysosomal route and harbors certain late endosomal markers but is devoid of lysosomal enzymes (Clemens et al., 2009). Acidification of the FCV is important for vacuolar escape and requires the host V-ATPase. Additionally, the host ubiquitin ligase CDC27 seems important to reach the cytosol, although the mechanism remains to be explored (Chong et al., 2008; Akimana et al., 2010). Furthermore, a subset of bacterial factors are involved in phagosomal escape, presumably by injecting some virulence factors such as IglI through a T6SS (Barker et al., 2009). Recently, Ramond et al. revealed that the Francisella glutamate transporter GadC plays a crucial role in phagosomal escape by neutralizing reactive oxygen species (ROS) production within the phagosome linking oxidative stress defense and phagosomal escape for this pathogen (Ramond et al., 2014).
Noteworthy, at late infection (12–18 h after its uptake into macrophages), Legionella is also released into the host cytosol prior to host cell lysis for bacterial dissemination (Molmeret et al., 2004). Recent studies have shown that Mycobacterium species can also escape from their phagosomal vacuole. In dendritic cells, a large portion of M. tuberculosis and M. leprae were found to reach the host cytosol in a T7SS-dependent manner after 2 days of infection (van der Wel et al., 2007). Furthermore, phagosomal escape of M. tuberculosis in macrophages was confirmed through the T7SS and especially requires the T7SS effector ESAT-6 (Simeone et al., 2012, 2015). Importantly, ESAT-6 has been directly implicated in vacuolar escape of the fish pathogen M. marinum by forming small pores into the vacuolar membrane (Smith et al., 2008).
Conclusion
Collectively, it emerges that Shigella and other intracellular bacteria have evolved sophisticated ways to destabilize their vacuole to reach the host cytoplasm. The classic view of a process driven entirely by bacterial effectors or toxins has been amended by more recent findings putting the subversion of host processes in the spotlight. Here, the modulation of PIPs through dedicated effectors, such as SopB in the case of Salmonella and IpgD for Shigella that correlates with altered Rab GTPase recruitment to the bacteria-containing vacuoles drives the fate of this compartment. The precise molecular mechanisms that underlie these events remain to be studied. It is likely that deciphering this will provide fundamental information on how cells regulate endomembrane integrity.
Author Contributions
All authors listed, have made substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank members of the “Dynamics of Host Pathogen Interactions (DIHP)” Research Unit and Guy Tran Van Nhieu (College de France) for fruitful discussions. We thank Allon Weiner for his contribution to the figure and his interest in the work. NM was supported by a fellowship from the Fondation pour la Recherche Medicale and a grant by the Region Ile de France (DIM-Malinf) to JE and Guy Tran Van Nhieu. JE is member of the LabEx consortium IBEID, and is supported by the Institut Pasteur CARNOT-MIE programme. JE also acknowledges support of an ERC starting grant for this work (Rupteffects, Nr. 261166).
References
Akimana, C., Al-Khodor, S., and Abu Kwaik, Y. (2010). Host factors required for modulation of phagosome biogenesis and proliferation of Francisella tularensis within the cytosol. PLoS ONE 5:e11025. doi: 10.1371/journal.pone.0011025
Allaoui, A., Ménard, R., Sansonetti, P. J., and Parsot, C. (1993). Characterization of the Shigella flexneri ipgD and ipgF genes, which are located in the proximal part of the mxi locus. Infect. Immun. 61, 1707–1714.
Bakowski, M. A., Braun, V., Lam, G. Y., Yeung, T., Heo, W., Do Meyer, T., et al. (2010). The phosphoinositide phosphatase SopB manipulates membrane surface charge and trafficking of the Salmonella-containing vacuole. Cell Host Microbe 7, 453–462. doi: 10.1016/j.chom.2010.05.011
Barker, J. R., Chong, A., Wehrly, T. D., Yu, J.-J., Rodriguez, S. A., Liu, J., et al. (2009). The Francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol. Microbiol. 74, 1459–1470. doi: 10.1111/j.1365-2958.2009.06947.x
Beuzón, C. R., Méresse, S., Unsworth, K. E., Ruíz-Albert, J., Garvis, S., Waterman, S. R., et al. (2000). Salmonella maintains the integrity of its intracellular vacuole through the action of SifA. EMBO J. 19, 3235–3249. doi: 10.1093/emboj/19.13.3235
Blocker, A., Gounon, P., Larquet, E., Niebuhr, K., Cabiaux, V., Parsot, C., et al. (1999). The tripartite type III secreton of Shigella flexneri inserts IpaB and IpaC into host membranes. J. Cell Biol. 147, 683–693.
Boal, F., Mansour, R., Gayral, M., Saland, E., Chicanne, G., Xuereb, J.-M., et al. (2015). TOM1 is a PI5P effector involved in the regulation of endosomal maturation. J. Cell Sci. 128, 815–827. doi: 10.1242/jcs.166314
Boucrot, E., Henry, T., Borg, J.-P., Gorvel, J.-P., and Méresse, S. (2005). The intracellular fate of Salmonella depends on the recruitment of kinesin. Science 308, 1174–1178. doi: 10.1126/science.1110225
Brombacher, E., Urwyler, S., Ragaz, C., Weber, S. S., Kami, K., Overduin, M., et al. (2009). Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-phosphate-binding effector protein of Legionella pneumophila. J. Biol. Chem. 284, 4846–4856. doi: 10.1074/jbc.M807505200
Brumell, J. H., and Scidmore, M. A. (2007). Manipulation of rab GTPase function by intracellular bacterial pathogens. Microbiol. Mol. Biol. Rev. 71, 636–652. doi: 10.1128/MMBR.00023-07
Brumell, J. H., Tang, P., Zaharik, M. L., and Finlay, B. B. (2002). Disruption of the Salmonella-containing vacuole leads to increased replication of Salmonella enterica serovar typhimurium in the cytosol of epithelial cells. Infect. Immun. 70, 3264–3270. doi: 10.1128/IAI.70.6.3264-3270.2002
Campbell-Valois, F.-X., Sachse, M., Sansonetti, P. J., and Parsot, C. (2015). Escape of actively secreting Shigella flexneri from ATG8/LC3-positive vacuoles formed during cell-to-cell spread is facilitated by IcsB and VirA. MBio 6, e02567–e02514. doi: 10.1128/mBio.02567-14
Carayol, N., and Tran Van Nhieu, G. (2013a). The inside story of Shigella invasion of intestinal epithelial cells. Cold Spring Harb. Perspect. Med. 3:a016717. doi: 10.1101/cshperspect.a016717
Carayol, N., and Tran Van Nhieu, G. (2013b). Tips and tricks about Shigella invasion of epithelial cells. Curr. Opin. Microbiol. 16, 32–37. doi: 10.1016/j.mib.2012.11.010
Chong, A., Wehrly, T. D., Nair, V., Fischer, E. R., Barker, J. R., Klose, K. E., et al. (2008). The early phagosomal stage of Francisella tularensis determines optimal phagosomal escape and Francisella pathogenicity island protein expression. Infect. Immun. 76, 5488–5499. doi: 10.1128/IAI.00682-08
Clemens, D. L., Lee, B.-Y., and Horwitz, M. A. (2009). Francisella tularensis phagosomal escape does not require acidification of the phagosome. Infect. Immun. 77, 1757–1773. doi: 10.1128/IAI.01485-08
Coburn, B., Sekirov, I., and Finlay, B. B. (2007). Type III secretion systems and disease. Clin. Microbiol. Rev. 20, 535–549. doi: 10.1128/CMR.00013-07
Cossart, P., and Roy, C. R. (2010). Manipulation of host membrane machinery by bacterial pathogens. Curr. Opin. Cell Biol. 22, 547–554. doi: 10.1016/j.ceb.2010.05.006
Creasey, E. A., and Isberg, R. R. (2012). The protein SdhA maintains the integrity of the Legionella-containing vacuole. Proc. Natl. Acad. Sci. U.S.A. 109, 3481–3486. doi: 10.1073/pnas.1121286109
Creasey, E. A., and Isberg, R. R. (2014). Maintenance of vacuole integrity by bacterial pathogens. Curr. Opin. Microbiol. 17, 46–52. doi: 10.1016/j.mib.2013.11.005
De Geyter, C., Vogt, B., Benjelloun-Touimi, Z., Sansonetti, P. J., Ruysschaert, J. M., Parsot, C., et al. (1997). Purification of IpaC, a protein involved in entry of Shigella flexneri into epithelial cells and characterization of its interaction with lipid membranes. FEBS Lett. 400, 149–154.
De Geyter, C., Wattiez, R., Sansonetti, P., Falmagne, P., Ruysschaert, J. M., Parsot, C., et al. (2000). Characterization of the interaction of IpaB and IpaD, proteins required for entry of Shigella flexneri into epithelial cells, with a lipid membrane. Eur. J. Biochem. 267, 5769–5776. doi: 10.1046/j.1432-1327.2000.01649.x
Derré, I., Pypaert, M., Dautry-Varsat, A., and Agaisse, H. (2007). RNAi screen in Drosophila cells reveals the involvement of the Tom complex in Chlamydia infection. PLoS Pathog. 3:e155. doi: 10.1371/journal.ppat.0030155
Dickenson, N. E., Choudhari, S. P., Adam, P. R., Kramer, R. M., Joshi, S. B., Middaugh, C. R., et al. (2013). Oligomeric states of the Shigella translocator protein IpaB provide structural insights into formation of the type III secretion translocon. Protein Sci. 22, 614–627. doi: 10.1002/pro.2245
Dumont, A., Boucrot, E., Drevensek, S., Daire, V., Gorvel, J.-P., Poüs, C., et al. (2010). SKIP, the host target of the Salmonella virulence factor SifA, promotes kinesin-1-dependent vacuolar membrane exchanges. Traffic 11, 899–911. doi: 10.1111/j.1600-0854.2010.01069.x
Dupont, N., Lacas-Gervais, S., Bertout, J., Paz, I., Freche, B., Van Nhieu, G. T., et al. (2009). Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe 6, 137–149. doi: 10.1016/j.chom.2009.07.005
Ehsani, S., Santos, J. C., Rodrigues, C. D., Henriques, R., Audry, L., Zimmer, C., et al. (2012). Hierarchies of host factor dynamics at the entry site of Shigella flexneri during host cell invasion. Infect. Immun. 80, 2548–2557. doi: 10.1128/IAI.06391-11
Fehon, R. G., McClatchey, A. I., and Bretscher, A. (2010). Organizing the cell cortex: the role of ERM proteins. Nat. Rev. Mol. Cell Biol. 11, 276–287. doi: 10.1038/nrm2866
Fernandez-Prada, C. M., Hoover, D. L., Tall, B. D., Hartman, A, B., Kopelowitz, J., and Venkatesan, M. M. (2000). Shigella flexneri IpaH(7.8) facilitates escape of virulent bacteria from the endocytic vacuoles of mouse and human macrophages. Infect. Immun. 68, 3608–3619.
Fratti, R. A., Chua, J., Vergne, I., and Deretic, V. (2003). Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc. Natl. Acad. Sci. U.S.A. 100, 5437–5442. doi: 10.1073/pnas.0737613100
Fredlund, J., and Enninga, J. (2014). Cytoplasmic access by intracellular bacterial pathogens. Trends Microbiol. 22, 128–137. doi: 10.1016/j.tim.2014.01.003
Guichard, A., Nizet, V., and Bier, E. (2014). RAB11-mediated trafficking in host–pathogen interactions. Nat. Rev. Microbiol. 12, 624–634. doi: 10.1038/nrmicro3325
Håkansson, S., Schesser, K., Persson, C., Galyov, E. E., Rosqvist, R., Homblé, F., et al. (1996). The YopB protein of Yersinia pseudotuberculosis is essential for the translocation of Yop effector proteins across the target cell plasma membrane and displays a contact-dependent membrane disrupting activity. EMBO J. 15, 5812–5823.
Hernandez, L. D., Hueffer, K., Wenk, M. R., and Galán, J. E. (2004). Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science 304, 1805–1807. doi: 10.1126/science.1098188
High, N., Mounier, J., Prévost, M. C., and Sansonetti, P. J. (1992). IpaB of Shigella flexneri causes entry into epithelial cells and escape from the phagocytic vacuole. EMBO J. 11, 1991–1999.
Hilbi, H., and Haas, A. (2012). Secretive bacterial pathogens and the secretory pathway. Traffic 13, 1187–1197. doi: 10.1111/j.1600-0854.2012.01344.x
Hilbi, H., Moss, J. E., Hersh, D., Chen, Y., Arondel, J., Banerjee, S., et al. (1998). Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J. Biol. Chem. 273, 32895–32900. doi: 10.1074/jbc.273.49.32895
Hoffmann, C., Finsel, I., Otto, A., Pfaffinger, G., Rothmeier, E., Hecker, M., et al. (2014). Functional analysis of novel Rab GTPases identified in the proteome of purified Legionella-containing vacuoles from macrophages. Cell. Microbiol. 16, 1034–1052. doi: 10.1111/cmi.12256
Hume, P. J., McGhie, E. J., Hayward, R. D., and Koronakis, V. (2003). The purified Shigella IpaB and Salmonella SipB translocators share biochemical properties and membrane topology. Mol. Microbiol. 49, 425–439. doi: 10.1046/j.1365-2958.2003.03559.x
Isberg, R. R., O'Connor, T. J., and Heidtman, M. (2009). The Legionella pneumophila replication vacuole: making a cosy niche inside host cells. Nat. Rev. Microbiol. 7, 13–24. doi: 10.1038/nrmicro1967
Keller, C., Mellouk, N., Danckaert, A., Simeone, R., Brosch, R., Enninga, J., et al. (2013). Single cell measurements of vacuolar rupture caused by intracellular pathogens. J. Vis. Exp. e50116. doi: 10.3791/50116
Kreibich, S., Emmenlauer, M., Fredlund, J., Rämö, P., Münz, C., Dehio, C., et al. (2015). Autophagy proteins promote repair of endosomal membranes damaged by the salmonella type three secretion system 1. Cell Host Microbe 18, 527–537. doi: 10.1016/j.chom.2015.10.015
Kumar, Y., and Valdivia, R. (2009). Leading a sheltered life: intracellular pathogens and maintenance of vacuolar compartments. Cell Host Microbe 5, 593–601. doi: 10.1016/j.chom.2009.05.014
Kumar, Y., and Valdivia, R. H. (2008). Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe 4, 159–169. doi: 10.1016/j.chom.2008.05.018
Lossi, N. S., Rolhion, N., Magee, A. I., Boyle, C., and Holden, D. W. (2008). The Salmonella SPI-2 effector SseJ exhibits eukaryotic activator-dependent phospholipase A and glycerophospholipid : cholesterol acyltransferase activity. Microbiology 154, 2680–2688. doi: 10.1099/mic.0.2008/019075-0
Machner, M. P., and Isberg, R. R. (2006). Targeting of host Rab GTPase function by the intravacuolar pathogen Legionella pneumophila. Dev. Cell 11, 47–56. doi: 10.1016/j.devcel.2006.05.013
Mallo, G. V., Espina, M., Smith, A. C., Terebiznik, M. R., Alemán, A., Finlay, B. B., et al. (2008). SopB promotes phosphatidylinositol 3-phosphate formation on Salmonella vacuoles by recruiting Rab5 and Vps34. J. Cell Biol. 182, 741–752. doi: 10.1083/jcb.200804131
Marcus, S. L., Wenk, M. R., Steele-Mortimer, O., and Finlay, B. B. (2001). A synaptojanin-homologous region of Salmonella typhimurium SigD is essential for inositol phosphatase activity and Akt activation. FEBS Lett. 494, 201–207. doi: 10.1016/S0014-5793(01)02356-0
Ménard, R., Sansonetti, P. J., and Parsot, C. (1993). Nonpolar mutagenesis of the ipa genes defines IpaB, IpaC, and IpaD as effectors of Shigella flexneri entry into epithelial cells. J. Bacteriol. 175, 5899–5906.
Mellouk, N., Weiner, A., Aulner, N., Schmitt, C., Elbaum, M., Shorte, S. L., et al. (2014). Shigella subverts the host recycling compartment to rupture its vacuole. Cell Host Microbe 16, 517–530. doi: 10.1016/j.chom.2014.09.005
Méresse, S., Unsworth, K. E., Habermann, A., Griffiths, G., Fang, F., Martínez-Lorenzo, M. J., et al. (2001). Remodelling of the actin cytoskeleton is essential for replication of intravacuolar Salmonella. Cell. Microbiol. 3, 567–577.
Molmeret, M., Bitar, D. M., Han, L., and Kwaik, Y. A. (2004). Disruption of the phagosomal membrane and egress of Legionella pneumophila into the cytoplasm during the last stages of intracellular infection of macrophages and Acanthamoeba polyphaga. Infect. Immun. 72, 4040–4051. doi: 10.1128/IAI.72.7.4040-4051.2004
Mounier, J., Laurent, V., Hall, A., Fort, P., Carlier, M. F., Sansonetti, P. J., et al. (1999). Rho family GTPases control entry of Shigella flexneri into epithelial cells but not intracellular motility. J. Cell Sci. 112(Pt 1), 2069–2080.
Niebuhr, K., Giuriato, S., Pedron, T., Philpott, D. J., Gaits, F., Sable, J., et al. (2002). Conversion of PtdIns(4,5)P(2) into PtdIns(5)P by the S. flexneri effector IpgD reorganizes host cell morphology. EMBO J. 21, 5069–5078. doi: 10.1093/emboj/cdf522
Niebuhr, K., Jouihri, N., Allaoui, A., Gounon, P., Sansonetti, P. J., and Parsot, C. (2000). IpgD, a protein secreted by the type III secretion machinery of Shigella flexneri, is chaperoned by IpgE and implicated in entry focus formation. Mol. Microbiol. 38, 8–19.
O'Connor, T. J., Boyd, D., Dorer, M. S., and Isberg, R. R. (2012). Aggravating genetic interactions allow a solution to redundancy in a bacterial pathogen. Science 338, 1440–1444. doi: 10.1126/science.1229556
Ohlson, M. B., Fluhr, K., Birmingham, C. L., Brumell, H., Miller, S. I., and Brumell, J. H. (2005). SseJ deacylase activity by Salmonella enterica serovar typhimurium promotes virulence in mice. Infect. Immun 73, 6249–6259. doi: 10.1128/IAI.73.10.6249-6259.2005
Paz, I., Sachse, M., Dupont, N., Mounier, J., Cederfur, C., Enninga, J., et al. (2010). Galectin-3, a marker for vacuole lysis by invasive pathogens. Cell. Microbiol. 12, 530–544. doi: 10.1111/j.1462-5822.2009.01415.x
Pendaries, C., Tronchère, H., Arbibe, L., Mounier, J., Gozani, O., Cantley, L., et al. (2006). PtdIns5P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. EMBO J. 25, 1024–1034. doi: 10.1038/sj.emboj.7601001
Pizarro-Cerdá, J., Kühbacher, A., and Cossart, P. (2012). Entry of Listeria monocytogenes in mammalian epithelial cells: an updated view. Cold Spring Harb. Perspect. Med. 2, 1–17. doi: 10.1101/cshperspect.a010009
Prada-Delgado, A., Carrasco-Marín, E., Peña-Macarro, C., Del Cerro-Vadillo, E., Fresno-Escudero, M., Leyva-Cobián, F., et al. (2005). Inhibition of Rab5a exchange activity is a key step for Listeria monocytogenes survival. Traffic 6, 252–265. doi: 10.1111/j.1600-0854.2005.00265.x
Radtke, A. L., Anderson, K. L., Davis, M. J., DiMagno, M. J., Swanson, J. A., and O'Riordan, M. X. (2011). Listeria monocytogenes exploits cystic fibrosis transmembrane conductance regulator (CFTR) to escape the phagosome. Proc. Natl. Acad. Sci. U.S.A. 108, 1633–1638. doi: 10.1073/pnas.1013262108
Rahman, M. S., Gillespie, J. J., Kaur, S. J., Sears, K. T., Ceraul, S. M., Beier-Sexton, M., et al. (2013). Rickettsia typhi possesses phospholipase A2 enzymes that are involved in infection of host cells. PLoS Pathog. 9:e1003399. doi: 10.1371/journal.ppat.1003399
Ramel, D., Lagarrigue, F., Pons, V., Mounier, J., Dupuis-Coronas, S., Chicanne, G., et al. (2011). Shigella flexneri infection generates the lipid PI5P to alter endocytosis and prevent termination of EGFR signaling. Sci. Signal. 4, ra61. doi: 10.1126/scisignal.2001619
Ramond, E., Gesbert, G., Rigard, M., Dairou, J., Dupuis, M., Dubail, I., et al. (2014). Glutamate utilization couples oxidative stress defense and the tricarboxylic acid cycle in francisella phagosomal escape. PLoS Pathog. 10:e1003893. doi: 10.1371/journal.ppat.1003893
Ray, K., Bobard, A., Danckaert, A., Paz-Haftel, I., Clair, C., Ehsani, S., et al. (2010). Tracking the dynamic interplay between bacterial and host factors during pathogen-induced vacuole rupture in real time. Cell. Microbiol. 12, 545–556. doi: 10.1111/j.1462-5822.2010.01428.x
Ray, K., Marteyn, B., Sansonetti, P. J., and Tang, C. M. (2009). Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat. Rev. Microbiol. 7, 333–340. doi: 10.1038/nrmicro2112
Rejman Lipinski, A., Heymann, J., Meissner, C., Karlas, A., Brinkmann, V., Meyer, T. F., et al. (2009). Rab6 and Rab11 regulate Chlamydia trachomatis development and golgin-84-dependent Golgi fragmentation. PLoS Pathog. 5:e1000615. doi: 10.1371/journal.ppat.1000615
Renesto, P., Dehoux, P., Gouin, E., Touqui, L., Cossart, P., and Raoult, D. (2003). Identification and characterization of a phospholipase D-superfamily gene in rickettsiae. J. Infect. Dis. 188, 1276–1283. doi: 10.1086/379080
Saarikangas, J., Zhao, H., and Lappalainen, P. (2010). Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinositides. Physiol. Rev. 90, 259–289. doi: 10.1152/physrev.00036.2009
Schroeder, N., Henry, T., de Chastellier, C., Zhao, W., Aude-Agnés, G., Gorvel, J. P., et al. (2010). The virulence protein SopD2 regulates membrane dynamics of Salmonella-containing vacuoles. PLoS Pathog. 6:e1001002. doi: 10.1371/journal.ppat.1001002
Senerovic, L., Tsunoda, S. P., Goosmann, C., Brinkmann, V., Zychlinsky, A., Meissner, F., et al. (2012). Spontaneous formation of IpaB ion channels in host cell membranes reveals how Shigella induces pyroptosis in macrophages. Cell Death Dis. 3, e384. doi: 10.1038/cddis.2012.124
Simeone, R., Bobard, A., Lippmann, J., Bitter, W., Majlessi, L., Brosch, R., et al. (2012). Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog. 8:e1002507. doi: 10.1371/journal.ppat.1002507
Simeone, R., Sayes, F., Song, O., Gröschel, M. I., Brodin, P., Brosch, R., et al. (2015). Cytosolic access of Mycobacterium tuberculosis: critical impact of phagosomal acidification control and demonstration of occurrence in vivo. PLOS Pathog. 11:e1004650. doi: 10.1371/journal.ppat.1004650
Singh, R., Jamieson, A., and Cresswell, P. (2008). GILT is a critical host factor for Listeria monocytogenes infection. Nature 455, 1244–1247. doi: 10.1038/nature07344
Smith, A. C. (2005). Interaction of the Salmonella-containing vacuole with the endocytic recycling system. J. Biol. Chem. 280, 24634–24641. doi: 10.1074/jbc.M500358200
Smith, J., Manoranjan, J., Pan, M., Bohsali, A., Xu, J., Liu, J., et al. (2008). Evidence for pore formation in host cell membranes by ESX-1-secreted ESAT-6 and its role in Mycobacterium marinum escape from the vacuole. Infect. Immun. 76, 5478–5487. doi: 10.1128/IAI.00614-08
Terebiznik, M. R., Vieira, O. V., Marcus, S. L., Slade, A., Yip, C. M., Trimble, W. S., et al. (2002). Elimination of host cell PtdIns(4,5)P(2) by bacterial SigD promotes membrane fission during invasion by Salmonella. Nat. Cell Biol. 4, 766–773. doi: 10.1038/ncb854
Tran Van Nhieu, G., Caron, E., Hall, A., and Sansonetti, P. J. (1999). IpaC induces actin polymerization and filopodia formation during Shigella entry into epithelial cells. EMBO J. 18, 3249–3262.
van der Wel, N., Hava, D., Houben, D., Fluitsma, D., van Zon, M., Pierson, J., et al. (2007). M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 129, 1287–1298. doi: 10.1016/j.cell.2007.05.059
Vergne, I., Chua, J., Lee, H.-H., Lucas, M., Belisle, J., and Deretic, V. (2005). Mechanism of phagolysosome biogenesis block by viable Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 102, 4033–4038. doi: 10.1073/pnas.0409716102
Weber, S., Wagner, M., and Hilbi, H. (2013). Live-cell imaging of phosphoinositide dynamics and membrane architecture during Legionella infection. MBio 5, e00839–13. doi: 10.1128/mBio.00839-13
Weber, S. S., Ragaz, C., and Hilbi, H. (2009). Pathogen trafficking pathways and host phosphoinositide metabolism. Mol. Microbiol. 71, 1341–1352. doi: 10.1111/j.1365-2958.2009.06608.x
Keywords: Shigella, vacuolar rupture, intracellular pathogens, membrane trafficking, Rab GTPases
Citation: Mellouk N and Enninga J (2016) Cytosolic Access of Intracellular Bacterial Pathogens: The Shigella Paradigm. Front. Cell. Infect. Microbiol. 6:35. doi: 10.3389/fcimb.2016.00035
Received: 18 January 2016; Accepted: 14 March 2016;
Published: 05 April 2016.
Edited by:
Guy T. V. Nhieu, Collège de France, FranceReviewed by:
Lee-Ann H. Allen, University of Iowa, USAHubert Hilbi, University of Zürich, Switzerland
Copyright © 2016 Mellouk and Enninga. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jost Enninga, am9zdC5lbm5pbmdhQHBhc3RldXIuZnI=