 Qing Zheng
Qing Zheng Zhi Li2†
Zhi Li2† Yueyun Ma
Yueyun Ma Xiaoke Hao
Xiaoke Hao- 1Department of Clinical Laboratory, Xijing Hospital, Fourth Military Medical University, Xi'an, China
- 2Department of Pharmacology, School of Pharmacy, Fourth Military Medical University, Xi'an, China
Airway epithelial cell is often the initial site of attack by pathogens, and cell death is commonly caused by internalization of Mycobacterium tuberculosis (Mtb). However, the mechanism of interaction between epithelial cells and Mtb is not well understood. In this study, we investigated the role of the heparin-binding hemagglutinin (HBHA) protein of Mtb in the function of epithelial cells. In particular, the autophagy of A549 cells was determined based on microtubule-associated protein 1 light chain 3 alpha (LC3) activity. Autophagosome formation was detected by Monodansylcadaverine (MDC) staining and immune fluorescence staining of LC3. Autophagy could be significantly suppressed by HBHA protein. In addition, the LDH assay results showed that HBHA treatment could induce death on A549 cells. To explore the form of cell death, we detected the activity of caspase-3 and LDH release of A549 cells in the presence or absence of caspase inhibitor Z-VAD-FMK. Results demonstrated that HBHA treatment could induce apoptosis of A549 cells. To further confirm these results, we constructed the recombinant Mycobacterium smegmatis (MS) expressing HBHA (rMS-HBHA) and explored the influence of rMS-HBHA on the function of A549 cells. rMS-HBHA infection significantly inhibited LC3 expression and the maturation of autophagosomes in A549 cells. Subsequently, we infected A549 cells with MS and detected the viability of intracellular MS by CFU counts. rMS-HBHA showed higher survival and replication capacity in A549 cells than those of the wild-type MS. Finally, infection of A549 cells with rMS-HBHA caused further apoptosis. These findings suggested that rMS-HBHA could inhibit autophagy, promote its survival and replication within A549 cells, and subsequently induce apoptosis on infected cells to facilitate infection.
Introduction
Tuberculosis (TB) remains a devastating disease with approximately 2 billion people infected worldwide and 1.2 million deaths in 2010 (Hawn et al., 2015). Approximately 36 million people will die from TB annually by 2020 (Moliva et al., 2015). The etiological agent of TB, that is, Mycobacterium tuberculosis (Mtb), is the most successful intracellular pathogen that can invade and replicate in many host cell types, including both phagocytic and nonphagocytic cells (Vir et al., 2014).
Alveolar epithelium is often the initial site of the lung's response against Mtb. It is composed of type I and type II pneumocytes, which form a cell layer that provides a barrier function (Lin et al., 1998; Chuquimia et al., 2013). Increasing evidence implicated that alveolar epithelium, particularly type II pneumocyte, plays an important role in both host cell defense and bacterial dissemination (Xiong et al., 2014; Fine-Coulson et al., 2015; Ryndak et al., 2015). Mycobacterium smegmatis (MS) takes advantage of macropinocytosis for entry into epithelial cells, and internalized MS are killed by A549 cells (Garcia-Perez et al., 2008). However, unlike MS, type II pneumocytes could provide a permissive position for Mtb to replicate considerably and ultimately help bacterial dissemination (Bermudez and Goodman, 1996; Ryndak et al., 2015). The mechanism of how Mtb destroys the defense system is still unknown.
Autophagy is an intracellular self-digestion process whereby cytoplasmic constituents are delivered to and degraded by lysosomes (Lamb et al., 2013). Upon sensing stress conditions, such as starvation, MTOR is inhibited, which is required to activate the ULK complex. The Beclin-1 complex is activated by sensing the activation of the ULK complex. Subsequently, ATG12–ATG5 and LC3II are generated, and the membrane is elongated to form a double-membraned vesicle, that is, the autophagosome. Finally, the autophagosome fuses with lysosome, thereby forming an autolysosome to digest the cargo (Chen et al., 2014). Autophagy is crucial for quality control, energy supply, and immune defense against invading bacterial and viral pathogens. This process can eliminate intracellular pathogens through inflammation regulation, antigen presentation, and microorganism capture and degradation (Deretic et al., 2013; Lamb et al., 2013). Autophagy could also kill intracellular pathogens, such as Mtb, on the basis of strong degradative and other antimicrobial properties distinct to autolysosomes (Bradfute et al., 2013). Our previous studies using transmission electron microscope (TEM) revealed that Mtb bacilli-containing compartments are surrounded with double membranes, which characterize the autophagic process in A549. In addition, induction of autophagy in A549 presents a protective role against Mtb infection. Mtb could cause further necrosis among LC3-silenced A549 than that among wild-type A549 (Guo et al., 2013). Therefore, autophagy maybe the main mechanism that defends against invasion of pathogen (Li et al., 2012; Thurston et al., 2012; Wileman, 2013). Although autophagy is an efficient mechanism for clearing pathogens, such as Mtb, the mechanisms by which Mtb avoid being killed by autophagy remain unknown. Identifying and understanding the role of Mtb proteins that are critical to this process are considerably significant and will help us understand whether host cell autophagy or Mtb proteins can be targeted by new therapeutics.
Heparin-binding hemagglutinin (HBHA), a major adhesin in Mtb (Esposito et al., 2011; Lebrun et al., 2012), is involved in the attachment of mycobacteria to epithelial cells and plays vital role in the dissemination of Mycobacterium from the site of primary infection (Locht et al., 2006; Esposito et al., 2011). Thus, Mycobacterium may use HBHA to inhibit autophagy and thereby facilitate infection. To confirm this hypothesis, we explored the role of the Mtb protein HBHA in regulating autophagy in host airway epithelial cells using the A549 cell line. To further validate our results, we used the fast-growing, nonpathogenic MS MC2155 strain (Snapper et al., 1990), which lacks the hbhA gene, to construct a recombinant MS strain that expressed HBHA (rMS-HBHA) (Delogu et al., 2004). Our results demonstrated that rMS-HBHA could inhibit autophagy, promote its survival and replication within A549 cells, and subsequently induce apoptosis of infected cells to facilitate infection.
Materials and Methods
Cells and Culture
The human non-small-cell lung carcinoma A549 cell line was obtained from The Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The cells were grown in modified RPMI-1640 medium (HyClone, USA) supplemented with 10% heat-inactivated fetal bovine serum (Gibco, New Zealand) at 37°C in a humidified incubator with a 5% CO2 atmosphere.
Bacteria Strains
The wild-type strain of M. smegmatis MC2155 strain was obtained from the Department of Clinical Laboratory, Xijing Hospital, Fourth Military Medical University. Cells were grown in 7H9/7H10 Middlebrook (BD, USA) broth supplemented with 0.05% Tween 80, OADC (BD, USA), and 0.2% glycerol (v/v). Cells were grown at 37°C with continuous agitation (220 rpm).
Expression and Purification of HBHA Purified Protein
To produce recombinant HBHA (rHBHA) in Escherichia coli, the corresponding genes were PCR amplified using M. tuberculosis H37Rv (ATCC27294) DNA as the template and the following oligonucleotide primers: F primer (5′-CACGGATCCATGGCTGAAAACTCGAACAT-3′) and R primer (5′-CTGAAGCTTACTACTTCTGGGTGACCTTC-3′). The forward and reverse primers contained the underlined BamHI and HindIII restriction sites, respectively. The PCR products were digested with BamHI and HindIII and cloned into the PQE80L vector (Laboratory Animal Center, The Fourth Military Medical University), and the resulting clones were sequenced (Sangon Biotech). E. coli strain BL21 (DE3), which expresses His-tagged protein, was grown in Luria–Bertani broth supplemented with 100 μg/ml ampicillin. After induction with 0.3 mM isopropy-β-D-thiogalactoside for 5 h, the cells were lysed by sonication for 30 min. The rEC-HBHA was purified by heparin–Sepharose chromatography and further purified using a His-GraviTrap purification kit (GE Healthcare, USA) in accordance with the manufacturer's instructions.
Construction of Recombinant rMS-HBHA and rMS-HBHA-GFP
The fast-growing, nonpathogenic M. smegmatis MC2155 strain (Snapper et al., 1990), which lacks the hbhA gene, was electroporated to obtain a mycobacteria recombinant strain that could be easily manipulated in most laboratories (Delogu et al., 2004). The shuttle plasmid vector pMV261 or pMN437, which contains GFP promoter and the full-length hbhA gene, was transformed through electroporation and grown in 7H9 broth containing kanamycin (15 μg/ml) and hygromycin B (20 μg/ml). Such vector was obtained from Dr. Babak Javid of the Medical College of Tsinghua University. E. coli strain BL21 (Takara, Japan) was used for transformation and protein expression and cultivated under standard conditions. The purified protein of HBHA was prepared by our laboratory. The hbhA gene was excised using the enzymes BamHI and HindIII and cloned at the same site of the mycobacterial shuttle vector pMV261. Alternatively, the hbhA gene was excised using the enzymes ClaI and HindIII, cloned at the same site of the pMN437 vector, and electroporated (2.5 kV, 25 μF, and 1000 Ω) for the preparation of M. smegmatis competent cells using standard techniques (Bardarov et al., 1997).
Monodansylcadaverine (MDC) Staining of Autophagic Vacuoles
To analyze the formation of autophagosome, A549 cells were starved for 90 min and subsequently treated with HBHA (8 μg/ml) for 90 min or 3-MA (100 μg/ml), an inhibitor of autophagy, for 4 h. For the infection assay, A549 cells were infected with MS-GFP and rMS-HBHA-GFP at the multiplicity of infection (MOI) of 10:1 for 18 h. Afterward, the A549 cells were washed three times with PBS and treated with 50 μM MDC in an incubator for 15 min. The cells were again washed three times with PBS and then immediately observed under a laser confocal microscope (FV10i, Olympus, Tokyo, Japan).
Immune Fluorescence Detection of LC3
The A549 cells were cultured in glass bottom cell culture dish (NEST, Hong Kong, China) and starved for 90 min. Afterward, A549 cells were treated with HBHA (8 μg/ml) for 90 min or 3-MA (100 μg/ml) for 4 h. The cells were washed three times with PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. Cells were treated with 0.3% Triton-100 20 min at room temperature to increase permeability. Subsequently, they were blocked with heat-inactivated fetal bovine serum (Gibco, New Zealand) for 30 min at room temperature. The cells were incubated with polyclonal antiLC3 antibody at 4°C overnight. After washing with PBS, cells were incubated with antirabbit Cy3 fluorescent secondary antibody (1:500 final dilution; BBI Life Sciences, China), and they were observed by laser confocal microscopy (FV10i, Olympus, Tokyo, Japan).
Western Blot Analysis
To detect the expression of the key proteins of autophagy, A549 cells were starved for 90 min and treated with HBHA of different concentrations for 90 min or with rapamycin or rapamycin plus 3-MA (100 μg/ml) for 4 h. To confirm the effect of HBHA on the autophagic flux of A549 cells, cells were treated with 350 nM bafilomycin A1 (BAF A1; Abcam, USA) to prevent lysosomal degradation. For caspase-3 detection, A549 cells were treated with HBHA for 18 h in the presence or absence of caspase inhibitor Z-VAD-FMK (20 μM). The cells were then lysed in cell lysis buffer with phenylmethylsulfonyl fluoride, phosphatase inhibitor, and protease inhibitor according to the instructions of the manufacturer of the protein extraction kit. The protein concentration in the lysate was quantified using a BCA protein assay kit. Equal amounts of protein from each sample were separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA). Subsequent to incubation in blocking buffer (LICOR, Odyssey, USA) for 1 h, the membranes were incubated with monoclonal primary antibodies against ATG5/LC3B/Beclin-1 (Abcam, USA)/Caspase-3 (GeneTex, USA) HBHA (antiHBHA was prepared by our laboratory) and β-actin (Abcam, USA) overnight at 4°C. The membranes were then incubated with a horseradish peroxidase-conjugated goat antirabbit IgG secondary antibody (LICOR, Odyssey, USA) or antimouse IgG secondary antibody (LICOR, Odyssey, USA) for 2 h. The bands were detected using a dual-color infrared laser (LICOR, Odyssey, USA), and the protein levels were quantitated by densitometry using Gel-Pro Analyzer software (Media Cybernetics, Inc., Rockville, MD, USA).
LDH Detection by Enzyme Labeling
The A549 cells were seeded at a density of 1 × 106 cells/well in six-well plates and incubated with HBHA of different concentrations for 24 h in the presence or absence of rapamycin or 3-MA. Alternatively, A549 cells were infected with MS or rMS-HBHA at the MOI of 10:1 for 24 h. Afterward, the culture supernatant was collected and added to a 96-well plate. The caspase inhibitor Z-VAD-FKM (20 μM) was added into cells 1 h prior to HBHA treatment or infection. Specific RIPK1 inhibitor Necrostatin-1 (Nec-1) (Sigma) was used at 30 μM. A LDH cytotoxicity kit was then used according to the manufacturer's instructions. Specifically, dinitrophenylhydrazine and sodium hydroxide solution were detected at 450 nm using a microplate reader.
Detection of Bacterial CFU in A549 Cells Infected with MS and rMS
For the assay, A549 cells were seeded onto 24-well plates at a density of 4 × 105 cells/well. The cells were infected with MS or rMS-HBHA at the MOI of 10:1 for 3 h in the presence or absence of rapamycin or 3-MA at 37°C in 5% CO2. For caspase inhibition, Z-VAD-FKM was added into cells 1 h prior to infection. Amikacin (200 mg/ml) was then added for 3 h to kill extracellular bacteria. At indicated time points, the cells were washed three times with basal RPMI 1640 medium, and viable intracellular bacteria were released by incubation with 0.5 ml of 0.1% Triton X-100 in sterile water for 10 min. The samples were mixed vigorously with 0.5 ml of 7H9 broth. Serial 10-fold dilutions of lysates were prepared in 7H9 broth and plated on 7H10 agar for determination of CFU numbers.
Statistical Analysis
All experiments were repeated at least three times. Statistical analyses were performed using SPSS 17.0. Data are expressed as the mean ± standard deviation and were analyzed using one-way ANOVA. P < 0.05 was considered to indicate a statistically significant difference.
Results
HBHA Inhibited LC3 and Beclin-1 Expression in A549 Cells
To evaluate the effect of HBHA on the autophagy of A549 cells, Western blot assay was performed to detect the expression of LC3 and Beclin-1. LC3 and Beclin-1, which were starved for 1 h, were significantly induced in A549. However, when HBHA was added after 90 min, the expression of LC3 and Beclin-1 was suppressed in a dose-dependent manner (Figures 1A–D). In particular, 8 μg/ml HBHA could remarkably inhibit the expression of starved LC3 and Beclin-1, and the ATG5 expression was not affected by HBHA (Figure 1C). Subsequently, to measure the effect of HBHA on the autophagic flux of A549 cells, the cells were treated with BAF A1 to prevent lysosomal degradation. Compared with BAF A1 alone, HBHA treatment with BAF A1 could decrease LC3II levels. This result confirmed that HBHA inhibited the synthesis of autophagy-related membranes (Figures 1E,F). When rapamycin or 3-MA was used to induce or inhibit autophagy, the LC3 expression could be suppressed by 3-MA. Similar to 3-MA, the LC3 expression was inhibited in a dose-dependent manner when HBHA was added (Figures 1G,H).
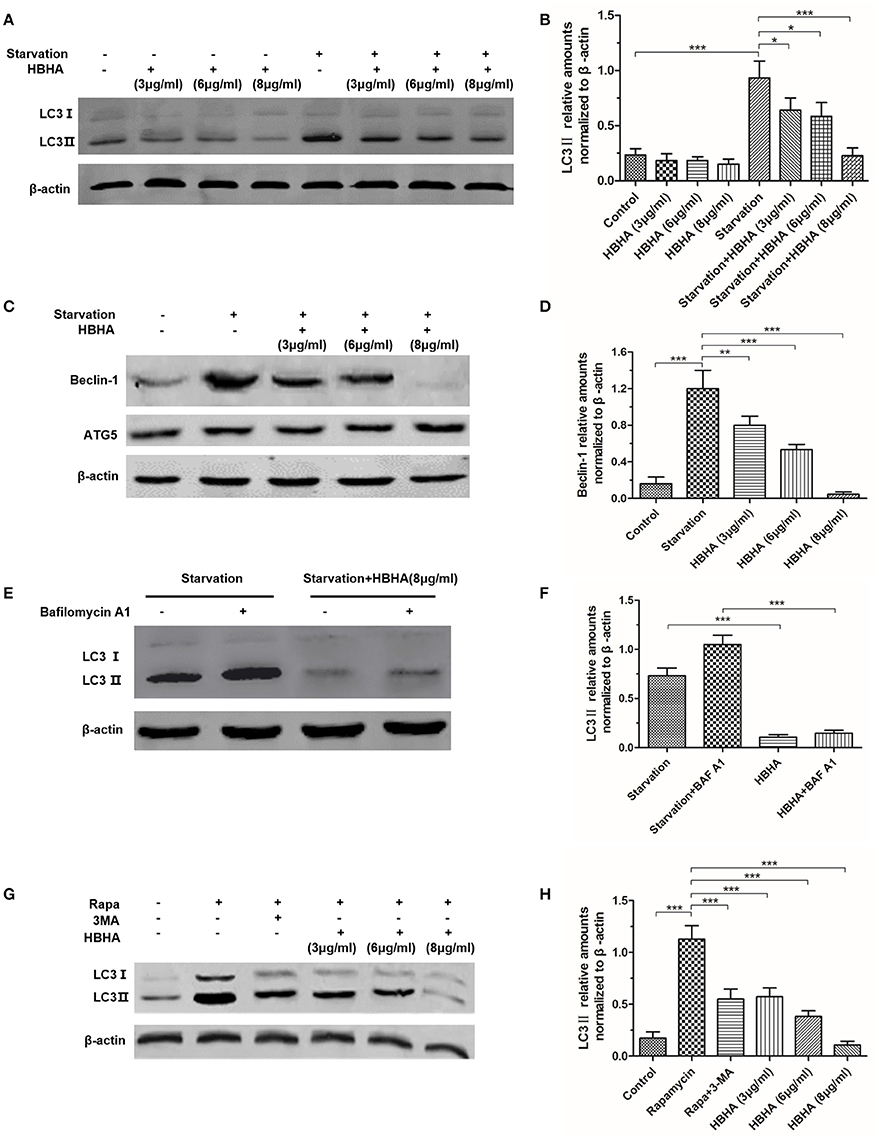
Figure 1. Heparin-binding hemagglutinin (HBHA) inhibited the expression of LC3 and Beclin-1 in A549 cells. (A–D) A549 cells were starved for 1 h, and HBHA proteins of different concentrations were subsequently added to the cells for 90 min. LC3 (A) and ATG5/Beclin-1 (C) expression was detected by Western blot. The intensities of LC3II (B) and Beclin-1 (D) bands were normalized to the intensity of β-actin. (*P < 0.05, **P < 0.01, ***P < 0.001 vs. starvation group in one-way ANOVA, n = 3). (E) To measure the effect of HBHA on the autophagic flux of A549 cells, the cells were treated with bafilomycin A1 (BAF A1) to prevent lysosomal degradation. LC3 expression was detected by Western blot. (F) The intensities of LC3II bands were normalized to the intensity of β-actin. (***P < 0.001 in one-way ANOVA, n = 3). (G) A549 cells were treated with rapamycin alone or rapamycin plus 3-MA or HBHA of different concentrations. Afterward, LC3 expression was detected by Western blot. (H) The intensities of LC3II bands were normalized to the intensity of β-actin. (***P < 0.001 vs. rapamycin group in one-way ANOVA, n = 3).
HBHA Inhibited the Formation of Autophagosome in A549 Cells
To further explore the effect of HBHA on autophagy, we observed the formation of autophagosomes using MDC staining and immunofluorescence staining of LC3 in A549 cells. The number of MDC foci increased after starvation, but it could be reversed by 3-MA. Similarly, HBHA decreased the number of MDC foci (Figures 2A,B), and the number of LC3 puncta decreased significantly after HBHA addition (Figures 2C,D). Taken together, these results demonstrated that autophagy maturation could be considerably suppressed by HBHA.
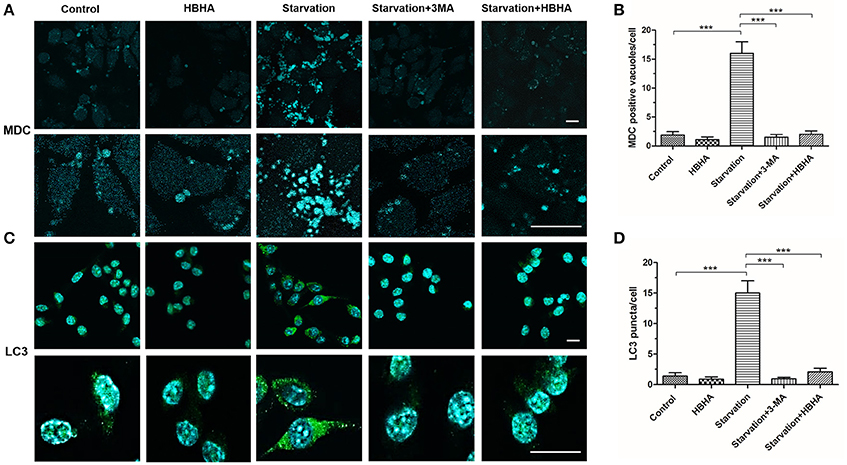
Figure 2. HBHA inhibited the maturation of autophagosome in A549 cells. (A) A549 cells were starved for 1 h and subsequently treated with HBHA (8 μg/ml) for 90 min or 3-MA (100 μg/ml) for 4 h. The cells were then stained with monodansylcadaverine (MDC), and MDC-labeled autophagic vacuoles were detected by confocal microscopy. Scale bars: 20 μm. (B) MDC-positive autophagic vacuoles were quantified in each group. (***P < 0.001 vs. starvation group in one-way ANOVA). (C) Immune fluorescence of LC3 puncta in A549 cells was detected by confocal microscopy. Scale bars: 20 μm. (D) LC3 puncta were quantified in each group. (***P < 0.001 vs. starvation group in one-way ANOVA).
rMS-HBHA Inhibited Autophagy among Starved A549 Cells
We transferred HBHA into MS MC2155 to construct recombinant rMS-HBHA. To confirm that HBHA was successfully expressed in rMS-HBHA, the total proteins of MS and rMS-HBHA were extracted. Western blot analysis was performed to detect the expression of HBHA protein using anti-HBHA antibody. The results showed that HBHA was not expressed in the parent strain MS MC2155, but expressed in the rMS-HBHA (Figure 3A). To further confirm the inhibition role of HBHA on autophagy, A549 cells were infected with MS or rMS-HBHA. Western blot analysis results showed that rMS-HBHA inhibited the expression of starvation-induced LC3, but the wild-type strain did not (Figures 3B,C). We constructed GFP-expressing MS and rMS-HBHA, infected A549, and performed MDC staining to examine autophagosome formation. The number of MDC foci increased after starvation, and rMS-HBHA significantly decreased the number of MDC foci (Figures 3D,E). These results suggested that rMS-HBHA inhibited the maturation of autophagy and consistent with those observed with pure HBHA protein.
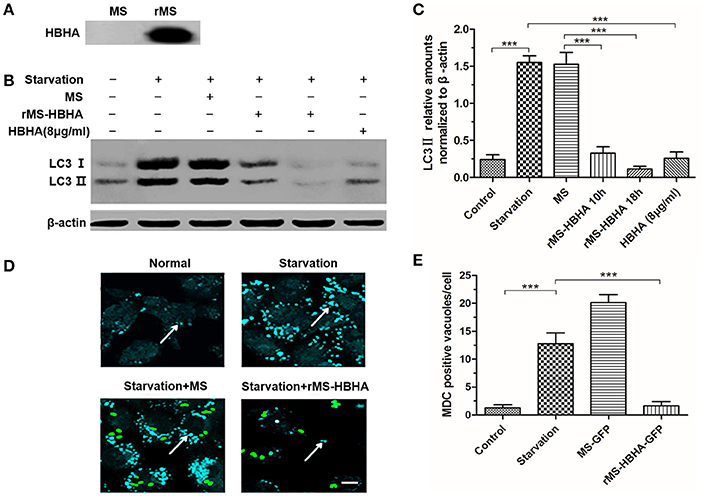
Figure 3. Recombinant Mycobacterium smegmatis (MS) expressing HBHA (rMS-HBHA) inhibited autophagy among starved A549 cells. (A) Total proteins of MC2155 and rMS-HBHA were extracted, and Western blot was carried out to detect HBHA using anti-HBHA antibody. (B) A549 cells were infected with the wild-type strain MC2155 or rMS-HBHA at the multiplicity of infection (MOI) of 10:1 for indicated time or treated with HBHA (8 μg/ml) for 90 min. The expression levels of LC3 were detected by Western blot. (C) The intensities of LC3II bands were normalized to the intensity of β-actin. (***P < 0.001 in one-way ANOVA, n = 3). (D) A549 cells were infected with GFP-expressing strains, and autophagosomes were stained with MDC. The white arrows indicate autophagosomes. (E) MDC-positive autophagic vacuoles were quantified in each group. (***P < 0.001 vs. starvation group in one-way ANOVA).
Inhibition of Autophagy Could Promote Survival of rMS-HBHA within A549 Cells
To determine whether autophagy inhibition could promote survival of rMS-HBHA within A549 cells, A549 cells were infected with WT MS or rMS-HBHA at the MOI of 10:1. Cells were lysed at 1, 10, or 18 h post-infection, and lysates were diluted and plated on agar plates to determine the number of viable intracellular bacteria. MS and rMS-HBHA showed no significant difference in growth curve (Figure 4A). However, rMS-HBHA showed higher survival and replication capacity within A549 cells than those of wild-type MS (Figure 4B). Autophagy activation in MS-infected A549 cells by rapamycin could decrease the number of intracellular MS, but showed no significant effect on rMS-HBHA (Figure 4C). Moreover, autophagy inhibition in MS-infected A549 cells by 3-MA could increase the number of intracellular MS (Figure 4D). These findings suggested that HBHA may enhance the capacity of MS to infect and survive within A549 cells through inhibition of autophagy.
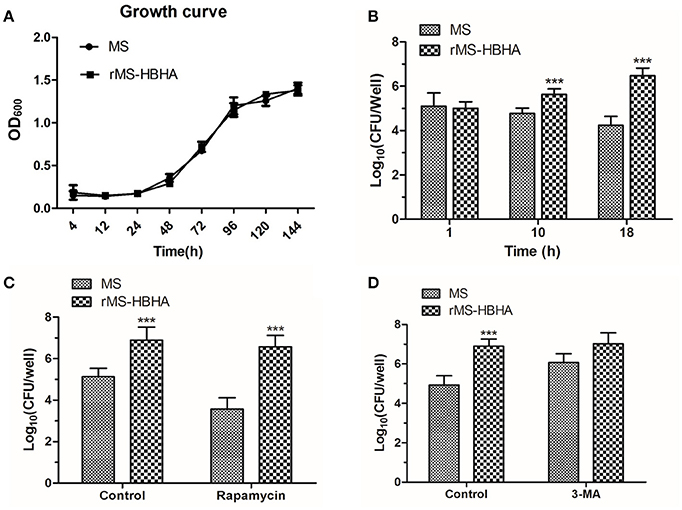
Figure 4. Inhibition of autophagy could promote survival of rMS-HBHA within A549 cells. (A) Growth curves of MS and rMS-HBHA were measured by OD600 absorbance. (B) A549 cells were infected with MS or rMS-HBHA at the MOI of 10:1. The cells were lysed at 1, 10, or 18 h post-infection, and lysates were diluted and plated on agar plates to determine the number of viable intracellular bacteria. (***P < 0.001 in two-way ANOVA, n = 3) (C) A549 cells were infected with MS or rMS-HBHA at the MOI of 10:1 and treated with rapamycin. Viable intracellular bacteria were measured at 18 h post-infection. (***P < 0.001 in one-way ANOVA, n = 3) (D) A549 cells were infected with MS or rMS-HBHA at the MOI of 10:1 and treated with 3-MA. Viable intracellular bacteria were measured at 18 h post-infection. (***P < 0.001 in one-way ANOVA, n = 3).
HBHA Protein and rMS-HBHA Infection Could Induce Cell Death of A549
To determine the fate of infected A549 cells, A549 cells were treated with HBHA of different concentrations or infected with MS or rMS-HBHA at the MOI of 10:1 for 24 h. The culture supernatant was collected to measure the released LDH. We found that HBHA protein treatment could induce large amount of cell death (Figure 5A). Similarly, rMS-HBHA infection could induce great number of cell death but not wild-type MS (Figure 5B). Activation of autophagy in rMS-HBHA-infected A549 cells by rapamycin showed no effect on cell death (Figure 5C). Furthermore, inhibition of autophagy in MS-infected A549 cells by 3-MA could slightly increase cell death but showed no effect on rMS-HBHA-infected A549 cells (Figure 5D). These results suggested that HBHA could promote cell death in infected A549 cells.
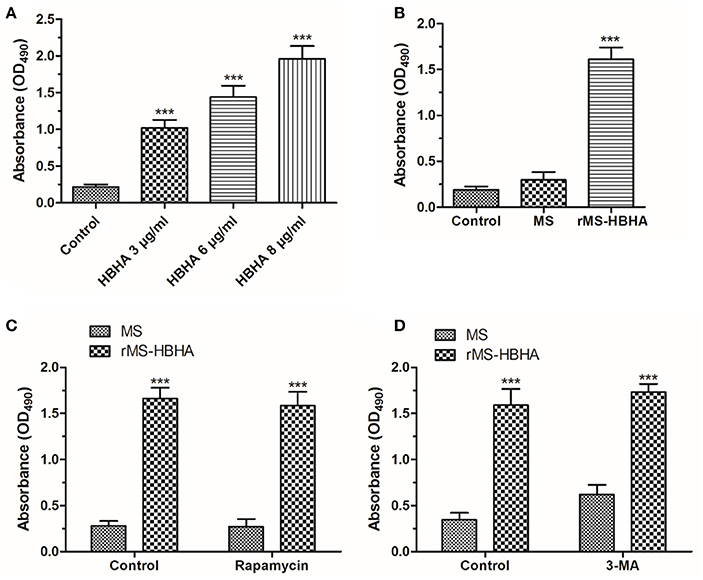
Figure 5. HBHA protein and rMS-HBHA infection could induce cell death on A549. (A) A549 cells were treated with HBHA of different concentrations for 24 h, and LDH release was detected. (***P < 0.001 vs. control group in one-way ANOVA, n = 3). (B) A549 cells were infected with MS or rMS-HBHA at the MOI of 10:1 for 24 h, and LDH release was detected. (***P < 0.001 vs. control group in one-way ANOVA, n = 3). (C) A549 cells were infected with MS or rMS-HBHA and treated with rapamycin, and LDH release was detected. (***P < 0.001 in one-way ANOVA, n = 3) (D) A549 cells were infected with MS or rMS-HBHA and treated with 3-MA, and LDH release was detected. (***P < 0.001 in one-way ANOVA, n = 3).
HBHA Treatment Induced Apoptosis of A549 Cells through Activation of Caspase-3
To investigate the form of HBHA-induced cell death, the RIPK1 inhibitor Nec-1 and caspase inhibitor Z-VAD-FMK was used. HBHA- or rMS-HBHA infection-induced cell death was not affected after Nec-1 treatment (Figures 6A,B). These results suggested necroptosis was not involved in this process. However, HBHA- or rMS-HBHA infection-induced cell death was significantly attenuated after Z-VAD-FMK treatment (Figures 6C,D). This result suggested that apoptosis may participate in this process. To further confirm the results, the cleaved-caspase-3 activity was detected by Western blot. HBHA treatment could increase the expression of cleaved-caspase-3 in a dose-dependent manner, and treatment with Z-VAD-FMK could inhibit the activation of caspase-3 (Figures 6E,F). These results indicated that HBHA protein treatment or rMS-HBHA infection could induce apoptosis of A549 cells through activation of caspase-3.
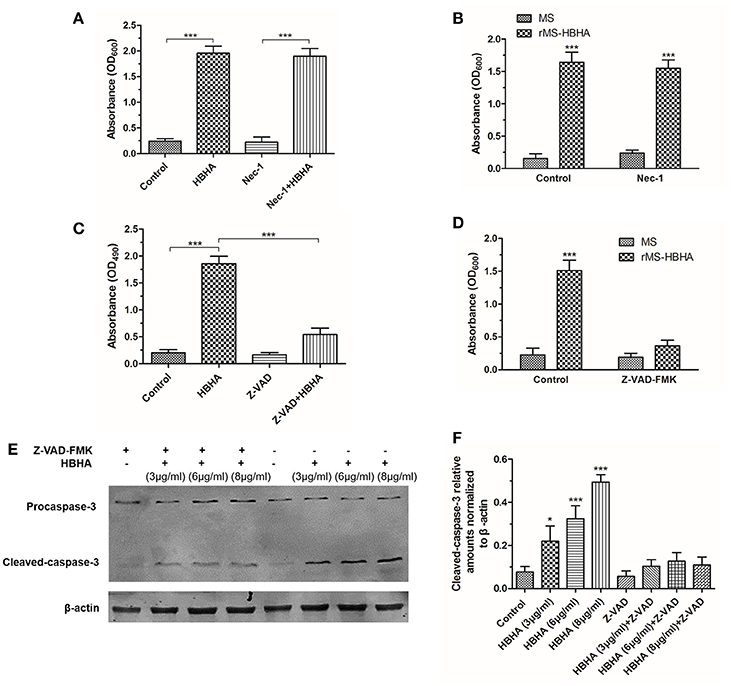
Figure 6. HBHA treatment induced apoptosis on A549 cells through activation of caspase-3. (A) A549 cells were treated with HBHA (8 μg/ml) for 24 h in the presence or absence of RIPK1 inhibitor Nec-1 (30 μM), and LDH release was detected. (***P < 0.001 in one-way ANOVA, n = 3). (B) A549 cells were infected with MS or rMS-HBHA in the presence or absence of Nec-1, and LDH release was detected. (***P < 0.001 in one-way ANOVA, n = 3) (C) A549 cells were treated with HBHA (8 μg/ml) for 24 h in the presence or absence of caspase inhibitor Z-VAD-FMK (20 μM), and LDH release was detected. (***P < 0.001 in one-way ANOVA, n = 3). (D) A549 cells were infected with MS or rMS-HBHA in the presence or absence of Z-VAD-FMK, and LDH release was detected. (***P < 0.001 in one-way ANOVA, n = 3) (E) A549 cells were treated with HBHA at different concentrations for 18 h in the presence or absence of Z-VAD-FMK. Caspase-3 expression was detected by Western blot. (F) The intensities of cleaved-caspase-3 bands were normalized to the intensity of β-actin. (*P < 0.05, ***P < 0.001 vs. starvation group in one-way ANOVA, n = 3).
Discussion
Among most of the Mtb virulence factors, HBHA is particularly important in the infection process and Mtb immune evasion (Sechi et al., 2006). The amount of HBHA protein in alveolar type II epithelial cells increases considerably at 6 h post-infection, and the bacteria break through the cells after 8 h (de Lima et al., 2009), thereby indicating that HBHA facilitates the dispersion and replication of Mtb in lung epithelial cells. By contrast, HBHA-deficient Mtb mutant strains are significantly impaired in their ability to disseminate from the lungs to other tissues, which suggested that HBHA is essential for the infection of lung epithelial cells and extrapulmonary dissemination of Mtb (Pethe et al., 2001; Parra et al., 2004; Temmerman et al., 2005; Locht et al., 2006; Kohama et al., 2008).
Autophagy plays key roles in immune defense against invading pathogens. Autophagy also presents a protective role against Mtb infection (Kawamura, 2006; Rovetta et al., 2014). Thus, autophagy inhibition may facilitate Mtb infection. The present study is the first to report that HBHA protein could significantly inhibit LC3 expression and autophagosome formation in A549 cells, which indicated that autophagy could be suppressed by HBHA (Figures 1, 2). To further confirm this phenomenon, we constructed rHBHA protein-expressing MS. HBHA protein only exists in Mtb and BCG and not in MS (Zhao et al., 2012). Therefore, recombinant MS is an appropriate strain for observing HBHA protein function. The HBHA expression in MS exerted no effect on the growth of bacterium (Figure 4A), but significantly inhibited the expression of LC3 and maturation of autophagosome in A549 (Figure 3), eventually leading to attenuated clearance of MS by the cells. Consequently, the number and survival rate of intracellular bacteria increased significantly because of the reduced capacity to eliminate bacteria from infected cells (Figure 4B).
Virulent Mtb induces necrosis of infected macrophages by inhibiting the repair process of the plasma membrane. This event leads to cellular lysis and reinforces spreading to adjacent infection sites (Chen et al., 2006, 2008; Divangahi et al., 2009). Recent reports suggested that a high intracellular burden of virulent Mtb induced macrophage cell death via a new apoptotic pathway involved in bacterial escape and extracellular replication (O'Sullivan et al., 2007). Similarly, our results demonstrated that rMS-HBHA could induce cell apoptosis of A549 cells through activation of caspase-3 (Figures 5, 6). This phenomenon may facilitate bacterial escape from lung epithelial cells and dissemination to the adjacent cells. The schematic model of the role of HBHA during mycobacterial infection was shown in Figure 7.
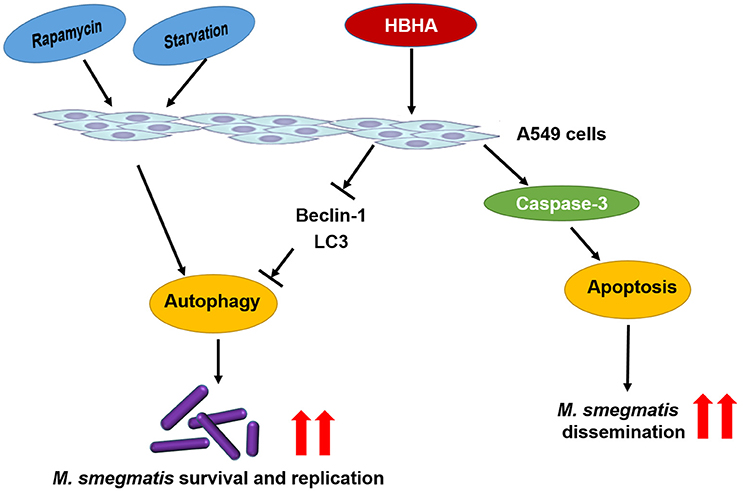
Figure 7. Schematic model of the role of HBHA during mycobacterial infection. HBHA inhibited autophagy in mycobacterial infected A549 cells, thereby promoted intracellular bacterial survival and replication. Subsequently, HBHA induced apoptosis on A549 cells through activation of caspase-3 which may facilitate bacterial escape from lung epithelial cells and dissemination to the adjacent cells.
Induction of autophagy in macrophages is an effective mechanism to enhance intracellular killing of Mtb, and the ability of pathogen to inhibit this process is considerably important for its survival (Deretic et al., 2006; Songane et al., 2012). Beijing strains could resist autophagic killing by host cells compared with H37Rv and a strain belonging to the East African Indian genotype (Haque et al., 2015). The virulent strain H37Rv presents remarkably increased ability to inhibit autophagy flux than those of avirulent strains H37Ra and BCG, depending on virulence regulators PhoP, ESAT-6, and ESX-1 system, which controlled the secretin of ESAT-6 (Chandra et al., 2015). PE_PGRS47 (Rv2741) and ESAT-6/CFP10 are inhibitors of autophagosome formation in macrophages (Zhang et al., 2012; Saini et al., 2016). Nevertheless, Mtb used HBHA as a support when suppressing autophagy for survival and dissemination. However, whether HBHA can inhibit macrophage autophagy in macrophage and subsequently trigger apoptosis to allow bacterium to avoid macrophage killing should be further verified experimentally.
Conclusion
Our study demonstrated that HBHA could inhibit autophagy of epithelial cells, facilitate MS intracellular survival, and promote its infection.
Author Contributions
QZhe, YM, and XH: Designed the experiments. QZhe, ZL, SZ, QZha, LZ, LY, and XF: Performed the experiments and analyzed the data. QZhe and YM: Wrote the paper. All authors reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81371857, 30872358, and 81000713) and National Key Instruments Foundation (2012YQ03026107).
References
Bardarov, S., Kriakov, J., Carriere, C., Yu, S., Vaamonde, C., McAdam, R. A., et al. (1997). Conditionally replicating mycobacteriophages: a system for transposon delivery to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 94, 10961–10966. doi: 10.1073/pnas.94.20.10961
Bermudez, L. E., and Goodman, J. (1996). Mycobacterium tuberculosis invades and replicates within type II alveolar cells. Infect. Immun. 64, 1400–1406.
Bradfute, S. B., Castillo, E. F., Arko-Mensah, J., Chauhan, S., Jiang, S., Mandell, M., et al. (2013). Autophagy as an immune effector against tuberculosis. Curr. Opin. Microbiol. 16, 355–365. doi: 10.1016/j.mib.2013.05.003
Chandra, P., Ghanwat, S., Matta, S. K., Yadav, S. S., Mehta, M., Siddiqui, Z., et al. (2015). Mycobacterium tuberculosis inhibits RAB7 recruitment to selectively modulate autophagy flux in macrophages. Sci. Rep. 5:16320. doi: 10.1038/srep16320
Chen, M., Divangahi, M., Gan, H., Shin, D. S., Hong, S., Lee, D. M., et al. (2008). Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J. Exp. Med. 205, 2791–2801. doi: 10.1084/jem.20080767
Chen, M., Gan, H., and Remold, H. G. (2006). A mechanism of virulence: virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. J. Immunol. 176, 3707–3716. doi: 10.4049/jimmunol.176.6.3707
Chen, P., Cescon, M., and Bonaldo, P. (2014). Autophagy-mediated regulation of macrophages and its applications for cancer. Autophagy 10, 192–200. doi: 10.4161/auto.26927
Chuquimia, O. D., Petursdottir, D. H., Periolo, N., and Fernandez, C. (2013). Alveolar epithelial cells are critical in protection of the respiratory tract by secretion of factors able to modulate the activity of pulmonary macrophages and directly control bacterial growth. Infect. Immun. 81, 381–389. doi: 10.1128/IAI.00950-12
de Lima, C. S., Marques, M. A., Debrie, A. S., Almeida, E. C., Silva, C. A., Brennan, P. J., et al. (2009). Heparin-binding hemagglutinin (HBHA) of Mycobacterium leprae is expressed during infection and enhances bacterial adherence to epithelial cells. FEMS Microbiol. Lett. 292, 162–169. doi: 10.1111/j.1574-6968.2009.01488.x
Delogu, G., Bua, A., Pusceddu, C., Parra, M., Fadda, G., Brennan, M. J., et al. (2004). Expression and purification of recombinant methylated HBHA in Mycobacterium smegmatis. FEMS Microbiol. Lett. 239, 33–39. doi: 10.1016/j.femsle.2004.08.015
Deretic, V., Saitoh, T., and Akira, S. (2013). Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 13, 722–737. doi: 10.1038/nri3532
Deretic, V., Singh, S., Master, S., Harris, J., Roberts, E., Kyei, G., et al. (2006). Mycobacterium tuberculosis inhibition of phagolysosome biogenesis and autophagy as a host defence mechanism. Cell. Microbiol. 8, 719–727. doi: 10.1111/j.1462-5822.2006.00705.x
Divangahi, M., Chen, M., Gan, H., Desjardins, D., Hickman, T. T., Lee, D. M., et al. (2009). Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 10, 899–906. doi: 10.1038/ni.1758
Esposito, C., Marasco, D., Delogu, G., Pedone, E., and Berisio, R. (2011). Heparin-binding hemagglutinin HBHA from Mycobacterium tuberculosis affects actin polymerisation. Biochem. Biophys. Res. Commun. 410, 339–344. doi: 10.1016/j.bbrc.2011.05.159
Fine-Coulson, K., Giguere, S., Quinn, F. D., and Reaves, B. J. (2015). Infection of A549 human type II epithelial cells with Mycobacterium tuberculosis induces changes in mitochondrial morphology, distribution and mass that are dependent on the early secreted antigen, ESAT-6. Microbes Infect. 17, 689–697. doi: 10.1016/j.micinf.2015.06.003
Garcia-Perez, B. E., Hernandez-Gonzalez, J. C., Garcia-Nieto, S., and Luna-Herrera, J. (2008). Internalization of a non-pathogenic mycobacteria by macropinocytosis in human alveolar epithelial A549 cells. Microb. Pathog. 45, 1–6. doi: 10.1016/j.micpath.2008.01.009
Guo, X. G., Ji, T. X., Xia, Y., and Ma, Y. Y. (2013). Autophagy protects type II alveolar epithelial cells from Mycobacterium tuberculosis infection. Biochem. Biophys. Res. Commun. 432, 308–313. doi: 10.1016/j.bbrc.2013.01.111
Haque, M. F., Boonhok, R., Prammananan, T., Chaiprasert, A., Utaisincharoen, P., Sattabongkot, J., et al. (2015). Resistance to cellular autophagy by Mycobacterium tuberculosis Beijing strains. Innate Immun. 21, 746–758. doi: 10.1177/1753425915594245
Hawn, T. R., Shah, J. A., and Kalman, D. (2015). New tricks for old dogs: countering antibiotic resistance in tuberculosis with host-directed therapeutics. Immunol. Rev. 264, 344–362. doi: 10.1111/imr.12255
Kohama, H., Umemura, M., Okamoto, Y., Yahagi, A., Goga, H., Harakuni, T., et al. (2008). Mucosal immunization with recombinant heparin-binding haemagglutinin adhesin suppresses extrapulmonary dissemination of Mycobacterium bovis bacillus Calmette-Guerin (BCG) in infected mice. Vaccine 26, 924–932. doi: 10.1016/j.vaccine.2007.12.005
Lamb, C. A., Yoshimori, T., and Tooze, S. A. (2013). The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 14, 759–774. doi: 10.1038/nrm3696
Lebrun, P., Raze, D., Fritzinger, B., Wieruszeski, J. M., Biet, F., Dose, A., et al. (2012). Differential contribution of the repeats to heparin binding of HBHA, a major adhesin of Mycobacterium tuberculosis. PLoS ONE 7:e32421. doi: 10.1371/journal.pone.0032421
Li, L., Wang, L., Xiao, R., Zhu, G., Li, Y., Liu, C., et al. (2012). The invasion of tobacco mosaic virus RNA induces endoplasmic reticulum stress-related autophagy in HeLa cells. Biosci. Rep. 32, 171–186. doi: 10.1042/BSR20110069
Lin, Y., Zhang, M., and Barnes, P. F. (1998). Chemokine production by a human alveolar epithelial cell line in response to Mycobacterium tuberculosis. Infect. Immun. 66, 1121–1126.
Locht, C., Hougardy, J. M., Rouanet, C., Place, S., and Mascart, F. (2006). Heparin-binding hemagglutinin, from an extrapulmonary dissemination factor to a powerful diagnostic and protective antigen against tuberculosis. Tuberculosis (Edinb). 86, 303–309. doi: 10.1016/j.tube.2006.01.016
Moliva, J. I., Turner, J., and Torrelles, J. B. (2015). Prospects in Mycobacterium bovis Bacille Calmette et Guerin (BCG) vaccine diversity and delivery: why does BCG fail to protect against tuberculosis? Vaccine 33, 5035–5041. doi: 10.1016/j.vaccine.2015.08.033
O'Sullivan, M. P., O'Leary, S., Kelly, D. M., and Keane, J. (2007). A caspase-independent pathway mediates macrophage cell death in response to Mycobacterium tuberculosis infection. Infect. Immun. 75, 1984–1993. doi: 10.1128/IAI.01107-06
Parra, M., Pickett, T., Delogu, G., Dheenadhayalan, V., Debrie, A. S., Locht, C., et al. (2004). The mycobacterial heparin-binding hemagglutinin is a protective antigen in the mouse aerosol challenge model of tuberculosis. Infect. Immun. 72, 6799–6805. doi: 10.1128/IAI.72.12.6799-6805.2004
Pethe, K., Alonso, S., Biet, F., Delogu, G., Brennan, M. J., Locht, C., et al. (2001). The heparin-binding haemagglutinin of M. tuberculosis is required for extrapulmonary dissemination. Nature 412, 190–194. doi: 10.1038/35084083
Rovetta, A. I., Pena, D., Hernandez Del Pino, R. E., Recalde, G. M., Pellegrini, J., Bigi, F., et al. (2014). IFNG-mediated immune responses enhance autophagy against Mycobacterium tuberculosis antigens in patients with active tuberculosis. Autophagy 10, 2109–2121. doi: 10.4161/15548627.2014.981791
Ryndak, M. B., Singh, K. K., Peng, Z., and Laal, S. (2015). Transcriptional profile of Mycobacterium tuberculosis replicating in type II alveolar epithelial cells. PLoS ONE 10:e0123745. doi: 10.1371/journal.pone.0123745
Saini, N. K., Baena, A., Ng, T. W., Venkataswamy, M. M., Kennedy, S. C., Kunnath-Velayudhan, S., et al. (2016). Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47. Nat. Microbiol. 1:16133. doi: 10.1038/nmicrobiol.2016.133
Sechi, L. A., Ahmed, N., Felis, G. E., Dupre, I., Cannas, S., Fadda, G., et al. (2006). Immunogenicity and cytoadherence of recombinant heparin binding haemagglutinin (HBHA) of Mycobacterium avium subsp. paratuberculosis: functional promiscuity or a role in virulence? Vaccine 24, 236–243. doi: 10.1016/j.vaccine.2005.11.030
Snapper, S. B., Melton, R. E., Mustafa, S., Kieser, T., and Jacobs, W. R. Jr. (1990). Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol. Microbiol. 4, 1911–1919. doi: 10.1111/j.1365-2958.1990.tb02040.x
Songane, M., Kleinnijenhuis, J., Netea, M. G., and van Crevel, R. (2012). The role of autophagy in host defence against Mycobacterium tuberculosis infection. Tuberculosis (Edinb). 92, 388–396. doi: 10.1016/j.tube.2012.05.004
Temmerman, S. T., Place, S., Debrie, A. S., Locht, C., and Mascart, F. (2005). Effector functions of heparin-binding hemagglutinin-specific CD8+ T lymphocytes in latent human tuberculosis. J. Infect. Dis. 192, 226–232. doi: 10.1086/430930
Thurston, T. L., Wandel, M. P., von Muhlinen, N., Foeglein, A., and Randow, F. (2012). Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 482, 414–418. doi: 10.1038/nature10744
Vir, P., Gupta, D., Agarwal, R., and Verma, I. (2014). Interaction of alveolar epithelial cells with CFP21, a mycobacterial cutinase-like enzyme. Mol. Cell. Biochem. 396, 187–199. doi: 10.1007/s11010-014-2154-8
Wileman, T. (2013). Autophagy as a defence against intracellular pathogens. Essays Biochem. 55, 153–163. doi: 10.1042/bse0550153
Xiong, X., Zhang, H. M., Wu, T. T., Xu, L., Gan, Y. L., Jiang, L. S., et al. (2014). Titer dynamic analysis of D29 within MTB-infected macrophages and effect on immune function of macrophages. Exp. Lung Res. 40, 86–98. doi: 10.3109/01902148.2013.873841
Zhang, L., Zhang, H., Zhao, Y., Mao, F., Wu, J., Bai, B., et al. (2012). Effects of Mycobacterium tuberculosis ESAT-6/CFP-10 fusion protein on the autophagy function of mouse macrophages. DNA Cell Biol. 31, 171–179. doi: 10.1089/dna.2011.1290
Keywords: HBHA, A549, Mycobacterium tuberculosis, Mycobacterium smegmatis, autophagy, apoptosis
Citation: Zheng Q, Li Z, Zhou S, Zhang Q, Zhou L, Fu X, Yang L, Ma Y and Hao X (2017) Heparin-binding Hemagglutinin of Mycobacterium tuberculosis Is an Inhibitor of Autophagy. Front. Cell. Infect. Microbiol. 7:33. doi: 10.3389/fcimb.2017.00033
Received: 21 November 2016; Accepted: 23 January 2017;
Published: 07 February 2017.
Edited by:
Suzana P. Salcedo, MMSB-Lyon, FranceReviewed by:
Mathias Faure, Claude Bernard University Lyon 1, FranceMaurizio Renna, University of Cambridge, UK
Copyright © 2017 Zheng, Li, Zhou, Zhang, Zhou, Fu, Yang, Ma and Hao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yueyun Ma, Y21ibWF5eUBmbW11LmVkdS5jbg==
Xiaoke Hao, aGFveGtnQGZtbXUuZWR1LmNu
†These authors have contributed equally to this work.