 Dongsheng Han
Dongsheng Han Fei Yu
Fei Yu Hui Tang3
Hui Tang3- 1Clinical Medical Examination Center, Northern Jiangsu People's Hospital, Yangzhou, China
- 2Key Laboratory of Clinical In Vitro Diagnostic Techniques of Zhejiang Province, Department of Clinical Laboratory, First Affiliated Hospital, College of Medicine, Zhejiang University, Hangzhou, China
- 3Department of Pathology and Pathophysiology, Institute for Brain Research, Collaborative Innovation Center for Brain Science, Huazhong University of Science and Technology, Wuhan, China
In China, V. parahaemolyticus has been a leading cause of foodborne outbreaks and bacterial infectious diarrhea since the 1990s, and most infections have been associated with the pandemic V. parahaemolyticus O3:K6 and its serovariants. However, a comprehensive overview of the sero-prevalence and genetic diversity of the pandemic V. parahaemolyticus clone in China is lacking. To compensate for this deficiency, pandemic isolates in both clinical and environmental Chinese samples collected from multiple studies were analyzed in this study. Surprisingly, as many as 27 clinical pandemic serovariants were identified and were widely distributed across nine coastal provinces and two inland provinces (Beijing and Sichuan). O3:K6, O4:K68, and O1:KUT represented the predominant clinical serovars. Only four environmental pandemic serovariants had previously been reported, and they were spread throughout Shanghai (O1:KUT, O3:K6), Jiangsu (O3:K6, O4:K48), Zhejiang (O3:K6), and Guangdong (O4:K9). Notably, 24 pandemic serovariants were detected within a short time frame (from 2006 to 2012). The pandemic isolates were divided into 15 sequence types (STs), 10 of which fell within clonal complex (CC) 3. Only three STs (ST3, ST192, and ST305) were identified in environmental isolates. Substantial serotypic diversity was mainly observed among isolates within pandemic ST3, which comprised 21 combinations of O/K antigens. The pandemic O3:K6 serotype showed a high level of sequence diversity, which was shared by eight different STs (ST3, ST227, ST431, ST435, ST487, ST489, ST526, and ST672). Antimicrobial susceptibility testing revealed that most isolates shared similar antibiotic susceptibility profiles. They were resistant to ampicillin but sensitive to most other drugs that were tested. In conclusion, the high levels of serotypic and genetic diversity of the pandemic clone suggest that the involved regions are becoming important reservoirs for the emergence of novel pandemic strains. We underscore the need for routine monitoring to prevent pandemic V. parahaemolyticus infection, which includes monitoring antimicrobial responses to avoid excessive misuse of antibiotics. Further investigations are also needed to delineate the specific mechanisms underlying the possible seroconversion of pandemic isolates.
Introduction
Vibrio parahaemolyticus, a Gram-negative bacterium, is a natural inhabitant of estuarine and coastal environments. In humans, this pathogen is a globally important cause of acute gastroenteritis. It's a multi-serotype pathogenic bacteria, and can be classified into 13 O serotypes and 71 K serotypes based on somatic (O) antigens and capsular (K) antigens (Han et al., 2008). Since 1997, the pandemic isolates, including serotype O3:K6 and its serovariants, have spread globally by either sporadic diarrhea or contaminated food-related outbreaks (Nair et al., 2007; Chowdhury et al., 2013).
All the pandemic isolates share the following specific genetic markers: the presence of the thermostable direct hemolysin(tdh) gene, the absence of the TDH-related hemolysin(trh) gene, and a distinctive toxRS sequence (toxRS/new), which can be amplified by a group-specific PCR (GS-PCR) (Matsumoto et al., 2000). Until 2007, 22 serotypes had been traced to the pandemic clone based on these characteristics (Nair et al., 2007). In recent years, an increasing number of pandemic serotypes have been reported to be widely distributed in countries across four continents (Asia, Europe, the Americas and Africa) (Ansaruzzaman et al., 2008; Ottaviani et al., 2010; Powell et al., 2013; Li et al., 2014; Velazquez-Roman et al., 2014; Guerrero et al., 2017), suggesting that these pandemic serotypes pose a mounting public health threat. This threat calls for higher surveillance of the pandemic clone to reduce illnesses.
In China, V. parahaemolyticus has been the leading cause of foodborne outbreaks and bacterial infectious diarrhea since the 1990s, especially in coastal regions (Gao et al., 2016; Li et al., 2016). The pandemic serotype O3:K6 was first documented as the dominant serotype in 2002 and was proven to be a pandemic clone in 2008 (Vongxay et al., 2008). During the period of 2007–2012, approximately 56% of the clinical isolates in southern coastal areas of China had pandemic characteristics (Li et al., 2014). In one of our multi-center active surveillance programs, we found that 63.3% of the tested isolates were pandemic isolates in southeastern China from 2009 to 2013 (Chen et al., 2016).
The above findings indicate that the pandemic clone of V. parahaemolyticus plays an important role in causing infectious diseases in China. However, although laboratory-based surveillance for acute infectious diarrhea has been established in several coastal regions (Yu et al., 2011; Zhang et al., 2014; Li et al., 2015), a nationwide study or review of the distribution of infections caused by the pandemic isolates of V. parahaemolyticus has not been conducted in this country. Thus, a full understanding of the spread of this unique clone is needed to prevent outbreaks and sporadic illnesses in China.
In this study, we identified Chinese pandemic isolates of V. parahaemolyticus mostly from published literatures, and re-analyzed the sero-prevalence and genetic diversity of these pandemic isolates as a whole. We isolated some of these pandemic isolates in our active studies of diarrheal infection, and mainly focused on their antimicrobial responses in this study, which has not been shown previously. Overall, our intention is to generate a comprehensive overview of the spread of pandemic Vibrio parahaemolyticus O3:K6 and its serovariants in China since the emergence of this clone.
Materials and Methods
Datasets Utilized in the Present Study
To identify as many available pandemic isolates of V. parahaemolyticus as possible, we conducted a comprehensive search of several databases, including the US National Library of Medicine, PubMed, Elsevier, Springer, and China National Knowledge Infrastructure. We searched for all relevant Chinese studies using combinations of the following terms (through October 1, 2016): “Vibrio parahaemolyticus,” “pandemic clone,” “pandemic strains,” “pandemic isolates,” “O3:K6 serotype,” and “O3:K6 clone.” Additional eligible studies were identified from references cited in the relevant articles. The full text of each potentially relevant paper was scrutinized, and a total of 290 representative clinical and environmental V. parahaemolyticus isolates (toxRS/new+, tdh+ and trh−) were extracted from 16 studies and selected as the research subjects of this investigation.
Among the 290 pandemic isolates, 120 are from our laboratory, including 98 ST3 isolates, 21 ST88 isolates, and one ST672 isolate. Most of the isolates include information about the sampling area, year of isolation, source, serotype and multilocus sequence typing. Details on the individual isolates are summarized in Additional file 1: Table S1.
We grouped these 290 pandemic isolates according to the integrity of background information (Figure 1), and then carried out the re-analysis of the spreading of pandemic V. parahaemolyticus O3:K6 and its serovariants.
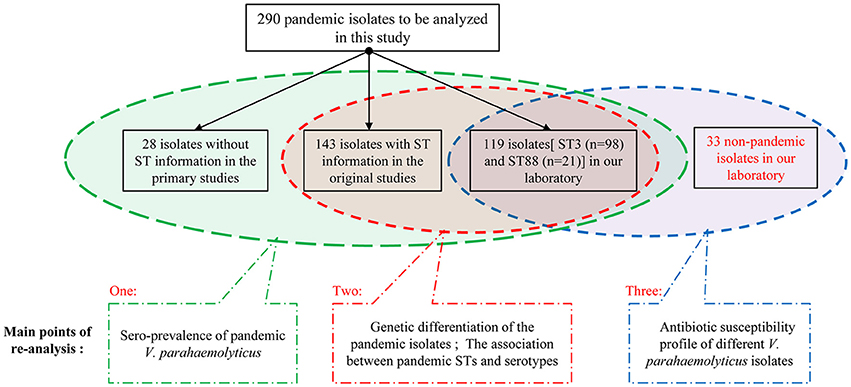
Figure 1. The grouping of the collected isolates and the main points of re-analysis in this study.
Assignment to Clonal Complexes
Various typing methods have been used to distinguish V. parahaemolyticus isolates for epidemiological investigations (Marshall et al., 1999; Gonzalez-Escalona et al., 2008; Lüdeke et al., 2015). The high accuracy and repeatability of current sequencing technology, the ability to compare DNA sequences universally and the ability to share data among laboratories make multi-locus sequence typing (MLST) a complete, robust, and reliable typing method. In recent years, scientists have begun using whole genome sequencing (WGS) to analyze historical collections of isolates, providing new insights for understanding population dynamics among different V. parahaemolyticus isolates. However, many laboratories are not currently using WGS, as data are not easily shared among laboratories. For this reason, we used MLST to assign clonal complexes for the pandemic isolates collected in this study.
The MLST scheme used internal fragments of the seven house-keeping genes [recA(729bp), dnaE(557bp), gyrB(592bp), dtdS(458bp), pntA(430bp), pyrC(493bp), and tnaA(423bp)]. The standard amplification protocol was published on the V. parahaemolyticus MLST web site (http://pubmlst.org/vparahaemolyticus/). The allele designations and sequence types (STs) of all the selected isolates had been determined based on the variation of the seven genes. Based on the defined STs, all the pandemic isolates were compared using global optimal eBURST analysis (goeBURST) version 1.2.1 (http://www.phyloviz.net/goeburst/). Clonal complexes were conservatively defined as a cluster of STs, in which all STs were linked as single-locus variants (SLVs, two STs differing from each other at a single locus) to at least one other ST (Feil et al., 2004). The singleton STs corresponded to STs differing from the others by three or more of the seven loci (Esteves et al., 2015).
Antimicrobial Susceptibility Testing
We selected the pandemic isolates [ST3 (n = 98) and ST88 (n = 21)] detected in our laboratory from the 290 isolates to be analyzed in this study and other 33 non-pandemic isolates identified in our laboratory to conduct antimicrobial susceptibility testing with 20 antimicrobial agents (Figure 1). The testing was performed using the disk diffusion method according to the Clinical and Laboratory Standards Institute (CLSI, 2010). E. coli ATCC25922 and Staphylococcus aureus ATCC25923 were employed as bacterial strains for quality control. Characterization of the resistance and susceptibility profile of the isolates was determined by measuring inhibitory zone, and then compared with the interpretative chart (Table 1).
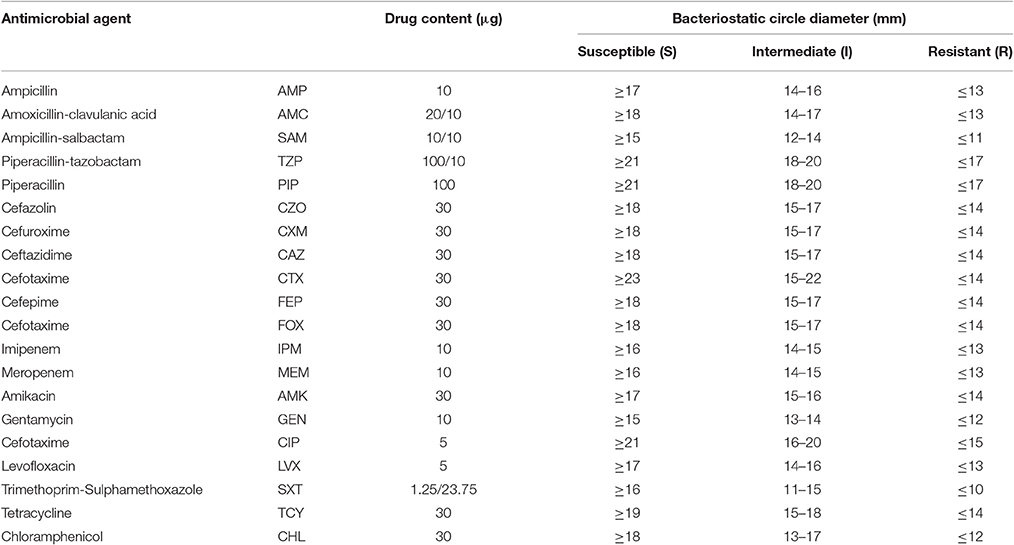
Table 1. The interpreted results of drug susceptibility testing.
Results
Sero-Prevalence of Pandemic V. parahaemolyticus
A comprehensive map of the dissemination of the pandemic serotypes detected in China was generated according to a detailed review (Figure 1). The pandemic serotypes were highly abundant and variable, with 27 clinical and four environmental pandemic serotypes identified in nine coastal provinces and two inland provinces (Beijing and Sichuan) (Figure 2). The most widely disseminated serotype of clinical isolates was O3:K6 (in 11 provinces), followed by O4:K68 (in eight provinces), O1:KUT (in five provinces), O1:K25 (in four provinces), and O1:K36 (in four provinces). The four environmental serotypes were identified in Shanghai (O1:KUT, O3:K6), Jiangsu (O3:K6, O4:K48), Zhejiang (O3:K6), and Guangdong (O4:K9). In addition, the sources of the environmental O3:K6 isolates were diverse in Jiangsu province (Table 2).
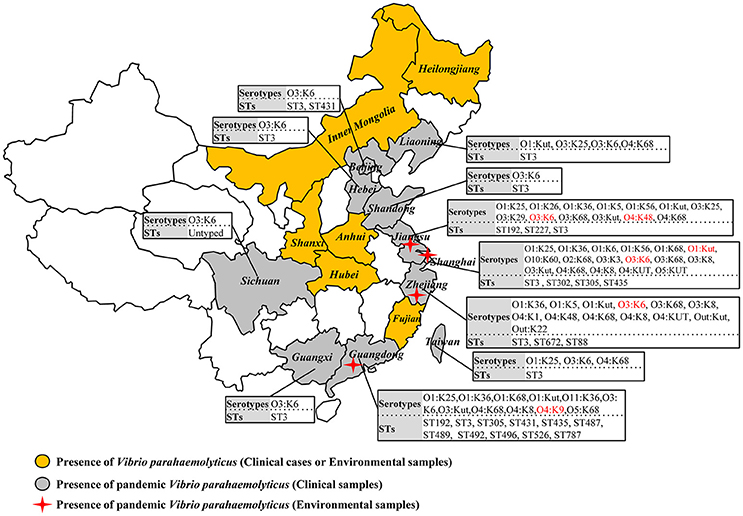
Figure 2. Map showing the sero-prevalence and sequence diversity of clinical and environmental pandemic O3:K6 and its serovariants of V. parahaemolyticus found in Chinese samples.
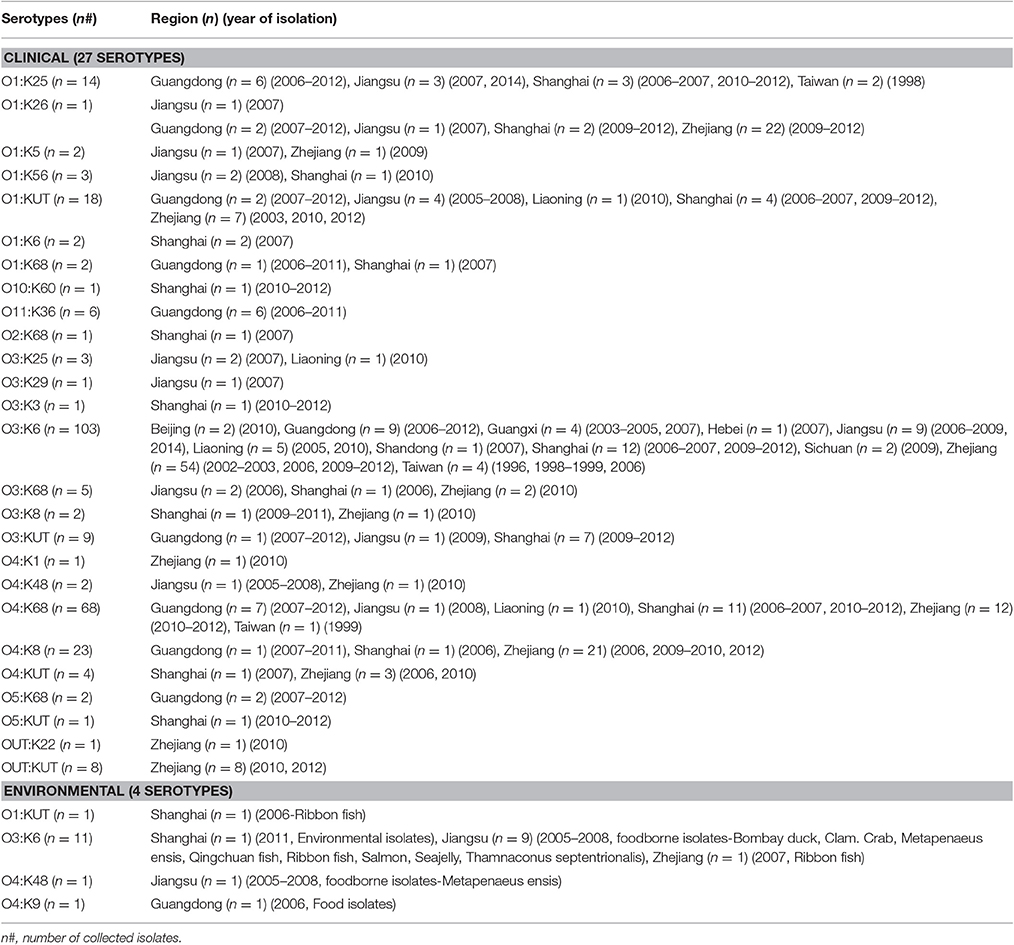
Table 2. Sero-prevalence of pandemic V. parahaemolyticus isolates from Chinese samples.
When sorting the isolates by the year of isolation, we found that only the O3:K6, O1:K25, O4:K68, and O1:KUT4 serotypes had been reported prior to 2005. However, from 2006 to 2012, as many as 24 more pandemic serotypes were detected (Table 3). This may be a significant period for the spreading of pandemic serotypes in China, but underreporting and possible study deviation should also be considered. As these factors directly affect the accuracy of our conjecture. O3:K6, first discovered in 1996, was detected each year from 2002 to 2012.
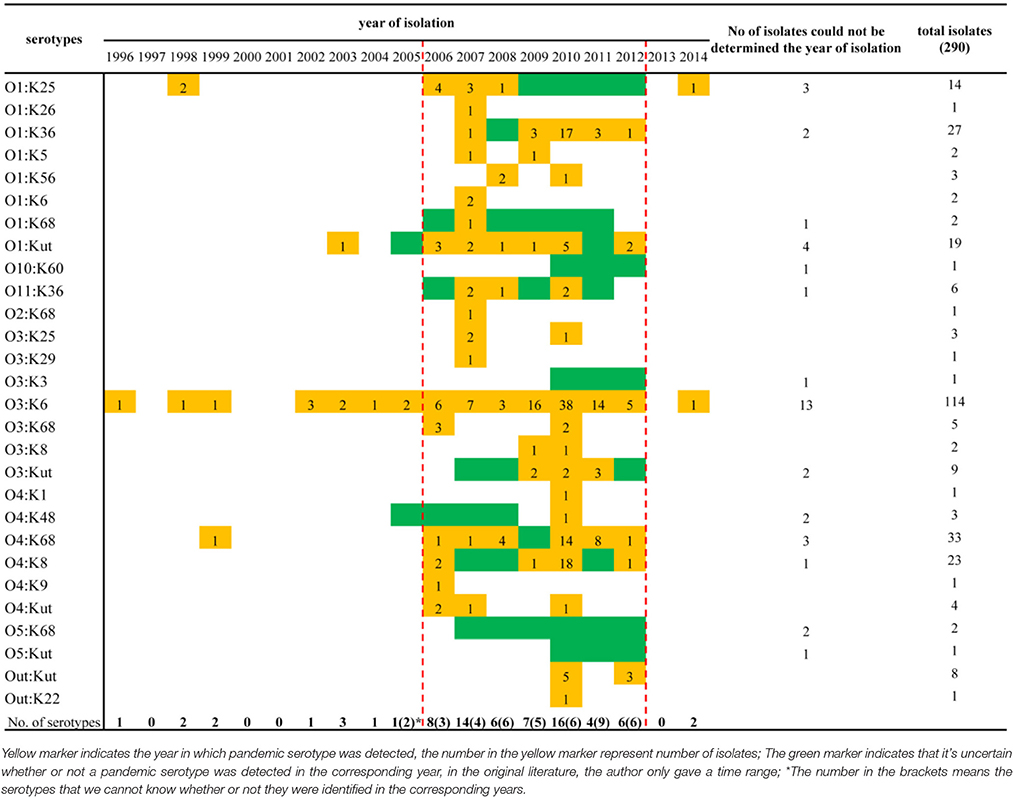
Table 3. Chronology of appearance of pandemic V. parahaemolyticus serotypes.
Genetic Differentiation of the Pandemic Isolates
Among the 290 collected pandemic isolates, 28 isolates was not typed by MLST analysis in their original studies, the other 262 isolates were involved the MLST analysis in the current study (Figure 1). These pandemic isolates exhibited 15 STs, revealing high genetic diversity (Table 4). The sequence variation of the isolates in Guangdong (11 STs) was significantly higher than that in other provinces (Figure 1). ST3 was the only sequence type that caused a wide range of infections in as many as ten provinces. Only three sequence types (ST3, ST192, and ST305) had ever been identified in environmental isolates.
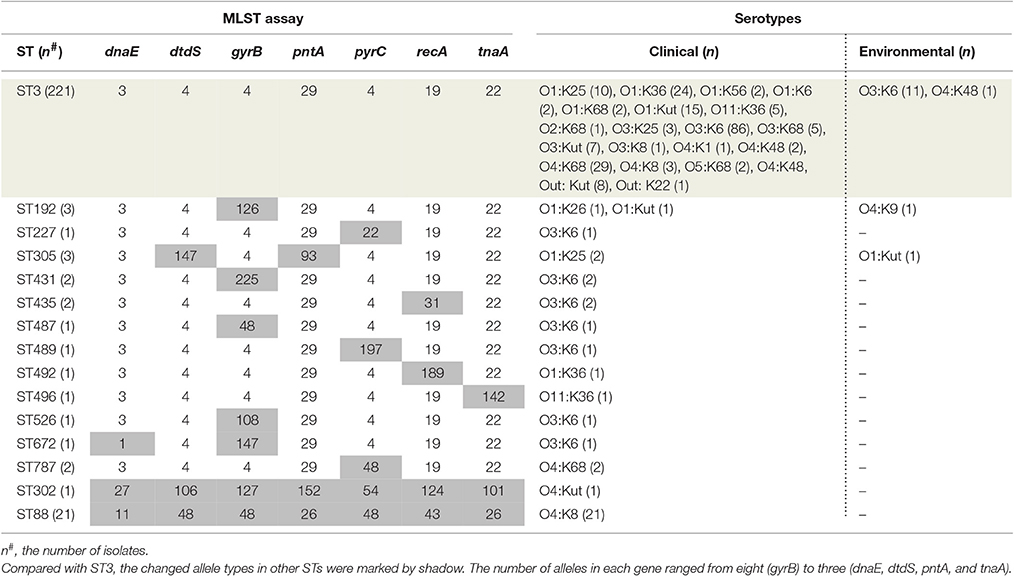
Table 4. Sequence types, allele profiles, and serotypes of pandemic V. paraheamolyticus isolates.
Ten of the 15 pandemic STs could be classified in CC3 (Figure 3). ST305 and ST672 were DLVs of ST3. Pandemic ST88 was found only in Zhejiang province during 2010–2012 and it belongs to CC345, not CC3. ST302 originated from in the Shanghai province and was identified as a singleton with no relationship to other STs.
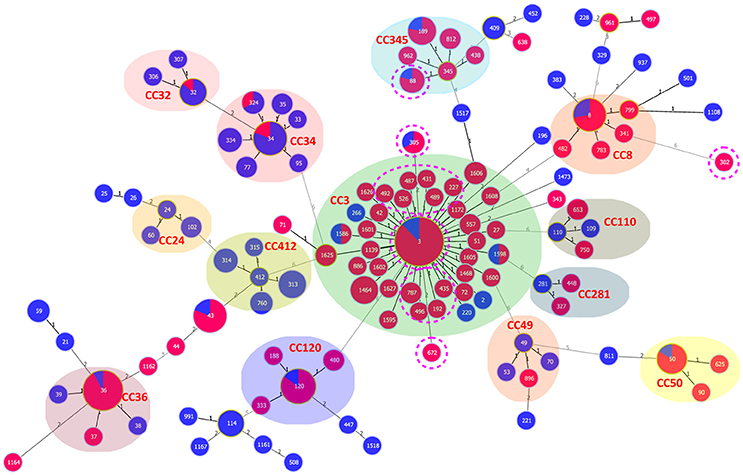
Figure 3. goeBURST full MST of the STs shows the clonal diversity of Chinese clinical and environmental pandemic V. parahaemolyticus isolates: clinical (red) and environmental (blue). The pandemic STs in China are denoted by pink dotted circles. Other STs were selected from the public MLST database (https://pubmlst.org/vparahaemolyticus/) to help us analyze the cluster relationship of pandemic STs in this study. The number of different alleles is presented between STs connected via a line. The circle size varies according to the frequency of STs. Each shaded area represents a unique clone complex.
The Association between Pandemic STs and Serotypes
The pandemic isolates within ST3 comprised 21 clinical and two environmental serotypes (O3:K6 and O4:K48), thus exhibiting high serotypic diversity (Table 4). ST192 included isolates that belong to O1:K26 (clinical), and O1: KUT (clinical) and O4:K9 (environmental) serotypes. ST305 consisted of two serotypes, O1:K25 (clinical) and O4:Kut (environmental). The remaining STs consisted of a single serotype. From another perspective, the pandemic O3:K6 serotype was shared by eight different STs (ST3, ST227, ST431, ST435, ST487, ST489, ST526, and ST672). O1:Kut isolates were divided into three STs (ST3, ST192, and ST305). Other serotypes were clustered in no more than two different pandemic STs.
Antibiotic Susceptibility Profile of Different Pandemic Isolates
The results of antimicrobial susceptibilities of the 98 pandemic ST3 isolates, 21 pandemic ST88 isolates and 33 non-pandemic isolates are shown in Table S2. The results indicate similar antimicrobial profiles between isolates within different STs. In other words, there were not obvious differences in their resistance spectrums (Figure 4). Specifically, the isolates were mostly resistant to ampicillin (AMP) (95.2% of ST88, 85.7% of ST3, and 84.9% of other STs), and showed intermediate resistance to cefazolin (CZO), Amikacin (AMK), and Cefuroxime (CXM). It is reassuring that the isolates were susceptible to the majority of antibiotic tested, and all of them were susceptible to levofloxacin (LVX), meropenem (MEM), imipenem (IPM), cefepime (FEP), ceftazidime (CAZ), and tetracyclines (TCY). Additionally, there was no multidrug resistant bacteria (MDR) found.
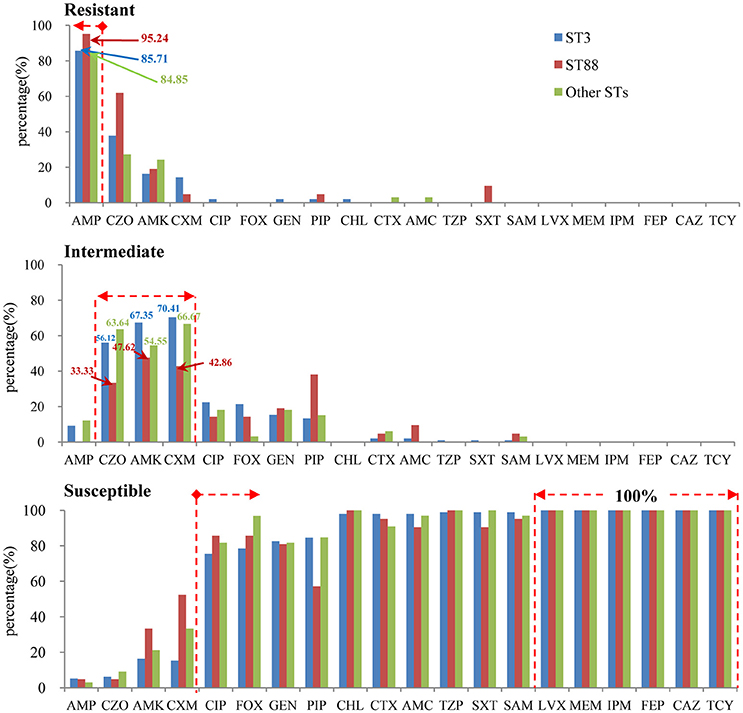
Figure 4. The antimicrobial profiles within different STs (pandemic: ST3 and ST88, non-pandemic: other STs).
Discussion
As an emerging public health concern, pandemic V. parahaemolyticus infection has attracted wide attention from scientists (Jun et al., 2014; Velazquez-Roman et al., 2014; de Jesús Hernández-Díaz et al., 2015). The present study has provided an overview of the prevalence of pandemic isolates of V. parahaemolyticus in both clinical and environmental samples collected from multiple Chinese studies. We demonstrated that these pandemic isolates showed high serotypic and genetic diversity. The O3:K6 pandemic isolates (persistent for 11 years from 2002 to 2012) spread across 11 provinces indicate that the pandemic clone has been endemically established in China. Continued monitoring of antibiotic resistance patterns in pandemic isolates is urgently needed to avoid the excessive misuse of antibiotics, although most of the isolates tested in this study only showed high resistance to ampicillin.
The serovariants of this pandemic clone were abundant and variable. From 1996 to 2007, up to 22 pandemic serovariants were identified around the world (Nair et al., 2007), suggesting that the pandemic isolates could easily adapt new serotypes to survive in new environments. In this study, we found that 27 pandemic clinical serotypes and four environmental serotypes have been identified in the collected Chinese isolates. This is a significant number when compared to the number of serotypes found in other countries or regions (Nair et al., 2007; Pazhani et al., 2014; Velazquez-Roman et al., 2014), although the regional persistence of O3:K6 pandemic isolates has also been discovered in many other countries, such as Peru (Gil et al., 2007), Chile (Ansede-Bermejo et al., 2010), Japan (Okuda et al., 1997), India (Pazhani et al., 2014), and Thailand (Mala et al., 2016). To the best of our knowledge, no study has explained why serotypes of the pandemic clone are so diverse in China. Researchers around the world have made some progress in finding the cause of such serodiversity of the pandemic clone. Such research has determined that the O- and K-antigens are mostly mutated concurrently by horizontal gene transfer (HGT) (Okura et al., 2008), and it is speculated that new serovariants have emerged from the pandemic O3:K6 strains via replacement of the putative O and K antigen gene clusters (Okura et al., 2008; Harth et al., 2009; Chen et al., 2010). This is important for the survival of the pandemic clone in the face of changing external environments and host immunological resistance.
The analyzed isolates in this study were distributed in regions where the differences of temperature and other environmental factors are vast. Therefore, we suspect that the strains must adapt to different living environments in the transmission process by altering their biological properties more frequently. Making serological changes may be one of the most efficient ways for this to be accomplished. However, the specific mechanism of serotype conversion is not yet known. The highest priority is currently to continuously monitor the emergence of new serovariants to prevent the pandemic strains from causing outbreaks along the coastline and spreading to other countries and regions.
MLST is known to provide greater resolution than stereotyping. In a previous study, we provided an extensive review of isolates found in Chinese patients, and the results indicated a high degree of genetic diversity and a complicated population structure of V. parahaemolyticus (Han et al., 2015). With the present MLST study, we intended to demonstrate genetic similarities or differences among the pandemic isolates identified from different sources. As expected, we found that most of the pandemic STs were classified into the same clonal complex (CC3). However, some molecular divergence was also found. All the pandemic ST88 in Zhejiang province were assigned to CC345 (not CC3), and ST302, which originated in Shanghai province, was identified as a singleton with no relationship to other pandemic STs. Singletons do not seem to belong to the same clone as other pandemic isolates, but they do share the pandemic traits (toxRS/new+, tdh+, and trh−).
Chen et al. demonstrated the isolates of pandemic ST302 were clustered with other pandemic isolates based on other molecular typing methods, such as enterobacterial repetitive intergenic consensus sequence PCR (ERIC-PCR) (Chen et al., 2012). Similarly, in our pulsed field gel electrophoresis (PFGE) analysis (data not shown), the pandemic isolates of ST88 and ST3 shared the same PFGE profile. Thus, we observed that current typing and clustering methods may lead to controversial results, making it difficult to draw conclusions.
Therefore, a combined application of several molecular typing techniques should be considered in epidemiological investigations of V. parahaemolyticus pandemic isolates. As explained before (in Materials and Methods section), WGS is the best way to accurately portray the evolution and population structure of V. parahaemolyticus isolates at the molecular level (Cui et al., 2015; Haendiges et al., 2015), but the high cost limits its popularity in the analysis of large quantities of specimens.
Another important aspect of this study was the investigation of the antimicrobial susceptibility of different pandemic isolates. Our results revealed similar antibiotic susceptibility profiles in pandemic ST3, ST88, and non-pandemic isolates. This finding was similar to that in the work of Elmahdi et al. (2016). They concluded that the sampling location or month in which the samples were collected did not significantly impact V. parahaemolyticus resistance patterns because isolates from both environmental and clinical sources shared similar antibiotic resistance profiles. Unsurprisingly, the majority of the isolates tested in this study showed ampicillin resistance, which is very common in V. parahaemolyticus isolates recovered from different sources (Sun et al., 2013; de Jesús Hernández-Díaz et al., 2015; Elmahdi et al., 2016; Mala et al., 2016). This result suggests that ampicillin should have a negligible role in the treatment of V. parahaemolyticus infection. In fact, a survey conducted in the United States showed that very high V. parahaemolyticus ampicillin resistance could be traced as far back as 1978 (Blake et al., 1979).
In contrast, most of the isolates tested were sensitive to the majority of antibiotics tested, and all isolates were susceptible to LVX, MEM, IPM, FEP, and CAZ. This result suggests that these drugs can be used as an alternative antibiotic therapy. It must be noted that recently isolated V. parahaemolyticus strains, including pandemic strains, have displayed resistance to multiple antibiotics (Jun et al., 2012, 2014), which increases concerns about possible antibiotic treatment failure. Although we did not discover any multidrug resistant bacteria (MDR) isolates in this study, continued monitoring of pandemic strain susceptibility to antibiotic resistance is urgently needed to avoid the excessive misuse of antibiotics used to treat infections that pose threats to public health.
Our findings represent a comprehensive review of the pandemic V. parahaemolyticus O3:K6 and its serovariants by thoroughly assessing an extensive collection of clinical and environmental pandemic isolates from multiple Chinese studies. High levels of serotypic and genetic diversity in the pandemic clone are found, which suggests that the involved regions are becoming important reservoirs for the emergence of novel pandemic strains, which makes the clinical management of the infection and its prevention potentially challenging. Thus, we underscore the need for routine clinical and environmental monitoring to prevent pandemic V. parahaemolyticus infection and dissemination, including monitoring of antimicrobial response even though most current antimicrobial agents in routine use are effective. The mechanism in which the isolates undergo seroconversion with pandemic genetic marks warrants extended investigation.
Author Contributions
Conceived and designed the experiments: DH and CH. Performed the experiments: DH, FY, and HT. Analyzed the data: DH, CW, and PZ. Contributed reagents/materials/analysis tools: DH and CR. Wrote the paper: DH.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was jointly supported by grant No. 81400899 from the National Natural Science Foundation of China, grant No. yz2016127 from the Science and Technology Foundation Program of Yangzhou city, and two research projects (No. yzucms201603 and No. yzucms201608) of Northern Jiangsu People's Hospital, Yangzhou, China.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcimb.2017.00188/full#supplementary-material
References
Ansaruzzaman, M., Chowdhury, A., Bhuiyan, N. A., Sultana, M., Safa, A., Lucas, M., et al. (2008). Characteristics of a pandemic clone of O3:K6 and O4:K68 Vibrio parahaemolyticus isolated in Beira, Mozambique. J. Med. Microbiol. 57(Pt 12), 1502–1507. doi: 10.1099/jmm.0.2008/004275-0
Ansede-Bermejo, J., Gavilan, R. G., Trinanes, J., Espejo, R. T., and Martinez-Urtaza, J. (2010). Origins and colonization history of pandemic Vibrio parahaemolyticus in South America. Mol. Ecol. 19, 3924–3937. doi: 10.1111/j.1365-294X.2010.04782.x
Blake, P. A., Merson, M. H., Weaver, R. E., Hollis, D. G., and Heublein, P. C. (1979). Disease caused by a marine Vibrio. Clinical characteristics and epidemiology. N. Engl. J. Med. 300, 1–5. doi: 10.1056/nejm197901043000101
CLSI (2010). Methods for Antimicrobial Dilution and Dusk Susceptibility Testing of Infrequently Isolated or Fastidious Bacteria; Approved Guideline, 2nd Edn. M45–2A. Wayne, PA: Clinical and Laboratory Standards Institute (CLSI).
Chen, W., Xie, Y., Xu, J., Wang, Q., Gu, M., Yang, J., et al. (2012). Molecular typing of Vibrio parahaemolyticus isolates from the middle-east coastline of China. Int. J. Food Microbiol. 153, 402–412. doi: 10.1016/j.ijfoodmicro.2011.12.001
Chen, Y., Chen, X., Yu, F., Wu, M., Wang, R., Zheng, S., et al. (2016). Serology, virulence, antimicrobial susceptibility and molecular characteristics of clinical Vibrio parahaemolyticus strains circulating in southeastern China from 2009 to 2013. Clin. Microbiol. Infect. 22, 258–259. doi: 10.1016/j.cmi.2015.11.003
Chen, Y., Dai, J., Morris, J. J. G., and Johnson, J. A. (2010). Genetic analysis of the capsule polysaccharide (K antigen) and exopolysaccharide genes in pandemic Vibrio parahaemolyticus O3:K6. BMC Microbiol. 10:274. doi: 10.1186/1471-2180-10-274
Chowdhury, G., Ghosh, S., Pazhani, G. P., Paul, B. K., Maji, D., Mukhopadhyay, A. K., et al. (2013). Isolation and characterization of pandemic and nonpandemic strains of Vibrio parahaemolyticus from an outbreak of diarrhea in North 24 Parganas, West Bengal, India. Foodborne Pathog. Dis. 10, 338–342. doi: 10.1089/fpd.2012.1340
Cui, Y., Yang, X., Didelot, X., Guo, C., Li, D., Yan, Y., et al. (2015). Epidemic clones, Oceanic gene pools, and Eco-LD in the free living Marine pathogen Vibrio parahaemolyticus. Mol. Biol. Evol. 32, 1396–1410. doi: 10.1093/molbev/msv009
de Jesús Hernández-Díaz, L., Leon-Sicairos, N., Velazquez-Roman, J., Flores-Villaseñor, H. C., Guadron-Llanos, A. M., Martinez-Garcia, J. J., et al. (2015). A pandemic Vibrio parahaemolyticus O3:K6 clone causing most associated diarrhea cases in the Pacific Northwest coast of Mexico. Front. Microbiol. 6:221. doi: 10.3389/fmicb.2015.00221
Elmahdi, S., DaSilva, L. V., and Parveen, S. (2016). Antibiotic resistance of Vibrio parahaemolyticus and Vibrio vulnificus in various countries: a review. Food Microbiol. 57, 128–134. doi: 10.1016/j.fm.2016.02.008
Esteves, K., Mosser, T., Aujoulat, F., Hervio-Heath, D., Monfort, P., and Jumas-Bilak, E. (2015). Highly diverse recombining populations of Vibrio cholerae and Vibrio parahaemolyticus in French Mediterranean coastal lagoons. Front. Microbiol. 6:708. doi: 10.3389/fmicb.2015.00708
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P., and Spratt, B. G. (2004). eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186, 1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004
Gao, L., Deng, Y. Q., Chen, C., Ke, C. W., Li, B. S., Long, Y. Y., et al. (2016). Population relationship of Vibrio parahaemolyticus isolates derived from aquaculture ponds, a Seafood market, restaurants, and clinical samples. Foodborne Pathog. Dis. 13, 333–342. doi: 10.1089/fpd.2015.2098
Gil, A. I., Miranda, H., Lanata, C. F., Prada, A., Hall, E. R., Barreno, C. M., et al. (2007). O3:K6 serotype of Vibrio parahaemolyticus identical to the global pandemic clone associated with diarrhea in Peru. Int. J. Infect. Dis. 11, 324–328. doi: 10.1016/j.ijid.2006.08.003
Gonzalez-Escalona, N., Martinez-Urtaza, J., Romero, J., Espejo, R. T., Jaykus, L. A., and DePaola, A. (2008). Determination of molecular phylogenetics of Vibrio parahaemolyticus strains by multilocus sequence typing. J. Bacteriol. 190, 2831–2840. doi: 10.1128/JB.01808-07
Guerrero, A., Lizárraga-Partida, M. L., Gómez, G., Licea-Navarro, A. F., and Revilla-Castellanos, V. J. (2017). Genetic analysis of Vibrio parahaemolyticus O3:K6 strains that have been isolated in Mexico since 1998. PLoS ONE 12:e0169722. doi: 10.1371/journal.pone.0169722
Haendiges, J., Timme, R., Allard, M. W., Myers, R. A., Brown, E. W., and Gonzalez-Escalona, N. (2015). Characterization of Vibrio parahaemolyticus clinical strains from Maryland (2012–2013) and comparisons to a locally and globally diverse V. parahaemolyticus strains by whole-genome sequence analysis. Front. Microbiol. 6:125. doi: 10.3389/fmicb.2015.00125
Han, D., Tang, H., Ren, C., Wang, G., Zhou, L., and Han, C. (2015). Prevalence and genetic diversity of clinical Vibrio parahaemolyticus isolates from China, revealed by multilocus sequence typing scheme. Front. Microbiol. 6:291. doi: 10.3389/fmicb.2015.00291
Han, H., Wong, H. C., Kan, B., Guo, Z., Zeng, X., Yin, S., et al. (2008). Genome plasticity of Vibrio parahaemolyticus: microevolution of the ‘pandemic group’. BMC Genomics 9:570. doi: 10.1186/1471-2164-9-570
Harth, E., Matsuda, L., Hernandez, C., Rioseco, M. L., Romero, J., Gonzalez-Escalona, N., et al. (2009). Epidemiology of Vibrio parahaemolyticus outbreaks, southern Chile. Emerg. Infect. Dis. 15, 163–168. doi: 10.3201/eid1502.071269
Jun, J. W., Kim, J. H., Choresca, C. J., Shin, S. P., Han, J. E., Han, S. Y., et al. (2012). Isolation, molecular characterization, and antibiotic susceptibility of Vibrio parahaemolyticus in Korean seafood. Foodborne Pathog. Dis. 9, 224–231. doi: 10.1089/fpd.2011.1018
Jun, J. W., Shin, T. H., Kim, J. H., Shin, S. P., Han, J. E., Heo, G. J., et al. (2014). Bacteriophage therapy of a Vibrio parahaemolyticus infection caused by a multiple-antibiotic-resistant O3:K6 pandemic clinical strain. J. Infect. Dis. 210, 72–78. doi: 10.1093/infdis/jiu059
Lüdeke, C. H. M., Gonzalez-Escalona, N., Fischer, M., and Jones, J. L. (2015). Examination of clinical and environmental Vibrio parahaemolyticus isolates by multi-locus sequence typing (MLST) and multiple-locus variable-number tandem-repeat analysis (MLVA). Front. Microbiol. 6:564. doi: 10.3389/fmicb.2015.00564
Li, B., Luo, J., Tan, H., Ke, B., He, D., Ke, C., et al. (2015). Phenotypic and phylogenetic analysis of Vibrio parahaemolyticus isolates recovered from diarrhea cases in Guangdong Province, China. Int. J. Food Microbiol. 200, 13–17. doi: 10.1016/j.ijfoodmicro.2014.12.009
Li, J., Xue, F., Yang, Z., Zhang, X., Zeng, D., Chao, G., et al. (2016). Vibrio parahaemolyticus strains of pandemic serotypes identified from clinical and environmental samples from Jiangsu, China. Front. Microbiol. 7:787. doi: 10.3389/fmicb.2016.00787
Li, Y., Xie, X., Shi, X., Lin, Y., Qiu, Y., Mou, J., et al. (2014). Vibrio parahaemolyticus, Southern Coastal Region of China, 2007–2012. Emerg. Infect. Dis. 20, 685–688. doi: 10.3201/eid2004.130744
Mala, W., Alam, M., Angkititrakul, S., Wongwajana, S., Lulitanond, V., Huttayananont, S., et al. (2016). Serogroup, virulence, and molecular traits of Vibrio parahaemolyticus isolated from clinical and cockle sources in northeastern Thailand. Infect. Genet. Evol. 39, 212–218. doi: 10.1016/j.meegid.2016.01.006
Marshall, S., Clark, C. G., Wang, G., Mulvey, M., Kelly, M. T., and Johnson, W. M. (1999). Comparison of molecular methods for typing Vibrio parahaemolyticus. J. Clin. Microbiol. 37, 2473–2478.
Matsumoto, C., Okuda, J., Ishibashi, M., Iwanaga, M., Garg, P., Rammamurthy, T., et al. (2000). Pandemic spread of an O3:K6 clone of Vibrio parahaemolyticus and emergence of related strains evidenced by arbitrarily primed PCR and toxRS sequence analyses. J. Clin. Microbiol. 38, 578–585.
Nair, G. B., Ramamurthy, T., Bhattacharya, S. K., Dutta, B., Takeda, Y., and Sack, D. A. (2007). Global dissemination of Vibrio parahaemolyticus serotype O3:K6 and its serovariants. Clin. Microbiol. Rev. 20, 39–48. doi: 10.1128/CMR.00025-06
Okuda, J., Ishibashi, M., Hayakawa, E., Nishino, T., Takeda, Y., Mukhopadhyay, A. K., et al. (1997). Emergence of a unique O3:K6 clone of Vibrio parahaemolyticus in Calcutta, India, and isolation of strains from the same clonal group from Southeast Asian travelers arriving in Japan. J. Clin. Microbiol. 35, 3150–3155.
Okura, M., Osawa, R., Tokunaga, A., Morita, M., Arakawa, E., and Watanabe, H. (2008). Genetic analyses of the putative O and K antigen gene clusters of pandemic Vibrio parahaemolyticus. Microbiol. Immunol. 52, 251–264. doi: 10.1111/j.1348-0421.2008.00027.x
Ottaviani, D., Leoni, F., Rocchegiani, E., Canonico, C., Potenziani, S., Santarelli, S., et al. (2010). Prevalence, serotyping and molecular characterization of Vibrio parahaemolyticus in mussels from Italian growing areas, Adriatic Sea. Environ. Microbiol. Rep. 2, 192–197. doi: 10.1111/j.1758-2229.2010.00134.x
Pazhani, G. P., Bhowmik, S. K., Ghosh, S., Guin, S., Dutta, S., Rajendran, K., et al. (2014). Trends in the Epidemiology of Pandemic and Non-pandemic Strains of Vibrio parahaemolyticus Isolated from Diarrheal Patients in Kolkata, India. PLoS Negl. Trop. Dis. 8:e2815. doi: 10.1371/journal.pntd.0002815
Powell, A., Baker-Austin, C., Wagley, S., Bayley, A., and Hartnell, R. (2013). Isolation of Pandemic Vibrio parahaemolyticus from UK water and shellfish produce. Microb. Ecol. 65, 924–927. doi: 10.1007/s00248-013-0201-8
Sun, Y. F., Fan, Y. Y., Huang, Y. F., Wang, M., and Lu, X. X. (2013). Antimicrobial resistance and virulence characteristics of Vibrio parahaemolyticus. Zhonghua Yi Xue Za Zhi, 93, 2733–2735.
Velazquez-Roman, J., León-Sicairos, N., de Jesus Hernández-Díaz, L., and Canizalez-Roman, A. (2014). Pandemic Vibrio parahaemolyticus O3:K6 on the American continent. Front. Cell. Infect. Microbiol. 3:110. doi: 10.3389/fcimb.2013.00110
Vongxay, K., Pan, Z., Zhang, X., Wang, S., Cheng, S., Mei, L., et al. (2008). Occurrence of pandemic clones of Vibrio parahaemolyticus isolates from seafood and clinical samples in a Chinese coastal province. Foodborne Pathog. Dis. 5, 127–134. doi: 10.1089/fpd.2007.0045
Yu, Y., Hu, W., Wu, B., Zhang, P., Chen, J., Wang, S., et al. (2011). Vibrio parahaemolyticus isolates from Southeastern Chinese coast are genetically diverse with circulation of clonal complex 3 strains since 2002. Foodborne Pathog. Dis. 8, 1169–1176. doi: 10.1089/fpd.2011.0865
Keywords: Vibrio parahaemolyticus, multilocus sequence typing, pandemic clone, gastroenteritis, genetic diversity
Citation: Han D, Yu F, Tang H, Ren C, Wu C, Zhang P and Han C (2017) Spreading of Pandemic Vibrio parahaemolyticus O3:K6 and Its Serovariants: A Re-analysis of Strains Isolated from Multiple Studies. Front. Cell. Infect. Microbiol. 7:188. doi: 10.3389/fcimb.2017.00188
Received: 04 March 2017; Accepted: 02 May 2017;
Published: 18 May 2017.
Edited by:
Dongsheng Zhou, Beijing Institute of Microbiology and Epidemiology, ChinaReviewed by:
Xiaohui Zhou, University of Connecticut, United StatesHuaiqi Jing, Chinese Center for Disease Control and Prevention, China
Meiying Yan, National Institute for Communicable Disease Control and Prevention, China
Copyright © 2017 Han, Yu, Tang, Ren, Wu, Zhang and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dongsheng Han, aGFuZHMxMTAzQDE2My5jb20=
†These authors have contributed equally to this work.