Abstract
Neutrophils are the most abundant leukocytes in human blood and the first line of defense after bacteria have breached the epithelial barriers. After migration to a site of infection, neutrophils engage and expose invading microorganisms to antimicrobial peptides and proteins, as well as reactive oxygen species, as part of their bactericidal arsenal. Ideally, neutrophils ingest bacteria to prevent damage to surrounding cells and tissues, kill invading microorganisms with antimicrobial mechanisms, undergo programmed cell death to minimize inflammation, and are cleared away by macrophages. Staphylococcus aureus (S. aureus) is a prevalent Gram-positive bacterium that is a common commensal and causes a wide range of diseases from skin infections to endocarditis. Since its discovery, S. aureus has been a formidable neutrophil foe that has challenged the efficacy of this professional assassin. Indeed, proper clearance of S. aureus by neutrophils is essential to positive infection outcome, and S. aureus has developed mechanisms to evade neutrophil killing. Herein, we will review mechanisms used by S. aureus to modulate and evade neutrophil bactericidal mechanisms including priming, activation, chemotaxis, production of reactive oxygen species, and resolution of infection. We will also highlight how S. aureus uses sensory/regulatory systems to tailor production of virulence factors specifically to the triggering signal, e.g., neutrophils and defensins. To conclude, we will provide an overview of therapeutic approaches that may potentially enhance neutrophil antimicrobial functions.
Introduction
Polymorphonuclear leukocytes (PMNs or neutrophils) are the first line of defense against bacterial pathogens that have breached epithelial barriers. Within minutes of bacterial invasion, neutrophils respond to soluble factors including chemokines and cytokines and are recruited to the site of infection where they ingest microbes. Subsequently, neutrophils expose microorganisms to antimicrobial proteins, peptides, and reactive oxygen species to kill the invading pathogen. This is a delicate process that must eliminate the pathogen while controlling excessive inflammation. Concurrently, neutrophils secrete cytokines and chemokines to continue to recruit neutrophils and enhance other host responses to infection. Finally, neutrophil death is essential for proper resolution of infection and must be regulated to minimize bystander damage while continuing to signal if more immune response is needed or if tissue repair should begin. These potent mechanisms are effective at eliminating most fungal and bacterial microorganisms. However, successful pathogens have developed strategies to disrupt various neutrophil functions to cause infection.
S. aureus is a highly-adaptable Gram-positive pathogen estimated to colonize 50–60% of the population (Wertheim et al., 2005; Gorwitz et al., 2008). It is also a leading cause of infections ranging from superficial skin abscesses to life-threatening diseases, including septicemia and necrotizing pneumonia (Klevens et al., 2007; Kobayashi et al., 2015). The ability of S. aureus to cause human disease is based in part on its ability to evade the innate immune response, thereby circumventing rapid elimination. Many factors contribute to S. aureus pathogenesis. These include production of numerous toxins, such as the barrel forming two-component toxins capable of directly lysing host immune cells (Menestrina et al., 2003), and tissue destroying enzymes including protease, lipase, and hyaluronidase, as well as many surface proteins and adhesins linked to virulence (Lowy, 1998). In this review, we will focus on evasion strategies used by S. aureus to disrupt neutrophil functions essential for bacterial clearance. First, we will highlight virulence factors produced by S. aureus to alter neutrophil priming, activation, chemotaxis, and adhesion. Then, we will discuss strategies used by S. aureus to subvert neutrophil killing by antimicrobial peptides and proteins and reactive oxygen species. Additionally, we will examine recent literature investigating mechanisms used by S. aureus to modulate neutrophil cell death programs. Finally, we will highlight the reciprocal communication between S. aureus and the neutrophil emphasizing sensing and adaptive responses used by S. aureus to recognize and respond to neutrophil challenge. The review will conclude with an overview of potential therapeutic approaches aimed at disrupting bacterial sensing and signaling to decrease production of virulence factors during neutrophil interaction and discuss putative immunotherapies to boost immune responses to S. aureus while limiting inflammatory damage caused by neutrophils.
Strategies used by S. aureus to disrupt neutrophil priming, activation, chemotaxis and adhesion
Neutrophils are initially recruited to a site of infection by following chemokine gradients in a process termed chemotaxis. Taking cues from activated endothelium, neutrophils slow their movement through blood vessels by selectin-mediated tethering to the endothelium followed by complete movement arrest through interaction with integrins on the endothelium. Extravasation from the blood vessels through the endothelial barrier is required for neutrophils to access interstitial fluid and migrate via a chemotactic gradient to the site of infection where ingestion of bacteria can take place. For detailed reviews of neutrophil chemotaxis, adhesion to the epithelium, and transmigration, please refer to (Kolaczkowska and Kubes, 2013; de Oliveira et al., 2016). Herein, we will focus on virulence factors produced by S. aureus to inhibit specific neutrophil receptors from binding host and bacterial derived ligands, which results in impaired neutrophil priming, activation, chemotaxis, and adhesion to the endothelium.
Neutrophil priming: a potential target of S. aureus?
Priming refers to the ability of a primary agonist to enhance a neutrophil's response to a secondary stimulus (Swain et al., 2002). There are many known neutrophil priming agents including: complement components C3a and C5a (Skjeflo et al., 2014), interferon-γ (IFN-γ) (Edwards et al., 1988), interleukin-8 (IL-8) (Mitchell et al., 2003), and tumor-necrosis factor-α (TNF-α; Rainard et al., 2000). Bacterial derived products such as N-formyl methionyl peptide, formyl-methionyl-leucyl phenylalanine (fMLF), peptidoglycan, and S. aureus cytolytic toxins also demonstrate an ability to prime neutrophils (Elbim et al., 1994; El-Benna et al., 2008; Clarke et al., 2010; Malachowa et al., 2012). The ability of these agents to prime neutrophils is typically not universal in that concentration and neutrophil response can vary drastically (Swain et al., 2002). Primed neutrophil responses influence many neutrophil functions including increases in adhesion, phagocytosis, superoxide production, and degranulation (Ellis and Beaman, 2004). It can also influence neutrophil apoptosis (Wright et al., 2013). Thus, priming can set the stage for subsequent neutrophil-pathogen interactions and influences outcome of this interaction.
Not much is known about the impact of S. aureus on neutrophil priming. Earlier studies investigated priming of neutrophils with conditioned medium from peripheral blood mononuclear cells (PBMC) challenged with killed S. aureus (Ferrante et al., 1989; Bates et al., 1991). These studies demonstrated increased neutrophil staphylocidal activity following priming with conditioned media from PBMCs challenged with killed S. aureus vs. priming with medium from unstimulated PBMCs. This enhanced bactericidal activity was shown to be dependent on TNF-α produced by PBMCs in response to killed S. aureus (Ferrante et al., 1989, 1993; Bates et al., 1991). These studies suggest that the response of PBMCs and resident cells to S. aureus may strongly impact the outcome of neutrophil-S. aureus interactions. Supporting this idea are studies demonstrating that S. aureus promotes production of IFN-γ and this production has been linked to poorer outcome of infection in mouse models (Watkins et al., 2011, 2013). IFN-γ impacts neutrophil function in many ways, including priming of oxidative burst and degranulation mechanisms (Ellis and Beaman, 2004). Using a murine wound model of S. aureus infection, IFN-γ-mediated CXC chemokine production by T cells promoted a robust recruitment of neutrophils that resulted in elevated S. aureus burdens at the infectious foci (McLoughlin et al., 2008). Subsequent studies using a mouse peritonitis model also demonstrated IFN-γ was associated with higher bacterial burdens but that the source of IFN-γ was neutrophils (Watkins et al., 2013). Potentially, priming by IFN-γ causes overactivation and inflammation in neutrophils making them less effective at clearing S. aureus. The role of IFN-γ and neutrophil bactericidal activity against S. aureus warrants further investigation especially considering that IFN-γ is used to treat patients with chronic granulomatous disease (Gallin et al., 1991). CGD patients receiving recombinant IFN-γ show a decrease in S. aureus infections (Gallin et al., 1991). Furthermore, in vitro studies have demonstrated that priming of human neutrophils with IFN-γ increased bactericidal activity against S. aureus (Edwards et al., 1988). Thus, more studies are needed to determine under what conditions IFN-γ promotes an effective neutrophil response against clinically relevant strains of S. aureus. Clearly, source, timing and amount of IFN-γ play an important role in outcome of S. aureus-neutrophil interactions.
S. aureus produces hemolysins, and bi-component leukocidins that directly impact immune cell function (Seilie and Bubeck Wardenburg, 2017). Many of these toxins act by forming pores in the membrane of immune cells causing lysis. Among these, the bi-component toxins leukotoxin GH (LukGH, also known as LukAB) and Panton-Valentine leukocidin (PVL) have the ability to cause neutrophil pore-formation when present at high concentrations. However, at sublytic concentrations, these factors can promote enhanced binding, uptake and killing of S. aureus through priming of neutrophils (Graves et al., 2012). This is also consistent with reports that have demonstrated the ability of PVL to enhance neutrophil superoxide, granule exocytosis, and release of leukotriene B4 and IL-8 (König et al., 1995; Colin and Monteil, 2003). In addition, low doses of alpha-toxin, a single component toxin encoded by hla, has been shown to promote transcriptional activity leading to production of inflammatory mediators that can prime neutrophils including IL-8 (Dragneva et al., 2001).
As mentioned above, most of the studies thus far have not directly investigated how modulation of known neutrophil priming agents (such as cytokines and chemokines) by S. aureus actually influences neutrophil function. Also, the impact of S. aureus toxins and immunomodulatory proteins on neutrophil priming has not been thoroughly investigated. Taken together, targeting neutrophil priming may be very important for S. aureus to gain an edge on evading the neutrophil. However, additional studies are needed to understand the impact of S. aureus on neutrophil priming.
Key factors used by S. aureus to disrupt neutrophil activation, chemotaxis and adhesion
In contrast to our understanding of how S. aureus may impact priming, there is an abundance of data that highlight how specific S. aureus factors impact neutrophil activation, chemotaxis and adhesion. In this section, we highlight individual virulence factors that have been studied for their ability to disrupt these key mechanisms (Figure 1).
Figure 1
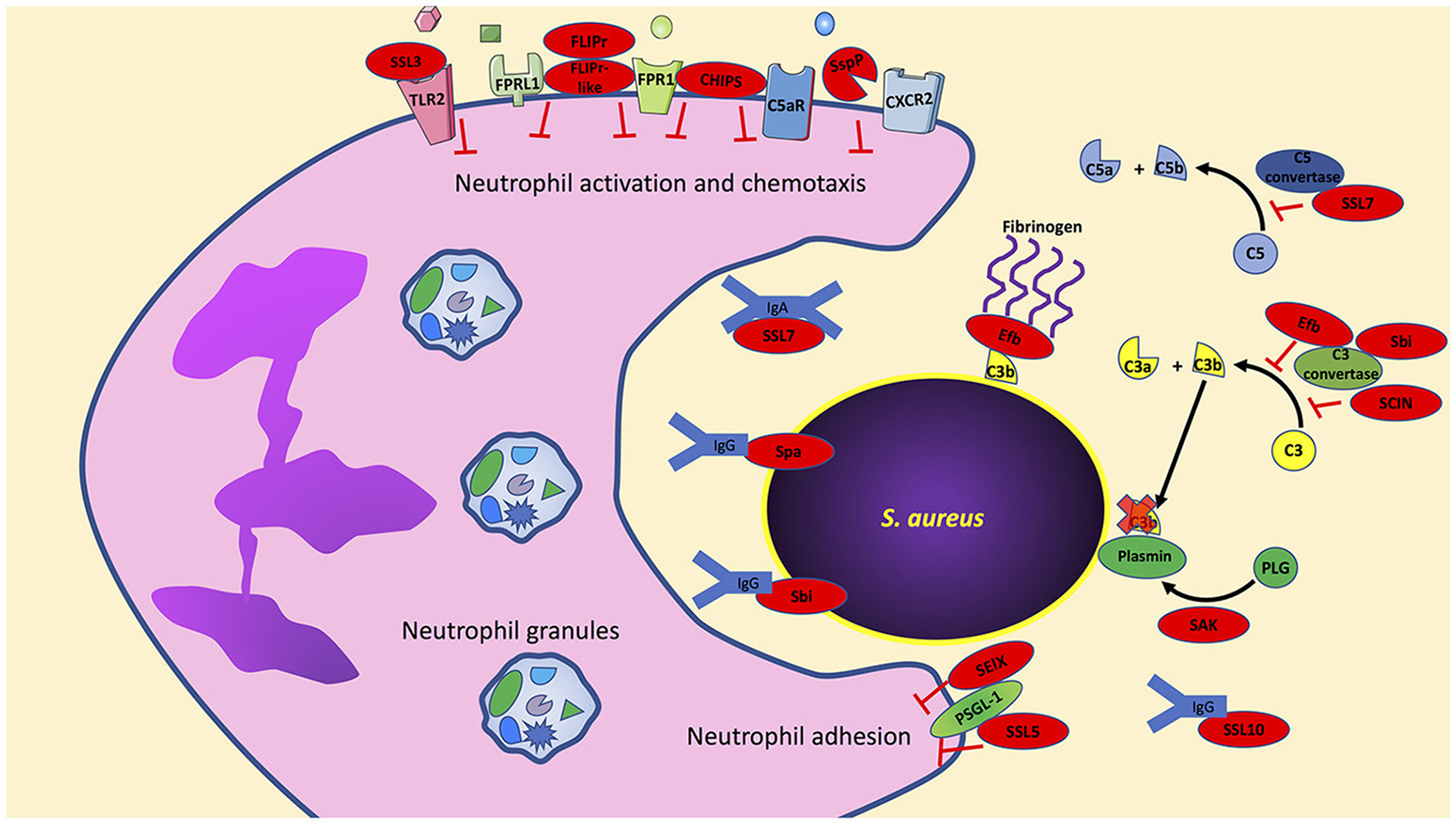
S. aureus has an arsenal of virulence factors to inhibit neutrophil activation, chemotaxis, and phagocytosis. Multiple virulence factors produced by S. aureus target key host effector proteins, for example, Efb, Sbi, and SCIN target the complement protein C3 convertase to prevent formation of C3a and C3b. Further studies are needed to determine if the production of these virulence factors are truly redundant or if they have multiple host targets as is the case with Sbi, which also targets immunoglobulins. Bacterial components indicated in red. PLG, plasminogen; SAK, staphylokinase; SspP, staphopain A.
CHIPS
The chemotaxis inhibitory protein of S. aureus (CHIPS) is a 14.1-kDa exoprotein that inhibits neutrophil migration and activation (De Haas et al., 2004). CHIPS prevents neutrophils from responding to both host- and bacteria-derived chemoattractants. Neutrophil activation by host-derived C5a is inhibited by CHIPS binding to the C5a receptor (C5aR). Similarly, neutrophil detection of bacteria-derived formylated peptides is inhibited by CHIPS binding to the formyl peptide receptor (FPR). CHIPS shows high affinity to both C5aR and FPR with KD-values of 1.1 ± 0.2 and 35.4 ± 7.7 nM, respectively; the affinity of C5aR and FPR for their natural ligands is within the CHIPS binding affinity to C5aR and FPR (Falk et al., 1982; Huey and Hugli, 1985; Postma et al., 2004). While C5aR and FPR are G-protein coupled receptors (GPCRs), the active blocking domain in CHIPS is distinct for inhibiting C5aR and FPR (Haas et al., 2005). By using FITC-labeled CHIPS, Postma et al. demonstrated that CHIPS is not internalized following binding to neutrophil C5aR and FPR and its activity is ATP and cell-signaling independent (Postma et al., 2004). It has also been demonstrated that CHIPS binds to C5aR and FPR extracellularly and does not act as an agonist since receptor binding does not induce calcium mobilization (De Haas et al., 2004). It was further demonstrated that intravenously administered CHIPS was able to inhibit mouse neutrophil influx following intraperitoneal administration of C5a despite a 30-fold lower affinity of CHIPS for mouse C5aR (De Haas et al., 2004). Finally, neutralizing antibodies to CHIPS have been observed in human sera isolated from both normal donor controls and S. aureus infected samples, implying CHIPS plays an active role during staphylococcal infection (Wright et al., 2007).
FLIPr and FLIPr-like
Neutrophils express formyl peptide receptor-like-1 (FPRL1). This receptor is activated by diverse peptides and proteins including: the synthetic peptides Trp-Lys-Tyr-Met-Val-D-Met- NH2 (WKYMVm), and L-conformer WKYMVM, lipoxin A4, the T21/DP107 leucine zipper-like domain of the HIV-1 envelope protein gp41, serum amyloid A, the mitochondrial peptide fragment MYFINILTL, the antimicrobial peptide LL-37, and prion peptide fragments (Fiore et al., 1994; Su et al., 1999a,b; Chiang et al., 2000; De Yang et al., 2000; Christophe et al., 2001; Le et al., 2001). Activation of FPRL1 on neutrophils leads to intracellular calcium mobilization and chemotaxis, while superoxide production through FPRL1 is ligand dependent (Bae et al., 2003). S. aureus produces two proteins that bind FPRL1 and inhibit its activation. FPRL1 inhibitory protein (FLIPr) is a secreted protein that binds and antagonizes both FPRL1 and FPR, but FPRL1 antagonism by FLIPr is much stronger than on FPR (Prat et al., 2006). Similarly, a second FPRL1 antagonist termed FLIPr-like shares 73% overall homology with FLIPr (Prat et al., 2009). FLIPr and FLIPr-like show similar inhibitory activity toward FPRL1 following stimulation with the FPRL1 agonist MMK-1; however, FLIPr-like antagonism toward FPR is 100-fold stronger than that of FLIPr following stimulation with fMLF (Prat et al., 2009). As expected, neutrophil exposure to purified FLIPr and FLIPr-like completely inhibits neutrophil chemotaxis toward the FPRL1 agonist MMK-1 (Prat et al., 2006, 2009).
FLIPr and FLIPr-like have also been shown to bind different FcγR isoforms to block IgG binding (Stemerding et al., 2013). Stemerding et al. showed that FLIPr preferentially binds to FcγR class II receptors while FLIPr-like can bind to FcγR cla ss I, II, and III receptors. As expected, neutrophil pretreatment with FLIPr or FLIPr-like significantly reduced phagocytosis of S. aureus opsonized with purified IgG (Stemerding et al., 2013). However, it should be noted that phagocytic inhibition of neutrophils toward S. aureus opsonized with human serum by FLIPr and FLIPr-like was only observed at low serum concentrations (< 1% serum). At higher serum concentrations (1–10% serum), FLIPr and FLIPr-like do not inhibit neutrophil-mediated phagocytosis of opsonized S. aureus. Under these higher serum conditions, neutrophil phagocytosis of opsonized S. aureus is likely mediated by complement receptors, which are not antagonized by FLIPr and FLIPr-like proteins. It follows that experiments using neutrophils treated with FLIPr and FLIPr-like had reduced uptake of S. aureus only when opsonized with complement inactivated serum thus showing the specific inhibition of FLIPr and FLIPr-like toward serum immunoglobulins (Stemerding et al., 2013).
Staphopain A
The CXCR2 chemokine receptor (also known as IL8RB) is highly expressed on human neutrophils and has high specificity for the potent neutrophil chemoattractants CXCL1, CXCL2, and CXCL7, as well as CXCL8 (IL-8, which can also bind to CXCR1) (Yoshimura et al., 1987; Baggiolini et al., 1989; Ben-Baruch et al., 1997; Ritzman et al., 2010). S. aureus infection induces CXCL1, CXCL2, CXCL8 production resulting in neutrophil recruitment (Mempel et al., 2003; Sasaki et al., 2003; Olaru and Jensen, 2010). To prevent neutrophil recruitment induced by CXCR2 ligands, S. aureus secretes the cysteine protease staphopain A which cleaves the N-terminus of CXCR2 required for ligand binding (Laarman et al., 2012). Neutrophils pretreated with staphopain A blocked calcium mobilization following stimulation with CXCL1 and CXCL7. Importantly, these chemokines only bind CXCR2 (and not CXCR1). In contrast, staphopain A did not inhibit neutrophil calcium mobilization following stimulation with CXCL8 since it can also signal through CXCR1. The impacts of staphopain A in vivo are lacking since staphopain A is specific for human CXCR2 and does not cleave murine CXCR2 (Laarman et al., 2012).
SElX and SSL5
Neutrophils express P-selectin glycoprotein ligand 1 (PSGL-1) on the cellular surface to bind P- and E-selectin on activated endothelial cells or platelets and L-selectin expressed on leukocytes (Moore et al., 1995; Guyer et al., 1996; Hidalgo et al., 2007; Huo and Xia, 2009; Stadtmann et al., 2013). PSGL-1 binding to selectins tethers neutrophils to activated endothelial cells and is a first step in the process of transmigration. S. aureus secretes two proteins that inhibit PSGL-1 binding to selectins. The staphylococcal enterotoxin-like toxin X (SElX) is a highly conserved superantigen that binds glycosylated PSGL-1 on neutrophils and inhibits binding to P-selectin (Wilson et al., 2011; Fevre et al., 2014). Similarly, the staphylococcal superantigen-like 5 (SSL5) protein directly binds PSGL-1 on neutrophils inhibiting rolling on endothelial cells (Bestebroer et al., 2007). In addition to binding PSGL-1, SSL5 also binds to other GPCRs but only inhibits the ligands that require the N-terminal domain of their respective receptors for activation (Bestebroer et al., 2009). Thus, SSL5 pretreatment of neutrophils inhibited activation induced by C3a, C5a, CXCL1, and CXCL8 (Bestebroer et al., 2009).
SSL3
Toll-like receptor 2 (TLR2) recognizes staphylococcal peptidoglycan-associated lipoproteins (Fournier and Philpott, 2005; Kurokawa et al., 2009). TLR2 can also discriminate between diacylated and triacylated lipoproteins by associating with TLR6 or TLR1, respectively (Takeuchi et al., 2001, 2002). The recognition of staphylococcal lipoproteins such as the staphylococcal iron transporter C (SitC) are exclusively dependent on TLR2 to induce immune activation (Stoll et al., 2005; Kurokawa et al., 2009; Müller et al., 2010). The importance of TLR2 during S. aureus infection is highlighted by TLR2−/− mice that show increased susceptibility and nasal colonization to S. aureus compared to wild-type mice (Takeuchi et al., 2000; González-Zorn et al., 2005; Hoebe et al., 2005). In addition, TLR2−/− mice infected with S. aureus show alterations in cytokines that modulate neutrophil function including TNF-α and IL-1β (Knuefermann et al., 2004). Engagement of TLR2 influences many neutrophil functions including: adhesion molecule expression, reactive oxygen species production (following stimulation with fMLF), and modulates CXCL8, and chemokine receptor expression (Sabroe et al., 2003). S. aureus produces the staphylococcal superantigen-like 3 protein that binds TLR2 to inhibit activation in neutrophils and other cell types expressing TLR2 (Bardoel et al., 2012; Yokoyama et al., 2012). Crystal structures showed that SSL3 binding to TLR2 reduced the lipopeptide binding pocket by ~50%, which inhibited binding of the TLR2 agonist Pam2CSK4 (Koymans et al., 2015). The same study demonstrated SSL3 can bind a preformed TLR2- Pam2CSK4 complex and this blocked TLR2-TLR1 and TLR2-TLR6 heterodimerization thereby inhibiting downstream signaling.
Strategies used by S. aureus to inhibit neutrophil phagocytosis
Phagocytosis is a process by which neutrophils, and other phagocytes, ingest particles from their extracellular environment including bacteria and host cells. In neutrophils, this sequestration results in the formation of an intracellular compartment termed the phagosome following invagination of the cellular membrane. Neutrophil granules fuse with the phagosome to release antimicrobial peptides and proteins, as well as produce reactive oxygen species to kill invading microorganisms. Neutrophils bind to pathogen-associated molecular patterns on the S. aureus surface to initiate the phagocytic process. Furthermore, binding of immunoglobulins to S. aureus and complement activation enhances phagocytosis, as well as engages different neutrophil receptors resulting in branching downstream signaling. S. aureus employs strategies and produces a wide range of virulence factors to disrupt neutrophil phagocytosis. Herein, we will highlight select strategies used by S. aureus to disrupt neutrophil phagocytosis. Since a complete review on the strategies used by S. aureus to avoid phagocytosis is outside the scope of this publication, we recommend previous detailed reviews focusing on immunoglobulin and complement evasion by S. aureus to inhibit phagocytosis (Lambris et al., 2008; Serruto et al., 2010; van Kesse et al., 2014).
Capsule synthesis
The production of capsular polysaccharide by S. aureus has been proposed as an antiphagocytic evasion strategy but its actual role in inhibiting neutrophil phagocytosis remains controversial. Indeed, previous studies have shown that S. aureus strains producing high levels of capsular polysaccharide withstand neutrophil killing better than microencapsulated strains (Xu et al., 1992; Thakker et al., 1998). However, it should be noted that under these experimental conditions neutrophil killing of S. aureus was correlated to the ability of neutrophils to associate with bacteria without clearly showing ingestion. Furthermore, the experimental conditions used suspended instead of adherent neutrophils which can greatly influence neutrophil ingestion of S. aureus (Lu et al., 2014). Also, an intraperitoneal S. aureus infection model showed no differences in neutrophil intracellular staphylococcal survival between a highly and microencapsulated strain (Gresham et al., 2000). Notably, the predominant clinical isolate USA300 is unencapsulated, further questioning the role of capsule synthesis in S. aureus pathogenicity (Montgomery et al., 2008; Carrel et al., 2015).
Complement inhibition
Human pathogens have developed strategies to evade complement, thus inhibiting immune recognition, cytokine production, and neutrophil uptake (Lambris et al., 2008; Serruto et al., 2010). S. aureus produces several virulence factors that target different machinery in the complement system. The staphylococcal complement inhibitor (SCIN), is a 9.8-kDa exoprotein that specifically binds and inhibits human C3 convertases (Rooijakkers et al., 2005a, 2006). SCIN binding to C3 convertases does not prevent C3 binding but completely inhibits generation of C3b and thus opsonization of S. aureus by C3b (Rooijakkers et al., 2005a; Ricklin et al., 2009; Garcia et al., 2010). In addition, SCIN stabilizes C3 convertases by competing with factor H which accelerates decay of C3 convertases (Ricklin et al., 2009). In vitro, recombinant SCIN can significantly inhibit neutrophil phagocytosis of human serum opsonized S. aureus resulting in increased bacterial survival (Rooijakkers et al., 2005a). In vivo studies with SCIN are lacking since it is human specific, but SCIN is immunogenic since a limited study with 80 healthy people and 20 individuals with recurring staphylococcal infections showed all produced antibodies to SCIN (Rooijakkers et al., 2005a).
S. aureus is also equipped with virulence factors that target complement without direct binding to C3 convertase. Staphylokinase is a secreted protein that binds human plasminogen converting it into its active form plasmin (Parry et al., 2000; Mölkänen et al., 2002; Rooijakkers et al., 2005b). Plasmin, a serine protease, is bound to the S. aureus surface and degrades C3 convertase-dependent C3b to prevent deposition on the bacterial surface. Human neutrophils show decreased phagocytic activity toward human serum opsonized S. aureus (depleted of IgG and IgM to rule out immunoglobulin mediated phagocytosis) and pretreated with recombinant staphylokinase (Rooijakkers et al., 2005b). In addition, S. aureus secretes the extracellular fibrinogen-binding (Efb) protein that binds to C3 to prevent cleavage to C3b and can also directly bind C3b deposited on the extracellular bacterial membrane (Lee et al., 2004; Ko et al., 2013). As its name implies, the extracellular fibrinogen-binding protein also contains a fibrinogen binding domain that recruits fibrinogen and inhibits neutrophil phagocytosis (Ko et al., 2013). Complement receptor recognition of C3b is blocked by a thick layer of fibrinogen linked by Efb on the bacterial surface (Ko et al., 2013). Ex vivo studies with human blood exposed to Efb and S. aureus showed a significant reduction in neutrophil phagocytosis compared to S. aureus alone. Similarly, supernatants from wild-type S. aureus reduced neutrophil phagocytosis of opsonized S. aureus compared to supernatants from Efb-deficient S. aureus in the presence of fibrinogen. Finally, neutrophils from mice intraperitoneally infected with GFP-expressing wild-type S. aureus or an Efb-deficient mutant showed significantly higher ingestion of the Efb mutant compared to the wild-type (Ko et al., 2013). While this observation suggests that sufficient levels of Efb are produced by S. aureus to have an in vivo effect, it should be noted that intraperitoneal injection was performed with S. aureus grown to late exponential phase without washing culture toxins. Thus, increased and unknown levels of Efb were likely present from the beginning giving an advantage to S. aureus to prevent neutrophil phagocytosis.
Inhibition of immunoglobulin recognition
Complement and immunoglobulins function as opsonins to enhance S. aureus recognition and ingestion by neutrophils. While S. aureus produces virulence factors that inhibit complement activation and deposition on the bacterial surface, the efficacy of these virulence factors in vivo is controversial since immunoglobulin opsonization plays a redundant role in opsonophagocytosis. In fact, opsonophagocytosis assays investigating the role of S. aureus virulence factors that disrupt complement must be done in the absence of immunoglobulins to observe an inhibitory effect on phagocytosis because S. aureus produces virulence factors that inhibit opsonization by immunoglobulins. Protein A, encoded by the spa gene, is one of the better characterized virulence factors produced by S. aureus that inhibits neutrophil phagocytosis. Protein A is a 42-kDa secreted and membrane bound protein that binds the constant Fcγ region of IgG thus preventing proper engagement of Fcγ receptors on neutrophils and antigen recognition (Forsgren and Sjöquist, 1966; Kronvall et al., 1970; Sjödahl, 1977). Indeed, early studies clearly showed that purified protein A inhibited neutrophil phagocytosis of S. aureus independent of capsule polysaccharide on the bacterial surface (Dossett et al., 1969; King and Wilkinson, 1981). More recent studies have demonstrated protein A induces B cell proliferation and production of VH3 serum IgG and IgM (Pauli et al., 2014; Kim et al., 2016). Although abundant, these antibodies do not provide protection against S. aureus. Generation of protein A variants that were unable to bind immunoglobulin induce a protective antibody response presumably via allowing Fc mediated uptake of opsonized S. aureus (Falugi et al., 2013). These data suggest that mechanisms of neutrophil recognition and uptake of S. aureus are likely critical to resolution of infection.
S. aureus produces a second binding protein of immunoglobulin (termed Sbi) that also binds IgG to inhibit neutrophil phagocytosis (Jacobsson and Frykberg, 1995; Zhang et al., 1998, 1999; Smith et al., 2011). Human neutrophils can phagocytose significantly more opsonized Sbi-deficient S. aureus compared to wild-type, and Sbi plays a protective role in ex vivo whole human blood killing assays (Smith et al., 2011). Interestingly, Sbi binds to lipoteichoic acid in the bacterial cell surface to remain anchored, while it can also exist in a secreted form that binds complement protein C3 to induce its degradation away from the bacterial surface (Burman et al., 2008; Upadhyay et al., 2008). Sbi also plays another immunomodulatory role that was demonstrated to impact neutrophil function. In a mouse model of peritonitis Sbi induced production of IL-6 and CXCL-1, which resulted in neutrophil recruitment and subsequently an exacerbated inflammatory response (Gonzalez et al., 2015).
In addition to Sbi, S. aureus secretes other proteins that bind both immunoglobulins and complement proteins to disrupt neutrophil phagocytosis. The 23-kDa staphylococcal superantigen-like 7 (SSL7) exoprotein binds to human IgA with high affinity (KD of 1.1 nM) and inhibits FcαR binding on the neutrophil surface (Langley et al., 2005). Using an ex vivo whole blood infection model it was demonstrated that binding of IgA by SSL7 reduced neutrophil phagocytosis of S. aureus (Bestebroer et al., 2010). SSL7 also binds to the C5 complement protein (KD of 18 nM) and inhibits C5a generation in an IgA binding-dependent manner, as well as formation of the C5b-9 membrane attack complex (Langley et al., 2005; Bestebroer et al., 2010; Laursen et al., 2010; Lorenz et al., 2013). In vivo mouse studies showed that administration of purified SSL7 inhibited neutrophil recruitment following exposure to heat-killed S. aureus. Thus, SSL7 serves diverse functions that include inhibiting neutrophil phagocytosis, recruitment, and complement activation (Bestebroer et al., 2010; Lorenz et al., 2013). Another staphylococcal superantigen-like protein termed SSL10 also binds to IgG (KD of 220 nM) and to the complement protein C1q (Itoh et al., 2010). SSL10 significantly inhibited neutrophil ingestion of IgG-opsonized S. aureus by blocking binding of FcγR (Patel et al., 2010).
Taken together, it is clear that S. aureus produces virulence factors that decrease neutrophil phagocytosis via production of physical barriers (capsule polysaccharide), inhibition of complement activation leading to decreased deposition of opsonins, and disruption of immunoglobulin binding to bacterial antigens. However, the efficacy of these virulence factors has been difficult to determine. Neutrophil-mediated phagocytosis of S. aureus is efficient in the absence of opsonins. In vitro studies have shown that adherent neutrophils rapidly ingest both opsonized and unopsonized S. aureus (Lu et al., 2014). Thus, neutrophils contain pattern-recognition receptors that are sufficient for phagocytosis of S. aureus independent of opsonins. Clearly, the redundancy in neutrophil receptor-mediated phagocytosis mechanisms compounds studies and strongly suggests the combination of S. aureus virulence factors are needed to thwart the innate ability of neutrophils to recognize this pathogen.
Evasion strategies used by S. aureus to survive neutrophil killing
S. aureus is clearly decorated to avoid ingestion by neutrophils. Additionally, S. aureus is loaded with mechanisms to disarm potent neutrophil bactericidal mechanisms. Soon after initiation of the phagocytic process, and possibly before complete sealing of the phagosome, neutrophil granules mobilize to fuse and release antimicrobial agents into the phagosome containing S. aureus (Flannagan et al., 2009). Following phagocytosis, neutrophils expose S. aureus to antimicrobial peptides, proteins, and reactive oxygen species to degrade essential bacterial proteins and disrupt homeostasis resulting in bacterial death (Figure 2). In this section, we will highlight strategies used by S. aureus to avoid oxidative and non-oxidative mechanisms of neutrophil killing. Although we separate S. aureus evasion of oxidative and non-oxidative killing, it is important to remember that these mechanisms likely have synergistic antimicrobial activity in a physiological setting.
Figure 2
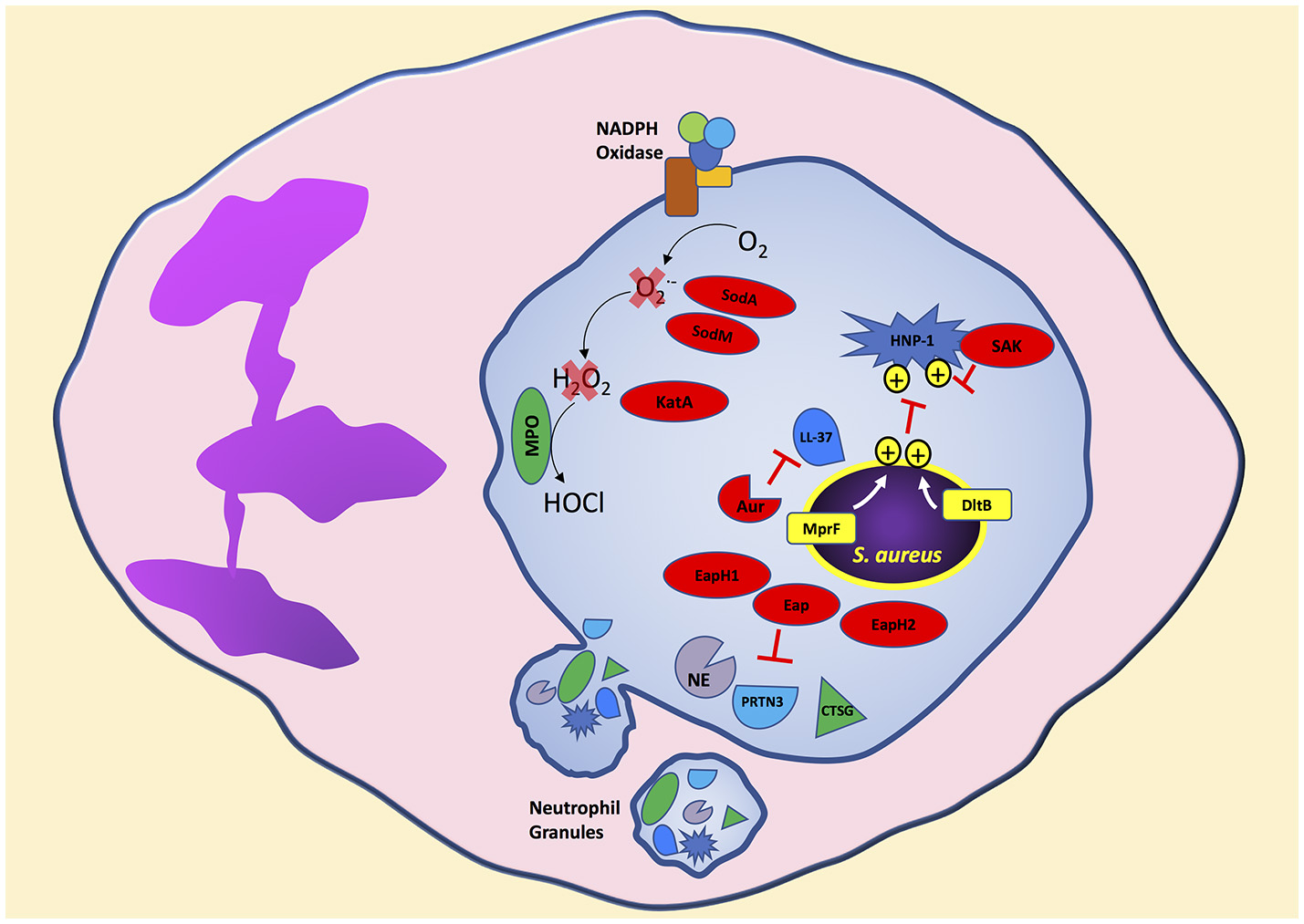
S. aureus produces virulence factors that target different neutrophil bactericidal mechanisms following phagocytosis. Cationic antimicrobial peptides are ineffective toward S. aureus due to the presence of positive charges on the bacterial surface transferred by MprF and DltB. S. aureus secretes virulence factors that degrade antimicrobial proteins and enzymes released into the neutrophil phagosome. In addition, neutrophil reactive oxygen species production is decreased by S. aureus virulence factors that degrade intermediate reactive oxygen species like superoxide and hydrogen peroxide to reduce the formation of the highly bactericidal chemical agent hypochlorous acid. Bacterial components indicated in red. Aur, aureolysin; PRTN3, proteinase 3; CTSG, cathepsin G; SAK, staphylokinase.
S. aureus evasion of non-oxidative killing
The dlt operon in S. aureus, containing genes dltABCD is involved in activation and transfer of D-alanine, and increases tolerance to cationic antimicrobial peptides (AMPs), including human neutrophil peptide-1 (HNP-1, also known as alpha-defensin), by incorporation of positively charged D-alanine into teichoic acids (Peschel et al., 1999; Collins et al., 2002). Teichoic acids are highly negatively charged by deprotonized phosphate groups that electrostatically interact with cationic peptides. Thus, a S. aureus dlt mutant strain retains a negatively charged teichoic acid backbone and is significantly more susceptible to killing by cationic peptides (Peschel et al., 1999). In vitro, S. aureus dlt mutants are highly susceptible to neutrophil non-oxidative killing mechanisms whereas wild-type S. aureus is resistant and mainly susceptible to neutrophil oxidative killing (Collins et al., 2002). The importance of the dlt operon to increase S. aureus survival was observed in an in vivo mouse model of sepsis, which showed increased host mortality infected with wild-type S. aureus compared to the non-lethal S. aureus dlt mutant (Collins et al., 2002). Similarly, the mprF gene in S. aureus encoding lysylphosphatidylglycerol (LPG) synthetase confers resistance to cationic AMPs by transferring the positively charged L-lysine to the negatively charged lipid phosphatidylglycerol creating LPG (Peschel et al., 2001; Oku et al., 2004; Ernst et al., 2009). Electrostatic repulsion between cationic AMPs and the attached lysine prevents binding of cationic AMPs and disruption of the bacterial membrane. Wild-type S. aureus is more resistant to killing by human neutrophils compared to a S. aureus mprF mutant (Peschel et al., 2001). Thus, S. aureus modifies its cell surface as an evasion strategy by increasing electrostatic repulsion of neutrophil antimicrobial peptides leading to decreased bacterial killing.
In addition to cell membrane modifications to decrease efficacy of antimicrobial peptides by electrostatic repulsion, S. aureus produces proteins that directly bind, inhibit, and degrade antimicrobial peptides. Staphylokinase, which activates plasminogen to the active serine protease plasmin leading to degradation of IgG and C3b (Rooijakkers et al., 2005b), also binds HNP-1 and inhibits its bactericidal activity (Jin et al., 2004). The cathelicidin LL-37, which has potent staphylococcal bactericidal activity, is degraded by the S. aureus secreted metalloprotenaise aureolysin (Sieprawska-Lupa et al., 2004). Thus, S. aureus strains producing aureolysin are significantly more resistant to LL-37 than aureolysin-negative strains. Furthermore, the extracellular adherence protein (Eap) and the Eap-homologs 1 and 2 (EapH1 and EapH2) are neutrophil serine protease inhibitors (Harraghy et al., 2005). Purified Eap, EapH1, or EapH2 inhibit the activity of neutrophil elastase, proteinase 3, and cathepsin G which are found in neutrophil azurophilic granules and are bactericidal to S. aureus (Papayannopoulos et al., 2010; Stapels et al., 2014).
S. aureus evasion of oxidative killing
Neutrophil activation leads to assembly of the NADPH oxidase resulting in production of reactive oxygen species (ROS; DeLeo and Quinn, 1996; DeLeo et al., 1999; Bréchard and Tschirhart, 2008; Nunes et al., 2013). Assembly of the membrane and cytoplasmic NADPH oxidase components results in electrons shuttled from NADPH to oxygen producing superoxide. Spontaneous and myeloperoxidase (MPO) catalyzed dismutation of superoxide produces hydrogen peroxide, which in the presence of MPO and chloride results in the highly bactericidal agent hypochlorous acid/hypochlorite anion (Kettle et al., 2007; Klebanoff et al., 2013). Neutrophil ingestion exposes S. aureus to concentrated ROS in the phagosome resulting in oxidation and chlorination of bacterial and host proteins (Green et al., 2014). Inhibition of neutrophil ROS production by diphenyleneiodonium significantly increases S. aureus survival following neutrophil phagocytosis even with functioning non-oxidative killing mechanisms (Hampton and Winterbourn, 1995; Hampton et al., 1996). Individuals with chronic granulomatous disease carry mutations in components of NADPH oxidase preventing formation of superoxide and hydrogen peroxide and suffer from recurring bacterial infections including S. aureus (Lekstrom-Himes and Gallin, 2000; Assari, 2006). Thus, uninhibited neutrophil ROS production is essential for S. aureus clearance.
Bacterial pathogens have developed strategies to inhibit ROS killing (Flannagan et al., 2009). S. aureus produces several enzymes in response to neutrophils to degrade and detoxify ROS. The ROS chain of production begins with the formation of superoxide from oxygen. S. aureus produces two superoxide dismutases, encoded by sodA and sodM, that convert superoxide into hydrogen peroxide and molecular oxygen (Karavolos et al., 2003). Superoxide dismutase activity assessed on nondenaturing polyacrylamide gel electrophoresis from the S. aureus cytoplasm shows three distinct bands consisting of SodA and SodM homodimers, and a SodA-SodM hybrid (Clements et al., 1999; Valderas and Hart, 2001). Wild-type S. aureus is more resistant to superoxide stress compared to isogenic sodA, or sodM, or sodAsodM-deficient strains (Karavolos et al., 2003). In a mouse subcutaneous model of infection, significantly higher wild-type S. aureus colony forming units were recovered from the site of infection compared to single or double sod mutant strains suggesting a role for superoxide dismutase in virulence (Karavolos et al., 2003). The expression of sodA and sodM is increased under oxidative stress and regulated by the transcriptional regulator SarA (Ballal and Manna, 2009). Interestingly, S. aureus uses multiple regulatory systems to respond to oxidative stress. KatA, the only catalase encoded by S. aureus that degrades hydrogen peroxide to water and oxygen, is co-regulated by the ferric uptake repressor (Fur) and the peroxide response regulator (Horsburgh et al., 2001b). As expected, katA deficient S. aureus is more sensitive to killing by hydrogen peroxide compared to wild-type S. aureus (Horsburgh et al., 2001a). Furthermore, the alkyl hydroperoxide reductase (AhpC) increases S. aureus resistance to the organic-hydroperoxide cumene hydroperoxide and its absence increases S. aureus resistance to hydrogen peroxide by a compensatory increase in katA expression (Cosgrove et al., 2007). However, the significance of KatA in surviving neutrophil ROS production is unclear since studies have shown no significant differences in neutrophil killing of wild-type S. aureus compared to an isogenic katA deficient strain (Cosgrove et al., 2007).
Superoxide dismutase and catalase protect S. aureus from ROS through enzymatic degradation. S. aureus also produces virulence factors that inhibit ROS killing by different mechanisms. The iconic yellow-golden pigment of S. aureus is a result of the antioxidant carotenoid named staphyloxanthin and synthesized by genes encoding crtM and crtN (Wieland et al., 1994; Pelz et al., 2005). The production of staphyloxanthin increases S. aureus resistance to killing by hydrogen peroxide and singlet oxygen (Liu et al., 2005). In addition, staphyloxanthin increases S. aureus survival during exposure to neutrophil ROS; in contrast, staphyloxanthin does not confer protection during exposure to neutrophils from patients with chronic granulomatous disease or neutrophils pretreated with diphenyleneiodonium which have deficiencies in ROS production (Liu et al., 2005). Following exposure to neutrophil ROS, surviving bacteria undergo repair of oxidized proteins to maintain homeostasis. S. aureus encodes four methionine sulfoxide reductases that play a role in reducing oxidized methionine residues (Singh, 2003; Schöneich, 2005; Singh et al., 2015). Deletion of the methionine sulfoxide reductases msrA1 and msrB increases S. aureus susceptibility to exogenous hydrogen peroxide and hypochlorous acid. Furthermore, a msrA1 and msrB double mutant strain of S. aureus is more susceptible to neutrophil killing compared to wild-type (Pang et al., 2014). Finally, the SaeR/S TCS also plays a role in regulating virulence factors that decrease neutrophil hydrogen peroxide and hypochlorous acid production following S. aureus phagocytosis (Guerra et al., 2016). The SaeR/S-regulated virulence factors that decrease neutrophil ROS remain unknown and are an active area of research, but are independent of superoxide dismutase and catalase activity since their expression is not regulated by SaeR/S (Rogasch et al., 2006; Voyich et al., 2009; Nygaard et al., 2010; Sun et al., 2010).
Human neutrophils extrude DNA decorated with antimicrobial proteins termed neutrophil extracellular traps (NETs) to ensnare and kill bacteria as a terminal cell fate pathway that has been named NETosis and is dependent on ROS (Brinkmann et al., 2004; Fuchs et al., 2007; Galluzzi et al., 2012). Neutrophil exposure to S. aureus is a potent inducer of NETs (Pilsczek et al., 2010). However, S. aureus escapes NETs by secreting nuclease (nuc), an SaeR/S-regulated factor (Berends et al., 2010; Olson et al., 2013). Furthermore, the nuclease products from DNA degradation 2′-deoxyadenosine-3′-monophosphate and 2′-deoxyadenosine-5′-monophosphate are converted by the S. aureus exoprotein adenosine synthase (adsA) into 2′-deoxyadenosine (dAdo), which induces caspase-3-mediated apoptosis in macrophages (Thammavongsa et al., 2013). A murine intravenous infection model showed macrophages are unable to diffuse into kidney abscesses containing neutrophils and wild-type S. aureus; however, infection with a nuc or adsA deficient S. aureus strain allowed macrophages to efficiently infiltrate the neutrophil abscess (Thammavongsa et al., 2013).
S. aureus modulation of neutrophil fate
Neutrophils undergo an apoptotic differentiation program in response to bacterial pathogens to limit host damage caused by a prolonged inflammatory response (Kobayashi et al., 2003). For excellent reviews on the mechanisms of neutrophil apoptosis and how bacterial pathogens modulate neutrophil fate, we recommend (Kennedy and Deleo, 2009; Rigby and DeLeo, 2012; Kobayashi et al., 2017). S. aureus produces pore-forming toxins that lyse neutrophils, other leukocytes, and red blood cells and this lytic activity promotes an intense inflammatory response. While these toxins clearly play a role in lysing neutrophils and other immune cells, their role if any beyond neutrophil cytolysis is unknown. Notable exceptions include Panton-Valentine Leukocidin (PVL) and LukGH which have been shown to prime neutrophils at sublytic concentrations and promote neutrophil extracellular trap formation, respectively (Graves et al., 2012; Malachowa et al., 2013). In addition, neutrophils exposed to S. aureus have been shown to undergo cell death with intact phagosomes undermining the role of cytolytic toxins in S. aureus escape from neutrophils (Kobayashi et al., 2010). For reviews on the lytic properties of these toxins and their targets, please refer to (DuMont and Torres, 2014; Reyes-Robles and Torres, 2016). Herein, we will highlight recent studies suggesting S. aureus modulates neutrophil fate to disrupt proper neutrophil clearance following phagocytosis of S. aureus independent of lytic activity.
Following phagocytosis, neutrophils are ingested by macrophages in a process called efferocytosis to remove spent neutrophils and limit inflammation (Martin et al., 2014). S. aureus inhibits macrophage efferocytosis of S. aureus laden neutrophils by upregulating the “don't eat me” signal CD47 (Greenlee-Wacker et al., 2014). Interestingly, macrophage production of cytokines that modulate neutrophil fate was altered and inflammasome activation was reduced in response to neutrophils harboring S. aureus compared to S. aureus alone. In vitro, human neutrophils containing S. aureus do not undergo the classical apoptotic death pathway as they fail to activate caspase-3, as well as caspase-2, -8, and -9. Instead, neutrophil death in this in vitro model in response to S. aureus is dependent on receptor-interacting protein 1 (RIP-1) which is a hallmark of programmed cell death or necroptosis. Studies by Zurek et al. showed that the SaeR/S TCS plays a role in modulating neutrophil fate by inhibiting IL-8 production and NF-κB activation (Greenlee-Wacker et al., 2014; Zurek et al., 2015). Programmed neutrophil death in response to wild-type S. aureus was accelerated compared to exposure to a saeR/S deletion mutant.
In addition, ROS production is implicated in programmed neutrophil death leading to NET formation (NETosis) and as discussed above S. aureus inhibits neutrophil ROS production (Fuchs et al., 2007; Akong-Moore et al., 2012). Interestingly, Pilsczek et al. described a NET forming mechanism that did not require neutrophil lysis and is ROS independent. Instead, DNA-containing vesicles budding from the nuclear membrane are released while maintaining neutrophil plasma membrane integrity (Pilsczek et al., 2010). Thus, these anuclear neutrophils may still retain phagocytic and chemotactic activity, as previously observed (Malawista et al., 1989). Further studies are needed to determine if inhibition of neutrophil ROS production by S. aureus modulates NETosis and to better elucidate ROS independent and dependent NET formation and NETosis. However, it is clear that S. aureus alters the neutrophil death program through a variety of mechanisms that impact not only cellular signaling by the neutrophil but clearance via efferocytosis.
The other side of the equation: the ability of S. aureus to sense neutrophils
As reviewed above, the neutrophil has many mechanisms to sense and respond to S. aureus and likewise individual S. aureus factors have evolved to disrupt every step of the neutrophil response to the invading pathogen. However, the ability of S. aureus to sense the host has been underestimated. The S. aureus genome consists of 16 two-component systems (TCS) that sense environmental stimuli and regulate gene expression accordingly (Kawada-Matsuo et al., 2011). Of these 16 TCSs, SaeR/S is recognized as a major contributor to S. aureus pathogenesis and neutrophil evasion (Voyich et al., 2009; Figure 3). Geiger et al. was the first to demonstrate that the TCS SaeR/S contained an upstream promoter element that could be activated by human neutrophil products H2O2 and alpha-defensin (Geiger et al., 2008). SaeR/S target genes are differentially regulated in response to whole blood, neutrophils and neutrophil components (H2O2, alpha-defensin, and calprotectin; Voyich et al., 2005; Palazzolo-Ballance et al., 2007; Malachowa et al., 2011; Flack et al., 2014; Zurek et al., 2014; Cho et al., 2015). The nine amino acid extracellular loop of SaeS plays an important role in sensing different neutrophil-derived stimuli (Flack et al., 2014; Liu et al., 2015). Highly conserved residues M31, W32, and F33 on SaeS are essential to the appropriate activation of sae-target genes. Residue M31 is essential to the activation of sae regulon and plays an important role in sensing neutrophil-derived stimuli including HNP-1. Strains with mutations in aromatic residues W32 and F33 have disrupted normal basal signaling of SaeS in the absence of inducing signals, yet both mutant kinases have appropriate activation of effector genes following exposure to neutrophil-derived stimuli (Flack et al., 2014). This posits that response regulators turn-on only subsets of genes based on the host stimulus. Such observations support a unique hypothesis that emerging strains of bacteria are not more virulent because they harbor new virulence factors but are better able to sense and respond to their human hosts.
Figure 3
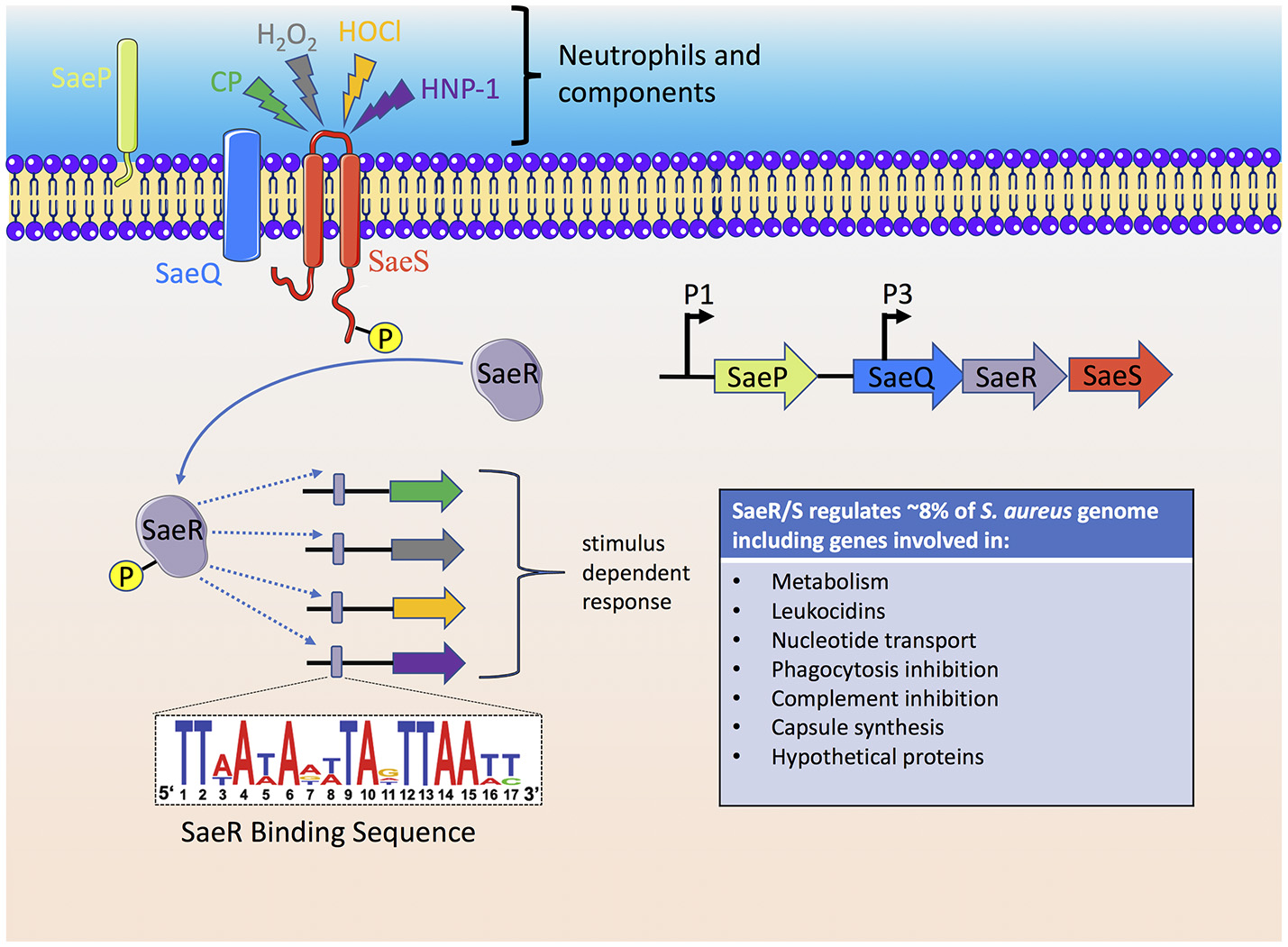
The S. aureus SaeR/S two-component system senses neutrophils and neutrophil components to activate an anti-host immune response. Activation of the SaeS histidine kinase results in autophosphorylation which is subsequently transferred to the SaeR response regulator. A promoter binding region recognized by phosphorylated SaeR (SaeR binding sequence) initiates gene transcription of SaeR/S-regulated virulence factors that target neutrophil antimicrobial mechanisms, cell fate and cellular signaling. The immediate genes transcribed by SaeR/S are dependent on the stimulus, i.e., the SaeR/S transcriptional profile is dynamic and specific to the stimulus. Since bacterial sensory-regulatory systems activate multiple virulence factors, therapeutic approaches to inhibit bacterial sensing, and activation is an active area of research. CP, calprotectin.
SaeR/S is the best studied for its role in sensing the neutrophil but additional studies are needed to understand just how S. aureus senses the neutrophil in vivo. Certainly other regulatory systems play a role either directly or potentially by responding to the activation of SaeR/S, however, these studies are currently limited. Considering HNP-1 is recognized early following phagocytosis, the recognition of HNP-1 is likely very important for S. aureus adaptation to the neutrophil environment. Interestingly, the recognition of HNP-1 by SaeS has not been demonstrated to influence the genes in S. aureus directly correlated with combating the antimicrobial capacity of antimicrobial peptides [e.g., dlt and mprF are regulated by the GraRS two-component system (Yang et al., 2012)] but instead appears to ready S. aureus for combating neutrophil reactive oxygen species and for producing cytolytic toxins. Additionally, the GraRS system recognizes cationic peptides including LL-37, and polymyxin B but does not recognize HNP-1 implying a complex interplay of sensory systems to combat the neutrophil (Yang et al., 2012). Another interesting two-component gene regulatory system is WalK/R. Studies have demonstrated this system is essential for S. aureus viability (Dubrac and Msadek, 2004; Dubrac et al., 2007). However, studies using a strain of S. aureus with a constitutively active WalR response regulator demonstrated constitutive expression caused up-regulation of virulence genes that are known to be SaeR-regulated including coa, hla hlgA, hlgB, hlgC, and sbi (Delauné et al., 2012). Although the exact mechanism of communication between WalR and SaeR/S is unknown data suggest WalR influences the SaeR/S system indirectly. This is inferred since deletion of saeR/S in a strain that constitutively expressed WalR showed the impact of WalR on the virulence factors during growth in vitro was through SaeR/S (Delauné et al., 2012). However, in mouse models of infection constitutive expression of WalR in S. aureus caused increased neutrophil recruitment and enhanced bacterial clearance compared to wild-type S. aureus. Constitutive expression of WalR decreasing virulence in vivo was independent of SaeR/S and linked to WalR dependent peptidoglycan release promoting an inflammatory environment that recruited neutrophils (Delaune et al). Currently the stimulus recognized by WalK is unknown. Understanding what stimulus(i) activates this system may reveal how WalK/R and SaeR/S regulation of virulence are linked. Additionally, the fatty acid kinase VfrB, that lacks any traditional DNA-binding domain, has been shown to influence gene expression of well-defined SaeR/S-dependent genes (Bose et al., 2014; Krute et al., 2016). However, when a vfrB mutant strain was exposed to the known SaeS-activation stimulus HNP-1, this kinase no longer impacted virulence genes regulated by SaeR/S (Krute et al., 2016). These data further emphasize the importance of defining virulence regulation in physiologically relevant environments and highlight the complexity of virulence gene regulation in S. aureus.
The challenge of investigating neutrophil—S. aureus interactions In vivo
Investigating the neutrophil-S. aureus interactions in vivo has been challenging. The relevance of using the murine immune system to mimic interactions between S. aureus and the human immune system is heavily debated (Buer and Balling, 2003; Kim et al., 2014; Montgomery et al., 2014; Reizner et al., 2014; Tseng et al., 2015). Interestingly, though HNP-1 is recognized as the most prominent trigger of saeR/S, murine neutrophils are deficient in its production (Inaba et al., 2010). Not only do they differ in the production of HNP-1 but notable differences are observed in chemokine production, cell trafficking, and susceptibility to secreted toxins (Johnston et al., 1990; Singer and Sansonetti, 2004; Parker and Prince, 2012; Tseng et al., 2015). Often the importance of single S. aureus virulence genes is highlighted in models of infection; however, canonical S. aureus toxins such as HlgA, HlgC, and LukF-PVL have extremely limited lytic activity on murine cells (Liu, 2009; Baba-Moussa et al., 2011; Parker and Prince, 2012; Vandenesch et al., 2012). While deficiencies in the murine model are apparent it remains the most prominent and cost effective method to explore relationships in vivo between S. aureus and the host immune system. To that end, advances are being made toward creating more effective murine models that retain benefits such as availability of working with immune-deficient lines while displaying similar disease characteristics as observed in human hosts (Montgomery et al., 2014; Tseng et al., 2015). Tseng et al. developed a “humanized” murine model wherein NOD/SCID/IL2rynull mice are engrafted with human CD34+ umbilical cord blood cells leading to the production of human immune cells within mice (Tseng et al., 2015). These mice demonstrated susceptibility to S. aureus infections at concentrations similar to what is observed in humans as well as increased susceptibility to LukF-PVL. In future efforts, humanized murine models such as this may be key to understanding how S. aureus regulates its pathogenesis in the context of a more relevant host environment and may be critical to advance research into effective therapeutics.
Potential of therapeutics to augment neutrophil function against S. aureus
Vaccine development to prevent or reduce S. aureus infections has not been successful (Fowler and Proctor, 2014). Antibody based vaccines targeting individual virulence factors have not conferred significant protection against S. aureus, and in some cases have even increased mortality (Fowler et al., 2013). As discussed above, S. aureus produces virulence factors that target specific neutrophil receptors to inhibit neutrophil functions. However, S. aureus virulence factors show redundancy in the neutrophil functions they inhibit. For example, multiple virulence factors target the complement pathway and immunoglobulin recognition to inhibit neutrophil phagocytosis and neutralizing one of these virulence factors is not likely to have a significant effect in vivo. Thus, vaccine approaches that simultaneously target multiple S. aureus virulence factors are needed and encouragingly some are in the pipeline (Torre et al., 2015; Frenck et al., 2016).
Targeting S. aureus sensory/regulatory systems has great potential for therapeutic and vaccine development. Theoretically, chemical inhibitors and neutralizing antibodies of sensory/regulatory systems could inhibit S. aureus from sensing its environment and producing virulence factors that disrupt host antimicrobial functions. The SaeR/S TCS is an attractive candidate for vaccine development since SaeR/S-regulated factors disrupt key neutrophil functions that include production of ROS, cytokine expression, and cell fate (Watkins et al., 2011, 2013; Nygaard et al., 2013; Zurek et al., 2014; Guerra et al., 2016). Many of the best studied immunomodulatory factors and cytolytic proteins that target neutrophil function are regulated by SaeR/S including Sbi, Efb, HlgABC, Hla, LukG/H (LukA/B), and PVL (Nygaard et al., 2010). Thus, inhibiting SaeS from sensing its environment has the potential to prevent expression of multiple virulence factors in response to host signals. In this line of thinking, targeting S. aureus quorum sensing via inhibition of Agr has been proposed as a therapeutic approach to inhibit production of virulence of factors (Clatworthy et al., 2007; Park et al., 2007; Cegelski et al., 2008; Kaufmann et al., 2008; Njoroge and Sperandio, 2009; Rutherford and Bassler, 2012). For a review on the S. aureus Agr regulatory system please refer to (Painter et al., 2014). In brief, findings have identified synthetic peptides, small molecules, and antibodies that disrupt S. aureus Agr-dependent quorum sensing either by blocking the sensor histidine kinase AgrC from recognizing its cognate signal autoinducing peptide (AIP), by inhibiting the AgrA response regulator from binding DNA, or by antibody interference of AIP (Park et al., 2007; Kaufmann et al., 2008; Kirchdoerfer et al., 2011; Tal-Gan et al., 2013; Broderick et al., 2014; Murray et al., 2014; Sully et al., 2014). Using this approach, reductions in mouse dermonecrosis following infection with S. aureus, increased bacterial clearance, and reduced hemolytic activity have been reported (Tal-Gan et al., 2013; Sully et al., 2014). In addition, the development of materials with quorum sensing inhibitors to apply during a skin infection is an active area of research and showing promising results (Broderick et al., 2014; Kratochvil et al., 2017). However, studies are needed to fully elucidate the effects of inhibiting sensory/regulatory systems since this can also increase the production of negatively regulated virulence factors and the impact on cross talk between regulatory systems that might result in compensation by one system at the loss of another, is not known.
Modulation of the inflammatory response may be another attractive target for therapeutics that might increase the likelihood of an effective neutrophil response. However, additional research is needed to identify key inflammatory mediators that resolve S. aureus infection and to understand those that exacerbate disease. Studies have identified IL-17 as a key determinant of proper host defense during S. aureus cutaneous infection (Cho et al., 2010). Murphy et al. identified γδ T cells as the major source of IL-17 during peritonitis caused by S. aureus and identified a subset of memory γδ T cells that enhanced IL-17 production to protect the host during subsequent S. aureus infection (Murphy et al., 2014). Thus, immunotherapies that enhance host bactericidal functions by neutralizing detrimental inflammatory responses, increasing beneficial cytokines at the site of infection, or stimulating the expansion of protective T cell subpopulations in response to S. aureus show therapeutic potential. Further research is also needed to understand site-specific cytokine requirements to mount an effective host response to S. aureus infection.
Lastly, as discussed above, the use of mouse models has undeniably increased our understanding of S. aureus-host interactions. However, due to the specificity of many of the S. aureus virulence factors toward human cells, improved models of disease are needed like the “humanized” mouse as discussed above. With improved models and advances in understanding neutrophil-S. aureus interactions, perhaps future reviews will be able to document neutrophils as the clear winner in this epic immune battle.
In conclusion, the ability of S. aureus to survive a neutrophil encounter is thought to contribute significantly to the virulence of this pathogen. This is exemplified by the observed increase in susceptibility to S. aureus infections in individuals suffering from defects that alter normal neutrophil function, such as chronic granulomatous disease, leukocyte adhesion deficiency, and neutropenia (Bodey et al., 1966; Pincus et al., 1976; Dale et al., 1979; Lekstrom-Himes and Gallin, 2000). It is clear that neutrophil antimicrobial activity is essential to eliminate S. aureus. However, studies have also demonstrated that increased neutrophil numbers at the site of infection can exacerbate disease (Gresham et al., 2000; McLoughlin et al., 2008) and that modulation of the neutrophil inflammatory response can significantly impact outcome of infection (Watkins et al., 2013; Zurek et al., 2015). Clearly more studies are needed to define the neutrophil responses that resolve a S. aureus infection. Finding the right balance of controlling inflammation while maintaining an effective neutrophil antimicrobial response will be key to the design of an effective therapeutic.
Statements
Author contributions
FG and JV wrote, edited, prepared manuscript. TB, DP, and ES wrote sections and contributed equally.
Funding
This work was supported by the U.S. National Institutes of Health (Grants NIH-R01A1090046, NIH-PAR98-072, and NIH-RR020185 for a fellowship award to FG and DP), as well as funds from the Montana State University Agriculture Experiment Station.
Acknowledgments
We would like to acknowledge the Servier Medical Art Powerpoint image bank by Servier (http://www.servier.com/Powerpoint-image-bank), which is licensed under a Creative Commons Attribution 3.0 Unported Licence, that was modified to create figures.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Akong-Moore K. Chow O. A. Von Kö Ckritz-Blickwede M. Nizet V. Wehkamp J. Fischer-Bosch M. (2012). Influences of chloride and hypochlorite on neutrophil extracellular trap formation. PLoS ONE7:e42984. 10.1371/journal.pone.0042984
2
Assari T. (2006). Chronic granulomatous disease; fundamental stages in our understanding of CGD. Med. Immunol.5:4. 10.1186/1476-9433-5-4
3
Baba-Moussa L. Sina H. Scheftel J. M. Moreau B. Sainte-Marie D. Kotchoni S. O. et al . (2011). Staphylococcal Panton-Valentine leucocidin as a major virulence factor associated to furuncles. PLoS ONE6:e25716. 10.1371/journal.pone.0025716
4
Bae Y.-S. Park J. C. He R. Ye R. D. Kwak J.-Y. Suh P.-G. et al . (2003). Differential signaling of formyl peptide receptor-like 1 by Trp-Lys-Tyr-Met-Val-Met-CONH2 or lipoxin A4 in human neutrophils. Mol. Pharmacol.64, 721–730. 10.1124/mol.64.3.721
5
Baggiolini M. Walz A. Kunkel S. L. (1989). Neutrophil-activating peptide-1/interleukin 8, a novel cytokine that activates neutrophils. J. Clin. Invest.84, 1045–1049. 10.1172/JCI114265
6
Ballal A. Manna A. C. (2009). Regulation of superoxide dismutase (sod) genes by SarA in Staphylococcus aureus. J. Bacteriol.191, 3301–3310. 10.1128/JB.01496-08
7
Bardoel B. W. Vos R. Bouman T. Aerts P. C. Bestebroer J. Huizinga E. G. et al . (2012). Evasion of Toll-like receptor 2 activation by staphylococcal superantigen-like protein 3. J. Mol. Med.90, 1109–1120. 10.1007/s00109-012-0926-8
8
Bates E. J. Ferrante A. Beard L. J. (1991). Characterization of the major neutrophil-stimulating activity present in culture medium conditioned by Staphylococcus aureus-stimulated mononuclear leucocytes. Immunology72, 448–450.
9
Ben-Baruch A. Grimm M. Bengali K. Evans G. A. Chertov O. Wang J. M. et al . (1997). The differential ability of IL-8 and neutrophil-activating peptide-2 to induce attenuation of chemotaxis is mediated by their divergent capabilities to phosphorylate CXCR2 (IL-8 receptor B). J. Immunol.158, 5927–5933.
10
Berends E. T. Horswill A. R. Haste N. M. Monestier M. Nizet V. Von Köckritz-Blickwede M. (2010). Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun.2, 576–586. 10.1159/000319909
11
Bestebroer J. Aerts P. C. Rooijakkers S. H. Pandey M. K. Köhl J. Van Strijp J. A. G. et al . (2010). Functional basis for complement evasion by staphylococcal superantigen-like 7. Cell. Microbiol.12, 1506–1516. 10.1111/j.1462-5822.2010.01486.x
12
Bestebroer J. Poppelier M. J. Ulfman L. H. Lenting P. J. Denis C. V. Van Kessel K. P. M. et al . (2007). Staphylococcal superantigen-like 5 binds PSGL-1 and inhibits P-selectin-mediated neutrophil rolling. Blood109, 2936–2943. 10.1182/blood-2006-06-015461
13
Bestebroer J. Van Kessel K. P. Azouagh H. Walenkamp A. M. Boer I. G. J. Romijn R. A. et al . (2009). Staphylococcal SSL5 inhibits leukocyte activation by chemokines and anaphylatoxins. Blood113, 328–337. 10.1182/blood-2008-04-153882
14
Bodey G. P. Buckley M. Sathe Y. S. Freireich E. J. (1966). Quantitative relationships between circulating leukocytes and infection in patients with acute leukemia. Ann. Intern. Med.64, 328–340. 10.7326/0003-4819-64-2-328
15
Bose J. L. Daly S. M. Hall P. R. Bayles K. W. (2014). Identification of the Staphylococcus aureus vfrAB Operon, a novel virulence factor regulatory locus. Infect. Immun.82, 1813–1822. 10.1128/IAI.01655-13
16
Bréchard S. Tschirhart E. J. (2008). Regulation of superoxide production in neutrophils: role of calcium influx. J. Leukoc. Biol.84, 1223–1237. 10.1189/jlb.0807553
17
Brinkmann V. Reichard U. Goosmann C. Fauler B. Uhlemann Y. Weiss D. S. et al . (2004). Neutrophil extracellular traps kill bacteria. Science303, 1532–1535. 10.1126/science.1092385
18
Broderick A. H. Stacy D. M. Tal-Gan Y. Kratochvil M. J. Blackwell H. E. Lynn D. M. (2014). Surface coatings that promote rapid release of peptide-based AgrC inhibitors for attenuation of quorum sensing in Staphylococcus aureus. Adv. Healthc. Mater.3, 97–105. 10.1002/adhm.201300119
19
Buer J. Balling R. (2003). Mice, microbes and models of infection. Nat. Rev. Genet.4, 195–205. 10.1038/nrg1019
20
Burman J. D. Leung E. Atkins K. L. O'Seaghdha M. N. Lango L. Bernado P. et al . (2008). Interaction of human complement with Sbi, a staphylococcal immunoglobulin-binding protein: Indications of a novel mechanism of complement evasion by Staphylococcus aureus. J. Biol. Chem.283, 17579–17593. 10.1074/jbc.M800265200
21
Carrel M. Perencevich E. N. David M. Z. (2015). USA300 Methicillin-Resistant Staphylococcus aureus, United States, 2000-2013. Emerging Infect. Dis.21, 1973–1980. 10.3201/eid2111.150452
22
Cegelski L. Marshall G. R. Eldridge G. R. Hultgren S. J. (2008). The biology and future prospects of antivirulence therapies. Nat. Rev. Microbiol.6, 17–27. 10.1038/nrmicro1818
23
Chiang N. Fierro I. M. Gronert K. Serhan C. N. (2000). Activation of lipoxin A(4) receptors by aspirin-triggered lipoxins and select peptides evokes ligand-specific responses in inflammation. J. Exp. Med.191, 1197–1208. 10.1084/jem.191.7.1197
24
Cho H. Jeong D.-W. Liu Q. Yeo W. S. Vogl T. Skaar E. P. et al . (2015). Calprotectin increases the activity of the SaeRS two component system and murine mortality during Staphylococcus aureus infections. PLoS Pathog.11:e1005026. 10.1371/journal.ppat.1005026
25
Cho J. S. Pietras E. M. Garcia N. C. Ramos R. I. Farzam D. M. Monroe H. R. et al . (2010). IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J. Clin. Invest.120, 1762–1773. 10.1172/JCI40891
26
Christophe T. Karlsson A. Dugave C. Rabiet M. J. Boulay F. Dahlgren C. (2001). The synthetic peptide Trp-Lys-Tyr-Met-Val-Met-NH2 specifically activates neutrophils through FPRL1/Lipoxin A4 receptors and is an agonist for the orphan monocyte-expressed chemoattractant receptor FPRL2. J. Biol. Chem.276, 21585–21593. 10.1074/jbc.M007769200
27
Clarke T. B. Davis K. M. Lysenko E. S. Zhou A. Y. Yu Y. Weiser J. N. (2010). Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nat. Med.16, 228–231. 10.1038/nm.2087
28
Clatworthy A. E. Pierson E. Hung D. T. (2007). Targeting virulence: a new paradigm for antimicrobial therapy. Nat. Chem. Biol.3, 541–548. 10.1038/nchembio.2007.24
29
Clements M. O. Watson S. P. Foster S. J. (1999). Characterization of the major superoxide dismutase of Staphylococcus aureus and its role in starvation survival, stress resistance, and pathogenicity. J. Bacteriol.181, 3898–3903.
30
Colin D. A. Monteil H. (2003). Control of the oxidative burst of human neutrophils by Staphylococcal Leukotoxins control of the oxidative burst of human neutrophils by Staphylococcal Leukotoxins. Infect. Immun.71, 3724–3729. 10.1128/IAI.71.7.3724-3729.2003
31
Collins L. V. Kristian S. A. Weidenmaier C. Faigle M. Van Kessel K. P. Van Strijp J. A. G. et al . (2002). Staphylococcus aureus strains lacking D-alanine modifications of teichoic acids are highly susceptible to human neutrophil killing and are virulence attenuated in mice. J. Infect. Dis.186, 214–219. 10.1086/341454
32
Cosgrove K. Coutts G. Jonsson I.-M. Tarkowski A. Kokai-Kun J. F. Mond J. J. et al . (2007). Catalase (KatA) and alkyl hydroperoxide reductase (AhpC) have compensatory roles in peroxide stress resistance and are required for survival, persistence, and nasal colonization in Staphylococcus aureus. J. Bacteriol.189, 1025–1035. 10.1128/JB.01524-06
33
Dale D. C. Guerry D. Wewerka J. R. Bull J. M. Chusid M. J. (1979). Chronic neutropenia. Medicine58, 128–144. 10.1097/00005792-197903000-00002
34
De Haas C. J. C. Veldkamp K. E. Peschel A. Weerkamp F. Van Wamel W. J. Heezius E. C. et al . (2004). Chemotaxis Inhibitory Protein of Staphylococcus aureus, a Bacterial Antiinflammatory Agent. J. Exp. Med.3687900, 687–695. 10.1084/jem.20031636
35
de Oliveira S. Rosowski E. E. Huttenlocher A. (2016). Neutrophil migration in infection and wound repair: going forward in reverse. Nat. Rev. Immunol.16, 378–391. 10.1038/nri.2016.49
36
De Yang Chen Q. Schmidt A. P. Anderson G. M. Wang J. M. Wooters J. et al . (2000). LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J. Exp. Med.192, 1069–1074. 10.1084/jem.192.7.1069
37
Delauné A. Dubrac S. Blanchet C. Poupel O. Mäder U. Hiron A. et al . (2012). The WalKR system controls major staphylococcal virulence genes and is involved in triggering the host inflammatory response. Infect. Immun.80, 3438–3453. 10.1128/IAI.00195-12
38
DeLeo F. R. Allen L. A. Apicella M. Nauseef W. M. (1999). NADPH oxidase activation and assembly during phagocytosis. J. Immunol.163, 6732–6740.
39
DeLeo F. R. Quinn M. T. (1996). Assembly of the phagocyte NADPH oxidase: molecular interaction of oxidase proteins. J. Leukoc. Biol.60, 677–691.
40
Dossett J. H. Kronvall G. Williams R. C. Quie P. G. (1969). Antiphagocytic effects of staphylococcal protein A. J. Immunol.103, 1405–1410.
41
Dragneva Y. Anuradha C. D. Valeva A. Hoffmann A. Bhakdi S. Husmann M. (2001). Subcytocidal attack by staphylococcal alpha-toxin activates NF-kappaB and induces interleukin-8 production. Infect. Immun.69, 2630–2635. 10.1128/IAI.69.4.2630-2635.2001
42
Dubrac S. Msadek T. (2004). Identification of genes controlled by the essential YycG / YycF two-component system of Staphylococcus aureus. J. Bacteriol.186, 1175–1181. 10.1128/JB.186.4.1175-1181.2004
43
Dubrac S. Boneca I. G. Poupel O. Msadek T. (2007). New insights into the WalK/WalR (YycG/YycF) essential signal transduction pathway reveal a major role in controlling cell wall metabolism and biofilm formation in Staphylococcus aureus. J. Bacteriol.189, 8257–8269. 10.1128/JB.00645-07
44
DuMont A. L. Torres V. J. (2014). Cell targeting by the Staphylococcus aureus pore-forming toxins: It's not just about lipids. Trends Microbiol.22, 21–27. 10.1016/j.tim.2013.10.004
45
Edwards S. W. Say J. E. Hughes V. (1988). Gamma interferon enhances the killing of Staphylococcus aureus by human neutrophils. J. Gen. Microbiol.134, 37–42. 10.1099/00221287-134-1-37
46
El-Benna J. Dang P. M. Gougerot-Pocidalo M. A. (2008). Priming of the neutrophil NADPH oxidase activation: role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin. Immunopathol.30, 279–289. 10.1007/s00281-008-0118-3
47
Elbim C. Bailly S. Chollet-Martin S. Hakim J. Gougerot-Pocidalo M. A. (1994). Differential priming effects of proinflammatory cytokines on human neutrophil oxidative burst in response to bacterial N-formyl peptides. Infect. Immun.62, 2195–201.
48
Ellis T. N. Beaman B. L. (2004). Interferon-gamma activation of polymorphonuclear neutrophil function. Immunology112, 2–12. 10.1111/j.1365-2567.2004.01849.x
49
Ernst C. M. Staubitz P. Mishra N. N. Yang S. J. Hornig G. Kalbacher H. et al . (2009). The bacterial defensin resistance protein MprF consists of separable domains for lipid lysinylation and antimicrobial peptide repulsion. PLoS Pathog.5, 1–9. 10.1371/journal.ppat.1000660
50
Falk W. Harvath L. Leonard E. J. (1982). Only the chemotactic subpopulation of human blood monocytes expresses receptors for the chemotactic peptide N-formylmethionyl-leucyl-phenylalanine. Infect. Immun.36, 450–454.
51
Falugi F. Kim H. K. Missiakas D. M. (2013). Role of Protein A in the evasion of host adaptive immune responses. MBio4, 1–9. 10.1128/mBio.00575-13
52
Ferrante A. Harvey D. P. Bates E. J. (1989). Staphylococcus aureus-stimulated mononuclear leucocyte-conditioned medium increases the neutrophil bactericidal activity, and augments oxygen radical production and degranulation in response to the bacteria. Clin. Exp. Immunol.78, 366–371.
53
Ferrante A. Martin A. J. Bates E. J. Goh D. H. Harvey D. P. Parsons D. et al . (1993). Killing of Staphylococcus aureus by tumor necrosis factor-alpha-activated neutrophils. The role of serum opsonins, integrin receptors, respiratory burst, and degranulation. J. Immunol.151, 4821–4828.
54
Fevre C. Bestebroer J. Mebius M. M. de Haas C. J. van Strijp J. A. G. Fitzgerald J. R. et al . (2014). Staphylococcus aureus proteins SSL6 and SElX interact with neutrophil receptors as identified using secretome phage display. Cell. Microbiol.16, 1646–1665. 10.1111/cmi.12313
55
Fiore S. Maddox J. F. Perez H. D. Serhan C. N. (1994). Identification of a human cDNA encoding a functional high affinity lipoxin A4 receptor. J. Exp. Med.180, 253–260. 10.1084/jem.180.1.253
56
Flack C. E. Zurek O. W. Meishery D. D. Pallister K. B. Malone C. L. Horswill A. R. et al . (2014). Differential regulation of staphylococcal virulence by the sensor kinase SaeS in response to neutrophil-derived stimuli. Proc. Natl. Acad. Sci. U.S.A.111, E2037–E2045. 10.1073/pnas.1322125111
57
Flannagan R. S. Cosío G. Grinstein S. (2009). Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol.7, 355–366. 10.1038/nrmicro2128
58
Forsgren A. Sjöquist J. (1966). “Protein A” from S. aureus. I. Pseudo-immune reaction with human gamma-globulin. J. Immunol.97, 822–827.
59
Fournier B. Philpott D. J. (2005). Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev.18, 521–540. 10.1128/CMR.18.3.521-540.2005
60
Fowler V. G. Allen K. B. Moreira E. D. Moustafa M. Isgro F. Boucher H. W. et al . (2013). Effect of an investigational vaccine for preventing Staphylococcus aureus infections after cardiothoracic surgery: a randomized trial. JAMA309, 1368–1378. 10.1001/jama.2013.3010
61
Fowler V. G. Proctor R. A. (2014). Where does a Staphylococcus aureus vaccine stand?Clin. Microbiol. Infect.20, 66–75. 10.1111/1469-0691.12570
62
Frenck R. W. Buddy Creech C. Sheldon E. A. Seiden D. J. Kankam M. K. Baber J. et al . (2016). Safety, tolerability, and immunogenicity of a 4-antigen Staphylococcus aureus vaccine (SA4Ag): results from a first-in-human randomised, placebo-controlled phase 1/2 study. Vaccine35, 375–384. 10.1016/j.vaccine.2016.11.010
63
Fuchs T. A. Abed U. Goosmann C. Hurwitz R. Schulze I. Wahn V. et al . (2007). Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol.176, 231–241. 10.1083/jcb.200606027
64
Gallin J. I. Malech H. L. Melnick D. A. Weening R. S. Roos D. (1991). A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. The International Chronic Granulomatous Disease Cooperative Study Group. N. Engl. J. Med.324, 509–516. 10.1056/NEJM199102213240801
65
Galluzzi L. Vitale I. Abrams J. M. Alnemri E. S. Baehrecke E. H. Blagosklonny M. V. et al . (2012). Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ.19, 107–120. 10.1038/cdd.2011.96
66
Garcia B. L. Ramyar K. X. Tzekou A. Ricklin D. McWhorter W. J. Lambris J. D. et al . (2010). Molecular basis for complement recognition and inhibition determined by crystallographic studies of the Staphylococcal Complement Inhibitor (SCIN) Bound to C3c and C3b. J. Mol. Biol.402, 17–29. 10.1016/j.jmb.2010.07.029
67
Geiger T. Goerke C. Mainiero M. Kraus D. Wolz C. (2008). The virulence regulator sae of Staphylococcus aureus: promoter activities and response to phagocytosis-related signals. J. Bacteriol.190, 3419–3428. 10.1128/JB.01927-07
68
Gonzalez C. D. Ledo C. Giai C. Garófalo A. Gómez M. I. (2015). The Sbi protein contributes to Staphylococcus aureus inflammatory response during systemic infection. PLoS ONE10:e0131879. 10.1371/journal.pone.0131879
69
González-Zorn B. Senna J. P. Fiette L. Shorte S. Testard A. Chignard M. et al . (2005). Bacterial and host factors implicated in nasal carriage of methicillin-resistant Staphylococcus aureus in mice. Infect. Immun.73, 1847–1851. 10.1128/IAI.73.3.1847-1851.2005
70
Gorwitz R. J. Kruszon-Moran D. McAllister S. K. McQuillan G. McDougal L. K. Fosheim G. E. et al . (2008). Changes in the prevalence of nasal colonization with Staphylococcus aureus in the United States, 2001-2004. J. Infect. Dis.197, 1226–1234. 10.1086/533494
71
Graves S. F. Kobayashi S. D. Braughton K. R. Whitney A. R. Sturdevant D. E. Rasmussen D. L. et al . (2012). Sublytic concentrations of Staphylococcus aureus Panton-Valentine leukocidin alter human PMN gene expression and enhance bactericidal capacity. J. Leukoc. Biol.92, 361–374. 10.1189/jlb.1111575
72
Green J. N. Kettle A. J. Winterbourn C. C. (2014). Protein chlorination in neutrophil phagosomes and correlation with bacterial killing. Free Radic. Biol. Med.77, 49–56. 10.1016/j.freeradbiomed.2014.08.013
73
Greenlee-Wacker M. C. Rigby K. M. Kobayashi S. D. Porter A. R. DeLeo F. R. Nauseef W. M. (2014). Phagocytosis of Staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J. Immunol.192, 4709–4717. 10.4049/jimmunol.1302692
74
Gresham H. D. Lowrance J. H. Caver T. E. Wilson B. S. Cheung A. L. Lindberg F. P. (2000). Survival of Staphylococcus aureus inside neutrophils contributes to infection. J. Immunol.164, 3713–3722. 10.4049/jimmunol.164.7.3713
75
Guerra F. E. Addison C. B. de Jong N. W. Azzolino J. Pallister K. B. van Strijp J. A. G. et al . (2016). Staphylococcus aureus SaeR/S-regulated factors reduce human neutrophil reactive oxygen species production. J. Leukoc. Biol.100, 1–6. 10.1189/jlb.4VMAB0316-100RR
76
Guyer D. A. Moore K. L. Lynam E. B. Schammel C. M. Rogelj S. McEver R. P. et al . (1996). P-selectin glycoprotein ligand-1 (PSGL-1) is a ligand for L-selectin in neutrophil aggregation. Blood88, 2415–2421.
77
Haas P. J. De Haas C. J. Poppelier M. J. Van Kessel K. P. Van Strijp J. A. Dijkstra K. et al . (2005). The structure of the C5a receptor-blocking domain of chemotaxis inhibitory protein of Staphylococcus aureus is related to a group of immune evasive molecules. J. Mol. Biol.353, 859–872. 10.1016/j.jmb.2005.09.014
78
Hampton M. B. Winterbourn C. C. (1995). Modification of neutrophil oxidant production with diphenyleneiodonium and its effect on bacterial killing. Free Radic. Biol. Med.18, 633–639. 10.1016/0891-5849(94)00181-I
79
Hampton M. Kettle A. Winterbourn C. (1996). Involvement of superoxide and myeloperoxidase in oxygen-dependent killing of Staphylococcus aureus by neutrophils. Infect. Immun.64, 3512–3517.
80
Harraghy N. Kormanec J. Wolz C. Homerova D. Goerke C. Ohlsen K. et al . (2005). sae is essential for expression of the staphylococcal adhesins Eap and Emp. Microbiology151, 1789–1800. 10.1099/mic.0.27902-0
81
Hidalgo A. Peired A. J. Wild M. K. Vestweber D. Frenette P. S. (2007). Complete identification of E-Selectin Ligands on neutrophils reveals distinct functions of PSGL-1, ESL-1, and CD44. Immunity26, 477–489. 10.1016/j.immuni.2007.03.011
82
Hoebe K. Georgel P. Rutschmann S. Du X. Mudd S. Crozat K. et al . (2005). CD36 is a sensor of diacylglycerides. Nature433, 523–527. 10.1038/nature03253
83
Horsburgh M. J. Clements M. O. Crossley H. Ingham E. Foster S. J. (2001a). PerR controls oxidative stress resistance and iron storage proteins and is required for virulence in Staphylococcus aureus. Infect. Immun.69, 3744–3754. 10.1128/IAI.69.6.3744-3754.2001
84
Horsburgh M. J. Ingham E. Foster S. J. (2001b). In Staphylococcus aureus, fur is an interactive regulator with PerR, contributes to virulence, and Is necessary for oxidative stress resistance through positive regulation of catalase and iron homeostasis. J. Bacteriol.183, 468–475. 10.1128/JB.183.2.468-475.2001
85
Huey R. Hugli T. E. (1985). Characterization of a C5a receptor on human polymorphonuclear leukocytes (PMN). J. Immunol.135, 2063–2068.
86
Huo Y. Xia L. (2009). P-selectin Glycoprotein Ligand-1 Plays a crucial role in the selective recruitment of Leukocytes into the Atherosclerotic arterial wall. Trends Cardiovasc. Med.19, 140–145. 10.1016/j.tcm.2009.07.006
87
Inaba Y. Ashida T. Ito T. Ishikawa C. Tanabe H. Maemoto A. et al . (2010). Expression of the antimicrobial peptide alpha-defensin/cryptdins in intestinal crypts decreases at the initial phase of intestinal inflammation in a model of inflammatory bowel disease, IL-10-deficient mice. Inflamm. Bowel Dis.16, 1488–1495. 10.1002/ibd.21253
88
Itoh S. Hamada E. Kamoshida G. Yokoyama R. Takii T. Onozaki K. et al . (2010). Staphylococcal superantigen-like protein 10 (SSL10) binds to human immunoglobulin G (IgG) and inhibits complement activation via the classical pathway. Mol. Immunol.47, 932–938. 10.1016/j.molimm.2009.09.027
89
Jacobsson K. Frykberg L. (1995). Cloning of ligand-binding domains of bacterial receptors by phage display. Biotechniques18, 878–885.
90
Jin T. Bokarewa M. Foster T. Mitchell J. Higgins J. Tarkowski A. (2004). Staphylococcus aureus resists human defensins by production of staphylokinase, a novel bacterial evasion mechanism. J. Immunol.172, 1169–1176. 10.4049/jimmunol.172.2.1169
91
Johnston S. C. Dustin M. L. Hibbs M. L. Springer T. A. (1990). On the species specificity of the interaction of LFA-1 with intercellular adhesion molecules. J. Immunol.145, 1181–1187.
92
Karavolos M. H. Horsburgh M. J. Ingham E. Foster S. J. (2003). Role and regulation of the superoxide dismutases of Staphylococcus aureus. Microbiology149, 2749–2758. 10.1099/mic.0.26353-0
93
Kaufmann G. F. Park J. Janda K. D. (2008). Bacterial quorum sensing: a new target for anti-infective immunotherapy. Expert Opin. Biol. Ther.8, 719–724. 10.1517/14712598.8.6.719
94
Kawada-Matsuo M. Yoshida Y. Nakamura N. Komatsuzawa H. (2011). Role of two-component systems in the resistance of Staphylococcus aureus to antibacterial agents. Virulence2, 427–430. 10.4161/viru.2.5.17711
95
Kennedy A. D. Deleo F. R. (2009). Neutrophil apoptosis and the resolution of infection. Immunol. Res.43, 25–61. 10.1007/s12026-008-8049-6
96
Kettle A. J. Anderson R. F. Hampton M. B. Winterbourn C. C. (2007). Reactions of superoxide with myeloperoxidase. Biochemistry46, 4888–4897. 10.1021/bi602587k
97
Kim H. K. Falugi F. Missiakas D. M. Schneewind O. (2016). Peptidoglycan-linked protein A promotes T cell-dependent antibody expansion during Staphylococcus aureus infection. Proc. Natl. Acad. Sci. U.S.A.113, 5718–5723. 10.1073/pnas.1524267113
98
Kim H. K. Missiakas D. Schneewind O. (2014). Mouse models for infectious diseases caused by Staphylococcus aureus. J. Immunol. Methods410, 88–99. 10.1016/j.jim.2014.04.007
99
King B. F. Wilkinson B. J. (1981). Binding of human immunoglobulin G to protein A in encapsulated Staphylococcus aureus. Infect. Immun.33, 666–672.
100
Kirchdoerfer R. N. Garner A. L. Flack C. E. Mee J. M. Horswill A. R. Janda K. D. et al . (2011). Structural basis for ligand recognition and discrimination of a quorum-quenching antibody. J. Biol. Chem.286, 17351–17358. 10.1074/jbc.M111.231258
101
Klebanoff S. J. Kettle A. J. Rosen H. Winterbourn C. C. Nauseef W. M. (2013). Myeloperoxidase: a front-line defender against phagocytosed microorganisms. J. Leukoc. Biol.93, 185–198. 10.1189/jlb.0712349
102
Klevens R. M. Morrison M. A. Nadle J. Petit S. Gershman K. Ray S. et al . (2007). Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA298, 1763–1771. 10.1001/jama.298.15.1763
103
Knuefermann P. Sakata Y. Baker J. S. Huang C. H. Sekiguchi K. Hardarson H. S. et al . (2004). Toll-like receptor 2 mediates Staphylococcus aureus-induced myocardial dysfunction and cytokine production in the heart. Circulation110, 3693–3698. 10.1161/01.CIR.0000143081.13042.04
104
Ko Y. P. Kuipers A. Freitag C. M. Jongerius I. Medina E. van Rooijen W. J. et al . (2013). Phagocytosis Escape by a Staphylococcus aureus protein that connects complement and coagulation proteins at the bacterial surface. PLoS Pathog.9:e1003816. 10.1371/journal.ppat.1003816
105
Kobayashi S. D. Braughton K. R. Palazzolo-Ballance A. M. Kennedy A. D. Sampaio E. Kristosturyan E. et al . (2010). Rapid neutrophil destruction following phagocytosis of Staphylococcus aureus. J. Innate Immun.2, 560–575. 10.1159/000317134
106
Kobayashi S. D. Braughton K. R. Whitney A. R. Voyich J. M. Schwan T. G. Musser J. M. et al . (2003). Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc. Natl. Acad. Sci. U.S.A.100, 10948–10953. 10.1073/pnas.1833375100
107
Kobayashi S. D. Malachowa N. DeLeo F. R. (2015). Pathogenesis of Staphylococcus aureus abscesses. Am. J. Pathol.185, 1518–1527. 10.1016/j.ajpath.2014.11.030
108
Kobayashi S. D. Malachowa N. DeLeo F. R. (2017). Influence of microbes on neutrophil life and death. Front. Cell. Infect. Microbiol. 7:159. 10.3389/fcimb.2017.00159
109
Kolaczkowska E. Kubes P. (2013). Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol.13, 159–175. 10.1038/nri3399
110
König B. Prévost G. Piémont Y. König W. (1995). Effects of Staphylococcus aureus leukocidins on inflammatory mediator release from human granulocytes. J. Infect. Dis.171, 607–613. 10.1093/infdis/171.3.607
111
Koymans K. J. Feitsma L. J. Brondijk T. H. Aerts P. C. Lukkien E. Lossl P. et al . (2015). Structural basis for inhibition of TLR2 by staphylococcal superantigen-like protein 3 (SSL3). Proc. Natl. Acad. Sci. U.S.A.112, 11018–11023. 10.1073/pnas.1502026112
112
Kratochvil M. J. Yang T. Blackwell H. E. Lynn D. M. (2017). Nonwoven polymer nanofiber coatings that inhibit quorum sensing in Staphylococcus aureus: toward new nonbactericidal approaches to infection control. ACS Infect. Dis.3, 271–280. 10.1021/acsinfecdis.6b00173
113
Kronvall G. Grey H. M. Williams R. C. (1970). Protein A reactivity with mouse immunoglobulins. Structural relationship between some mouse and human immunoglobulins. J. Immunol.105, 1116–1123.
114
Krute C. N. Rice K. C. Bose J. L. (2017). VfrB is a key activator of the Staphylococcus aureus SaeRS two-component system. J. Bacteriol.199, e00828–16. 10.1128/JB.00828-16
115
Kurokawa K. Lee H. Roh K. B. Asanuma M. Kim Y. S. Nakayama H. et al . (2009). The triacylated ATP binding cluster transporter substrate-binding lipoprotein of Staphylococcus aureus functions as a native ligand for toll-like receptor 2. J. Biol. Chem.284, 8406–8411. 10.1074/jbc.M809618200
116
Laarman A. J. Mijnheer G. Mootz J. M. van Rooijen W. J. Ruyken M. Malone C. L. et al . (2012). Staphylococcus aureus Staphopain A inhibits CXCR2-dependent neutrophil activation and chemotaxis. EMBO J.31, 3607–3619. 10.1038/emboj.2012.212
117
Lambris J. D. Ricklin D. Geisbrecht B. V. (2008). Complement evasion by human pathogens. Nat. Rev. Microbiol.6, 132–142. 10.1038/nrmicro1824
118
Langley R. Wines B. Willoughby N. Basu I. Proft T. Fraser J. D. (2005). The Staphylococcal Superantigen-Like Protein 7 Binds IgA and Complement C5 and Inhibits IgA-Fc RI binding and serum killing of bacteria. J. Immunol.174, 2926–2933. 10.4049/jimmunol.174.5.2926
119
Laursen N. S. Gordon N. Hermans S. Lorenz N. Jackson N. Wines B. et al . (2010). Structural basis for inhibition of complement C5 by the SSL7 protein from Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A.107, 3681–3686. 10.1073/pnas.0910565107
120
Le Y. Yazawa H. Gong W. Yu Z. Ferrans V. J. Murphy P. M. et al . (2001). The neurotoxic prion peptide fragment PrP(106-126) is a chemotactic agonist for the G protein-coupled receptor formyl peptide receptor-like 1. J. Immunol.166, 1448–1451. 10.4049/jimmunol.166.3.1448
121
Lee L. Y. Höök M. Haviland D. Wetsel R. A. Yonter E. O. Syribeys P. et al . (2004). Inhibition of complement activation by a secreted Staphylococcus aureus Protein. J. Infect. Dis.190, 571–579. 10.1086/422259
122
Lekstrom-Himes J. A. Gallin J. I. (2000). Immunodeficiency diseases caused by defects in phagocytes. N. Engl. J. Med.343, 1703–1714. 10.1056/NEJM200012073432307
123
Liu G. Y. (2009). Molecular pathogenesis of Staphylococcus aureus infection. Pediatr. Res.65, 71–77. 10.1203/PDR.0b013e31819dc44d
124
Liu G. Y. Essex A. Buchanan J. T. Datta V. Hoffman H. M. Bastian J. F. et al . (2005). Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J. Exp. Med.202, 209–215. 10.1084/jem.20050846
125
Liu Q. Cho H. Yeo W.-S. Bae T. (2015). The extracytoplasmic linker peptide of the sensor protein SaeS tunes the kinase activity required for Staphylococcal Virulence in response to host signals. PLoS Pathog.11:e1004799. 10.1371/journal.ppat.1004799
126
Lorenz N. Clow F. Radcliff F. J. Fraser J. D. (2013). Full functional activity of SSL7 requires binding of both complement C5 and IgA. Immunol. Cell Biol.91, 469–476. 10.1038/icb.2013.28
127
Lowy F. D. (1998). Staphylococcus aureus infections. N. Engl. J. Med.339, 520–532. 10.1056/NEJM199808203390806
128
Lu T. Porter A. R. Kennedy A. D. Kobayashi S. D. DeLeo F. R. (2014). Phagocytosis and killing of Staphylococcus aureus by human neutrophils. J. Innate Immun.6, 639–649. 10.1159/000360478
129
Malachowa N. Kobayashi S. D. Braughton K. R. Whitney A. R. Parnell M. J. Gardner D. J. et al . (2012). Staphylococcus aureus leukotoxin GH promotes inflammation. J. Infect. Dis.206, 1185–1193. 10.1093/infdis/jis495
130
Malachowa N. Kobayashi S. D. Freedman B. Dorward D. W. DeLeo F. R. (2013). Staphylococcus aureus leukotoxin GH promotes formation of neutrophil extracellular traps. J. Immunol.191, 6022–6029. 10.4049/jimmunol.1301821
131
Malachowa N. Whitney A. R. Kobayashi S. D. Sturdevant D. E. Kennedy A. D. Braughton K. R. et al . (2011). Global changes in Staphylococcus aureus gene expression in human blood. PLoS ONE6:e18617. 10.1371/journal.pone.0018617
132
Malawista S. E. Van Blaricom G. Breitenstein M. G. (1989). Cryopreservable neutrophil surrogates. Stored cytoplasts from human polymorphonuclear leukocytes retain chemotactic, phagocytic, and microbicidal function. J. Clin. Invest.83, 728–732. 10.1172/JCI113939
133
Martin C. J. Peters K. N. Behar S. M. (2014). Macrophages clean up: efferocytosis and microbial control. Curr. Opin. Microbiol.17, 17–23. 10.1016/j.mib.2013.10.007
134
McLoughlin R. M. Lee J. C. Kasper D. L. Tzianabos A. O. (2008). IFN-gamma regulated chemokine production determines the outcome of Staphylococcus aureus infection. J. Immunol.181, 1323–1332. 10.4049/jimmunol.181.2.1323
135
Mempel M. Voelcker V. Köllisch G. Plank C. Rad R. Gerhard M. et al . (2003). Toll-Like Receptor expression in human keratinocytes: Nuclear Factor κB controlled gene activation by Staphylococcus aureus is Toll-Like Receptor 2 but Not Toll-Like Receptor 4 or platelet activating factor receptor dependent. J. Invest. Dermatol.121, 1389–1396. 10.1111/j.1523-1747.2003.12630.x
136
Menestrina G. Dalla Serra M. Comai M. Coraiola M. Viero G. Werner S. et al . (2003). Ion channels and bacterial infection: the case of beta-barrel pore-forming protein toxins of Staphylococcus aureus. FEBS Lett.552, 54–60. 10.1016/S0014-5793(03)00850-0
137
Mitchell G. B. Albright B. N. Caswell J. L. (2003). Effect of interleukin-8 and granulocyte colony-stimulating factor on priming and activation of bovine neutrophils. Infect. Immun.71, 1643–1649. 10.1128/IAI.71.4.1643-1649.2003
138
Mölkänen T. Tyynelä J. Helin J. Kalkkinen N. Kuusela P. (2002). Enhanced activation of bound plasminogen on Staphylococcus aureus by staphylokinase. FEBS Lett.517, 72–78. 10.1016/S0014-5793(02)02580-2
139
Montgomery C. P. Boyle-Vavra S. Adem P. V. Lee J. C. Husain A. N. Clasen J. et al . (2008). Comparison of virulence in community-associated methicillin-resistant Staphylococcus aureus Pulsotypes USA300 and USA400 in a rat model of Pneumonia. J. Infect. Dis.198, 561–570. 10.1086/590157
140
Montgomery C. P. Daniels M. Zhao F. Alegre M. L. Chong A. S. Daum R. S. (2014). Protective immunity against recurrent Staphylococcus aureus skin infection requires antibody and interleukin-17A. Infect. Immun.82, 2125–2134. 10.1128/IAI.01491-14
141
Moore K. L. Patel K. D. Bruehl R. E. Fugang L. Johnson D. A. Lichenstein H. S. et al . (1995). P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. J. Cell Biol.128, 661–671. 10.1083/jcb.128.4.661
142
Müller P. Müller-Anstett M. Wagener J. Gao Q. Kaesler S. Schaller M. et al . (2010). The Staphylococcus aureus lipoprotein SitC colocalizes with Toll-like receptor 2 (TLR2) in murine keratinocytes and elicits intracellular TLR2 accumulation. Infect. Immun.78, 4243–4250. 10.1128/IAI.00538-10
143
Murphy A. G. O'Keeffe K. M. Lalor S. J. Maher B. M. Mills K. H. McLoughlin R. M. (2014). Staphylococcus aureus infection of mice expands a population of memory γδ T cells that are protective against subsequent infection. J. Immunol.192, 3697–3708. 10.4049/jimmunol.1303420
144
Murray E. J. Crowley R. C. Truman A. Clarke S. R. Cottam J. A. Jadhav G. P. et al . (2014). Targeting Staphylococcus aureus quorum sensing with nonpeptidic small molecule inhibitors. J. Med. Chem.57, 2813–2819. 10.1021/jm500215s
145
Njoroge J. Sperandio V. (2009). Jamming bacterial communication: new approaches for the treatment of infectious diseases. EMBO Mol. Med.1, 201–210. 10.1002/emmm.200900032
146
Nunes P. Demaurex N. Dinauer M. C. (2013). Regulation of the NADPH oxidase and associated ion fluxes during phagocytosis. Traffic14, 1118–1131. 10.1111/tra.12115
147
Nygaard T. K. Pallister K. B. Ruzevich P. Griffith S. Vuong C. Voyich J. M. (2010). SaeR binds a consensus sequence within virulence gene promoters to advance USA300 pathogenesis. J. Infect. Dis.201, 241–254. 10.1086/649570
148
Nygaard T. K. Pallister K. B. Zurek O. W. Voyich J. M. (2013). The impact of α-toxin on host cell plasma membrane permeability and cytokine expression during human blood infection by CA-MRSA USA300. J. Leukoc. Biol.94, 971–979. 10.1189/jlb.0213080
149
Oku Y. Kurokawa K. Ichihashi N. Sekimizu K. (2004). Characterization of the Staphylococcus aureus mprF gene, involved in lysinylation of phosphatidylglycerol. Microbiology150, 45–51. 10.1099/mic.0.26706-0
150
Olaru F. Jensen L. E. (2010). Staphylococcus aureus stimulates neutrophil targeting chemokine expression in keratinocytes through an autocrine IL-1alpha signaling loop. J. Invest. Dermatol.130, 1866–1876. 10.1038/jid.2010.37
151
Olson M. E. Nygaard T. K. Ackermann L. Watkins R. L. Zurek O. W. Pallister K. B. et al . (2013). Staphylococcus aureus nuclease is a SaeR/S-dependent virulence factor. Infect. Immun. 10.1128/iai.01242-12
152
Painter K. L. Krishna A. Wigneshweraraj S. Edwards A. M. (2014). What role does the quorum-sensing accessory gene regulator system play during Staphylococcus aureus bacteremia?Trends Microbiol.22, 676–685. 10.1016/j.tim.2014.09.002
153
Palazzolo-Ballance A. M. Reniere M. L. Braughton K. R. Sturdevant D. E. Otto M. Kreiswirth B. N. et al . (2007). Neutrophil microbicides induce a pathogen survival response in community-associated methicillin-resistant Staphylococcus aureus. J. Immunol.180, 500–509. 10.4049/jimmunol.180.1.500
154
Pang Y. Y. Schwartz J. Bloomberg S. Boyd J. M. Horswill A. R. Nauseef W. M. (2014). Methionine sulfoxide reductases protect against oxidative stress in Staphylococcus aureus encountering exogenous oxidants and human neutrophils. J. Innate Immun.6, 353–364. 10.1159/000355915
155
Papayannopoulos V. Metzler K. D. Hakkim A. Zychlinsky A. (2010). Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol.191, 677–691. 10.1083/jcb.201006052
156
Park J. Jagasia R. Kaufmann G. F. Mathison J. C. Ruiz D. I. Moss J. A. et al . (2007). Infection control by antibody disruption of bacterial quorum sensing signaling. Chem. Biol.14, 1119–1127. 10.1016/j.chembiol.2007.08.013
157
Parker D. Prince A. (2012). Immunopathogenesis of Staphylococcus aureus pulmonary infection. Semin. Immunopathol.34, 281–297. 10.1007/s00281-011-0291-7
158
Parry M. A. Zhang X. C. Bode W. (2000). Molecular mechanisms of plasminogen activation: bacterial cofactors provide clues. Trends Biochem. Sci.25, 53–59. 10.1016/S0968-0004(99)01521-2
159
Patel D. Wines B. D. Langley R. J. Fraser J. D. (2010). Specificity of staphylococcal superantigen-like protein 10 toward the human IgG1 Fc domain. J. Immunol.184, 6283–6292. 10.4049/jimmunol.0903311
160
Pauli N. T. Kim H. K. Falugi F. Huang M. Dulac J. Henry Dunand C. et al . (2014). Staphylococcus aureus infection induces protein A-mediated immune evasion in humans. J. Exp. Med.211, 2331–2339. 10.1084/jem.20141404
161
Pelz A. Wieland K. P. Putzbach K. Hentschel P. Albert K. Götz F. (2005). Structure and biosynthesis of staphyloxanthin from Staphylococcus aureus. J. Biol. Chem.280, 32493–32498. 10.1074/jbc.M505070200
162
Peschel A. Jack R. W. Otto M. Collins L. V. Staubitz P. Nicholson G. et al . (2001). Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J. Exp. Med.193, 1067–1076. 10.1084/jem.193.9.1067
163
Peschel A. Otto M. Jack R. W. Kalbacher H. Jung G. Götz F. (1999). Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J. Biol. Chem.274, 8405–8410. 10.1074/jbc.274.13.8405
164
Pilsczek F. H. Salina D. Poon K. K. Fahey C. Yipp B. G. Sibley C. D. et al . (2010). A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol.185, 7413–7425. 10.4049/jimmunol.1000675
165
Pincus S. H. Boxer L. A. Stossel T. P. (1976). Chronic neutropenia in childhood. Analysis of 16 cases and a review of the literature. Am. J. Med.61, 849–861. 10.1016/0002-9343(76)90409-5
166
Postma B. Poppelier M. J. van Galen J. C. Prossnitz E. R. van Strijp J. A. G. de Haas C. J. et al . (2004). Chemotaxis inhibitory protein of Staphylococcus aureus binds specifically to the C5a and formylated peptide receptor. J. Immunol.172, 6994–7001. 10.4049/jimmunol.172.11.6994
167
Prat C. Bestebroer J. de Haas C. J. van Strijp J. A. van Kessel K. P. (2006). A new staphylococcal anti-inflammatory protein that antagonizes the formyl peptide receptor-like 1. J. Immunol.177, 8017–8026. 10.4049/jimmunol.177.11.8017
168
Prat C. Haas P.-J. Bestebroer J. de Haas C. J. C. van Strijp J. A. G. van Kessel K. P. M. (2009). A homolog of formyl peptide receptor-like 1 (FPRL1) inhibitor from Staphylococcus aureus (FPRL1 inhibitory protein) that inhibits FPRL1 and FPR. J. Immunol.183, 6569–6578. 10.4049/jimmunol.0801523
169
Rainard P. Riollet C. Poutrel B. Paape M. J. (2000). Phagocytosis and killing of Staphylococcus aureus by bovine neutrophils after priming by tumor necrosis factor-alpha and the des-arginine derivative of C5a. Am. J. Vet. Res.61, 951–959. 10.2460/ajvr.2000.61.951
170
Reizner W. Hunter J. G. O'Malley N. T. Southgate R. D. Schwarz E. M. Kates S. L. (2014). A systematic review of animal models for Staphylococcus aureus osteomyelitis. Eur. Cell. Mater.27, 196–212. 10.22203/eCM.v027a15
171
Reyes-Robles T. Torres V. J. (2016). Staphylococcus aureus Pore-Forming Toxins. Curr. Top. Microbiol. Immunol.6, 23–27. 10.1007/82_2016_16
172
Ricklin D. Tzekou A. Garcia B. L. Hammel M. McWhorter W. J. Sfyroera G. et al . (2009). A molecular insight into complement evasion by the staphylococcal complement inhibitor protein family. J. Immunol.183, 2565–2574. 10.4049/jimmunol.0901443
173
Rigby K. M. DeLeo F. R. (2012). Neutrophils in innate host defense against Staphylococcus aureus infections. Semin. Immunopathol.34, 237–259. 10.1007/s00281-011-0295-3
174
Ritzman A. M. Hughes-Hanks J. M. Blaho V. A. Wax L. E. Mitchell W. J. Brown C. R. (2010). The chemokine receptor CXCR2 ligand KC (CXCL1) mediates neutrophil recruitment and is critical for development of experimental lyme arthritis and carditis. Infect. Immun.78, 4593–4600. 10.1128/IAI.00798-10
175
Rogasch K. Rühmling V. Pané-Farré J. Höper D. Weinberg C. Fuchs S. et al . (2006). Influence of the two-component system SaeRS on global gene expression in two different Staphylococcus aureus strains. J. Bacteriol.188, 7742–7758. 10.1128/JB.00555-06
176
Rooijakkers S. H. M. Ruyken M. Roos A. Daha M. R. Presanis J. S. Sim R. B. et al . (2005a). Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat. Immunol.6, 920–927. 10.1038/ni1235
177
Rooijakkers S. H. Ruyken M. van Roon J. van Kessel K. P. van Strijp J. A. van Wamel W. J. (2006). Early expression of SCIN and CHIPS drives instant immune evasion by Staphylococcus aureus. Cell. Microbiol.8, 1282–1293. 10.1111/j.1462-5822.2006.00709.x
178
Rooijakkers S. H. van Wamel W. J. Ruyken M. van Kessel K. P. van Strijp J. A. (2005b). Anti-opsonic properties of staphylokinase. Microbes Infect.7, 476–484. 10.1016/j.micinf.2004.12.014
179
Rutherford S. T. Bassler B. L. (2012). Bacterial quorum sensing: its role in virulence and possibilities for its control. Cold Spring Harb. Perspect. Med.2, 1–26. 10.1101/cshperspect.a012427
180
Sabroe I. Prince L. R. Jones E. C. Horsburgh M. J. Foster S. J. Vogel S. N. et al . (2003). Selective roles for Toll-like receptor (TLR)2 and TLR4 in the regulation of neutrophil activation and life span. J. Immunol.170, 5268–5275. 10.4049/jimmunol.170.10.5268
181
Sasaki T. Kano R. Sato H. Nakamura Y. Watanabe S. Hasegawa A. (2003). Effects of staphylococci on cytokine production from human keratinocytes. Br. J. Dermatol.148, 46–50. 10.1046/j.1365-2133.2003.05017.x
182
Schöneich C. (2005). Methionine oxidation by reactive oxygen species: reaction mechanisms and relevance to Alzheimer's disease. Biochim. Biophys. Acta1703, 111–119. 10.1016/j.bbapap.2004.09.009
183
Seilie E. S. Bubeck Wardenburg J. (2017). Staphylococcus aureus pore-forming toxins: the interface of pathogen and host complexity. Semin. Cell Dev. Biol. [Epub ahead of print]. 10.1016/j.semcdb.2017.04.003
184
Serruto D. Rappuoli R. Scarselli M. Gros P. van Strijp J. A. (2010). Molecular mechanisms of complement evasion: learning from staphylococci and meningococci. Nat. Rev. Microbiol.8, 393–399. 10.1038/nrmicro2366
185
Sieprawska-Lupa M. Mydel P. Krawczyk K. Wójcik K. Puklo M. Lupa B. et al . (2004). Degradation of human antimicrobial peptide LL-37 by Staphylococcus aureus-derived proteinases. Antimicrob. Agents Chemother.48, 4673–4679. 10.1128/AAC.48.12.4673-4679.2004
186
Singer M. Sansonetti P. J. (2004). IL-8 is a key chemokine regulating neutrophil recruitment in a new mouse model of Shigella-induced colitis. J. Immunol.173, 4197–4206. 10.4049/jimmunol.173.6.4197
187
Singh V. K. (2003). Multiple methionine sulfoxide reductase genes in Staphylococcus aureus: expression of activity and roles in tolerance of oxidative stress. Microbiology149, 2739–2747. 10.1099/mic.0.26442-0
188
Singh V. K. Vaish M. Johansson T. R. Baum K. R. Ring R. P. Singh S. et al . (2015). Significance of four methionine sulfoxide reductases in Staphylococcus aureus. PLoS ONE10:e0117594. 10.1371/journal.pone.0117594
189
Sjödahl J. (1977). Structural studies on the four repetitive Fc-binding regions in protein A from Staphylococcus aureus. Eur. J. Biochem.78, 471–490. 10.1111/j.1432-1033.1977.tb11760.x
190
Skjeflo E. W. Christiansen D. Espevik T. Nielsen E. W. Mollnes T. E. (2014). Combined inhibition of complement and CD14 efficiently attenuated the inflammatory response induced by Staphylococcus aureus in a human whole blood model. J. Immunol.192, 2857–2864. 10.4049/jimmunol.1300755
191
Smith E. J. Visai L. Kerrigan S. W. Speziale P. Foster T. J. (2011). The Sbi protein is a multifunctional immune evasion factor of Staphylococcus aureus. Infect. Immun.79, 3801–3809. 10.1128/IAI.05075-11
192
Stadtmann A. Germena G. Block H. Boras M. Rossaint J. Sundd P. et al . (2013). The PSGL-1-L-selectin signaling complex regulates neutrophil adhesion under flow. J. Exp. Med.210, 2171–2180. 10.1084/jem.20130664
193
Stapels D. A. Ramyar K. X. Bischoff M. von Kockritz-Blickwede M. Milder F. J. Ruyken M. et al . (2014). Staphylococcus aureus secretes a unique class of neutrophil serine protease inhibitors. Proc. Natl. Acad. Sci. U.S.A.111, 13187–13192. 10.1073/pnas.1407616111
194
Stemerding A. M. Köhl J. Pandey M. K. Kuipers A. Leusen J. H. Boross P. et al . (2013). Staphylococcus aureus formyl peptide receptor-like 1 inhibitor (FLIPr) and its homologue FLIPr-like are potent FcγR antagonists that inhibit IgG-mediated effector functions. J. Immunol.191, 353–362. 10.4049/jimmunol.1203243
195
Stoll H. Dengjel J. Nerz C. Götz F. (2005). Staphylococcus aureus deficient in lipidation of prelipoproteins is attenuated in growth and immune activation. Infect. Immun.73, 2411–2423. 10.1128/IAI.73.4.2411-2423.2005
196
Su S. B. Gao J. Gong W. Dunlop N. M. Murphy P. M. Oppenheim J. J. et al . (1999a). T21/DP107, A synthetic leucine zipper-like domain of the HIV-1 envelope gp41, attracts and activates human phagocytes by using G-protein-coupled formyl peptide receptors. J. Immunol.162, 5924–5930.
197
Su S. B. Gong W. Gao J. L. Shen W. Murphy P. M. Oppenheim J. J. et al . (1999b). A seven-transmembrane, G protein-coupled receptor, FPRL1, mediates the chemotactic activity of serum amyloid A for human phagocytic cells. J. Exp. Med.189, 395–402. 10.1084/jem.189.2.395
198
Sully E. K. Malachowa N. Elmore B. O. Alexander S. M. Femling J. K. Gray B. M. et al . (2014). Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance. PLoS Pathog.10:e1004174. 10.1371/journal.ppat.1004174
199
Sun F. Li C. Jeong D. Sohn C. He C. Bae T. (2010). In the Staphylococcus aureus two-component system sae, the response regulator SaeR binds to a direct repeat sequence and DNA binding requires phosphorylation by the sensor kinase SaeS. J. Bacteriol.192, 2111–2127. 10.1128/JB.01524-09
200
Swain S. D. Rohn T. T. Quinn M. T. (2002). Neutrophil priming in host defense: role of oxidants as priming agents. Antioxid. Redox Signal.4, 69–83. 10.1089/152308602753625870
201
Takeuchi O. Hoshino K. Akira S. (2000). Cutting Edge: TLR2-Deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J. Immunol.165, 5392–5396. 10.4049/jimmunol.165.10.5392
202
Takeuchi O. Kawai T. Mühlradt P. F. Morr M. Radolf J. D. Zychlinsky A. et al . (2001). Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol.13, 933–940. 10.1093/intimm/13.7.933
203
Takeuchi O. Sato S. Horiuchi T. Hoshino K. Takeda K. Dong Z. et al . (2002). Cutting edge: role of Toll-Like Receptor 1 in mediating immune response to microbial lipoproteins. J. Immunol.169, 10–14. 10.4049/jimmunol.169.1.10
204
Tal-Gan Y. Stacy D. M. Foegen M. K. Koenig D. W. Blackwell H. E. (2013). Highly potent inhibitors of quorum sensing in Staphylococcus aureus revealed through a systematic synthetic study of the group-III autoinducing peptide. J. Am. Chem. Soc.135, 7869–7882. 10.1021/ja3112115
205
Thakker M. Park J. S. Carey V. Lee J. C. (1998). Staphylococcus aureus serotype 5 capsular polysaccharide is antiphagocytic and enhances bacterial virulence in a murine bacteremia model. Infect. Immun.66, 5183–5189.
206
Thammavongsa V. Missiakas D. M. Schneewind O. (2013). Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science342, 863–866. 10.1126/science.1242255
207
Torre A. Bacconi M. Sammicheli C. Galletti B. Laera D. Fontana M. R. et al . (2015). Four-component Staphylococcus aureus vaccine 4C-staph enhances fc?? receptor expression in neutrophils and monocytes and mitigates S. aureus infection in neutropenic mice. Infect. Immun.83, 3157–3163. 10.1128/IAI.00258-15
208
Tseng C. W. Biancotti J. C. Berg B. L. Gate D. Kolar S. L. Müller S. et al . (2015). Increased susceptibility of humanized NSG mice to panton-valentine leukocidin and Staphylococcus aureus skin infection. PLoS Pathog.11, 1–17. 10.1371/journal.ppat.1005292
209
Upadhyay A. Burman J. D. Clark E. A. Leung E. Isenman D. E. Van Den Elsen J. M. et al . (2008). Structure-function analysis of the C3 binding region of Staphylococcus aureus immune subversion protein Sbi. J. Biol. Chem.283, 22113–22120. 10.1074/jbc.M802636200
210
Valderas M. W. Hart M. E. (2001). Identification and characterization of a second superoxide dismutase gene (sodM) from Staphylococcus aureus. J. Bacteriol.183, 3399–3407. 10.1128/JB.183.11.3399-3407.2001
211
van Kesse K. P. Bestebroer J. van Strijp J. A. (2014). Neutrophil-mediated phagocytosis of Staphylococcus aureus. Front. Immunol.5:467. 10.3389/fimmu.2014.00467
212
Vandenesch F. Lina G. Henry T. (2012). Staphylococcus aureus Hemolysins, bi-component Leukocidins, and Cytolytic Peptides: a redundant arsenal of membrane-damaging virulence factors?Front. Cell. Infect. Microbiol.2:12. 10.3389/fcimb.2012.00012
213
Voyich J. M. Braughton K. R. Sturdevant D. E. Whitney A. R. Saïd-Salim B. Porcella S. F. et al . (2005). Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J. Immunol.175, 3907–3919. 10.4049/jimmunol.175.6.3907
214
Voyich J. M. Vuong C. DeWald M. Nygaard T. K. Kocianova S. Griffith S. et al . (2009). The SaeR/S gene regulatory system is essential for innate immune evasion by Staphylococcus aureus. J. Infect. Dis.199, 1698–1706. 10.1086/598967
215
Watkins R. L. Pallister K. B. Voyich J. M. (2011). The SaeR/S gene regulatory system induces a pro-inflammatory cytokine response during Staphylococcus aureus infection. PLoS ONE6:e19939. 10.1371/journal.pone.0019939
216
Watkins R. L. Zurek O. W. Pallister K. B. Voyich J. M. (2013). The SaeR/S two-component system induces interferon-gamma production in neutrophils during invasive Staphylococcus aureus infection. Microbes Infect.15, 749–754. 10.1016/j.micinf.2013.05.004
217
Wertheim H. F. Melles D. C. Vos M. C. van Leeuwen W. van Belkum A. Verbrugh H. A. et al . (2005). The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect. Dis.5, 751–762. 10.1016/S1473-3099(05)70295-4
218
Wieland B. Feil C. Gloria-Maercker E. Thumm G. Lechner M. Bravo J. M. et al . (1994). Genetic and biochemical analyses of the biosynthesis of the yellow carotenoid 4,4'-diaponeurosporene of Staphylococcus aureus. J. Bacteriol.176, 7719–7726. 10.1128/jb.176.24.7719-7726.1994
219
Wilson G. J. Seo K. S. Cartwright R. A. Connelley T. Chuang-Smith O. N. Merriman J. A. et al . (2011). A novel core genome-encoded superantigen contributes to lethality of community-associated MRSA necrotizing pneumonia. PLoS Pathog.7:e1002271. 10.1371/journal.ppat.1002271
220
Wright A. J. Higginbottom A. Philippe D. Upadhyay A. Bagby S. Read R. C. et al . (2007). Characterisation of receptor binding by the chemotaxis inhibitory protein of Staphylococcus aureus and the effects of the host immune response. Mol. Immunol.44, 2507–2517. 10.1016/j.molimm.2006.12.022
221
Wright H. L. Thomas H. B. Moots R. J. Edwards S. W. (2013). RNA-Seq reveals activation of both common and cytokine-specific pathways following neutrophil priming. PLoS ONE8:e58598. 10.1371/journal.pone.0058598
222
Xu S. Arbeit R. D. Lee J. C. (1992). Phagocytic killing of encapsulated and microencapsulated Staphylococcus aureus by human polymorphonuclear leukocytes. Infect. Immun.60, 1358–1362.
223
Yang S. J. Bayer A. S. Mishra N. N. Meehl M. Ledala N. Yeaman M. R. et al . (2012). The Staphylococcus aureus two-component regulatory system, grars, senses and confers resistance to selected cationic antimicrobial peptides. Infect. Immun.80, 74–81. 10.1128/IAI.05669-11
224
Yokoyama R. Itoh S. Kamoshida G. Takii T. Fujii S. Tsuji T. et al . (2012). Staphylococcal superantigen-like protein 3 binds to the toll-like receptor 2 extracellular domain and inhibits cytokine production induced by Staphylococcus aureus, cell wall component, or lipopeptides in murine macrophages. Infect. Immun.80, 2816–2825. 10.1128/IAI.00399-12
225
Yoshimura T. Matsushima K. Tanaka S. Robinson E. A. Appella E. Oppenheim J. J. et al . (1987). Purification of a human monocyte-derived neutrophil chemotactic factor that has peptide sequence similarity to other host defense cytokines. Proc. Natl. Acad. Sci. U.S.A.84, 9233–9237. 10.1073/pnas.84.24.9233
226
Zhang L. Jacobsson K. Strom K. Lindberg M. Frykberg L. (1999). Staphylococcus aureus expresses a cell surface protein that binds both IgG and beta2-glycoproteinI. Microbiology145(Pt 1), 177–183. 10.1099/13500872-145-1-177
227
Zhang L. Jacobsson K. Vasi J. Lindberg M. Frykberg L. (1998). A second IgG-binding protein in Staphylococcus aureus. Microbiology144(Pt 4), 985–991. 10.1099/00221287-144-4-985
228
Zurek O. W. Nygaard T. K. Watkins R. L. Pallister K. B. Torres V. J. Horswill A. R. et al . (2014). The role of innate immunity in promoting SaeR/S-Mediated Virulence in Staphylococcus aureus. J. Innate Immun.6, 21–30. 10.1159/000351200
229
Zurek O. W. Pallister K. B. Voyich J. M. (2015). Staphylococcus aureus Inhibits Neutrophil-derived IL-8 to promote cell death. J. Infect. Dis.212, 1–5. 10.1093/infdis/jiv124
Summary
Keywords
host-pathogen interactions, innate immunity, immune evasion, host defense, phagocytosis, chemotaxis, Staphylococcus aureus
Citation
Guerra FE, Borgogna TR, Patel DM, Sward EW and Voyich JM (2017) Epic Immune Battles of History: Neutrophils vs. Staphylococcus aureus. Front. Cell. Infect. Microbiol. 7:286. doi: 10.3389/fcimb.2017.00286
Received
30 March 2017
Accepted
12 June 2017
Published
30 June 2017
Volume
7 - 2017
Edited by
Lee-Ann H. Allen, University of Iowa, United States
Reviewed by
Francis Alonzo, Loyola University Chicago, United States; Alison Criss, University of Virginia, United States
Updates
Copyright
© 2017 Guerra, Borgogna, Patel, Sward and Voyich.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jovanka M. Voyich jovanka@montana.edu
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.