 Leili Jia
Leili Jia Jing Xie1†
Jing Xie1† Ligui Wang
Ligui Wang- 1Institute of Disease Control and Prevention of Chinese People's Liberation Army, Beijing, China
- 2Center for Disease Control and Prevention of Chinese People's Armed Police Forces, Beijing, China
- 3State Key Laboratory for Infectious Disease Prevention and Control, National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, China
Influenza virus infection remains one of the largest disease burdens on humans. Influenza-associated bacterial co-infections contribute to severe disease and mortality during pandemic and seasonal influenza episodes. The mechanisms of severe morbidity following influenza-bacteria co-infections mainly include failure of an antibacterial immune response and pathogen synergy. Moreover, failure to resume function and tolerance might be one of the main reasons for excessive mortality. In this review, recent advances in the study of mechanisms of severe disease, caused by bacterial co-infections following influenza virus pathogenesis, are summarized. Therefore, understanding the synergy between viruses and bacteria will facilitate the design of novel therapeutic approaches to prevent mortality associated with bacterial co-infections.
Introduction
Influenza viruses are responsible for an average of 400,000 deaths per year globally (Simonsen et al., 1997; Bakaletz, 2004; King et al., 2017; Tansey, 2017). During previous influenza pandemics (H1N1, 1918; H2N2, 1957; H3N2, 1968; H1N1, 2009) and seasonal epidemics, many influenza-related deaths actually occurred due to bacterial co-infections (Guarner et al., 2006; Taubenberger and Morens, 2008; Weiser, 2010; Klein et al., 2016; McDanel et al., 2016; Shah et al., 2016). Since the 1950s, researchers have increasingly focused on concomitant infections with influenza viruses and a range of bacterial agents (Table 1).
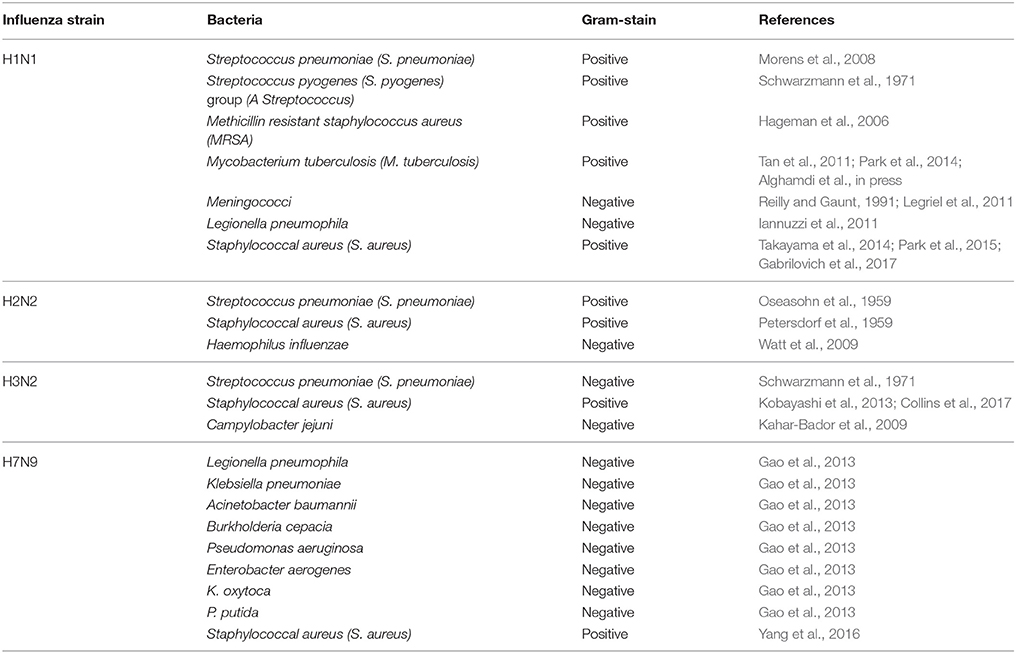
Table 1. Bacterial co-infection with influenza.
Mortality incidence can be affected by several factors, one of which being the order of co-infections. Although it is difficult to distinguish the order in which bacterial and influenza infection occurs in a clinical setting, laboratory data have shown that mortality is associated with this sequence. Specifically, mortality incidence peaks when bacterial infections occur 3–7 days after an established influenza infection (Jamieson et al., 2013). In this review, we mainly address the mechanisms of severe morbidity and mortality associated with bacterial co-infections following influenza infection.
Influenza Infection Increases Host Susceptibility to Bacteria
In both humans and mice, influenza virus titers in the lung reach a peak 3–5 days after primary infection is established. Thereafter, the virus clearance begins, with almost complete resolution of infection between 10 and 12 days (Metzger and Sun, 2013). In general, the influenza virus preferentially replicates in epithelial cells and induces the most lung tissue damage at approximately day 6 (Nugent and Pesanti, 1983). This pathology is partly responsible for the observed increase in susceptibility to opportunistic bacterial pathogens, as epithelial cell damage and increased receptor availability enable invading bacteria to adhere and grow.
Influenza Affects the Antibacterial Innate Immune Response
Typically, the respiratory tract immune system is strictly controlled to prevent inflammation in response to innocuous antigens or commensal bacteria. When harmful pathogens colonize the respiratory tract, the local immune system becomes activated to eliminate the threat. It was found that typical mice can effectively clear up to 105 pneumococci within 4–12 h (Sun and Metzger, 2008). However, with influenza infection onset, several processes occur that might impact the antibacterial innate immune response, rendering both the upper airways and lungs susceptible to subsequent bacterial infiltration, leading to increased bacterial load and mortality (Hillyer et al., 2004; Ishikawa et al., 2016). These processes include inhibition by type I interferons (IFNs) and depletion of alveolar macrophages.
Influenza-Induced Type I IFNs Might Interfere with Antibacterial Responses
The influenza non-structural protein 1 (NS1) is produced by infected cells and can modulate innate immune pathways including IFN signaling (Hale et al., 2008; Bucasas et al., 2013). Besides playing a central role in the host antiviral response (Theofilopoulos et al., 2005), type I IFNs can also disrupt lung immune responses to bacteria (Kukavica-Ibrulj et al., 2009; Techasaensiri et al., 2010; Kimaro et al., 2013; Lee et al., 2015).
In general, all cells are equipped with specific receptors, known as pattern-recognition receptors, to detect the presence of pathogens such as viruses and bacteria. The Toll-like receptor family comprises this class of receptors and includes receptors to viral and bacterial products. Type I IFNs are produced following the recognition of influenza nucleic acids by these receptors (Tian et al., 2012), which probably functions by suppressing the normal phagocytic activity and early innate responses of macrophages and neutrophils, which would normally help to clear the bacteria from the lungs (Sun and Metzger, 2008; Shahangian et al., 2009).
Furthermore, type I IFNs can inhibit Type 17 T cells (Kudva et al., 2011; Nakamura et al., 2011), which play an important role in clearance of pulmonary pathogenic bacteria. The immunity of Type 17 T cells depends on IL-17, IL-22, and IL-23. However, type I IFNs can decrease these cytokines. Meanwhile, type I IFNs can decrease the production CCL2, which is required for macrophage recruitment (Nakamura et al., 2011). Interestingly, the antibacterial innate immune response is recovered until type I IFN levels return to baseline (Lee et al., 2015).
Influenza Viruses Deplete Alveolar Macrophages
Alveolar macrophages are vital for the first cellular line of defense against inhaled antigens, and account for >90% of all cells in the bronchoalveolar lavage fluid of uninfected respiratory tissue (Vermaelen and Pauwels, 2004). However, airway-resident alveolar macrophages are specifically targeted by influenza viruses during the primary stages of infection (Ghoneim et al., 2013). These depleted alveolar macrophages can be replaced over next 2 weeks by the proliferation and differentiation of macrophages of other classes. Therefore, there is a window of primary susceptibility to bacterial infection (Douek et al., 2009). For example, pneumococcal colonization density increases in mice 1 week after inoculation of influenza due to the absence of macrophages, which are necessary to clear the infection upon single-agent inoculation (Zhang et al., 2009). Moreover, influenza infection can inhibit G-CSF secretion; this decrease in G-CSF might reduce myeloperoxidase activity. Interestingly, the digestion ability of phagocytized bacteria of Neutrophils dependents on the activity of myeloperoxidase (Anderson et al., 1985; Ishikawa et al., 2016).
Influenza Viruses Help to Provide More Binding Receptors and Sites for Bacteria
After influenza infection is established, most viral subtypes replicate in the mucosal epithelial cells of the upper respiratory tract, providing more receptors for bacteria (Hatta et al., 2007; Nakamura et al., 2011). However, some viral subtypes can target both upper and lower respiratory tract tissues (Shinya et al., 2006; Childs et al., 2009; Maines et al., 2009; Munster et al., 2009). In particular, when viral infection precedes the presence of bacteria, access to otherwise inaccessible receptors in the lower respiratory tract might be available to invading bacteria (McCullers and Bartmess, 2003). Here, we consider three main mechanisms associated with this phenomenon.
First, influenza virus proteins can contribute. Neuraminidase, present on the envelope of the influenza virus, is responsible for sialidase activity, required by the virus for budding. After neuraminidase cleaves sialic acid from the termini of glycochains, cryptic receptors on host cells become exposed, and bacteria such as pneumococci can adhere (Foster and Hook, 1998; McCullers and Tuomanen, 2001). Furthermore, disrupted sialylated mucins can provide decoy receptors for bacteria (Plotkowski et al., 1986, 1993). For example, substantial numbers of epithelial cells can be destroyed by virulent viruses such as the mouse-adapted influenza strain PR8, resulting in exposed sites for bacteria to attach in the tracheobronchial tree (Plotkowski et al., 1986; McCullers and Rehg, 2002). Interestingly, neuraminidase proteins are not restricted to viruses. Some bacteria like Streptococcus pneumoniae also provide neuraminidases to access receptors and inhibit host defenses by cleaving sialic acids from protective mucins, allowing efficient infection of host lungs (Camara et al., 1991).
Second, the host inflammatory response to influenza infections can provide additional receptors. The host inflammatory response can alter not only the regulatory state, but also the surface display of multiple proteins, to facilitate pneumococcal invasion (Cundell and Tuomanen, 1994; Miller et al., 2007).
Third, adherence sites might also be provided during wound recovery in the airways (Plotkowski et al., 1993; de Bentzmann et al., 1996; Martin and Leibovich, 2005). There are some differences in terms of the wound recovery between common infection and co-infection. During co-infection with complex pathogens, reduced damage tolerance will occur; however, as a result, repair efficiency will decrease compared to that with a typical infection. This is also one of the causes of increased mortality with co-infection. Although some progress has been made, many potential mechanisms still need to be elucidated. Apical receptors including asialylated glycans (for example, GalNacβ1-Gal) or α5β1 integrins can be expressed on the surfaces of injured cells or those in an intermediate state of differentiation. Bacteria such as Staphylococcus aureus or Pseudomonas aeruginosa can adhere to receptors (Puchelle et al., 2006). Furthermore, bacteria such as S. pneumoniae, Haemophilus influenzae, or S. aureus can bind to exposed areas of incomplete healing via traditional adhesins. These exposed areas can be covered by basement membrane elements such as fibrin and fibrinogen deposition, laminin, or type I and IV collagen. This phenomenon has been observed in the clinics; patients can be easily infected by bacteria while recovering from primary illness (Louria et al., 1959; Peteranderl et al., 2017). In addition, many bacterial virulence factors can attach to elements of the extracellular matrix or basement membrane (Foster and Hook, 1998; McCullers and Tuomanen, 2001).
Bacterial Co-infections Following Influenza Infection Results in Morbidity and Mortality
Influenza Causes Substantial Lung Epithelial Damage
The viral cytotoxin PB1-F2, not present on all subtypes of influenza, is a non-structural protein encoded by an alternative reading frame on genomic segment 2 of influenza A. PB1-F2 is encoded by a small, variable open reading frame in PB1 that exists in most influenza viruses. This protein is capable of activating AP-1 transcription factors via ERK1/2 kinase, and was confirmed to be a determinant of virulence. The length of PB1-F2 differs according to subtype; full-length PB1-F2 is 90 aa (amino acids). However, that of the A/Puerto Rico/8/24 strain was found to be 87 aa. Most avian influenza viruses have complete PB1-F2-encoding genes. After 1947, PB1-F2 of the H1N1 subtype was found to be cleaved at position 57, and that of the classical swine H1N1 subtype was determined to be cleaved at 11, 25, and 34 aa, which results in reduced viral pathogenicity. Since 1968, variations in the PB1-encoding gene of the H3N2 subtype gradually stabilized, and PB1-F2 truncation gradually represented a novel evolutionary feature. The 2009 H1N1 strain did not show any significant antigenic and pathogenic variation. Patient infection after obvious gastrointestinal symptoms is not associated with viral virulence, but might be caused by individual-specific properties. When viral infection is established, PB1-F2 can induce apoptosis, mediated by mitochondrial permeabilization (McAuley et al., 2007, 2010; Alymova et al., 2011; Leymarie et al., 2013), ultimately providing nutrients to invading opportunistic bacteria, following cytopathic damage and disruption of surfactant in the lungs. Consequently, inhaled or commensal bacteria can become overgrown, adversely affecting host survival (Loosli et al., 1975). In 2016, observations (Sun et al., 2016) showed that effective antibiotic treatment of clinical post-influenza bacteria depends on nicotinamide adenine dinucleotide phosphate oxidase 2 (Nox2), and that a balance exists between Nox2-dependent antibacterial immunity and inflammation. However, influenza infection might disrupt this balance and increase susceptibility to bacterial infection.
Synergism during Influenza/Bacterial Co-infections
Both influenza and bacteria contribute to the immunopathogenicity of co-infection. For example, expression of PB1-F2 has been associated with excessive inflammatory responses, which can lead to increased cellular infiltration of the lungs and airways, together with a cytokine storm (Conenello et al., 2007; McAuley et al., 2007, 2010). Interestingly, it was proposed that nascent hemagglutinin can be cleaved, from its primary state to a fusion-active complex, by bacterial proteases from S. aureus (Tashiro et al., 1987), which might increase influenza viral titers and spread.
Likewise, bacterial cytotoxins such as pneumolysin and Panton-Valentine leukocidin (PVL), can also contribute to immunopathogenicity (Rogolsky, 1979; Tuomanen et al., 1995; Loffler et al., 2013; Wolf et al., 2014). Bacterial components that lead to exacerbated cell death, associated with pore formation or enhanced inflammatory signaling, might synergize with influenza virulence factors (Boulnois et al., 1991). In addition, multiple innate immune mechanisms that involve pathogen recognition receptors also generate inflammatory responses to influenza and/or bacteria (Koppe et al., 2012; Ramos and Fernandez-Sesma, 2012). These inflammatory pathways work together, leading to synergistic activation of the immune system and increased mortality (Joyce et al., 2009; Bucasas et al., 2013; Kimaro et al., 2013).
Moreover, certain pathogenic virulence factors are only evident with the presence of a co-pathogen, as is the case for a mouse model of influenza and H. influenzae co-infection (Wong et al., 2013). However, it has not been possible to identify synergistic pathogenicity genes that facilitate co-infections using traditional virulence screens during single-agent infections (Bellinghausen et al., 2016).
Influenza Compromises Host Tolerance
The concept of tolerance used here is different from “immunological tolerance,” which is described as a state of unresponsiveness to self-antigens. Tolerance, in the current context, can reduce the negative impact of pathogen outgrowth and immunopathology, but can be compromised by the influenza virus, resulting in mortality even in the context of effective resistance.
For example, recent observations (Jamieson et al., 2013) demonstrated that influenza can compromise tolerance to tissue damage and result in mortality when mice were co-infected with Legionella pneumophila. It was shown that lethal synergy can be independent of impaired resistance to either influenza or L. pneumophila. Notably, this is different from previously discussed co-infections with influenza and opportunistic bacterial pathogens. Moreover, during the 1918 influenza outbreak, it was observed that human mortality was not directly related to infection rates (Shanks and Brundage, 2012). This study also demonstrated that lethal synergy between the influenza and L. pneumophila is unlikely to be due to failed immune resistance to either agent (Mendel et al., 1998).
Discussion
The mechanisms of enhanced mortality following influenza-associated bacterial co-infections not only include failed antibacterial resistance and synergistic immunopathogenicity, but also failed tolerance. As a relatively new concept in animal immunity, tolerance has been largely overlooked and deserves further consideration regarding its effect on co-infection (Medzhitov et al., 2012).
Upon infection by a virus and/or bacterium, the host can protect itself through two distinct strategies, resistance and tolerance. Resistance is based on pathogen detection and elimination, whereas tolerance can restrict harm caused by the pathogen burden. However, virus/bacterial co-infections results in some difficulty in the treatment of either infection. For example, it is unknown whether antiviral treatments that viral load have any effect on concurrent bacterial infection (McCullers, 2004; Naguib et al., 2017). Similarly, the efficacy of treatments to counteract host inflammatory responses to bacterial co-infection is unknown (Kudva et al., 2011; Arduin et al., 2017). Therefore, distinguishing between failed resistance and failed tolerance might be of vital importance for the selection of therapeutic approaches to treat the primary problem.
Considering that mortality during co-infection can be decreased when bacterial infection occurs before influenza challenge, albeit through unknown mechanisms (McCullers and Rehg, 2002; Wang et al., 2013), it is possible to develop novel recombinant vaccines that include both influenza and bacterial antigens. Ideally, this vaccine would elicit cross-reactive antibody responses to both influenza and bacteria. Furthermore, such vaccines would represent an appealing alternative to classical inactivated vaccines.
Therefore, understanding the mechanisms involved in the synergy between viral and bacterial co-infection will facilitate the design of novel therapeutic approaches for the prevention of elevated mortality associated with bacterial co-infections following influenza infection (Boianelli et al., 2016; McDonald et al., 2017; Smith, 2017).
Author Contributions
All author participated in developing the hypothesis and collaborated in writing and reviewing of the article.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Qiao Li for reviewing the manuscript and advice. This work was supported by grants from the National Key Research and Development Program of China (Nos. 2016YFC1200905, 2016YFC1200700) and National Natural Science Foundation of China (No. 20873999).
References
Alghamdi, A. N., Mahfouz, M. E., Hamdi, F. A., Al Aboud, A. D., Al-Laylah, T. Z., Alotaibi, M. I., et al. (in press). Sequence analysis of haemagglutinin neuraminidase of H1N1 strain from a patient coinfected with Mycobacterium tuberculosis. Mol. Cell. Probes. doi: 10.1016/j.mcp.2017.05.002. Available online at: http://www.sciencedirect.com/science/article/pii/S0890850817300427?via%3Dihub
Alymova, I. V., Green, A. M., van de Velde, N., McAuley, J. L., Boyd, K. L., Ghoneim, H. E., et al. (2011). Immunopathogenic and antibacterial effects of H3N2 influenza A virus PB1-F2 map to amino acid residues 62, 75, 79, and 82. J. Virol. 85, 12324–12333. doi: 10.1128/JVI.05872-11
Anderson, N. V., DeBowes, R. M., Nyrop, K. A., and Dayton, A. D. (1985). Mononuclear phagocytes of transport-stressed horses with viral respiratory tract infection. Am. J. Vet. Res. 46, 2272–2277.
Arduin, H., Domenech, D. C. M., Guillemot, D., Watier, L., and Opatowski, L. (2017). An agent-based model simulation of influenza interactions at the host level: insight into the influenza-related burden of pneumococcal infections. BMC Infect. Dis. 17:382. doi: 10.1186/s12879-017-2464-z
Bakaletz, L. O. (2004). Developing animal models for polymicrobial diseases. Nat. Rev. Microbiol. 2, 552–568. doi: 10.1038/nrmicro928
Bellinghausen, C., Gulraiz, F., Heinzmann, A. C., Dentener, M. A., Savelkoul, P. H., Wouters, E. F., et al. (2016). Exposure to common respiratory bacteria alters the airway epithelial response to subsequent viral infection. Respir. Res. 17:68. doi: 10.1186/s12931-016-0382-z
Boianelli, A., Sharma-Chawla, N., Bruder, D., and Hernandez-Vargas, E. A. (2016). Oseltamivir PK/PD Modeling and simulation to evaluate treatment strategies against influenza-pneumococcus coinfection. Front. Cell. Infect. Microbiol. 6:60. doi: 10.3389/fcimb.2016.00060
Boulnois, G. J., Paton, J. C., Mitchell, T. J., and Andrew, P. W. (1991). Structure and function of pneumolysin, the multifunctional, thiol-activated toxin of Streptococcus pneumoniae. Mol. Microbiol. 5, 2611–2616. doi: 10.1111/j.1365-2958.1991.tb01969.x
Bucasas, K. L., Mian, A. I., Demmler-Harrison, G. J., Caviness, A. C., Piedra, P. A., Franco, L. M., et al. (2013). Global gene expression profiling in infants with acute respiratory syncytial virus broncholitis demonstrates systemic activation of interferon signaling networks. Pediatr. Infect. Dis. J. 32, e68–e76. doi: 10.1097/inf.0b013e318278b4b3
Camara, M., Mitchell, T. J., Andrew, P. W., and Boulnois, G. J. (1991). Streptococcus pneumoniae produces at least two distinct enzymes with neuraminidase activity: cloning and expression of a second neuraminidase gene in Escherichia coli. Infect. Immun. 59, 2856–2858.
Childs, R. A., Palma, A. S., Wharton, S., Matrosovich, T., Liu, Y., Chai, W., et al. (2009). Receptor-binding specificity of pandemic influenza A (H1N1) 2009 virus determined by carbohydrate microarray. Nat. Biotechnol. 27, 797–799. doi: 10.1038/nbt0909-797
Collins, L. F., Anderson, B. D., and Gray, G. C. (2017). A case of influenza A (H3N2) complicated by community-acquired pneumonia and death in a young healthy adult during the 2013-2014 Season. Front. Public Health 5:1. doi: 10.3389/fpubh.2017.00001
Conenello, G. M., Zamarin, D., Perrone, L. A., Tumpey, T., and Palese, P. (2007). A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 3, 1414–1421. doi: 10.1371/journal.ppat.0030141
Cundell, D. R., and Tuomanen, E. I. (1994). Receptor specificity of adherence of Streptococcus pneumoniae to human type-II pneumocytes and vascular endothelial cells in vitro. Microb. Pathog. 17, 361–374. doi: 10.1006/mpat.1994.1082
de Bentzmann, S., Plotkowski, C., and Puchelle, E. (1996). Receptors in the Pseudomonas aeruginosa adherence to injured and repairing airway epithelium. Am. J. Respir. Crit. Care Med. 154, S155–S162. doi: 10.1164/ajrccm/154.4
Douek, D. C., Roederer, M., and Koup, R. A. (2009). Emerging concepts in the immunopathogenesis of AIDS. Annu. Rev. Med. 60, 471–484. doi: 10.1146/annurev.med.60.041807.123549
Foster, T. J., and Hook, M. (1998). Surface protein adhesins of Staphylococcus aureus. Trends Microbiol. 6, 484–488. doi: 10.1016/S0966-842X(98)01400-0
Gabrilovich, M. I., Huff, M. D., McMillen, S. M., and Quinter, C. (2017). Severe necrotizing tracheobronchitis from panton-valentine leukocidin-positive MRSA pneumonia complicating influenza A-H1N1-09. J. Bronchology. Interv. Pulmonol. 24, 63–66. doi: 10.1097/LBR.0000000000000314
Gao, H. N., Lu, H. Z., Cao, B., Du, B., Shang, H., Gan, J. H., et al. (2013). Clinical findings in 111 cases of influenza A (H7N9) virus infection. N. Engl. J. Med. 368, 2277–2285. doi: 10.1056/NEJMoa1305584
Ghoneim, H. E., Thomas, P. G., and McCullers, J. A. (2013). Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J. Immunol. 191, 1250–1259. doi: 10.4049/jimmunol.1300014
Guarner, J., Paddock, C. D., Shieh, W. J., Packard, M. M., Patel, M., Montague, J. L., et al. (2006). Histopathologic and immunohistochemical features of fatal influenza virus infection in children during the 2003-2004 season. Clin. Infect. Dis. 43, 132–140. doi: 10.1086/505122
Hageman, J. C., Uyeki, T. M., Francis, J. S., Jernigan, D. B., Wheeler, J. G., Bridges, C. B., et al. (2006). Severe community-acquired pneumonia due to Staphylococcus aureus, 2003-04 influenza season. Emerg. Infect. Dis. 12, 894–899. doi: 10.3201/eid1206.051141
Hale, B. G., Randall, R. E., Ortin, J., and Jackson, D. (2008). The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 89, 2359–2376. doi: 10.1099/vir.0.2008/004606-0
Hatta, M., Hatta, Y., Kim, J. H., Watanabe, S., Shinya, K., Nguyen, T., et al. (2007). Growth of H5N1 influenza A viruses in the upper respiratory tracts of mice. PLoS Pathog. 3, 1374–1379. doi: 10.1371/journal.ppat.0030133
Hillyer, J. F., Schmidt, S. L., and Christensen, B. M. (2004). The antibacterial innate immune response by the mosquito Aedes aegypti is mediated by hemocytes and independent of Gram type and pathogenicity. Microb. Infect. 6, 448–459. doi: 10.1016/j.micinf.2004.01.005
Iannuzzi, M., De Robertis, E., Piazza, O., Rispoli, F., Servillo, G., and Tufano, R. (2011). Respiratory failure presenting in H1N1 influenza with Legionnaires disease: two case reports. J. Med. Case Rep. 5:520. doi: 10.1186/1752-1947-5-520
Ishikawa, H., Fukui, T., Ino, S., Sasaki, H., Awano, N., Kohda, C., et al. (2016). Influenza virus infection causes neutrophil dysfunction through reduced G-CSF production and an increased risk of secondary bacteria infection in the lung. Virology 499, 23–29. doi: 10.1016/j.virol.2016.08.025
Jamieson, A. M., Pasman, L., Yu, S., Gamradt, P., Homer, R. J., Decker, T., et al. (2013). Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science 340, 1230–1234. doi: 10.1126/science.1233632
Joyce, E. A., Popper, S. J., and Falkow, S. (2009). Streptococcus pneumoniae nasopharyngeal colonization induces type I interferons and interferon-induced gene expression. BMC Genomics 10:404. doi: 10.1186/1471-2164-10-404
Kahar-Bador, M., Nathan, A. M., Soo, M. H., Mohd, N. S., AbuBakar, S., Lum, L. C., et al. (2009). Fatal influenza A (H3N2) and Campylobacter jejuni coinfection. Singapore Med. J. 50, e112–e113. Available online at: http://smj.sma.org.sg/5003/5003cr9.pdf
Kimaro, M. S., Peret, T. C., Kumar, N., Romero-Steiner, S., Dunning, H. J., Ishmael, N., et al. (2013). Transcriptional adaptation of pneumococci and human pharyngeal cells in the presence of a virus infection. BMC Genomics 14:378. doi: 10.1186/1471-2164-14-378
King, J. C., Schweinle, J. E., Hatchett, R. J., Gao, Y., Lichenstein, R., and Zhou, J. (2017). Surges of advanced medical support associated with influenza outbreaks. Epidemiol. Infect. 145, 2409–2416. doi: 10.1017/S095026881700111X
Klein, E. Y., Monteforte, B., Gupta, A., Jiang, W., May, L., Hsieh, Y. H., et al. (2016). The frequency of influenza and bacterial coinfection: a systematic review and meta-analysis. Influenza Respir. Viruses 10, 394–403. doi: 10.1111/irv.12398
Kobayashi, S. D., Olsen, R. J., LaCasse, R. A., Safronetz, D., Ashraf, M., Porter, A. R., et al. (2013). Seasonal H3N2 influenza A virus fails to enhance Staphylococcus aureus co-infection in a non-human primate respiratory tract infection model. Virulence 4, 707–715. doi: 10.4161/viru.26572
Koppe, U., Suttorp, N., and Opitz, B. (2012). Recognition of Streptococcus pneumoniae by the innate immune system. Cell. Microbiol. 14, 460–466. doi: 10.1111/j.1462-5822.2011.01746.x
Kudva, A., Scheller, E. V., Robinson, K. M., Crowe, C. R., Choi, S. M., Slight, S. R., et al. (2011). Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J. Immunol. 186, 1666–1674. doi: 10.4049/jimmunol.1002194
Kukavica-Ibrulj, I., Hamelin, M. E., Prince, G. A., Gagnon, C., Bergeron, Y., Bergeron, M. G., et al. (2009). Infection with human metapneumovirus predisposes mice to severe pneumococcal pneumonia. J. Virol. 83, 1341–1349. doi: 10.1128/JVI.01123-08
Lee, B., Robinson, K. M., McHugh, K. J., Scheller, E. V., Mandalapu, S., Chen, C., et al. (2015). Influenza-induced type I interferon enhances susceptibility to gram-negative and gram-positive bacterial pneumonia in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, L158–L167. doi: 10.1152/ajplung.00338.2014
Legriel, S., Merceron, S., Tattevin, P., Mouvier, M. A., Marque-Juillet, S., Le Monnier, A., et al. (2011). Favorable outcome after life-threatening meningococcal disease complicating influenza A(H1N1) infection. Infection 39, 477–480. doi: 10.1007/s15010-011-0134-4
Leymarie, O., Jouvion, G., Herve, P. L., Chevalier, C., Lorin, V., Lecardonnel, J., et al. (2013). Kinetic characterization of PB1-F2-mediated immunopathology during highly pathogenic avian H5N1 influenza virus infection. PLoS ONE 8:e57894. doi: 10.1371/journal.pone.0057894
Loffler, B., Niemann, S., Ehrhardt, C., Horn, D., Lanckohr, C., Lina, G., et al. (2013). Pathogenesis of Staphylococcus aureus necrotizing pneumonia: the role of PVL and an influenza coinfection. Expert Rev. Anti. Infect. Ther. 11, 1041–1051. doi: 10.1586/14787210.2013.827891
Loosli, C. G., Stinson, S. F., Ryan, D. P., Hertweck, M. S., Hardy, J. D., and Serebrin, R. (1975). The destruction of type 2 pneumocytes by airborne influenza PR8-A virus; its effect on surfactant and lecithin content of the pneumonic lesions of mice. Chest 67, 7S–14S. doi: 10.1378/chest.67.2
Louria, D. B., Blumenfeld, H. L., Ellis, J. T., Kilbourne, E. D., and Rogers, D. E. (1959). Studies on influenza in the pandemic of 1957-1958. II. Pulmonary complications of influenza. J. Clin. Invest. 38, 213–265. doi: 10.1172/JCI103791
Maines, T. R., Jayaraman, A., Belser, J. A., Wadford, D. A., Pappas, C., Zeng, H., et al. (2009). Transmission and pathogenesis of swine-origin 2009 A(H1N1) influenza viruses in ferrets and mice. Science 325, 484–487. doi: 10.1126/science.1177238
Martin, P., and Leibovich, S. J. (2005). Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol. 15, 599–607. doi: 10.1016/j.tcb.2005.09.002
McAuley, J. L., Hornung, F., Boyd, K. L., Smith, A. M., McKeon, R., Bennink, J., et al. (2007). Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microb. 2, 240–249. doi: 10.1016/j.chom.2007.09.001
McAuley, J. L., Chipuk, J. E., Boyd, K. L., Van De Velde, N., Green, D. R., and McCullers, J. A. (2010). PB1-F2 proteins from H5N1 and 20 century pandemic influenza viruses cause immunopathology. PLoS Pathog. 6:e1001014. doi: 10.1371/journal.ppat.1001014
McCullers, J. A., and Bartmess, K. C. (2003). Role of neuraminidase in lethal synergism between influenza virus and Streptococcus pneumoniae. J. Infect. Dis. 187, 1000–1009. doi: 10.1086/368163
McCullers, J. A., and Rehg, J. E. (2002). Lethal synergism between influenza virus and Streptococcus pneumoniae: characterization of a mouse model and the role of platelet-activating factor receptor. J. Infect. Dis. 186, 341–350. doi: 10.1086/341462
McCullers, J. A., and Tuomanen, E. I. (2001). Molecular pathogenesis of pneumococcal pneumonia. Front. Bio Sci. 6, D877–D889. doi: 10.2741/A649
McCullers, J. A. (2004). Effect of antiviral treatment on the outcome of secondary bacterial pneumonia after influenza. J. Infect. Dis. 190, 519–526. doi: 10.1086/421525
McDanel, J. S., Perencevich, E. N., Storm, J., Diekema, D. J., Herwaldt, L., Johnson, J. K., et al. (2016). Increased mortality rates associated with Staphylococcus aureus and influenza co-infection, Maryland and Iowa, USA(1). Emerg. Infect. Dis. 22, 1253–1256. doi: 10.3201/eid2207.151319
McDonald, H. I., Thomas, S. L., Millett, E., Quint, J., and Nitsch, D. (2017). Do influenza and pneumococcal vaccines prevent community-acquired respiratory infections among older people with diabetes and does this vary by chronic kidney disease? A cohort study using electronic health records. BMJ Open Diab. Res. Care 5:e000332. doi: 10.1136/bmjdrc-2016-000332
Medzhitov, R., Schneider, D. S., and Soares, M. P. (2012). Disease tolerance as a defense strategy. Science 335, 936–941. doi: 10.1126/science.1214935
Mendel, D. B., Tai, C. Y., Escarpe, P. A., Li, W., Sidwell, R. W., Huffman, J. H., et al. (1998). Oral administration of a prodrug of the influenza virus neuraminidase inhibitor GS 4071 protects mice and ferrets against influenza infection. Antimicrob. Agents Chemother. 42, 640–646.
Metzger, D. W., and Sun, K. (2013). Immune dysfunction and bacterial coinfections following influenza. J. Immunol. 191, 2047–2052. doi: 10.4049/jimmunol.1301152
Miller, M. L., Gao, G., Pestina, T., Persons, D., and Tuomanen, E. (2007). Hypersusceptibility to invasive pneumococcal infection in experimental sickle cell disease involves platelet-activating factor receptor. J. Infect. Dis. 195, 581–584. doi: 10.1086/510626
Morens, D. M., Taubenberger, J. K., and Fauci, A. S. (2008). Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J. Infect. Dis. 198, 962–970. doi: 10.1086/591708
Munster, V. J., de Wit, E., van den Brand, J. M., Herfst, S., Schrauwen, E. J., Bestebroer, T. M., et al. (2009). Pathogenesis and transmission of swine-origin 2009 A(H1N1) influenza virus in ferrets. Science 325, 481–483. doi: 10.1126/science.1177127
Naguib, M. M., Grund, C., Arafa, A. S., Abdelwhab, E. M., Beer, M., and Harder, T. C. (2017). Heterologous post-infection immunity against Egyptian avian influenza virus (AIV) H9N2 modulates the course of subsequent infection by highly pathogenic AIV H5N1, but vaccination immunity does not. J. Gen. Virol. 98, 1169–1173. doi: 10.1099/jgv.0.000767
Nakamura, S., Davis, K. M., and Weiser, J. N. (2011). Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J. Clin. Invest. 121, 3657–3665. doi: 10.1172/JCI57762
Nugent, K. M., and Pesanti, E. L. (1983). Tracheal function during influenza infections. Infect. Immun. 42, 1102–1108.
Oseasohn, R., Adelson, L., and kaji, M. D. (1959). Clinicopathologic study of thirty-three fatal cases of Asian influenza. N. Engl. J. Med. 260, 509–518. doi: 10.1056/NEJM195903122601101
Park, Y., Chin, B. S., Han, S. H., Yun, Y., Kim, Y. J., Choi, J. Y., et al. (2014). Pandemic influenza (H1N1) and Mycobacterium tuberculosis co-infection. Tuberc. Respir. Dis. 76, 84–87. doi: 10.4046/trd.2014.76.2.84
Park, S. S., Kim, S. H., Kim, M., Kim, J. W., Ko, Y. M., Kim, S.-K., et al. (2015). A case of severe pseudomembranous tracheobronchitis complicated by co-infection of influenza A (H1N1) and Staphylococcus aureus in an immunocompetent patient. Tuberc. Respir. Dis. (Seoul). 78, 366–370. doi: 10.4046/trd.2015.78.4.366
Peteranderl, C., Sznajder, J. I., Herold, S., and Lecuona, E. (2017). Inflammatory responses regulating Alveolar Ion transport during pulmonary infections. Front. Immunol. 8:446. doi: 10.3389/fimmu.2017.00446
Petersdorf, R. G., Fusco, J. J., Harter, D. H., and Albrink, W. S. (1959). Pulmonary infections complicating Asian influenza. AMA. Arch. Intern. Med. 103, 262–272. doi: 10.1001/archinte.1959.00270020090010
Plotkowski, M. C., Puchelle, E., Beck, G., Jacquot, J., and Hannoun, C. (1986). Adherence of type I Streptococcus pneumoniae to tracheal epithelium of mice infected with influenza A/PR8 virus. Am. Rev. Respir. Dis. 134, 1040–1044. doi: 10.1164/arrd.1986.134.5.1040
Plotkowski, M. C., Bajolet-Laudinat, O., and Puchelle, E. (1993). Cellular and molecular mechanisms of bacterial adhesion to respiratory mucosa. Eur. Respir. J. 6, 903–916.
Puchelle, E., Zahm, J. M., Tournier, J. M., and Coraux, C. (2006). Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 3, 726–733. doi: 10.1513/pats.200605-126SF
Ramos, I., and Fernandez-Sesma, A. (2012). Cell receptors for influenza a viruses and the innate immune response. Front. Microbiol. 3:117. doi: 10.3389/fmicb.2012.00117
Reilly, S., and Gaunt, P. N. (1991). Influenza A and meningococcal disease. Lancet 338, 1143–1144. doi: 10.1016/0140-6736(91)91999-B
Schwarzmann, S. W., Adler, J. L., Sullivan, R. J., and Marine, W. M. (1971). Bacterial pneumonia during the Hong Kong influenza epidemic of 1968-1969. Arch. Intern. Med. 127, 1037–1041. doi: 10.1001/archinte.1971.00310180053006
Shah, N. S., Greenberg, J. A., McNulty, M. C., Gregg, K. S., Riddell, J. T., Mangino, J. E., et al. (2016). Bacterial and viral co-infections complicating severe influenza: Incidence and impact among 507 U.S. patients, 2013-14. J. Clin. Virol. 80, 12–9. doi: 10.1016/j.jcv.2016.04.008
Shahangian, A., Chow, E. K., Tian, X., Kang, J. R., Ghaffari, A., Liu, S. Y., et al. (2009). Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Invest. 119, 1910–1920. doi: 10.1172/JCI35412
Shanks, G. D., and Brundage, J. F. (2012). Pathogenic responses among young adults during the 1918 influenza pandemic. Emerg. Infect. Dis. 18, 201–207. doi: 10.3201/eid1802.102042
Shinya, K., Ebina, M., Yamada, S., Ono, M., Kasai, N., and Kawaoka, Y. (2006). Avian flu: influenza virus receptors in the human airway. Nature 440, 435–436. doi: 10.1038/440435a
Simonsen, L., Clarke, M. J., Williamson, G. D., Stroup, D. F., Arden, N. H., and Schonberger, L. B. (1997). The impact of influenza epidemics on mortality: introducing a severity index. Am. J. Public Health 87, 1944–1950. doi: 10.2105/AJPH.87.12.1944
Smith, A. M. (2017). Quantifying the therapeutic requirements and potential for combination therapy to prevent bacterial coinfection during influenza. J. Pharmacokinet. Pharmacodyn. 44, 81–93. doi: 10.1007/s10928-016-9494-9
Sun, K., and Metzger, D. W. (2008). Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat. Med. 14, 558–564. doi: 10.1038/nm1765
Sun, K., Yajjala, V. K., Bauer, C., Talmon, G. A., Fischer, K. J., Kielian, T., et al. (2016). Nox2-derived oxidative stress results in inefficacy of antibiotics against post-influenza S. aureus pneumonia. J. Exp. Med. 213, 1851–1864. doi: 10.1084/jem.20150514
Takayama, Y., Yano, H., Nojima, Y., Nakano, R., Okamoto, R., Hirakata, Y., et al. (2014). Influence of prior pandemic A(H1N1)2009 virus infection on invasion of MDCK cells by community-associated methicillin-resistant Staphylococcus aureus. J. Infect. Chemother. 20, 71–73. doi: 10.1016/j.jiac.2013.07.012
Tan, C. K., Kao, C. L., Shih, J. Y., Lee, L. N., Hung, C. C., Lai, C. C., et al. (2011). Coinfection with Mycobacterium tuberculosis and pandemic H1N1 influenza A virus in a patient with lung cancer. J. Microbiol. Immunol. Infect. 44, 316–318. doi: 10.1016/j.jmii.2010.03.001
Tashiro, M., Ciborowski, P., Klenk, H. D., Pulverer, G., and Rott, R. (1987). Role of Staphylococcus protease in the development of influenza pneumonia. Nature 325, 536–537. doi: 10.1038/325536a0
Taubenberger, J. K., and Morens, D. M. (2008). The pathology of influenza virus infections. Annu. Rev. Pathol. 3, 499–522. doi: 10.1146/annurev.pathmechdis.3.121806.154316
Techasaensiri, B., Techasaensiri, C., Mejias, A., McCracken, G. J., and Ramilo, O. (2010). Viral coinfections in children with invasive pneumococcal disease. Pediatr. Infect. Dis. J. 29, 519–523.
Theofilopoulos, A. N., Baccala, R., Beutler, B., and Kono, D. H. (2005). Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 23, 307–336. doi: 10.1146/annurev.immunol.23.021704.115843
Tian, X., Xu, F., Lung, W. Y., Meyerson, C., Ghaffari, A. A., Cheng, G., et al. (2012). Poly I:C enhances susceptibility to secondary pulmonary infections by gram-positive bacteria. PLoS ONE 7:e41879. doi: 10.1371/journal.pone.0041879
Tuomanen, E. I., Austrian, R., and Masure, H. R. (1995). Pathogenesis of pneumococcal infection. N. Engl. J. Med. 332, 1280–1284. doi: 10.1056/NEJM199505113321907
Vermaelen, K., and Pauwels, R. (2004). Accurate and simple discrimination of mouse pulmonary dendritic cell and macrophage populations by flow cytometry: methodology and new insights. Cytometry A 61, 170–177. doi: 10.1002/cyto.a.20064
Wang, J., Li, F., Sun, R., Gao, X., Wei, H., Li, L. J., et al. (2013). Bacterial colonization dampens influenza-mediated acute lung injury via induction of M2 alveolar macrophages. Nat. Commun. 4:2106. doi: 10.1038/ncomms3106
Watt, J. P., Wolfson, L. J., O'Brien, K. L., Henkle, E., Deloria-Knoll, M., McCall, N., et al. (2009). Burden of disease caused by Haemophilus influenzae type b in children younger than 5 years: global estimates. Lancet 374, 903–911. doi: 10.1016/S0140-6736(09)61203-4
Weiser, J. N. (2010). The pneumococcus: why a commensal misbehaves. J. Mol. Med. 88, 97–102. doi: 10.1007/s00109-009-0557-x
Wolf, A. I., Strauman, M. C., Mozdzanowska, K., Williams, K. L., Osborne, L. C., Shen, H., et al. (2014). Pneumolysin expression by streptococcus pneumoniae protects colonized mice from influenza virus-induced disease. Virology 462–463, 254–265. doi: 10.1016/j.virol.2014.06.019
Wong, S. M., Bernui, M., Shen, H., and Akerley, B. J. (2013). Genome-wide fitness profiling reveals adaptations required by Haemophilus in coinfection with influenza A virus in the murine lung. Proc. Natl. Acad. Sci. U.S.A. 110, 15413–15418. doi: 10.1073/pnas.1311217110
Yang, M., Gao, H., Chen, J., Xu, X., Tang, L., Yang, Y., et al. (2016). Bacterial coinfection is associated with severity of avian influenza A (H7N9), and procalcitonin is a useful marker for early diagnosis. Diagn. Microbiol. Infect. Dis. 84, 165–169. doi: 10.1016/j.diagmicrobio.2015.10.018
Keywords: co-infection, influenza, bacteria, resumption of function, tolerance, mortality
Citation: Jia L, Xie J, Zhao J, Cao D, Liang Y, Hou X, Wang L and Li Z (2017) Mechanisms of Severe Mortality-Associated Bacterial Co-infections Following Influenza Virus Infection. Front. Cell. Infect. Microbiol. 7:338. doi: 10.3389/fcimb.2017.00338
Received: 09 May 2017; Accepted: 10 July 2017;
Published: 03 August 2017.
Edited by:
Dongsheng Zhou, Beijing Institute of Microbiology and Epidemiology, ChinaReviewed by:
ShiXing Tang, Southern Medical University, ChinaJianjun Zhou, Shanghai East Hospital, China
Xuhu Mao, Southwest Hospital, China
Copyright © 2017 Jia, Xie, Zhao, Cao, Liang, Hou, Wang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ligui Wang, d2FuZ2xpZ3VpMTk4M0AxMjYuY29t
Zhenjun Li, bGl6aGVuanVuQGljZGMuY24=
†These authors have contributed equally to this work.