 Tingting Feng
Tingting Feng Kezhen Wang
Kezhen Wang Jianfeng Dai
Jianfeng Dai- Jiangsu Key Laboratory of Infection and Immunity, Institute of Biology and Medical Sciences, Soochow University, Suzhou, China
Dengue is a mosquito-borne viral disease that rapidly spread in tropic and subtropic area in recent years. DEAD (Glu-Asp-Ala-Glu)-box RNA helicases have been reported to play important roles in viral infection, either as cytosolic sensors of viral nucleic acids or as essential host factors for the replication of different viruses. In this study, we reported that DDX25, a DEAD-box RNA helicase, plays a proviral role in DENV infection. The expression levels of DDX25 mRNA and protein were upregulated in DENV infected cells. During DENV infection, the intracellular viral loads were significantly lower in DDX25 silenced cells and higher in DDX25 overexpressed cells. Meanwhile, the expression level of type I interferon (IFN) was increased in DDX25 siRNA treated cells during viral infection. Consistent with the in vitro findings, the Ddx25-transgenic mice have an increased susceptibility to lethal vesicular stomatitis virus (VSV) virus challenge. The viremia was significantly higher while the anti-viral cytokine levels were lower in Ddx25-transgenic mice. Further, DDX25 modulated RIG-I signaling pathway and blocked IFNβ production, by interrupting IFN regulatory factor 3 (IRF3) and NFκB activation. Thus, DDX25 is a novel negative regulator of IFN pathway and facilitates RNA virus infection.
Introduction
Dengue virus (DENV) is a mosquito-borne viral pathogen, which is mainly transmitted by Aedes aegypti and Aedes albopicuts. Dengue imposes a significant burden on human health around the world and is endemic in more than 100 countries in tropical and subtropical areas, especially in Southeast Asia, the Americas, the Western Pacific, Africa, and Eastern Mediterranean regions (Guzman and Harris, 2015). Currently, there is no effective treatment for DENV infections. The first DENV vaccine was recently licensed for use after several decades of efforts, unfortunately, it confers only partial protection to all DENV serotypes (Gan, 2014; Thisyakorn and Thisyakorn, 2014).
Viruses have limited genetic capacity and as such rely on cellular factors to complete their life cycle. Thus, viruses interact with cellular proteins to acquire activities not encoded in the viral genome, to evade host immune defenses, and to manipulate cellular pathways to facilitate their replication (Khadka et al., 2011). However, while viruses attempt to hijack host cell machinery, the host cells attempt to halt, or slow down viral efficacy. DENV infection would normally activate the host antiviral response, in which the type I interferon pathway is crucial for host natural immunity. RNA helicases of DEAD-box protein family modulate the RNA structure and are crucial in many fundamental biological processes (de la Cruz et al., 1999; Rocak and Linder, 2004; Jankowsky, 2010). In addition to the traditional functions in RNA metabolism, DEAD-box RNA helicases have been reported as mediators of anti-viral innate immunity, or essential host factors for viral replication (Steimer and Klostermeier, 2012; Fullam and Schroder, 2013). Thus, DEAD-box RNA helicases family members play either a proviral or antiviral role during viral infection.
DDX3 is one of the first host cell DEAD-box RNA helicases identified as flavivirus cofactor, which is important for replication of viral RNA (Ariumi et al., 2007). For example, DDX3 colocalizes with NS3 near the nucleus during West Nile virus infection (Chahar et al., 2013). Another cellular helicase, DDX1 can directly bind to poly (I:C) (Zhang et al., 2011a). DDX21 and DHX36 are located downstream of DDX1 and both DDX21 and DHX36 interact with the downstream protein TIR-domain-containing-adapter-inducing interferon-β (TRIF). This suggests that DDX1 senses dsRNA and then triggers signaling via DDX21 and DHX36 to TRIF (Zhang et al., 2011a). DDX1 has also been reported to be important for the human immunodeficiency virus type 1 (HIV-1) replication as it binds to and serves as a cofactor of the HIV-1 Rev protein (Fang et al., 2004). DDX41 binds to STING (stimulator of interferon genes), which are located on the endoplasmic reticulum membrane (Zhang et al., 2011b; Parvatiyar et al., 2012).
In our previous study, 40 genes of DEAD-box family were screened using the RNAi approach to identify the putative host factors for DENV infection. Our result showed host DDX family members, DDX3X and DDX25, played an antiviral and a proviral role respectively during DENV infection (Li G. et al., 2015). DDX25, also named as Gonadotropin-regulated testicular RNA helicase (GRTH), is well-known as an essential testis-specific protein for completion of spermatogenesis in mouse model (Kavarthapu and Dufau, 2015). The expression profile and the role of DDX25 during viral infection has not been reported yet. We here reported that DDX25 interrupts the IFN-signaling pathway by inhibiting IRF3 and NFκB activation, which negatively regulated IFNβ activity, thereby facilitates RNA virus infection.
Materials and Methods
Ethics Statements and Mice
C57BL/6 adult wild type mice (ages 6–8 week) were purchased from Shanghai Laboratory Animal Center (Shanghai, China). C57BL/6-Ddx25 transgenic mice (Ddx25-Tg) were produced by Shanghai Biomodel Organism Science & Technology Development Co., Ltd. (Shanghai, China). Microinjections and mouse transgenesis experiments were performed by inserting CAG-Ddx25-IRES-Luciferase-polyA plasmid into the genome of fertilized egg of C57BL/6 mice using a PiggyBAC Transposon system. Mice were genotyped by tails snips and PCR at 3 weeks of age. The genotyping primers were TCCAAGGGGCACCGAAGTCACCAA(forward), and GCGCCGGGCCTTTCTTTATGTTTT(reverse).
All mice were housed in specific pathogen free facilities in accordance with the Guide of National Animal Care and Use committee. All animal experiments were approved by the Institutional Laboratory Animal Care and Use Committee of Soochow University.
Virus, Tissues, Cell Culture, and Infection
DENV-2 virus (DENV New Guinea C stain) were propagated in mosquito C6/36 cells (ATCC® CRL-1660). Sendai virus (SeV) was propagated in 10 days old embryonated chicken eggs (Bejing Laboratory Animal Research Center, Beijing, China), and the virus titer was detected by hemagglutination assay using chicken red blood cells (BeNa Culture Collection, Bejing, China). The VSV and VSV-green fluorescent protein (GFP) virus that expresses GFP as a non-structural protein was provided by Dr. Chunsheng Dong (Soochow University). The VSV-GFP virus was grown on a Vero E6 cell in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) containing 2% fetal bovine serum, titrated in Vero E6 cells.
The HEK293T cells were cultured in Dulbecco's modified eagle medium supplemented with fetal bovine serum (10%) and penicillin/streptomycin (1%). Cells were maintained at 37°C in a 5% CO2 laboratory incubator that was routinely cleaned and decontaminated. HEK293T cells were infected with DENV, SeV, or VSV-GFP at a multiplicity of infection (MOI) of 1, unless otherwise stated.
For in vivo VSV intranasal infections, 1 × 106 pfu of VSV in 50 μl of endotoxin-free PBS were inhaled by isoflurane-anesthetized C57BL/6 wild type and Ddx25-Tg mice, with PBS-only as control. Thereafter, mice were monitored daily for weight loss and symptoms of disease.
Human tumor tissue samples were obtained from the First Affiliated Hospital of Soochow University (Suzhou, China) under institutional guidelines and used in gene expression analysis.
Plasmid Constructs
Recombinant plasmid for DDX25 expression was constructed using standard protocols by inserting the DDX25 open reading frame into the pcDNA3.1 vector. Reporter plasmids NFκB-luc and pRL-TK were purchased from Clontech (USA) and used for dual luciferase reporter assays. Luciferase reporter plasmids IFNβ-luc, IRF3-luc, and ISRE-luc were kindly provided by Dr. Rongtuan Lin, McGill University, Canada (Zhao et al., 2007).
siRNA and Transfections
Transfections of HEK293T cells with plasmid DNA and siRNA were conducted using Lipofectamine 2000 (Invitrogen, USA) according to the manual of the manufacturer. The siRNA sequences for human DDX25 gene were target I-GCAGCTAATTCACTCTTAA, target II-GCAATGTTAAGCAGAGTTA, and target III-GCCACCAGGTGTCTTTGTT, respectively (RiboBio Co., Guangzhou, China). RNAi efficiency was confirmed through quantitative reverse transcription polymerase chain reaction (qRT-PCR). Then, the cells were infected with DENV at an MOI of 1 for 48 h (except for the cases noted in the text) to test the influence of DDX25 silencing on DENV replication.
TCID50 Assay and Viral Growth Kinetics
The cell-free supernatants were collected and the titers of DENV and VSV were assayed with a TCID50 assay according to standard protocols on Vero cells (ATCC® CCL-81) as described previously (Wang et al., 2016). The viral replication levels inside cells, in terms of the transcript levels of the DENV-2 envelop gene (E) or VSV glycoprotein gene (G), were quantified by qRT-PCR and normalized to β-actin gene.
RNA Isolation and Real-Time PCR
Total RNA was isolated using an RNA extraction Kit (Omega, Netherlands) and reverse-transcribed into cDNA using the first strand cDNA Synthesis Kit (Takara, Japan). qRT-PCR was performed using a SYBR Green with gene-specific primers (Applied Biosystems, USA) and normalized to β-actin gene. (oligo-primer sequences for qRT-PCR of this study were shown in Table S1 in the Supplemental Materials).
Luciferase Reporter Assays
For luciferase reporter assays, 70% confluent HEK293T cells were transfected with 10 ng of pRL-TK reporter plasmid (herpes simplex virus thymidine kinase promoter driving Renilla luciferase, internal control), 100 ng of IFNβ luciferase reporter plasmid (firefly luciferase, experimental reporter), 50 ng of IFNβ activators (RIG-IN, MAVS, TBK1, IKKε, IKKα, NFκB, or IRF3), as well as either 100 ng of recombinant expressing plasmids or siRNAs [Vector, DDX25, 50 nM negative control (N.C.), or DDX25 siRNA]. For measuring the activation of transcription factor NFκB and IRF3, NFκB and IRF3 responsive element specific reporter plasmids were used in the luciferase reporter assays. Subconfluent HEK293T cells were transfected with 10 ng of pRL-TK reporter, 100 ng of NFκB (pNFκB-Luc), or IRF3 (pPRD(III–I)-Luc) luciferase reporter plasmid, various doses of recombinant expression plasmids (Vector or DDX25), along with 50 ng of expression plasmids of RIG-IN. At 24 h post-transfection, cells were infected with DENV-2 at an MOI of 1 and incubated further for 24 h. Luciferase activity was measured using a Promega Dual Glow Kit according to the instructions of the manufacturer (Promega, USA).
Immunofluorescence Microscopy
HEK293T cells were transfected with siRNA (N.C. or DDX25 siRNA) using Lipofectamine 2000 to examine the effect of DDX25 silencing on DENV infection. At 24 h post-transfection, cells were infected with DENV-2 at an MOI of 1. Cells were fixed in 1% paraformaldehyde and permeabilized with 1% Triton X100 at 48 h post-infection. DENV envelope proteins were probed with mouse anti-DENV E antibody (Santa Cruz, USA) and stained with FITC-labeled anti-mouse IgG (Jackson ImmunoResearch). Cell nuclei were stained with DAPI. Cells were then examined using a fluorescence microscope.
Western Blot
HEK293T Cells were transfected with pcDNA-DDX25 (or pcDNA3.1 vector) and infected with DENV-2 at an MOI of 10. At 24 h post-infection, cell lysates were subjected to SDS-PAGE and transferred onto a PVDF membrane for western blotting. Non-reducing native PAGE was performed to detect the dimerization of IRF3. Briefly, cell lysates were prepared in a native sample buffer without SDS and electrophoresed on a 10% non-reducing polyacrylamide gel without SDS.
The following primary antibodies were used for western blotting: anti-human Actin polyclonal Ab (Proteintech, USA), His-tag polyclone antibody (GenScript, USA), IRF3 polyclonal antibody (Biolegend, USA), anti-IRF3 (phospho S386) Ab (Cell Signaling, USA). HRP-conjugated donkey anti-rabbit IgG and rabbit anti mouse IgG mAb (Biolegend, USA) were used as secondary antibodies. The signals were detected using an ECL detection system (Merck Millipore Ltd., USA).
Enzyme-Linked Immunosorbent Assay (ELISA)
The protein concentrations of IFNβ and TNFα in VSV infected mouse serum samples were measured by ELISA Kits according to the instructions of the manufacturer (R&D Systems).
Histopathology
Two days post VSV infection, wild type and Ddx25-Tg mice were sacrificed and the lung tissues were fixed in 10% formalin and paraffin embedded. Hematoxylin and eosin (H&E) staining was conducted for histopathology.
Statistical Analysis
Statistical significances were calculated with an unpaired two tailed Student's t-test and Log-rank (Mantel-Cox) Test (for survival data only) using Prism 5 software (GraphPad).
Results
DDX25 Is Upregulated upon DENV Infection
In mice, Ddx25 was reported to be highly expressed in testis and critical for the process of spermatogenesis (Sheng et al., 2003). Contrastingly, by searching the gene expression database, we have found that human DDX25 mRNA was expressed in almost all the human tissues (data from database of GTEx, BioGPS, Illumina Human BodyMap, and SAGE). Human DDX25 proteins are also detected in multiple tissues, such as testis, spleen, tonsil, ovary, and retina, as well as various cell lines including A549, HEK293T, SNB-75, JUNKAT, and H1734 cells (The Human Integrated Protein Expression Database, Integrated protein expression data from ProteomicsDB, PaxDb, MaxQB, and MOPED).
We further confirmed the mRNA expression profile of human DDX25 gene by qRT-PCR. As shown in Figure 1A, DDX25 mRNA is expressed in a wide range of human tissues. With the exception of breast tissue, DDX25 is expressed in all seven tested tissues, with the highest expression in the kidney (Figure 1A).
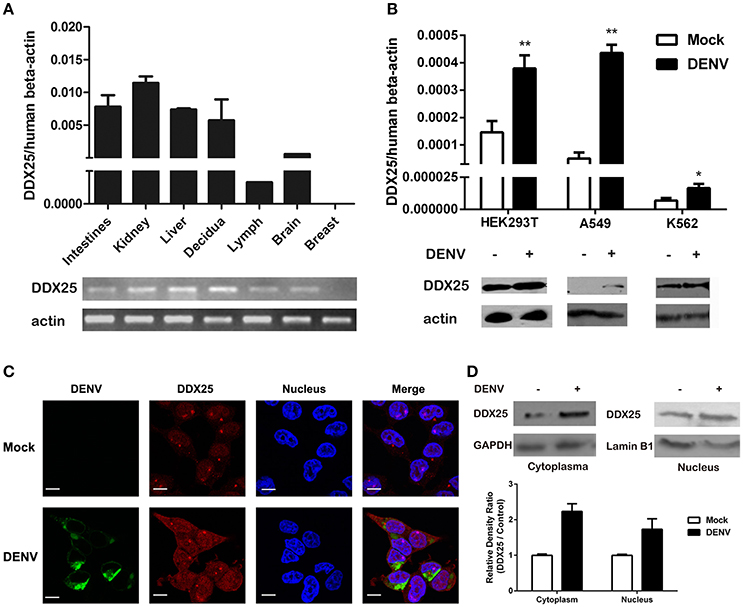
Figure 1. Human DDX25 is ubiquitously expressed and upregulated after DENV infection. (A) qRT-PCR and electrophoresis analysis of DDX25 expression in various human tissues. Human β-actin was used as an internal control. (B) The qRT-PCR and immunoblots analysis of DDX25 expression in HEK293T, A549, and K562 cells before and after DENV infection. (C) Microscopic images of immunofluorescence staining for expression and subcellular distribution of DDX25 in HEK293T with or without DENV infection. (MOI of 1 for 24 h). Scale bars, 10 μm. (D) Immunoblots analysis of DDX25 expression in the cytoplasm and nucleus before and after DENV infection. The relative gray density of immunoblots bands (DDX25/β-actin) was analyzed by ImageJ software. Results were expressed as the mean + the SEM. *p < 0.05 and **p < 0.01 (t-test). Representative results from at least three independent experiments.
We further noted an increased DDX25 expression in human cell line HEK293T, A549, and K562 during DENV infection. Consistently, the protein level of DDX25 was also significantly induced in cells infected with DENV (Figure 1B). We further investigated the cellular distribution of DDX25 in HEK293T cells before and after DENV infection. Interestingly, DDX25 expression was upregulated both in cytoplasm and nucleus (Figures 1C,D). These data demonstrate that DDX25 may have a low basal level of expression in multiple tissues and cells, and can be upregulated upon DENV infection.
DDX25 Promotes Virus Replication
Our previous studies of RNAi screening against DDX family demonstrated that DDX25 could be a proviral factor for DENV infection in HEK293T cells (Li G. et al., 2015). To confirm the role of endogenous DDX25 in DENV replication, endogenous DDX25 expression was silenced using RNAi approach. SiRNAs that targeting DDX25 significantly reduced DDX25 mRNA expression compared with cells transfected with scramble siRNA (N.C.) (Figure 2A). Cell viability was not affected after siRNA transfection, as tested through Promega Cell Viability Assay (Figure 2B). Silencing DDX25 with three individual or pooled siRNAs significantly impaired DENV replication at 24 h post-infection (Figure 2C) [The intracellular viral loads were determined by measuring the transcript levels of the DENV envelop gene (E) and normalized to human β-actin]. Further experiment confirmed that the DENV viral load were decreased by ~2 to 4-fold (p < 0.05) in DDX25-silenced cells compared with control cells after DENV infection at 12, 24, and 48 h, respectively (Figure 2D). Similar results were observed in DDX25 silenced K562 cells infected with DENV (Data not shown). In line with this, immunofluorescence assay showed DENV viral burden [as determined by staining of viral envelop (E) protein] were decreased in DDX25 silenced cells (Figure 2E). Conversely, we noted a ~4-fold increase in viral load in DDX25 overexpressing HEK293T cell at 48 h post-infection (Figure 2F). Consistently, the titers of DENV in cell supernatants, as determined by TCID50 assay, were significantly higher in DDX25 overexpressed cells (Figure 2G). Moreover, we observed elevated viral loads in DDX25 overexpressing HEK293T cells after infection with VSV-GFP virus (Figures 2H–J). Overall, these data suggest that DDX25 could promote DENV and VSV replication.
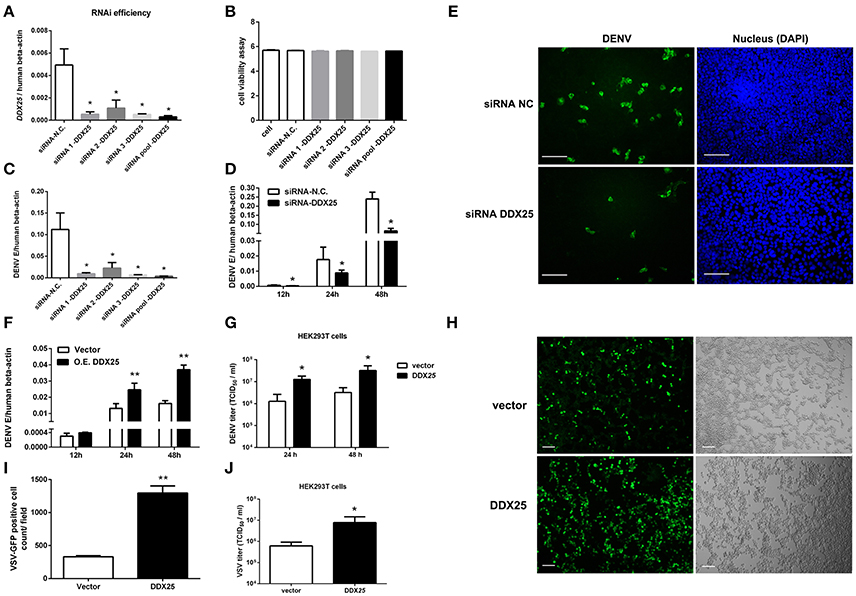
Figure 2. DDX25 promotes virus replication. (A) The RNAi efficiency of three individual or pooled siRNAs that targeting DDX25 were tested in transfected HEK293T cells by qRT-PCR. (B) RNAi of DDX25 showed no cytotoxic effect to HEK 293T cells (Measured by Cell Viability assay, Promega). (C) DENV viral loads in DDX25 siRNAs (or NC siRNA) treated HEK293T cells at 24 h post DENV infection (MOI of 1). The viral burdens were analyzed by measuring the virus E gene copy using qRT-PCR, and normalized to human β-actin. (D) DENV viral loads in DDX25 siRNA (or NC siRNA) treated HEK293T cells at 12, 24, and 48 h post DENV infection (MOI of 1). (E) Microscopic images of immunofluorescence staining for DENV E antigen in DDX25 siRNA (or NC siRNA) treated HEK293T after DENV infection for 24 h (MOI of 1). Scale bars, 100 μm. (F) qRT-PCR analysis of DENV viral loads in HEK293T cells with or without DDX25 overexpression at 12, 24, and 48 h post DENV infection. (G) Viral titers in supernatants of DENV infected HEK293T cells determined by TCID50 assay on Vero cells. (H,I) Fluorescence microscopy analysis of VSV-GFP infection of HEK293T cells transfected with vector or DDX25 plasmid. Viral infection rates were indicated with GFP signal and quantified with ImageJ software. Scale bars, 100 μm. (J) VSV titers in supernatants of HEK293T cells infected with VSV-GFP (MOI of 1) at 24 h. Results were expressed as the mean + the SEM. *p < 0.05 and **p < 0.01 (t-test). Representative results from at least three independent experiments.
DDX25 Regulates IFNβ Production
The aforementioned results clearly demonstrate the importance of DDX25 in promoting DENV infection in vitro. We next evaluated whether DDX25 influence the immune response to viral infection. The mRNA expression of IFNβ was increased by 1.5- and 1.4-fold at 24 and 48 h post DENV infection respectively, in DDX25 silenced cells compared with controls (Figure 3A). DDX25 silencing also enhanced the IFNβ promoter-driven luciferase (IFNβ-Luc) expression in HEK293T cells infected with other RNA viruses, such as VSV and sendai virus (SeV) (Figure 3B). Conversely, we noted a decreased IFNβ mRNA level on 24 h in DDX25-overexpressed HEK293T cells post DENV infection (Figure 3C). While, at 48 h post-infection, the IFNβ mRNA level was increased in DDX25 overexpressed cells (Figure 3C). We hypothesized that this was due to the expansion of DENV burdens in those cells at the late time point, which induced more IFNs that DDX25 could not fully suppressed. Consistent with the qPCR results, overexpression of DDX25 could decrease the transcription of IFNβ-Luc reporter after DENV (Figure 3D) and other RNA virus infection (Figure 3E). These results suggest that DDX25 negatively regulates innate immune-signaling processes and suppress type I IFN production during virus infection.
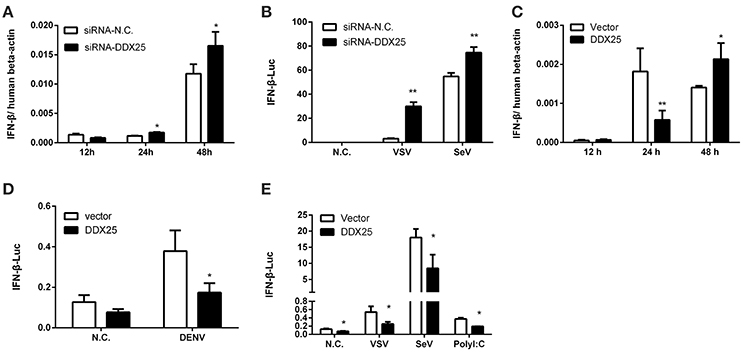
Figure 3. DDX25 negatively regulates IFNβ production. (A) qRT-PCR analysis of the mRNA levels of IFNβ in HEK293T cells with DDX25 RNAi during DENV infection for 12, 24, and 48 h. (B) VSV and SeV induced IFNβ transcriptional activities were enhanced in DDX25 silenced HEK293T cells compared to controls. (C) qRT-PCR analysis of the mRNA levels of IFNβ in control and DDX25 overexpressed HEK293T cells during DENV infection for 12, 24, and 48 h. (D,E) Luciferase reporter assay of IFNβ transcriptional activity in DDX25 overexpressed HEK293T cells during DENV (D), VSV, SeV, and Poly I:C (E) stimulation. Results were expressed as the mean + the SEM. *p < 0.05 and **p < 0.01 (t-test). Representative results from at least three independent experiments.
DDX25 Transgenic Mice Were More Susceptible to RNA Virus Infection
DDX25 null mice are sterile due to a defect in spermiogenesis (Tsai-Morris et al., 2004). So we constructed Ddx25-Tg mice that overexpress DDX25 to identify the role of Ddx25 in virus infection in vivo (Figure 4A). Since immunocompetent wild-type mice do not show dengue hemorrhagic fever-like symptoms upon DENV infection, we used a VSV-infection mouse model to evaluate the importance of DDX25 in RNA viral infection in vivo. Consistent with the in vitro data, the Ddx25 mRNA in total blood cells was upregulated after VSV virus infection in wild type mice (Figure 4B). Wild-type (W.T.) and Ddx25-Tg mice were infected intranasally with a lethal dose of VSV (1 × 106 pfu). All infected Ddx25-Tg mice show increased body weight loss compared with wide type controls (Figure 4C). Approximately 90% of Ddx25-Tg mice died within 10 days post-infection (Figure 4D), while only 40% of the wide type control mice died due to the VSV infection. This suggested that Ddx25-Tg mice are more susceptible to VSV infection. The viremia, in terms of the copy number of VSV glycoprotein gene (VSV-G) in blood cells and infectious viral particles in serum, were significantly higher in Ddx25-Tg mice compared with W.T. mice at day 1 and day 3 after infection. We also found that VSV titers were higher in the liver, kidney, and spleen of Ddx25-Tg mice than in those organs of W.T. mice (Figure 4E). Meanwhile, the mRNA expression of Ifnα, Ifnβ, Ifnγ, Tnfα, and selective chemokines (Cxcl1, Cxcl2, and Ccl5) by leukocytes was lower in the blood of Ddx25-Tg mice on day 1 post-infection (Figure 4F). The IFNβ and TNFα proteins in the serum were also significantly lower in Ddx25-Tg mice compared with those in controls (Figure 4G). While, Ifnα and Ifnβ expression on day 3 post-infection were increased in Ddx25-Tg mice, this may be the secondary effect of the increased viremia (Figure 4H). The protein levels of IFNβ and TNFα were consistent with their mRNA levels (Figure 4I). Histological analysis by H&E staining indicated that the bronchial lumen and alveolar both exhibit healthy integrity in the non-infected W.T. and Ddx25-Tg mice. However, the W.T. mice infected virus displayed mucosal edema, inflammatory cell infiltration (submucosal and mucosal layers), as well as the increased number of eosinophils (EOS) in the airway walls. Compared to the virus infected W.T. mice group, a significantly increased inflammatory cell infiltration and serious immunopathological tissue damage were observed in VSV infected Ddx25-Tg mice (Figure 4J). These data suggested that DDX25 could promote RNA virus infection and suppress Ifnβ and other cytokines production in vivo.
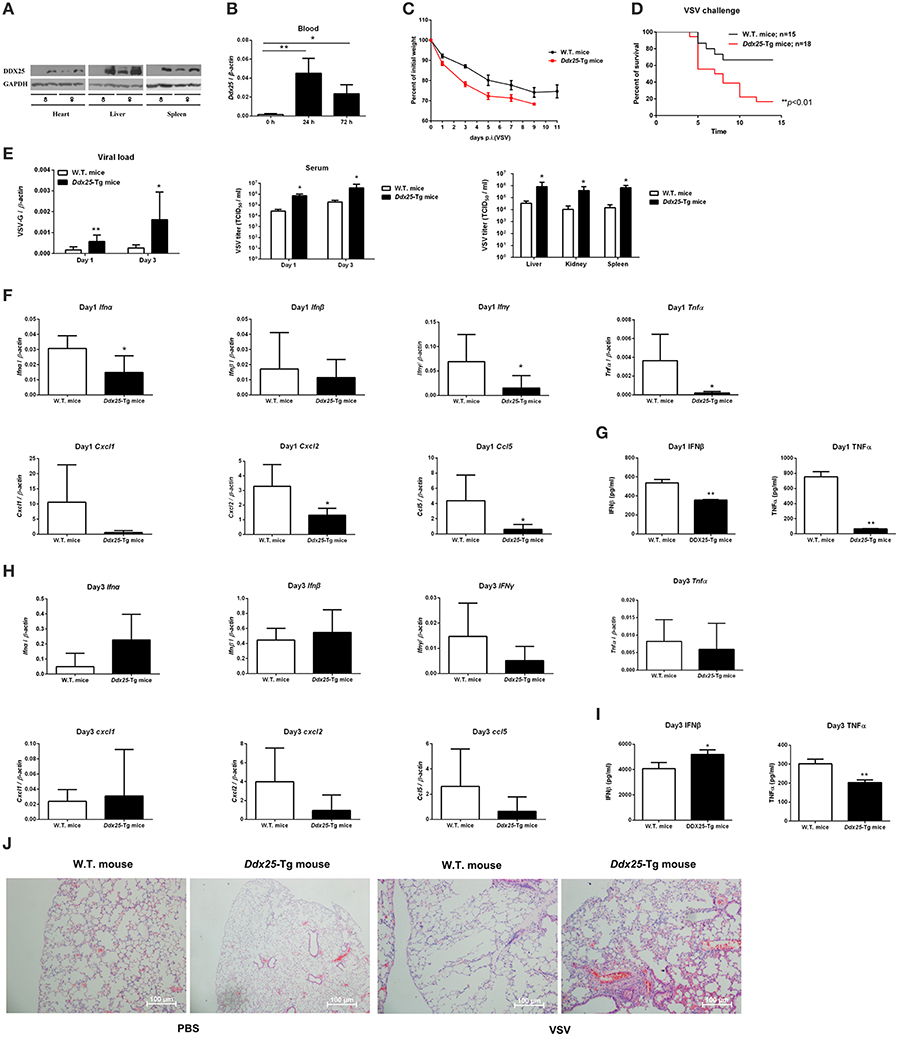
Figure 4. Exacerbated VSV pathogenesis in Ddx25 transgenic mice. Seven to eight weeks old, sex-matched, C57BL/6 (W.T.), and Ddx25-Tg mice were challenged with VSV (1 × 106 pfu/mouse). (A) Immunoblots of DDX25 in various tissues of W.T. and Ddx25-Tg mice, suggesting the evaluated expression of DDX25 in Ddx25-Tg mice. (B) qRT-PCR quantification of Ddx25 mRNA in whole blood cells of wild type (W.T.) mice during VSV infection for 0, 24, and 72 h. (C,D) Changes in body weight (C) and survival rate (D) of W.T., Ddx25-Tg mice infected VSV, as monitored daily until days 11 and 14 post-infection, respectively. (E) VSV titers in the blood and tissue lysates of liver, kidney, and spleen from infected mice were measured via qRT-PCR and TCID50 assay. (F–I) The expression levels of the selective cytokines/chemokines (Ifnα, Ifnβ, Ifnγ, Cxcl1, Cxcl2, Ccl5, and Tnfα) in total leukocytes of W.T. and Ddx25-Tg mice (N = 10 each) at day 1 (F,G) and day 3 (H,I) post-infection. Expression levels of mRNAs were determined by qRT-PCRs (F,H). Protein levels of IFNβ and TNFα in serums at day 1 (G) and day 3 (I) were determined by ELISA. Results were expressed as the mean + the SEM. *p < 0.05 and **p < 0.01 (t-test). Representative results from at least three independent experiments. (J) H&E staining of lung tissues of W.T. and Ddx25-Tg mice at 2 days post mock infection (PBS) or VSV infection. Scale bars, 100 μm.
DDX25 Inhibits IFN Production by Interrupting IRF3 and NFκB Activation
We next sought to understand how DDX25 suppresses of IFNβ induction. Particularly, RNA recognition by RIG-I emanates an activation signal that is passed on sequentially to mitochondrial antiviral signaling protein (MAVS), TBK1, IKKε, IRF3, and IRF7 (Sharma et al., 2003; Liu et al., 2015; Yoneyama et al., 2015). Therefore, the action point of DDX25 can be determined by assessing its suppressive effects on a series of transducer proteins. As the first step, we chose to evaluate the impact of DDX25 on the IFN-inducing activity of RIG-I-signaling pathway components, including the active caspase recruitment domain (CARD) containing form of RIG-I (RIG-IN), MDA5, MAVS, TBK1 kinase, and the active form of IRF3 (IRF3/5D) (Lin et al., 1998), or IKKα and p65. DDX25-expressing plasmids, together with one of the IFN pathway activators, RIG-IN, MDA5, MAVS, TBK1, IRF3/5D, IKKα, or p65, were co-transfected into HEK293T cells. All the expression constructs of IFN pathway activators induced an IFNβ-Luc or NFκB-Luc reporter activity. Interestingly, DDX25 was found to potently inhibit IFNβ-Luc activity induced by RIG-IN, MDA5, MAVS, TBK1, and IRF3/5D (Figure 5A), as well as NFκB-Luc reporter activity induced by RIG-IN, IKKα and p65 (Figure 5B). These results indicate that DDX25 inhibits the IFN antiviral response at or downstream of IRF3 and NFκB.
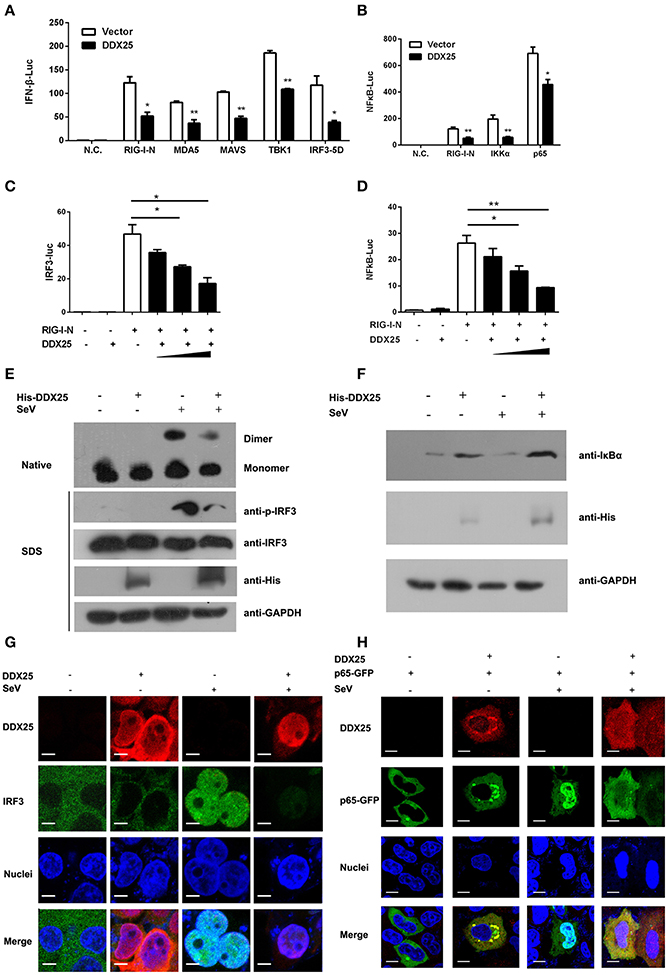
Figure 5. DDX25 interrupts the IFN-signaling pathway by blocking IRF3 and NFκB activation. (A) Overexpression of DDX25 inhibits RIG-IN, MDA-5, MAVS, TBK1, and IRF3-5D directed IFNβ promoter activation. IFNβ-Luc assay was performed in vector or DDX25 transfected HEK293T cells. IFNβ promoter activations were driven by co-expression of distinct RIG-I signaling pathway molecules. (B) Overexpression of DDX25 inhibits IG-IN, IKKα, and NFκB p65 induced NFκB promoter activation. NFκB promoter was activated by transfecting RIG-IN, IKKα, or NFκB p65, and measured by a NFκB-Luc. (C,D) DDX25 suppresses RIG-IN directed IRF3-Luc (C) and NFκB-Luc (D) in a dose dependent manner. Results are expressed as the mean + the SEM. *p < 0.05 and **p < 0.01 (t-test). Representative results from at least three independent experiments. (E) Overexpression of DDX25 impaired SeV induced IRF3 phosphorylation and dimerization. (F) Overexpression of DDX25 suppressed SeV induced IκB degradation. (G,H) Nucleic translocation of IRF3 (G) and NFκB p65 (H) in SeV stimulated HEK293T cells were impaired by DDX25. Immunofluorescence assay were performed in control or DDX25 overexpressed HEK293T cells with or without SeV infection. IRF3, DDX25 were probed with specific primary antibodies followed by TRITC or FITC labeled secondary antibodies. NFκB p65 was visualized by a fused GFP protein. Scale bars, 10 μm.
To further validate the step in which DDX25 inhibits IRF3 and NFκB activation, IRF3 and NFκB-specific luciferases activity were induced by coexpression of RIG-IN. In the presence of DDX25, the IRF3 and NFκB activities induced by RIG-IN were reduced in a DDX25 dose-dependent manner (Figures 5C,D), which imply that DDX25 inhibits both IRF3 and NFκB transcriptional activity directly.
IRF3 and NFκB activation requires the association of distinct signaling molecules (Baeuerle and Baichwal, 1997; Barnes and Karin, 1997). Phosphorylation of IRF3 induces a conformational change, leading to IRF3 dimerization and nuclear translocation. While activation of the NFκB is initiated by the signal-induced degradation of IκB proteins. IκB proteins mask the nuclear localization signals (NLS) of NFκB proteins and keep them sequestered in an inactive state in the cytoplasm of unstimulated cells. To investigate whether IRF3 and NFκB activation were directly or indirectly affected by DDX25, we examined the protein level of IκBα and phosphorylation of IRF3 in the DDX25 overexpressed cells. Western blot results revealed that SeV induced the phosphorylation of IRF3 and the degradation of IκBα was inhibited by DDX25 (Figures 5E,F). Complementary to phosphorylation, SeV infection induced the dimerization of IRF3 and is also inhibited by DDX25 overexpression (Figure 5E). These results suggest that DDX25 inhibits IRF3 phosphorylation and NFκB activation.
To strengthen our existing data, immunofluorescence was carried out to investigate whether DDX25 prevented the nuclear translocation of IRF3 and NFκB p65 subunit. In mock-treated HeLa cells, IRF3, and p65 localized exclusively to the cytoplasm. SeV infection induced nuclear translocation of IRF3 and p65 in cells, while ectopic expression of DDX25 impaired the nuclear translocation of IRF3 and p65 (Figures 5G,H). Taken together, these results indicate that DDX25 inhibits the host antiviral response, by repressing the IFN antiviral response by suppressing IRF3 and NFκB transcriptional activity, and thereby inhibited the host antiviral response.
Discussion
Previous work suggested that GRTH/DDX25 is highly expressed in testis and essential for completion of spermatogenesis in mouse mode (Mendelson, 2013). In this report, we further suggested that DDX25 is expressed in multiple human tissues and cell lines. Strikingly, we observed an increased production of DDX25 both in vivo and in vitro during viral infection. We further discovered DDX25 promotes the replication of RNA viruses, including DENV, VSV (Figures 2G,H), and Zika virus (ZIKV) (data not shown). In vivo data also demonstrates that DDX25 promotes RNA virus replication. As such, DDX25 has additional function for viral infection in human cells and mouse models.
The IFN system is triggered after viral infection via cellular recognition of viral components. Mammalian cells utilize multiple pattern recognition receptors, e.g., TLRs, RLRs, NLRs, or cGAS/STING, to detect the incoming viral particles in the cytoplasm or inside specific cellular compartments and trigger intracellular signaling pathways, which lead to the induction of IFN through the action of the transcription factor IRF3 (Chattopadhyay and Sen, 2016). Viral infection activates IRF3 by causing phosphorylation of its specific serine residues and its translocation to the nucleus, where it binds to the promoters of the target genes (Lin et al., 1998; Sato et al., 1998). On the other hand, NFκB is rapidly activated after exposure to pathogens (Rahman and McFadden, 2011). Once IκB is degraded, the NFκB complex is translocated to the nucleus where it functions as a transcription factor for numerous effector genes including type I IFN. Our data here indicated that DDX25 may negatively regulate these processes. Recently, the DEAD/H-box helicase family members have been drawing more and more attention not only for their powerful capacities in detecting invading pathogen-associated molecular patterns (PAMPs) as direct sensors (Fullam and Schroder, 2013; Mitoma et al., 2013), but also for their key roles involved in viral replication (Wang et al., 2009; Fullam and Schroder, 2013; Yasuda-Inoue et al., 2013; Zhou et al., 2013). RIG-I (DDX58) is reported to be a key sensor for viral RNA mediated innate immune signaling processes (Schmidt et al., 2012). Several other RNA helicases, such as DDX19 (Li J. et al., 2015), DDX41 (Jiang et al., 2017), DHX9 and DHX36 (Kim et al., 2010), are also implicated in the regulation of host defense processes. DDX3X and DDX24 function downstream of nucleic acid recognition to affect multi-protein signaling complexes required for efficient primary innate immune gene transcription (Ma et al., 2013; Li G. et al., 2015).
To evade the antiviral response of host cells, many viruses utilize the host factors to interfere with IRF3 or NFκB signaling by sophisticated mechanisms (Rahman and McFadden, 2011; Ye et al., 2013). Here, we demonstrate that DDX25 interfered with the activation of IRF3 and NFκB induced by DENV and other RNA viruses. Our findings suggested that DDX25 alters the phosphorylation level of IRF3 and the degradation of IκBα mediated by SeV.
DDX25 plays a negative regulatory role for IFNβ during DENV replication, confirming that inhibition of nuclear-cytoplasmic protein transport is a strategy for a virus to prevent the antiviral response of host cells. In summary, our data demonstrated that DDX25 interferes with the nuclear translocation of IRF3 and NFκB induced by RNA virus. Via this mechanism, DDX25 suppresses the induction of type I IFN in host cells during viral infection. These findings reveal a novel strategy for DENV to utilize a host factor to evade the host innate immune response and provide us new insight into the function of DDX25.
Author Contributions
TF and JD designed the experiments and analyzed the data. TF, TS, GL, KW, and WP performed the experiments. TF and JD wrote the manuscript with all the authors contributing to writing, discussion, and agreeing with the conclusion presented in the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31300714, 31400737, 81471571, 81271792, and 31500700), Suzhou Science and Technology Development Project (SNG201607), China Postdoctoral Science Foundation (2013M541725), the Priority Academic Program Development of Jiangsu Higher Education Institutions, Program for Changjiang Scholars and Innovative Research Team in University (PCSIRT), and Jiangsu Natural Science Foundation (BK20140322).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcimb.2017.00356/full#supplementary-material
References
Ariumi, Y., Kuroki, M., Abe, K., Dansako, H., Ikeda, M., Wakita, T., et al. (2007). DDX3 DEAD-box RNA helicase is required for hepatitis C virus RNA replication. J. Virol. 81, 13922–13926. doi: 10.1128/JVI.01517-07
Baeuerle, P. A., and Baichwal, V. R. (1997). NF-kappa B as a frequent target for immunosuppressive and anti-inflammatory molecules. Adv. Immunol. 65, 111–137. doi: 10.1016/S0065-2776(08)60742-7
Barnes, P. J., and Karin, M. (1997). Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 336, 1066–1071. doi: 10.1056/NEJM199704103361506
Chahar, H. S., Chen, S., and Manjunath, N. (2013). P-body components LSM1, GW182, DDX3, DDX6 and XRN1 are recruited to WNV replication sites and positively regulate viral replication. Virology 436, 1–7. doi: 10.1016/j.virol.2012.09.041
Chattopadhyay, S., and Sen, G. C. (2016). RIG-I-like receptor-induced IRF3 mediated pathway of apoptosis (RIPA): a new antiviral pathway. Protein Cell 8, 165–168. doi: 10.1007/s13238-016-0334-x
de la Cruz, J., Kressler, D., and Linder, P. (1999). Unwinding RNA in Saccharomyces cerevisiae: DEAD-box proteins and related families. Trends Biochem. Sci. 24, 192–198. doi: 10.1016/S0968-0004(99)01376-6
Fang, J., Kubota, S., Yang, B., Zhou, N., Zhang, H., Godbout, R., et al. (2004). A DEAD box protein facilitates HIV-1 replication as a cellular co-factor of Rev. Virology 330, 471–480. doi: 10.1016/j.virol.2004.09.039
Fullam, A., and Schroder, M. (2013). DExD/H-box RNA helicases as mediators of anti-viral innate immunity and essential host factors for viral replication. Biochim. Biophys. Acta 1829, 854–865. doi: 10.1016/j.bbagrm.2013.03.012
Gan, V. C. (2014). Dengue: moving from current standard of care to state-of-the-art treatment. Curr. Treat. Options Infect. Dis. 6, 208–226. doi: 10.1007/s40506-014-0025-1
Guzman, M. G., and Harris, E. (2015). Dengue. Lancet 385, 453–465. doi: 10.1016/S0140-6736(14)60572-9
Jankowsky, E. (2010). RNA helicases at work: binding and rearranging. Trends Biochem. Sci. 36, 19–29. doi: 10.1016/j.tibs.2010.07.008
Jiang, Y., Zhu, Y., Liu, Z. J., and Ouyang, S. (2017). The emerging roles of the DDX41 protein in immunity and diseases. Protein Cell 8, 83–89. doi: 10.1007/s13238-016-0303-4
Kavarthapu, R., and Dufau, M. L. (2015). Germ cell nuclear factor (GCNF/RTR) regulates transcription of gonadotropin-regulated testicular RNA helicase (GRTH/DDX25) in testicular germ cells–the androgen connection. Mol. Endocrinol. 29, 1792–1804. doi: 10.1210/me.2015-1198
Khadka, S., Vangeloff, A. D., Zhang, C., Siddavatam, P., Heaton, N. S., Wang, L., et al. (2011). A physical interaction network of dengue virus and human proteins. Mol. Cell. Proteomics 10:M111.012187. doi: 10.1074/mcp.M111.012187
Kim, T., Pazhoor, S., Bao, M., Zhang, Z., Hanabuchi, S., Facchinetti, V., et al. (2010). Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 107, 15181–15186. doi: 10.1073/pnas.1006539107
Li, G., Feng, T., Pan, W., Shi, X., and Dai, J. (2015). DEAD-box RNA helicase DDX3X inhibits DENV replication via regulating type one interferon pathway. Biochem. Biophys. Res. Commun. 456, 327–332. doi: 10.1016/j.bbrc.2014.11.080
Li, J., Hu, L., Liu, Y., Huang, L., Mu, Y., Cai, X., et al. (2015). DDX19A senses viral RNA and mediates NLRP3-dependent inflammasome activation. J. Immunol. 195, 5732–5749. doi: 10.4049/jimmunol.1501606
Lin, R., Heylbroeck, C., Pitha, P. M., and Hiscott, J. (1998). Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 18, 2986–2996. doi: 10.1128/MCB.18.5.2986
Liu, S., Cai, X., Wu, J., Cong, Q., Chen, X., Li, T., et al. (2015). Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347:aaa2630. doi: 10.1126/science.aaa2630
Ma, Z., Moore, R., Xu, X., and Barber, G. N. (2013). DDX24 negatively regulates cytosolic RNA-mediated innate immune signaling. PLoS Pathog. 9:e1003721. doi: 10.1371/journal.ppat.1003721
Mendelson, C. R. (2013). GRTH: a key to understanding androgen-mediated germ cell signaling. Endocrinology 154, 1967–1969. doi: 10.1210/en.2013-1395
Mitoma, H., Hanabuchi, S., Kim, T., Bao, M., Zhang, Z., Sugimoto, N., et al. (2013). The DHX33 RNA helicase senses cytosolic RNA and activates the NLRP3 inflammasome. Immunity 39, 123–135. doi: 10.1016/j.immuni.2013.07.001
Parvatiyar, K., Zhang, Z., Teles, R. M., Ouyang, S., Jiang, Y., Iyer, S. S., et al. (2012). The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat. Immunol. 13, 1155–1161. doi: 10.1038/ni.2460
Rahman, M. M., and McFadden, G. (2011). Modulation of NF-kappaB signalling by microbial pathogens. Nat. Rev. Microbiol. 9, 291–306. doi: 10.1038/nrmicro2539
Rocak, S., and Linder, P. (2004). DEAD-box proteins: the driving forces behind RNA metabolism. Nat. Rev. Mol. Cell Biol. 5, 232–241. doi: 10.1038/nrm1335
Sato, M., Tanaka, N., Hata, N., Oda, E., and Taniguchi, T. (1998). Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-beta gene. FEBS Lett. 425, 112–116. doi: 10.1016/S0014-5793(98)00210-5
Schmidt, A., Rothenfusser, S., and Hopfner, K. P. (2012). Sensing of viral nucleic acids by RIG-I: from translocation to translation. Eur. J. Cell Biol. 91, 78–85. doi: 10.1016/j.ejcb.2011.01.015
Sharma, S., tenOever, B. R., Grandvaux, N., Zhou, G. P., Lin, R., and Hiscott, J. (2003). Triggering the interferon antiviral response through an IKK-related pathway. Science 300, 1148–1151. doi: 10.1126/science.1081315
Sheng, Y., Tsai-Morris, C. H., and Dufau, M. L. (2003). Cell-specific and hormone-regulated expression of gonadotropin-regulated testicular RNA helicase gene (GRTH/Ddx25) resulting from alternative utilization of translation initiation codons in the rat testis. J. Biol. Chem. 278, 27796–27803. doi: 10.1074/jbc.M302411200
Steimer, L., and Klostermeier, D. (2012). RNA helicases in infection and disease. RNA Biol. 9, 751–771. doi: 10.4161/rna.20090
Thisyakorn, U., and Thisyakorn, C. (2014). Latest developments and future directions in dengue vaccines. Ther. Adv. Vaccines 2, 3–9. doi: 10.1177/2051013613507862
Tsai-Morris, C. H., Sheng, Y., Lee, E., Lei, K. J., and Dufau, M. L. (2004). Gonadotropin-regulated testicular RNA helicase (GRTH/Ddx25) is essential for spermatid development and completion of spermatogenesis. Proc. Natl. Acad. Sci. U.S.A. 101, 6373–6378. doi: 10.1073/pnas.0401855101
Wang, H., Kim, S., and Ryu, W. S. (2009). DDX3 DEAD-Box RNA helicase inhibits hepatitis B virus reverse transcription by incorporation into nucleocapsids. J. Virol. 83, 5815–5824. doi: 10.1128/JVI.00011-09
Wang, K., Wang, J., Sun, T., Bian, G., Pan, W., Feng, T., et al. (2016). Glycosphingolipid GM3 is indispensable for dengue virus genome replication. Int. J. Biol. Sci. 12 872–883. doi: 10.7150/ijbs.15641
Yasuda-Inoue, M., Kuroki, M., and Ariumi, Y. (2013). Distinct DDX DEAD-box RNA helicases cooperate to modulate the HIV-1 Rev function. Biochem. Biophys. Res. Commun. 434, 803–808. doi: 10.1016/j.bbrc.2013.04.016
Ye, J., Zhu, B., Fu, Z. F., Chen, H., and Cao, S. (2013). Immune evasion strategies of flaviviruses. Vaccine 31 461–471. doi: 10.1016/j.vaccine.2012.11.015
Yoneyama, M., Onomoto, K., Jogi, M., Akaboshi, T., and Fujita, T. (2015). Viral RNA detection by RIG-I-like receptors. Curr. Opin. Immunol. 32, 48–53. doi: 10.1016/j.coi.2014.12.012
Zhang, Z., Kim, T., Bao, M., Facchinetti, V., Jung, S. Y., Ghaffari, A. A., et al. (2011a). DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity 34, 866–878. doi: 10.1016/j.immuni.2011.03.027
Zhang, Z., Yuan, B., Bao, M., Lu, N., Kim, T., and Liu, Y. J. (2011b). The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 12, 959–965. doi: 10.1038/ni.2091
Zhao, T., Yang, L., Sun, Q., Arguello, M., Ballard, D. W., Hiscott, J., et al. (2007). The NEMO adaptor bridges the nuclear factor-kappaB and interferon regulatory factor signaling pathways. Nat. Immunol. 8, 592–600. doi: 10.1038/ni1465
Keywords: DDX25, interferon, innate immune response, dengue virus, IRF3, NFκB
Citation: Feng T, Sun T, Li G, Pan W, Wang K and Dai J (2017) DEAD-Box Helicase DDX25 Is a Negative Regulator of Type I Interferon Pathway and Facilitates RNA Virus Infection. Front. Cell. Infect. Microbiol. 7:356. doi: 10.3389/fcimb.2017.00356
Received: 19 April 2017; Accepted: 25 July 2017;
Published: 04 August 2017.
Edited by:
Shelton S. Bradrick, University of Texas Medical Branch, United StatesReviewed by:
Kui Li, University of Tennessee Health Science Center, United StatesAlan G. Goodman, Washington State University, United States
Copyright © 2017 Feng, Sun, Li, Pan, Wang and Dai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianfeng Dai, ZGFpamlhbmZlbmdAc3VkYS5lZHUuY24=