 Zuzana Krocova
Zuzana Krocova Ales Macela
Ales Macela Klara Kubelkova
Klara Kubelkova- Department of Molecular Pathology and Biology, Faculty of Military Health Sciences, University of Defence, Hradec Kralove, Czechia
The intracellular bacterial pathogen Francisella tularensis causes serious infectious disease in humans and animals. Moreover, F. tularensis, a highly infectious pathogen, poses a major concern for the public as a bacterium classified under Category A of bioterrorism agents. Unfortunately, research has so far failed to develop effective vaccines, due in part to the fact that the pathogenesis of intracellular bacteria is not fully understood and in part to gaps in our understanding of innate immune recognition processes leading to the induction of adaptive immune response. Recent evidence supports the concept that immune response to external stimuli in the form of bacteria is guided by the primary interaction of the bacterium with the host cell. Based on data from different Francisella models, we present here the basic paradigms of the emerging innate immune recognition concept. According to this concept, the type of cell and its receptor(s) that initially interact with the target constitute the first signaling window; the signals produced in the course of primary interaction of the target with a reacting cell act in a paracrine manner; and the innate immune recognition process as a whole consists in a series of signaling windows modulating adaptive immune response. Finally, the host, in the strict sense, is the interacting cell.
Introduction
The mammalian immune system defends against a variety of microbial pathogens. The innate and the adaptive immune responses closely collaborate in developing the stage for protective immunity against microorganisms. Early recognition of invading microorganisms is provided by germline-encoded pattern recognition receptors (PRRs) that recognize conserved microbial components known as pathogen-associated molecular patterns (PAMPs). One of the best-characterized PRRs is the still-growing family of Toll-like receptors (TLRs), which are type I integral membrane proteins recognizing such PAMPs as lipopolysaccharide (TLR4), bacterial lipoproteins (TLR2), flagellin (TLR5), and/or CpG DNA (TLR9). The members of this PRRs family are located at cell surface membranes (TLR5, TLR11, TLR4, and the heterodimers of TLR2–TLR1 or TLR2–TLR6), binding to their respective ligands at the cell surface. Others (TLR3, TLR7–TLR8, TLR9, and TLR13) are expressed on endosomal membranes, where they sense microbial and host-derived nucleic acids. TLR4 localizes to both the plasma membrane and the endosomes (see, for example, O'Neill et al., 2013). Ligand-induced dimerization of TLRs leads to signaling by almost all TLRs (except TLR3) using the adaptor protein myeloid differentiation primary response gene 88 (MyD88). MyD88 activates transcription factor NF-κB signaling via serine-threonine kinases IRAK1 and IRAK2 (Warner and Núñez, 2013). For some TLRs, other adaptor proteins are needed to assemble the receptor signaling pathway. Mal (also known as TIR adaptor protein—TIRAP) is necessary to recruit Myd88 to TLR2 and TLR4 to ensure signaling via IRAKs. In the case of TLR4, the MyD88-dependent or MyD88-independent TRIF/TRAM (TIR domain-containing adaptor inducing IFN-β/TRIF-related adaptor molecule) signaling pathways can be activated by lipopolysaccharide (LPS) of Gram-negative bacteria. Ligation of LPS to TLR4 is facilitated by lipopolysaccharide binding protein (LBP) and CD14 (Lu et al., 2008).
Both macrophages (Mϕ) and dendritic cells (DC), which act as a dominant phagocytic and antigen processing and presentation component of the immune system, are equipped, in addition to TLRs, with numerous membrane-bound and cytosolic receptors that can detect microbes. Among them are complement receptors, C-type lectin receptors (CR), and Fcγ receptors (FcγRs) at the cell membrane, as well as cytosolic nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) or interferon-inducible protein, also known as absent in melanoma 2 (AIM2). NLRs and AIM2 constitute the pattern-recognition components of inflammasomes, which sense nucleotide sequences appearing in the cytosol (Kim et al., 2016; Man et al., 2016a). Upon binding a ligand, NLRs as well as AIM2 assemble multiprotein complexes called inflammasomes, which drive pyroptosis and proteolytic cleavage of the proinflammatory cytokines pro-IL-1β and pro-IL-18. The NLRs and/or AIM2 proteins recruit the inflammasome adaptor protein ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain), which in turn interacts with pro-caspase-1 leading to its activation. Once activated, caspase-1 promotes maturation of the proinflammatory cytokines interleukin (IL)-1β and IL-18 (Jin and Xiao, 2015; Xiao, 2015). The general scheme of signaling pathways associated with ligation of PRRs is presented in Figure 1.
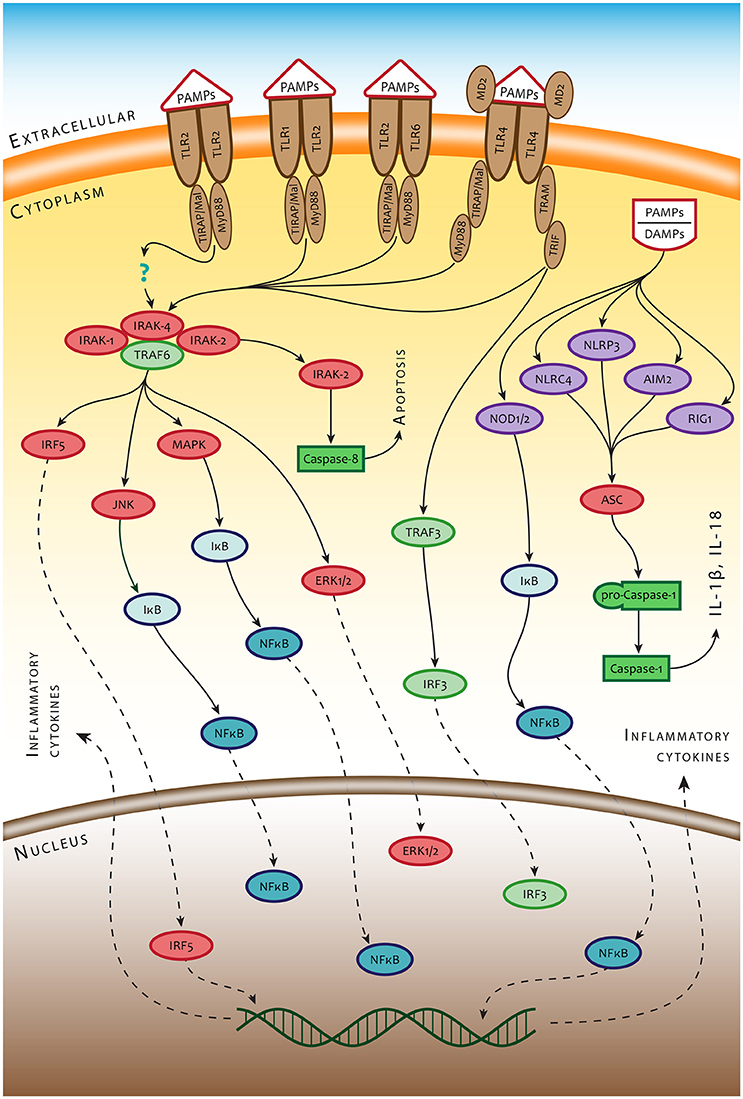
Figure 1. Simplified scheme of innate immune recognition mediated by PRRs. TLR2/TLR1, TLR2/TLR6 heterodimers, and TLR2 homodimer are controlled mainly by the MyD88-dependent signaling pathway and/or the TRIF-dependent signaling pathway using sorting adaptors TIRAP/Mal and TRAM. MyD88 recruits IRAK4, IRAK1, IRAK2 and TRAF6 and induces inflammatory responses by activating NF-κB, MAPK, and IRF5. TRIF recruits TRAF6 and TRAF3, which leads to activation of MAPK and NF-κB. The signals from cell surface PRRs control the ultimate fate of the cell and production of intercellular signals inducing inflammatory response to infection. A different set of PRRs and amplification mechanisms operate in detecting bacteria inside the cytosol. Bacterial small nucleic acids secreted into the cytosol and bacterial mRNA are recognized by RNA-sensing RIG-1 or DNA-sensing Aim2 and NLRP3. Such structural components of bacteria as, for example, flagellin or peptidoglycan are recognized by NLRC4 and NOD1/2 receptors, respectively. Recognition of intracytosolic bacterial nucleic acids activates inflammasome(s) through the adaptor molecule ASC, which leads, in turn, to activation of caspase 1 and production of IL-1 beta (IL-1β) and IL18.
Serious infectious diseases in humans and animals caused by intracellular bacteria pose a major concern for the public because, to date, researchers have failed to develop effective vaccines. The reasons lie in the complicated pathogenesis and incomplete understanding of the innate immune recognition processes controlling the generation of immune responses. Knowledge regarding both the innate immune recognition of pathogens and the outfit of pathogens enabling the avoidance of defensive reactions by host cells at the beginning and, subsequently, of the whole host immune system response are the keys to constructing efficient prophylactic tools. Recent evidence supports the concept that the immune response to external stimuli in the form of bacteria is guided by the primary interaction of the bacterium with the host cell. In this review, we provide the basic paradigms of the innate immune recognition concept arising from analyses of data obtained from the various Francisella models.
Francisella tularensis—Etiological Agent of Tularemia
Francisella tularensis (F. tularensis) is one of the most virulent microorganisms currently known. Francisellae are Gram-negative intracellular bacteria causing the zoonotic systemic disease tularemia (Carvalho et al., 2014). Although, the severity of illness varies greatly depending upon which Francisella subspecies induces the disease, the taxonomy of the genus Francisella is in fact somewhat uncertain. Currently, there are four recognized species: F. endosymbionts, F. philomiragia, F. novicida, and F. tularensis with three subspecies (tularensis, holarctica, and mediasiatica; Duncan et al., 2013). The majority of tularemia cases are caused by Type A F. tularensis subsp. tularensis found exclusively in North America and Type B F. tularensis subsp. holarctica found throughout the northern hemisphere. A big unknown has long been the taxonomy of F. novicida, which is frequently used as a model microorganism to study the pathogenesis of Francisella infections. Based on genomic, virulence, pathogenic, clinical, and finally ecological differences, between F. novicida and F. tularensis, it was recently suggested that F. novicida and F. tularensis be maintained as separate species (Kingry and Petersen, 2014).
The morbidity and mortality of infection caused by different F. tularensis strains vary also according to the gateway of infection. The most dangerous is the pneumonic form of tularemia, followed by gastrointestinal, ulceroglandular, and oculoglandular forms. Francisella invades and replicates within phagocytic cell types, such as Mϕ and DC, as well as structural tissue cells, included hepatocytes, alveolar type II cells, or endothelial cells. For this reason, F. tularensis has been occasionally called a promiscuous intracellular pathogen (Hall et al., 2008). F. tularensis has been previously shown to infect and replicate in Mϕ, both in vitro and in vivo (Thorpe and Marcus, 1964a,b; Nutter and Myrvik, 1966; Fortier et al., 1994). The attenuated F. tularensis type B live vaccine strain (LVS) replicates exponentially in mouse and human DC (Bosio and Dow, 2005; Ben Nasr et al., 2006), and the strain F. tularensis Type A Schu S4 efficiently infects and replicates in human myeloid DC (Chase et al., 2009). The CD11b(high) Mϕ, DC, monocytes, and alveolar type II cells in murine lung were shown to be infected after intranasal infection with several strains of F. tularensis (Hall et al., 2008). Murine peritoneal Mϕ (F4/80+), neutrophils (Gr-1+CD11b+), and surprisingly almost all B1a B cells (CD19+CD5+CD11b+) have also been shown to be infected at different frequency after experimental intraperitoneal infection induced by LVS (Plzakova et al., 2014).
The prophylaxis of tularemia infection is still problematic. The only vaccine, the live vaccine strain (LVS), is not authorized for human use. The current effort to construct a new F. tularensis vaccine is focused on developing both live attenuated and subunit vaccines. Live attenuated vaccine candidates are constructed by deleting genes involved mainly in metabolic and/or virulence pathways, which genes are necessary for F. tularensis intracellular replication and in vivo survival (Marohn and Barry, 2013). Subunit vaccine construction is oriented to Francisella molecular components that induce some degree of protection against lethal respiratory changes, for example surface proteins or lipoproteins administered with appropriate adjuvants or incorporated into liposomes (Putzova et al., 2016). A substantial challenge for vaccine development is to ascertain why Francisella seems to be immunologically silent for several days post infection. Vitally needed, therefore, is knowledge regarding host–pathogen interaction in general, and particularly during early stages of the innate immune response that modulate the induction, regulation, and expression of the adaptive immune response.
Recognition at the Host Cell Membrane
As a Gram-negative bacterium, F. tularensis has LPS as a dominant component of its cellular surface. Similar to those of other Gram-negative bacteria, F. tularensis LPS is composed of lipid A, which anchors the LPS to the outer membrane, a core oligosaccharide attached to lipid A, 3-deoxy-D-manno-octulosonic acid (Kdo), and an O-polysaccharide (also known as O-antigen) which contains a varying number of tetrasaccharide repeating units (Gunn and Ernst, 2007). Francisella LPS also has many unusual characteristics, however, and these lead to unexpected consequences during innate immune recognition (Okan and Kasper, 2013). The LPS of gram-negative bacteria is generally recognized by TLR4/MD2, the PRR at the surface of a host cell, and induces a strong proinflammatory response (Maeshima and Fernandez, 2013; Park and Lee, 2013). One therefore could assume that TLR4 will be the dominant PRR recognizing F. tularensis at the cell membrane. Purified F. tularensis LPS has been shown, however, not to have an agonistic or antagonistic effect on the Escherichia coli LPS-induced activation of J774 cells and to have relatively weak endotoxic activity (Sandström et al., 1992; Ancuta et al., 1996; Telepnev et al., 2003; Hajjar et al., 2006). This has been somewhat surprising, because in a previous study the authors reported that TLR4-defective mice (C3H/HeJ strain) were more susceptible than wild-type mice to intradermal infection with LVS (Macela et al., 1996). The limited ability of F. tularensis LPS to signal via TLR4 might depend on some structural properties of the lipid A moiety, most likely related to the number and length of the acyl chain substituents and absence of phosphate moieties (Dueñas et al., 2006; Maeshima and Fernandez, 2013). In parallel, it was demonstrated that TLR4 does not contribute to resistance of mice to airborne type A F. tularensis infection or intradermal infection caused by LVS (Chen et al., 2004, 2005). Other studies also have demonstrated the inability of F. tularensis LPS to act as either TLR agonists or antagonists (Ancuta et al., 1996; Hajjar et al., 2006). Nevertheless, some constituents of the bacterial body alone can function as TLR4 agonists. For example, the recombinant F. tularensis heat shock protein DnaK induced maturation of murine bone marrow-derived DC (demonstrated by an up-regulation of costimulatory molecules CD40, CD80, and CD86) and activated the production of proinflammatory cytokines (IL-6, TNF-alpha, and IL-12 p40, as well as low levels of IL-10) in a TLR4-dependent manner (Ashtekar et al., 2008). These finding may explain the observation (Macela et al., 1996) that TLR4-defective mice are more susceptible than wild-type mice to intradermal infection with LVS. Thus, TLR4 might, to some extent, be engaged in Francisella recognition at the cell membrane and, as a coreceptor, could modulate TLR2 signaling pathways downstream.
During the first decade of the twenty-first century, there appeared increasing evidence suggesting that TLR2 is involved in the recognition of F. tularensis on the surface of mouse innate immune system cells. TLR2 after ligation recognizes lipid-containing PAMPs such as lipoteichoic acid and di- and tri-acylated cysteine-containing lipopeptides, lipoarabinomannan from mycobacteria, or zymosan from yeast. The specific recognition of ligands by TLR2 is realized either in the form of TLR2 homodimer or as a heterodimer with TLR1 (recognizes tri-acylated lipopeptides) or TLR6 (recognizes the di-acylated ligand; Botos et al., 2011). Within Francisella models, TLR2–/– mice had impaired bacterial clearance from livers, lungs, and spleens after intranasal challenge with a sublethal dose of F. tularensis LVS. Moreover, infected TLR2–/– mice succumbed to a 10-fold lower challenge dose than did wild-type mice (Malik et al., 2006). Further studies documented that TLR2 of the mouse Mϕ and DC plays a significant role in the recognition of F. tularensis and functional activation of their antigen presenting function and controls the proinflammatory cytokine gene transcription (Katz et al., 2006; Li et al., 2006; Malik et al., 2006; Cole et al., 2007). DC from TLR2-deficient mice failed to produce IL-12p70 and did not co-stimulate liver lymphocytes for IFN−γ production in response to viable F. tularensis organisms (Hong et al., 2007). TLR2-dependent signaling appears to some extent to control F. tularensis infection and modulate inflammatory responses monitored by expression of proinflammatory cytokines and chemokines, such as TNF-α, IL-1β, IL-6, Mϕ inflammatory protein 1α, and Mϕ inflammatory protein 2 (Katz et al., 2006; Li et al., 2006; Malik et al., 2006; Abplanalp et al., 2009). TLR2 signaling seems to be dependent on new bacterial protein synthesis because the TLR2 agonist activity was abrogated when F. tularensis LVS organisms were heat- or formalin-killed or treated with chloramphenicol (Cole et al., 2007).
A substantial number of studies have identified TLR2 signaling as a critical event during the host innate immune response to F. tularensis infection. The data may indicate that, in a Francisella model, the TLR2 alone (perhaps as a homodimer—see for example Zheng et al., 2015; Udgata et al., 2016), rather than TLR2/TLR1 or TLR2/TLR6 heterodimers, play a critical role in F. tularensis recognition at the surface of immunocompetent cells (Abplanalp et al., 2009). Furthermore, a TLR2-independent pathway for activation of macrophages has also been identified (Hong et al., 2007). Contrary to the majority of information, some data suggest that signals utilizing the adaptor protein MyD88 without the involvement of TLR2 are essential for controlling resistance to intradermal challenge with F. tularensis LVS but not for intra-macrophage bacterial multiplication (Collazo et al., 2006). Adaptor protein MyD88 is used by almost all TLRs (except TLR3) to activate the transcription factor NF-κB (Lord et al., 1990). TIRAP/Mal (TIR domain-containing adaptor protein/MyD88-adaptor-like), an adaptor protein closely related to MyD88, is necessary to recruit Myd88 to TLR2 as well as to TLR4 (Horng et al., 2001, 2002). In the context of other Francisella studies it was demonstrated that the molecular complex of TLR2/MyD88 (signaling through IRAKs; Arancibia et al., 2007) is indispensable for NF-κB activation initiating macrophage proinflammatory cytokine production and, subsequently, protective innate immune responses in mice following challenge with attenuated as well as virulent F. tularensis strains (Collazo et al., 2006; Abplanalp et al., 2009; Cole et al., 2010; Russo et al., 2013).
Recognition of Francisellae at the host cell membrane is a fundamental step in its life cycle because it facilitates bacterial entry into host cells. This event is not exclusively a matter of TLRs. Such receptors as C-type lectin receptors, complement receptors (CR), and Fc-gamma receptors (FcγR) were shown in a specific situation to recognize either unopsonized or opsonized bacteria. The presence of natural opsonins in an experimental setup has a major role in the early phases of host–pathogen interactions and alters the intracellular fate of bacteria (Dai et al., 2013). Several models of Francisella–host cell interaction have identified the receptors engaged in the Francisella uptake at the host cells surface. The mannose receptor, one of the C-type lectin receptors (Balagopal et al., 2006; Schulert and Allen, 2006), the complement receptors CR3 (CD11b/CD18) in the case of Mϕ (Clemens et al., 2005; Balagopal et al., 2006; Geier and Celli, 2011), CR4 (CD11c/CD18) in the case of DC (Ben Nasr et al., 2006), and CR1/2 in the case of B cells (Plzakova et al., 2015), the scavenger receptor A (SRA) (Pierini, 2006; Geier and Celli, 2011), FcγRs (Balagopal et al., 2006; Geier and Celli, 2011), and surface-exposed nucleolin with its bacterial ligand EF-Tu (Barel et al., 2008, 2010; Barel and Charbit, 2014) are involved in internalization of Francisellae into host cells.
Opsonization of F. tularensis Schu S4 strain with fresh serum or purified antibodies reoriented the interaction of bacteria with mouse bone marrow-derived Mϕ from the mannose receptor to the complement receptor CR3, the scavenger receptor A (SRA), and the FcγR (Geier and Celli, 2011). Experimental data demonstrated that opsonization of bacteria prior to engulfment by phagocytes substantially changes the intracellular fate of the bacteria and modulates parameters of the host APCs response to infection. CR3-mediated uptake of Francisellae negatively modulated maturation of the early Francisella-containing phagosome (FCP) and minimize phagosomal escape, whereas FcγR-dependent phagocytosis was associated with intensive superoxide production in the early FCP, a rapid, FcγR-mediated, NADPH oxidase-dependent oxidative burst, and restricted phagosomal escape (Geier and Celli, 2011). Serum opsonins modulate maturation of human monocyte-derived immature DC and change their cytokine production profile in favor of IL-10 at the expense of IL-12 production (Ben Nasr et al., 2006). Efficient attachment and uptake of the highly virulent Type A F. tularensis subsp. tularensis strain Schu S4 by human monocyte-derived macrophages (hMDMs) require complement C3 opsonization and CR3. A complex cascade of events ending in uptake of Francisellae by phagocytes can initiate natural IgM binding to surface capsular and/or O-Ag polysaccharides of F. tularensis, a process activating classical complement cascade via C1q and promoting C3 opsonization of the bacterium and phagocytosis via CRs in a phagocyte-specific manner. CR1 (CD35) and CR3 (CD11b/CD18) have been observed to act in concert for phagocytosis of opsonized F. tularensis by human neutrophils, whereas CR3 and CR4 (CD11c/CD18) mediated uptake by hMDMs (Schwartz et al., 2012b). However, the CR3 engagement in an efficient uptake of Francisellae by hMDMs, in parallel, initiated CR3–TLR2 crosstalk leading to down-regulation of TLR2-dependent proinflammatory responses by inhibiting MAPK activation through outside–in signaling (Dai et al., 2013). Thus, such complex activation of several receptor signaling pathways influences the result of host–microbe interaction.
Taken together, the entry of Francisellae into host cells can be realized either by uptake of unopsonized or opsonized bacteria. In real in vivo situations, uring natural infections, however, the uptake of opsonized bacteria is probably much more frequent than contact of host cells with unopsonized microbes. This latter mode of cell infection comes into play only during phagocytosis of whole infected cells by bystander phagocytes or during cytosolic transfer between macrophages via the process known as trogocytosis (Bourdonnay and Henry, 2016; Steele et al., 2016). Engagement of opsonophagocytic receptors alters the intracellular trafficking of Francisella by modulating the phagocytic pathways that restrict phagosomal escape and intracellular proliferation. Both can impact profoundly the final fate of Francisella in a host by modulating the intracellular recognition of Francisella in a cytosolic compartment of a host cell. These facts should be taken into account when designing in vitro experimental cell infection systems.
Innate Recognition at Intracellular Compartments
Uptake of Francisella by host cells is dependent on actin polymerization and functional microtubules in both phagocytic and nonphagocytic cells (Clemens and Horwitz, 2007; Lindemann et al., 2007). The mechanism of Francisella entry into host cells is dependent on the host cell type and the conditions under which the interaction with host cells takes place. One of the specific forms of entry is the formation of asymmetric, spacious pseudopod loops around live or killed bacteria (Clemens et al., 2005). More general mechanisms include macropinocytosis that had been demonstrated for the entry of Francisella LVS into type II alveolar epithelial cells or, rarely, during infection of macrophages with LVS (Clemens et al., 2005; Bradburne et al., 2013). Early after interaction with host cell surface receptors Francisella is enclosed in a phagosome. There is no information available as to whether the bacteria are recognized during this stage of intracellular trafficking in spite of the fact that phagosomal PRRs recognize bacterial molecular markers (for example, phagosomal TLR9 has the capacity to recognize bacterial CpG motifs). The only information on intracellular colocalization of Francisella and TLR2 and MyD88 within macrophages suggests that Francisella LVS initiates signaling through TLR2 both at the cell surface and within the phagosome (Cole et al., 2007). The original engagement of the cell membrane PRRs and opsonophagocytic receptors on the cell surface, through which the mutual interaction of Francisella with a host cell is realized, produces the signals that dictate the fate of Francisella during intracellular trafficking (see, for example, Geier and Celli, 2011). Through modulation of phagosome biogenesis, Francisella escapes from its initial phagosome into the cytosol of a host macrophage.
Escape from the phagosome is the second fundamental step in the Francisella life cycle. This event is very dynamic, and experimental data has documented that phagosomal escape occurs within several tens of minutes to several hours post infection, depending upon the experimental setup (Golovliov et al., 2003; Clemens et al., 2004; Santic et al., 2005, 2008; Checroun et al., 2006). The cytosolic compartment of infected cells enables the proliferation of Francisellae. Once in the cytosol, however, the bacterial load is monitored by intracellular cytosolic DNA sensors, such as DNA-dependent activator of IFN-regulatory sensor (Takaoka et al., 2007), RIG-I (Ablasser et al., 2009), and/or AIM2 (Fernandes-Alnemri et al., 2009; Hornung et al., 2009; Jones et al., 2010), or by cytosolic nuclear oligomerization domain (NOD)-like receptors (NLRs) (Franchi et al., 2008, 2009). Both types of sensors are critical for innate defense by recognizing conserved structures of microorganisms. Upon sensing adequate ligands, these cytosolic PRRs trigger oligomerization of the inflammasome complex. Once completed, inflammasomes interact with 45-kDa pro-caspase 1, which undergoes auto-proteolytic processing that results in active caspase 1 (Thornberry et al., 1992; Miller et al., 1993; Ayala et al., 1994; Wilson et al., 1994). Subsequent cleavage of pro-IL-1β and pro-IL-18 into their mature forms is critical for the host response to infection and is accompanied by caspase-1-dependent inflammatory cell death—pyroptosis (Man and Kanneganti, 2015).
Most information related to cytosolic recognition of Francisella has originated from F. novicida experimental models. Mice lacking AIM2, ASC, or caspase-1 are highly susceptible to infection and exhibit an increased bacterial burden compared with wild-type mice (Mariathasan et al., 2005; Fernandes-Alnemri et al., 2010; Jones et al., 2010). Macrophages from mice lacking AIM2 cannot sense cytosolic double-stranded DNA and fail to trigger inflammasome assembly. Immunofluorescence microscopy of macrophages infected with Francisella further revealed striking colocalization of bacterial DNA with endogenous AIM2 and inflammasome adaptor ASC (Jones et al., 2010). For the intracytosolic recognition of Francisella, therefore, the assembly of inflammasome and the accessibility of bacterial DNA or other molecular components of the bacteria are key events. In the case of AIM2 inflammasome and Francisella, the critical role seems to be the integration of innate immune signaling. TLR2 signaling through MyD88 and NF-κB in macrophages infected with F. novicida contributes to the rapid induction of inflammasome assembly and inflammasome functional activation (Jones and Weiss, 2011). How the recognizable bacterial components in the cytosol are produced is still under investigation. They can be produced directly in the cytosol or, alternatively, can originate from dead, partially destroyed bacteria released into the cytosol after fragmentation of the phagosomal membrane by live, fully active bacterial partners entrapped in a phagosome together with dead ones. For direct destruction of bacteria residing in the cytosol it can be argued that assembly and activation of the AIM2 inflammasome during infection with F. novicida requires transcription factor IRF1. The DNA sensor cGAS and its adaptor STING (Sun et al., 2013; Wu et al., 2013) induce type I interferon-dependent expression of IRF1. The IRF1 subsequently modulates the expression of guanylate-binding proteins (GBPs) that can ensure intracellular killing of bacteria and mediate cytosolic release of ligands for recognition by the AIM2 inflammasome (Man et al., 2015). Moreover, the interferon-inducible protein IRGB10 participates in the cytosolic destructive process of F. novicida by a mechanism requiring guanylate-binding proteins (Man et al., 2016b). Just because they are recognized by cytosolic DNA sensors does not necessarily mean that the bacteria will be killed; their product, cyclic dinucleotides, is sensed by STING directly and can initiate the innate immune response (see below).
Evidence of type I IFN involvement in the process of cytosolic recognition of Francisella by the inflammasome documents the need for multifold signal integration during adaptation of cells to recognize intracellular pathogens during primary interaction with host cells (Henry et al., 2007). Type I IFN signaling was observed to be necessary for activation of the inflammasome during infection with F. novicida. Production of type I IFN was coupled with recognition of cytosolic F. novicida. The process of F. novicida recognition was dependent on IRF-3 signaling and independent of signaling from RIG-I, MDA5, Nod1/2, and inflammasome adaptors. It was also independent of TLR signaling, which evidently demonstrated the intracytosolic recognition process (Henry et al., 2007). Production of type I IFN by BMDM after infection with F. tularensis LVS was observed to be dependent on STING, also known as MITA, MPYS, ERIS, and TMEM173. STING functions as both a direct cytosolic DNA sensor and an adaptor protein utilizing different molecular mechanisms (Ishikawa and Barber, 2008; Ishikawa et al., 2009). Downstream, STING activate transcription factors STAT6 and IRF3 through kinase TBK1 (Burdette and Vance, 2013). Signaling utilizing STING by cultured macrophages infected with LVS was required for type I IFN production, however, in parallel; a STING-dependent as well as a STING-independent signaling pathway were activated in in vivo Francisella infection models (Jin et al., 2011). The cyclic GMP-AMP synthase cGAS (Sun et al., 2013), as well as IFI214, a murine homolog of IFI16 (Unterholzner et al., 2010; Veeranki and Choubey, 2012), act as cytosolic DNA sensors and seem to be involved in the sensing of intracytosolic Francisella DNA; both contribute to STING-dependent type I IFN response to high concentrations of cytosolic dsDNA (Storek et al., 2015). Moreover, as a direct innate immune sensor, STING alone can recognize cyclic dinucleotides (c-di-AMP and c-di-GMP), which are produced by bacteria, and mediate type I IFN cell response (Burdette et al., 2011; Barker et al., 2013).
To recapitulate and summarize the studies on intracytosolic recognition of Francisella, there are multiple intracytosolic signaling pathways that can, alone or in tandem, ensure the recognition of Francisellae localized in cytosolic compartment of a host cell. Reasons for these findings may be several. Unterholzner (2013) summarizes possible answers to the question of why so many receptors recognize DNA localized in the cytosol (nucleus) and induce an interferon response. First, receptors may have redundant functions. Further, DNA receptors may differ in their ligand specificity, different DNA receptors operate in different cell types, and/or receptors may act sequentially over time. Finally, some of the proposed DNA sensors may not be receptors. For our model of the intracellular pathogen Francisella, it can be concluded that at least two basic intracytosolic recognition processes are indispensable for expression of a proinflammatory response to infection. First, and in general, it is such recognition which results in the production of type I IFN needed for activation of IRFs. The second process is the expression of components and their assembly into inflammasome, by which the Francisella is recognized and the production of proinflammatory IL-1β and IL-18 cytokines is ensured.
Signaling Windows Concept—Spatiotemporal Network of Cellular Hosts
The emerging concept of signaling windows of innate immune recognition is based on the idea that there exist functional cellular immune response modules that temporarily, in spatiotemporal configuration, regulate innate immune recognition and sequentially modulate induction, regulation, and expression of the adaptive immune response. Intracellular pathogens such as Francisella are ideal models to construct realistic scenarios of innate immune recognition. Utilizing integrated approaches and analyzing the fate of mutual host cell–pathogen interaction, recent complex dynamic studies on immune cell networking have attempted to overcome the traditional static view on induction of immune cell signaling during induction of the immune response (see, for example, Nunes-Alves et al., 2014; Budak et al., 2015; Hotson et al., 2016; Rothchild et al., 2016). The Francisella experimental infection models provide sufficient information for attempting to construct the architecture of innate immune signaling windows. The basic paradigms of the signaling window concept define, in the strict sense, a cell as a decisive host of microbes and accept the idea of functional cellular modules of immune responses. The concept encompasses several basic assumptions: (1) There is a “jumble” of cellular hosts within a multicellular organism infected by bacteria. (2) Cellular hosts create a four-dimensional net in the host organism. (3) Cellular hosts recognize and respond to interaction with the bacterium on spatiotemporal levels. (4) The host cell with which a microbe originally interacts forms the first signaling window. (5) Every spatiotemporal level opens a new signaling window. (6) The interplay among individual signaling windows ensures paracrine cytokine messages induced by microbial challenge. (7) Signaling windows integrate innate immune recognition signals. (8) PRRs cross-inhibition and interference of host and microbe signals influence the result of host–microbe interaction.
Macrophages and DC, which have been most utilized for studies on innate immune recognition, are not the only cells that recognize Francisellae in the context of the innate immune response. Moreover, TLRs are not the only cell surface receptors engaged in the innate immune recognition. Francisella infects phagocytic as well as nonphagocytic cells. Along with macrophages and DC, neutrophils (Löfgren et al., 1983; Hall et al., 2008), B cells (Plzakova et al., 2014), endothelial cells (Forestal et al., 2003), epithelial cells (Gentry et al., 2007; Lindemann et al., 2007), and/or hepatocytes (Law et al., 2011; Rennert et al., 2016) are all potential primary targets of Francisella. All these are permissive, and all are significant producers of signals representing the first signaling window. Neutrophils utilize a combination of NADPH oxidase-derived reactive oxygen species (ROS), antimicrobial peptides, and degradative enzymes to kill engulfed microorganisms for innate host defense (Kennedy and DeLeo, 2009). Phagocytosis of microorganisms leads very quickly to neutrophil apoptosis, which is known also as phagocytosis-induced cell death (Kobayashi et al., 2003). Tested strains of Francisella, however, inhibit the respiratory burst and profoundly prolong neutrophil lifespan (Schwartz et al., 2012a). In parallel, at the gene level, Francisella infection of neutrophils enhances expression of such neutrophil-specific survival factors as cdk2 or cdk7, cytokine and chemokine genes that promote inflammation as well as neutrophil survival (Il1b, Il1rn, Il6, osm, pbef1, cxcl1, 0ccl4, cxcr4). Francisella also has been shown to significantly affect expression of genes associated with cytosolic pattern recognition systems and inflammasome activation, as well as with early induction of NLRP3 and NOD2 followed by down-regulation of AIM2, NAIP, PYCARDI, and NLRP1. As in other cells that may be regarded as providing the cellular background of the first signaling window, Francisella escapes from the phagosome into the neutrophil cytosol (McCaffrey and Allen, 2006), where it might be recognized by inflammasomes or NOD-like receptors. B cells are engaged in the strong early protective response against F. tularensis LVS (Culkin et al., 1997). As early as 12 h post infection, peritoneal CD19(+) cells produce IFN-γ, IL-1β, IL-4, IL-6, IL-12, IL-17, IL-23, and TNF-α (Plzakova et al., 2014). Mice deficient in mature B cells and antibodies (B-cell knockout mice) actually control primary sublethal infection but are 100-fold less well protected against a secondary lethal challenge (Elkins et al., 1999). Direct contact and entry of Francisella into B cells, depending on a given B cell subset, are mediated by B cell receptors (BCRs) with or without complement receptor CR1/2. In the B-1a cell subset, BCRs alone can ensure the internalization process, whereas BCRs on B-1b and B-2 cells require co-signaling from the coreceptor containing CR1/2 in order to initiate F. tularensis engulfment (Plzakova et al., 2015). The production of IL-1β by infected B cells suggests early cytosolic innate immune recognition after interaction of Francisella and B cell. At some stage of dissemination into various organs, Francisella must overcome the endothelial barrier in the microvasculature by one of three well-known mechanisms: transcellular, paracellular, or the so-called Trojan horse mechanism (i.e., crossing the barrier using infected phagocytes). To achieve this, Francisella readily adheres to the endothelial cell surface and uses PilE4 (type IV pili subunit) to interact with ICAM-1 molecule, adhere to the endothelial surface, and cross the endothelial barrier in vivo as well as in vitro (Bencurova et al., 2015). Endothelial as well as epithelial cells and hepatocytes produce a variety of cytokines at various levels after interaction with bacteria. This is just a reason for modulation of the functional profile of cells subsequently interacting with Francisellae, and in the suggested concept it corresponds to opening of the secondary signaling windows.
As part of innate immune response, moreover, unconventional T cell subsets might be engaged in innate immune recognition and constitute a bridge between innate and adaptive immune responses. This might be achieved through unconventionally presented byproducts or intermediates of bacterial metabolism secreted by the bacteria early after interaction with the host cells. Human Vγ9/Vδ2 T cells recognize specifically, in a non-MHC restricted manner, microbial isoprenoid precursor (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate (HMB-PP), which is an intermediate of the microbial non-mevalonate pathway of isoprenoid biosynthesis utilized by most pathogenic Gram-negative and Gram-positive bacteria (Eberl and Jomaa, 2003; Eberl et al., 2003; Heuston et al., 2012). This molecule can function as a danger signal because HMB-PP is not present in higher eukaryotes, including humans (Sicard and Fournie, 2000; Morita et al., 2007). One of the consequences of the human Vγ9/Vδ2 T cells–pathogen interaction, and, in a broader sense, of their activation, is the rapid acquisition of antigen presenting cell characteristics that are reminiscent of mature DC (Moser and Eberl, 2011; Tyler et al., 2015), and in such way they can function as a primary signaling window. Concerning human tularemia, Vγ9/Vδ2 T cells expand after infection and comprise on average 30% of human peripheral blood T lymphocytes, thus suggesting some role in control of F. tularensis infection (Poquet et al., 1998). Experiments with co-culture of human Vγ9/Vδ2 T cells isolated from healthy donors with the THP-1 human monocyte cell line infected with F. tularensis has demonstrated the ability of Vγ9/Vδ2 T cells to recognize infection, to produce a whole range of cytokines and chemokines (including IL-1β, IL-6, and IFN-γ, but not IL-10), and to limit bacterial proliferation in the culture (Rowland et al., 2012). Another unconventional T cell subset, mucosa-associated invariant T (MAIT) cells, also responds very early and intensively to F. tularensis infection. A murine in vivo model of sublethal F. tularensis LVS pulmonary infection demonstrated robust expansion of MAIT cells in the lungs during the early acute phase of infection. The MAIT cells recognized vitamin B metabolites in association with evolutionarily conserved MHC-related protein 1 (Kjer-Nielsen et al., 2012), which possesses a unique antigen-binding cleft producing an antigen presenting function on this cell type (Huang et al., 2005). Because vitamin B biosynthesis pathways are unique to bacteria and yeast, MAIT cells may also constitute one of the primary signaling windows.
Knowing that the severity and time course of tularemia is, at least in an experimental setup, dependent on the route of infection, we can accept the view that these differences are due, at least in part, to the various types of original cellular hosts that convey primary interaction with the bacteria. Thus, the initial signals originating from different infected cell types might dictate the outcome of primary host cell–bacterium interaction, which in turn affects all subsequent events during the induction of immune responses. The cells that interact with the Francisellae in the first sequence create a timeframe for modulating the functional profile of cells that will interact with the bacterium in a secondary order. The intercellular contact is mediated by an integrated cytokine message produced by originally infected cellular “senders.” To our knowledge, the lifetime of infected cells in the case of macrophages is from 12 to 18 h post infection, depending on the experimental model (Libich, 1981). The transient bacteremia contributing to spreading Francisellae over the body begins 24–48 h post infection, which is thus the timeframe for changing the microenvironment for as yet uninfected cells. Cellular “receivers” located in a given microenvironment can thus respond accordingly. We hypothesize that the modulated functional profile of a secondarily reacting cell will be dependent on the type of cell that initiates the cytokine messages as well as on the expressed cell surface receptors able to recognize the cytokine messages and simultaneously (or subsequently) recognize incoming bacterium. The cytokine response of a cellular “receiver” of the same cell type as the cellular “sender,” whether infected or not, would necessarily be different from the message produced by the originally infected cell. Moreover, in the case of antigen presenting cells reacting in the secondary order, the recognition, handling, and processing the interacting bacteria might be quite different from the processes of the primary infected cells and could have a profound impact on the expression of the adaptive immune response. An example is the paracrine action of type I IFN, which changes the transcriptional response to innate immune recognition of Francisellae by infected primary human monocyte-derived macrophages and primary murine peritoneal macrophages but not by murine bone marrow-derived macrophages. This type I IFN-dependent modulated response of infected cells is synergistic with TLR2 transcriptional responses, partially TLR2-independent, but strictly MyD88-dependent, thus suggesting the supplementary action of co-receptor(s) (Richard et al., 2016). Alternatively, it could demonstrate the modulated function of cells reacting to Francisellae as a receiver of signals generated by cells infected in the primary order. An example can be seen in the signaling through IL-1 receptor of cells infected with Francisellae in a secondary order (Figure 2).
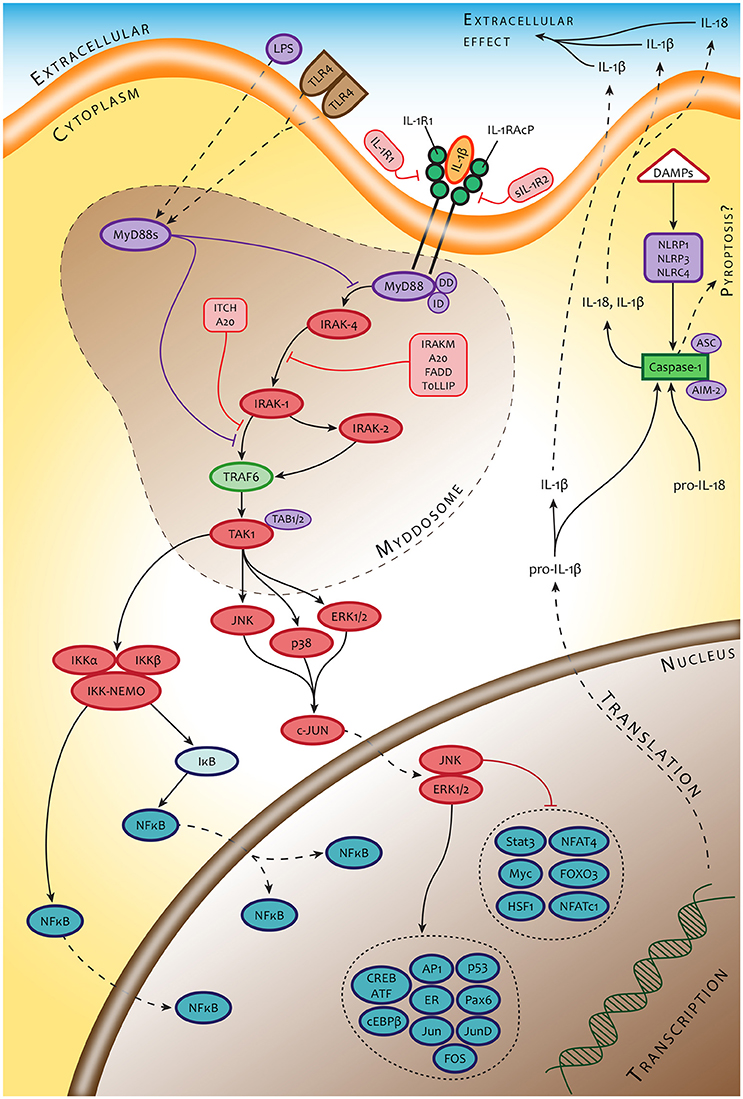
Figure 2. An example of signaling pathways of cells interacting with Francisella in secondary order. The signaling pathways of these cells are modulated by signals originating from a primary responding cell (IL-1β) or collateral signals from invading microbes (LPS).
Thus, in our opinion, the data from the literature indicate that the differences in severity of tularemia depending on the route of infection can originate from differences in the local microenvironments and types of cells that are first infected with Francisella. As an example, the difference in LD50 after aerogenic and subcutaneous F. tularensis infection of mice is greater than six logarithms. Route of infection dominates over genetic background of recipients in the severity of illness. In the case of genetic background, the difference in LD50 for susceptible and resistant mice is less than three logarithms (Fortier et al., 1994). The reason for these differences can be seen in the different primary cellular hosts of Francisellae. Langerhans cells are primarily infected in dermis whereas alveolar macrophages are predominantly infected in the lungs. Such conclusion can also be deduced by comparing intranasal- vs. intradermal-induced murine tularemia (Roberts et al., 2014), which may be an example testifying to the concept proposed above.
The basic experiments with individual cell types and opsonizing bacteria have demonstrated not only the importance of responding cell type to primary interaction with Francisella but also the importance of type of expressed surface membrane receptor involved in the primary contact with the bacterium. Complement factors, such as ubiquitous opsonins in mammals, are critical factors mediating adhesion and subsequently internalization of Francisellae into host cells. The co-signaling from different surface receptors engaged in the adhesion of the bacterium modulates the response of host cells. There is an obvious dichotomy in macrophage response to interaction with C3 opsonized or unopsonized microbes, especially in relation to inflammatory response due to the interference of signaling pathways (Dai et al., 2013). Moreover, Francisella itself modulates the signaling pathways of infected cells. The data demonstrate, for example, that F. tularensis fails to induce production of proinflammatory cytokines or IFN-γ, inhibits increased expression of activation markers, including MHCII, on the surface of professional APCs, and suppresses activation of the inflammasome during early Francisella infection via targeting of TLR2-dependent signaling (Bosio and Dow, 2005; Bosio et al., 2007; Parsa et al., 2008; Chase et al., 2009; Dotson et al., 2013). There is uncertainty in the interpretation of these results, however, because much of this data was generated using different experimental setups and in different time proportions. Moreover, the majority of these studies were based on entirely artificial conditions not corresponding to in vivo situations (natural opsonization of bacteria and very high multiplicity, which is quite exceptional for natural infection). Some conclusions have been generalized according to the final fate of the eukaryotic host cell/organism–pathogen interaction using a traditional, more or less static experimental arrangement. To solve the general problem of innate immune recognition's involvement in the process of protective immunity induction, analyses are needed of the so-called social network of immune cells that participate in the early stages of infection.
To conclude the possible sequence of events during innate immune recognition of Francisella according to the emerging concept of signaling windows, we can formulate a minimum of four basic assumptions: (1) The first batch of signals, resulting from natural recognition of Francisella inside the multicellular organism, depends on the type of interacting cell and its surface receptor(s), which mediate(s) primary pathogen conjunction with the host cell. (2) Francisella–host cell interaction at the single-cell level corresponds to the concept of crosstalk between innate immune receptors and integrates the signals into a prototypic signaling response corresponding to a particular cell type. (3) During the process of Francisella innate immune recognition, cells form a four-dimensional signaling network represented by signaling windows. This means that the concept reflects the spatiotemporal changes in the function of the individual cells that are engaged in immune recognition, which are caused by changing microenvironment over time. Integrated signals at the level of signaling windows generate a new signal for “opening” the additional signaling window. (4) Interference of host and microbe signals at a single-cell level can subvert and reprogram PPRs-mediated innate immune responses.
Conclusions and Prospects
Innate immune response constitutes the first line of defense against bacterial infection. Data collected from germ-free as well as specific pathogen free animal models of microbial pathogenesis demonstrate the dependency of induce and adaptive immunity on primary interaction between a microorganism and the host cell that the microbe first encounters. The innate immune recognition process plays a dominant role along with the intrinsic characteristics of the microorganism and host. The epigenetic reprogramming of innate immune cells, creating the hierarchy of immune response functional modules, is critical for inducing and regulating expression of the adaptive immune response. This process is extremely dynamic and the populations of individual cell types, whether infected or uninfected, change their functional characteristics depending on the local microenvironment modulated by the cells that were originally infected even at a distant place. If this is the case, then not all the cells of the same cell type in the body will respond to infection in an identical way at a given time. From this point of view, the cell, with its functional and secretion profile, rather than the host organism in its entirety, seems to be the primary microbe host (Kubelkova et al., 2016). In the case of in vitro Francisella models, the literature presents quite different conclusions concerning the outcome of Francisella innate immune recognition. Some studies provide evidence that F. tularensis LVS represses inflammasome activation, while other studies have demonstrated that F. tularensis LVS increases mRNA levels of proinflammatory cytokines and this is followed by increased protein secretion. Moreover, data from experiments with host-adapted Francisella LVS isolated from infected macrophages explain this heterogeneity of results by a “stealthy mode” of intra-host lifestyle (for a review, see Holland et al., 2016).
The host cells as well as invading Francisellae are affected by their “historical memory” and mutually generate, at any given time, an immediate microenvironment that should be respected in the generation of new infection models. In our experience, for example, germ-free mice infected subcutaneously with F. tularensis reacted differently to attenuated and virulent strains in comparison with specific-pathogen free mice. These mice, not having had contact with bacteria in ontogeny, can demonstrate the unique primary reaction of their immune systems to a pathogenic bacterium and can help us to understand the processes that lead to the establishment of full-fledged protective immunity. To understand the innate immune response to infection we need to obtain multidimensional data sets providing comprehensive, cell-specific, and time-structured information on epigenetic reprogramming of innate immune cells that can provide us with the logic of interplay among immune cells.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by a Ministry of Defense of the Czech Republic—long-term organization development plan Medical Aspects of Weapons of Mass Destruction of the Faculty of Military Health Sciences, University of Defense.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors would like to thank Martin Ondrej (University of Defence) for providing us with graphical cooperation on the manuscript figures.
Abbreviations
AIM2, absent in melanoma 2; BCR, B cell receptor; CR, complement receptor; hMDMs, human monocyte-derived macrophages; LPS, lipopolysaccharide; LVS, live vaccine strain; MAIT cells, mucosa-associated invariant T cells; MyD88, myeloid differentiation primary response protein; NLRs, nucleotide-binding and oligomerization domain (NOD)-like receptors; PAMPs, pathogen-associated molecular patterns; PRRs, pattern recognition receptors; TLRs, Toll-like receptors.
References
Ablasser, A., Bauernfeind, F., Hartmann, G., Latz, E., Fitzgerald, K. A., and Hornung, V. (2009). RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 10, 1065–1072. doi: 10.1038/ni.1779
Abplanalp, A. L., Morris, I. R., Parida, B. K., Teale, J. M., and Berton, M. T. (2009). TLR-dependent control of Francisella tularensis infection and host inflammatory responses. PLoS ONE 4:e7920. doi: 10.1371/journal.pone.0007920
Ancuta, P., Pedron, T., Girard, R., Sandström, G., and Chaby, R. (1996). Inability of the Francisella tularensis lipopolysaccharide to mimic or to antagonize the induction of cell activation by endotoxins. Infect. Immun. 64, 2041–2046.
Arancibia, S. A., Beltrán, C. J., Aguirre, I. M., Silva, P., Peralta, A. L., Malinarich, F., et al. (2007). Toll-like receptors are key participants in innate immune responses. Biol. Res. 40, 97–112. doi: 10.4067/S0716-97602007000200001
Ashtekar, A. R., Zhang, P., Katz, J., Deivanayagam, C. C., Rallabhandi, P., Vogel, S. N., et al. (2008). TLR4-mediated activation of dendritic cells by the heat shock protein DnaK from Francisella tularensis. J. Leukoc. Biol. 84, 1434–1446. doi: 10.1189/jlb.0308215
Ayala, J. M., Yamin, T. T., Egger, L. A., Chin, J., Kostura, M. J., and Miller, D. K. (1994). IL-1 beta-converting enzyme is present in monocytic cells as an inactive 45-kDa precursor. J. Immunol. 153, 2592–2599.
Balagopal, A., MacFarlane, A. S., Mohapatra, N., Soni, S., Gunn, J. S., and Schlesinger, L. S. (2006). Characterization of the receptor-ligand pathways important for entry and survival of Francisella tularensis in human macrophages. Infect. Immun. 74, 5114–5125. doi: 10.1128/IAI.00795-06
Barel, M., and Charbit, A. (2014). Detection of the interaction between host and bacterial proteins: eukaryotic nucleolin interacts with Francisella elongation factor Tu. Methods Mol. Biol. 1197, 123–139. doi: 10.1007/978-1-4939-1261-2_7
Barel, M., Hovanessian, A. G., Meibom, K., Briand, J. P., Dupuis, M., and Charbit, A. (2008). A novel receptor - ligand pathway for entry of Francisella tularensis in monocyte-like THP-1 cells: interaction between surface nucleolin and bacterial elongation factor Tu. BMC Microbiol. 8:145. doi: 10.1186/1471-2180-8-145
Barel, M., Meibom, K., and Charbit, A. (2010). Nucleolin, a shuttle protein promoting infection of human monocytes by Francisella tularensis. PLoS ONE 5:e14193. doi: 10.1371/journal.pone.0014193
Barker, J. R., Koestler, B. J., Carpenter, V. K., Burdette, D. L., Waters, C. M., Vance, R. E., et al. (2013). STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. MBio 4:e00018–e00013. doi: 10.1128/mBio.00018-13
Bencurova, E., Kovac, A., Pulzova, L., Gyuranecz, M., Mlynarcik, P., Mucha, R., et al. (2015). Deciphering the protein interaction in adhesion of Francisella tularensis subsp. holarctica to the endothelial cells. Microb. Pathog. 81, 6–15. doi: 10.1016/j.micpath.2015.03.007
Ben Nasr, A., Haithcoat, J., Masterson, J. E., Gunn, J. S., Eaves-Pyles, T., and Klimpel, G. R. (2006). Critical role for serum opsonins and complement receptors CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in phagocytosis of Francisella tularensis by human dendritic cells (DC): uptake of Francisella leads to activation of immature DC and intracellular survival of the bacteria. J. Leukoc. Biol. 80, 774–786. doi: 10.1189/jlb.1205755
Bosio, C. M., Bielefeldt-Ohmann, H., and Belisle, J. T. (2007). Active suppression of the pulmonary immune response by Francisella tularensis Schu4. J. Immunol. 178, 4538–4547. doi: 10.4049/jimmunol.178.7.4538
Bosio, C. M., and Dow, S. W. (2005). Francisella tularensis induces aberrant activation of pulmonary dendritic cells. J. Immunol. 175, 6792–6801. doi: 10.4049/jimmunol.175.10.6792
Botos, I., Segal, D. M., and Davies, D. R. (2011). The structural biology of Toll-like receptors. Structure 19, 447–459. doi: 10.1016/j.str.2011.02.004
Bradburne, C. E., Verhoeven, A. B., Manyam, G. C., Chaudhry, S. A., Chang, E. L., Thach, D. C., et al. (2013). Temporal transcriptional response during infection of type II alveolar epithelial cells with Francisella tularensis Live Vaccine Strain (LVS) supports a general host suppression and bacterial uptake by macropinocytosis. J. Biol. Chem. 288, 10780–10791. doi: 10.1074/jbc.M112.362178
Budak, G., Eren Ozsoy, O., Aydin Son, Y., Can, T., and Tuncbag, N. (2015). Reconstruction of the temporal signaling network in Salmonella-infected human cells. Front. Microbiol. 6:730. doi: 10.3389/fmicb.2015.00730
Burdette, D. L., Monroe, K. M., Sotelo-Troha, K., Iwig, J. S., Eckert, B., Hyodo, M., et al. (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518. doi: 10.1038/nature10429
Burdette, D. L., and Vance, R. E. (2013). STING and the innate immune response to nucleic acids in the cytosol. Nat. Immunol. 14, 19–26. doi: 10.1038/ni.2491
Carvalho, C. L., Lopes de Carvalho, I., Zé-Zé, L., Núncio, M. S., and Duarte, E. L. (2014). Tularaemia: a challenging zoonosis. Comp. Immunol. Microbiol. Infect. Dis. 37, 85–96. doi: 10.1016/j.cimid.2014.01.002
Chase, J. C., Celli, J., and Bosio, C. M. (2009). Direct and indirect impairment of human dendritic cell function by virulent Francisella tularensis Schu S4. Infect. Immun. 77, 180–195. doi: 10.1128/IAI.00879-08
Checroun, C., Wehrly, T. D., Fischer, E. R., Hayes, S. F., and Celli, J. (2006). Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc. Natl. Acad. Sci. U.S.A. 103, 14578–14583. doi: 10.1073/pnas.0601838103
Chen, W., KuoLee, R., Shen, H., Bùsa, M., and Conlan, J. W. (2004). Toll-like receptor 4 (TLR4) does not confer a resistance advantage on mice against low-dose aerosol infection with virulent type A Francisella tularensis. Microb. Pathog. 37, 185–191. doi: 10.1016/j.micpath.2004.06.010
Chen, W., Kuolee, R., Shen, H., Bùsa, M., and Conlan, J. W. (2005). Toll-like receptor 4 (TLR4) plays a relatively minor role in murine defense against primary intradermal infection with Francisella tularensis LVS. Immunol. Lett. 97, 151–154. doi: 10.1016/j.imlet.2004.10.001
Clemens, D. L., and Horwitz, M. A. (2007). Uptake and intracellular fate of Francisella tularensis in human macrophages. Ann. N.Y. Acad. Sci. 1105, 160–186. doi: 10.1196/annals.1409.001
Clemens, D. L., Lee, B. Y., and Horwitz, M. A. (2004). Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect. Immun. 72, 3204–3217. doi: 10.1128/IAI.72.6.3204-3217.2004
Clemens, D. L., Lee, B. Y., and Horwitz, M. A. (2005). Francisella tularensis enters macrophages via a novel process involving pseudopod loops. Infect. Immun. 73, 5892–5902. doi: 10.1128/IAI.73.9.5892-5902.2005
Cole, L. E., Laird, M. H., Seekatz, A., Santiago, A., Jiang, Z., Barry, E., et al. (2010). Phagosomal retention of Francisella tularensis results in TIRAP/Mal-independent TLR2 signaling. J. Leukoc. Biol. 87, 275–281. doi: 10.1189/jlb.0909619
Cole, L. E., Shirey, K. A., Barry, E., Santiago, A., Rallabhandi, P., Elkins, K. L., et al. (2007). Toll-like receptor 2-mediated signaling requirements for Francisella tularensis live vaccine strain infection of murine macrophages. Infect. Immun. 75, 4127–4137. doi: 10.1128/IAI.01868-06
Collazo, C. M., Sher, A., Meierovics, A. I., and Elkins, K. L. (2006). Myeloid differentiation factor-88 (MyD88) is essential for control of primary in vivo Francisella tularensis LVS infection, but not for control of intra-macrophage bacterial replication. Microbes Infect. 8, 779–790. doi: 10.1016/j.micinf.2005.09.014
Culkin, S. J., Rhinehart-Jones, T., and Elkins, K. L. (1997). A novel role for B cells in early protective immunity to an intracellular pathogen, Francisella tularensis strain LVS. J. Immunol. 158, 3277–3284.
Dai, S., Rajaram, M. V., Curry, H. M., Leander, R., and Schlesinger, L. S. (2013). Fine tuning inflammation at the front door: macrophage complement receptor 3-mediates phagocytosis and immune suppression for Francisella tularensis. PLoS Pathog. 9:e1005504. doi: 10.1371/journal.ppat.1003114
Dotson, R. J., Rabadi, S. M., Westcott, E. L., Bradley, S., Catlett, S. V., Banik, S., et al. (2013). Repression of inflammasome by Francisella tularensis during early stages of infection. J. Biol. Chem. 288, 23844–23857. doi: 10.1074/jbc.M113.490086
Dueñas, A. I., Aceves, M., Orduña, A., Díaz, R., Sánchez Crespo, M., and García-Rodríguez, C. (2006). Francisella tularensis LPS induces the production of cytokines in human monocytes and signals via Toll-like receptor 4 with much lower potency than E. coli LPS. Int. Immunol. 18, 785–795. doi: 10.1093/intimm/dxl015
Duncan, D. D., Vogler, A. J., Wolcott, M. J., Li, F., Sarovich, D. S., Birdsell, D. N., et al. (2013). Identification and typing of Francisella tularensis with a highly automated genotyping assay. Lett. Appl. Microbiol. 56, 128–134. doi: 10.1111/lam.12022
Eberl, M., Hintz, M., Reichenberg, A., Kollas, A. K., Wiesner, J., and Jomaa, H. (2003). Microbial isoprenoid biosynthesis and human gammadelta T cell activation. FEBS Lett. 544, 4–10. doi: 10.1016/S0014-5793(03)00483-6
Eberl, M., and Jomaa, H. (2003). A genetic basis for human gammadelta T-cell reactivity towards microbial pathogens. Trends Immunol. 24, 407–409. doi: 10.1016/S1471-4906(03)00170-4
Elkins, K. L., Bosio, C. M., and Rhinehart-Jones, T. R. (1999). Importance of B cells, but not specific antibodies, in primary and secondary protective immunity to the intracellular bacterium Francisella tularensis live vaccine strain. Infect. Immun. 67, 6002–6007.
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J., and Alnemri, E. S. (2009). AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513. doi: 10.1038/nature07710
Fernandes-Alnemri, T., Yu, J. W., Juliana, C., Solorzano, L., Kang, S., Wu, J., et al. (2010). The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol. 11, 385–393. doi: 10.1038/ni.1859
Forestal, C. A., Benach, J. L., Carbonara, C., Italo, J. K., Lisinski, T. J., and Furie, M. B. (2003). Francisella tularensis selectively induces proinflammatory changes in endothelial cells. J. Immunol. 171, 2563–2570. doi: 10.4049/jimmunol.171.5.2563
Fortier, A. H., Green, S. J., Polsinelli, T., Jones, T. R., Crawford, R. M., Leiby, D. A., et al. (1994). Life and death of an intracellular pathogen: Francisella tularensis and the macrophage. Immunol. Ser. 60, 349–361.
Franchi, L., Park, J. H., Shaw, M. H., Marina-Garcia, N., Chen, G., Kim, Y. G., et al. (2008). Intracellular NOD-like receptors in innate immunity, infection and disease. Cell. Microbiol. 10, 1–8. doi: 10.1111/j.1462-5822.2007.01059.x
Franchi, L., Warner, N., Viani, K., and Nuñez, G. (2009). Function of Nod-like receptors in microbial recognition and host defense. Immunol. Rev. 227, 106–128. doi: 10.1111/j.1600-065X.2008.00734.x
Geier, H., and Celli, J. (2011). Phagocytic receptors dictate phagosomal escape and intracellular proliferation of Francisella tularensis. Infect. Immun. 79, 2204–2214. doi: 10.1128/IAI.01382-10
Gentry, M., Taormina, J., Pyles, R. B., Yeager, L., Kirtley, M., Popov, V. L., et al. (2007). Role of primary human alveolar epithelial cells in host defense against Francisella tularensis infection. Infect. Immun. 75, 3969–3978. doi: 10.1128/IAI.00157-07
Golovliov, I., Baranov, V., Krocova, Z., Kovarova, H., and Sjöstedt, A. (2003). An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect. Immun. 71, 5940–5950. doi: 10.1128/IAI.71.10.5940-5950.2003
Gunn, J. S., and Ernst, R. K. (2007). The structure and function of Francisella lipopolysaccharide. Ann. N.Y. Acad. Sci. 1105, 202–218. doi: 10.1196/annals.1409.006
Hajjar, A. M., Harvey, M. D., Shaffer, S. A., Goodlett, D. R., Sjöstedt, A., Edebro, H., et al. (2006). Lack of in vitro and in vivo recognition of Francisella tularensis subspecies lipopolysaccharide by Toll-like receptors. Infect. Immun. 74, 6730–6738. doi: 10.1128/IAI.00934-06
Hall, J. D., Woolard, M. D., Gunn, B. M., Craven, R. R., Taft-Benz, S., Frelinger, J. A., et al. (2008). Infected-host-cell repertoire and cellular response in the lung following inhalation of Francisella tularensis Schu S4, LVS, or U112. Infect. Immun. 76, 5843–5852. doi: 10.1128/IAI.01176-08
Henry, T., Brotcke, A., Weiss, D. S., Thompson, L. J., and Monack, D. M. (2007). Type I interferon signaling is required for activation of the inflammasome during Francisella infection. J. Exp. Med. 204, 987–994. doi: 10.1084/jem.20062665
Heuston, S., Begley, M., Gahan, C. G., and Hill, C. (2012). Isoprenoid biosynthesis in bacterial pathogens. Microbiology 158, 1389–1401. doi: 10.1099/mic.0.051599-0
Holland, K. M., Rosa, S. J., and Hazlett, K. R. (2016). Francisella tularensis- immune cell activator, suppressor, or stealthy evader: the evolving view from the petri dish. J. Bioterror. Biodef. 7:144. doi: 10.4172/2157-2526.1000144
Hong, K. J., Wickstrum, J. R., Yeh, H. W., and Parmely, M. J. (2007). Toll-like receptor 2 controls the gamma interferon response to Francisella tularensis by mouse liver lymphocytes. Infect. Immun. 75, 5338–5345. doi: 10.1128/IAI.00561-07
Horng, T., Barton, G. M., Flavell, R. A., and Medzhitov, R. (2002). The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature 420, 329–333. doi: 10.1038/nature01180
Horng, T., Barton, G. M., and Medzhitov, R. (2001). TIRAP: an adapter molecule in the Toll signaling pathway. Nat. Immunol. 2, 835–841. doi: 10.1038/ni0901-835
Hornung, V., Ablasser, A., Charrel-Dennis, M., Bauernfeind, F., Horvath, G., Caffrey, D. R., et al. (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518. doi: 10.1038/nature07725
Hotson, A. N., Gopinath, S., Nicolau, M., Khasanova, A., Finck, R., Monack, D., et al. (2016). Coordinate actions of innate immune responses oppose those of the adaptive immune system during Salmonella infection of mice. Sci. Signal. 9, ra4. doi: 10.1126/scisignal.aaa9303
Huang, S., Gilfillan, S., Cella, M., Miley, M. J., Lantz, O., Lybarger, L., et al. (2005). Evidence for MR1 antigen presentation to mucosal-associated invariant T cells. J. Biol. Chem. 280, 21183–21193. doi: 10.1074/jbc.M501087200
Ishikawa, H., and Barber, G. N. (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. doi: 10.1038/nature07317
Ishikawa, H., Ma, Z., and Barber, G. N. (2009). STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792. doi: 10.1038/nature08476
Jin, L., Hill, K. K., Filak, H., Mogan, J., Knowles, H., Zhang, B., et al. (2011). MPYS is required for IRF3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers c-di-AMP and c-di-GMP. J. Immunol. 187, 2595–2601. doi: 10.4049/jimmunol.1100088
Jin, T., and Xiao, T. S. (2015). Activation and assembly of the inflammasomes through conserved protein domain families. Apoptosis 20, 151–156. doi: 10.1007/s10495-014-1053-5
Jones, C. L., and Weiss, D. S. (2011). TLR2 signaling contributes to rapid inflammasome activation during F. novicida infection. PLoS ONE 6:e20609. doi: 10.1371/journal.pone.0020609
Jones, J. W., Kayagaki, N., Broz, P., Henry, T., Newton, K., O'Rourke, K., et al. (2010). Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. U.S.A. 107, 9771–9776. doi: 10.1073/pnas.1003738107
Katz, J., Zhang, P., Martin, M., Vogel, S. N., and Michalek, S. M. (2006). Toll-like receptor 2 is required for inflammatory responses to Francisella tularensis LVS. Infect. Immun. 74, 2809–2816. doi: 10.1128/IAI.74.5.2809-2816.2006
Kennedy, A. D., and DeLeo, F. R. (2009). Neutrophil apoptosis and the resolution of infection. Immunol. Res. 43, 25–61. doi: 10.1007/s12026-008-8049-6
Kim, Y. K., Shin, J. S., and Nahm, M. H. (2016). NOD-like receptors in infection, immunity, and diseases. Yonsei Med. J. 57, 5–14. doi: 10.3349/ymj.2016.57.1.5
Kingry, L. C., and Petersen, J. M. (2014). Comparative review of Francisella tularensis and Francisella novicida. Front. Cell. Infect. Microbiol. 4:35. doi: 10.3389/fcimb.2014.00035
Kjer-Nielsen, L., Patel, O., Corbett, A. J., Le Nours, J., Meehan, B., Liu, L., et al. (2012). MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 491, 717–723. doi: 10.1038/nature11605
Kobayashi, S. D., Braughton, K. R., Whitney, A. R., Voyich, J. M., Schwan, T. G., Musser, J. M., et al. (2003). Bacterial pathogens modulate an apoptosis differentiation program in human neutrophils. Proc. Natl. Acad. Sci. U.S.A. 100, 10948–10953. doi: 10.1073/pnas.1833375100
Kubelkova, K., Benuchova, M., Kozakova, H., Sinkora, M., Krocova, Z., Pejchal, J., et al. (2016). Gnotobiotic mouse model's contribution to understanding host-pathogen interactions. Cell. Mol. Life Sci. 73, 3961–3969. doi: 10.1007/s00018-016-2341-8
Law, H. T., Lin, A. E. S., Kim, Y., Quach, B., Nano, F. E., and Guttman, J. A. (2011). Francisella tularensis uses cholesterol and clathrin-based endocytic mechanisms to invade hepatocytes. Sci. Rep. 1:192. doi: 10.1038/srep00192
Li, H., Nookala, S., Bina, X. R., Bina, J. E., and Re, F. (2006). Innate immune response to Francisella tularensis is mediated by TLR2 and caspase-1 activation. J. Leukoc. Biol. 80, 766–773. doi: 10.1189/jlb.0406294
Lindemann, S. R., McLendon, M. K., Apicella, M. A., and Jones, B. D. (2007). An in vitro model system used to study adherence and invasion of Francisella tularensis live vaccine strain in nonphagocytic cells. Infect. Immun. 75, 3178–3182. doi: 10.1128/IAI.01811-06
Löfgren, S., Tärnvik, A., Bloom, G. D., and Sjöberg, W. (1983). Phagocytosis and killing of Francisella tularensis by human polymorphonuclear leukocytes. Infect. Immun. 39, 715–720.
Lord, K. A., Hoffman-Liebermann, B., and Liebermann, D. A. (1990). Nucleotide sequence and expression of a cDNA encoding MyD88, a novel myeloid differentiation primary response gene induced by IL6. Oncogene 5, 1095–1097.
Lu, Y. C., Yeh, W. C., and Ohashi, P. S. (2008). LPS/TLR4 signal transduction pathway. Cytokine 42, 145–151. doi: 10.1016/j.cyto.2008.01.006
Macela, A., Stulik, J., Hernychova, L., Kroca, M., Krocova, Z., and Kovarova, H. (1996). The immune response against Francisella tularensis live vaccine strain in Lps(n) and Lps(d) mice. FEMS Immunol. Med. Microbiol. 13, 235–238. doi: 10.1111/j.1574-695X.1996.tb00243.x
Maeshima, N., and Fernandez, R. C. (2013). Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front. Cell. Infect. Microbiol. 3:3. doi: 10.3389/fcimb.2013.00003
Malik, M., Bakshi, C. S., Sahay, B., Shah, A., Lotz, S. A., and Sellati, T. J. (2006). Toll-like receptor 2 is required for control of pulmonary infection with Francisella tularensis. Infect. Immun. 74, 3657–3662. doi: 10.1128/IAI.02030-05
Man, S. M., and Kanneganti, T. D. (2015). Regulation of inflammasome activation. Immunol. Rev. 265, 6–21. doi: 10.1111/imr.12296
Man, S. M., Karki, R., and Kanneganti, T. D. (2016a). AIM2 inflammasome in infection, cancer, and autoimmunity: role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 46, 269–280. doi: 10.1002/eji.201545839
Man, S. M., Karki, R., Malireddi, R. K., Neale, G., Vogel, P., Yamamoto, M., et al. (2015). The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat. Immunol. 16, 467–475. doi: 10.1038/ni.3118
Man, S. M., Karki, R., Sasai, M., Place, D. E., Kesavardhana, S., Temirov, J., et al. (2016b). IRGB10 liberates bacterial ligands for sensing by the AIM2 and Caspase-11-NLRP3 inflammasomes. Cell 167, 382–396.e17. doi: 10.1016/j.cell.2016.09.012
Mariathasan, S., Weiss, D. S., Dixit, V. M., and Monack, D. M. (2005). Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J. Exp. Med. 202, 1043–1049. doi: 10.1084/jem.20050977
Marohn, M. E., and Barry, E. M. (2013). Live attenuated tularemia vaccines: recent developments and future goals. Vaccine 31, 3485–3491. doi: 10.1016/j.vaccine.2013.05.096
McCaffrey, R. L., and Allen, L.-A. H. (2006). Francisella tularensis LVS evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J. Leukoc. Biol. 80, 1224–1230. doi: 10.1189/jlb.0406287
Miller, D. K., Ayala, J. M., Egger, L. A., Raju, S. M., Yamin, T. T., Ding, G. J., et al. (1993). Purification and characterization of active human interleukin-1 beta-converting enzyme from THP.1 monocytic cells. J. Biol. Chem. 268, 18062–18069.
Morita, C. T., Jin, C., Sarikonda, G., and Wang, H. (2007). Nonpeptide antigens, presentation mechanisms, and immunological memory of human Vγ2Vδ2 T cells: discriminating friend from foe through the recognition of prenyl pyrophosphate antigens. Immunol. Rev. 215, 59–76. doi: 10.1111/j.1600-065X.2006.00479.x
Moser, B., and Eberl, M. (2011). γδ T-APCs: a novel tool for immunotherapy? Cell. Mol. Life Sci. 68, 2443–2452. doi: 10.1007/s00018-011-0706-6
Nunes-Alves, C., Booty, M. G., Carpenter, S. M., Jayaraman, P., Rothchild, A. C., and Behar, S. M. (2014). In search of a new paradigm for protective immunity to TB. Nat. Rev. Microbiol. 12, 289–299. doi: 10.1038/nrmicro3230
Nutter, J. E., and Myrvik, Q. N. (1966). In vitro interactions between rabbit alveolar macrophages and Pasteurella tularensis. J. Bacteriol. 92, 645–651.
Okan, N. A., and Kasper, D. L. (2013). The atypical lipopolysaccharide of Francisella. Carbohydr. Res. 378, 79–83. doi: 10.1016/j.carres.2013.06.015
O'Neill, L. A., Golenbock, D., and Bowie, A. G. (2013). The history of Toll-like receptors - redefining innate immunity. Nat. Rev. Immunol. 13, 453–460. doi: 10.1038/nri3446
Park, B. S., and Lee, J. O. (2013). Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 45:e66. doi: 10.1038/emm.2013.97
Parsa, K. V., Butchar, J. P., Rajaram, M. V., Cremer, T. J., Gunn, J. S., Schlesinger, L. S., et al. (2008). Francisella gains a survival advantage within mononuclear phagocytes by suppressing the host IFNgamma response. Mol. Immunol. 45, 3428–3437. doi: 10.1016/j.molimm.2008.04.006
Pierini, L. M. (2006). Uptake of serum-opsonized Francisella tularensis by macrophages can be mediated by class A scavenger receptors. Cell. Microbiol. 8, 1361–1370. doi: 10.1111/j.1462-5822.2006.00719.x
Plzakova, L., Krocova, Z., Kubelkova, K., and Macela, A. (2015). Entry of Francisella tularensis into murine B Cells: the role of B cell receptors and complement receptors. PLoS ONE 10:e0132571. doi: 10.1371/journal.pone.0132571
Plzakova, L., Kubelkova, K., Krocova, Z., Zarybnicka, L., Sinkorova, Z., and Macela, A. (2014). B cell subsets are activated and produce cytokines during early phases of Francisella tularensis LVS infection. Microb. Pathog. 75C, 49–58. doi: 10.1016/j.micpath.2014.08.009
Poquet, Y., Kroca, M., Halary, F., Stenmark, S., Peyrat, M. A., Bonneville, M., et al. (1998). Expansion of Vγ9Vδ2 T cells is triggered by Francisella tularensis-derived phosphoantigens in tularemia but not after tularemia vaccination. Infect. Immun. 66, 2107–2114.
Putzova, D., Senitkova, I., and Stulik, J. (2016). Tularemia vaccines. Folia Microbiol. 61, 495–504. doi: 10.1007/s12223-016-0461-z
Rennert, K., Otto, P., Funke, H., Huber, O., Tomaso, H., and Mosig, A. S. (2016). A human macrophage-hepatocyte co-culture model for comparative studies of infection and replication of Francisella tularensis LVS strain and subspecies holarctica and mediasiatica. BMC Microbiol. 16:2. doi: 10.1186/s12866-015-0621-3
Richard, K., Vogel, S. N., and Perkins, D. J. (2016). Type I interferon licenses enhanced innate recognition and transcriptional responses to Franciscella tularensis live vaccine strain. Innate Immun. 22, 363–372. doi: 10.1177/1753425916650027
Roberts, L. M., Tuladhar, S., Steele, S. P., Riebe, K. J., Chen, C. J., Cumming, R. I., et al. (2014). Identification of early interactions between Francisella and the host. Infect. Immun. 82, 2504–2510. doi: 10.1128/IAI.01654-13
Rothchild, A. C., Sissons, J. R., Shafiani, S., Plaisier, C., Min, D., Mai, D., et al. (2016). MiR-155-regulated molecular network orchestrates cell fate in the innate and adaptive immune response to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 113, E6172–E6181. doi: 10.1073/pnas.1608255113
Rowland, C. A., Hartley, M. G., Flick-Smith, H., Laws, T. R., Eyles, J. E., and Oyston, P. C. (2012). Peripheral human γδ T cells control growth of both avirulent and highly virulent strains of Francisella tularensis in vitro. Microbes Infect. 14, 584–589. doi: 10.1016/j.micinf.2012.02.001
Russo, B. C., Brown, M. J., and Nau, G. J. (2013). MyD88-dependent signaling prolongs survival and reduces bacterial burden during pulmonary infection with virulent Francisella tularensis. Am. J. Pathol. 183, 1223–1232. doi: 10.1016/j.ajpath.2013.06.013
Sandström, G., Sjöstedt, A., Johansson, T., Kuoppa, K., and Williams, J. C. (1992). Immunogenicity and toxicity of lipopolysaccharide from Francisella tularensis LVS. FEMS Microbiol. Immunol. 5, 201–210. doi: 10.1111/j.1574-6968.1992.tb05902.x
Santic, M., Asare, R., Skrobonja, I., Jones, S., and Abu Kwaik, Y. (2008). Acquisition of the vacuolar ATPase proton pump and phagosome acidification are essential for escape of Francisella tularensis into the macrophage cytosol. Infect. Immun. 76, 2671–2677. doi: 10.1128/IAI.00185-08
Santic, M., Molmeret, M., Klose, K. E., Jones, S., and Kwaik, Y. A. (2005). The Francisella tularensis pathogenicity island protein IglC and its regulator MglA are essential for modulating phagosome biogenesis and subsequent bacterial escape into the cytoplasm. Cell. Microbiol. 7, 969–979. doi: 10.1111/j.1462-5822.2005.00526.x
Schulert, G. S., and Allen, L. A. (2006). Differential infection of mononuclear phagocytes by Francisella tularensis: role of the macrophage mannose receptor. J. Leukoc. Biol. 80, 563–571. doi: 10.1189/jlb.0306219
Schwartz, J. T., Barker, J. H., Kaufman, J., Fayram, D. C., McCracken, J. M., and Allen, L. A. (2012a). Francisella tularensis inhibits the intrinsic and extrinsic pathways to delay constitutive apoptosis and prolong human neutrophil lifespan. J. Immunol. 188, 3351–3363. doi: 10.4049/jimmunol.1102863
Schwartz, J. T., Barker, J. H., Long, M. E., Kaufman, J., McCracken, J., and Allen, L.-A. H. (2012b). Natural IgM mediates complement-dependent uptake of Francisella tularensis by human neutrophils via CR1 and CR3 in nonimmune serum. J. Immunol. 189, 3064–3077. doi: 10.4049/jimmunol.1200816
Sicard, H., and Fournie, J. J. (2000). Metabolic routes as targets for immunological discrimination of host and parasite. Infect. Immun. 68, 4375–4377. doi: 10.1128/IAI.68.8.4375-4377.2000
Steele, S., Radlinski, L., Taft-Benz, S., Brunton, J., and Kawula, T. H. (2016). Trogocytosis-associated cell to cell spread of intracellular bacterial pathogens. Elife 5:e10625. doi: 10.7554/eLife.10625
Storek, K. M., Gertsvolf, N. A., Ohlson, M. B., and Monack, D. M. (2015). cGAS and Ifi204 cooperate to produce type I IFNs in response to Francisella infection. J. Immunol. 194, 3236–3245. doi: 10.4049/jimmunol.1402764
Sun, L., Wu, J., Du, F., Chen, X., and Chen, Z. J. (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. doi: 10.1126/science.1232458
Takaoka, A., Wang, Z., Choi, M. K., Yanai, H., Negishi, H., Ban, T., et al. (2007). DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505. doi: 10.1038/nature06013
Telepnev, M., Golovliov, I., Grundström, T., Tärnvik, A., and Sjöstedt, A. (2003). Francisella tularensis inhibits Toll-like receptor-mediated activation of intracellular signalling and secretion of TNF-alpha and IL-1 from murine macrophages. Cell. Microbiol. 5, 41–51. doi: 10.1046/j.1462-5822.2003.00251.x
Thornberry, N. A., Bull, H. G., Calaycay, J. R., Chapman, K. T., Howard, A. D., Kostura, M. J., et al. (1992). A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356, 768–774. doi: 10.1038/356768a0
Thorpe, B. D., and Marcus, S. (1964a). Phagocytosis and intracellular fate of Pasteurella tularensis. II. In vitro studies with rabbit alveolar and guinea pig alveolar and peritoneal mononuclear phagocytes. J. Immunol. 93, 558–565.
Thorpe, B. D., and Marcus, S. (1964b). Phagocytosis and intracellular fate of Pasteurella tularensis. I. In vitro studies with rabbit peritoneal mononuclear phagocytes. J. Immunol. 92, 657–663.
Tyler, C. J., Doherty, D. G., Moser, B., and Eberl, M. (2015). Human Vγ9/Vδ2 T cells: innate adaptors of the immune system. Cell. Immunol. 296, 10–21. doi: 10.1016/j.cellimm.2015.01.008
Udgata, A., Qureshi, R., and Mukhopadhyay, S. (2016). Transduction of functionally contrasting signals by two Mycobacterial PPE proteins downstream of TLR2 receptors. J. Immunol. 197, 1776–1787. doi: 10.4049/jimmunol.1501816
Unterholzner, L. (2013). The interferon response to intracellular DNA: why so many receptors? Immunobiology 218, 1312–1321. doi: 10.1016/j.imbio.2013.07.007
Unterholzner, L., Keating, S. E., Baran, M., Horan, K. A., Jensen, S. B., Sharma, S., et al. (2010). IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004. doi: 10.1038/ni.1932
Veeranki, S., and Choubey, D. (2012). Interferon-inducible p200-family protein IFI16, an innate immune sensor for cytosolic and nuclear double-stranded DNA: regulation of subcellular localization. Mol. Immunol. 49, 567–571. doi: 10.1016/j.molimm.2011.11.004
Warner, N., and Núñez, G. (2013). MyD88: a critical adaptor protein in innate immunity signal transduction. J. Immunol. 190, 3–4. doi: 10.4049/jimmunol.1203103
Wilson, K. P., Black, J. A., Thomson, J. A., Kim, E. E., Griffith, J. P., Navia, M. A., et al. (1994). Structure and mechanism of interleukin-1 beta converting enzyme. Nature 370, 270–275. doi: 10.1038/370270a0
Wu, J., Sun, L., Chen, X., Du, F., Shi, H., Chen, C., et al. (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830. doi: 10.1126/science.1229963
Xiao, T. S. (2015). The nucleic acid-sensing inflammasomes. Immunol. Rev. 265, 103–111. doi: 10.1111/imr.12281
Keywords: innate immunity, intracellular bacteria, immune recognition, Francisella tularensis, signaling windows concept, spatiotemporal network
Citation: Krocova Z, Macela A and Kubelkova K (2017) Innate Immune Recognition: Implications for the Interaction of Francisella tularensis with the Host Immune System. Front. Cell. Infect. Microbiol. 7:446. doi: 10.3389/fcimb.2017.00446
Received: 11 April 2017; Accepted: 29 September 2017;
Published: 16 October 2017.
Edited by:
Anders Sjöstedt, Umeå University, SwedenReviewed by:
Lee-Ann H. Allen, University of Iowa, United StatesJacqueline Sharon, Boston University, United States
Copyright © 2017 Krocova, Macela and Kubelkova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Klara Kubelkova, a2xhcmEua3ViZWxrb3ZhQHVub2IuY3o=