 Osmel Fleitas Martínez1,2
Osmel Fleitas Martínez1,2 Pietra Orlandi Rigueiras2
Pietra Orlandi Rigueiras2 Állan da Silva Pires2
Állan da Silva Pires2 William Farias Porto2,3,4
William Farias Porto2,3,4 Osmar Nascimento Silva3
Osmar Nascimento Silva3 Cesar de la Fuente-Nunez5,6,7,8,9*
Cesar de la Fuente-Nunez5,6,7,8,9* Octavio Luiz Franco1,2,3*
Octavio Luiz Franco1,2,3*- 1Programa de Pós-Graduação em Patologia Molecular, Universidade de Brasília, Brasília, Brazil
- 2Centro de Análises Proteômicas e Bioquímicas, Universidade Católica de Brasília, Brasília, Brazil
- 3S-Inova Biotech, Programa de Pós-Graduação em Biotecnologia, Universidade Católica Dom Bosco, Campo Grande, Brazil
- 4Porto Reports, Brasília, Brazil
- 5Synthetic Biology Group, MIT Synthetic Biology Center, Massachusetts Institute of Technology, Cambridge, MA, United States
- 6Research Laboratory of Electronics, Massachusetts Institute of Technology, Cambridge, MA, United States
- 7Department of Biological Engineering, Department of Electrical Engineering and Computer Science, Massachusetts Institute of Technology, Cambridge, MA, United States
- 8Broad Institute of MIT and Harvard, Cambridge, MA, United States
- 9The Center for Microbiome Informatics and Therapeutics, Cambridge, MA, United States
Faced with the global health threat of increasing resistance to antibiotics, researchers are exploring interventions that target bacterial virulence factors. Quorum sensing is a particularly attractive target because several bacterial virulence factors are controlled by this mechanism. Furthermore, attacking the quorum-sensing signaling network is less likely to select for resistant strains than using conventional antibiotics. Strategies that focus on the inhibition of quorum-sensing signal production are especially attractive because the enzymes involved are expressed in bacterial cells but are not present in their mammalian counterparts. We review here various approaches that are being taken to interfere with quorum-sensing signal production via the inhibition of autoinducer-2 synthesis, PQS synthesis, peptide autoinducer synthesis, and N-acyl-homoserine lactone synthesis. We expect these approaches will lead to the discovery of new quorum-sensing inhibitors that can help to stem the tide of antibiotic resistance.
Introduction
The increase in bacterial resistance to antimicrobial compounds and the spread of drug-resistant pathogens have become serious threats to human health. Currently, most antimicrobial compounds target essential bacterial physiological processes, thereby exerting a strong selective pressure on bacteria and facilitating the emergence and dissemination of resistant strains (Munguia and Nizet, 2017). Therapeutic strategies that circumvent the emergence and spread of multidrug-resistant pathogens are, therefore, urgently needed.
New attractive approaches for generating new therapeutics have focused on interfering with bacterial virulence factors, specifically, interfering with compounds synthesized by pathogens that facilitate colonization of the host and subsequent infection (Kong et al., 2016; Vale et al., 2016; Dickey et al., 2017; Munguia and Nizet, 2017). Because interference with virulence factors does not aim to eradicate the bacteria, it does not exert a strong selective pressure on the bacteria and probably decelerates the emergence and dissemination of resistant mutant strains (Gutierrez et al., 2009; Sully et al., 2014; Daly et al., 2015; Quave et al., 2015). However, the emergence of anti-virulence drug-resistant pathogens has been reported (Maeda et al., 2012; García-Contreras et al., 2013, 2015). Anti-virulence therapy appears all the more advantageous if we also consider that the production of virulence factors is under the control of regulatory mechanisms (e.g., quorum-sensing), and it should be possible to interfere with these mechanisms, consequently affecting the production of multiple virulence factors (Dickey et al., 2017; Defoirdt, 2018).
Quorum-sensing networks allow bacterial communication through the action of small diffusible autoinducer molecules (AI). These AI molecules comprise a diversity of molecular species such as autoinducer-2 (AI-2), acylated homoserine lactones (acyl-HSLs), oligopeptides, the Pseudomonas quinolone signal molecule (PQS), diffusible signal factor (DSF), γ-butyrolactone, 2-(2-hydroxyphenyl)-thiazole-4-carbaldehyde (IQS) among others (Guo et al., 2013; LaSarre and Federle, 2013; Pereira et al., 2013). Quorum-sensing systems operate in a cell density-dependent fashion, allowing the increase of AI concentration when cell density increases. After the AI concentration reaches a certain threshold, it triggers signaling events that modulate the expression of genes related to bacterial physiology, virulence, and biofilm formation (Papenfort and Bassler, 2016).
Interference with quorum-sensing systems has been envisioned as a suitable strategy to address the multi-drug resistance problem (Hirakawa and Tomita, 2013; Defoirdt, 2018). In this regard, a great diversity of compounds that interfere with quorum-sensing systems have been reported, as well as tools for their discovery (Jian and Li, 2013; Quave and Horswill, 2013; Nandi, 2016; Ali et al., 2017; Asfour, 2018). Strategies for inhibiting quorum sensing systems are designed mainly to interfere with the biosynthesis of AI, extracellular accumulation of the AI, and signal detection (LaSarre and Federle, 2013; Reuter et al., 2016; Singh et al., 2016; Haque et al., 2018). One of the most thoroughly explored strategies so far is interference with the extracellular accumulation of the signal. This interference can be achieved by using enzymes that degrade the signal or modify it, the use of antibodies that sequester the signal, as well as by synthetic polymers that sequester the signal (Fetzner, 2015; Daly et al., 2017; Ma et al., 2018). Interference in signal detection implies the use of compounds that interfere with the signal binding to the receptor (Singh et al., 2016; Wang and Muir, 2016; Kim et al., 2018). Other quorum-quenching strategies involve interfering with transcription factors binding to DNA and inhibiting the synthesis of the quorum-sensing signal (Gutierrez et al., 2009; Baldry et al., 2016; Scoffone et al., 2016; Greenberg et al., 2018).
The bacterial enzymes involved in quorum-sensing signal biosynthesis may be an attractive target for the development of anti-virulence agents because these enzymes are absent in mammals (Sun et al., 2004; Christensen et al., 2013; Pereira et al., 2013; Chan et al., 2015; Ji et al., 2016). Moreover, the inhibition of some of these enzymes could affect the production of more than one signal (Singh et al., 2006; Gutierrez et al., 2007, 2009; LaSarre and Federle, 2013). Experimental evidence suggests that dysfunctional AI-producing enzymes could turn pathogens less virulent for the host than pathogens expressing wild-type enzymes (Gallagher et al., 2002; Déziel et al., 2005; Kim et al., 2010; Komor et al., 2012). Thus, inhibiting the biosynthesis of the quorum-sensing signal could be a suitable strategy for developing anti-virulence agents. Because signal biosynthesis inhibition has emerged as an especially attractive way to perturb quorum-sensing networks, this strategy is emphasized in this review. The array of quorum-sensing signal biosynthesis inhibitors that have been developed, their main targets, the effects of these inhibitors on pathogen virulence, and new approaches for quorum-sensing signal biosynthesis inhibition will be summarized.
Inhibition of Autoinducer-2 Synthesis
AI-2 compounds have been claimed as “universal” signal molecules involved in inter- and intra-bacterial species communication. This is supported by the fact that luxS gene homologs are widely distributed among bacterial genomes [luxS encodes the S-ribosylhomocysteine lyase (LuxS) enzyme, which synthesizes AI-2] (Pereira et al., 2013; Pérez-Rodríguez et al., 2015; Kaur et al., 2018). Moreover, some bacteria that are unable to produce AI-2 (e. g., Pseudomonas aeruginosa and Riemerella anatipestifer) respond to AI-2 external supply, and AI-2 mediates the interaction between polymicrobial biofilm members (Han et al., 2015; Li et al., 2015; Laganenka and Sourjik, 2017). In addition to regulation of biofilm formation, AI-2 has been linked to the regulation of pathogen virulence factors production, colonization capacity, persistence, and adaption to host environment (Armbruster et al., 2009; Li et al., 2017; Ma Y. et al., 2017). Therefore, interference with AI-2 production could be used as a strategy to attenuate pathogen virulence. Two main enzymes participate in AI-2 biosynthesis: Methylthioadenosine/S- adenosylhomocysteine nucleosidase (MTA/SAH nucleosidase) and LuxS. Both enzymes are involved in the activated methyl cycle, and they therefore influence bacterial metabolism. Strategies focused on inhibiting AI-2 production have, therefore, targeted these enzymes (Lebeer et al., 2007; Parveen and Cornell, 2011; LaSarre and Federle, 2013; Pereira et al., 2013).
Methylthioadenosine/S-Adenosylhomocysteine Nucleosidase Inhibitors
MTA/SAH nucleosidase has been identified in several bacterial species but is absent from mammalian cells (Sun et al., 2004). It is also linked to the acyl-HSLs biosynthesis pathway; therefore, MTA/SAH nucleosidase inhibition could interfere with the production of these quorum-sensing signals (Singh et al., 2006; Gutierrez et al., 2007, 2009). In addition, MTA/SAH nucleosidase appears to influence pathogens' capacity to produce biofilms (Bao et al., 2015; Han et al., 2017). Therefore, MTA/SAH nucleosidase could be an excellent choice as a target for the development of new quorum-sensing inhibitors. However, caution must be exercised, because the inhibition of MTA/SAH nucleosidase could result in the accumulation of S-adenosyl-homocysteine (SAH) and 5-methylthioadenosine (MTA) which, if present at high levels, could inhibit reactions catalyzed by polyamine synthases and S-adenosylmethionine dependent methyltransferases, interfering with bacterial growth (Heurlier et al., 2009; Parveen and Cornell, 2011). Vibrio cholerae MTA/SAH nucleosidase mutants with impaired growth have been reported (Silva et al., 2015). Nevertheless, experimental evidence has demonstrated that it is possible to inhibit MTA/SAH nucleosidase activity without severely affecting bacterial growth and without inducing resistance toward inhibitors (Gutierrez et al., 2009). In addition, Bourgeois et al. (2018) observed that a Salmonella enterica serovar Typhimurium ΔmetJ mutant strain, which was defective in methionine metabolism, presented elevated intracellular MTA levels without affecting bacterial growth (Bourgeois et al., 2018). In a S. aureus pfs mutant strain (pfs encodes the MTA/SAH nucleosidase), growth was not impaired in nutrient-rich conditions but it was affected in zebrafish embryos (Bao et al., 2013). MTA is also a substrate of the human enzyme MTA phosphorylase, but the structural differences between the human and bacterial enzymes (in the purine, ribose and 5′-alkylthio binding sites) make it possible to develop MTA structural analogs as inhibitors that are selective for MTA/SAH nucleosidase (Lee et al., 2004; Guo et al., 2013; Figure 1).
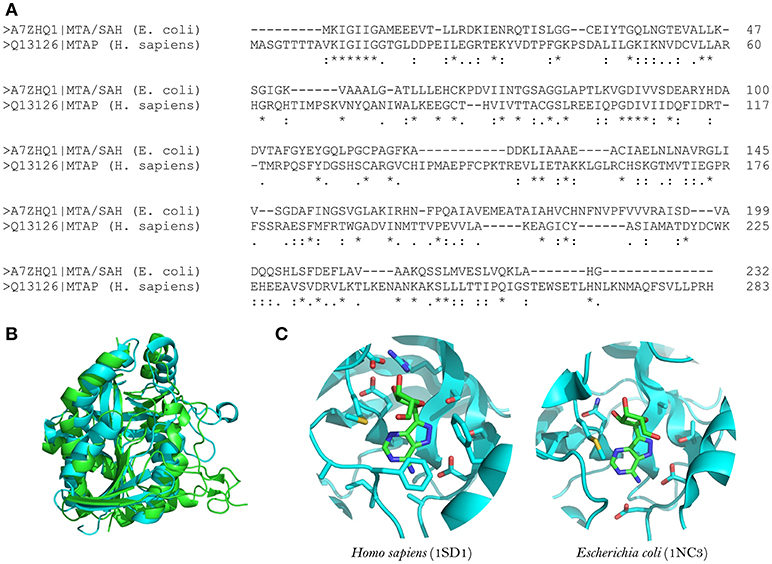
Figure 1. Structural comparison between human MTA phosphorylase and E. coli MTA/SAH nucleosidase. (A) Sequence alignment between E. coli MTA/SAH nucleosidase (MTAN; Uniprot ID: A7ZHQ1) and human MTA phosphorylase (MTAN; Uniprot ID: Q13126). (B) Structural alignment between MTAN (green) and MTAP (blue). (C) Active site comparison between MTAN (PDB ID: 1SD1, left) and MTAP (PDB ID: 1NC3, right) with formycin. The structures were retrieved from PDB, and the visualization was done by using Pymol v.1.6.
The structures of MTA/SAH nucleosidase homologs in several bacterial species have been resolved; these species include Escherichia coli, Helicobacter pylori, Streptococcus pneumoniae, Staphylococcus aureus, S. enterica, V. cholerae, Brucella melitensis, and more recently Aeromonas hydrophila (Lee et al., 2003; Singh et al., 2006; Siu et al., 2008; Ronning et al., 2010; Haapalainen et al., 2013; Kang et al., 2014; Thomas et al., 2015; Xu et al., 2017). Basically, the MTA/SAH nucleosidase is a homodimer that contains two active sites in which specific sub-sites (purine, ribose and 5′-alkylthio binding sites) are involved in the interactions with the substrate (Ronning et al., 2010). MTA/SAH nucleosidase removes adenine from SAH, MTA, and 5′-deoxyadenosine yielding S-ribosyl-L-homocysteine (SRH), S-methyl-5′-thioribose, and 5′-deoxyribose, respectively. In Neisseria meningitidis and H. pylori, this reaction takes place through an early transition state while in other bacteria such as Klebsiella pneumoniae, E. coli, S. aureus, and S. pneumoniae it occurs through a late dissociative transition state (Singh et al., 2005b; Gutierrez et al., 2007). In line with this, most of the MTA/SAH nucleosidase inhibitors that have been developed are transition state analogs (Singh et al., 2005a,b, 2006; Gutierrez et al., 2007, 2009).
Several of these transition state analogs have been tested on E. coli and S. pneumoniae MTA/SAH nucleosidases (Singh et al., 2005a, 2006). The analogs were mainly based on 5′-thio-Immucillin-A and 5′-thio-DADMe-Immucillin-A derivate compounds, in which diverse chemical groups (aromatic, cycloalkyl, halogenated aliphatic and hydrophobic groups) were incorporated at the 5′-thio position. The 5′-thio-DADMe-Immucillin-A-derived analogs behaved as more potent MTA/SAH nucleosidase inhibitors than 5′-thio-Immucillin-A-derived analogs, because these analogs mimic the late dissociative transition state through which the enzymatic reaction in these bacterial species takes place (Singh et al., 2005a, 2006). Moreover, 5′-thio-DADMe-Immucillin-A-based inhibitors showed V. cholerae N16961 and E. coli O157: H7 cellular MTA/SAH nucleosidase inhibition. The inhibitors interfered with AI production without affecting bacterial growth. Although the inhibitors reach their intracellular target, a significant diffusion barrier was observed. For both V. cholerae and E. coli, the inhibition of AI-2 production by 5′-butylthio-DADMe-Immucillin-A was sustained over several bacterial generations, suggesting that bacterial resistance had not emerged toward the MTA/SAH nucleosidase inhibitor. The 5′-butylthio-DADMe-Immucillin-A inhibitor reduced biofilm production in both species of bacteria (Gutierrez et al., 2009). However, Silva et al. (2015) recently showed that V. cholerae N16961 was able to form biofilm when treated with 5′-methylthio-DADMe-Immucillin-A, although a high percentage of MTA/SAH nucleosidase inhibition was reached. In addition, in two MTA/SAH nucleosidase mutant strains biofilm production was similar to the wild-type strain (V. cholerae N16961) whereas the growth rate and swarming motility were lower than the wild-type strain (Silva et al., 2015).
In another line of research, novel inhibitors of S. enterica MTA/SAH nucleosidase based on transition state analogs were designed. Interestingly, S. enterica MTA/SAH nucleosidase presented an elongated 5′-alkylthio-binding pocket. In that case, the design of novel inhibitors involved adding elongated 5′-alkylthio groups to the DADMe-Imm-A core to fill this site. The new inhibitors were 2-hydroxyethylthio-DADMe-Imm-A, 3-hydroxypropylthio-DADMe-Imm-A, 4-hydroxybutylthio-DADMe-Imm-A and 2-(2-hydroxyethoxy)ethylthio-DADMe-Imm-A, all of which showed dissociation constants in the pM range (Haapalainen et al., 2013). Recently, the putative Mycobacterium tuberculosis MTA/SAH nucleosidase (Rv0091) was expressed and characterized kinetically, showing a preference for 5′-deoxyadenosine as the substrate in comparison with MTA and SAH. For Rv0091, DADMe-Imm-A inhibitors consisting of derivatized analogs with long alkyl groups at the C5′ position exerted potent inhibitory activity (Namanja-Magliano et al., 2016). Additionally, when using 5′-deoxyalkyl- and 5′-alkylthio-DADMe-Immucillin-A transition state analogs it was observed that for the 5′-deoxyalkyl-DADMe-Immucillin-A analogs, shorter 5′-alkyl-substituents led to the most potent inhibition, in contrast to 5′-alkylthio-DADMe-Immucillin-A analogs, in which longer 5′-alkyl-substituents led to the most potent inhibition. These inhibitors did not affect the growth of M. tuberculosis or Mycobacterium smegmatis; however, they showed an antimicrobial effect on H. pylori (due to the involvement of H. pylori MTA/SAH nucleosidase in the menaquinone biosynthesis pathway). The authors suggested that Rv0091 plays a role in 5′-deoxyadenosine recycling but is not essential for the growth of M. tuberculosis or M. smegmatis (Namanja-Magliano et al., 2017).
Furthermore, the MTA/SAH nucleosidase has been suggested to influence the virulence of pathogens in an AI-2-independent fashion. Based on mouse infection models and zebrafish embryo infection models, Bao et al. (2013) demonstrated that a S. aureus NCTC 8325 pfs mutant strain displayed attenuated virulence in vivo. In addition, a luxS mutant was as virulent as the isogenic wild-type S. aureus NCTC8325 strain, suggesting that the effects of pfs deletion on S. aureus virulence were independent of the AI-2-based quorum sensing pathway. The attenuated virulence of the pfs mutant strain was associated with reduced proliferation in vivo. Additionally, in vitro analysis showed reduced extracellular protease activity in the pfs mutant strain linked to reduced sspABC operon transcription and aur gene transcription (Bao et al., 2013). Another study showed that S. aureus NCTC 8325 pfs mutant strain displayed reduced biofilm formation in vitro by AI-2-independent mechanisms. The pfs deletion reduced the transcription of autolysis-related genes atlE and lytM in the mutant strain; therefore, autolysis-dependent extracellular DNA release in the pfs mutant, and consequently biofilm formation, was affected (Bao et al., 2015).
The experimental findings reviewed above suggest that MTA/SAH nucleosidase could influence pathogen virulence via quorum-sensing-independent or –dependent mechanisms (Gutierrez et al., 2009; Bao et al., 2013, 2015; Silva et al., 2015). Therefore, for MTA/SAH nucleosidase inhibitor evaluation studies, precise experimental designs aimed at distinguishing by which mechanism (i.e., quorum sensing-independent and/or –dependent) the pathogen's virulence is affected are warranted. Moreover, most of the studies about MTA/SAH nucleosidase inhibition have been focused on designing inhibitors and evaluating their inhibitory activity via enzymatic assays using purified enzymes, but data about the real impact that such inhibitors have on pathogens virulence is scarce (Singh et al., 2005a,b, 2006; Gutierrez et al., 2007; Haapalainen et al., 2013; Namanja-Magliano et al., 2016).
Therefore, it is essential to perform studies that evaluate the effects of MTA/SAH nucleosidase inhibitors on pathogen virulence gene expression as well as testing the effectiveness of these to attenuate pathogens in vivo. Immucillin-based inhibitors appear to be a reasonable option as quorum quenching agents. This class of inhibitors has been used as antiviral, antibacterial (specifically in H. pylori), anti-malarial, and antineoplastic agents. Some of them are in clinical trials or have been approved for use in humans. Immucillin-based inhibitors are chemically stable, specific, and it is possible to chemically modify them to gain bioavailability without severely affecting their inhibitory activity (Longshaw et al., 2010; Evans et al., 2018).
S-Ribosylhomocysteine Lyase Inhibitors
The enzyme S-ribosylhomocysteine lyase (LuxS) is a potential target for the development of new therapeutic agents because it is present in numerous bacterial species but not in mammals (Pereira et al., 2013; Pérez-Rodríguez et al., 2015; Kaur et al., 2018). In addition, LuxS also appears to modulate bacterial biofilm formation based on results obtained with luxS mutant bacteria (Hardie and Heurlier, 2008; Kang et al., 2017; Ma R. et al., 2017; Zuberi et al., 2017). However, through which mechanisms luxS influences biofilm formation is under debate. LuxS could influence biofilm formation in an AI-2-dependent fashion, in which the expression of genes associated with bacterial adherence and biofilm matrix production may be modulated through AI-2-mediated signaling (Hardie and Heurlier, 2008; Duanis-Assaf et al., 2016; Ma R. et al., 2017; Velusamy et al., 2017; Pang et al., 2018). In addition, AI-2 may promote single and mixed species biofilm formation through chemotaxis-mediated aggregation events (Laganenka et al., 2016; Laganenka and Sourjik, 2017). However, other findings suggest that luxS also influences biofilm formation in an AI-2 signaling-independent fashion, probably involving the activated methyl cycle, fimbriation modulation and biofilm-associated gene expression modulation (Niu et al., 2013; Hu et al., 2018; Yadav et al., 2018).
Furthermore, LuxS may influence the virulence of pathogens during the host infection process. Recently, Yadav et al. (2018) demonstrated using a rat model of otitis media that S. pneumoniae D39ΔluxS mutant strain had decreased capacity for host colonization in comparison with the wild-type strain. Similarly, in a murine model D39ΔluxS displayed reduced capacity of nasopharynx colonization as well as reduced dissemination toward lung and blood. Interestingly, when AI-2 was administered to the D39ΔluxS infected mice, the D39ΔluxS mutant became as virulent as the wild-type strain without AI-2 treatment, suggesting that attenuated virulence in D39ΔluxS was associated with impaired AI-2 production and signaling (Trappetti et al., 2017). Moreover, in mice dual-infected with wild-type Borrelia burgdorferi and a luxS mutant strain, a higher wild-type bacterial load was observed than luxS mutant bacterial load in distal tissues from infection site, suggesting attenuated virulence for the luxS mutant (Arnold et al., 2015). However, using a pneumonic plague mouse model Fitts et al. (2016) observed that a Yersinia pestis CO92 ΔluxS mutant was as virulent as the wild-type CO92 strain. In addition, deletion of luxS from a ΔrbsAΔlsrA strain (attenuated virulence) turns it into the ΔrbsAΔlsrAΔluxS mutant, which was also as virulent as the wild-type strain (Fitts et al., 2016).
Taking into consideration the experimental evidence mentioned above, implementation of therapeutic strategies focused on LuxS inhibition may turn out to be complex and more difficult than initially envisioned. The effects of luxS on biofilm formation and virulence appear be dependent on bacterial species and genetic background (Bao et al., 2013; Fitts et al., 2016; Ma Y. et al., 2017; Trappetti et al., 2017; Velusamy et al., 2017; Hu et al., 2018). Also, LuxS could influence gene expression in an AI-2 signaling-independent fashion (Pereira et al., 2013). Moreover, LuxS inhibition could facilitate the accumulation of toxic compounds that disturb bacterial viability, and it could therefore exert selective pressure on the pathogen (Heurlier et al., 2009). In addition, the effects of luxS suppression on bacterial growth could be dependent on environmental stress conditions (Park et al., 2017). LuxS-independent pathways could also be involved in AI-2 formation (Tavender et al., 2008). However, in vivo studies have shown that it is possible to attenuate pathogen virulence via LuxS inhibition (Zhang et al., 2009; Sun and Zhang, 2016).
LuxS is a homodimer metalloenzyme that catalyzes the SRH cleavage through a proposed mechanism involving two isomerization steps (aldo-ketose and keto-ketose isomerization) followed by a β-elimination step to yielding L-homocysteine and the enol form of 4,5 dihydroxy-2,3-pentanedione (DPD) (Zhu et al., 2003, 2006; Pei and Zhu, 2004; Figure 2). The DPD spontaneously cyclizes and reorganizes into various furanone molecules that constitute the AI-2 (LaSarre and Federle, 2013). In this regard, most of the strategies for directly inhibiting LuxS are centered on substrate analogs that compete with the natural substrate for binding to the enzyme active site and interfere with any of the mechanistic steps that LuxS catalyzes (Alfaro et al., 2004; Wnuk et al., 2008; Malladi et al., 2011; Figure 2).
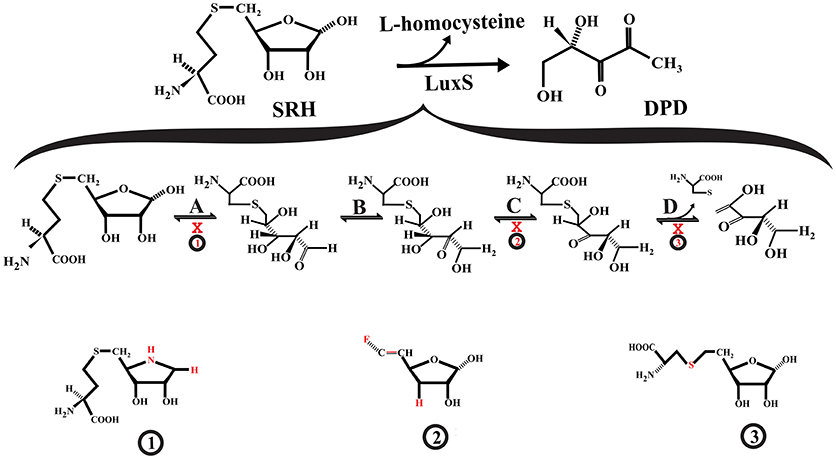
Figure 2. Interference with S-ribosylhomocysteine lyase (LuxS) activity. (A) S-ribosyl-homocysteine (SRH) ribose ring opening. (B) The opened SRH molecule undergoes aldose-ketose isomerization yielding 2-keto intermediate. (C) The 2-keto intermediate is transformed into 3-keto intermediate via ketose-ketose isomerization. (D) The 3-keto intermediate suffers a β-elimination reaction releasing L-homocysteine and the enol form of DPD. The SRH analog S-(1-Amino-1,4-anhydro-1,5-dideoxy-D-ribitol-5-yl)-L-homocysteine (1) may act as a competitive inhibitor that not suffer ring opening, affecting the aldose-keto isomerization. The 3,5,6-trideoxy-6-fluoro-D-erythro-hex-5-enofuranose (2) may interfere with the formation of the 3-keto intermediate. The S-homoribosyl-L-cysteine inhibitor (3) may interfere with the β-elimination step. The red X indicates inhibition.
Some of the reported LuxS competitive inhibitors are SRH analogs that contain a modified C1 at the ribose or [4-aza]ribose, which could affect the initial ring opening and the subsequent isomerization [e.g., S-anhydroribosyl-L-homocysteine, and S-(1-Amino-1,4-anhydro-1,5-dideoxy-D-ribitol-5-yl)-L-homocysteine] (Alfaro et al., 2004; Malladi et al., 2011; Figure 2, compound 1). Another potential LuxS inhibitor that could affect the ring opening is the SRH analog S-(1,5-Dideoxy-4-thio-D-ribofuranos-5-yl)-L-homocysteine (Sobczak et al., 2015). Moreover, compounds that interfere with the tautomerization/isomerization steps during the catalytic cycle act as competitive inhibitors of LuxS. Some of these compounds are SRH analogs in which the hydroxyl group at ribose C3 position has been removed or modified, making it a non-enolizable hydroxyl group and therefore interfering with the formation of the 3-ketone intermediate (e.g., 3,5,6-trideoxy-6-fluoro-D-erythro-hex-5-enofuranose) (Wnuk et al., 2008; Figure 2, compound 2). In addition, analogs of enediolate intermediates in which the enediolate moiety was substituted by a planar hydroxamate group act as powerful reversible competitive inhibitors of LuxS (Shen et al., 2006).
Inhibitors of the LuxS catalytic β-elimination step targeted the carbon (C5)-sulfur bond or the hydrogen atom at C4 position in the SRH substrate. In the inhibitor S-homoribosyl-L-cysteine, the carbon (C5)-sulfur bond was replaced by a C5-C6 carbon-carbon bond, which interferes with the cleavage of the carbon-sulfur bond (Alfaro et al., 2004; Figure 2, compound 3). Recently, Chbib et al. (2016) synthesized 4-C-Alkyl/aryl-SRH analogs that potentially could inhibit LuxS at the β-elimination step. The substitution of the hydrogen atom at C4 position by alkyl/aryl groups should prevent the abstraction of the C4-proton, which is important for the β-elimination step (Chbib et al., 2016). Interestingly, these authors suggested that the 4-C-Alkyl/aryl-SRH analogs also could inhibit LuxS by interfering with the dimerization of the enzyme. Previously, SRH analogs that carried alkyl/aryl groups at the C3 position of the homocysteine moiety of SRH were suggested as LuxS dimerization inhibitors (Liu, 2012).
Indeed, LuxS inhibitors [e.g., S-xylosylhomocysteine and S-(3-deoxy-3-fluoroxylosyl) homocysteine] that exerted a time-dependent inhibition of LuxS have been identified. They act as slow-binding inhibitors of improved potency, similarly to the halogenated S-[3-Bromo-3,5-dideoxy-D-ribofuranose-5-yl]-L-homocysteine and S-[3-fluoro-3,5-dideoxy-D-ribofuranos-5-yl]-L-homocysteine analogs (Gopishetty et al., 2009; Wnuk et al., 2009). The time-dependent inhibition exerted by these halogenated analogs was produced by the enzyme-catalyzed elimination of halide ions (Gopishetty et al., 2009). Equally, time-dependent inhibition of LuxS was observed for the S-[4-Amino-4,5-dideoxy-α/β-D-ribofuranos-5-yl]-L-homocysteine hemiaminal analog. This time-dependent inhibition was suggested as a result from the formation of ketone intermediates that bind to the LuxS active site with a higher affinity than the ribose natural substrate (Malladi et al., 2011).
Molecules that covalently modify LuxS also mediate its inhibition. Along these lines, halogenated furanones have been shown to inactivate LuxS. Specifically, it was observed that furanones that contain a vinyl monobromide moiety inhibit LuxS in a concentration-dependent manner. Mechanistic studies showed that LuxS was inactivated by covalent modification (Zang et al., 2009). Recently, it was hypothesized that the 2-deoxy-2-propylthiol-S-ribosylhomocysteine potentially could inhibit LuxS via covalent modification (Chbib, 2017).
Most of these luxS inhibitors have been tested in vitro using enzymatic assays with the purified LuxS enzyme. However, if these inhibitors are able to inhibit luxS in vivo with the consequent attenuation of pathogen virulence still needs to be investigated.
In addition to the SRH substrate analogs described above, other LuxS inhibitors are substrate non-analogs. Using phage display, representative peptide sequences that bind to LuxS were found. Of these, only the peptide TNRHNPHHLHHV inhibited LuxS and, then, only weakly, showing that there is not necessarily a correlation between peptide binding to LuxS and enzyme inhibition (Han and Lu, 2009). However, two LuxS-derived peptides have been described [peptide 5411 (MHTLEHLFAGFM) and 5906 (MLFAGFM)] that acted as potential LuxS inhibitors and mediated the attenuation of Edwardsiella tarda TX1 virulence in vivo (Zhang et al., 2009). The expression of the inhibitory peptides in TX1 strain (via plasmids) affected AI-2 production, reduced biofilm formation, and reduced esrA and orf26 gene expression. Additionally, the virulence of TX1 strain was attenuated in infected Japanese flounder fish. Fish infected with the TX1 strain could be attenuated either by means of a commensal Pseudomonas sp. strain expressing the peptide 5411 or by directly expressing such peptide in tissues of infected fish (Zhang et al., 2009). Recent findings demonstrated that peptide 5906 production by E. coli DH5α/p5906 or by fish tissues attenuated E. tarda pathogenesis in Japanese flounder. In addition, the pathogenesis of A. hydrophila AH1 and V. harveyi T4 in infected fish was attenuated by E. coli DH5α/p5906. However, this attenuation was moderate in comparison with the attenuation that E. tarda underwent. It has been suggested that differences in the LuxS sequences of these pathogens could be responsible for the observed differences in the attenuation levels (Sun and Zhang, 2016). All these findings suggest that LuxS-derived peptides have the potential to act as AI-2-based quorum sensing system inhibitors in several bacterial species and that engineered commensal bacteria to produce LuxS-derived peptides could be a feasible strategy for quorum quenching in vivo.
To disturb LuxS functionality, the design of small molecule inhibitors has been the most exploited strategy. However, interference with the luxS gene expression represents an alternative approach to impairing LuxS function. The development of the Clustered Regularly Interspaced Short Palindromic Repeats-Cas 9 (CRISPR-Cas9) genome-editing technology now permits the expression of genes to be modulated with a high specificity and reduced off-target effect. This technology has been proposed as a promising method for fighting against antimicrobial resistance (Greene, 2018). Recently, Kang et al. (2017), using CRISPR-Cas9 genome-editing technology, obtained E. coli SE15ΔluxS mutant clones from the E. coli SE15 clinical strain isolated from the indwelling catheter of a patient suffering from urinary tract infections. E. coli SE15ΔluxS clones showed reduced biofilm formation in comparison to the wild-type strain. Moreover, in E. coli SE15ΔluxS mutant the expression of the genes mqsR, pgaB, pgaC, csgE, and csgF (involved in biofilm formation) was down-regulated (Kang et al., 2017). This work showed that by using CRISPR-Cas9 genome-editing technology it is possible to disturb the luxS gene expression and consequently impair one of the mechanisms of pathogenicity (biofilm formation) employed by pathogens. However, if this approach will be effective in vivo remains to be seen. Nevertheless, the feasibility of CRISPR-Cas9 genome-editing technology for attenuating the virulence of pathogens in vivo has been demonstrated. In this respect, the use of CRISPR-Cas9 phagemid vectors attenuated the virulence of enterohaemorrhagic E. coli in a Galleria mellonella infection model and S. aureus in a mouse skin colonization model (Bikard et al., 2014; Citorik et al., 2014).
An alternative CRISPR-Cas9-based approach, namely the CRISPR interference (CRISPRi) approach, has been applied successfully for luxS attenuation in clinical bacterial isolates (Zuberi et al., 2017). This system is based on the use of a Cas-9 DNA endonuclease that is not catalytically active (dCas-9) but can be directed toward the target gene by a small guide RNA and repress the expression of the target gene via interfering with the transcriptional process (Qi et al., 2013). Using a CRISPRi approach that targeted the luxS gene of the E. coli clinical strain AK-117 (isolated from urinary catheters), Zuberi et al. (2017) obtained three luxS knockdown strains (AK-LV1, AK-LV2, and AK-LV3) that were metabolically actives but with impaired biofilm formation capacity (Zuberi et al., 2017).
It has been described that in E. coli the CyaR small RNA regulated luxS gene expression negatively via post-transcriptional binding to the luxS mRNA 5′end (including the ribosome binding site), consequently, CyaR small RNA expression reduced AI-2 production (De Lay and Gottesman, 2009). Moreover, Zhang and Sun (2012) using an antisense RNA interference approach, impaired luxS expression in Edwardsiella ictaluri J901 strain, yielding the luxS-defective E. ictaluri J901Ri strain. E. ictaluri J901Ri showed lower AI-2 production, reduced biofilm formation, and down-regulated expression of orf26, esrA, eseB, eseD, eihA, and wbiT genes in comparison to E. ictaluri J901C control strain. Moreover, E. ictaluri J901Ri-infected zebrafish group showed lower accumulated mortality than E. ictaluri J901C- infected group, and E. ictaluri J901Ri infectivity on ZF4 cells was reduced in comparison to the control strain (Zhang and Sun, 2012). Therefore, it was possible that using an RNA interference approach attenuated the E. ictaluri J901 virulence. Based on an antisense RNA interference approach that targeted the luxS gene, Zhang et al. (2008), attenuated the virulence of the pathogen E. tarda. The E. tarda TX1/pJR18 strain, which contained a plasmid (pJR18) that constitutively expressed the luxS antisense RNA, showed lower AI-2 production, reduced biofilm formation and reduced expression of esrA and orf26 genes in comparison to the control E. tarda TX1/pJRA strain. Furthermore, in E. tarda TX1/pJR18-infected Japanese flounder group the accumulated mortality, the bacteria recovered from blood and kidney, and orf 26 and esrA expression were lower than the E. tarda TX1/pJRA-infected group (Zhang et al., 2008). Based on all these findings, it could be envisioned that antisense oligonucleotide-based inhibition could be a feasible strategy for the development of new luxS inhibitors.
Inhibition of PQS Synthesis
Pseudomonas aeruginosa produces 2-heptyl-3-hydroxy-4(1H)-quinolone, which is commonly known as Pseudomonas quinolone signal (PQS) and acts as a quorum-sensing signal molecule (Déziel et al., 2004). Among the proteins involved at PQS synthesis, several of them are encoded by the pqsABCDE operon. The first step in the PQS biosynthesis pathway involves the formation of anthraniloyl-coenzyme A from anthranilate catalyzed by an anthranilate CoA ligase (PqsA). Subsequently, two condensation reactions take place. First, anthraniloyl-coenzyme A condenses with malonyl-CoA to form (2-aminobenzoyl) acetate with the participation of the proteins PqsD and PqsE. Second, the (2-aminobenzoyl) acetate condenses with octanoate through the PqsB and PqsC catalytic activity, producing 2-heptyl-4(1H)-quinolone (HHQ). Finally, HHQ is hydroxylated by PqsH FAD-dependent monooxygenase to form PQS (Déziel et al., 2004; Dulcey et al., 2013). Both HHQ and PQS act as signaling molecules.
Several enzyme-catalyzed reactions in the PQS biosynthesis pathway are being targeted for interference with PQS production (Sahner et al., 2013; Hinsberger et al., 2014; Ji et al., 2016; Maura et al., 2017). Anthranilate-CoA ligase (PqsA) constitutes an attractive target for developing drugs because the ortholog enzyme is absent in humans. PqsA catalyzes the conversion of anthranilate to anthraniloyl-coenzyme A in a reaction that involves an anthranilyl-AMP intermediate, which has been targeted in the design of PqsA inhibitors (Ji et al., 2016).
Recently, Ji et al. (2016) designed and evaluated the inhibitory activity of several sulfonyladenosine compounds on PqsA. These small molecules mimic the anthranilyl-AMP intermediate. The anthranilyl-AMS and anthranilyl-AMSN compounds were the most potent PqsA inhibitors that were found to reduce HHQ and PQS quinolone production by P. aeruginosa strain PA14; while salicyl-AMS, salicyl-AMSN, and benzoyl-AMS inhibitors were less potent. The authors suggested that differences in cell penetration, stability, and/or target specificity could be responsible for the variations in potency observed in inhibiting HHQ and PQS quinolone production (Ji et al., 2016). Other types of PqsA inhibitors that have been studied are the substrate analogs. Challenging the P. aeruginosa strain PAO1 culture with the anthranilate analog methyl-anthranilate was observed to inhibit the production of PQS and to decrease activity of the virulence factor elastase in a concentration-dependent manner. Interestingly, the methyl-anthranilate treatment did not affect the growth of cultures (Calfee et al., 2001). Moreover, other anthranilic acid analogs, specifically halogenated anthranilic acid analogs, exerted inhibitory activity on the production 4-hydroxy-2-alkylquinolines (HAQs) in P. aeruginosa and Burkholderia thailandensis without significantly disturbing bacterial growth (Lesic et al., 2007). The treatment of P. aeruginosa with several of these halogenated analogs represses the expression of HAQ biosynthetic operons pqsA-E and phnAB as well as the virulence factors pyocyanin (phz ABCDEFG, phzH, phzM, and phzS), hydrogen cyanide (hcnABC), chitinase (chiC), lectins (lecA and lecB), and elastase (lasB). Interestingly, these compounds are also effective in vivo, as they limited the virulence of P. aeruginosa in mice, delayed mortality in the treated animals, reduced the production of HHQ, and prevented systemic dissemination of the bacteria (Lesic et al., 2007). Furthermore, Coleman et al. (2008) tested the inhibitory activity of several anthranilate analogs on PQS production in bacterial cultures as well as on PqsA activity. Most of the chloro- and fluoro-anthranilate derivatives inhibited the production of PQS in P. aeruginosa culture and were PqsA substrates. Additionally, the anthranilonitrile, 5-nitroanthranilonitrile, methylanthranilate, and 3-fluoro-O-anisidine analogs did not behave as PqsA substrates but inhibited the production of PQS (Coleman et al., 2008).
Another protein involved in the PQS biosynthesis pathway that has been considered for the development of anti-virulence drugs is PqsD. PqsD forms homodimers in solution and structurally is similar to E. coli β-ketoacyl-ACP synthase III (FabH), showing the Cys-His-Asn catalytic triad typical of FabH-like enzymes (Bera et al., 2009). PqsD catalyzes the formation of 2-aminobenzoylacetyl-CoA in the PQS biosynthesis pathway using as substrates anthraniloyl-CoA and malonyl-CoA. Initially PqsD forms an anthraniloyl-PqsD intermediate via Cys 112 in the PqsD active site; subsequently, a condensation reaction takes place with malonyl-CoA. Given the structural similarity between PqsD and FabH-like enzymes, it has been suggested that FabH inhibitors potentially could inhibit PqsD (Pistorius et al., 2011; Dulcey et al., 2013).
Along the same lines, Pistorius et al. (2011) demonstrated that the well-established FabH inhibitors, such as 2-(4-bromo-3-diethylsulfamoyl-benzoylamino)-benzoic acid and 2-[(2- phenoxybiphenyl-4-carbonyl) amino] benzoic acid, had IC50 in the micromolar range; therefore, they exerted a modest inhibitory activity toward PqsD (Pistorius et al., 2011). Subsequently, introducing modifications in the 2-(4-bromo-3-diethylsulfamoyl-benzoylamino)-benzoic acid inhibitor yielded a series of sulfonamide-substituted benzamidobenzoic acids that inhibited PqsD. It was suggested that the binding of these compounds within the anthraniloyl-CoA channel of PqsD (involving hydrogen bonds, π-stackings, and hydrophobic interactions) hinders the access of substrate to the catalytic site; the compounds are therefore acting as entropy-driven channel-blocker inhibitors (Weidel et al., 2013). Moreover, Hinsberger et al. (2014) identified PqsD inhibitors with preferential selectivity to PqsD over RNA polymerase. These inhibitors were derivatives of benzamidobenzoic acid. The selectivity to PqsD was favored by introducing modifications on the benzamidobenzoic acid scaffold. The development of PqsD inhibitors that minimally affect the activity of RNA polymerase is desirable, because compounds that hinder this activity could exert selective pressure on the targeted bacteria (Hinsberger et al., 2014).
Using a ligand-based approach, Storz et al. (2012), identified several PqsD inhibitors that consisted of PqsD substrates and transition state analogs. The most potent inhibitor was (2-nitrophenyl)phenyl methanol, which contained a (2-nitrophenyl)methanol core rigidified by an unsubstituted phenyl moiety. This molecule inhibited the production of HHQ and PQS by P. aeruginosa PA14 cultures and reduced the biovolume of biofilm formed by this bacterial strain. At a concentration of 250 μM, this molecule did not affect bacterial growth or have a toxic effect on human THP-1 macrophages (Storz et al., 2012). A subsequent study showed that the inhibition of PqsD by (2-nitrophenyl)phenyl methanol was time-dependent (Storz et al., 2013). Subsequently, Storz et al. (2014) synthesized and evaluated several (2-nitrophenyl)methanol derivatives with improved in vitro PqsD inhibition. However, most of these derivatives did not show similar potency in inhibiting HHQ production in a pqsH-deficient P. aeruginosa PA14 strain. The derivative that contained an ethyl group at the methanol moiety, as well as those which contained heteroaromatic pentacycles, strongly inhibited PqsD activity in cells, even though these derivatives were not among the best PqsD inhibitors in vitro. Therefore, for (2-nitrophenyl)methanol derivatives, improved in vitro PqsD inhibition does not necessarily mean improved inhibitory activity in cellulo (Storz et al., 2014).
Other PqsD inhibitors that have been reported are compounds of the aryl-ureidothiophene-2-carboxylic acid class. These compounds were predicted to bind to the substrate channel of PqsD via their aryloxy-moiety pointed toward the bottom of the pocket and thereby block the binding of the substrate, anthraniloyl-CoA (Sahner et al., 2013). Moreover, the chemical structure combination of ureidothiophene-2-carboxylic acids with (2-nitrophenyl)methanol inhibitors yielded some derivatives with improved PqsD inhibitory activity when activity was measured in a cell-free enzyme assay. However, these compounds were ineffective in reducing HHQ production in a whole-cell P. aeruginosa assay. Ureidothiophene-2-carboxylic acid-based inhibitors were suggested to be expelled by efflux pumps in P. aeruginosa; if this were found to be the case, they would not be suitable for development as quorum-quenching strategies (Sahner et al., 2015). Based on the similarity between PqsD and the chalcone synthase (CHS2) expressed in alfalfa (Medicago sativa), Allegretta et al. (2015) developed new PqsD inhibitors from substrates of CHS2. These substrate analogs contained a catechol core that was important for inhibitory activity. Apparently, these compounds inhibited PqsD by blocking the enzyme substrate channel. Several of these inhibitors reduced the production of HHQ in bacterial cultures without affecting bacterial growth (Allegretta et al., 2015). Recently, Thomann et al. (2016) introduced an innovative and original strategy for quenching the PQS quorum-sensing system in P. aeruginosa. This strategy was based on the development of a dual-inhibitor compound that simultaneously inhibited both the PQS transcriptional regulator (PqsR) and PqsD. This compound acted as a dual inhibitor that affected the production of the virulence factors, pyocyanin and pyoverdine, but without affecting bacterial growth. Additionally, this compound reduced biofilm formation by P. aeruginosa and boosted the anti-bacterial activity of ciprofloxacin under biofilm conditions. Importantly, the dual inhibitor increased, in a dose-dependent manner and without cytotoxic effects, the survival rate of G. mellonella larvae challenged with lethal doses of P. aeruginosa (Thomann et al., 2016).
Inhibitors of the heterodimeric enzyme PqsBC have also been described. PqsBC participates in the PQS biosynthesis pathway by catalyzing the condensation of 2-aminobenzoyl acetate and octanoyl-CoA to form HHQ. Drees et al. (2016) demonstrated that 2-aminoacetophenone (secondary metabolite) acts as a competitive inhibitor of PqsBC and also inhibits HHQ production by Pseudomonas putida KT2440 (Drees et al., 2016). Moreover, PqsBC synthetic inhibitors more potent than 2-aminoacetophenone were recently described. This class of compounds were benzamide-benzimidazole derivatives and acted as dual inhibitors (acting simultaneously on PqsR and PqsBC). These PqsBC synthetic inhibitors attenuated P. aeruginosa PA14 virulence during infection of human lung epithelial cells and mouse macrophages. Some of the dual inhibitors reduced bacterial meropenem tolerance, specifically, the dual inhibitors with high anti-PqsR activity (Maura et al., 2017). Dual inhibitors with high anti-PqsR activity block 2-aminoacetophenone production more potently than dual inhibitors with low anti-PqsR activity, of particular interest because 2-aminoacetophenone has been associated with bacterial tolerance to antibiotics (Maura et al., 2017). Moreover, using two selective inhibitors to PqsBC, Allegretta et al. (2017) showed that in inhibitor-treated P. aeruginosa PA14 cells the 2-aminoacetophenone levels were higher than in non-treated bacteria. Consequently, the treatment with one of the PqsBC inhibitors favored P. aeruginosa PA14 tolerance to meropenem (Allegretta et al., 2017).
Inhibition of Autoinducer Peptide Synthesis
In important Gram-positive pathogens including S. aureus, Enteroccocus faecalis, Listeria monocytogenes, Clostridium difficile, Clostridium botulinum, Clostridium perfringens, Bacillus cereus, Streptococcus pyogenes and others, the control of virulence factor expression is associated with peptide-based quorum sensing systems (Gray et al., 2013; Jimenez and Federle, 2014; Le and Otto, 2015; Singh et al., 2016; Ali et al., 2017). The S. aureus accessory gene regulator (agr) system and E. faecalis fsr quorum-sensing system are the most extensively characterized peptide-based quorum sensing systems (Gray et al., 2013; Ali et al., 2017; Tan et al., 2018).
The S. aureus agr-system controls the expression of several virulence factors, including RNAIII, δ-hemolysin, and phenol soluble modulins (PSMs). Transcription of the agr operon produces the RNA II and RNA III transcripts. Specifically, RNA II translation produces the proteins AgrA, AgrB, AgrC, and AgrD, which are the structural components of the agr-system, while RNAIII is involved in the post-transcriptional control of virulence factors expression and encodes δ-hemolysin (Tan et al., 2018). AgrD is the precursor of the autoinducer peptides (AIP) (AIP-I, AIP-II, AIP-III, and AIP-IV). AgrB is an endopeptidase involved in AIP maturation. The AgrB endopeptidase and the type I signal peptidase SpsB remove the C-terminal tail and N-terminal leader segment of AgrD, respectively, producing the thiolactone AIPs (LaSarre and Federle, 2013; Tan et al., 2018). The proteins AgrC/AgrA constitute a two-component system that is involved in AIP signaling. After AIPs are secreted, they bind to the histidine kinase AgrC receptor, which autophosphorylates with the subsequent transference of the phosphoryl group to the response regulator AgrA rendering phosphorylated AgrA. Phosphorylated AgrA forms a dimer, which at low concentration acts as a transcription factor that preferentially binds to the P2 promoter, triggering the production of RNAII transcripts. Consequently, the production of the agr components increases in an autocatalytic way (Wang and Muir, 2016; Tan et al., 2018). After phosphorylated AgrA accumulates to a threshold level, it binds to the P3 promoter stimulating the production of RNAIII transcripts. Additionally, phosphorylated AgrA binds to the psmα and psmβ promoters, stimulating the production of PSMs (Gray et al., 2013; Tan et al., 2018). Because the agr system operates as a positively regulated auto-loop system, in principle, it is possible to disturb AIP production through interfering with any step of the circuit.
In a study performed by Kavanaugh et al. (2007), which focused on the identification of peptidases involved in AIP biosynthesis in S. aureus, the type I signal peptidase SpsB was identified as having a role in the S. aureus AIP biosynthesis pathway. Specifically, two SpsB inhibitors [(P+1) and NIF] were developed that consisted of peptides that mimic the N-terminal cleavage site of AgrD. The inhibitor NIF showed improved stability and stronger inhibition of quorum sensing in comparison to inhibitor P+1 (Kavanaugh et al., 2007). On the other hand, it has been reported that ambuic acid (a secondary fungal metabolite) inhibits the production of AIP in S. aureus as well as the biosynthesis of GBAP in E. faecalis and the putative cyclic peptide pheromones LsrD698 and LsrD826 in Listeria innocua (Nakayama et al., 2009). Later Todd et al. (2016), using an ultraperformance liquid chromatography coupled to mass spectrometry (UPLC-MS) platform, showed that ambuic acid suppresses AIP-I production by a clinical isolate of methicillin-resistant S. aureus (MRSA), in a dose-dependent manner (Todd et al., 2016). More recently, an MRSA strain was genetically manipulated to constitutively produce AIP-I without quorum-sensing control. Ambuic acid was found to effectively inhibit the biosynthesis of AIP-I by this strain. Interestingly, in vivo experiments in a murine model of intradermal MRSA challenge verified that ambuic acid attenuates MRSA pathogenesis and mediates quorum quenching in vivo. Moreover, ambuic acid proved effective in AIP biosynthesis inhibition of several pathogens besides S. aureus, i.e., Staphylococcus saprophyticus, L. monocytogenes, and E. faecalis, but did not affect commensal bacteria such as Staphylococcus lugdunensis and some Staphylococcus epidermidis strains. This selectivity is a desirable characteristic for therapeutic agents (Todd et al., 2017). In sum, all this evidence showed the potential of ambuic acid as an anti-virulence therapeutic agent.
Another target for the agr-system inhibition is the response regulator protein AgrA, which acts as agr operon transcriptional factor. The impairing of AgrA functionality might perturb the agr operon transcription and consequently AIP production as well as agr-controlled virulence factors production. Several chemical compounds including 2-(4-methylphenyl)-1,3-thiazole-4-carboxylic acid, 9H-xanthene-9-carboxylic acid, 4-phenoxyphenol, savirin, ω-hydroxyemodin, biaryl hydroxyketones, and norlichexanthone appear to act by blocking the binding of AgrA to the agr operon promoters via direct interaction with the AgrA C-terminal DNA binding domain (Leonard et al., 2012; Sully et al., 2014; Daly et al., 2015; Baldry et al., 2016; Greenberg et al., 2018). Moreover, other compounds like naphthalene derivatives and biaryl compounds potentially could bind to the AgrA N-terminal phosphoryl-binding pocket, interfering with ArgA phosphorylation and binding to DNA (Khodaverdian et al., 2013).
Recently, some of these AgrA inhibitors have shown be promising in attenuating bacterial virulence in vivo. In this regard, ω-hydroxyemodin (a polyhidroxyanthraquinone) inhibited in vitro all the S. aureus agr-system types (I–IV). Consequently, ω-hydroxyemodin treatment reduced the RNAIII, psmα and hla transcription without bactericidal and cytotoxic-associated effects. In addition, ω-hydroxyemodin inhibited the S. epidermidis agr-system and attenuated the virulence of S. aureus in a mouse skin and soft tissue infection model, apparently via disruption of the arg-system, facilitating the bacterial clearance by the host immune system (Daly et al., 2015). Previously, Sully et al. (2014), using an airpouch skin infection model and a dermonecrosis model, described S. aureus-attenuated virulence by savirin via agr-system disruption and improved host immune response. The treatment of S. aureus USA300 strain LAC with savirin down-regulated the expression of several agr-regulated virulence factors, including RNAIII, V8 protease, serine proteases, lipase, staphopain, PMSβ1, PMSα, PVL, and others, whereas it up-regulated the expression of Spa, SdrD and fibrinogen-binding protein. In addition, savirin treatment reduced α-hemolysin, protease and lipase activity. Clinical isolates (comprising the agr-systems I, II, III, and IV) treated with savirin down-regulated psmα transcript levels, and α-hemolysin activity was reduced in several MRSA and MSSA clinical isolates. Interestingly, resistance or tolerance to savirin were not observed, and the S. epidermidis agr system was not significantly disturbed (Sully et al., 2014). Moreover, two biaryl hydroxyketones (F12 and F19) have been reported that downregulated hla, psmα and RNAIII expression in the MRSA USA300 strain. The methicillin-resistant Staphylococcus epidermidis (MRSE) strain treated with F19 down-regulated AtlE, psmα and RNAIII transcript levels. F-19 protected monocyte and macrophage cells from the lysis caused by several Gram-positive pathogens. Importantly, in an MRSA wound infection model, compound F-19 potentiated β-lactam and fluoroquinolone antibiotic activity, whereas in an MRSA bacteremia/sepsis model, F-19 alone and in combination with cephalothin protected the animals from a lethal infection with MRSA (Greenberg et al., 2018). In a previous study, F-12 and F-19 treatment increased the survival time of MRSA-infected larvae, as well as when they were used in combination with β-lactam antibiotics. In addition, in mice, F-12 stimulated the healing of MRSA-infected wounds (Kuo et al., 2015). Antisense oligonucleotides that target the agrA mRNA have also been used to inhibit AgrA activity. Recently, antisense oligonucleotides against agrA mRNA were used as a strategy to quench the agr-system in a community-associated MRSA strain (CA-MRSA USA300 LAC). The antisense oligonucleotide treatment affected the agrA expression as well as the expression of several virulence factors, including psmα, psmβ, hla, and pvl. The CA-MRSA USA300 LAC strain virulence in a mouse subcutaneous infection model was attenuated by antisense oligonucleotide treatment (Da et al., 2017).
Disruption of the agr-system via interference with AgrC activity, in principle, could also influence AIP biosynthesis. The most exploited strategy to develop AgrC inhibitors is based on producing structural modifications in native AIP scaffold to yield AIP structural analogs (Singh et al., 2016; Wang and Muir, 2016). Some of the recently developed AgrC inhibitors are amide-bridged AIP-III analogs, in which the thioester bond was replaced by an amide bond conferring higher hydrolytic stability and solubility in aqueous media on them than their precursors. The introduction of the amide bridge did not severely affect the inhibitory potency of the lactam analogs toward S. aureus AgrC (type I-IV) (Tal-Gan et al., 2016). Simplified AIP-II peptidomimetics were developed from a truncated AIP-II by Vasquez et al. (2017). Some of these peptidomimetics were pan-group S. aureus AgrC inhibitors; however, the most soluble mimetic in aqueous media (a desirable characteristic for the inhibitors) did not show a potent inhibitory activity toward S. aureus AgrC (group III-IV) in comparison with the parental peptide (truncated AIP-II), but displayed an inhibitory activity similar to the parental peptide toward S. aureus group I, which is one of the main etiologic agents in human infections (Vasquez et al., 2017). Moreover, Karathanasi et al. (2018) described linear synthetic peptidomimetics that interfered with the S. aureus agr-system through competitive binding to the AgrC receptor (Karathanasi et al., 2018). Recently, other AgrC inhibitors described were S. epidermidis AIP and S. lugdunensis AIP analogs (Gordon et al., 2016; Yang et al., 2016). Furthermore, secondary metabolites such as WS9326A, WS9326B, and cochimicin II/III from actinomycetes, avellanin from Hamigera ingelheimensis, ngercheumicins and solonamides from Photobacterium sp. strain S2753 probably influence the AgrC activity via competitive inhibition (Mansson et al., 2011; Kjaerulff et al., 2013; Desouky et al., 2015; Igarashi et al., 2015; Wang and Muir, 2016). Solonamide B showed to be effective in a mouse model for atopic dermatitis to attenuate S. aureus virulence via δ-toxin-induced immunopathologic response inhibition (Baldry et al., 2018).
Inhibition of N-Acyl-Homoserine Lactone Synthesis
In Gram-negative bacteria, quorum-sensing systems based on acyl-HSLs as signal molecules are the most common. The category of acyl-HSLs (also known as autoinducer-1) comprises more than 30 different molecules that share a common structural scaffold, consisting of an acyl chain linked to a homoserine lactone ring. The acyl chains vary in length (4-18 carbons), oxidation state, and degree of saturation (LaSarre and Federle, 2013; Chan et al., 2015). The acyl-HSLs are biosynthesized mainly by the acyl-HSL synthases belonging to the Lux I family (Lux I-type acyl-HSL synthases). These synthases use as substrates S-adenosyl-L-methionine (SAM) and acylated acyl-carrier protein (acyl-ACP) and yield the respective acyl-HSL, the holo-ACP, and MTA, as products (Chung et al., 2011). The Lux I-type acyl-HSLs synthases are present in hundreds of bacterial species, and enzymes from different bacterial species may share conserved regions. Lux I-type acyl-HSLs synthases are not present in Eukarya, making them a potential target for the development of quorum-sensing inhibitors (Chan et al., 2015; Papenfort and Bassler, 2016).
The synthesis of butyryl-HSL is mediated by P. aeruginosa RhlI synthase using as substrates butyryl-ACP and SAM. An early study by Parsek et al. (1999) showed that the end products, MTA, and holo-ACP, and the SAM substrate analogs SAH, S-adenosylcysteine, and sinefungin, act as RhlI synthase inhibitors (Parsek et al., 1999). Another acyl-HSL synthase present in P. aeruginosa for which inhibitors have been reported is LasI. In a study by Lidor et al. (2015), the compound (z)-5-octylidenethiazolidine-2, 4-dione (TZD-C8) was found to inhibit biofilm formation by P. aeruginosa PAO1 in a dose-dependent manner, as well as to induce the downregulation of the expression of the pqsABCDE operon and the lasI gene. Therefore, potentially TZD-C8 could perturb both the quorum-sensing system based on PQS and that based on 3-oxo-C12-HSL. The in vitro swarming motility and PQS production of the bacteria were also affected. In silico evaluation of the interaction between TZD-C8 and LasI suggested that the inhibitory activity of TZD-C8 could result from its binding to the LasI activity pocket (Lidor et al., 2015). In the Gram-negative bacterium Burkholderia glumae the quorum-sensing signal octanoyl-L-HSL (C8-HSL) is synthesized by the acyl-HSL synthase TofI. Chung et al. (2011) found the TofI inhibitor J8-C8 from a library of acyl-HSL analogs. This compound reduced the production of C8-HSL by B. glumae BGR1 cells. In addition, J8-C8 inhibited C8-HSL synthesis in a dose-dependent manner and, together with MTA, had a synergistic inhibitory effect on TofI. X-ray crystal structure analyses showed that J8-C8 binds to the acyl-ACP binding site on TofI, specifically the binding site for the acyl chain, while MTA independently binds to the binding site for SAM (Chung et al., 2011). Moreover, Christensen et al. (2013) reported five compounds that inhibited Burkholderia mallei BmaI1 synthase and YspI synthase from Y. pestis, which is phylogenetically distant from B. mallei BmaI1 synthase. Additionally, two of the five compounds were found to reduce the production of octanoyl-HSL without affecting bacterial growth. The most potent compound [3-(4-methylpiperazin-1-yl)(pyridin-2-yl)methyl-2-phenyl-1H-indol-1-ol] acted as a noncompetitive inhibitor of BmaI1 synthase, and some analogs of this compound showed inhibitory activity. One interesting finding in this study was that one of the five inhibitory compounds selected was the cephalosporin antibiotic cefatrizine, suggesting that cephalosporin antibiotics may inhibit acyl-HSL synthases (Christensen et al., 2013).
It has been reported that thioether analogs of the thioester acyl-substrates of acyl-HSL synthase inhibit the enzyme. Specifically, octyl-ACP noncompetitively inhibited B. mallei BmaI1 synthase while the isopentyl-CoA competitively inhibited the Bradyrhizobium japonicum BjaI-synthase (Christensen et al., 2014). Recently, new diketopiperazine derivatives have been described that inhibit Burkholderia cenocepacia CepI acyl-HSL synthase. The most potent of these derivatives [(3S)-3-Benzyl-6-(3,6-dioxocyclohexa-1,4-dien-1-yl)piperazine-2,5-dione] acted as a non-competitive inhibitor toward both C8-ACP and SAM substrates. This finding was also supported by molecular docking analysis, which showed several high-affinity contact sites for the inhibitor on the CepI structure, but none of these sites was the SAM- and acyl substrate-binding site. Besides, some of these diketopiperazine derivative compounds did not exert an antimicrobial effect on B. cenocepacia J2315 but did interfere with the production of virulence factors such as proteases and siderophores. Furthermore, they perturbed biofilm formation, protected Caenorhabditis elegans nematodes against infection with B. cenocepacia J2315, and had low toxicity for HeLa cells (Scoffone et al., 2016). Recently, based on comparative proteomics approach, Buroni et al. (2018) demonstrated that B. cenocepacia J2315 treated with (3S)-3-benzyl-6-(3,6-dioxocyclohexa-1,4-dien-1-yl)piperazine-2,5-dione displayed a protein expression pattern quite similar to B. cenocepacia ΔcepI mutant (B. cenocepacia J2315 with deleted cepI gene). Interestingly, both the inhibitor-treated strain and the ΔcepI mutant overexpressed the giant cable pilus protein CblA, which has been associated with B. cenocepacia virulence in vivo. In addition, using site-directed mutagenesis and enzymatic activity inhibition approaches it was observed that the pocket around the Ser 41 residue on the CepI structure appears to be the inhibitor binding site (Buroni et al., 2018).
Quorum-quenching agents of plant origin have been identified as inhibitors of acyl-HSL synthases. Chang et al. (2014) identified salicylic acid, tannic acid, and trans-cinnamaldehyde as potential acyl-HSL synthase inhibitors. Subsequently, it was demonstrated that trans-cinnamaldehyde was an RhlI-specific inhibitor and did not affect the growth of P. aeruginosa. Molecular docking analysis of trans-cinnamaldehyde suggested that this inhibition might be mediated by the occupation of the substrate-binding pocket on the synthase (Chang et al., 2014). Recently, other plant-derived compounds have been identified as acyl-HSL synthase inhibitors. It was observed that carvacrol and eugenol reduce biofilm formation, the activity of plant cell wall degrading enzymes, the expression of quorum-sensing-related genes, and virulence in the phytopathogens Pectobacterium carotovorum subsp. brasiliense Pcb1692 and Pectobacterium aroidearum PC1. Based on docking of these compounds to the computational models of ExpR (regulatory protein) and ExpI (acyl-HSL synthase), the mechanism of action of these quorum-quenching agents was suggested to involve direct interaction with ExpI/ExpR proteins and the consequent inhibition of acyl-HSL production (Joshi et al., 2016).
Even though most of the strategies for inhibiting signal production have targeted the acyl-HSL synthases, it is possible that other enzymes linked to the signal biosynthesis pathway could also be targeted. In this respect, it has been reported that triclosan inhibited the P. aeruginosa enoyl-acyl carrier protein reductase (FabI) in vitro. This inhibition reduced the production of butyryl-HSL because FabI supplies the butyryl-ACP necessary for RhlI synthase-mediated butyryl-HSL synthesis (Hoang and Schweizer, 1999). Since FabI is involved in the metabolism of the fatty acids (an essential process for the bacteria), inhibitory agents directed toward it could potentially exert selective pressure on the bacteria with the subsequent emergence of resistant mutants. In fact, resistance to triclosan by P. aeruginosa PAO1 has been reported; this resistance results from active efflux pumps and a triclosan-resistant enoyl-acyl carrier protein reductase (FabV) (Chuanchuen et al., 2003; Zhu et al., 2010; Huang et al., 2016).
Future Directions
The book “Sun Tzu on The Art of War” postulated that “In the practical art of war, the best thing of all is to take the enemy's country whole and intact; to shatter and destroy it is not so good. So, too, it is better to recapture an army entire than to destroy it, to capture a regiment, a detachment or a company entire than to destroy them” (Giles, 2000). In this light, the war of science against bacterial pathogens should not exclusively focus on novel bactericidal agents (which would destroy the enemy), but should also consider antivirulence factors (which would trap the enemy, leaving it without weapons and/or communication systems), allowing the human or animal host to subsequently eliminate the pathogens. Because quorum sensing is a critical process for controlling collective traits including lifestyle and biofilm formation, the synthetic modulators of quorum sensing seem to be the key to manipulating bacterial behavior on demand. This is particularly so in the case of pathogenic bacteria, whose virulence factors include quorum sensing mechanisms (Papenfort and Bassler, 2016).
In this Review, we have summarized the main targets for quorum sensing signal biosynthesis inhibition. The control of bacterial behavior by small molecules has been viewed as a promising strategy for the control of biofilms; and despite the differences among species, quorum sensing plays a crucial role in the infectious process. Although therapies that affect quorum sensing are less likely to select for resistance in comparison with traditional antibiotics, some cases reported in the literature show that bacteria can become resistant to quorum-sensing inhibitors (Defoirdt et al., 2010; Kalia et al., 2014; García-Contreras et al., 2016). Nevertheless, the selective pressure exerted by traditional antibiotics is higher than that of the quorum-sensing inhibitors; therefore, the latter may have longer functional lives and greater utility in treating bacterial infections than the former, which have been, in many cases, rendered ineffective by resistance. To date, few clinical trials of molecules that inhibit quorum sensing have been conducted (Papenfort and Bassler, 2016); therefore, it is still too early to assess the therapeutic potential of these molecules. Efforts to determine mechanisms of resistance and to screen for more effective inhibitors, as well as studies focusing on the in vivo application of such molecules, could lead to the next generation of antimicrobial agents.
Author Contributions
OF, OS, CFN, and OLF contributed conception and design of the review. OF, PR, ÁP, WP, and OS wrote the manuscript. PR and ÁP made the figures. OLF and CFN revised the manuscript. All authors read and approved the submitted version.
Funding
This work was supported by CAPES, CNPq, FAPDF, FUNDECT, UCB, and UCDB. OS holds a postdoctoral scholarship from the National Council of Technological and Scientific Development (CNPq) and Fundação de Apoio ao Desenvolvimento do Ensino, Ciência e Tecnologia do Estado de Mato Grosso do Sul (FUNDECT)—Brazil [300583/2016-8]. This work was supported by the Ramon Areces Foundation (to CFN).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Alfaro, J. F., Zhang, T., Wynn, D. P., Karschner, E. L., and Zhou, Z. S. (2004). Synthesis of LuxS inhibitors targeting bacterial cell– cell communication. Org. Lett. 6, 3043–3046. doi: 10.1021/ol049182i
Ali, L., Goraya, M. U., Arafat, Y., Ajmal, M., Chen, J. L., and Yu, D. (2017). Molecular mechanism of quorum-sensing in Enterococcus faecalis: its role in virulence and therapeutic approaches. Int. J. Mol. Sci. 18:E960. doi: 10.3390/ijms18050960
Allegretta, G., Maurer, C. K., Eberhard, J., Maura, D., Hartmann, R. W., Rahme, L., et al. (2017). In-depth profiling of MvfR-regulated small molecules in Pseudomonas aeruginosa after quorum sensing inhibitor treatment. Front. Microbiol. 8:924. doi: 10.3389/fmicb.2017.00924
Allegretta, G., Weidel, E., Empting, M., and Hartmann, R. W. (2015). Catechol-based substrates of chalcone synthase as a scaffold for novel inhibitors of PqsD. Eur. J. Med. Chem. 90, 351–359. doi: 10.1016/j.ejmech.2014.11.055
Armbruster, C. E., Hong, W., Pang, B., Dew, K.E., Juneau, R. A., Byrd, M. S., et al. (2009). LuxS promote biofilms maturation and persistence of nontypeable Haemophilus influenzae in vivo via modulation of lipooligosccharides on bacterial surface. Infect. Immun. 77, 4081–4019. doi: 10.1128/IAI.00320-09
Arnold, W. K., Savage, C. R., Antonicello, A. D., and Stevenson, B. (2015). Apparent role for Borrelia burgdorferi luxS during mammalian infection. Infect. Immun. 83:1347. doi: 10.1128/IAI.00032-15
Asfour, H. Z. (2018). Anti-quorum sensing natural compounds. J. Microsc. Ultrastruct. 6, 1–10. doi: 10.4103/JMAU.JMAU_10_18
Baldry, M., Nakamura, Y., Nakagawa, S., Frees, D., Matsue, H., Núñez, G., et al. (2018). Application of an agr-specific antivirulence compound as therapy for Staphylococcus aureus-induced inflammatory skin disease. J. Infect. Dis. 218, 1009–1013. doi: 10.1093/infdis/jiy259
Baldry, M., Nielsen, A., Bojer, M. S., Zhao, Y., Friberg, C., Ifrah, D., et al. (2016). Norlichexanthone reduces virulence gene expression and biofilm formation in Staphylococcus aureus. PLoS ONE 11:e0168305. doi: 10.1371/journal.pone.0168305
Bao, Y., Li, Y., Jiang, Q, Zhao, L., Xue, T., Hu, B., et al. (2013). Methylthioadenosine/S-adenosylhomocysteine nucleosidase (Pfs) of Staphylococcus aureus is essential for the virulence independent of LuxS/AI-2 system. Int. J. Med. Microbiol. 303, 190–200. doi: 10.1016/j.ijmm.2013.03.004
Bao, Y., Zhang, X., Jiang, Q., Xue, T., and Sun, B. (2015). Pfs promotes autolysis-dependent release of eDNA and biofilm formation in Staphylococcus aureus. Med. Microbiol. Immunol. 204, 215–226. doi: 10.1007/s00430-014-0357-y
Bera, A. K., Atanasova, V., Robinson, H., Eisenstein, E., Coleman, J. P., Pesci, E. C., et al. (2009). Structure of PqsD, a Pseudomonas quinolone signal biosynthetic enzyme, in complex with anthranilate. Biochemistry 48, 8644–8655. doi: 10.1021/bi9009055
Bikard, D., Euler, C. W., Jiang, W., Nussenzweig, P. M., Goldberg, G. W., Duportet, X., et al. (2014). Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat Biotechnol. 32, 1146–1150. doi: 10.1038/nbt.3043
Bourgeois, J. S., Zhou, D., Thurston, T. L. M., Gilchrist, J. J., and Ko, D. C. (2018). Methylthioadenosine suppresses Salmonella virulence. Infect. Immun. 86:e00429. doi: 10.1128/IAI.00429-18
Buroni, S., Scoffone, V. C., Fumagalli, M., Makarov, V., Cagnone, M., Trespidi, G., et al. (2018). Investigating the mechanism of action of diketopiperazines inhibitors of the Burkholderia cenocepacia quorum sensing synthase CepI: a site-directed mutagenesis study. Front. Pharmacol. 9:836. doi: 10.3389/fphar.2018.00836
Calfee, M. W., Coleman, J. P., and Pesci, E. C. (2001). Interference with Pseudomonas quinolone signal synthesis inhibits virulence factor expression by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 98, 11633–11637. doi: 10.1073/pnas.201328498
Chan, K. G., Liu, Y.C., and Chang, C.Y. (2015). Inhibiting N-acyl-homoserine lactone synthesis and quenching Pseudomonas quinolone quorum sensing to attenuate virulence. Front. Microbiol. 6:1173. doi: 10.3389/fmicb.2015.01173
Chang, C. Y., Krishnan, T., Wang, H., Chen, Y., Yin, W. F., Chong, T. M., et al. (2014). Non-antibiotic quorum sensing inhibitors acting against N-acyl homoserine lactone synthase as druggable target. Sci. Rep. 4:7245. doi: 10.1038/srep07245
Chbib, C. (2017). Synthesis of isomeric analogues of S-ribosylhomocysteine analogues with homocysteine unit attached to C2 of ribose. Bioorg. Med. Chem. Lett. 27, 1681–1685. doi: 10.1016/j.bmcl.2017.03.004
Chbib, C., Sobczak, A. J., Mudgal, M., Gonzalez, C., Lumpuy, D., Nagaj, J., et al. (2016). S-Ribosylhomocysteine analogues modified at the ribosyl C-4 position. J. Sulphur Chem. 37, 307–327. doi: 10.1080/17415993.2015.1137921
Christensen, Q. H., Brecht, R. M., Dudekula, D., Greenberg, E. P., and Nagarajan, R. (2014). Evolution of acyl-substrate recognition by a family of acyl-homoserine lactone synthases. PLoS ONE 9:e112464. doi: 10.1371/journal.pone.0112464
Christensen, Q. H., Grove, T. L., Booker, S. J., and Greenberg, E. P. (2013). A high-throughput screen for quorum-sensing inhibitors that target acyl-homoserine lactone synthases. Proc. Natl. Acad. Sci. U.S.A. 110, 13815–13820. doi: 10.1073/pnas.1313098110
Chuanchuen, R., Karkhoff-Schweizer, R. R., and Schweizer, H. P. (2003). High-level triclosan resistance in Pseudomonas aeruginosa is solely a result of efflux. Am. J. Infect. Control 31, 124–127. doi: 10.1067/mic.2003.11
Chung, J., Goo, E., Yu, S., Choi, O., Lee, J., Kim, J., et al. (2011). Small-molecule inhibitor binding to an N-acyl-homoserine lactone synthase. Proc. Natl. Acad. Sci. U.S.A. 108, 12089–12094. doi: 10.1073/pnas.1103165108
Citorik, R. J., Mimee, M., and Lu, T. K. (2014). Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 32, 1141–1145. doi: 10.1038/nbt.3011
Coleman, J. P., Hudson, L. L., McKnight, S. L., Farrow, J. M. III., Calfee, M. W., Lindsey, C. A., et al. (2008). Pseudomonas aeruginosa PqsA is an anthranilate-coenzyme A ligase. J. Bacteriol. 190:1247. doi: 10.1128/JB.01140-07
Da, F., Yao, L., Su, Z., Hou, Z., Li, Z., Xue, X., et al. (2017). Antisense locked nucleic acids targeting agr inhibit quorum sensing and pathogenesis of community-associated methicillin-resistant Staphylococccus aureus. J. Appl. Microbiol. 122, 257–267. doi: 10.1111/jam.13321
Daly, S. M., Elmore, B. O., Kavanaugh, J. S., Triplett, K. D., Figueroa, M., Raja, H. A., et al. (2015). ω-Hydroxyemodin limits Staphylococcus aures quorum sensing-mediated pathogenesis and immflamation. Antimicrob. Agents Chemother. 59, 2223–2235. doi: 10.1128/AAC.04564-14
Daly, S. M., Joyner, J. A., Triplett, K. D., Elmore, B. O., Pokhrel, S., Frietze, K. M., et al. (2017). VLP-based vaccine induces immune control of Staphylococcus aureus virulence regulation. Sci. Rep. 7:637. doi: 10.1038/s41598-017-00753-0
De Lay, N., and Gottesman, S. (2009). The Crp-activated small non-coding regulatory RNA CyaR (RyeE) links nutritional status to group behavior. J. Bacteriol. 191, 461–476. doi: 10.1128/JB.01157-08
Defoirdt, T. (2018). Quorum-sensing systems as targets for antivirulence therapy. Trends Microbiol. 26, 313–328. doi: 10.1016/j.tim.2017.10.005
Defoirdt, T., Boon, N., and Bossier, P. (2010). Can bacteria evolve resistance to quorum sensing disruption? PLoS Pathog. 6:e1000989. doi: 10.1371/journal.ppat.1000989
Desouky, S. E., Shojima, A., Singh, R. P., Matsufuji, T., Igarashi, Y., Suzuki, T., et al. (2015). Cyclodepsipeptides produced by actinomycetes inhibit cyclic-peptide-mediated quorum sensing in Gram-positive bacteria. FEMS Microbiol. Lett. 362:fvn109. doi: 10.1093/femsle/fnv109
Déziel, E., Gopalan, S., Tampakaki, A. P., Lépine, F., Padfield, K. E., Saucier, M., et al. (2005). The contribution of MvfR to Pseudomonas aeruginosa pathogenesis and quorum sensing circuitry regulation: multiple quorum sensing-regulated genes are modulated without affecting lasRI, rhlRI or the production of N-acyl-l-homoserine lactones. Mol. Microbiol. 55, 998–1014. doi: 10.1111/j.1365-2958.2004.04448.x
Déziel, E., Lépine, F., Milot, S., He, J., Mindrinos, M. N., Tompkins, R. G., et al. (2004). Analysis of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines (HAQs) reveals a role for 4-hydroxy-2-heptylquinoline in cell-to-cell communication. Proc. Natl. Acad. Sci. U.S.A. 101, 1339–1344. doi: 10.1073/pnas.0307694100
Dickey, S. W., Cheung, G. Y. C., and Otto, M. (2017). Different drugs for bad bugs: antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discov. 16, 457–471. doi: 10.1038/nrd.2017.23
Drees, S. L., Li, C., Prasetya, F., Saleem, M., Dreveny, I., Williams, P., et al. (2016). PqsBC, a condensing enzyme in the biosynthesis of the Pseudomonas aeruginosa quinolone signal CRYSTAL STRUCTURE, INHIBITION, AND REACTION MECHANISM. J. Biol. Chem. 291, 6610–6624. doi: 10.1074/jbc.M115.708453
Duanis-Assaf, D., Steinberg, D., Chai, Y., and Shemesh, M. (2016). The LuxS based quorum sensing governs lactose induced biofilm formation by Bacillus subtilis. Front. Microbiol. 6:1517. doi: 10.3389/fmicb.2015.01517
Dulcey, C. E., Dekimpe, V., Fauvelle, D. A., Milot, S., Groleau, M. C., Doucet, N., et al. (2013). The end of an old hypothesis: the Pseudomonas signaling molecules 4-hydroxy-2-alkylquinolines derive from fatty acids, not 3-ketofatty acids. Chem. Biol. 20, 1481–1491. doi: 10.1016/j.chembiol.2013.09.021
Evans, G. B., Tyler, P. C., and Scharmm, V. L. (2018). Immucillins in infectious diseases. ACS Infect. Dis. 4, 107–117. doi: 10.1021/acsinfecdis.7b00172
Fetzner, S. (2015). Quorum quenching enzymes. J. Biotechnol. 201, 2–14. doi: 10.1016/j.jbiotec.2014.09.001
Fitts, E. C., Andersson, J. A., Kirtley, M. L., Sha, J., Erova, T. E., Chauhan, S., et al. (2016). New insights into Autoinducer-2 signaling as a virulence regulator in a mouse model of pneumonic plague. mSphere 1:e00342-16. doi: 10.1128/mSphere.00342-16
Gallagher, L. A., McKnight, S. L., Kuznetsova, M. S., Pesci, E. C., and Manoil, C. (2002). Functions required for extracellular quinolone signaling by Pseudomonas aeruginosa. J. Bacteriol. 184, 6472–6480. doi: 10.1128/JB.184.23.6472-6480.2002
García-Contreras, R., Maeda, T., and Wood, T. K. (2016). Can resistance against quorum-sensing interference be selected? ISME J. 10, 4–10. doi: 10.1038/ismej.2015.84
García-Contreras, R., Martínez-Vázquez, M., Velázquez-Guadarrama, N., Villegas-Pañeda, A. G., Hashimoto, T., Maeda, T., et al. (2013). Resistance to the quorum-quenching compounds brominated furanone C-30 and 5-fluorouracil in Pseudomonas aeruginosa clinical isolates. Pathog. Dis. 68, 8–11. doi: 10.1111/2049-632X.12039
García-Contreras, R., Peréz-Eretza, B., Jasso-Chávez, R., Lira-Silva, E., Roldán-Sánchez, J. A., González-Valdez, A., et al. (2015). High variability in quorum quenching and growth inhibition by furanone C-30 in Pseudomonas aeruginosa clinical isolates from cystic fibrosis patients. Pathog. Dis. 73:ftv040. doi: 10.1093/femspd/ftv040
Giles, L. (2000). Sun Tzu on The Art of War: The Oldest Military Treatise in the World. Leicester: Allandale Online Publishing, 8. Available online at: https://sites.ualberta.ca/~enoch/Readings/The_Art_Of_War.pdf
Gopishetty, B., Zhu, J., Rajan, R., Sobczak, A. J., Wnuk, S. F., Bell, C. E., et al. (2009). Probing the catalytic mechanism of S-ribosylhomocysteinase (LuxS) with catalytic intermediates and substrate analogues. J. Am. Chem. Soc. 131, 1243–1250. doi: 10.1021/ja808206w
Gordon, C. P., Olson, S. D., Lister, J. L., Kavanaugh, J. S., and Horswill, A. R. (2016). Truncated autoinducing peptides as antagonists of Staphylococcus lugdunensis quorum sensing. J. Med. Chem. 59, 8879–8888. doi: 10.1021/acs.jmedchem.6b00727
Gray, B., Hall, P., and Gresham, H. (2013). Targeting agr- and agr-like quorum sensing systems for development of common therapeutics to treat multiple gram-positive bacterial infections. Sensor 13, 5130–5166. doi: 10.3390/s130405130
Greenberg, M., Kuo, D., Jankowsky, E., Long, L., Hager, C., Bandi, K., et al. (2018). Small molecule AgrA inhibitors F12 and F19 act as antivirulence agents against Gram-positive pathogens. Sci. Rep. 8:14578. doi: 10.1038/s41598-018-32829-w
Greene, A. C. (2018). CRISPR-based antibacterials: transforming bacterial defense into offense. Trends Biotechnol. 36, 127–130. doi: 10.1016/j.tibtech.2017.10.021
Guo, M., Gamby, S., Zheng, Y., and Sintim, H. O. (2013). Small molecule inhibitors of AI-2 signaling in bacteria: state-of-the-art and future perspectives for anti-quorum sensing agents. Int. J. Mol. Sci. 14, 17694–17728. doi: 10.3390/ijms140917694
Gutierrez, J. A., Crowder, T., Rinaldo-Matthis, A., Ho, M. C., Almo, S. C., and Schramm, V. L. (2009). Transition state analogs of 5′-methylthioadenosine nucleosidase disrupt quorum sensing. Nat. Chem. Biol. 5, 251–257. doi: 10.1038/nchembio.153
Gutierrez, J. A., Luo, M., Singh, V., Li, L., Brown, R. L., Norris, G. E., et al. (2007). Picomolar inhibitors as transition-state probes of 5′-methylthioadenosine nucleosidases. ACS Chem. Biol. 2, 725–734. doi: 10.1021/cb700166z
Haapalainen, A. M., Thomas, K., Tyler, P. C., Evans, G. B., Almo, S. C., and Schramm, V. L. (2013). Salmonella enterica MTAN at 1.36 Å resolution: a structure-based design of tailored transition state analogs. Structure 6, 963–974. doi: 10.1016/j.str.2013.04.009
Han, T., Li, Y., Shan, Q., Liang, W., Hao, W., Li, Y., et al. (2017). Characterization of S-adenosylhomocysteine/Methylthioadenosine nucleosidase on secretion of AI-2 and biofilm formation of Escherichia coli. Microb. Pathog. 108, 78–84. doi: 10.1016/j.micpath.2017.05.015
Han, X., Liu, L., Fan, G., Zhang, Y., Xu, D., Zuo, J., et al. (2015). Riemerella anatipestifer lacks luxS, but can uptake exogenous autoinducer-2 to regulate biofilm formation. Res. Microbiol. 166, 486–483. doi: 10.1016/j.resmic.2015.06.004
Han, X., and Lu, C. (2009). Biological activity and identification of a peptide inhibitor of LuxS from Streptococcus suis serotype 2. FEMS Microbiol. Lett. 294, 16–23. doi: 10.1111/j.1574-6968.2009.01534.x
Haque, S., Ahmad, F., Dar, S. A., Jawed, A., Mandal, R. K., Wahid, M., et al. (2018). Developments in strategies for quorum sensing virulence factor inhibition to combat bacterial drug resistance. Microb. Pathog. 121, 293–302. doi: 10.1016/j.micpath.2018.05.046
Hardie, K. R., and Heurlier, K. (2008). Establishing bacterial communities by ‘word of mouth': LuxS and autoinducer 2 in biofilm development. Nat. Rev. Microbiol. 6, 635–643. doi: 10.1038/nrmicro1916
Heurlier, K., Vendeville, A., Halliday, N., Green, A, Winzer, K., Tang, C. M., et al. (2009). Growth deficiencies of Nisseria meningitidis pfs and luxS mutants are not due to inactivaction of quorum sensing. J. Bacteriol. 191:1293. doi: 10.1128/JB.01170-08
Hinsberger, S., de Jong, J. C., Groh, M., Haupenthal, J., and Hartmann, R. W. (2014). Benzamidobenzoic acids as potent PqsD inhibitors for the treatment of Pseudomonas aeruginosa infections. Eur. J. Med. Chem. 76, 343–351. doi: 10.1016/j.ejmech.2014.02.014
Hirakawa, H., and Tomita, H. (2013). Interference of bacterial cell-to-cell communication: a new concept of antimicrobial chemotherapy breaks antibiotic resistance. Front. Microbiol. 4:114. doi: 10.3389/fmicb.2013.00114
Hoang, T. T., and Schweizer, H. P. (1999). Characterization of Pseudomonas aeruginosa enoyl-acyl carrier protein reductase (FabI): a target for the antimicrobial triclosan and its role in acylated homoserine lactone synthesis. J. Bacteriol. 181, 5489–5497.
Hu, X., Wang, Y., Gao, L., Jiang, W., Lin, W., Niu, C., et al. (2018). The impairment of methyl metabolism from luxS mutation of Streptococcus mutans. Front. Microbiol. 9:404. doi: 10.3389/fmicb.2018.00404
Huang, Y. H., Lin, J. S., Ma, J. C., and Wang, H. H. (2016). Functional characterization of triclosan-resistant enoyl-acyl-carrier protein reductase (FabV) in Pseudomonas aeruginosa. Front. Microbiol. 7:1903. doi: 10.3389/fmicb.2016.01903
Igarashi, Y., Gohda, F., Kadoshima, T., Fukuda, T., Hanafusa, T., Shojima, A., et al. (2015). Avellanin C, an inhibitor of quorum-sensing signaling in Staphylococcus aureus, from Hamigera ingelheimensis. J. Antibiot. 68, 707–710. doi: 10.1038/ja.2015.50
Ji, C., Sharma, I., Pratihar, D., Hudson, L. L., Maura, D., Guney, T., et al. (2016). Designed small-molecule inhibitors of the anthranilyl-CoA synthetase PqsA block quinolone biosynthesis in Pseudomonas aeruginosa. ACS Chem. Biol. 11, 3061–3067. doi: 10.1021/acschembio.6b00575
Jian, T., and Li, M. (2013). Quorum sensing inhibitors: a patent review. Expert Opin. Ther. Pat. 23, 867–894. doi: 10.1517/13543776.2013.779674
Jimenez, J. C., and Federle, M. J. (2014). Quorum sensing in group A Streptococcus. Front. Cell. Infect. Microbiol. 4:127. doi: 10.3389/fcimb.2014.00127
Joshi, J. R., Khazanov, N., Senderowitz, H., Burdman, S., Lipsky, A., and Yedidia, I. (2016). Plant phenolic volatiles inhibit quorum sensing in pectobacteria and reduce their virulence by potential binding to ExpI and ExpR proteins. Sci. Rep. 6:38126. doi: 10.1038/srep38126
Kalia, V. C., Wood, T. K., and Kumar, P. (2014). Evolution of resistance to quorum-sensing inhibitors. Microb. Ecol. 68, 13–23. doi: 10.1007/s00248-013-0316-y
Kang, S., Kim, J., Hur, J. K., and Lee, S. S. (2017). CRISPR-based genome editing of clinically important Escherichia coli SE15 isolated from indwelling urinary catheters of patients. J. Med. Microbiol. 66, 18–25. doi: 10.1099/jmm.0.000406
Kang, X., Zhao, Y., Jiang, D., Li, X., Wang, X., Wu, Y., et al. (2014). Crystal structure and biochemical studies of Brucella melitensis 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. Biochem. Biophys. Res. Commun. 446, 965–970. doi: 10.1016/j.bbrc.2014.03.045
Karathanasi, G., Bojer, M. S., Baldry, M., Johannessen, B. A., Wolff, S., Greco, I., et al. (2018). Linear peptidomimetics as potent antagonists of Staphylococcus aureus agr quorum sensing. Sci Rep. 8:3562. doi: 10.1038/s41598-018-21951-4
Kaur, A., Capalash, N., and Sharma, P. (2018). Quorum sensing in thermophiles: prevalence of autoinducer-2 system. BMC Microbiol. 18:62. doi: 10.1186/s12866-018-1204-x
Kavanaugh, J. S., Thoendel, M., and Horswill, A. R. (2007). A role for type I signal peptidase in Staphylococcus aureus quorum sensing. Mol. Microbiol. 65, 780–798. doi: 10.1111/j.1365-2958.2007.05830.x
Khodaverdian, V., Pesho, M., Truitt, B., Bollinger, L., Patel, P., Nithianantham, S., et al. (2013). Discovery of antivirulence agents against methicillin-resistant Staphylococcus aures. Antimicrob. Agents Chemother. 57, 3645–3652. doi: 10.1128/AAC.00269-13
Kim, B., Park, J. S., Choi, H. Y., Yoon, S. S., and Kim, W. G. (2018). Terrein is an inhibitor of quorum sensing and c-di-GMP in Pseudomonas aeruginosa: a connection between quorum sensing and c-di-GMP. Sci. Rep. 8:8617. doi: 10.1038/s41598-018-26974-5
Kim, K., Kim, Y. U., Koh, B. H., Hwang, S. S., Kim, S. H., Lépine, F., et al. (2010). HHQ and PQS, two Pseudomonas aeruginosa quorum-sensing molecules, down-regulate the innate immune responses through the nuclear factor-κB pathway. Immunology 129, 578–588. doi: 10.1111/j.1365-2567.2009.03160.x
Kjaerulff, L., Nielsen, A., Mansson, M., Gram, L., Larsen, T. O., Ingmer, H., et al. (2013). Identification of four new agr quorum sensing-interfering cyclodepsipeptides from a marine Photobacterium. Mar. Drugs. 11, 5051–5062. doi: 10.3390/md11125051
Komor, U., Bielecki, P., Loessner, H., Rohde, M., Wolf, K., Westphal, K., et al. (2012). Biofilm formation by Pseudomonas aeruginosa in solid murine tumors–a novel model system. Microbes Infect. 14, 951–958. doi: 10.1016/j.micinf.2012.04.002
Kong, C., Neoh, H. M., and Nathan, S. (2016). Targeting Staphylococcus aureus toxins: a potential form of anti-virulence therapy. Toxins 8:E72. doi: 10.3390/toxins8030072
Kuo, D., Yu, G., Hoch, W., Gabay, D., Long, L., Ghannoum, M., et al. (2015). Novel quorum-quenching agents promote methicillin-resistant Staphylococcus aureus (MRSA) wound healing and sensitize MRSA to β-lactam antibiotics. Antimicrob. Agents Chemother. 59, 1512–1518. doi: 10.1128/AAC.04767-14
Laganenka, L., Colin, R., and Sourjik, V. (2016). Chemotaxis towards autoinducer 2 mediates autoaggregation in Escherichia coli. Nat. Commun. 7:12984. doi: 10.1038/ncomms12984
Laganenka, L., and Sourjik, V. (2017). Autoinducer 2-dependent Escherichia coli biofilm formation is enhanced in a dual-species co-culture. Appl. Environ. Microbiol. 84:AEM02638–17. doi: 10.1128/AEM.02638-17
LaSarre, B., and Federle, M. J. (2013). Exploiting quorum sensing to confuse bacterial pathogens. Microbiol. Mol. Biol. Rev. 77, 73–111. doi: 10.1128/MMBR.00046-12
Le, K. Y., and Otto, M. (2015). Quorum-sensing regulation in staphylococci-an overview. Front. Microbiol. 6:1174. doi: 10.3389/fmicb.2015.01174
Lebeer, S., De Keersmaecker, S. C., Verhoeven, T. L., Fadda, A. A., Marchal, K., and Vanderleyden, J. (2007). Functional analysis of luxS in the probiotic strain Lactobacillus rhamnosus GG reveals a central metabolic role important for growth and biofilm formation. J. Bacteriol. 189:860. doi: 10.1128/JB.01394-06
Lee, J. E., Cornell, K. A., Riscoe, M. K., and Howell, P. L. (2003). Structure of Escherichia coli 5′-Methylthioadenosine/S-Adenosylhomocysteine nucleosidase inhibitor complexes provide insight into the conformational changes required for substrate binding and catalysis. J. Biol. Chem. 278, 8761–8770. doi: 10.1074/jbc.M210836200
Lee, J. E., Settembre, E. C., Cornell, K. A., Riscoe, M. K., Sufrin, J. R., Ealick, S. E., et al. (2004). Structural comparison of MTA phosphorylase and MTA/AdoHcy nucleosidase explains substrate preferences and identifies regions exploitable for inhibitor design. Biochemistry 43, 5159–5169. doi: 10.1021/bi035492h
Leonard, P. G., Bezar, I. F., Sidote, D. J., and Stock, A. M. (2012). Identification of a hydrophobic cleft in the LytTR domain of AgrA as a locus for small molecule interactions that inhibit DNA binding. Biochemestry 51, 10035–10043. doi: 10.1021/bi3011785
Lesic, B., Lépine, F., Déziel, E., Zhang, J., Zhang, Q., Padfield, K., et al. (2007). Inhibitors of pathogen intercellular signals as selective anti-infective compounds. PLoS Pathog. 3:e126. doi: 10.1371/journal.ppat.0030126
Li, H., Li, X., Song, C., Zhang, Y., Wang, Z., Liu, Z., et al. (2017). Autoinducer-2 facilitates Pseudomonas aeruginosa PAO1 pathogenicity in vitro and in vivo. Front. Microbiol. 8:1944. doi: 10.3389/fmicb.2017.01944
Li, H., Li, X., Wang, Z., Fu, Y., Ai, Q., Dong, Y., et al. (2015). Autoinducer-2 regulates Pseudomonas aeruginosa PAO1 biofilm formation and virulence production in a dose-dependent manner. BMC Microbiol. 15:192. doi: 10.1186/s12866-015-0529-y
Lidor, O., Al-Quntar, A., Pesci, E. C., and Steinberg, D. (2015). Mechanistic analysis of a synthetic inhibitor of the Pseudomonas aeruginosa LasI quorum-sensing signal synthase. Sci. Rep. 5:16569. doi: 10.1038/srep16569
Liu, R. (2012). Synthesis of S-Ribosyl-l-Homocysteine and Analogs Modified at the Homocysteine-C3 Position. Master's thesis, San Fransisco, CA, University of San Fransisco.
Longshaw, A. I., Adanitsch, F., Gutierrez, J. A., Evans, G. B., Tyler, P. C., and Schramm, V. L. (2010). Design and synthesis of potent “sulfur-free” transition state analogue inhibitors of 5′-methylthioadenosine nucleosidase and 5′-methylthioadenosine phosphorylase. J. Med. Chem. 53, 6730–6746. doi: 10.1021/jm100898v
Ma, L., Feng, S., Fuente-Núñez, C., Hancock, R. E. W., and Lu, X. (2018). Development of moleculary imprinted polymers to block quorum sensing and inhibit bacterial biofilm formation. ACS Appl. Mater. Interfaces. 10, 18450–18457. doi: 10.1021/acsami.8b01584
Ma, R., Qiu, S., Jiang, Q., Sun, H., Xue, T., Cai, G., et al. (2017). AI-2 quorum sensing negatively regulates rbf expression and biofilm formation in Staphylococccus aureus. Int. J. Med. Microbiol. 307, 257–267. doi: 10.1016/j.ijmm.2017.03.003
Ma, Y., Hao, L., Ke, H., Liang, Z., Ma, J., Liu, Z., et al. (2017). LuxS/AI-2 in Streptococcus agalactiae reveals a key role in acid tolerance and virulence. Res. Vet. Sci. 115, 501–507. doi: 10.1016/j.rvsc.2017.07.032
Maeda, T., García-Contreras, R., Pu, M., Sheng, L., Garcia, L.R., Tomás, M., et al. (2012). Quorum quenching quandary: resistance to antivirulence compounds. ISME J. 6, 493–501. doi: 10.1038/ismej.2011.122
Malladi, V. L., Sobczak, A. J., Meyer, T. M., Pei, D., and Wnuk, S. F. (2011). Inhibition of LuxS by S-ribosylhomocysteine analogues containing a [4-aza] ribose ring. Bioorg. Med. Chem. 19, 5507–5519. doi: 10.1016/j.bmc.2011.07.043
Mansson, M., Nielsen, A., Kjaerulff, L., Gotfredsen, C. H., Wietz, M., Ingmer, H., et al. (2011). Inhibition of virulence gene expression in Staphylococcus aureus by novel depsipeptides from a marine photobacterium. Mar. Drugs. 9, 2537–2552. doi: 10.3390/md9122537
Maura, D., Drees, S. L., Bandyopadhaya, A., Kitao, T., Negri, M., Starkey, M., et al. (2017). Polypharmacology approaches against the Pseudomonas aeruginosa MvfR regulon and their application in blocking virulence and antibiotic tolerance. ACS Chem. Biol. 12, 1435–1443. doi: 10.1021/acschembio.6b01139
Munguia, J., and Nizet, V. (2017). Pharmacological targeting of the host–pathogen interaction: alternatives to classical antibiotics to combat drug-resistant superbugs. Trends Pharmacol. Sci. 38, 473–488. doi: 10.1016/j.tips.2017.02.003
Nakayama, J., Uemura, Y., Nishiguchi, K., Yoshimura, N., Igarashi, Y., and Sonomoto, K. (2009). Ambuic acid inhibits the biosynthesis of cyclic peptide quormones in gram-positive bacteria. Antimicrob. Agents Chemother. 53, 580–586. doi: 10.1128/AAC.00995-08
Namanja-Magliano, H. A., Evans, G. B., Harijan, R. K., Tyler, P. C., and Schramm, V. L. (2017). Transition state analogue inhibitors of 5′-deoxyadenosine/5′-methylthioadenosine nucleosidase from Mycobacterium tuberculosis. Biochemistry 56, 5090–5098. doi: 10.1021/acs.biochem.7b00576
Namanja-Magliano, H. A., Stratton, C. F., and Schramm, V. L. (2016). Transition state structure and inhibition of Rv0091, a 5′-Deoxyadenosine/5′-methylthioadenosine nucleosidase from Mycobacterium tuberculosis. ACS Chem. Biol. 11, 1669–1676. doi: 10.1021/acschembio.6b00144
Nandi, S. (2016). Recent advances in ligand and structure based screening of potent quorum sensing inhibitors against antibiotic resistance induced bacterial virulence. Recent Pat. Biotechnol. 10, 195–216. doi: 10.2174/1872208310666160728104450
Niu, C., Robbins, C. M., Pittman, K. J., Osborn, J. L., Stubblefield, B. A., Simmons, R. B., et al. (2013). LuxS influences Escherichia coli biofilm formation through autoinducer-2-dependent and autoinducer-2-independent modalities. FEMS Microbiol. Ecol. 83, 778–791. doi: 10.1111/1574-6941.12034
Pang, B., Armbruster, C. E., Foster, G., Learman, B. S., Gandhi, U., and Swords, W. E. (2018). Autoinducer 2 (AI-2) production by nontypeable Haemophilus influenzae 86-028NP promotes expression of a predicted glycosyltransferase that is a determinant of biofilm maturation, prevention of dispersal, and persistence in vivo. Infect. Immun. 86:e00506-18. doi: 10.1128/IAI.00506-18
Papenfort, K., and Bassler, B. L. (2016). Quorum sensing signal–response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 14, 576–588. doi: 10.1038/nrmicro.2016.89
Park, H., Lee, K., Yeo, S., Shin, H., and Holzapfel, W. H. (2017). Autoinducer-2 quorum sensing influences viability of Escherichia coli O157:H7 under osmotic and in vitro gastrointestinal stress conditions. Front. Microbiol. 8:1077. doi: 10.3389/fmicb.2017.01077
Parsek, M. R., Val, D. L., Hanzelka, B. L., Cronan, J. E Jr., and Greenberg, E. P. (1999). Acyl homoserine-lactone quorum-sensing signal generation. Proc. Natl. Acad. Sci. U.S.A. 96, 4360–4365. doi: 10.1073/pnas.96.8.4360
Parveen, N., and Cornell, K. A. (2011). Methylthioadenosine/S-adenosylhomocysteine nucleosidase, a critical enzyme for bacterial metabolism. Mol. Microbiol. 79, 7–20. doi: 10.1111/j.1365-2958.2010.07455.x
Pei, D., and Zhu, J. (2004). Mechanism of action of S-ribosylhomocysteinase. Curr. Opin. Chem. Biol. 8, 492–497. doi: 10.1016/j.cbpa.2004.08.003
Pereira, C. S., Thompson, J. A., and Xavier, K. B. (2013). AI-2-mediated signalling in bacteria. FEMS Microbiol. Rev. 37, 156–181. doi: 10.1111/j.1574-6976.2012.00345.x
Pérez-Rodríguez, I., Bolognini, M., Ricci, J., Bini, E., and Vetriani, C. (2015). From deep-sea volcanoes to human pathogens: a conserved quorum-sensing signal in Epsilonproteobacteria. ISME J. 9, 1222–1234. doi: 10.1038/ismej.2014.214
Pistorius, D., Ullrich, A., Lucas, S., Hartmann, R. W., Kazmaier, U., and Müller, R. (2011). Biosynthesis of 2-Alkyl-4 (1H)-Quinolones in Pseudomonas aeruginosa: potential for therapeutic interference with pathogenicity. Chembiochem 12, 850–853. doi: 10.1002/cbic.201100014
Qi, L. S., Larson, M. H., Gilbert, L. A., Doudna, J. A., Weissman, J. S., Arkin, A. P., et al. (2013). Repurposing CRISPR as an RNA guide plataform for sequence-specific control of gene expression. Cell 152, 1173–1183. doi: 10.1016/j.cell.2013.02.022
Quave, C. L., and Horswill, A. R. (2013). Flipping the switch: tools for detecting small molecules inhibitors of staphylococcal virulence. Front. Microbiol. 5:706. doi: 10.3389/fmicb.2014.00706
Quave, C. L., Lyles, J. T., Kavanaugh, J. S., Nelson, K., Parlet, C. P., Crosby, H. A., et al. (2015). Castanea sativa (European Chestnut) leaf extract rich in ursene and oleanene derivatives block Sthaphylococcus aureus virulence and pathogenesis without detectble resistance. PLoS ONE 10:e0136486. doi: 10.1371/journal.pone.0136486
Reuter, K., Steinbach, A., and Helms, V. (2016). Interfering with bacterial quorum sensing. Perspect. Med. Chem. 8, 1–15. doi: 10.4137/PMC.S13209
Ronning, D. R., Iacopelli, N. M., and Mishra, V. (2010). Enzyme–ligand interactions that drive active site rearrangements in the Helicobacter pylori 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. Protein Sci. 19, 2498–2510. doi: 10.1002/pro.524
Sahner, J. H., Brengel, C., Storz, M. P., Groh, M., Plaza, A., Müller, R., et al. (2013). Combining in silico and biophysical methods for the development of Pseudomonas aeruginosa quorum sensing inhibitors: an alternative approach for structure-based drug design. J. Med. Chem. 56, 8656–8664. doi: 10.1021/jm401102e
Sahner, J. H., Empting, M., Kamal, A., Weidel, E., Groh, M., Börger, C., et al. (2015). Exploring the chemical space of ureidothiophene-2-carboxylic acids as inhibitors of the quorum sensing enzyme PqsD from Pseudomonas aeruginosa. Eur. J. Med. Chem. 96, 14–21. doi: 10.1016/j.ejmech.2015.04.007
Scoffone, V. C., Chiarelli, L. R., Makarov, V., Brackman, G., Israyilova, A., Azzalin, A., et al. (2016). Discovery of new diketopiperazines inhibiting Burkholderia cenocepacia quorum sensing in vitro and in vivo. Sci. Rep. 6:32487. doi: 10.1038/srep32487
Shen, G., Rajan, R., Zhu, J., Bell, C. E., and Pei, D. (2006). Design and synthesis of substrate and intermediate analogue inhibitors of S-ribosylhomocysteinase. J. Med. Chem. 49, 3003–3011. doi: 10.1021/jm060047g
Silva, A. J., Parker, W. B., Allan, P. W., Ayala, J. C., and Benitez, J. A. (2015). Role of methylthioadenosine /S-adenosylhomocysteine nucleosidase in Vibrio cholerae in cellular communication and biofilm development. Biochem. Biophys. Res. Commun. 461, 65–69. doi: 10.1016/j.bbrc.2015.03.170
Singh, R.P., Desouky, S. E., and Nakayama, J. (2016). Quorum quenching strategy targeting Gram-positive pathogenic bacteria. Adv. Exp. Med. Biol. 901, 109–130. doi: 10.1007/5584_2016_1
Singh, V., Evans, G. B., Lenz, D. H., Mason, J. M., Clinch, K., Mee, S., et al. (2005a). Femtomolar transition state analogue inhibitors of 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase from Escherichia coli. J. Biol. Chem. 280, 18265–18273. doi: 10.1074/jbc.M414472200
Singh, V., Lee, J. E., Núñez, S., Howell, P. L., and Schramm, V. L. (2005b). Transition state structure of 5 ′-methylthioadenosine/S-adenosylhomocysteine nucleosidase from Escherichia coli and its similarity to transition state analogues. Biochemistry 44, 11647–11659. doi: 10.1021/bi050863a
Singh, V., Shi, W., Almo, S. C., Evans, G. B., Furneaux, R. H., Tyler, P. C., et al. (2006). Structure and inhibition of a quorum sensing target from Streptococcus pneumoniae. Biochemistry 45, 12929–12941. doi: 10.1021/bi061184i
Siu, K. K., Lee, J. E., Smith, G. D., Horvatin-Mrakovcic, C., and Howell, P. L. (2008). Structure of Staphylococcus aureus 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 64, 343–350. doi: 10.1107/S1744309108009275
Sobczak, A. J., Chbib, C., and Wnuk, S. F. (2015). S-ribosylhomocysteine analogs containing a [4-thio] ribose ring. Carbohydr. Res. 415, 39–47. doi: 10.1016/j.carres.2015.07.005
Storz, M. P., Allegretta, G., Kirsch, B., Empting, M., and Hartmann, R. W. (2014). From in vitro to in cellulo: structure–activity relationship of (2-nitrophenyl) methanol derivatives as inhibitors of PqsD in Pseudomonas aeruginosa. Org. Biomol. Chem. 12, 6094–6104. doi: 10.1039/C4OB00707G
Storz, M. P., Brengel, C., Weidel, E., Hoffmann, M., Hollemeyer, K., Steinbach, A., et al. (2013). Biochemical and biophysical analysis of a chiral PqsD inhibitor revealing tight-binding behavior and enantiomers with contrary thermodynamic signatures. ACS Chem. Biol. 8, 2794–2801. doi: 10.1021/cb400530d
Storz, M. P., Maurer, C. K., Zimmer, C., Wagner, N., Brengel, C., de Jong, J. C., et al. (2012). Validation of PqsD as an anti-biofilm target in Pseudomonas aeruginosa by development of small-molecule inhibitors. J. Am. Chem. Soc. 134, 16143–16146. doi: 10.1021/ja3072397
Sully, E. K., Malachowa, N., Elmore, B. O., Alexander, S. M., Femling, J. K., Gray, B. M., et al. (2014). Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promote host defense with minimal impacto on the resistance. PLoS Pathog. 10:e1004174. doi: 10.1371/journal.ppat.1004174
Sun, B., and Zhang, M. (2016). Analysis of the antibacterial effect of an Edwardsiella tarda LuxS inhibitor. Springerplus 5:92. doi: 10.1186/s40064-016-1733-4
Sun, J., Daniel, R., Wagner-Döbler, I., and Zeng, A. P. (2004). Is autoinducer-2 a universal signal for interspecies communication: a comparative genomic and phylogenetic analysis of the synthesis and signal transduction pathways. BMC Evol. Biol. 4:36. doi: 10.1186/1471-2148-4-36
Tal-Gan, Y., Ivancic, M., Cornilescu, G., Yang, T, and Blackwell, HE. (2016). Highly stable, amide bridged autoinducing peptides analogues that strongly inhibit the AgrC quorum sensing receptor in Staphylococcus aureus. Angew. Chem. Int. Ed Engl. 55, 8913–8917. doi: 10.1002/anie.201602974
Tan, L., Li, S. R., Jiang, B., Hu, X. M., and Li, S. (2018). Therapeutic targeting of the Staphylococcus aureus accessory gene regulator (agr) system. Front. Microbiol. 9:55. doi: 10.3389/fmicb.2018.00055
Tavender, T. J., Halliday, N. M., Hardie, K. R., and Winzer, K. (2008). LuxS-independent formation of AI-2 from ribulose-5-phosphate. BMC Microbiol. 8:98. doi: 10.1186/1471-2180-8-98
Thomann, A., de Mello Martins, A. G., Brengel, C., Empting, M., and Hartmann, R. W. (2016). Application of dual inhibition concept within looped autoregulatory systems toward antivirulence agents against Pseudomonas aeruginosa infections. ACS Chem. Biol. 11, 1279–1286. doi: 10.1021/acschembio.6b00117
Thomas, K., Cameron, S. A., Almo, S. C., Burgos, E. S., Gulab, S. A., and Schramm, V. L. (2015). Active site and remote contributions to catalysis in methylthioadenosine nucleosidases. Biochemistry 54, 2520–2529. doi: 10.1021/bi501487w
Todd, D. A., Parlet, C. P., Crosby, H. A., Malone, C. L., Heilmann, K. P., Horswill, A. R., et al. (2017). Signal biosynthesis inhibition with ambuic acid as a strategy to target antibiotic-resistant infections. Antimicrob. Agents Chemother. 61:e00263-17. doi: 10.1128/AAC.00263-17
Todd, D. A., Zich, D. B., Ettefagh, K. A., Kavanaugh, J. S., Horswill, A. R., and Cech, N. B. (2016). Hybrid Quadrupole-Orbitrap mass spectrometry for quantitative measurement of quorum sensing inhibition. J. Microbiol. Methods 127, 89–94. doi: 10.1016/j.mimet.2016.05.024
Trappetti, C., McAllister, J. L., Chen, A., Wang, H., Paton, W. A., Oggioni, R. M., et al. (2017). Autoinducer 2 signaling via the phosphotransferase FruA drives galactose utilization by Streptococcus pneumoniae, resulting in hypervirulence. MBio 8:e02269-16. doi: 10.1128/mBio.02269-16
Vale, P. F., McNally, L., Doeschl-Wilson, A., King, K. C., Popat, R., Domingo-Sananes, M. R., et al. (2016). Beyond killing: can we find new ways to manage infection? Evol Med Public Health 2016, 148–157. doi: 10.1093/emph/eow012
Vasquez, J. K., Tal-Gan, Y., Cornilescu, G., Tyler, K. A., and Blackwell, H. E. (2017). Simplified AIP-II peptidomimetics are potent inhibitors of Staphylococcus aureus AgrC quorum sensing receptors. Chembiochem 18, 413–423. doi: 10.1002/cbic.201600516
Velusamy, S. K., Sampathkumar, V., Godboley, D., and Fine, D. H. (2017). Survival of an Aggregatibacter actinomycetemcomitans quorum sensing luxS mutant in the mouths of Rhesus monkeys: insights into ecological adaptation. Mol. Oral Microbiol. 32, 432–442. doi: 10.1111/omi.12184
Wang, B., and Muir, T. W. (2016). Regulation of virulence in Staphylococcus aureus: molecular mechanisms and remaining puzzles. Cell Chem Biol. 23, 214–224. doi: 10.1016/j.chembiol.2016.01.004
Weidel, E., de Jong, J. C., Brengel, C., Storz, M. P., Braunshausen, A., Negri, M., et al. (2013). Structure optimization of 2-benzamidobenzoic acids as PqsD inhibitors for Pseudomonas aeruginosa infections and elucidation of binding mode by SPR, STD NMR, and molecular docking. J. Med. Chem. 56, 6146–6155. doi: 10.1021/jm4006302
Wnuk, S. F., Lalama, J., Garmendia, C. A., Robert, J., Zhu, J., and Pei, D. (2008). S-Ribosylhomocysteine analogues with the carbon-5 and sulfur atoms replaced by a vinyl or (fluoro) vinyl unit. Bioorg. Med. Chem. 16, 5090–5102. doi: 10.1016/j.bmc.2008.03.028
Wnuk, S. F., Robert, J., Sobczak, A. J., Meyers, B. P., Malladi, V. L., Zhu, J., et al. (2009). Inhibition of S-ribosylhomocysteinase (LuxS) by substrate analogues modified at the ribosyl C-3 position. Bioorg. Med. Chem. 17, 6699–6706. doi: 10.1016/j.bmc.2009.07.057
Xu, Y., Wang, L., Chen, J., Zhao, J., Fan, S., Dong, Y., et al. (2017). Structural and functional analyses of Periplasmic 5′-Methylthioadenosine/S-Adenosylhomocysteine nucleosidase from Aeromonas hydrophila. Biochemistry 56, 5347–5355. doi: 10.1021/acs.biochem.7b00691
Yadav, M.K., Vidal, J. E., Go, Y. Y., Kim, S. H., Chae, S. W., and Song, J. J. (2018). The LuxS/AI-2 quorum sensing system of Streptococcus pneumoniae is required to cause disease, and to regulate virulence-and metabolism-related genes in a rat model of middle ear infection. Front. Cell. Infect. Microbiol. 8:138. doi: 10.3389/fcimb.2018.00138
Yang, T., Tal-Gan, Y., Paharik, A. E., Horswill, A. R., and Blackwell, H. E. (2016). Structure-function analyses of a Staphylococcus epidermidis autoinducing peptide reveals motifs critical for AgrC-type receptor modulation. ACS Chem. Biol. 11, 1982–1991. doi: 10.1021/acschembio.6b00120
Zang, T., Lee, B. W., Cannon, L. M., Ritter, K. A., Dai, S., Ren, D., et al. (2009). A naturally occurring brominated furanone covalently modifies and inactivates LuxS. Bioorg. Med. Chem. Lett. 19, 6200–6204. doi: 10.1016/j.bmcl.2009.08.095
Zhang, M., Jiao, X. D., Hu, Y. H., and Sun, L. (2009). Attenuation of Edwardsiella tarda virulence by small peptides that interfere with LuxS/autoinducer type 2 quorum sensing. Appl. Environ. Microbiol. 75:3882. doi: 10.1128/AEM.02690-08
Zhang, M., Sun, K., and Sun, L. (2008). Regulation of autoinducer 2 production and luxS expression in a pathogenic Edwardsiella tarda strain. Microbiology 154, 2060–2069. doi: 10.1099/mic.0.2008/017343-0
Zhang, M., and Sun, L. (2012). Edwardsiella ictaluri LuxS: activity, expression, and involvement in pathogenicity. Pol. J. Microbiol. 61, 263–271.
Zhu, J., Hu, X., Dizin, E., and Pei, D. (2003). Catalytic mechanism of S-ribosylhomocysteinase (LuxS): direct observation of ketones intermediates by 13C NMR spectroscopy. J. Am Chem Soc. 125, 13379–81. doi: 10.1021/ja0369663
Zhu, J., Knottenbelt, S., Kirk, M. L., and Pei, D. (2006). Catalytic mechanism of S-ribosylhomocysteinase: ionization state of active-site residues. Biochemistry 45, 12195–12203. doi: 10.1021/bi061434v
Zhu, L., Lin, J., Ma, J., Cronan, J. E., and Wang, H. (2010). Triclosan resistance of Pseudomonas aeruginosa PAO1 is due to FabV, a triclosan-resistant enoyl-acyl carrier protein reductase. Antimicrob. Agents Chemother. 54, 689–698. doi: 10.1128/AAC.01152-09
Keywords: virulence, antibiotic resistance, quorum sensing, quorum-sensing inhibition, anti-virulence therapy
Citation: Fleitas Martínez O, Rigueiras PO, Pires ÁS, Porto WF, Silva ON, de la Fuente-Nunez C and Franco OL (2019) Interference With Quorum-Sensing Signal Biosynthesis as a Promising Therapeutic Strategy Against Multidrug-Resistant Pathogens. Front. Cell. Infect. Microbiol. 8:444. doi: 10.3389/fcimb.2018.00444
Received: 21 August 2018; Accepted: 12 December 2018;
Published: 05 February 2019.
Edited by:
Rodolfo García-Contreras, National Autonomous University of Mexico, MexicoReviewed by:
Hanne Ingmer, University of Copenhagen, DenmarkYael González Tinoco, Ensenada Center for Scientific Research and Higher Education (CICESE), Mexico
Copyright © 2019 Fleitas Martínez, Rigueiras, Pires, Porto, Silva, de la Fuente-Nunez and Franco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cesar de la Fuente-Nunez, Y2Z1ZW50ZUBtaXQuZWR1
Octavio Luiz Franco, b2NmcmFuY29AZ21haWwuY29t