 Poshmaal Dhar
Poshmaal Dhar Julie McAuley
Julie McAuley- 1Faculty of Health, School of Medicine, Deakin University, Geelong, VIC, Australia
- 2Department of Microbiology and Immunology, Peter Doherty Institute, University of Melbourne, Melbourne, VIC, Australia
The family of cell surface (cs-) mucins are constitutively expressed at the cell surface by nearly all epithelial cells, beneath the gel-mucin layer. All cs-mucin family members have structural features that enable them to act as a releasable decoy barrier to mucosal pathogens, by providing ligands for pathogen binding and the ability to shed the bound extracellular domain. Due to the towering structure of cs-mucins at the surface, binding of mucosal pathogens can also sterically block binding to underlying cellular receptors. The cytoplasmic tail domain of cs-mucins are capable of initiating signal transduction cascades and due to their conservation across species, may play an important biological role in cellular signaling. MUC1 is one of the most extensively studied of the cs-mucin family. With respect to its physiological function in the mucosal environment, MUC1 has been demonstrated to play a dynamic role in protection of the host from infection by a wide variety of pathogens and to regulate inflammatory responses to infection. This review briefly summarizes the current knowledge and new findings regarding the structural features relating to the function of MUC1, its role as a protective barrier against pathogen invasion and mechanisms by which this cs-mucin regulates inflammation.
Introduction
To access its receptor on the host cell, invading pathogens must first circumvent the protective mucus layer, which is composed of a network of mucins. Mucins are of two main types, secreted and cell bound. Secretory mucins are synthesized and extruded into the lumen by goblet cells. Cell bound mucins are tethered to the apical cell surface (cs-) of most epithelia and are likely to be the first point of direct contact between host tissues and organisms that penetrate the secreted mucus layer (Hattrup and Gendler, 2008). The first mucin to be characterized structurally was MUC1 and has since been largely studied for its aberrant expression and role in cancer (Taylor-Papadimitriou et al., 1999). However, there is burgeoning evidence that MUC1 plays a dynamic role in the host mucosal barrier to infection. Importantly, MUC1 has been shown to not only provide a physical barrier, limiting infection and colonization, it has also been demonstrated to play a significant role as a modulator of pathogen-induced inflammation (McAuley et al., 2007; Guang et al., 2010; Ng et al., 2016).
Similar to all of the cs-mucin family, MUC1 consists of a large, extracellular, O-glycosylated polypeptide backbone that extends 200–500 nm above the apical surface; a transmembrane domain that can be cleaved and enables shedding of the extracellular domain; and a cytoplasmic tail capable of signaling (Figure 1A; Hattrup and Gendler, 2008; Sheng et al., 2012). The MUC1 extracellular domain (-ED) is extensively O-glycosylated toward the N-terminus (Figure 1A; Hanisch et al., 1998; Perez-Vilar and Hill, 1999; Silverman et al., 2003) and offers many adhesion sites for microbes (reviewed in Hooper and Gordon, 2001). The extracellular region that is proximal to the cell membrane is N-glycosylated which affects folding, secretion and localization of MUC1 (Parry et al., 2006). The glycosylation pattern is influenced by the glycosyl transferases involved in the process of glycosylation, as well as the physiological state and location of the cell (Lloyd et al., 1996; Hanisch and Müller, 2000; Parry et al., 2006).
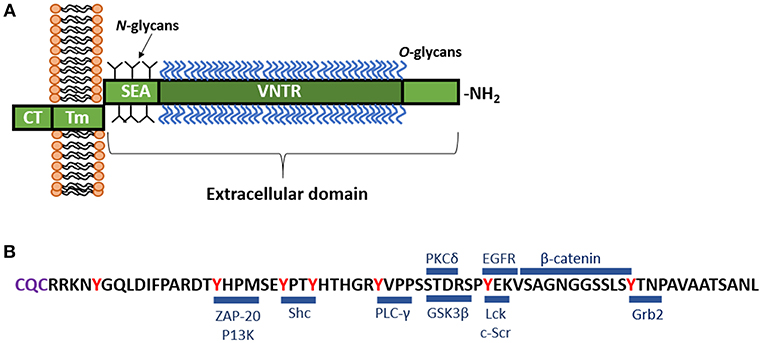
Figure 1. (A) Schematic representation of MUC1. MUC1 consists of a large, extracellular, O-glycosylated polypeptide backbone that extends 200–500 nm above the apical surface and consists of the variable number tandem repeat region (VNTR). The VNTR which is composed of between 25 and 125 repeats of a conserved 20 amino acid sequence. This extracellular domain is heavily 0-glycosylated toward the N-terminal, while closer to the cell membrane, MUC1 displays N-glycans. Next to the VNTR lies the sea-urchin sperm protein, enterokinase and agrin (SEA) domain, which is non-covalently linked to the transmembrane (Tm) region and can undergo autoproteolysis in response to a range of stimuli. The Tm region, which is 28 amino acids in length, tethers MUC1 to the cell membrane. Immediately underlying the Tm region is a 72 amino acid long cytoplasmic tail (-CT), which lies inside the cell. The MUC1-CT is known to interact with multiple signaling cascades. (B) Amino acid sequence of the MUC1-CT and known binding partners. The MUC1-CT contains 7 tyrosine residues (shown in red), along with serine and threonine residues, which serve as docking sites for kinases and other adaptor proteins. Blue bars mark the known binding sites of specific cellular signaling proteins including: β-catenin (Yamamoto et al., 1997); ZAP-70, zeta chain-associated protein kinase-70 (Li et al., 2004); P13K, phosphoinositide 3-kinase (Kato et al., 2007); Shc, Src homology 2 domain containing protein (Li et al., 2001a); PLC-γ, phospholipase C-γ (Wang et al., 2003); GSK3β, glycogen synthase kinase 3β (Li et al., 1998); PKCδ, protein kinase C-δ (Ren et al., 2002); Lck, lymphocyte-specific protein tyrosine kinase (Li et al., 2004); EGFR, epidermal growth factor receptor (Li et al., 2001b); Grb2, growth factor receptor-bound protein 2 (Pandey et al., 1995). The C-terminal end of the CT contains the motif CQC (purple) which has been demonstrated to be essential for localization of MUC1 at the cell membrane (Leng et al., 2007).
Characteristic of MUC1 is the presence of a variable number tandem repeat (VNTR) region in the extracellular polypeptide backbone. The MUC1 VNTR consists of 20–125 copies of 20 amino acids GSTAPPAHGVTSAPDTRPAP (Swallow et al., 1987; Ligtenberg et al., 1991; Vos et al., 1991). The VNTR of MUC1 is followed by the sea-urchin sperm protein, enterokinase and agrin (SEA) domain which possess the GSVVV motif, where auto-proteolysis occurs inducing self-cleavage of the molecule. This enables the extracellular domain to be shed from the cell surface, while the transmembrane and cytoplasmic domains remain tethered to the cell membrane (Parry et al., 2001). Proteases, particularly TNF-α converting enzyme, MMP-14, and ADAM17 are capable of inducing release of the MUC1 extracellular domain by acting on the cleavage site in the SEA module (Thathiah et al., 2003; Brayman et al., 2004; Thathiah and Carson, 2004; Macao et al., 2006). Upon cleavage of the SEA module, the extrusion of MUC1 extracellular domain into the lumen provides a mechanism by which the mucin-bound pathogen can be released from the cell and excreted from the body via the mucocilary escalator. As such, this cell surface mucin has been referred to as a releasable decoy barrier to infection (Lindén et al., 2009).
The cytoplasmic tail (-CT) of MUC1 is highly conserved throughout most species (Spicer et al., 1991, 1995; Vos et al., 1991). This region possesses seven tyrosine residues, 4 of which can be phosphorylated by kinases and initiate signal transduction cascades (Figure 1B; Wang et al., 2003). The presence of extracellular EGF-like domains and phosphorylation sites in the cytoplasmic tail suggest MUC1 is able to have a functional role in signaling cascades (Sheng et al., 2012), enabling recycling of the MUC1-ED for re-glycosylation after degradation in the lumen, or as a result of microbial interaction (Altschuler et al., 2000).
Gastrointestinal Expression of MUC1 Role as a Releasable Decoy Barrier to Infection
Campylobacter jejuni is an intestinal pathogen that is a major cause of gastroenteritis in humans (Altekruse et al., 1999). Using an in vivo model of infection, MUC1 expression at the epithelial surface was shown to limit gastrointestinal and systemic spread of C. jejuni and reduce intestinal inflammation in mice (McAuley et al., 2007). Interestingly, MUC1 deficiency was not found to have an effect on systemic infections caused by another Gram-negative intestinal pathogen, Salmonella typhimurium. However, S. typhimurium targets Microfold (M) cells, which do not secrete mucins and have a thinner glycocalyx (Sansonetti and Phalipon, 1999), while C. jejuni binds intestinal mucins and targets goblet cell thecae in vivo (Ruiz-Palacios et al., 2003). In a recent study, MUC1 has been shown to serve as an adhesion receptor for another gastrointestinal pathogen, enteroaggregative Escherichia coli (EAEC) and knockdown of MUC1 expression in colonic cancer cell line was also associated with reduced EAEC binding (Boll et al., 2017).
Helicobacter pylori are highly motile bacteria that colonize the mucus layer lining the human stomach. H. pylori infection is associated with a myriad of pathologies, including gastritis, peptic ulcer disease and severe diseases such as gastric mucosa-associated lymphoid tissue lymphoma and gastric adenocarcinoma (Wotherspoon et al., 1993; Uemura et al., 2001; Malfertheiner et al., 2009). Numerous epidemiological studies have shown an association between MUC1 VNTR polymorphisms and susceptibility for both H. pylori-induced gastritis and gastric cancer. These studies have revealed individuals with short MUC1 alleles are at a higher risk of H. pylori-pathologies which represents a clinical risk factor (Garcia et al., 1997; Silva et al., 2001, 2003; Vinall et al., 2002). The MUC1 extracellular domain contains the antigens Lewisb, sialyl Lewisa, and sialyl Lewisx, which serve as potential binding sites for the H. pylori adhesins BabA and SabA (Lindén et al., 2009). Proving MUC1 as a releasable decoy, H. pylori binding to MUC1-ED triggered shedding via self-cleavage at the SEA domain, resulting in removal of the bacteria from the cell surface (Lindén et al., 2009). Using murine models of H. pylori infection in MUC1-deficient mice (Muc1−/−), epithelial expression of MUC1 was shown to inhibit the adhesion of H. pylori to the gastric mucosae, restricting colonization of the stomach (McAuley et al., 2007; McGuckin et al., 2007; Lindén et al., 2009) and MUC1 expressed by monocytes reduced the severity of H. pylori-induced gastritis (Ng et al., 2016).
MUC1 Influence on the Ability for Pathogens to Infect the Respiratory Tract
Pseudomonas aeruginosa causes respiratory infections in humans, particularly in immunocompromised individuals, such as those with cystic fibrosis (Gellatly and Hancock, 2013). Similar to the enteric pathogens C. jejuni, EAEC and H. pylori, MUC1 serves as an adhesion ligand for the flagellin of P. aeruginosa (Lillehoj et al., 2002, 2015). Opposite to the effects of the MUC1 barrier to infection in the intestinal tract, studies reveal a reduced colonization of P. aeruginosa in lungs of Muc1−/− mice as compared to wild-type (WT) mice (Lu et al., 2006). In the murine P. aeruiginosa infection model, reduced colonization of P. aeruginosa in Muc1−/− mice was due to enhanced clearance via heightened early inflammatory responses to the infection, compared to that observed in WT mice (Lu et al., 2006). Interestingly, the host enzyme, Neuraminidase 1 (NEU1) has been implicated to impede MUC1-mediated protection against P. aeruginosa infection (Lillehoj et al., 2015). It was proposed that flagellin of P. aeruginosa engagement of NEU1 desialylates MUC1 and other cell surface receptors expressed by airway epithelial cells, unveiling underlying potential P. aeruginosa binding mediators, facilitating pathogenicity (Lillehoj et al., 2015). It is unknown whether this NEU1-mediated desialylation of MUC1 occurs in response to other common respiratory pathogens found to interact directly with this cs-mucin.
S. pneumoniae are extracellular pneumococcal pathogens that asymptomatically colonize the mucosal surface of the human nasopharynx and upper airways, especially children (Bogaert et al., 2004; Van Der Poll and Opal, 2009). Using the lung epithelial cell line A549 and CRISPR technology to knock-down MUC1 expression, the ability for S. pneumoniae to bind to epithelial cells was shown to be MUC1 dependent (Dhar et al., 2017). Furthermore, indicating MUC1 binding to S. pneumoniae may play an essential role in triggering phagocytosis, MUC1-deficient macrophages were shown to be inefficient at phagocytosing the pneumococci (Dhar et al., 2017). Dhar et al also demonstrated the inability for mice to express MUC1 was associated with enhanced pneumococcal disease in the murine S. pneumoniae infection model.
Influenza A virus (IAV) is a highly contagious respiratory pathogen that is a constant threat to the health of the human population. Recently, it was demonstrated that IAV closely associates with MUC1 and as MUC1 is sialylated, it has the potential to bind to virions, reducing the ability to infect host cells (McAuley et al., 2017). Similar to bacterial infection studies involving the gastrointestinal pathogens C. jejuni and H. pylori, as well as respiratory S. pneumoniae, Muc1−/− mice had heightened inflammatory disease than WT mice caused by the laboratory mouse adapted A/Puerto Rico/8/34 IAV and a 2009 H1N1 pandemic human IAV isolate (McAuley et al., 2017). Linking both barrier and immunomodulatory function of MUC1 during IAV infection, Muc1−/− mice reached maximal viral titers earlier than the WT mice, but also exhibited enhanced inflammatory responses to the infection when viral titers were equivalent. Implicating a role for MUC1 barrier and anti-inflammatory function for other respiratory viruses, Respiratory Syncytial Virus (RSV) and human metapneumovirus have both been shown to cause upregulation of MUC1 in a time-course dependent manner in A549 epithelial cells (Li et al., 2010; Baños-Lara Mdel et al., 2015). Li et al additionally showed that during RSV infection of A549 cells caused a negative feedback loop involving TNF production and MUC1 expression, indicating a role for MUC1 in regulating inflammatory disease (Li et al., 2010). Whether MUC1 plays a significant barrier or immunomodulatory role in vivo during RSV or pneumovirus infection is currently unknown.
MUC1 and Regulation of Inflammation During Infection
The innate immune system response mediates the first-line of defense to clear infection and leads to the recruitment of the adaptive arm of immunity. Epithelial and immune cells constitutively express three families of innate pattern recognition receptors (PRRs), which include Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), and NOD-like receptor proteins (NLRPs) that cooperate for early recognition of pathogens and triggering of host responses to infection. It is now clear that MUC1 plays an integral role in regulating inflammatory responses to infection and is a key modulator in controlling pathogen-induced inflammatory disease.
MUC1 as a Negative Regulator of TLR Signaling
The MUC1-CT has been demonstrated by many studies to play an important role in inhibiting TLR activation. Infection by Pseudomonas aeruginosa (Ueno et al., 2008) and respiratory syncytial virus (Li et al., 2010), induces TLR activation in airway epithelial cells and macrophages. This induced production of inflammatory mediators such as IL-8 (KC in the mouse) and TNFα, subsequently recruiting effector cells to the site of infection (Guang et al., 2010; Li et al., 2010; Kato et al., 2016). In turn, MUC1 is upregulated and suppresses TLR signaling, attenuating inflammation (reviewed in Kim and Lillehoj, 2008). It has also been demonstrated that following direct binding of P. aeruginosa flagellin to MUC1, the cytosolic epidermal growth factor receptor phosphorylates the tyrosine residues of the MUC1-CT, increasing MUC1-CT association with TLR5, competitively inhibiting recruitment of myeloid differentiation primary response gene 88 (MyD88) to TLR5, and thus blocks downstream signaling (Figure 2A; Kato et al., 2012). MUC1 restriction of the TLR5 pathway further limits downstream signaling cascades, resulting in downregulation of NF-κB-mediated cytokine production (Lu et al., 2006; Kato et al., 2012).
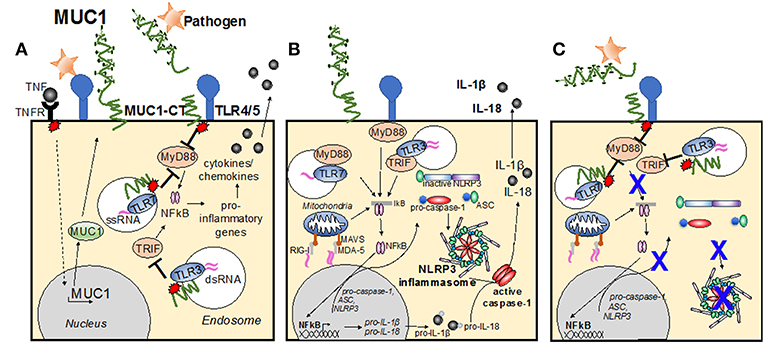
Figure 2. Modes of MUC1 control of cellular inflammatory signaling pathways. (A) Infection causes TLR activation, causing production of inflammatory mediators, such as TNF. In turn, MUC1 is upregulated and suppresses TLR signaling, attenuating inflammation. Direct binding of pathogen to MUC1-ED induces phosphorylation of the MUC1 cytoplasmic tail, increasing MUC1-CT association with TLRs at the cell surface, or within the endosome, competitively inhibiting recruitment of MyD88 and/or TRIF to the TLR and blocks downstream signaling. (B) Upon detection of PAMPs by PRRs at the cellular membrane and within the endosome, the TLR becomes activated and induces the NF-kB transcription pathway, resulting in upregulation of pro-IL-1β, pro-IL-18 and pro-caspase-1, ASC and NLRP3. A second stimulus then causes NLRP3 to nucleate ASC, inducing the characteristic speck-like structure and causes caspase-1 activation. Active caspase-1 cleaves the pro-IL-1β and pro-IL-18, creating the active cytokines IL-1β and IL-18, which are then secreted from the cell. (C) Activation of the MUC1-CT after interaction with pathogen causes recruitment to the TLR, and/or subsequent signaling that limits TLR activation of the signaling cascade and blocks upregulation of inflammasome components. Whether MUC1-CT interacts with NLRP3 directly remains to be determined.
Similar to its suppressive role of the TLR5 signaling pathway, phosphorylation of MUC1-CT has also been implicated as a suppressor of the TLR3-signaling cascade (Figure 2A). Using polyI:C as a model for TLR3 stimulation by double-stranded RNA and murine tracheal, human lung and human embryonic kidney epithelial cells, modified for their expression of MUC1, Kato et al (Kato et al., 2014) demonstrated that activated MUC1-CT co-localizes with TLR3, blocking Toll/IL-1 receptor-domain–containing adapter-inducing IFN-β (TRIF) docking and inhibiting downstream signaling. This reduced the level of apoptosis and production of inflammatory mediators by epithelial cells. Adding credence to the notion that MUC1-CT is a general inhibitor of the TLR pathway, Ueno et al (Ueno et al., 2008) utilized a variety of artificial TLR agonists, including Pam3Cys (TLR2), Poly I:C (TLR3), LPS (TLR4), loxoribine (TLR7), and CpG DNA (TLR9), to show that the presence of the MUC1-CT is required for the anti-inflammatory activity of MUC1 during TLR activation. In vitro infection studies with non-typeable Haemophilus influenzae also revealed that MUC1-CT inhibited the TLR2 signaling pathway (Kyo et al., 2012). Furthermore, links to anti-inflammatory cytokine IL-10 and type I interferon (IFN) correlating with increased MUC1 expression have also been made (Gaemers et al., 2001; Kato et al., 2014). The degree to which MUC1 inhibition of activated TLR pathways diminishes inflammatory responses to infection and the critical timing to which this occurs in vivo remains to be elucidated, however must play a significant role in controlling disease severity.
MUC1 Modulation of the NLRP3 Inflammasome Complex
The inflammasome pathway is now regarded as an essential component of host-defense against infection (Martinon et al., 2002). The inflammasome pathway enables the early detection of pathogens and induces production of inflammatory cytokines, subsequently inducing recruitment of effector cells. The complete mechanism of inflammasome activation requires two signals: an initial priming step upon detection of pathogen (or pathogen-associated molecules), followed by activation of intracellular pattern recognition receptors critical to the formation of the inflammasome complex. For the initial priming step, TLR activation and the associated NF-kB transcription pathway, triggers upregulation of pro-IL-1β, pro-IL-18, and pro-caspase-1, in addition to the inflammasome complex components which include a sensor protein, such as the NLRP, or the receptor protein known as absent in malenoma-2 (AIM2); and the adapter protein known as the apoptosis-associated speck-like protein containing a CARD (ASC) (Figure 2B). Thus, TLRs are the main priming signal for induction of the inflammasome pathway. As such, it is likely that MUC1 is an important inhibitor of the inflammasome pathway, by limiting TLR activation and halting upregulation of inflammasome components (Figure 2C).
Upon priming of signal 2, the NLRPs (or AIM2) cause ASC to nucleate. This causes the clustering of the ASC protein complex, forming a characteristic speck-like structure. The speck then induces oligomerization of pro-caspase 1, inducing it cleavage into the mature form of caspase-1 (Figure 2B; Yang et al., 1998). Active caspase-1 then cleaves pro-IL-1β and pro-IL-18, inducing their activation and secretion, which in turn causes recruitment of effector cells to the site of infection (Martinon et al., 2002).
Production of IL-1β in the stomach potently inhibited gastric acid secretion, reducing the permissible micro-environment enabling H. pylori colonization and spread (Wallace et al., 1991; Beales and Calam, 1998). Linking MUC1 as an important suppressor of NLRP3-inflammasome activation was the recent discovery that expression of MUC1 during H. pylori infection in mice and in vitro suppressed the production of gastric IL-1β (Ng et al., 2016). This was found to in-turn, limit H. pylori induced inflammation and resultant pathology of the gastric mucosa (Ng et al., 2016). Furthermore, compared to wildtype macrophages, stimulation of MUC1-deficient macrophages with two other NLRP3-activating bacteria, H. influenzae and S. aureus have also been associated with elevated levels of IL-1β (Ng and Sutton, 2016). Studies using purified ligands for different inflammasome complexes revealed that this MUC1 restriction is unique for the NLRP3 inflammasome only, as MUC1 deficiency did not have an effect on IL-1β production after stimulation with synthetic activators of other inflammasome receptors (NLRC4, AIM2, and NLRP1b) (Sutterwala et al., 2007; Ng and Sutton, 2016).
Complicating our understanding of MUC1-dependent regulation of the NLRP3 inflammasome, levels of IL-1β in the lungs of S. pneumoniae infection of Muc1−/− mice may have been attributed to by a lack of appropriate MUC1 signaling, as well as the production of pneumolysin, a toxin produced by S. pneumoniae that specifically induces NLRP3-inflammasome activation (McNeela et al., 2010; Witzenrath et al., 2011). Additionally, while influenza infected Muc1−/− mice exhibited heightened levels of inflammation (McAuley et al., 2017) and that the NLRP3-inflammasome is considered to be a critical host-response mechanism to the infection (Allen et al., 2009; Thomas et al., 2009), direct links of MUC1 regulation of the complex during influenza infection have yet to be established. Whether the role of MUC1 inhibition of NRLP3-inflammasome dependent inflammation is a general function in response to the majority of mucosal pathogens remains to be elucidated.
MUC1 as Clinical Markers for Inflammatory Disease
In addition to an association between MUC1 VNTR polymorphisms and susceptibility for both H. pylori-induced gastritis and gastric cancer (Garcia et al., 1997; Silva et al., 2001, 2003; Vinall et al., 2002), altered MUC1 expression levels and glycosylation patterns have been associated with patients with inflammatory bowel diseases such as ulcerative colitis and Crohn's disease (reviewed in Jass and Walsh, 2001). Chronic infections by opportunistic microflora and chronic inflammation of the gastrointestinal tract in those with gastritis, ulcerative colitis and Crohn's disease are hallmarks of the illness severity. While the role of aberrant glycosylation, altered MUC1-ED length and MUC1-regulation of inflammation is far from understood, it is likely that MUC1 does not function appropriately in these individuals and contributes significantly to their disease.
Indicative of a functional role for increased MUC1 expression in respiratory disorders, the sialylated carbohydrate antigen Krebs von den Lungen 6 [KL-6, now identified as human MUC1 (Hirasawa et al., 1997)], is a marker for interstitial lung disease. MUC1 is also elevated in serum taken from patients suffering cystic fibrosis, severe pneumonia, asthma exacerbations, and measles pneumonia (Imai et al., 2002; Ohnishi et al., 2002; Ohshimo et al., 2009). Sputum taken from patients suffering from chronic obstructive pulmonary disease (COPD) also contains significantly more MUC1 compared to samples taken from individuals with prolonged coughing (Ishikawa et al., 2011). With the exception of potential utilization of MUC1 as a diagnostic marker for respiratory disease severity (Ohnishi et al., 2002; Kubota and Haruta, 2006), there has been no study to-date assessing the mechanism(s) of overexpression of MUC1 and the increased vulnerability to inflammatory disease resulting from infection in patients with chronic respiratory conditions.
Concluding Remarks
Clearly, MUC1 plays a dynamic role in the mucosal barrier to infection and regulation of innate immune responses to control inflammation. Dysregulated and inappropriately glycosylated MUC1 has been correlated with many chronic epithelial diseases and cancers, suggesting when the general function of this glycoprotein is perturbed, normal homeostasis of tissue, cannot be properly maintained. Comprehensively unveiling MUC1-dependent pathways important for maintaining mucosal barrier integrity and modulation of inflammation will aid in our understanding of the development of chronic inflammatory diseases. This will pave the way for development of targeted treatments with the aim to improve mucosal barrier integrity and function, which will ultimately reduce the severity of inflammatory disease, particularly for those suffering chronic conditions.
Author Contributions
JM and PD contributed equally to the conception and design of the review, as well as development of the figures, manuscript text and revision. Both JM and PD have read and approved the submitted version.
Funding
This work was supported by the Australian Government National Health and Medical Research Council program grant 1071916.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Allen, I. C., Scull, M. A., Moore, C. B., Holl, E. K., Mcelvania-Tekippe, E., Taxman, D. J., et al. (2009). The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30, 556–565. doi: 10.1016/j.immuni.2009.02.005
Altekruse, S. F., Stern, N. J., Fields, P. I., and Swerdlow, D. L. (1999). Campylobacter jejuni–an emerging foodborne pathogen. Emerg. Infect.Dis. 5, 28–35. doi: 10.3201/eid0501.990104
Altschuler, Y., Kinlough, C. L., Poland, P. A., Bruns, J. B., Apodaca, G., Weisz, O. A., et al. (2000). Clathrin-mediated endocytosis of MUC1 is modulated by its glycosylation state. Mol. Biol. Cell 11, 819–831. doi: 10.1091/mbc.11.3.819
Baños-Lara Mdel, R., Piao, B., and Guerrero-Plata, A. (2015). Differential mucin expression by respiratory syncytial virus and human metapneumovirus infection in human epithelial cells. Mediators Inflamm. 2015:347292. doi: 10.1155/2015/347292
Beales, I. L., and Calam, J. (1998). Interleukin 1 beta and tumour necrosis factor alpha Inhibit acid secretion in cultured rabbit parietal cells by multiple pathways. Gut 42, 227–234. doi: 10.1136/gut.42.2.227
Bogaert, D., De Groot, R., and Hermans, P. W. (2004). Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet 4, 144–154. doi: 10.1016/S1473-3099(04)00938-7
Boll, E. J., Ayala-Lujan, J., Szabady, R. L., Louissaint, C., Smith, R. Z., Krogfelt, K. A., et al. (2017). Enteroaggregative Escherichia coli adherence fimbriae drive inflammatory cell recruitment via interactions with epithelial MUC1. mBio 8 e00717–17. doi: 10.1128/mBio.00717-17
Brayman, M., Thathiah, A., and Carson, D. D. (2004). MUC1: a multifunctional cell surface component of reproductive tissue epithelia. Reproduc. Biol. Endocrinol. 2:4. doi: 10.1186/1477-7827-2-4
Dhar, P., Ng, G. Z., Dunne, E. M., and Sutton, P. (2017). Mucin 1 protects against severe Streptococcus pneumoniae infection. Virulence 8, 1631–1642. doi: 10.1080/21505594.2017.1341021
Gaemers, I. C., Vos, H. L., Volders, H. H., Van Der Valk, S. W., and Hilkens, J. (2001). A stat-responsive element in the promoter of the episialin/MUC1 gene is involved in its overexpression in carcinoma cells. J. Biol. Chem. 276, 6191–6199. doi: 10.1074/jbc.M009449200
Garcia, E., Carvalho, F., Amorim, A., and David, L. (1997). MUC6 gene polymorphism in healthy individuals and in gastric cancer patients from northern Portugal. Cancer Epidemiol. Biomarkers Prevent. 6, 1071–1074.
Gellatly, S. L., and Hancock, R. E. (2013). Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog. Dis. 67, 159–173. doi: 10.1111/2049-632X.12033
Guang, W., Ding, H., Czinn, S. J., Kim, K. C., Blanchard, T. G., and Lillehoj, E. P. (2010). Muc1 cell surface mucin attenuates epithelial inflammation in response to a common mucosal pathogen. J. Biol. Chem. 285, 20547–20557. doi: 10.1074/jbc.M110.121319
Hanisch, F. G., Green, B. N., Bateman, R., and Peter-Katalinic, J. (1998). Localization of O-glycosylation sites of MUC1 tandem repeats by QTOF ESI mass spectrometry. J. Mass Spectrometry. 33, 358–362. doi: 10.1002/(SICI)1096-9888(199804)33:4<358::AID-JMS642>3.0.CO;2-3
Hanisch, F. G., and Müller, S. (2000). MUC1: the polymorphic appearance of a human mucin. Glycobiology. 10, 439–449. doi: 10.1093/glycob/10.5.439
Hattrup, C. L., and Gendler, S. J. (2008). Structure and function of the cell surface (tethered) mucins. Annu. Rev. Physiol. 70, 431–457. doi: 10.1146/annurev.physiol.70.113006.100659
Hirasawa, Y., Kohno, N., Yokoyama, A., Inoue, Y., Abe, M., and Hiwada, K. (1997). KL-6, a human MUC1 mucin, is chemotactic for human fibroblasts. Am. J. Respir. Cell Mol. Biol. 17, 501–507. doi: 10.1165/ajrcmb.17.4.2253
Hooper, L. V., and Gordon, J. I. (2001). Glycans as legislators of host-microbial interactions: spanning the spectrum from symbiosis to pathogenicity. Glycobiology. 11, 1R−10R. doi: 10.1093/glycob/11.2.1R
Imai, T., Takase, M., Takeda, S., and Kougo, T. (2002). Serum KL-6 levels in pediatric patients: reference values for children and levels in pneumonia, asthma, and measles patients. Pediatr. Pulmonol. 33, 135–141. doi: 10.1002/ppul.10044
Ishikawa, N., Hattori, N., Tanaka, S., Horimasu, Y., Haruta, Y., Yokoyama, A., et al. (2011). Levels of surfactant proteins A and D and KL-6 are elevated in the induced sputum of chronic obstructive pulmonary disease patients: a sequential sputum analysis. Respiration. 82, 10–18. doi: 10.1159/000324539
Jass, J. R., and Walsh, M. D. (2001). Altered mucin expression in the gastrointestinal tract: a review. J. Cell. Mol. Med. 5, 327–351. doi: 10.1111/j.1582-4934.2001.tb00169.x
Kato, K., Lillehoj, E. P., and Kim, K. C. (2014). MUC1 regulates epithelial inflammation and apoptosis by PolyI:C through inhibition of Toll/IL-1 receptor-domain-containing adapter-inducing IFN-beta (TRIF) recruitment to Toll-like receptor 3. Am. J. Respir. Cell. Mol. Biol. 51, 446–454. doi: 10.1165/rcmb.2014-0018OC
Kato, K., Lillehoj, E. P., Park, Y. S., Umehara, T., Hoffman, N. E., Madesh, M., et al. (2012). Membrane-tethered MUC1 mucin is phosphorylated by EGFR in airway epithelial cells and associates with TLR5 to inhibit recruitment of MyD88. J. Immunol. 188, 2014–2022. doi: 10.4049/jimmunol.1102405
Kato, K., Lu, W., Kai, H., and Kim, K. C. (2007). Phosphoinositide 3-kinase is activated by MUC1 but not responsible for MUC1-induced suppression of Toll-like receptor 5 signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L686–692. doi: 10.1152/ajplung.00423.2006
Kato, K., Uchino, R., Lillehoj, E. P., Knox, K., Lin, Y., and Kim, K. C. (2016). Membrane-tethered MUC1 mucin counter-regulates the phagocytic activity of macrophages. Am. J. Respir. Cell. Mol. Biol. 54, 515–523. doi: 10.1165/rcmb.2015-0177OC
Kim, K. C., and Lillehoj, E. P. (2008). MUC1 mucin: a peacemaker in the lung. Am. J. Respir. Cell. Mol. Biol. 39, 644–647. doi: 10.1165/rcmb.2008-0169TR
Kubota, M., and Haruta, T. (2006). The role of serum KL-6 measurement in common pediatric respiratory infections. J. Infect. Chemother. 12, 22–24. doi: 10.1007/s10156-005-0416-9
Kyo, Y., Kato, K., Park, Y. S., Gajghate, S., Umehara, T., Lillehoj, E. P., et al. (2012). Antiinflammatory role of MUC1 mucin during infection with nontypeable Haemophilus influenzae. Am.J. Respir. Cell Mol. Biol. 46, 149–156. doi: 10.1165/rcmb.2011-0142OC
Leng, Y., Cao, C., Ren, J., Huang, L., Chen, D., Ito, M., et al. (2007). Nuclear import of the MUC1-C oncoprotein is mediated by nucleoporin Nup62. J. Biol. Chem. 282, 19321–19330. doi: 10.1074/jbc.M703222200
Li, Q., Ren, J., and Kufe, D. (2004). Interaction of human MUC1 and beta-catenin is regulated by Lck and ZAP-70 in activated Jurkat T cells. Biochem. Biophys. Res. Commun. 315, 471–476. doi: 10.1016/j.bbrc.2004.01.075
Li, Y., Bharti, A., Chen, D., Gong, J., and Kufe, D. (1998). Interaction of glycogen synthase kinase 3beta with the DF3/MUC1 carcinoma-associated antigen and beta-catenin. Mol. Cell. Biol. 18, 7216–7224. doi: 10.1128/MCB.18.12.7216
Li, Y., Dinwiddie, D. L., Harrod, K. S., Jiang, Y., and Kim, K. C. (2010). Anti-inflammatory effect of MUC1 during respiratory syncytial virus infection of lung epithelial cells in vitro. Am. J. Physiol. Lung Cell Mol. Physiol. 298, L558–563. doi: 10.1152/ajplung.00225.2009
Li, Y., Kuwahara, H., Ren, J., Wen, G., and Kufe, D. (2001a). The c-Src tyrosine kinase regulates signaling of the human DF3/MUC1 carcinoma-associated antigen with GSK3 beta and beta-catenin. J. Biol. Chem. 276, 6061–6064. doi: 10.1074/jbc.C000754200
Li, Y., Ren, J., Yu, W., Li, Q., Kuwahara, H., Yin, L., et al. (2001b). The epidermal growth factor receptor regulates interaction of the human DF3/MUC1 carcinoma antigen with c-Src and beta-catenin. J. Biol. Chem. 276, 35239–35242. doi: 10.1074/jbc.C100359200
Ligtenberg, M. J., Gennissen, A. M., Vos, H. L., and Hilkens, J. (1991). A single nucleotide polymorphism in an exon dictates allele dependent differential splicing of episialin mRNA. Nucleic Acids Res. 19, 297–301. doi: 10.1093/nar/19.2.297
Lillehoj, E. P., Hyun, S. W., Liu, A., Guang, W., Verceles, A. C., Luzina, I. G., et al. (2015). NEU1 sialidase regulates membrane-tethered mucin (MUC1) ectodomain ahesiveness for Pseudomonas aeruginosa and decoy receptor release. J. Biol. Chem. 290, 18316–18331. doi: 10.1074/jbc.M115.657114
Lillehoj, E. P., Kim, B. T., and Kim, K. C. (2002). Identification of Pseudomonas aeruginosa. flagellin as an adhesin for Muc1 mucin. Am. J. Physiol. Lung Cell. Mol. Physiol. 282, L751–756. doi: 10.1152/ajplung.00383.2001
Lindén, S. K., Sheng, Y. H., Every, A. L., Miles, K. M., Skoog, E. C., Florin, T. H., et al. (2009). MUC1 limits Helicobacter pylori infection both by steric hindrance and by acting as a releasable decoy. PLoS Pathog. 5:e1000617. doi: 10.1371/journal.ppat.1000617
Lloyd, K. O., Burchell, J., Kudryashov, V., Yin, B. W., and Taylor-Papadimitriou, J. (1996). Comparison of O-linked carbohydrate chains in MUC-1 mucin from normal breast epithelial cell lines and breast carcinoma cell lines. J. Biol. Chem. 271, 33325–33334. doi: 10.1074/jbc.271.52.33325
Lu, W., Hisatsune, A., Koga, T., Kato, K., Kuwahara, I., Lillehoj, E. P., et al. (2006). Cutting edge: enhanced pulmonary clearance of Pseudomonas aeruginosa by Muc1 knockout mice. J. Immunol. 176, 3890–3894. doi: 10.4049/jimmunol.176.7.3890
Macao, B., Johansson, D. G., Hansson, G. C., and Härd, T. (2006). Autoproteolysis coupled to protein folding in the SEA domain of the membrane-bound MUC1 mucin. Nat. Struc. Mol. Biol. 13, 71–76. doi: 10.1038/nsmb1035
Malfertheiner, P., Chan, F. K., and McColl, K. E. (2009). Peptic ulcer disease. Lancet. 374, 1449–1461. doi: 10.1016/S0140-6736(09)60938-7
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 10, 417–426. doi: 10.1016/S1097-2765(02)00599-3
McAuley, J. L., Corcilius, L., Tan, H. X., Payne, R. J., McGuckin, M. A., and Brown, L. E. (2017). The cell surface mucin MUC1 limits the severity of influenza A virus infection. Mucosal Immunol. 10, 1581–1593. doi: 10.1038/mi.2017.16
McAuley, J. L., Linden, S. K., Png, C. W., King, R. M., Pennington, H. L., Gendler, S. J., et al. (2007). MUC1 cell surface mucin is a critical element of the mucosal barrier to infection. J. Clin. Invest. 117, 2313–2324. doi: 10.1172/JCI26705
McGuckin, M. A., Every, A. L., Skene, C. D., Linden, S. K., Chionh, Y. T., Swierczak, A., et al. (2007). Muc1 mucin limits both Helicobacter pylori colonization of the murine gastric mucosa and associated gastritis. Gastroenterology. 133, 1210–1218. doi: 10.1053/j.gastro.2007.07.003
McNeela, E. A., Burke, A., Neill, D. R., Baxter, C., Fernandes, V. E., Ferreira, D., et al. (2010). Pneumolysin activates the NLRP3 inflammasome and promotes proinflammatory cytokines independently of TLR4. PLoS Pathog. 6:e1001191. doi: 10.1371/journal.ppat.1001191
Ng, G. Z., Menheniott, T. R., Every, A. L., Stent, A., Judd, L. M., Chionh, Y. T., et al. (2016). The MUC1 mucin protects against Helicobacter pylori pathogenesis in mice by regulation of the NLRP3 inflammasome. Gut. 65, 1087–1099. doi: 10.1136/gutjnl-2014-307175
Ng, G. Z., and Sutton, P. (2016). The MUC1 mucin specifically inhibits activation of the NLRP3 inflammasome. Genes Immunity 17, 203–206. doi: 10.1038/gene.2016.10
Ohnishi, H., Yokoyama, A., Kondo, K., Hamada, H., Abe, M., Nishimura, K., et al. (2002). Comparative study of KL-6, surfactant protein-A, surfactant protein-D, and monocyte chemoattractant protein-1 as serum markers for interstitial lung diseases. Am. J. Respir. Crit. Care Med. 165, 378–381. doi: 10.1164/ajrccm.165.3.2107134
Ohshimo, S., Bonella, F., Grammann, N., Starke, K., Cui, A., Bauer, P. C., et al. (2009). Serum KL-6 as a novel disease marker in adolescent and adult cystic fibrosis. Sarcoidosis Vasc. Diffuse Lung Dis. 26, 47–53.
Pandey, P., Kharbanda, S., and Kufe, D. (1995). Association of the DF3/MUC1 breast cancer antigen with Grb2 and the Sos/Ras exchange protein. Cancer Res. 55, 4000–4003.
Parry, S., Hanisch, F. G., Leir, S. H., Sutton-Smith, M., Morris, H. R., Dell, A., et al. (2006). N-Glycosylation of the MUC1 mucin in epithelial cells and secretions. Glycobiology 16, 623–634. doi: 10.1093/glycob/cwj110
Parry, S., Silverman, H. S., McDermott, K., Willis, A., Hollingsworth, M. A., and Harris, A. (2001). Identification of MUC1 proteolytic cleavage sites in vivo. Biochem. Biophys. Res. Commun. 283, 715–720. doi: 10.1006/bbrc.2001.4775
Perez-Vilar, J., and Hill, R. L. (1999). The structure and assembly of secreted mucins. J. Biol. Chem. 274, 31751–31754. doi: 10.1074/jbc.274.45.31751
Ren, J., Li, Y., and Kufe, D. (2002). Protein kinase C delta regulates function of the DF3/MUC1 carcinoma antigen in beta-catenin signaling. J. Biol. Chem. 277, 17616–17622. doi: 10.1074/jbc.M200436200
Ruiz-Palacios, G. M., Cervantes, L. E., Ramos, P., Chavez-Munguia, B., and Newburg, D. S. (2003). Campylobacter jejuni binds intestinal H(O) antigen (Fuc alpha 1, 2Gal beta 1, 4GlcNAc), and fucosyloligosaccharides of human milk inhibit its binding and infection. J. Biol. Chem. 278, 14112–14120. doi: 10.1074/jbc.M207744200
Sansonetti, P. J., and Phalipon, A. (1999). M cells as ports of entry for enteroinvasive pathogens: mechanisms of interaction, consequences for the disease process. Semin. Immunol. 11, 193–203. doi: 10.1006/smim.1999.0175
Sheng, Y. H., Triyana, S., Wang, R., Das, I., Gerloff, K., Florin, T. H., et al. (2012). MUC1 and MUC13 differentially regulate epithelial inflammation in response to inflammatory and infectious stimuli. Mucosal Immunol. 6, 557–568. doi: 10.1038/mi.2012.98
Silva, F., Carvalho, F., Peixoto, A., Seixas, M., Almeida, R., Carneiro, F., et al. (2001). MUC1 gene polymorphism in the gastric carcinogenesis pathway. Eur. J. Hum. Genet. 9, 548–552. doi: 10.1038/sj.ejhg.5200677
Silva, F., Carvalho, F., Peixoto, A., Teixeira, A., Almeida, R., Reis, C., et al. (2003). MUC1 polymorphism confers increased risk for intestinal metaplasia in a Colombian population with chronic gastritis. Eur. J. Hum. Genetics. 11, 380–384. doi: 10.1038/sj.ejhg.5200978
Silverman, H. S., Sutton-Smith, M., Mcdermott, K., Heal, P., Leir, S. H., Morris, H. R., et al. (2003). The contribution of tandem repeat number to the O-glycosylation of mucins. Glycobiology. 13, 265–277. doi: 10.1093/glycob/cwg028
Spicer, A. P., Duhig, T., Chilton, B. S., and Gendler, S. J. (1995). Analysis of mammalian MUC1 genes reveals potential functionally important domains. Mamm. Genome. 6, 885–888. doi: 10.1007/BF00292441
Spicer, A. P., Parry, G., Patton, S., and Gendler, S. J. (1991). Molecular cloning and analysis of the mouse homologue of the tumor-associated mucin, MUC1, reveals conservation of potential O-glycosylation sites, transmembrane, and cytoplasmic domains and a loss of minisatellite-like polymorphism. J. Biol. Chem. 266, 15099–15109.
Sutterwala, F. S., Mijares, L. A., Li, L., Ogura, Y., Kazmierczak, B. I., and Flavell, R. A. (2007). Immune recognition of Pseudomonas aeruginosa. mediated by the IPAF/NLRC4 inflammasome. J. Exp. Med. 204, 3235–3245. doi: 10.1084/jem.20071239
Swallow, D. M., Gendler, S., Griffiths, B., Corney, G., Taylor-Papadimitriou, J., and Bramwell, M. E. (1987). The human tumour-associated epithelial mucins are coded by an expressed hypervariable gene locus PUM. Nature. 328, 82–84. doi: 10.1038/328082a0
Taylor-Papadimitriou, J., Burchell, J., Miles, D. W., and Dalziel, M. (1999). MUC1 and cancer. Biochim. Biophys. Acta Mol. Basis Dis. 1455, 301–313. doi: 10.1016/S0925-4439(99)00055-1
Thathiah, A., Blobel, C. P., and Carson, D. D. (2003). Tumor necrosis factor-alpha converting enzyme/ADAM 17 mediates MUC1 shedding. J. Biol. Chem. 278, 3386–3394. doi: 10.1074/jbc.M208326200
Thathiah, A., and Carson, D. D. (2004). MT1-MMP mediates MUC1 shedding independent of TACE/ADAM17. Biochem. J. 382, 363–373. doi: 10.1042/BJ20040513
Thomas, P. G., Dash, P., Aldridge, J. R. Jr., Ellebedy, A. H., Reynolds, C., Funk, A. J., et al. (2009). The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 30, 566–575. doi: 10.1016/j.immuni.2009.02.006
Uemura, N., Okamoto, S., Yamamoto, S., Matsumura, N., Yamaguchi, S., Yamakido, M., et al. (2001). Helicobacter pylori infection and the development of gastric cancer. N. Engl. J. Med. 345, 784–789. doi: 10.1056/NEJMoa001999
Ueno, K., Koga, T., Kato, K., Golenbock, D. T., Gendler, S. J., Kai, H., et al. (2008). MUC1 mucin is a negative regulator of toll-like receptor signaling. Am. J. Respir. Cell Mol. Biol. 38, 263–268. doi: 10.1165/rcmb.2007-0336RC
Van Der Poll, T., and Opal, S. M. (2009). Pathogenesis, treatment, and prevention of pneumococcal pneumonia. Lancet 374, 1543–1556. doi: 10.1016/S0140-6736(09)61114-4
Vinall, L. E., King, M., Novelli, M., Green, C. A., Daniels, G., Hilkens, J., et al. (2002). Altered expression and allelic association of the hypervariable membrane mucin MUC1 in Helicobacter pylori. gastritis. Gastroenterology. 123, 41–49. doi: 10.1053/gast.2002.34157
Vos, H. L., De Vries, Y., and Hilkens, J. (1991). The mouse episialin (Muc1) gene and its promoter: rapid evolution of the repetitive domain in the protein. Biochem. Biophys. Res. Commun. 181, 121–130. doi: 10.1016/S0006-291X(05)81390-7
Wallace, J. L., Cucala, M., Mugridge, K., and Parente, L. (1991). Secretagogue effects of interleukin-1 on gastric acid secretion. Am. J. Physiol. 261, G559–G564. doi: 10.1152/ajpgi.1991.261.4.G559
Wang, H., Lillehoj, E. P., and Kim, K. C. (2003). Identification of four sites of stimulated tyrosine phosphorylation in the MUC1 cytoplasmic tail. Biochem. Biophys. Res. Commun. 310, 341–346. doi: 10.1016/j.bbrc.2003.09.030
Witzenrath, M., Pache, F., Lorenz, D., Koppe, U., Gutbier, B., Tabeling, C., et al. (2011). The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J. Immunol. 187, 434–440. doi: 10.4049/jimmunol.1003143
Wotherspoon, A. C., Doglioni, C., Diss, T. C., Pan, L., Moschini, A., De Boni, M., et al. (1993). Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet 342, 575–577. doi: 10.1016/0140-6736(93)91409-F
Yamamoto, M., Bharti, A., Li, Y. Q., and Kufe, D. (1997). Interaction of the DF3/MUC1 breast carcinoma-associated antigen and beta-catenin in cell adhesion. J. Biol. Chem. 272, 12492–12494. doi: 10.1074/jbc.272.19.12492
Keywords: MUC1, Helicobacter pylori, Campylobacter jejuni, influenza A virus, Streptococcus pneumoniae, Pseudomonas aeruginosa, toll-like receptor, NLRP3-inflammasome
Citation: Dhar P and McAuley J (2019) The Role of the Cell Surface Mucin MUC1 as a Barrier to Infection and Regulator of Inflammation. Front. Cell. Infect. Microbiol. 9:117. doi: 10.3389/fcimb.2019.00117
Received: 05 February 2019; Accepted: 02 April 2019;
Published: 24 April 2019.
Edited by:
Nesrin Ozoren, Bogaziçi University, TurkeyReviewed by:
Kai Yang, Indiana University School of Medicine-Lafayette, United StatesLaurent Gorvel, INSERM U1068 Centre de Recherche en Cancérologie de Marseille, France
Copyright © 2019 Dhar and McAuley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Julie McAuley, am1jYXVsZXlAdW5pbWVsYi5lZHUuYXU=