 Sébastien Pomel
Sébastien Pomel Wei Mao
Wei Mao Tâp Ha-Duong
Tâp Ha-Duong Christian Cavé
Christian Cavé Philippe M. Loiseau
Philippe M. Loiseau- UMR 8076 CNRS BioCIS, Université Paris-Sud, Université Paris-Saclay, Châtenay-Malabry, France
Leishmaniases are neglected tropical diseases that threaten about 350 million people in 98 countries around the world. In order to find new antileishmanial drugs, an original approach consists in reducing the pathogenic effect of the parasite by impairing the glycoconjugate biosynthesis, necessary for parasite recognition and internalization by the macrophage. Some proteins appear to be critical in this way, and one of them, the GDP-Mannose Pyrophosphorylase (GDP-MP), is an attractive target for the design of specific inhibitors as it is essential for Leishmania survival and it presents significant differences with the host counterpart. Two GDP-MP inhibitors, compounds A and B, have been identified in two distinct studies by high throughput screening and by a rational approach based on molecular modeling, respectively. Compound B was found to be the most promising as it exhibited specific competitive inhibition of leishmanial GDP-MP and antileishmanial activities at the micromolar range with interesting selectivity indexes, as opposed to compound A. Therefore, compound B can be used as a pharmacological tool for the development of new specific antileishmanial drugs.
Introduction
Leishmaniases are vector-borne neglected tropical diseases caused by a protozoan parasite from the genus Leishmania and transmitted by hematophagous female phlebotomine sandflies. During its life cycle, the parasite alternates from a promastigote motile form within the phlebotome to an intracellular amastigote form in mammalian host macrophages. Leishmaniases can be classified in three main groups according to their clinical manifestations: cutaneous, which is the most common form, muco-cutaneous leading to nasal and oropharyngeal lesions and marked disfigurements, and visceral, the most severe form, always fatal in the absence of adequate treatment. These clinical manifestations can be provoked by several Leishmania species: for instance, L. major or L. mexicana will give rise to cutaneous leishmaniasis, and L. donovani or L. infantum visceral leishmaniasis. Only few drugs are currently available for the treatment of leishmaniases. Antimonials, which have been historically used since 1920s, generate a strong toxicity at cardiac, renal, and hepatic levels and select drug resistance. The other classical drugs, namely oral miltefosine, injectable liposomal amphotericin B, and paromomycin, display some deleterious effects and now represent a potential threat of drug resistance as well (Croft et al., 2006; Sundar and Singh, 2016; Ponte-Sucre et al., 2017). The development of new antileishmanial treatments is thus crucial in this context.
In order to overcome the limitations of the existing treatments, rational approaches have been used to develop new specific therapies for leishmaniases (Zulfiqar et al., 2017). Among the different strategies elaborated, the identification of new targets that are essential for parasite viability or virulence is an attractive approach for the development of specific antileishmanial compounds (Jiang et al., 1999; Burchmore et al., 2003; Jain and Jain, 2018). Indeed, these essential targets can be exploited by chemical screening in order to characterize inhibitor scaffolds whose specificities are optimized by pharmacomodulations, based on target three-dimensional structures. In this way, targets from Leishmania energy metabolism (i.e., glycolysis, folate or redox metabolism) were first intensively studied (Aronov et al., 1999; Chowdhury et al., 1999; Verlinde et al., 2001; Olin-Sandoval et al., 2010; Colotti et al., 2013; Leroux and Krauth-Siegel, 2016). Other biochemical pathways were also investigated, but the characterized inhibitors met some limitations such as parasite specificity, inhibitors synthesis cost and lack of in vivo activity (Croft and Coombs, 2003).
Targeting Membrane Glycoconjugate Metabolism
There are two main ways to impair parasite development within the host, considering proteins expressed in the amastigote form as therapeutic targets. The first one relies on targeting some biochemical pathways leading to an unbalanced metabolism, toxic for the parasite. Many proteins have been considered for this purpose (Aronov et al., 1999; Chowdhury et al., 1999; Verlinde et al., 2001; Olin-Sandoval et al., 2010). The second one considers that a relevant Achille's heel consists in avoiding macrophage-parasite interactions (Descoteaux et al., 1995; Descoteaux and Turco, 1999; Podinovskaia and Descoteaux, 2015; Lamotte et al., 2017). As host-Leishmania interactions mainly rely on glycoconjugate recognition, an inhibition of glycoconjugate biosynthesis could affect this molecular recognition, and therefore reduce parasite burden. Furthermore, as the glycosylation is a crucial pathway for macrophage infection (Descoteaux et al., 1995; Descoteaux and Turco, 1999; Pomel and Loiseau, 2013; Podinovskaia and Descoteaux, 2015), we hypothesize that an alteration of glycoconjugate structures would not easily select drug resistance.
Mannose-containing glycoconjugates represent a large proportion of the carbohydrates addressed at the surface of a eukaryotic cell and are involved in many biological processes such as intercellular recognition, adhesion or signaling (Varki, 2007; Colley et al., 2017). In Leishmania, a wide range of unusual mannose-containing glycoconjugates [e.g., GlycosylPhosphatidylInositol (GPI) anchors, LipoPhosphoGlycans (LPG), ProteoPhosphoGlycans (PPG) or GlycosylInositolPhosphoLipids (GIPLs)] are synthesized and are essential for parasite virulence (Descoteaux and Turco, 1999; Pomel and Loiseau, 2013). The biosynthesis of these glycoconjugates requires initially the conversion of mannose into GDP-mannose. The mannose moiety of this nucleotide sugar is then transferred into nascent glycoconjugates to allow mannosylation reaction. In eukaryotic cells, mannose can either be imported via membrane transporters or be generated from the reaction catalyzed by the PhosphoMannose Isomerase (PMI) on fructose-6-phosphate originating from glycolysis to produce mannose-6-phosphate (Figure 1A). In the mannosylation pathway, the PhosphoMannoMutase (PMM) converts mannose-6-phosphate in mannose-1-phosphate (Figure 1). The activated form of mannose, GDP-mannose, is then produced by the action of the GDP-Mannose Pyrophosphorylase (GDP-MP) according to the following reversible enzymatic reaction (Ning and Elbein, 2000):
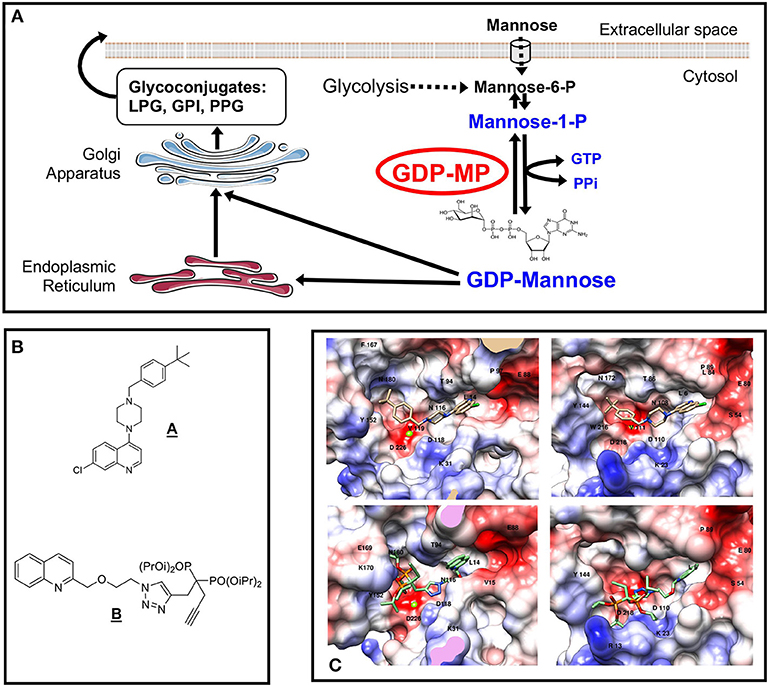
Figure 1. Mannose activation pathways and GDP-MP inhibitors. (A) Mannose activation pathways and glycoconjugate biosynthesis in Leishmania. The GDP-MP substrates and products are indicated in blue. The GDP-MP is circled in red. (B) Chemical structures of compounds A,B. (C) Docking analyses of compound A (top) and B (bottom) in LdGDP-MP (left) and hGDP-MP (right) catalytic sites. The protein surface is colored as a function of the charge density: red, white, and blue colors indicating negative, neutral, and positive area, respectively. Magnesium ion is represented by a green sphere. The amino acids that make contact with compound A,B in the catalytic sites are indicated in their one-letter code and number in the sequence (Adapted from Daligaux et al., 2016a; Mao et al., 2017).
The GDP-MP is a ubiquitous enzyme found in bacteria, fungi, plants, and animals and belonging to the family of nucleotidyl-transferases. In mammalian organisms, GDP-MP was mainly studied in swine (Szumilo et al., 1993; Ning and Elbein, 2000). The swine native enzyme is a complex of about 450 kDa with two distinct subunits: α (43 kDa) and β (37 kDa). In pig, as well as in human, the β subunit displays the enzymatic activity, while the α subunit would have a regulatory function (Szumilo et al., 1993; Ning and Elbein, 2000; Carss et al., 2013; Koehler et al., 2013). In human, α and β subunits share 32% identity. Mutations in the genes coding for α or β subunits in human lead to glycosylation disorders characterized notably by neurological deficits and muscular dystrophies (Carss et al., 2013; Koehler et al., 2013). Two β isoforms, named β1 and β2, have been characterized in the human genome, displaying 90 and 97% identity with the porcine β subunit, respectively. The human β2 isoform is strongly expressed in a wide range of tissues, in opposition to β1 which is only weakly expressed, especially in liver, heart, and kidney (Carss et al., 2013). Additionally, the β2 isoform shows a better homology with Leishmania mexicana GDP-MP, compared to β1 (49% for β2 vs. 46% for β1). In bacteria, GDP-MP are mostly dimeric, either mono- or bifunctional, the latter displaying both GDP-MP and PMI activities in separate domains of an individual enzyme (Shinabarger et al., 1991; May et al., 1994; Ning and Elbein, 1999; Wu et al., 2002; Asencion Diez et al., 2010; Pelissier et al., 2010; Akutsu et al., 2015). Unlike in other organisms, leishmanial GDP-MP has been shown to assemble as a hexamer of 240 kDa in several Leishmania species (Davis et al., 2004; Mao et al., 2017). As this hexamer can dissociate at low ionic strength conditions and at low protein concentration, a mixture of the three forms may be present in the reaction medium in vitro.
Both human and leishmanial GDP-MP have been reported to display a high substrate specificity (Mao et al., 2017), in agreement with previous studies performed in bacterial, trypanosomal, and swine GDP-MP (Ning and Elbein, 2000; Denton et al., 2010; Pelissier et al., 2010). The investigation of the mechanism of reaction has shown a sequential ordered mechanism in most bacterial GDP-MP like in some other nucleotidyl-transferases, with GTP fixation prior to mannose-1-phosphate (Barton et al., 2001; Zuccotti et al., 2001; Asencion Diez et al., 2010; Pelissier et al., 2010; Boehlein et al., 2013). However, leishmanial and human GDP-MP have been characterized by a sequential random mechanism (Mao et al., 2017), in which the substrate binding order is not defined, in agreement with a mammalian nucleotidyl-transferase (Persat et al., 1983), suggesting that the GDP-MP mechanism of reaction differs from bacteria to Leishmania and human.
A knockout of the gene encoding for GDP-MP in L. mexicana lead to an absence of development in the macrophage in vitro and to an absence of parasite persistence in vivo (Garami and Ilg, 2001; Stewart et al., 2005). These results show that GDP-MP is critical for amastigote survival and is therefore an interesting drug therapeutic target to be exploited for antileishmanial drug development. Likewise, GDP-MP has been described to be essential for cell integrity and survival in other microorganisms such as Trypanosoma brucei, Aspergillus fumigatus, or Candida albicans showing the biological validation as a potential therapeutic target of this enzyme in several kinetoplastids and fungi (Warit et al., 2000; Jiang et al., 2008; Denton et al., 2010). Additionally, a High-Throughput Screening (HTS) assay, allowed the selection of leishmanial GDP-MP inhibitors (Lackovic et al., 2010). From this study, the most potent inhibitor identified was a piperazinyl quinoline derivative (compound A; Figure 1B) demonstrating an in vitro activity on L. major GDP-MP and on intracellular parasite proliferation with IC50 values at 0.58 and 21.9 μM, respectively.
Computational and Target-Based Drug Design
A molecular model of the GDP-MP quaternary structure has been generated in L. mexicana, confirming the hexameric structure of the enzyme (Perugini et al., 2005). Based on this model, GDP-MP hexamers would be assembled by a contact between trimer structures in a head-to-head manner involving only the N-terminal end of the protein. These results are however in opposition to crystallography studies of other GDP-MP or nucleotidyl-transferases, showing a tail-to-tail arrangement of the C-terminal β-helices in their quaternary structures (Cupp-Vickery et al., 2005; Jin et al., 2005; Pelissier et al., 2010; Führing et al., 2015).
As no GDP-MP crystal could be obtained in Leishmania, molecular models of L. infantum and L. donovani GDP-MP were generated using distinct sequence alignment strategies and were compared with the human counterpart (Pomel et al., 2012; Daligaux et al., 2016a). Both analyses showed a structural conservation of a consensus sequence GXGXRXnK in leishmanial and human GDP-MP corresponding to a pyrophosphorylase signature motif, as well as the F(V)EKP sequence previously described to be part of the GDP-MP active site (Sousa et al., 2008). Interestingly, several specific residues have been identified in the catalytic site of both L. infantum and L. donovani GDP-MP compared to the human counterpart (Pomel et al., 2012; Daligaux et al., 2016a). Moreover, GDP-MP sequences share more than 85% of identity in the Leishmania genus. Therefore, the differences identified between the leishmanial and human catalytic sites could potentially be exploited to design specific antileishmanial agents.
The GDP-mannose, as a substrate or a product of the GDP-MP, has been selected as the basis for inhibitor design because of its steric volume presenting the maximum of interactions within the enzyme catalytic pocket (Mao et al., 2017). In this work, the chemical approach to design leishmanial GDP-MP inhibitors relied on the pharmacomodulation of the GDP-mannose from the analysis of enzyme molecular models, by substituting for example the mannose moiety by a phenyl group, the pyrophosphate by a triazole or a phosphonate, the ribose by an ether oxide group or a deoxyribose and the guanine by different heterocycles such as purine analogs or quinolines, especially two-substituted quinolines which have been previously described to display promising in vitro and in vivo antileishmanial activities (Fournet et al., 1993, 1994, 1996; Nakayama et al., 2005, 2007; Campos-Vieira et al., 2008; Loiseau et al., 2011). Therefore, the presence of two-substituted quinolines in these compounds designed could potentiate their antileishmanial activities through GDP-MP inhibition.
Cell-Free in vitro and in silico Evaluation of Compounds on Purified GDP-MPs
From the analysis of GDP-MP structural models, a library of 100 compounds was designed and synthesized (Daligaux et al., 2016b; Mao et al., 2017). These compounds were evaluated on recombinant GDP-MP purified from L. donovani (LdGDP-MP), L. mexicana (LmGDP-MP), and human (hGDP-MP). In this work, the hGDP-MP corresponded to the β2 subunit displaying the enzyme activity and showing the highest homology with leishmanial GDP-MP (see above). This evaluation allowed to identify compound B, a quinoline derivative substituted in position 2 with a methoxy-ethyl-triazol-butyn-diisopropylphosphonate group (Figure 1B), as a specific competitive inhibitor of LdGDP-MP with a Ki at 7 μM. In comparison, compound A, previously identified from a HTS (Lackovic et al., 2010), displayed a competitive inhibition of both LdGDP-MP and hGDP-MP with Ki values at 62 and 20 μM, respectively, reflecting a lower affinity for the leishmanial enzyme compared to the human counterpart.
A docking study of the identified competitive inhibitors on GDP-MP structural models showed that compound A binds to both LdGDP-MP and hGDP-MP with similar potency and binding modes: the quinoline, piperazine, and tert-butyl groups occupying the same position as the GDP-mannose nucleotide, ribose and mannose moieties, respectively, in both catalytic sites (Daligaux et al., 2016a; Figure 1C). In contrast, compound B was found to bind more strongly to LdGDP-MP compared to hGDP-MP, with the diisopropylphosphonate group located more deeply in the leishmanial enzyme catalytic pocket compared to the human one (Mao et al., 2017; Figure 1C). These in silico data are in agreement with the non-selective inhibition of both leishmanial and human GDP-MP by compound A and the specific competitive inhibition observed with compound B on LdGDP-MP.
Cellular in vitro Antileishmanial Activity and Cytotoxicity of Compounds A and B
Both compounds have been evaluated on L. donovani and L. mexicana axenic and intramacrophage amastigotes in two host cell models: the RAW264.7 macrophage cell line and primary Bone Marrow Derived Macrophages (BMDM; Mao et al., 2017). Compound A showed a moderate antileishmanial activity on both L. mexicana and L. donovani with IC50 values between 30 and 50 μM and between 12 and 28 μM on axenic and intramacrophage amastigotes, respectively (Mao et al., 2017; Table 1). These data are in agreement with the IC50 previously reported at 21.9 μM on L. major intramacrophage amastigotes by Lackovic et al. (2010). Moreover, this GDP-MP inhibitor showed some cytotoxicity on both RAW264.7 and BMDM macrophages, giving a low Selectivity Index (SI) in both host cell models. On the other hand, compound B exhibited a very interesting activity on L. donovani axenic amastigotes with an IC50 at the micromolar range (Mao et al., 2017; Table 1). However, it was inactive on L. mexicana axenic amastigotes, in line with the data obtained on the purified enzyme showing a specific competitive inhibition of LdGDP-MP. In L. donovani intramacrophage amastigotes, the activity of compound B was maintained with an IC50 at the micromolar range in both host cell models (Mao et al., 2017; Table 1). Interestingly, this compound was also active on L. mexicana intramacrophage amastigotes with IC50 values at 1.5 and 8.6 μM on RAW264.7 and BMDM cell models, respectively, suggesting that an additional mechanism of action, distinct from the parasite GDP-MP inhibition, may be involved. Moreover, no cytotoxicity was observed with compound B on BMDM, giving a promising SI above 94 and 12 in L. donovani and L. mexicana, respectively (Mao et al., 2017; Table 1). Nevertheless, some cytotoxicity was observed on RAW264.7 macrophages, giving a low SI on this cell model. These differences could be due to distinct mechanisms of drug uptake and accumulation between host cell models, the BMDM being closer to physiological and clinical conditions as they are primary macrophages.
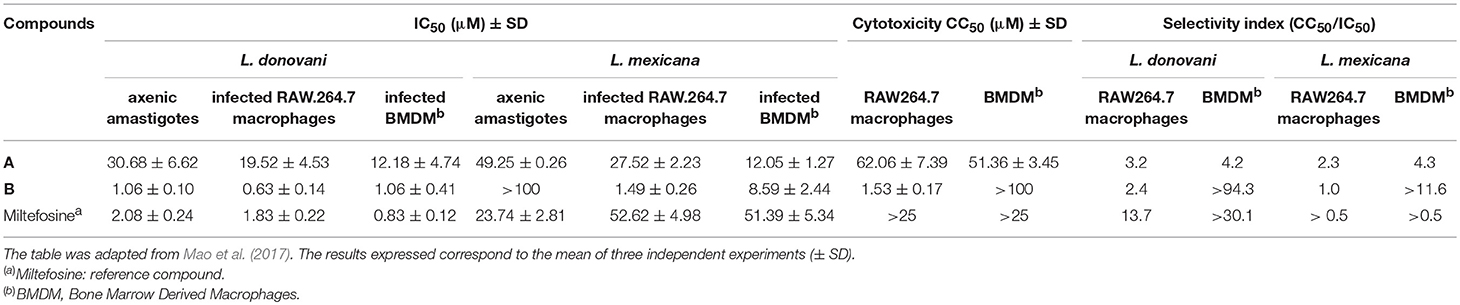
Table 1. Antileishmanial and cytotoxic activities of compounds A and B.
Conclusion and Future Directions
The mannose activation enzyme systems leading to GDP-mannose biosynthesis are essential for host-parasite interactions. Thus, GDP-MP, but also PMI and PMM, are interesting targets to be inhibited for impairing glycoconjugate biosynthesis. In this review, we focus on GDP-MP, this enzyme being responsible for GDP-mannose biosynthesis. GDP-MP is a druggable protein involved in the host-cell/parasite interactions, that has now been biologically and pharmacologically validated. Although ubiquitous, molecular modeling on both leishmanial and human GDP-MPs strongly suggests that specific inhibitors could be designed. From a rational design of 100 compounds based on leishmanial and human GDP-MP tertiary structural models, compound B appeared to be the most promising. In comparison with compound A which displayed a competitive inhibition of both leishmanial and human GDP-MP with moderate antileishmanial activities and a low SI, compound B showed a specific competitive inhibition of LdGDP-MP and an activity on both L. donovani and L. mexicana intramacrophage amastigotes at the micromolar range giving an interesting SI above 10 in the BMDM host cell model. Therefore, the in vivo antileishmanial activity of this compound should be analyzed in order to determine its potency for the treatment of leishmaniasis. Further investigations will address in vivo antileishmanial evaluation, pharmacokinetics, and pharmacodynamics of compound B to confirm its status as a hit. Furthermore, the pathways altered in the parasite by compound B could be investigated in future works through glycomics analysis in order to study the impact of this inhibitor on the membrane glycoconjugate composition. Pharmacomodulations of compound B would also allow to optimize its selectivity and affinity for the target in Leishmania. However, the large molecular volume of this compound required to fill GDP-MP catalytic pocket (Mao et al., 2017), as well as its high polarity, could present challenges for downstream optimization. In order to assess the relative importance of GDP-MP in the most pathogenic leishmanial species, comparative functional analyses should be performed to optimize the inhibitor strategy.
In conclusion, compound B can be considered as an original and interesting hit to be optimized proving that GDP-MP inhibition is a promising strategy to impair host-parasite interactions. However, the capacity of this specific metabolism alteration to prevent drug resistance emergence is still to be proved.
Author Contributions
SP wrote the manuscript. WM, TH-D, CC, and PL contributed to manuscript revision and read and approved the submitted version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to DIM Malinf (Région Île de France) for WM Ph.D. funding.
References
Akutsu, J., Zhang, Z., Morita, R., and Kawarabayasi, Y. (2015). Identification and characterization of a thermostable binfunctional enzyme with phosphomannose isomerase and sugar-1-phosphate nucleotidylyltransferase activities from a hyperthermophilic archeon, Pyrococcus horikoshii OT3. Extremophiles 19, 1077–1085. doi: 10.1007/s00792-015-0779-5
Aronov, A. M., Suresh, S., Buckner, F. S., Van Voorhis, W. C., Verlinde, C. L., Opperdoes, F. R., et al. (1999). Structure-based design of submicromolar, biologically active inhibitors of trypanosomatid glyceraldehyde-3-phosphate dehydrogenase. Proc. Natl. Acad. Sci. U.S.A. 96, 4273–4278. doi: 10.1073/pnas.96.8.4273
Asencion Diez, M. D., Demonte, A., Giacomelli, J., Garay, S., Rodrigues, D., Hofmann, B., et al. (2010). Functional characterization of GDP-mannose pyrophosphorylase from Leptospira interrogans serovar Copenhageni. Arch. Microbiol. 192, 103–114. doi: 10.1007/s00203-009-0534-3
Barton, W. A., Lesniak, J., Biggins, J. B., Jeffrey, P. D., Jiang, J., Rajashankar, K. R., et al. (2001). Structure, mechanism and engineering of a nucleotidylyltransferase as a first step toward glycorandomization. Nat. Struct. Biol. 8, 545–551. doi: 10.1038/88618
Boehlein, S. K., Shaw, J. R., Hwang, S. K., Stewart, J. D., and Curtis Hannah, L. (2013). Deciphering the kinetic mechanisms controlling selected plant ADP-glucose pyrophosphorylases. Arch. Biochem. Biophys. 535, 215–226. doi: 10.1016/j.abb.2013.04.003
Burchmore, R. J., Rodriguez-Contreras, D., McBride, K., Merkel, P., Barrett, M. P., Modi, G., et al. (2003). Genetic characterization of glucose transporter function in Leishmania mexicana. Proc. Natl. Acad. Sci. U.S.A. 100, 3901–3906. doi: 10.1073/pnas.0630165100
Campos-Vieira, N., Vacus, J., Fournet, A., Baudoin, R., Bories, C., Séon-Méniel, B., et al. (2008). Antileishmanial activity of a formulation of 2-n-propylquinoline by oral route in mice model. Parasite 18, 333–336. doi: 10.1051/parasite/2011184333
Carss, K. J., Stevens, E., Foley, A. R., Cirak, S., Riemersma, M., Torelli, S., et al. (2013). Mutations in GDP-mannose pyrophosphorylase B cause congenital and limb-girdle muscular dystrophies associated with hypoglycosylation of α-dystroglycan. Am. J. Hum. Genet. 93, 29–41. doi: 10.1016/j.ajhg.2013.05.009
Chowdhury, S. F., Villamor, V. B., Guerrero, R. H., Leal, I., Brun, R., Croft, S. L., et al. (1999). Design, synthesis, and evaluation of inhibitors of trypanosomal and leishmanial dihydrofolate reductase. J. Med. Chem. 42, 4300–4312. doi: 10.1021/jm981130+
Colley, K. J., Varki, A., and Kinoshita, T. (2017). “Chapter 4: cellular organization of glycosylation,” in Essentials in Glycobiology, 3rd Edn., eds A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, et al. (New York, NY: Cold Spring Harbor Laboratory Press), 41–49.
Colotti, G., Baiocco, P., Fiorillo, A., Boffi, A., Poser, E., Chiaro, F. D., et al. (2013). Structural insights into the enzymes of the trypanothione pathway: targets for antileishmanial drugs. Future Med. Chem. 5, 1861–1875. doi: 10.4155/fmc.13.146
Croft, S. L., and Coombs, G. H. (2003). Leishmaniasis – current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol. 19, 502–508. doi: 10.1016/j.pt.2003.09.008
Croft, S. L., Sundar, S., and Fairlamb, A. H. (2006). Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 19, 111–126. doi: 10.1128/CMR.19.1.111-126.2006
Cupp-Vickery, J. R., Igarashi, R. Y., and Meyer, C. R. (2005). Preliminary crystallographic analysis of ADP-glucose pyrophosphorylase from Agrobacterium tumefasciens. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 61, 266–268. doi: 10.1107/S1744309105002265
Daligaux, P., Bernadat, G., Tran, L., Cavé, C., Loiseau, P. M., Pomel, S., et al. (2016a). Comparative study of structural models of Leishmania donovani and human GDP-mannose pryphosphorylases. Eur. J. Med. Chem. 107, 109–118. doi: 10.1016/j.ejmech.2015.10.037
Daligaux, P., Pomel, S., Leblanc, K., Loiseau, P. M., and Cavé, C. (2016b). Simple and efficient synthesis of 5'-aryl-5'-deoxyguanosine analogs by azide-alkyne click reaction and their antileishmanial activities. Mol. Divers. 20, 507–519. doi: 10.1007/s11030-015-9652-9
Davis, A. J., Perugini, M. A., Smith, B. J., Stewart, J. D., Ilg, T., Hodder, A. N., et al. (2004). Properties of GDP-mannose pyrophosphorylase, a critical enzyme and drug target in Leishmania mexicana. J. Biol. Chem. 279, 12462–12468. doi: 10.1074/jbc.M312365200
Denton, H., Fyffe, S., and Smith, T. K. (2010). GDP-mannose pyrophosphorylase is essential in the bloodsteam form of Trypanosoma brucei. Biochem. J. 425, 603–614. doi: 10.1042/BJ20090896
Descoteaux, A., Luo, Y., Turco, S. J., and Beverley, S. M. (1995). A specialized pathway affecting virulence glycoconjugates of Leishmania. Science 269, 1869–1872. doi: 10.1126/science.7569927
Descoteaux, A., and Turco, S. J. (1999). Glycoconjugates in Leishmania infectivity. Biochim. Biophys. Acta 1455, 341–352. doi: 10.1016/S0925-4439(99)00065-4
Fournet, A., Barrios, A. A., Munoz, V., Hocquemiller, R., Cavé, A., and Bruneton, J. (1993). 2-substituted quinoline alkaloids as potential antileishmanial drugs. Antimicrob. Agents Chemother. 37, 859–863. doi: 10.1128/AAC.37.4.849
Fournet, A., Ferreira, M. E., Rojas de Arias, A., Torres de Ortiz, S., Fuentes, S., Nakayama, H., et al. (1996). In vivo efficacy of oral and intralesional administration of 2-substituted quinolines in experimental treatment of new world cutaneous leishmaniasis caused by Leishmania amazonensis. Antimicrob. Agents Chemother. 40, 2447–2451. doi: 10.1128/AAC.40.11.2447
Fournet, A., Gantier, J. C., Gautheret, A., Leysalles, L., Munos, M. H., Mayrargue, J., et al. (1994). The activity of 2-substituted quinoline alkaloids in BALB/c mice infected with Leishmania donovani. J. Antimicrob. Chemother. 33, 537–544. doi: 10.1093/jac/33.3.537
Führing, J. I., Cramer, J. T., Schneider, J., Baruch, P., Gerardy-Schahn, R., and Fedorov, R. (2015). A quaternary mechanism enables the complex biological functions of octameric human UDP-glucose pyrophosphorylase, a key enzyme in cell metabolism. Sci. Rep. 5:9618. doi: 10.1038/srep09618
Garami, A., and Ilg, T. (2001). Disruption of mannose activation in Leishmania mexicana: GDP-mannose pyrophosphorylase is required for virulence, but not viability. EMBO J. 20, 3657–3666. doi: 10.1093/emboj/20.14.3657
Jain, V., and Jain, K. (2018). Molecular targets and pathways for the treatment of visceral leishmaniasis. Drug Discov. Today 23, 161–170. doi: 10.1016/j.drudis.2017.09.006
Jiang, H., Ouyang, H., Zhou, H., and Jin, C. (2008). GDP-mannose pyrophosphorylase is essential for cell wall integrity, morphogenesis and viability of Aspergillus fumigatus. Microbiology 154, 2730–2739. doi: 10.1099/mic.0.2008/019240-0
Jiang, Y., Roberts, S. C., Jardim, A., Carter, N. S., Ariyanayagam, M., Fairlamb, A. H., et al. (1999). Ornithine decarboxylase gene deletion mutants of Leishmania donovani. J. Biol. Chem. 274, 3781–3788. doi: 10.1074/jbc.274.6.3781
Jin, X., Ballicora, M. A., Preiss, J., and Geiger, J. H. (2005). Crystal structure of potato tuber ADP-glucose pyrophosphorylase. EMBO J. 24, 694–704. doi: 10.1038/sj.emboj.7600551
Koehler, K., Malik, M., Mahmood, S., Giesselman, S., Beetz, C., Hennings, J. C., et al. (2013). Mutations in GMPPA cause a glycosylation disorder characterized by intellectual disability and autonomic dysfunction. Am. J. Hum. Genet. 93, 727–734. doi: 10.1016/j.ajhg.2013.08.002
Lackovic, K., Parisot, J. P., Sleebs, N., Baell, J. B., Debien, L., Watson, K. G., et al. (2010). Inhibitors of Leishmania GDP-mannose pyrophosphorylase identified by high-throughput screening of small-molecule chemical library. Antimicrob. Agents Chemother. 54, 1712–1719. doi: 10.1128/AAC.01634-09
Lamotte, S., Späth, G. F., Rachidi, N., and Prina, E. (2017). The enemy within: targeting host-parasite interaction for antileishmanial drug discovery. PLoS Negl. Trop. Dis. 11:e0005480. doi: 10.1371/journal.pntd.0005480
Leroux, A. E., and Krauth-Siegel, R. L. (2016). Thiol redox biology of trypanosomatids and potential targets for chemotherapy. Mol. Biochem. Parasitol. 206, 67–74. doi: 10.1016/molbiopara.2015.11.003
Loiseau, P. M., Gupta, S., Verma, A., Srivastava, S., Puri, S. K., Sliman, F., et al. (2011). In vitro activities of new 2-substituted quinolines against Leishmania donovani. Antimicrob. Agents Chemother. 55, 1777–1780. doi: 10.1128/AAC.01299-10
Mao, W., Daligaux, P., Lazar, N., Ha-Duong, T., Cavé, C., van Tilbeurgh, H., et al. (2017). Biochemical analysis of leishmanial and human GDP-mannose pyrophosphorylases and selection of inhibitors as new leads. Sci. Rep. 7:751. doi: 10.1038/s41598-017-00848-8
May, T. B., Shinabarger, D., Boyd, A., and Chakrbarty, A. M. (1994). Identification of amino acid residues involved in the activity of phosphomannose isomerase-guanosine 5′-diphospho-D-mannose pyrophosphorylase. A bifunctional enzyme in the alginate biosynthetic pathway of Pseudomonas aeruginosa. J. Biol. Chem. 269, 4872–4877.
Nakayama, H., Desrivot, J., Bories, C., Franck, X., Figadère, B., Hocquemiller, R., et al. (2007). In vitro and in vivo antileishmanial efficacy of a new nitriquinoline against Leishmania donovani. Biomed. Pharmacother. 61, 186–188. doi: 10.1016/j.biopha.2007.02.001
Nakayama, H., Loiseau, P. M., Bories, C., Torres de Ortiz, S., Schinini, A., Serna, E., et al. (2005). Efficacy of orally administered 2-substituted quinolines in experimental cutaneous and visceral leishmaniases. Antimicrob. Agents Chemother. 49, 4950–4956. doi: 10.1128/AAC.49.12.4950-4956.2005
Ning, B., and Elbein, A. D. (1999). Purification and properties of mycobacterial GDP-mannose pyrophosphorylase. Arch. Biochem. Biophys. 362, 339–345. doi: 10.1006/abbi.1998.1053
Ning, B., and Elbein, A. D. (2000). Cloning, expression and characterization of the pig liver GDP-mannose pyrophosphorylase. Evidence that GDP-mannose and GDP-Glc pyrophosphorylases are different proteins. Eur. J. Biochem. 267, 6866–6874. doi: 10.1046/j.1432-1033.2000.01781.x
Olin-Sandoval, V., Moreno-Sanchez, R., and Saavedra, E. (2010). Targeting trypanothione metabolism in trypanosomatid parasites. Curr. Drug Targets 11, 1614–1630. doi: 10.2174/1389450111009011614
Pelissier, M. C., Lesley, S. A., Kuhn, P., and Bourne, P. (2010). Structural insights into the catalytic mechanism of bacterial guanosine-diphospho-D-mannose pyrophosphorylase and its regulation by divalent ions. J. Biol. Chem. 285, 27468–27476. doi: 10.1074/jbc.M109.095182
Persat, F., Azzar, G., Martel, M. B., and Got, R. (1983). Properties of uridine diphosphate glucose pyrophosphorylase from Golgi apparatus of liver. Biochim. Biophys. Acta 749, 329–332. doi: 10.1016/0167-4838(83)90243-1
Perugini, M. A., Griffin, M. D., Smith, B. J., Webb, L. E., Davis, A. J., Handman, E., et al. (2005). Insight into the self-association of key enzymes from pathogenic species. Eur. Biophys. J. 34, 469–476. doi: 10.1007/s00249-005-0491-y
Podinovskaia, M., and Descoteaux, A. (2015). Leishmania and the macrophage: a multifaceted interaction. Future Microbiol. 10, 111–129. doi: 10.2217/fmb.14.103
Pomel, S., and Loiseau, P. M. (2013). “GDP-mannose: a key-point for target identification and drug design in kinetoplastids,” in Trypanosomatid Diseases: Molecular Routes to Drug Discoveries, eds T. Jäger, O. Koch, and L. Flohe (Weinheim: Wiley-VCH Verlag GmbH and Co. KGaA), 315–334.
Pomel, S., Rodrigo, J., Hendra, F., Cavé, C., and Loiseau, P. M. (2012). In silico analysis of a therapeutic target in Leishmania infantum: the guanosine-diphospho-D-mannose pyrophosphorylase. Parasite 19, 63–70. doi: 10.1051/parasite/2012191063
Ponte-Sucre, A., Gamarro, F., Dujardin, J. C., Barrett, M. P., Lopez-Velez, R., Garcia-Hernandez, R., et al. (2017). Drug resistance and treatment failure in leishmaniasis: a 21st century challenge. PLoS Negl. Trop. Dis. 11:e0006052. doi: 10.1371/journal.pntd.0006052
Shinabarger, D., Berry, A., May, T. B., Rothmel, R., Fialho, A., and Chakrabarty, A. M. (1991). Purification and characterization of phosphomannose isomerase-guanosine diphospho-D-mannose pyrophosphorylase. A bifunctional enzyme in the alginate biosynthetic pathway of Pseudomonas aeruginosa. J. Biol. Chem. 266, 2080–2088.
Sousa, S. A., Moreira, L. M., and Leitao, J. H. (2008). Functional analysis of the Burkholderia cenocepacia J2315 BceAJ protein with phosphomannose isomerase and GDP-mannose pyrophosphorylase activities. Appl. Microbiol. Biotechnol. 80, 1015–1022. doi: 10.1007/s00253-008-1612-1
Stewart, J., Curtis, J., Spurck, T. P., Ilg, T., Garami, A., Baldwin, T., et al. (2005). Charaterization of a Leishmania mexicana knockout lacking guanosine diphosphate-mannose pyrophosphorylase. Int. J. Parasitol. 35, 861–873. doi: 10.1016/j.ijpara.2005.03.008
Sundar, S., and Singh, A. (2016). Recent developments and future prospects in the treatment of visceral leishmaniasis. Ther. Adv. Infect. Dis. 3, 98–109. doi: 10.1177/2049936116646063
Szumilo, T., Drake, R. R., York, J. L., and Elbein, A. D. (1993). GDP-mannose pyrophosphorylase. Purification to homogeneity, properties, and utilization to prepare photoaffinity analogs. J. Biol. Chem. 268, 17943–17950.
Varki, A. (2007). Glycan-based interactions involving vertebrate sialic-acid recognizing proteins. Nature 446, 1023–1029. doi: 10.1038/nature05816
Verlinde, C. L., Hannaert, V., Blonski, C., Willson, M., Périé, J. J., Fothergill-Gilmore, L. A., et al. (2001). Glycolysis as a target for the design of new anti-trypanosome drugs. Drug Resist. Updat. 4, 50–65. doi: 10.1054/drup.2000.0177
Warit, S., Zhang, N., Short, A., Walmsley, R. M., Oliver, S. G., and Stateva, L. I. (2000). Glycosylation deficiency phenotypes resulting from depletion of GDP-mannose pyrophosphorylase in two yeast species. Mol. Microbiol. 36, 1156–1166. doi: 10.1046/j.1365-2958.2000.01944.x
Wu, B., Zhang, Y., Zheng, R., Guo, C., and Wang, P. G. (2002). Bifunctional phosphomannose isomerase/GDP-D-mannose pyrophosphorylase is the point of control for GDP-D-mannose biosynthesis in Helicobacter pylori. FEBS Lett. 519, 87–92. doi: 10.1016/S0014-5793(02)02717-5
Zuccotti, S., Zanardi, D., Rosano, C., Sturla, L., Tonetti, M., and Bolognesi, M. (2001). Kinetic and crystallographic analyses support a sequential ordered bi bi mechanism for Escherichia coli glucose-1-phosphate thymidilyltransferase. J. Mol. Biol. 31o3, 831–843. doi: 10.1006/jmbi.2001.5073
Keywords: GDP-mannose pyrophosphorylase, Leishmania, therapeutic target, inhibitors, drug development
Citation: Pomel S, Mao W, Ha-Duong T, Cavé C and Loiseau PM (2019) GDP-Mannose Pyrophosphorylase: A Biologically Validated Target for Drug Development Against Leishmaniasis. Front. Cell. Infect. Microbiol. 9:186. doi: 10.3389/fcimb.2019.00186
Received: 15 March 2019; Accepted: 15 May 2019;
Published: 31 May 2019.
Edited by:
Brice Rotureau, Institut Pasteur, FranceReviewed by:
Rosa M. Reguera, Universidad de León, SpainAlicia Ponte-Sucre, Central University of Venezuela, Venezuela
Andrea Ilari, Istituto di Biologia e Patologia Molecolari (CNR), Italy
Copyright © 2019 Pomel, Mao, Ha-Duong, Cavé and Loiseau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sébastien Pomel, c2ViYXN0aWVuLnBvbWVsQHUtcHN1ZC5mcg==