Abstract
The lifecycle of the causative agent of the severe secretory diarrheal disease cholera, Vibrio cholerae, is characterized by the transition between two dissimilar habitats, i.e., as a natural inhabitant of aquatic ecosystems and as a pathogen in the human gastrointestinal tract. Vibrio cholerae faces diverse stressors along its lifecycle, which require effective adaptation mechanisms to facilitate the survival fitness. Not surprisingly, the pathogen's transcriptome undergoes global changes during the different stages of the lifecycle. Moreover, recent evidence indicates that several of the transcription factors (i.e., ToxR, TcpP, and ToxT) and alternative sigma factors (i.e., FliA, RpoS, and RpoE) involved in transcriptional regulations along the lifecycle are controlled by regulated proteolysis. This post-translational control ensures a fast strategy by the pathogen to control cellular checkpoints and thereby rapidly respond to changing conditions. In this review, we discuss selected targets for regulated proteolysis activated by various stressors, which represent a key feature for fast adaptation of V. cholerae.
A Brief Survey of Regulatory Events Along Vibrio cholerae's Lifecycle
Vibrio cholerae spends much of its lifecycle outside of the host in estuarine and costal aquatic reservoirs with a geographical range from tropics to temperate waters world-wide. Along its interepidemic persistence in the aquatic reservoirs, V. cholerae faces temperature shifts, osmotic stress, bacterivorous predators and nutrient limitation (Lutz et al., 2013; List et al., 2018). Vibrio cholerae employs several strategies to cope with these numerous stressors. In particular, biofilm formation has been highlighted as a key factor for environmental survival and transmission of V. cholerae (comprehensively reviewed in Yildiz and Visick, 2009; Teschler et al., 2015). A central player of biofilm regulation is the transcriptional repressor HapR, which acts negatively on biofilm formation via repression of exopolysaccharide synthesis. Additionally, HapR is a quorum sensing key regulator affecting virulence factor expression and natural competence (Ng and Bassler, 2009). Since transcription of hapR is also activated by the alternative sigma factor RpoS, the pathways mentioned above are regulated by central physiological signals, like cell density, or carbon concentration. Biofilm-associated bacteria are generally better protected against host-derived stressors ranging from digestive enzymes, acidic pH to antimicrobial substances and exhibit hyperinfectivity in the murine model (Tamayo et al., 2010; Seper et al., 2011). Thus, biofilm clumps are a likely form by which clinically relevant V. cholerae initiate outbreaks (Colwell et al., 2003; Hall-Stoodley and Stoodley, 2005).
Upon oral ingestion, V. cholerae passages through the stomach to finally reach the small intestine, representing the primary site of colonization. Intestinal stimuli induce expression of virulence factors such as the toxin coregulated pilus (TCP) and the cholera toxin (CTX) (Childers and Klose, 2007; Matson et al., 2007). TCP represents the main colonization factor responsible for adherence to epithelial cells, while CTX constitutively activates adenylate cyclase of the host, causing a massive water efflux into the intestinal lumen known as rice-water stool (Sharp et al., 1971; Burns et al., 1983; Miller et al., 1987; Taylor et al., 1987; Herrington et al., 1988; Fishmann, 1990).
The expression of virulence factors is controlled by a complex regulatory cascade. It includes the membrane-bound transcription complexes ToxR/S and TcpP/H as well as the cytosolic transcription factor ToxT (Childers and Klose, 2007). Most of the virulence factors, e.g., CTX and TCP, are regulated by the ToxT-dependent pathway in response to intestinal stimuli, such as temperature and sodium bicarbonate (Thomson and Withey, 2014; Weber et al., 2014). In addition, ToxR can directly regulate several genes independently of ToxT. For example, ToxR inversely regulates the expression of the porins OmpU and OmpT, which plays an essential role to achieve bile resistance and full colonization fitness in vivo (Provenzano and Klose, 2000).
Once adapted to the intestinal conditions, V. cholerae starts to massively proliferate and the patient develops a severe secretory diarrhea, releasing the bacteria back into the aquatic environment. Transcriptional control of a defined set of genes at the late stage of infection facilitates the transition of V. cholerae into the aquatic reservoir (Schild et al., 2007). Under these conditions HapR and RpoS coordinate a drastic shift in the gene expression profile also known as “mucosal escape response” (Nielsen et al., 2006).
As highlighted above, adaptation to diverse conditions along the lifecycle is achieved by spatio-temporal induction of gene expression. However, termination of a regulatory pathway could be equally important to facilitate transition to the next stage of the lifecycle. An effective way to remove factors involved in gene transcription is regulated proteolysis, comprising directed degradation of defined effectors by specific proteases (Mahmoud and Chien, 2018). Indeed, regulated proteolysis has been reported as a control mechanism for several transcriptional effectors (i.e., FliA, ToxR, TcpP, ToxT, RpoS, and RpoE) along the lifecycle of V. cholerae (Figure 1 and Table 1), which will be discussed with an emphasis on the physiological impact and players involved.
Figure 1
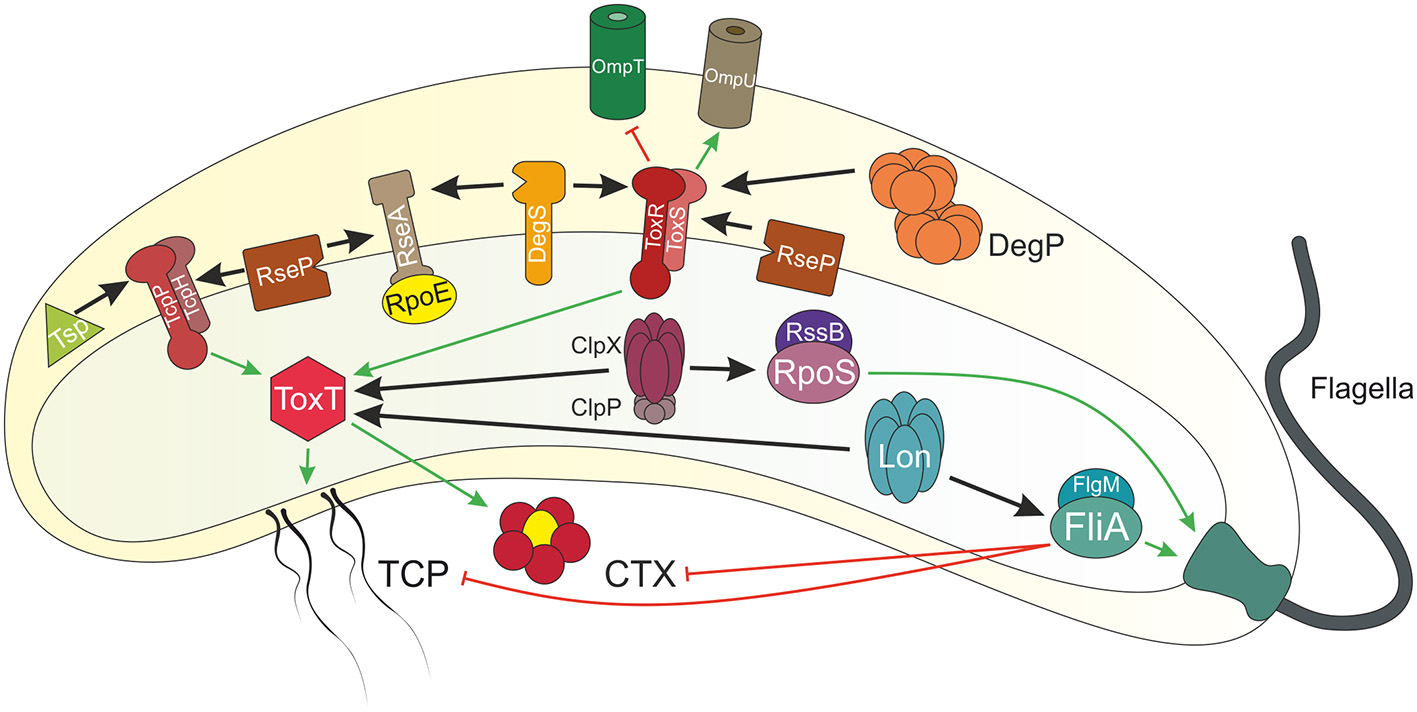
Overview of regulated proteolysis in V. cholerae. Shown is a V. cholerae cell with proteins illustrated by icons. Regulated proteolysis is indicated by black arrows. Transcriptional activation is highlighted in green and repression in red. At early stages of infection, the single polar flagellum of V. cholerae breaks by entering the viscous mucosal layer of the small intestine. Thereby, levels of the anti-sigma factor FlgM decreases within the cell and the alternative sigma factor FliA activates transcription of flagella biosynthesis genes. Besides of this repair mechanism, FliA also inhibits virulence genes expression, e.g., tcp and ctx. The AAA+ protease Lon degrades FliA in absence of FlgM to achieve full virulence at early stage infection. The membrane embedded transcriptional regulators and their respective partner proteins ToxRS and TcpPH activate toxT expression, which in turn encodes for the master regulator for transcription of the downstream genes tcp and ctx. Furthermore, ToxRS also coordinate the inverse regulation of the outer membrane porins OmpU and OmpT, in order to build up a resistance to bile salts. Both regulators are targets of regulated intramembrane proteolysis (RIP). ToxR periplasmic domain is cut by the site-1 proteases DegS and DegP, followed by site-2 protease RseP. Commensurately, Tsp mediates TcpP degradation as a site-1 protease, which in turn triggers RseP. The half-life of the virulence master regulator ToxT is controlled by the AAA+ proteases Lon and ClpXP. Additionally, the proteases DegS and RseP also act on the transmembrane anti-sigma factor RseA under envelope stress conditions, e.g., cellular or environmental changes, in order to release the alternative sigma factor RpoE to the cytosol, eventually to activate transcription of degP, rpoE itself or genes encoding for the T2SS. The alternative sigma factor RpoS is responsible to cope with starvation conditions, e.g., high (p)pGpp levels. At low levels of (p)pGpp, the anti-sigma factor RssB is bound to RpoS, leading to proteolysis mediated by ClpXP. At late stages of infection, RpoS is required to activate mucosal escape response by inducing the expression of chemotaxis and motility genes.
Table 1
| Target for proteolysis | Binding partners/(anti-)sigma factors | Protease(s) | Physiological role/regulated pathways | Trigger for proteolysis |
|---|---|---|---|---|
| FliA (σ28) | FlgM | Lon | Motility, virulence | Broken flagellum |
| ToxR | ToxS | DegS, DegP, RseP | Persistence, virulence | Alkaline pH in combination with starvation |
| TcpP | TcpH | Tsp, RseP | Virulence | Non-virulence-inducing conditions |
| ToxT | – | ClpXP, Lon | Virulence | High temperature, alkaline pH |
| RseA | RpoE (σE or σ24) | DegS, RseP | Envelope stress response | Misfolded periplasmic protein |
| RpoS (σS or σ38) | RssB | ClpXP | Motility, chemotaxis, biofilm | Non-starvation condition |
Examples for regulated proteolysis in V. cholerae (for details see text).
The Alternative Sigma Factor FliA (σ28) Is Degraded by Lon
In addition to virulence factor expression, flagella-dependent motility contributes to virulence of V. cholerae. The single polar flagellum is required to approach and penetrate the mucosal layer of the intestinal epithelium (Freter and Jones, 1976; Freter and O'Brien, 1981; Lee et al., 2001; Butler and Camilli, 2005). Entrance into the viscous mucosal layer puts substantial shear force on the rotating flagellum, which eventually breaks. As a result, the anti-sigma factor FlgM, usually bound to the alternative sigma factor FliA and preventing its association with the RNA polymerase, is released through the broken flagellar apparatus (Correa et al., 2004; Liu et al., 2008). Decreasing levels of FlgM result in derepression of FliA important for activation of flagella biosynthesis. Although this is an efficient feedback mechanism to sense damage of the flagellum and initiate its repair, it is dispensable in the in vivo setting once V. cholerae has penetrated through the mucosal layer. FliA inhibits virulence factor expression in V. cholerae by a so far unknown mechanism (Syed et al., 2009). Therefore, efficient removal of FliA from the cytosol is essential to allow full virulence expression in early stages of infection. A recent study demonstrated that FliA of V. cholerae is target for rapid proteolysis via the AAA+ (ATPase associated with a variety of cellular activities) protease Lon in the absence of its anti-sigma factor FlgM (Pressler et al., 2016). Under virulence-inducing conditions [i.e., in vitro cultivation using AKI conditions (Iwanaga and Yamamoto, 1985; Iwanaga et al., 1986)], the Lon-dependent proteolysis facilitates cholera toxin production in the presence of a damaged flagellum. Thus, the rapid removal of FliA via Lon provides a first molecular explanation for high virulence expression upon mucosal penetration at early stages of infection. Concordantly, FliA of Escherichia coli is also a target for Lon-mediated proteolysis and can be protected by the anti-sigma factor FlgM (Barembruch and Hengge, 2007). Thus, the FliA-FlgM-Lon feedback circuit could represent a conserved mechanism for correct flagella assembly and repair upon flagellar damage.
Regulated Proteolysis of ToxR Is Mediated by DegS, DegP and RseP
Vibrio cholerae persistence and virulence are coordinated by a complex network that has been historically referred as the “ToxR-regulon” (Matson et al., 2007). ToxR is a single component signal transduction regulator comprising the N-terminal winged helix-turn-helix domain promoting DNA-binding, a transmembrane domain and the C-terminal periplasmic sensor domain (Miller et al., 1987). ToxR binds multiple AT-rich promoter proximal regions termed ToxR-boxes and is involved in transcriptional control of more than 100 genes (Miller et al., 1987; Bina et al., 2003; Goss et al., 2013). Molecular activation mechanisms for the membrane bound transcription factor ToxR are limited. Although, it has been recently shown that the transcriptional activation by the ToxR-like protein CadC in E. coli and binding to its operator sites, follows a model termed diffusion and capture mechanism (Brameyer et al., 2019). ToxR and TcpP (discussed below), together with their respective co-transcribed interaction partners ToxS and TcpH, are both required for maximal toxT expression, whereas ToxRS mediates outer membrane porin (OMP) expression directly (Higgins and DiRita, 1994; Häse and Mekalanos, 1998; Bina et al., 2003; Childers and Klose, 2007). The two major porins of V. cholerae, OmpT and OmpU are inversely regulated by ToxR. The ompT expression is repressed, whereby ompU is strongly induced by ToxR under nutrient rich conditions or bile salts (i.e., sodium deoxycholate), facilitating resistance toward antimicrobial compounds (Miller and Mekalanos, 1988; Provenzano et al., 2000; Mathur et al., 2007; Lembke et al., 2018). Unlike the remainder of the regulon, ompT is the only verified gene negatively regulated by ToxR and is derepressed under nutrient limiting conditions (Li et al., 2000). In general, toxR is constitutively expressed, environmental and stress stimuli may modulate the expression of ToxR regulated genes (Miller and Mekalanos, 1988). Molecular activation mechanisms for ToxR transcriptional activity are largely unknown. The two periplasmic cysteine residues (Cys236 and Cys293) of ToxR influence ToxR regulated gene expression. More specifically, DsbAB-mediated intramolecular disulfide bond and homodimer formation increase ToxR transcription factor activity (Ottemann and Mekalanos, 1996; Fengler et al., 2012; Lembke et al., 2018). Interventions in these cysteine residues (e.g., cysteine to serine substitution, reducing conditions) decrease ToxR transcription factor activity and consequently abolish the ability of proper porin gene regulation, but does not affect toxT transcription (Fengler et al., 2012; Lembke et al., 2018). The cysteine-reduced ToxR is a trigger to stimulate site-1 mediated proteolysis by DegS and DegP, hence resulting in ToxR degradation, most effective in strains lacking toxS (Lembke et al., 2018). Regulated intramembrane proteolysis (RIP) control of ToxR seems to play a physiologically important role for V. cholerae to properly adapt to changing environmental conditions (Almagro-Moreno et al., 2015a,b). Upon transition into a dormant stage in presence of unfavorable stress conditions, e.g., alkaline pH and nutrient limitation, ToxR becomes a substrate for RIP by the site-2 protease RseP (YaeL), which belongs to the RpoE response system (see below). ToxS also plays a major role in protecting ToxR from proteolysis under these conditions at late stationary phase (Almagro-Moreno et al., 2015b). A point mutation in ToxS (ToxSL33S) even triggers ToxR proteolysis comprising several site-1 proteases, including DegS, DegP, VesC, and TapA (Almagro-Moreno et al., 2015b). The two major routes of ToxR proteolysis, one responding toward the redox state and the other being sensitive to an alkaline pH and starvation, can be inhibited by bile salts, which are present in the human gut and are also known to strengthen ToxRS interaction (Midgett et al., 2017; Lembke et al., 2018). The RIP of ToxR is a highly versatile 2-step process, leading to a clearance of ToxR molecules and eventually to a termination of ToxR dependent gene regulation.
TcpP Is a Substrate of Tsp and RseP Proteases
A second membrane-bound transcription factor that coordinates expression of toxT is TcpP. Like ToxR, TcpP is a bitopic protein containing a carboxy-terminal periplasmic domain and an amino-terminal cytoplasmic DNA-binding domain similar to transcription activators of the OmpR/PhoB-family (Martínez-Hackert and Stock, 1997). TcpP functions together with TcpH, a membrane protein that interacts with the periplasmic domain of TcpP. In order to activate transcription of toxT, ToxR recruits TcpP to the toxT promoter region through protein-protein interaction, where TcpP binds two pentameric repeats located between positions-53 and -38 relative to the toxT transcription start site (Krukonis and DiRita, 2003; Goss et al., 2010). TcpP levels in the bacterial cell are regulated both transcriptionally and post-transcriptionally. Upon entering the human intestine, environmental signals activate expression of tcpPH through AphA and AphB (Kovacikova and Skorupski, 1999; Skorupski and Taylor, 1999). Alternatively, under conditions that do not activate virulence gene expression, TcpP is degraded by RIP. The site-1 protease that first acts to cleave TcpP within its periplasmic domain is Tsp (tail-specific protease) (Teoh et al., 2015). Tsp is a serine protease that generally controls protein quality and gene regulation, and is rarely associated with RIP mechanisms. After the initial cleavage, TcpP becomes a substrate for the site-2 protease RseP, a membrane-localized metalloprotease that cuts within the transmembrane domain (Matson and DiRita, 2005). This cleavage and removal from the inner membrane inactivates TcpP, halting expression of toxT and downstream virulence genes. TcpP is normally protected from degradation through its interaction with TcpH under virulence-gene inducing conditions (Beck et al., 2004). Disruption of a periplasmic disulfide bond in TcpP results in instability of the protein, even in the presence of TcpH (Morgan et al., 2016). In addition, disruption of these periplasmic cysteines causes TcpH to also become unstable, suggesting a role for these intramolecular disulfide bonds in the TcpP-TcpH interaction (Morgan et al., 2016). Furthermore, transcriptionally active TcpP-homodimers are formed by an intermolecular disulfide bond via Cys207 in presence of the bile salt taurocholate (Yang et al., 2013). Heterodimers between TcpP and ToxR depend on the periplasmic thiol-disulfide-oxidoreductase DsbA and are enhanced by anaerobic growth conditions resulting in virulence gene induction (Fan et al., 2014). It should be noted that the outcome of ToxR and TcpP RIP is unusual in that it functions to inactivate a membrane-bound regulator and halt transcription. In the case of the RpoE-pathway (see below) and others, RIP results in transcriptional activation of downstream genes.
ClpXP and Lon Mediate ToxT Proteolysis
ToxT was identified as a central transcription factor, activating expression of important virulence genes, e.g., encoding for TCP and CTX (DiRita and Mekalanos, 1991). ToxT is a crucial checkpoint, thereby its own synthesis is under complex control, i.e., by endogenous and exogenous factors as reviewed elsewhere (Weber and Klose, 2011). Exogenous signals negatively control ToxT activity, such as bile-derived unsaturated fatty acids (Plecha and Withey, 2015), or positively, like sodium carbonate (Thomson and Withey, 2014). Additionally, a 5′ mRNA thermometer control element of toxT allows access of ribosomes to the Shine-Dalgarno sequence at 37°C, but not at 20°C (Weber et al., 2014). ToxT becomes a substrate for proteolysis during virulence gene expression, reducing ToxT protein half-life (Abuaita and Withey, 2011). Precise timing of ToxT activity is crucial for the colonization success, e.g., by determining the duration and intensity of virulence gene expression. This was best monitored under in vivo conditions (Lee et al., 1999) and by a microarray series performed on cells grown under virulence activating conditions in vitro analyzing 13 time points within a 6 h period (Kanjilal et al., 2010). ToxT is part of a positive forward feedback loop and therefore positively autoregulated (Yu and DiRita, 1999). ToxT proteolysis is one mechanism to terminate its activity, which is mediated by AAA+ proteases including Lon, ClpXP, and others (Abuaita and Withey, 2011). Proteolytic instability of ToxT is regulated via stressors like high temperature and alkaline pH, and depends on an unstructured region located at amino acid positions 100-109 (Abuaita and Withey, 2011; Thomson et al., 2015). Thereby, ToxT itself harbors a protease sensitive response domain, which may trigger proteolysis depending on the listed exogenous conditions.
The RpoE (σE or σ24)-dependent Stress Response Requires Proteolysis by DegS and RseP
The periplasmic protease/chaperone DegP and the membrane embedded proteases DegS and RseP are essential to react to environmental and cellular changes in Gram-negative bacteria (reviewed by Alba and Gross, 2004; Rowley et al., 2006). They are part of the envelope stress response mediated by the alternative sigma factor RpoE, first described in E. coli upon high temperature conditions, exposure to ethanol, or the overproduction of OMPs (Erickson and Gross, 1989; Wang and Kaguni, 1989; Mecsas et al., 1993). In absence of such stimuli, the N-terminal cytoplasmic portion of the integral membrane bound anti-sigma factor RseA captures RpoE, retaining it to the inner membrane (De Las Peñas et al., 1997; Missiakas et al., 1997; Campbell et al., 2003). The stepwise cleavage of RseA is characteristic of RIP (Ehrmann and Clausen, 2004). The site-1 protease DegS senses misfolded C-terminal portions of OMPs with its PDZ-domain (Walsh et al., 2003; Wilken et al., 2004). In V. cholerae, OmpU is the essential stress sensor for membrane damaging and misfolded periplasmic proteins, e.g., in presence of antimicrobial peptides to activate a RpoE-dependent resistance. The signal transduction is based on the exposure of OmpU C-terminal YDF motifs, which interact with the PDZ-domain of DegS and hence activate RpoE activity (Mathur et al., 2007). DegS is a member of the DegS/HtrA2-subfamily of oligomeric serine HtrA proteases that possesses an N-terminal transmembrane portion, a catalytic serine protease domain and a C-terminal PDZ-domain (Clausen et al., 2002). DegS is the only known protease targeting RseA in E. coli. Additionally, in V. cholerae DegS also finds the sulfide-thiol reduced ToxR as its substrate (Lembke et al., 2018). DegS is activated by the interaction between C-terminal OMP peptides and its PDZ-domain. Refolding of DegS is induced resulting in proteolytic activity and the cleavage of RseA at its periplasmic portion, which triggers a second cut by the site-2 protease RseP at the cytoplasmic portion of RseA. RseP is a zinc metalloprotease of the inner membrane, harboring highly conserved HEXXH and LDG motifs and a PDZ-domain (Rudner et al., 1999; Kanehara et al., 2001; Drew et al., 2002). Eventually, RpoE is liberated into the cytoplasm where it assembles into the RNA polymerase holoenzyme. The first identified RpoE-dependent promoter in V. cholerae is located upstream of the rpoErseABC operon and harbors high similarity with consensus sequences of RpoE-regulated promoters in E. coli (Kovacikova and Skorupski, 2002). Comparative microarray analyses of a rseA deletion strain and wild type (WT) indicate that degP is also under RpoE-control in V. cholerae (Ding et al., 2004). Additionally, in a degS deletion strain less RpoE is released from RseA and consequently the RpoE-response is decreased, resulting in significantly reduced transcription of degP compared to WT (Lembke et al., 2018).
DegP is a periplasmic heat-shock protein, which is highly conserved across species and can act as both chaperone and protease (Spiess et al., 1999). DegP belongs to the HtrA-family of PDZ-domain containing proteases (Kolmar et al., 1996; Krojer et al., 2008a). Its structure is formed by trimer subunits that assemble to proteolytically inactive hexamers (Krojer et al., 2002). In presence of unfolded protein substrates, active DegP builds up dodecamers or icosatetramers (Jiang et al., 2008; Krojer et al., 2008b).
The type II secretion system (T2SS) of V. cholerae, encoded by two different eps operons (Sandkvist, 2001), is required for the secretion of enzymes and cholera toxin into the environment (Korotkov et al., 2012). Interestingly, a deletion of eps genes causes outer membrane damage which in turn activates RpoE-dependent response (Sikora et al., 2007). Furthermore, RpoE is also responsible for expression of the T2SS in V. cholerae, essential for release of important effectors along the lifecycle like the CTX or biofilm adhesion factors (Zielke et al., 2014). Concordantly, rpoE deletion strains are significantly attenuated in the murine model compared to WT (Kovacikova and Skorupski, 2002).
The Alternative Sigma Factor RpoS (σS or σ38) Is Targeted by the ClpXP Protease
RpoS is a hallmark of a proteolysis-controlled regulator and was mainly characterized in E. coli to be responsible to counteract starvation conditions (Hengge-Aronis, 2002). Degradation of RpoS is under control of its specific proteolysis targeting factor, termed RssB (Muffler et al., 1996), which is activated by the kinase ArcB (Mika and Hengge, 2005) leading to a proteolytic complex comprising ClpXP, phosphorylated RssB, and RpoS (Becker et al., 1999; Zhou et al., 2001; Stüdemann et al., 2003). Anti-adaptor proteins, termed Ira (inhibitor of RssB activity) are identified to block RssB activity, thus stabilizing RpoS. Ira proteins (Battesti et al., 2013) respond to specific physiological stress conditions (Hryckowian et al., 2014), as well as to the accumulation of intracellular metabolites (Battesti et al., 2015). In V. cholerae the role of RpoS is less clear as a rpoS-mutant only exhibits minor defects in intestinal colonization (Yildiz and Schoolnik, 1998; Merrell et al., 2000). However, the “mucosal escape” of V. cholerae at late stages of the infection depends on RpoS regulated gene expression (Nielsen et al., 2006). This phenotype is marked by activation of chemotaxis and motility genes resulting in detachment from the mucosal surface and entrance into the gastrointestinal lumen (Nielsen et al., 2006). While flagellar motility is crucial to direct V. cholerae to the mucosal layer, the bacteria enter a non-motile state upon mucosal penetration (Liu et al., 2008). After infection progression, nutrients in the gut decline, thereby starvation and high cell density may trigger rpoS expression. RpoS activates chemotaxis and motility gene expression, subsequently resulting in the mucosal escape phenotype. Moreover, RpoS is responsible for biofilm dispersal in a hydrodynamic model (Müller et al., 2007) or for biofilm escape (Wurm et al., 2017).
Termination of the RpoS-program is less clear, but likely involves proteolytic control (Wurm et al., 2017). Under laboratory conditions, RpoS-levels can increase if bacteria are stressed by a shift from rich into poor nutrient conditions. The enhanced ppGpp concentrations activate rpoS transcription leading to high RpoS-levels, which in turn activate chemotaxis and motility gene expression (Wurm et al., 2017). Interestingly, as soon as ppGpp-levels decline, rpoS transcription stops and RpoS-proteolysis is subsequently activated by a RssB homolog and the ClpXP-protease complex (Wurm et al., 2017).
To date, rpoS expression control seems to be conserved in Enterobacteriacae and other Gram-negative bacteria (e.g., Vibrio and Pseudomonas). Stringent control, mRNA stability and ClpXP- or Lon-mediated proteolysis control rpoS expression levels. In contrast, RpoS-mediated responses are completely different in such species, leading to physiological changes like persistence, motility, and virulence (Schellhorn, 2014).
Concluding Remarks
While transcriptional regulation along the lifecycle of V. cholerae has been intensively studied, we are just beginning to identify and understand post-translational control elements. Recent reports highlight that regulated proteolysis of alternative sigma factors or transcriptional regulators is involved in blockage or termination of gene expression comprising virulence, transmission, and environmental persistence conditions.
Regulated proteolysis events during V. cholerae's lifecycle are summarized in Table 1. To conclude, FliA proteolysis ensures full virulence induction during initial colonization upon penetration of V. cholerae through the intestinal mucus. Meanwhile, the virulence cascade starts to operate, i.e., the ToxT positive forward loop regulation is activated. At some point during the end of the colonization phase and shortly before onset of mucosal escape, this virulence regulatory loop needs to be terminated via proteolysis of TcpP, ToxR, and ToxT (Figure 1 and Table 1). During colonization stage, RpoE is released via RIP of RseA and mediates host protective strategies. Regulated proteolysis maintains low RpoS levels during colonization, while starvation conditions inhibit RpoS proteolysis via RssB. Elevated RpoS levels at late infection stages subsequently initiate the mucosal escape mechanism.
Several important questions remain to be addressed to understand such complex post-translational regulation events. For example, what are the specific triggers initiating or preventing regulated proteolysis? What are the associated anti-proteolytic or targeting proteolysis factors, which protect proteins from degradation or attract key-proteases such as Lon, ClpXP, and DegS? With FlgM, RssB, RseA, ToxS, or TcpH probably only a fraction of such factors have been identified. Due to established intestinal infection and environmental persistence assays as well as its rapid proliferation requiring fast adaptation, V. cholerae is a valuable model to study and identify the proteolytic regulatory networks to gain deeper insights into the pathogen's bacterial physiology.
Statements
Author contributions
NP wrote parts of the RpoE chapter and RpoS chapter as well as designed Figure 1. ML wrote parts of the ToxR chapter. KP wrote parts of the introduction and FliA chapter. JM wrote the Tcp chapter. JR wrote parts of the RpoE, RpoS, ToxR chapter, and the ToxT chapter. SS was the coordinating author involved in writing and editing of all chapters as well as finalizing the manuscript for submission.
Funding
. The work was supported by the Austrian Science Fund (FWF) grants: W901 (DK Molecular Enzymology) to KP and SS, the doc.fund Molecular Metabolism to SS, P29405 to JR as well as P27654 to SS.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Abuaita B. H. Withey J. H. (2011). Termination of Vibrio cholerae virulence gene expression is mediated by proteolysis of the major virulence activator, ToxT. Mol. Microbiol.81, 1640–1653. 10.1111/j.1365-2958.2011.07798.x
2
Alba B. M. Gross C. A. (2004). Regulation of the Escherichia coli sigma-dependent envelope stress response. Mol. Microbiol.52, 613–619. 10.1111/j.1365-2958.2003.03982.x
3
Almagro-Moreno S. Kim T. K. Skorupski K. Taylor R. K. (2015a). Proteolysis of virulence regulator ToxR is associated with entry of Vibrio cholerae into a dormant state. PLoS Genet.11:e1005145. 10.1371/journal.pgen.1005145
4
Almagro-Moreno S. Root M. Z. Taylor R. K. (2015b). Role of ToxS in the proteolytic cascade of virulence regulator ToxR in Vibrio cholerae. Mol. Microbiol.98, 963–976. 10.1111/mmi.13170
5
Barembruch C. Hengge R. (2007). Cellular levels and activity of the flagellar sigma factor FliA of Escherichia coli are controlled by FlgM-modulated proteolysis. Mol. Microbiol.65, 76–89. 10.1111/j.1365-2958.2007.05770.x
6
Battesti A. Hoskins J. R. Tong S. Milanesio P. Mann J. M. Kravats A. et al . (2013). Anti-adaptors provide multiple modes for regulation of the RssB adaptor protein. Genes Dev.27, 2722–2735. 10.1101/gad.229617.113
7
Battesti A. Majdalani N. Gottesman S. (2015). Stress sigma factor RpoS degradation and translation are sensitive to the state of central metabolism. Proc. Natl. Acad. Sci. U.S.A.112, 5159–5164. 10.1073/pnas.1504639112
8
Beck N. A. Krukonis E. S. DiRita V. J. (2004). TcpH influences virulence gene expression in Vibrio cholerae by inhibiting degradation of the transcription activator TcpP. J. Bacteriol.186, 8309–8316. 10.1128/JB.186.24.8309-8316.2004
9
Becker G. Klauck E. Hengge-Aronis R. (1999). Regulation of RpoS proteolysis in Escherichia coli: the response regulator RssB is a recognition factor that interacts with the turnover element in RpoS. Proc. Natl. Acad. Sci. U.S.A.96, 6439–6444. 10.1073/pnas.96.11.6439
10
Bina J. Zhu J. Dziejman M. Faruque S. Calderwood S. Mekalanos J. (2003). ToxR regulon of Vibrio cholerae and its expression in vibrios shed by cholera patients. Proc. Natl. Acad. Sci. U.S.A.100, 2801–2806. 10.1073/pnas.2628026100
11
Brameyer S. Rösch T. C. El Andari J. Hoyer E. Schwarz J. Graumann P. L. et al . (2019). DNA-binding directs the localization of a membrane-integrated receptor of the ToxR family. Commun Biol2:4. 10.1038/s42003-018-0248-7
12
Burns D. L. Moss J. Vaughan M. (1983). Release of guanyl nucleotides from the regulatory subunit of adenylate cyclase. J. Biol. Chem.258, 1116–1120
13
Butler S. M. Camilli A. (2005). Going against the grain: chemotaxis and infection in Vibrio cholerae. Nat. Rev. Microbiol.3, 611–620. 10.1038/nrmicro1207
14
Campbell E. A. Tupy J. L. Gruber T. M. Wang S. Sharp M. M. Gross C. A. et al . (2003). Crystal structure of Escherichia coli sigmaE with the cytoplasmic domain of its anti-sigma RseA. Mol. Cell11, 1067–1078. 10.1016/S1097-2765(03)00148-5
15
Childers B. M. Klose K. E. (2007). Regulation of virulence in Vibrio cholerae: the ToxR regulon. Future Microbiol.2, 335–344. 10.2217/17460913.2.3.335
16
Clausen T. Southan C. Ehrmann M. (2002). The HtrA family of proteases: implications for protein composition and cell fate. Mol. Cell10, 443–455. 10.1016/S1097-2765(02)00658-5
17
Colwell R. R. Huq A. Islam M. S. Aziz K. M. Yunus M. Khan N. H. et al . (2003). Reduction of cholera in Bangladeshi villages by simple filtration. Proc. Natl. Acad. Sci. U.S.A.100, 1051–1055. 10.1073/pnas.0237386100
18
Correa N. E. Barker J. R. Klose K. E. (2004). The Vibrio cholerae FlgM homologue is an anti-sigma28 factor that is secreted through the sheathed polar flagellum. J. Bacteriol.186, 4613–4619. 10.1128/JB.186.14.4613-4619.2004
19
De Las Peñas A. Connolly L. Gross C. A. (1997). The sigmaE-mediated response to extracytoplasmic stress in Escherichia coli is transduced by RseA and RseB, two negative regulators of sigmaE. Mol. Microbiol.24, 373–385. 10.1046/j.1365-2958.1997.3611718.x
20
Ding Y. Davis B. M. Waldor M. K. (2004). Hfq is essential for Vibrio cholerae virulence and downregulates sigma expression. Mol. Microbiol.53, 345–35410.1111/j.1365-2958.2004.04142.x
21
DiRita V. J. Mekalanos J. J. (1991). Periplasmic interaction between two membrane regulatory proteins, ToxR and ToxS, results in signal transduction and transcriptional activation. Cell64, 29–3710.1016/0092-8674(91)90206-E
22
Drew D. Sjöstrand D. Nilsson J. Urbig T. Chin C. N. de Gier J. W. et al . (2002). Rapid topology mapping of Escherichia coli inner-membrane proteins by prediction and PhoA/GFP fusion analysis. Proc. Natl. Acad. Sci. U.S.A.99, 2690–2695. 10.1073/pnas.052018199
23
Ehrmann M. Clausen T. (2004). Proteolysis as a regulatory mechanism. Annu. Rev. Genet.38, 709–724. 10.1146/annurev.genet.38.072902.093416
24
Erickson J. W. Gross C. A. (1989). Identification of the sigma E subunit of Escherichia coli RNA polymerase: a second alternate sigma factor involved in high-temperature gene expression. Genes Dev.3, 1462–1471. 10.1101/gad.3.9.1462
25
Fan F. Liu Z. Jabeen N. Birdwell L. D. Zhu J. Kan B. (2014). Enhanced interaction of Vibrio cholerae virulence regulators TcpP and ToxR under oxygen-limiting conditions. Infect. Immun.82, 1676–1682. 10.1128/IAI.01377-13
26
Fengler V. H. Boritsch E. C. Tutz S. Seper A. Ebner H. Roier S. et al . (2012). Disulfide bond formation and ToxR activity in Vibrio cholerae. PLoS ONE7:e47756. 10.1371/journal.pone.0047756
27
Fishmann P. H. (1990). Mechanism of action of cholera toxin, in ADP-Ribosylating Toxins and G Proteins, eds MossJ.VaughanM. (Washington, DC: American Society for Microbiology, 127–137.
28
Freter R. Jones G. W. (1976). Adhesive properties of Vibrio cholerae: nature of the interaction with intact mucosal surfaces. Infect. Immun.14, 246–256.
29
Freter R. O'Brien P. C. (1981). Role of chemotaxis in the association of motile bacteria with intestinal mucosa: chemotactic responses of Vibrio cholerae and description of motile nonchemotactic mutants. Infect. Immun.34, 215–221.
30
Goss T. J. Morgan S. J. French E. L. Krukonis E. S. (2013). ToxR recognizes a direct repeat element in the toxT, ompU, ompT, and ctxA promoters of Vibrio cholerae to regulate transcription. Infect. Immun.81, 884–895. 10.1128/IAI.00889-12
31
Goss T. J. Seaborn C. P. Gray M. D. Krukonis E. S. (2010). Identification of the TcpP-binding site in the toxT promoter of Vibrio cholerae and the role of ToxR in TcpP-mediated activation. Infect. Immun.78, 4122–4133. 10.1128/IAI.00566-10
32
Hall-Stoodley L. Stoodley P. (2005). Biofilm formation and dispersal and the transmission of human pathogens. Trends Microbiol.13, 7–10. 10.1016/j.tim.2004.11.004
33
Häse C. C. Mekalanos J. J. (1998). TcpP protein is a positive regulator of virulence gene expression in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A.95, 730–734. 10.1073/pnas.95.2.730
34
Hengge-Aronis R. (2002). Signal transduction and regulatory mechanisms involved in control of the sigma(S) (RpoS) subunit of RNA polymerase. Microbiol. Mol. Biol. Rev.66, 373–395. 10.1128/MMBR.66.3.373-395.2002
35
Herrington D. A. Hall R. H. Losonsky G. Mekalanos J. J. Taylor R. K. Levine M. M. (1988). Toxin, Toxin-coregulated Pili and the toxR regulation are essential for Vibrio cholerae pathogenesis in humans. J. Exp. Med.168, 1487–1492. 10.1084/jem.168.4.1487
36
Higgins D. E. DiRita V. J. (1994). Transcriptional control of toxT, a regulatory gene in the ToxR regulon of Vibrio cholerae. Mol. Microbiol.14, 17–29. 10.1111/j.1365-2958.1994.tb01263.x
37
Hryckowian A. J. Battesti A. Lemke J. J. Meyer Z. C. Welch R. A. (2014). IraL is an RssB anti-adaptor that stabilizes RpoS during logarithmic phase growth in Escherichia coli and Shigella. MBio5:e01043–14. 10.1128/mBio.01043-14
38
Iwanaga M. Yamamoto K. (1985). New medium for the production of cholera toxin by Vibrio cholerae O1 biotype El Tor. J. Clin. Microbiol.22, 405–408.
39
Iwanaga M. Yamamoto K. Higa N. Ichinose Y. Nakasone N. Tanabe M. (1986). Culture conditions for stimulating cholera toxin production by Vibrio cholerae O1 El Tor. Microbiol. Immunol.30, 1075–1083. 10.1111/j.1348-0421.1986.tb03037.x
40
Jiang J. Zhang X. Chen Y. Wu Y. Zhou Z. H. Chang Z. et al . (2008). Activation of DegP chaperone-protease via formation of large cage-like oligomers upon binding to substrate proteins. Proc. Natl. Acad. Sci. U.S.A.105, 11939–11944. 10.1073/pnas.0805464105
41
Kanehara K. Akiyama Y. Ito K. (2001). Characterization of the yaeL gene product and its S2P-protease motifs in Escherichia coli. Gene281, 71–79. 10.1016/S0378-1119(01)00823-X
42
Kanjilal S. Citorik R. LaRocque R. C. Ramoni M. F. Calderwood S. B. (2010). A systems biology approach to modeling Vibrio cholerae gene expression under virulence-inducing conditions. J. Bacteriol.192, 4300–4310. 10.1128/JB.00182-10
43
Kolmar H. Waller P. R. Sauer R. T. (1996). The DegP and DegQ periplasmic endoproteases of Escherichia coli: specificity for cleavage sites and substrate conformation. J. Bacteriol.178, 5925–5929. 10.1128/jb.178.20.5925-5929.1996
44
Korotkov K. V. Sandkvist M. Hol W. G. (2012). The type II secretion system: biogenesis, molecular architecture and mechanism. Nat. Rev. Microbiol.10, 336–351. 10.1038/nrmicro2762
45
Kovacikova G. Skorupski K. (1999). A Vibrio cholerae LysR homolog, AphB, cooperates with AphA at the tcpPH promoter to activate expression of the ToxR virulence cascade. J. Bacteriol.181, 4250–4256.
46
Kovacikova G. Skorupski K. (2002). The alternative sigma factor sigma(E) plays an important role in intestinal survival and virulence in Vibrio cholerae. Infect. Immun.70, 5355–5362. 10.1128/IAI.70.10.5355-5362.2002
47
Krojer T. Garrido-Franco M. Huber R. Ehrmann M. Clausen T. (2002). Crystal structure of DegP (HtrA) reveals a new protease-chaperone machine. Nature416, 455–459. 10.1038/416455a
48
Krojer T. Pangerl K. Kurt J. Sawa J. Stingl C. Mechtler K. et al . (2008a). Interplay of PDZ and protease domain of DegP ensures efficient elimination of misfolded proteins. Proc. Natl. Acad. Sci. U.S.A.105, 7702–7707. 10.1073/pnas.0803392105
49
Krojer T. Sawa J. Schäfer E. Saibil H. R. Ehrmann M. Clausen T. (2008b). Structural basis for the regulated protease and chaperone function of DegP. Nature453, 885–890. 10.1038/nature07004
50
Krukonis E. S. DiRita V. J. (2003). DNA binding and ToxR responsiveness by the wing domain of TcpP, an activator of virulence gene expression in Vibrio cholerae. Mol. Cell12, 157–165. 10.1016/S1097-2765(03)00222-3
51
Lee S. H. Butler S. M. Camilli A. (2001). Selection for in vivo regulators of bacterial virulence. Proc. Natl. Acad. Sci. U.S.A.98, 6889–6894. 10.1073/pnas.111581598
52
Lee S. H. Hava D. L. Waldor M. K. Camilli A. (1999). Regulation and temporal expression patterns of Vibrio cholerae virulence genes during infection. Cell99, 625–634. 10.1016/S0092-8674(00)81551-2
53
Lembke M. Pennetzdorfer N. Tutz S. Koller M. Vorkapic D. Zhu J. et al . (2018). Proteolysis of ToxR is controlled by cysteine-thiol redox state and bile salts in Vibrio cholerae. Mol. Microbiol. 10, 796–810. 10.1111/mmi.14125
54
Li C. C. Crawford J. A. DiRita V. J. Kaper J. B. (2000). Molecular cloning and transcriptional regulation of ompT, a ToxR-repressed gene in Vibrio cholerae. Mol. Microbiol.35, 189–203. 10.1046/j.1365-2958.2000.01699.x
55
List C. Grutsch A. Radler C. Cakar F. Zingl F. G. Schild-Prüfert K. et al . (2018). Genes Activated by Vibrio cholerae upon exposure to Caenorhabditis elegans reveal the mannose-sensitive hemagglutinin to be essential for colonization. mSphere3:e00238–18. 10.1128/mSphereDirect.00238-18
56
Liu Z. Miyashiro T. Tsou A. Hsiao A. Goulian M. Zhu J. (2008). Mucosal penetration primes Vibrio cholerae for host colonization by repressing quorum sensing. Proc. Natl. Acad. Sci. U.S.A.105, 9769–9774. 10.1073/pnas.0802241105
57
Lutz C. Erken M. Noorian P. Sun S. McDougald D. (2013). Environmental reservoirs and mechanisms of persistence of Vibrio cholerae. Front. Microbiol.4:375. 10.3389/fmicb.2013.00375
58
Mahmoud S. A. Chien P. (2018). Regulated proteolysis in bacteria. Annu. Rev. Biochem.87, 677–696. 10.1146/annurev-biochem-062917-012848
59
Martínez-Hackert E. Stock A. M. (1997). Structural relationships in the OmpR family of winged-helix transcription factors. J. Mol. Biol.269, 301–312. 10.1006/jmbi.1997.1065
60
Mathur J. Davis B. M. Waldor M. K. (2007). Antimicrobial peptides activate the Vibrio cholerae sigmaE regulon through an OmpU-dependent signalling pathway. Mol. Microbiol.63, 848–858. 10.1111/j.1365-2958.2006.05544.x
61
Matson J. S. DiRita V. J. (2005). Degradation of the membrane-localized virulence activator TcpP by the YaeL protease in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A.102, 16403–16408. 10.1073/pnas.0505818102
62
Matson J. S. Withey J. H. DiRita V. J. (2007). Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect. Immun.75, 5542–5549. 10.1128/IAI.01094-07
63
Mecsas J. Rouviere P. E. Erickson J. W. Donohue T. J. Gross C. A. (1993). The activity of Sigma(E), an Escherichia coli heat-inducible sigma-factor, is modulated by expression of outer-membrane proteins. Genes Dev.7, 2618–2628. 10.1101/gad.7.12b.2618
64
Merrell D. S. Tischler A. D. Lee S. H. Camilli A. (2000). Vibrio cholerae requires rpoS for efficient intestinal colonization. Infect. Immun.68, 6691–6696. 10.1128/IAI.68.12.6691-6696.2000
65
Midgett C. R. Almagro-Moreno S. Pellegrini M. Taylor R. K. Skorupski K. Kull F. J. (2017). Bile salts and alkaline pH reciprocally modulate the interaction between the periplasmic domains of Vibrio cholerae ToxR and ToxS. Mol. Microbiol.105, 258–272. 10.1111/mmi.13699
66
Mika F. Hengge R. (2005). A two-component phosphotransfer network involving ArcB, ArcA, and RssB coordinates synthesis and proteolysis of sigmaS (RpoS) in E. coli. Genes Dev.19, 2770–2781. 10.1101/gad.353705
67
Miller V. L. Mekalanos J. J. (1988). A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J. Bacteriol.170, 2575–2583. 10.1128/jb.170.6.2575-2583.1988
68
Miller V. L. Taylor R. K. Mekalanos J. J. (1987). Cholera toxin transcriptional activator toxR is a transmembrane DNA binding protein. Cell48, 271–279. 10.1016/0092-8674(87)90430-2
69
Missiakas D. Mayer M. P. Lemaire M. Georgopoulos C. Raina S. (1997). Modulation of the Escherichia coli sigmaE (RpoE) heat-shock transcription-factor activity by the RseA, RseB and RseC proteins. Mol. Microbiol.24, 355–371. 10.1046/j.1365-2958.1997.3601713.x
70
Morgan S. J. French E. L. Thomson J. J. Seaborn C. P. Shively C. A. Krukonis E. S. (2016). Formation of an intramolecular periplasmic disulfide bond in TcpP protects TcpP and TcpH from degradation in Vibrio cholerae. J. Bacteriol.198, 498–509. 10.1128/JB.00338-15
71
Muffler A. Fischer D. Altuvia S. Storz G. Hengge-Aronis R. (1996). The response regulator RssB controls stability of the sigma(S) subunit of RNA polymerase in Escherichia coli. EMBO J.15, 1333–1339. 10.1002/j.1460-2075.1996.tb00475.x
72
Müller J. Miller M. C. Nielsen A. T. Schoolnik G. K. Spormann A. M. (2007). vpsA- and luxO-independent biofilms of Vibrio cholerae. FEMS Microbiol. Lett.275, 199–206. 10.1111/j.1574-6968.2007.00884.x
73
Ng W. L. Bassler B. L. (2009). Bacterial quorum-sensing network architectures. Annu. Rev. Genet.43, 197–222. 10.1146/annurev-genet-102108-134304
74
Nielsen A. T. Dolganov N. A. Otto G. Miller M. C. Wu C. Y. Schoolnik G. K. (2006). RpoS controls the Vibrio cholerae mucosal escape response. PLoS Pathog.2:e109. 10.1371/journal.ppat.0020109
75
Ottemann K. M. Mekalanos J. J. (1996). The ToxR protein of Vibrio cholerae forms homodimers and heterodimers. J. Bacteriol.178, 156–162. 10.1128/jb.178.1.156-162.1996
76
Plecha S. C. Withey J. H. (2015). Mechanism for inhibition of Vibrio cholerae ToxT activity by the unsaturated fatty acid components of bile. J. Bacteriol.197, 1716–1725. 10.1128/JB.02409-14
77
Pressler K. Vorkapic D. Lichtenegger S. Malli G. Barilich B. P. Cakar F. et al . (2016). AAA+ proteases and their role in distinct stages along the Vibrio cholerae lifecycle. Int. J. Med. Microbiol.306, 452–462. 10.1016/j.ijmm.2016.05.013
78
Provenzano D. Klose K. E. (2000). Altered expression of the ToxR-regulated porins OmpU and OmpT diminishes Vibrio cholerae bile resistance, virulence factor expression, and intestinal colonization. Proc. Natl. Acad. Sci. U.S.A.97, 10220–10224. 10.1073/pnas.170219997
79
Provenzano D. Schuhmacher D. A. Barker J. L. Klose K. E. (2000). The virulence regulatory protein ToxR mediates enhanced bile resistance in Vibrio cholerae and other pathogenic Vibrio species. Infect. Immun.68, 1491–1497. 10.1128/IAI.68.3.1491-1497.2000
80
Rowley G. Spector M. Kormanec J. Roberts M. (2006). Pushing the envelope: extracytoplasmic stress responses in bacterial pathogens. Nat. Rev. Microbiol.4, 383–394. 10.1038/nrmicro1394
81
Rudner D. Z. Fawcett P. Losick R. (1999). A family of membrane-embedded metalloproteases involved in regulated proteolysis of membrane-associated transcription factors. Proc. Natl. Acad. Sci. U.S.A.96, 14765–14770. 10.1073/pnas.96.26.14765
82
Sandkvist M. (2001). Biology of type II secretion. Mol. Microbiol.40, 271–283. 10.1046/j.1365-2958.2001.02403.x
83
Schellhorn H. E. (2014). Elucidating the function of the RpoS regulon. Future Microbiol.9, 497–507. 10.2217/fmb.14.9
84
Schild S. Tamayo R. Nelson E. J. Qadri F. Calderwood S. B. Camilli A. (2007). Genes induced late in infection increase fitness of Vibrio cholerae after release into the environment. Cell Host Microbe2, 264–277. 10.1016/j.chom.2007.09.004
85
Seper A. Fengler V. H. Roier S. Wolinski H. Kohlwein S. D. Bishop A. L. et al . (2011). Extracellular nucleases and extracellular DNA play important roles in Vibrio cholerae biofilm formation. Mol. Microbiol.82, 1015–1037. 10.1111/j.1365-2958.2011.07867.x
86
Sharp G. W. Hynie S. Lipson L. C. Parkinson D. K. (1971). Action of cholera toxin to stimulate adenyl cyclase. Trans. Assoc. Am. Physicians84, 200–211.
87
Sikora A. E. Lybarger S. R. Sandkvist M. (2007). Compromised outer membrane integrity in Vibrio cholerae Type II secretion mutants. J. Bacteriol.189, 8484–8495. 10.1128/JB.00583-07
88
Skorupski K. Taylor R. K. (1999). A new level in the Vibrio cholerae ToxR virulence cascade: AphA is required for transcriptional activation of the tcpPH operon. Mol. Microbiol.31, 763–771. 10.1046/j.1365-2958.1999.01215.x
89
Spiess C. Beil A. Ehrmann M. (1999). A temperature-dependent switch from chaperone to protease in a widely conserved heat shock protein. Cell97, 339–347. 10.1016/S0092-8674(00)80743-6
90
Stüdemann A. Noirclerc-Savoye M. Klauck E. Becker G. Schneider D. Hengge R. (2003). Sequential recognition of two distinct sites in sigma(S) by the proteolytic targeting factor RssB and ClpX. EMBO J.22, 4111–4120. 10.1093/emboj/cdg411
91
Syed K. A. Beyhan S. Correa N. Queen J. Liu J. Peng F. et al . (2009). The Vibrio cholerae flagellar regulatory hierarchy controls expression of virulence factors. J. Bacteriol.191, 6555–6570. 10.1128/JB.00949-09
92
Tamayo R. Patimalla B. Camilli A. (2010). Growth in a biofilm induces a hyperinfectious phenotype in Vibrio cholerae. Infect. Immun.78, 3560–3569. 10.1128/IAI.00048-10
93
Taylor R. K. Miller V. L. Furlong D. B. Mekalanos J. J. (1987). Use of phoA gene fusions to identify a pilus colonization factor coordinately regulated with cholera toxin. Proc. Natl. Acad. Sci. U.S.A.84, 2833–2837. 10.1073/pnas.84.9.2833
94
Teoh W. P. Matson J. S. DiRita V. J. (2015). Regulated intramembrane proteolysis of the virulence activator TcpP in Vibrio cholerae is initiated by the tail-specific protease (Tsp). Mol. Microbiol.97, 822–831. 10.1111/mmi.13069
95
Teschler J. K. Zamorano-Sánchez D. Utada A. S. Warner C. J. Wong G. C. Linington R. G. et al . (2015). Living in the matrix: assembly and control of Vibrio cholerae biofilms. Nat. Rev. Microbiol.13, 255–268. 10.1038/nrmicro3433
96
Thomson J. J. Plecha S. C. Withey J. H. (2015). A small unstructured region in Vibrio cholerae ToxT mediates the response to positive and negative effectors and ToxT proteolysis. J. Bacteriol.197, 654–668. 10.1128/JB.02068-14
97
Thomson J. J. Withey J. H. (2014). Bicarbonate increases binding affinity of Vibrio cholerae ToxT to virulence gene promoters. J. Bacteriol.196, 3872–3880. 10.1128/JB.01824-14
98
Walsh N. P. Alba B. M. Bose B. Gross C. A. Sauer R. T. (2003). OMP peptide signals initiate the envelope-stress response by activating DegS protease via relief of inhibition mediated by its PDZ domain. Cell113, 61–71. 10.1016/S0092-8674(03)00203-4
99
Wang Q. P. Kaguni J. M. (1989). A novel sigma-factor is involved in expression of the rpoh gene of Escherichia coli. J. Bacteriol.171, 4248–4253. 10.1128/jb.171.8.4248-4253.1989
100
Weber G. G. Klose K. E. (2011). The complexity of ToxT-dependent transcription in Vibrio cholerae. Indian J. Med. Res.133, 201–206.
101
Weber G. G. Kortmann J. Narberhaus F. Klose K. E. (2014). RNA thermometer controls temperature-dependent virulence factor expression in Vibrio cholerae. Proc. Natl. Acad. Sci. U.S.A.111, 14241–14246. 10.1073/pnas.1411570111
102
Wilken C. Kitzing K. Kurzbauer R. Ehrmann M. Clausen T. (2004). Crystal structure of the DegS stress sensor: how a PDZ domain recognizes misfolded protein and activates a protease. Cell117, 483–494. 10.1016/S0092-8674(04)00454-4
103
Wurm P. Tutz S. Mutsam B. Vorkapic D. Heyne B. Grabner C. et al . (2017). Stringent factor and proteolysis control of sigma factor RpoS expression in Vibrio cholerae. Int. J. Med. Microbiol.307, 154–165. 10.1016/j.ijmm.2017.01.006
104
Yang M. Liu Z. Hughes C. Stern A. M. Wang H. Zhong Z. et al . (2013). Bile salt-induced intermolecular disulfide bond formation activates Vibrio cholerae virulence. Proc. Natl. Acad. Sci. U.S.A.110, 2348–2353. 10.1073/pnas.1218039110
105
Yildiz F. H. Schoolnik G. K. (1998). Role of rpoS in stress survival and virulence of Vibrio cholerae. J. Bacteriol.180, 773–784.
106
Yildiz F. H. Visick K. L. (2009). Vibrio biofilms: so much the same yet so different. Trends Microbiol.17, 109–118. 10.1016/j.tim.2008.12.004
107
Yu R. R. DiRita V. J. (1999). Analysis of an autoregulatory loop controlling ToxT, cholera toxin, and toxin-coregulated pilus production in Vibrio cholerae. J. Bacteriol.181, 2584–2592.
108
Zhou Y. Gottesman S. Hoskins J. R. Maurizi M. R. Wickner S. (2001). The RssB response regulator directly targets sigma(S) for degradation by ClpX. Genes Dev.15, 627–637. 10.1101/gad.864401
109
Zielke R. A. Simmons R. S. Park B. R. Nonogaki M. Emerson S. Sikora A. E. (2014). The type II secretion pathway in Vibrio cholerae is characterized by growth phase-dependent expression of exoprotein genes and is positively regulated by sigmaE. Infect. Immun.82, 2788–2801. 10.1128/IAI.01292-13
Summary
Keywords
post-translational regulation, stressor, Lon, Clp, DegS, DegP YaeL, tail-specific protease
Citation
Pennetzdorfer N, Lembke M, Pressler K, Matson JS, Reidl J and Schild S (2019) Regulated Proteolysis in Vibrio cholerae Allowing Rapid Adaptation to Stress Conditions. Front. Cell. Infect. Microbiol. 9:214. doi: 10.3389/fcimb.2019.00214
Received
26 March 2019
Accepted
03 June 2019
Published
21 June 2019
Volume
9 - 2019
Edited by
Vincenzo Scarlato, University of Bologna, Italy
Reviewed by
Torsten Waldminghaus, University of Marburg, Germany; Eric Krukonis, University of Detroit Mercy, United States
Updates
Copyright
© 2019 Pennetzdorfer, Lembke, Pressler, Matson, Reidl and Schild.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefan Schild stefan.schild@uni-graz.at
This article was submitted to Molecular Bacterial Pathogenesis, a section of the journal Frontiers in Cellular and Infection Microbiology
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.