 Zhewei Sun1,2,3†Danying Zhou1,3†
Zhewei Sun1,2,3†Danying Zhou1,3† Xueya Zhang1,2,3Qiaoling Li1,2,3Hailong Lin1,2,3Wei Lu1,3Hongmao Liu1,2,3
Xueya Zhang1,2,3Qiaoling Li1,2,3Hailong Lin1,2,3Wei Lu1,3Hongmao Liu1,2,3 Junwan Lu1,3
Junwan Lu1,3 Xi Lin1,3
Xi Lin1,3 Kewei Li1,3
Kewei Li1,3 Teng Xu4
Teng Xu4 Qiyu Bao1,3*Hailin Zhang2,3*
Qiyu Bao1,3*Hailin Zhang2,3*- 1Key Laboratory of Medical Genetics of Zhejiang Province, Key Laboratory of Laboratory Medicine, Ministry of Education, School of Laboratory Medicine and Life Sciences, Wenzhou Medical University, Wenzhou, China
- 2The Second Affiliated Hospital and Yuying Children's Hospital, Wenzhou Medical University, Wenzhou, China
- 3Institute of Biomedical Informatics, Wenzhou Medical University, Wenzhou, China
- 4Institute of Translational Medicine, Baotou Central Hospital, Baotou, China
Staphylococcus caprae, Staphylococcus capitis, and Staphylococcus epidermidis belong to the “Epidermidis Cluster Group” (ECG) and are generally opportunistic pathogens. In this work, whole genome sequencing, molecular cloning and pan-genome analysis were performed to investigate the genetic characteristics of the resistance, virulence and genome structures of 69 ECG strains, including a clinical isolate (S. caprae SY333) obtained in this work. Two resistance genes (blaZ and aadD2) encoded on the plasmids pSY333-41 and pSY333-45 of S. caprae SY333 were confirmed to be functional. The bla region in ECG exhibited three distinct structures, and these chromosome- and plasmid-encoded bla operons seemed to follow two different evolutionary paths. Pan-genome analysis revealed their pan-genomes tend to be “open.” For the virulence-related factors, the genes involved in primary attachment were observed almost exclusively in S. epidermidis, while the genes associated with intercellular aggregation were observed more frequently in S. caprae and S. capitis. The type VII secretion system was present in all strains of S. caprae and some of S. epidermidis but not in S. capitis. Moreover, the isd locus (iron regulated surface determinant) was first found to be encoded on the genomes of S. caprae and S. capitis. These findings suggested that the plasmid and chromosome encoded bla operons of ECG species underwent different evolution paths, as well as they differed in the abundance of virulence genes associated with adherence, invasion, secretion system and immune evasion. Identification of isd loci in S. caprae and S. capitis indicated their ability to acquire heme as nutrient iron during infection.
Introduction
Coagulase-negative staphylococci (CoNS) commonly live on the human skin (Piette and Verschraegen, 2009; Becker et al., 2014). They often caused infectious diseases in specific groups of patients, such as those with neonates, neutropenia, and so on (Ma et al., 2011; Zong et al., 2011), and infections at metastatic sites, such as joints, heart and bones. The infections in these populations are often not easy to treat (Casey et al., 2007). The virulence properties of CoNS species are mainly related to their ability to form biofilms and produce colonizing biomaterials (Becker et al., 2014). At present, these species are considered as important bloodstream pathogens usually with multidrug resistance (May et al., 2014). As a member of CoNS, Staphylococcus caprae commonly colonizes the milk gland and skin of goats and occasionally causes goat mastitis (Watanabe et al., 2018). In addition, S. caprae causes human infections, such as acute otitis externa (Shuttleworth et al., 1997), peritonitis (Shin et al., 2011), urinary tract diseases (Kanda et al., 1991), endocarditis (Vandenesch et al., 1995), meningitis (Benedetti et al., 2008), and many cases of bacteremia. However, the reasons why S. caprae can cause hospital-acquired infections haven't been fully elucidated.
Based on the 16S rRNA, tuf (elongation factor Tu), rpoB (β-subunit of RNA polymerase) and dnaJ (heat shock protein 40) genes (Lamers et al., 2012), Lamers et al. proposed a new classification and classified the Staphylococcus species into 15 cluster groups. This finding revealed that S. caprae was a member of“Epidermidis Cluster Group” (ECG) with S. epidrmidis as the leading causative organism which included S. caprae, S. saccharolyticus, S. epidermidis, S. capitis subsp. urealyticus and S. capitis subsp. capitis. The ECG species is composed of the “medium”-pathogenic staphylococci which means that when they are isolated from clinical specimens, it's uncertain that whether the infections are really caused by them (Becker et al., 2014). As S. caprae, S. capitis and S. epidermidis all belong to ECG, they are thought to share the basic mechanisms causing various hospital-acquired infections (Watanabe et al., 2018). Therefore, evaluating genomic structure divergences in virulence factors and metabolism is required to elucidate the mechanism of infections caused by ECG.
The complete set of conserved genes in all studied strains were defined as the core genome, while the accessory genome represents the genes existing in part of the strains, and the pan-genome comprises all genes in the core genome and accessory genome (Nourdin-Galindo et al., 2017; Wu et al., 2018). The pan-genome reflects the diversity among the species, host/environment adaptations as well as the variety of pathogenic mechanisms (Tettelin et al., 2008). In pan-genome analysis, unlike classical comparative genome analysis, all the genes were encompassed in a studied phylogenetic clade or a certain species (Chen et al., 2018) and will provide a better understanding of ECG genome diversity and virulence capabilities.
In this work, the whole genome sequence of a clinical S. caprae strain isolated from a puncture fluid specimen was determined. Furthermore, a genomic comparison among recently available ECGs was performed. Specifically, we analyzed the pan-genomes of each ECG species, as well as the virulence factors, and for the first time, we identified novel isd loci (iron uptake system allowing bacteria to steal iron from host heme) present in S. caprae and S. capitis. These genomic analyses will clarify the genomic differences, evolutionary relationships and pathogenic potentials of the strains.
Materials and Methods
Bacterial Strains, Genome Sequencing, Gene Predictions, and Functional Annotations
S. caprae SY333 was isolated from a puncture fluid specimen of a 29-year-old woman with fever in Lishui Hospital, Zhejiang, China. The strain was identified by Vitek-60 microorganism autoanalysis system (BioMerieux Corporate, Craponne, France), and then verified by homologous comparisons of 16S rRNA gene of S. caprae SY333 with those of the bacteria available in the nucleotide database of the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov). Finally, the average nucleotide identity (ANI) was calculated to confirm the result.
The AxyPrep Bacterial Genomic DNA Miniprep kit (Axygen Scientific, Union City, CA, United States) was used to extract the genomic DNA of S. caprae SY333. Sequencing of the S. caprae SY333 genome was performed by a combination of technologies producing short (HiSeq 2500; Illumina) and long reads (MinION and PacBio RS II; Oxford Nanopore Technologies and Pacific Biosciences). Canu v1.8 (Koren et al., 2017) was used for initial assembly, and a hybrid assembly was subsequently performed using the Unicycler pipeline (Wick et al., 2017), with the contigs generated by Canu and all the sequenced reads (short and long reads) served as an input. The cyclization of final contigs was confirmed through the built-in tools of the unicycler. Other publicly available ECG genome sequences including 24 full genomes, and 44 draft genomes were downloaded from the NCBI public database (Table 1). Genes were predicted and annotated by using Prokka v1.14.0 (Seemann, 2014); furthermore, the predicted proteins were searched against the NCBI non-redundant (NCBI Resource Coordinators, 2016) and Swiss-Prot (UniProt Consortium, 2015) databases (Tatusov et al., 2003) using DIAMOND (Buchfink et al., 2015) with an e-value threshold of 1e-5. Annotation of the resistance genes was performed using ResFinder (Zankari et al., 2012) and Resistance Gene Identifier (RGI) software of Comprehensive Antibiotic Resistance Database version 4.0.3 (https://card.mcmaster.ca/) (McArthur et al., 2013) with an e-value threshold of 1e-10. CrisprCasFinder was employed to identify CRISPR/Cas system (Couvin et al., 2018).
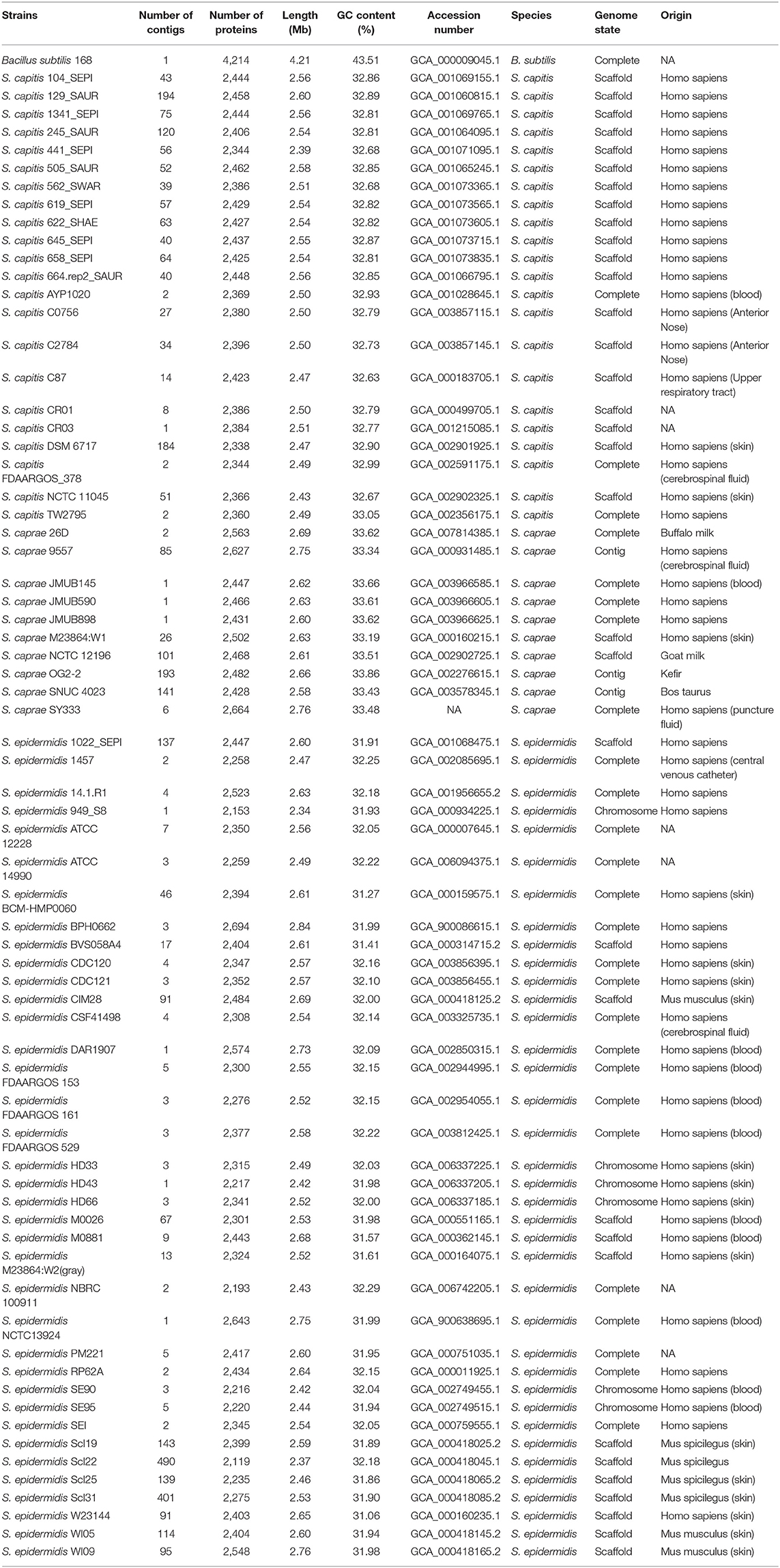
Table 1. Genomes used for phylogenetic and pan-genome analyses in this study.
Antimicrobial Susceptibility Testing and Cloning Experiments
The minimum inhibitory concentration (MIC) was detected by agar dilution method recommended by Clinical and Laboratory Standards Institute (CLSI). The result was interpreted following the CLSI breakpoint criteria for Staphylococcus (CLSI, 2019). Enterococcus faecalis ATCC 29212 and Escherichia coli ATCC 25922 were used as reference strains for quality control. The resistance gene sequences (aadD2 and blaZ) along with their promoter regions were PCR-amplified using the primers 5'-GCTCTAGAGCTTTCTATTATTGCAATGTGGAATTG-3' and 5'-CGGGATCCCGTCAAAATGGTATGCGTTTTGACACA-3' for aadD2, and 5'-CGGGATCCCGATTTAGCCATTTTGACACCTTCTTT-3' and 5'-CCAAGCTTGGTTAAAATTCCTTCATTACACTCTTGGCG-3' for blaZ, with each having a pair of flanking restriction endonuclease adapters (Xball and BamHI for aadD2, and BamHI and HindIII for blaZ). The PCR products were then eluted from agarose gel, digested with the corresponding restriction endonucleases, and ligated into the pAM401 and pUCP24 vectors, respectively. The recombinant plasmid (pAM401-aadD2) was transformed into E. faecalis JH2-2 via the calcium chloride method, and the transformants were cultured on brain heart infusion agar plates with chloramphenicol (16 μg/mL). The recombinant plasmid (pUCP24-blaZ) was extracted and further transformed into E. coli DH5α. The cloned resistance gene was verified by restriction endonuclease digestion and sequencing (Shanghai Sunny Biotechnology Co., Ltd., Shanghai, China). All plasmids and strains of this work were shown in Table 2.

Table 2. Bacteria and plasmids used in antimicrobial susceptibility testing and cloning experiments.
Phylogenetic and Phylogenomic Analyses
The ubiquitous, conserved single-copy genes, including atpD, recA, gyrA, gyrB, and ftsZ (Nourdin-Galindo et al., 2017; Chen et al., 2019), from each strain were used for phylogenetic analysis. Bacillus subtilis 168 was used as the outgroup. Initially, the nucleotide sequences of each gene was translated and then concatenated by a custom-derived shell script; Multi-FASTA alignment was performed using MAFFT v7.407 (Katoh and Standley, 2013), and the resulting alignment was used to infer the phylogeny by the maximum likelihood algorithm (ML) using RAxML version 8.2.12 (Stamatakis, 2014) under the substitution matrix LG, which was selected by ProtTest version 3.4 (Darriba et al., 2011).
For further verification, phylogenomic analysis (Comas et al., 2007) was conducted using 764 single-copy orthologous genes existing in all 69 ECG strains as well as Bacillus subtilis 168. These single-copy orthologous genes were identified using Orthofinder version 2.3.8 (Emms and Kelly, 2019). The methods of Multi-FASTA alignment and phylogeny inference were the same as those mentioned above.
Pan-Genome Inference and COG Functional Characterization
The pan-genomes of each ECG species were inferred with Roary version 3.12.0 (Page et al., 2015). The annotation files generated by Prokka were provided to Roary as an input. The gene presence/absence matrix produced by Roary was listed in Supplementary Table 1, and further analysis was based on this file. The gene accumulation curve was produced via ggplot2 (Wickham, 2009) using the results of Roary. COG categorization of each pan-genome was carried out using DIAMOND, and reference gene sequences provided by Roary were searched against the COG database. Only those hits with an e-value <1e-10, an identity higher than 40% and a coverage higher than 70% were considered significant (Nourdin-Galindo et al., 2017).
Identification of the Virulence Genes in ECG Genomes
BLASTX program was used to search all coding sequences of ECG strains against Virulence Factor Database (VFDB) (http://www.mgc.ac.cn/VFs/) (Chen et al., 2005). Compared with the virulent genes in the database at an e-value <1e-10, only those query genes with an identity higher than 40% and a coverage higher than 70% were considered as the potential virulence genes (Nourdin-Galindo et al., 2017). Functional annotations were performed based on the categories and subcategories presented in VFDB.
Sequence Analysis of bla Region and isd Locus
Only the complete ECG genome sequences were selected to perform comparative genomic analyses of the bla region and isd locus. Nucleotide sequence alignment and construction of the neighbor-joining phylogenetic tree of the bla operon were performed using the MAFFT program and MEGAX (Kumar et al., 2018) with a bootstrap of 1,000 replicates. Since the bla regions belonging to the same structure are similar to each other, comparative genomics analysis was performed using several representative sequences of these three structures (Figure 2), and the results were visualized via the gggenes package in Rstudio. The near-iron transporter (NEAT) domains and secretion signal of isd proteins were predicted online using InterProScan software by searching against the InterPro database (Hunter et al., 2009). Typical features including a signal peptide (predicted using the SignalP server at www.cbs.dtu.dk/services/SignalP/) at the N terminus, the LPXTG-motif (identified using a custom-derived script written in Python) close to the C terminus followed by a hydrophobic transmembrane segment (predicted using the TMHMM server at www.cbs.dtu.dk/services/TMHMM-2.0/) and several positively charged residues at the C terminus (manually checked) of putative cell-wall-anchored proteins (four Isd proteins and OrfA) in S. caprae and S. capitits (Bowden et al., 2005) were predicted. Phylogenetic analysis of isd NEAT domains was carried out using the method described above. Comparisons of the isd loci were carried out using BLASTN and BLASTP. Other bioinformatics tools were written using Python and Biopython (Cock et al., 2009).
Nucleotide Sequence Accession Number
The complete chromosome and five plasmids sequences of S. caprae SY333 (pSY333-92, pSY333-45, pSY333-41, pSY333-7, and pSY333-2) have been submitted to DDBJ/EMBL/GenBank under accession numbers CP051643, CP051644, CP051648, CP051645, CP051646, and CP051647, respectively.
Results and Discussion
General Features of the S. caprae SY333 Genome
The S. caprae SY333 genome consists of a circularly closed chromosome and five non-conjugative plasmids named as pSY333-92, pSY333-45, pSY333-41, pSY333-7, and pSY333-2. The chromosome of S. caprae SY333 is ~2.58 Mb in length with an average GC content of 33.72% and encodes 2,435 open reading frames (ORFs). These five plasmids (pSY333-92, pSY333-45, pSY333-41, pSY333-7, and pSY333-2) are all circular DNA sequences with 91,820, 44,854, 41,252, 7,385, and 1,983 bp in length, encoding 126, 56, 49, 9 and 1 ORFs, respectively. Staphylococcal plasmids range from just over 1.0 kb to >60.0 kb in size (Kwong et al., 2017), and the smaller plasmids (between 1.0 and 8.0 kb) generally replicate via a rolling-circle replication (RCR) mechanism that is hallmarked by the production of single-stranded intermediates during replication. The pSY333-2 harbors a single ORF which encodes a replication protein. Searching against NCBI nucleotide database showed that pSY333-2 shared a high sequence similarity (85% coverage and 83% identity) with a plasmid SAP108D (2,422 bp) from S. epidermidis and also encodes only one replication gene.
Up to date, there are only 10 genome sequences of S. caprae present in NCBI genome database, of which 6 are incomplete genome sequences. Among the 4 complete genome sequences, 3 (S. caprae JMUB145, JMUB590 and JMUB898) were isolated from human skin without any plasmid, and the other one (S. caprae 26D) was isolated from buffalo milk with a plasmid (carrying blaZ). The genome of S. caprae SY333 is the first one of a clinical S. caprae isolate carrying multiple plasmids, of which 2 are resistance plasmids.
Resistance Genes and Their Functions in S. caprae SY333
A total of 5 drug resistance genes associated with 3 antibiotics classes (β-lactams, aminoglycosides, and macrolides) were identified in the S. caprae SY333 genome, with 2 on the chromosome (norA and mgrA), 2 on pSY333-45 (aadD2 and msrA) and 1 on pSY333-41 (blaZ). Two resistance genes (aadD2 and blaZ) were cloned for functional evaluation. Compared with the control strain (JH2-2/pAM401), the cloned aadD2 gene increased 8- and >16-fold of the MIC levels to tobramycin and kanamycin, respectively. Meanwhile, the blaZ gene exhibited >512-, 128-, >256-, and >1024-fold increase in MIC levels in response to treatment with ampicillin, cephazolin, ceftazidime, and meropenem, respectively, compared with those of the control (DH5α/pUCP24) (Table 3). The in vitro susceptibility testing of S. caprae SY333 exhibited resistance to a number of antibiotics, including erythromycin and clarithromycin (macrolides), amikacin and azithromycin (aminoglycosides) and penicillin (β-lactam), according to CLSI breakpoint criteria for Staphylococcus. Moreover, the MIC level of roxithromycin against S. caprae SY333 was 128 μg/mL. Although there was no interpretation criteria of resistance breakpoint for roxithromycin, it was significantly higher than that for erythromycin (>8 μg/mL).
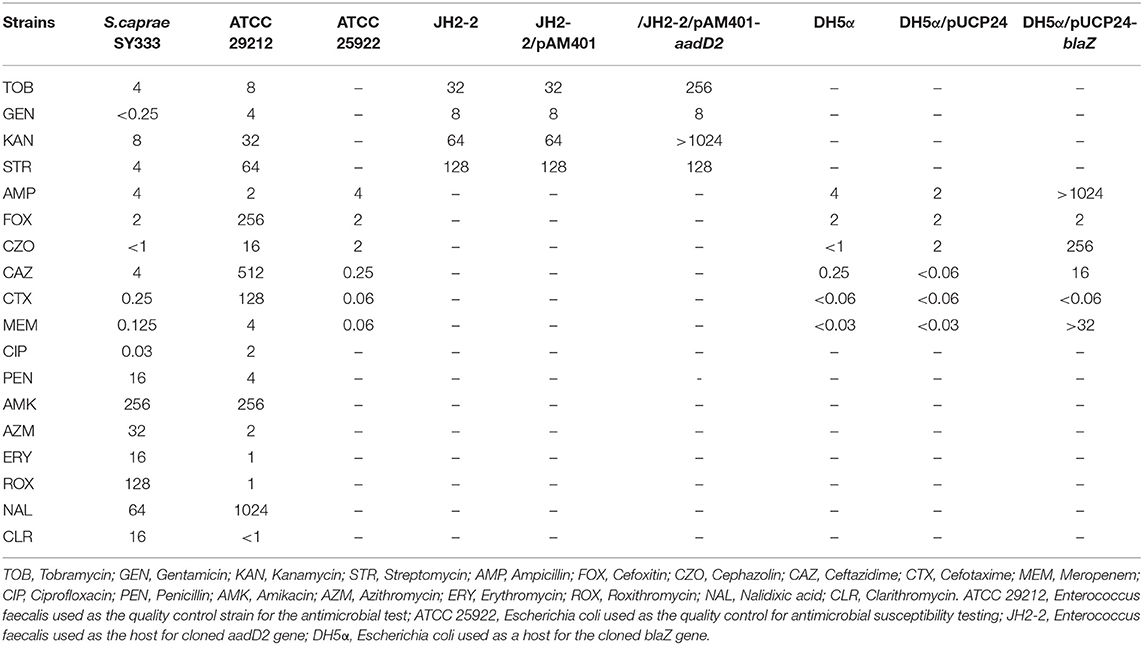
Table 3. MIC values of antibacterial drugs for all strains (μg/mL).
Two mechanisms have been reported to confer penicillin resistance in staphylococci (Olsen et al., 2006). The primary mechanism is the production of β-lactamase encoded by blaZ. The second is the expression of PBP2a, a penicillin-binding protein encoded by mecA. In this work, only one mechanism (blaZ) conferring penicillin resistance in the S. caprae SY333 genome was confirmed.
Comparison of bla Region in ECG Species
The bla operon (blaI-blaR1-blaZ) carried by a Tn552-like element in S. caprae SY333 is encoded on the plasmid pSY333-41. According to serotyping and different hydrolyzing substrate profiles, the β-lactamases expressed by blaZ could be divided into four types A, B, C, and D (Pereira et al., 2014). Among them, type B is usually encoded in the chromosome, while the other three types (A, C, and D) are generally encoded on the plasmids (Bagcigil et al., 2012). Phylogenetic analysis of plasmid-borne and chromosomally located bla operons (blaI-blaR1-blaZ) in ECG species identified two distinct clades (clade A and clade B, Figure 1). Clade A included a mixture of strains with bla operons encoded on either chromosomes or plasmids, while Clade B only included strains in which the bla operon was encoded on the chromosome (Figure 1).
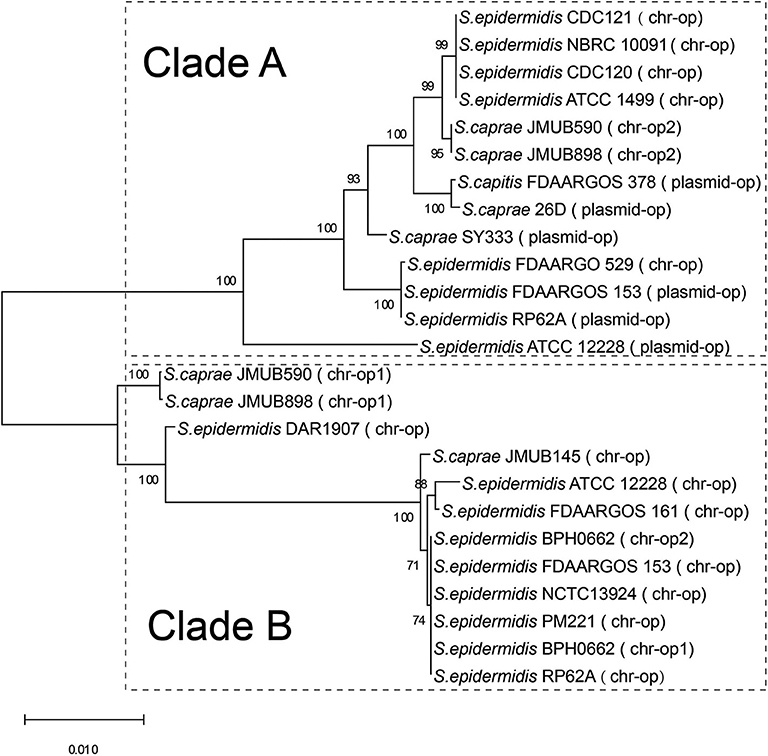
Figure 1. Phylogenetic relationships of bla operons among the complete genome sequences of ECG. The bla operon is abbreviated to op. chr-op1 indicates the first copy of the bla operon encoded on the chromosome, and chr-op2 indicates the second copy of the bla operon downstream of the first copy of the bacterium. Plasmid-op indicates the bla operon encoded on a plasmid.
Comparison of the bla region (the bla operon and its immediate surroundings) showed that there were three distinct structures: (1) the bla operon was associated with a Tn552-like element that is commonly located on the plasmid, for example, S. caprae SY333 (plasmid-op) (except for S. epidermidis FDARGOS_529); (2) two tyrosine recombinase genes xerC were located downstream of the blaZ gene, which was only encoded on the chromosome, for example, S. epidermidis ATCC 12228 (chr-op); (3) the bla operon had no significantly featured surroundings and was only located on the chromosome, for example, S. caprae JMUB898 (chr-op1) (Figure 2). The β-lactamase gene-related transposon Tn552 and its derivatives belong to a group of transposons targeting resolution sites (resL) and are almost ubiquitous in modern S. aureus isolates (Yui Eto et al., 2019). The above results indicated that the bla operon located on the chromosome and the plasmid had followed two distinct evolutionary paths, which has been confirmed by the hypothesis raised by Olsen et al. (2006). The appearance of the Tn552-like element in the chromosomes of S. epidermidis FDARGOS_529 also indicated that the bla operon-related region could be translocated by Tn552 between chromosomes and plasmids. The fact that those chromosome-encoded bla regions clustered together with those plasmid-encoded regions indicated that they may originate from the same ancestors (Figure 1).
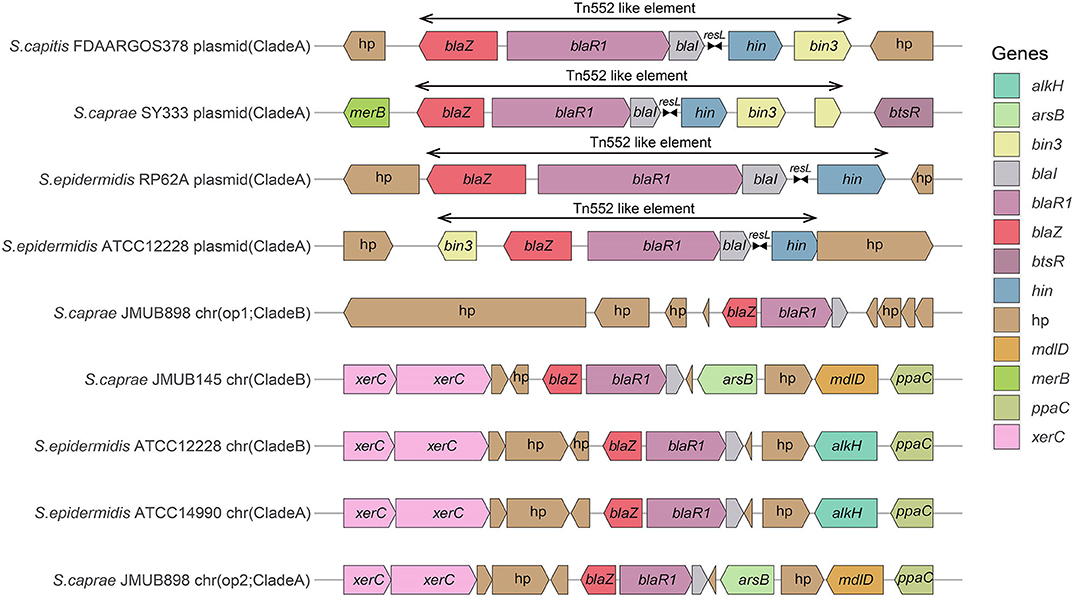
Figure 2. Comparison of the bla operons and their genetic environments. The genes are denoted by arrows and colored based on their assigned gene functions. The gene hp stands for hypothetical protein.
Phylogenetic Relationship Among ECG Species
To infer the phylogenetic relationship of the ECG strains, a phylogenetic tree using the five ubiquitously conserved core genes of the 70 strains (including Bacillus subtilis) was constructed. The resulting phylogenetic tree clearly grouped the 69 ECG strains into three distinct clades as expected. S. caprae was phylogenetically closer to S. capitis (Figure 3A). Through phylogenomics analyses of 764 concatenated genes, a tree similar to that reconstructed from five ubiquitously conserved core genes was obtained (Figure 3B). This inferred phylogenetic relationship among ECG species was highly consistent with a former report in which the phylogenetic relationship was reconstructed using single-nucleotide polymorphisms in 82 Staphylococcus genome sequences, including ECG species (Watanabe et al., 2018).
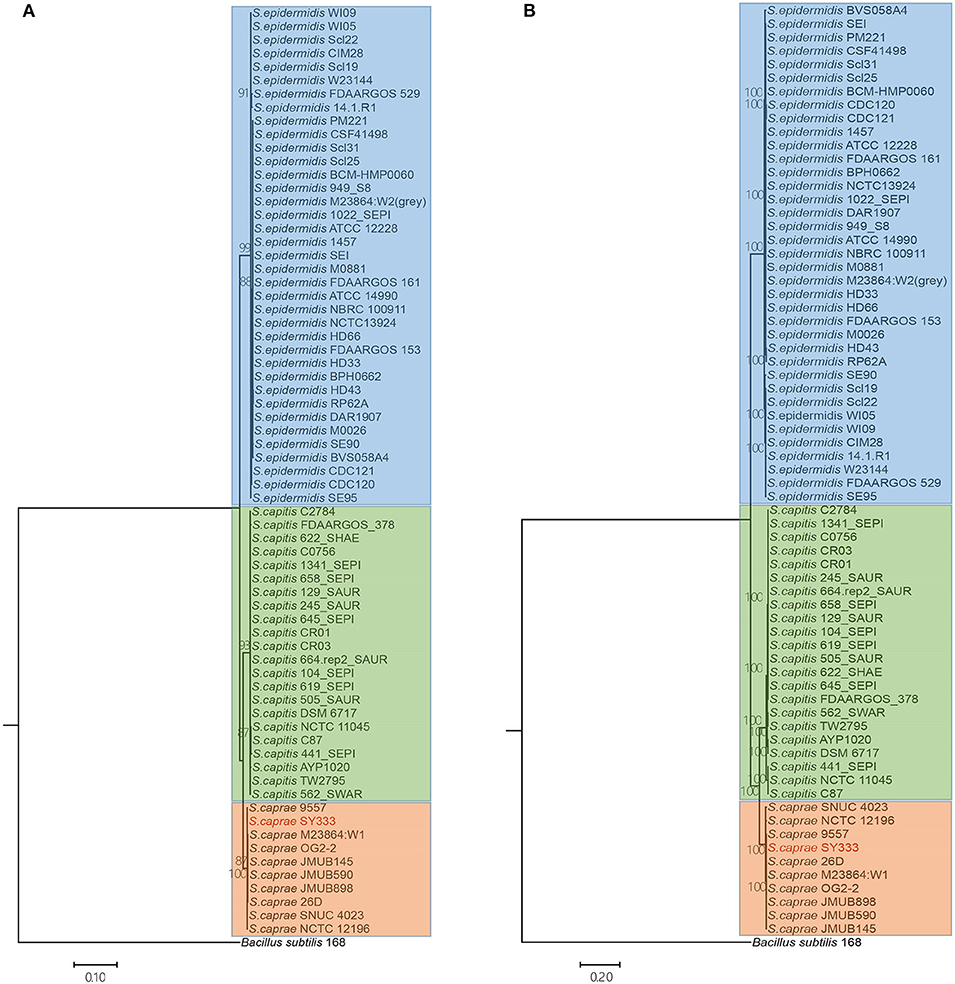
Figure 3. Phylogenetic and phylogenomics relationships of the housekeeping genes and single-copy orthologous genes of the ECG strains. (A) Phylogenetic relationship of ECG strains, according to the housekeeping genes shared by all complete genomes (i.e., atpD, recA, gyrA, gyrB and ftsZ). (B) Phylogenomics tree generated based on a total of 764 single-copy orthologous genes. Color shading indicates the species to which each stain belongs (blue: S. epidermids; green: S. capitis; orange: S. caprae). More information on strain characteristics is shown in Table 1.
Pan-Genome Inference and COG Functional Characterization
In order to compare the general genetic similarities and differences within the three species, the core and pan-genome of each species was determined. The result revealed that there were a total of 1,568 core genes, 2,788 accessory genes (genes of accessory genome present in at least two strains) and 2,263 unique genes (genes of accessory genome present in only one strain) among all S. epidermidis strains (Figure 4A). Similar to S. epidermidis, 1,789 core genes, 1,441 accessory genes, and 946 unique genes, and 2,065 core genes, 780 accessory genes and 1,008 unique genes were predicted among S. capitis strains and S. caprae strains, respectively.
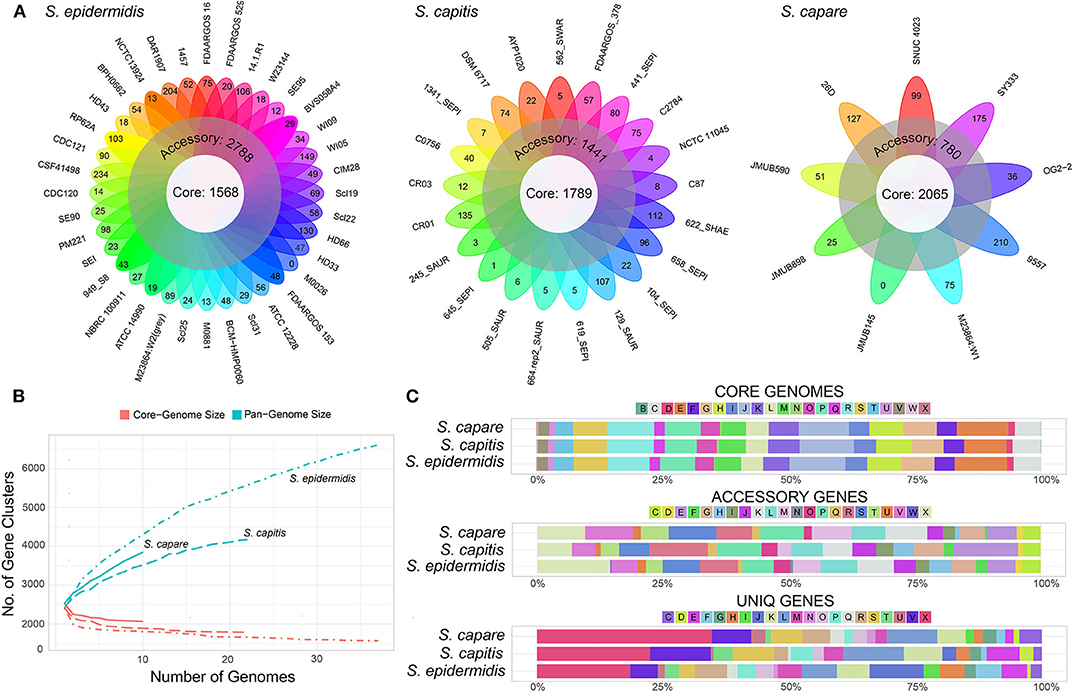
Figure 4. Representation of the pan-genomes and COG functional annotations of ECG species. (A) Comparative overview of ECG pan- and core-genomes. (B) Accumulation plots for ECG species pan- (blue) and core-genomes (red). The curves show the relationship among the pan-genomes, the core-genomes, and the number of genomes. As the number of genomes sequenced increased, pan-genomes also increased. (C) COG functional categories from the pan-genomes of ECG strains. COG categories are as follows: For cellular processes and signaling: D, cell cycle control, cell division and chromosome partitioning; M, cell wall/membrane/envelope biogenesis; N, cell motility; O, post-translational modification, protein turnover and chaperones; T, signal transduction mechanisms; U, intracellular trafficking, secretion and vesicular transport; V, defense mechanisms and Z, cytoskeleton. For information storage and processing: B, chromatin structure and dynamics; J, translation, ribosomal structure and biogenesis; K, transcription and L, replication, recombination and repair. For metabolism: C, energy production and conversion; E, amino acid transport and metabolism; F, nucleotide transport and metabolism; G, carbohydrate transport and metabolism; H, coenzyme transport and metabolism; I, lipid transport and metabolism; P, inorganic ion transport and metabolism and Q, secondary metabolite biosynthesis, transport and catabolism. X for mobilome or prophages. R for general function prediction only, and S for unknown function.
The rare faction curve (Figure 4B) showed that as genomes were sampled, the genes never observed before are continuously added at a fairly steady rate, causing the pan-genome size to increase, with no sign of getting stable soon. This tended to indicate that the pan-genomes of the three species are “open” (Medini et al., 2005; Diene et al., 2013). In this work, the number and diversity of the S. epidermidis strains were greater than those of both S. capitis and S. caprae. This is the main reason why the pan-genome size of S. epidermidis is larger than the other two species (Figure 4A). To some extent, the pan-genome state (“open” or “close”) for an organism partially depended on its capacity of acquiring exogenous DNA (Diene et al., 2013), especially for the species living in bacterial communities, such as those skin inhabitants [coagulase-negative staphylococci (CoNS)]. These species had a high horizontal gene transfer range and were most likely to have an open pan-genome (Georgiades and Raoult, 2010). Moreover, several systems which prevent horizontal gene transfer (HGT) could also influence the pan-genome state, for example, the concomitant identification of CRISPR/Cas, RM and T/AT loci that constitute specialized systems preventing HGT in S. lugdunensis result in a closed pan-genome which was in contrast to all other staphylococci studied to date (Argemi et al., 2018). So far, however, these systems have rarely been identified in other CoNS (Argemi et al., 2018). In this study, only 2 (S. capitis CR01 and S. capitis CR03) and 3 strains (S. epidermidis FDAARGOS_153, S. epidermidis M0881 and S. epidermidis RP62A) of S. capitis and S. epidermidis were confirmed to harbor complete CRISPR/Cas system (Type III), while such system was more common in S. lugdunensis. To some extent, these factors led to open pan-genomes of ECG species analyzed in this work.
Functional classification according to COG category showed that, the core genes of the three species were successfully assigned to 23 subcategories which were more than those of the accessory genes and unique genes, suggesting that the core genes of the three species have been intensively studied. The core gene category repartition was highly similar among the 3 species, and was found to be abundant in “Translation, ribosomal structure and biogenesis,” “Amino acid transport and metabolism,” and “Energy production and conversion” (Figure 4C). However, the functions of accessory and unique genes of the three species showed significant diversity which could result in the diverse biological characteristics of ECG species. Compared to the core genomes, the accessory and unique gene pools of the ECG species contained significant abundance of the genes belonging to the class “Mobilome or prophages.” These genes often appear to transfer laterally between strains and result in the transmission of virulence and resistance genes between strains, thus influencing bacterial pathogenicity (Jackson et al., 2011).
Virulence Genes in ECG Genomes
To analyze the different pathogenic potential within ECG, a total of 164 virulence genes from 69 ECG genomes were identified, and 57 (57/164, 34.8%) virulence genes coexisted in all ECG genomes. Interestingly, S. caprae carried the highest average number of virulence genes (128), and S. caprae SY333 carried the most virulence genes (134) among all 69 ECG genomes.
The virulence genes could be grouped into 8 categories according to the Virulence Factor Database, including “adherence and invasion factor,” “immune evasion,” “enzyme,” “toxin,” “stress protein,” “metabolic adaptation,” “metal uptake,” and “regulation and other surface component.” Remarkably, each species of ECG has its own specific virulence genes (Figure 5, Supplementary Figure 1). For S. caprae, the species-specific virulence genes were involved in “immune evasion” and “metal uptake”; for S. capitis, “metal uptake”; and for S. epidermidis, all categories except for “stress protein.” Most species-specific virulence genes in S. caprae belonged to “immune evasion,” especially those biosynthesis genes of capsular polysaccharide (cap locus), while in S. epidermidis, they mostly belonged to “secretion system.” In ECG genomes, most of the virulence genes were associated with adherence and immune evasion, indicating their strong ability to colonize their hosts and evade the innate immune system.
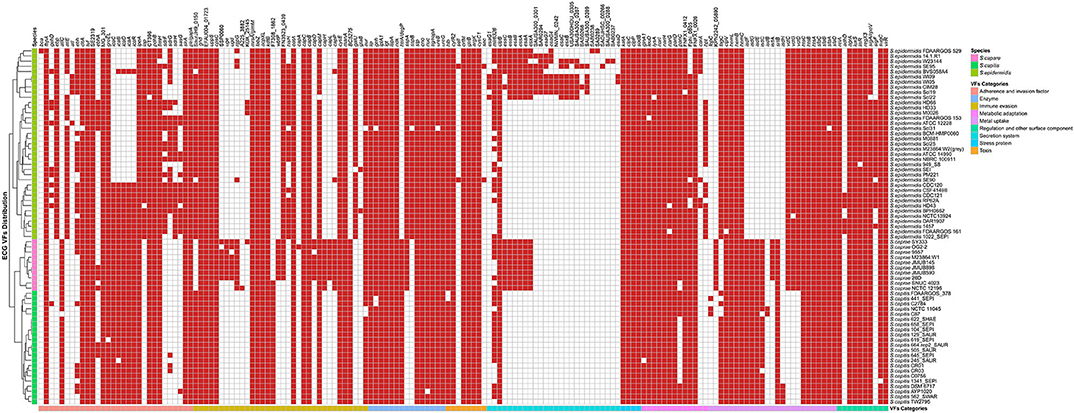
Figure 5. Distribution of virulence genes in ECG strains. The red square indicates the presence of the gene, while the white square indicates absence. The VF (virulence factor) categories and species are marked in the legend with different colors.
The genes involved in “enzyme,” “toxin,” “stress protein,” “metabolic adaptation,” and “regulation and other surface component” were relatively conserved in ECG genomes. Except for two toxin genes (sell and sec), which were only present in two strains of S. epidermidis (S. epidermidis SE90 and S. epidermidis SE95), the other toxin-related virulence factors were present in almost all strains of ECG. For secretion systems, the T6SS gene clpB and the T3SS gene mlr6326 were commonly found in the ECG, while EsaA, EssA, EssB, and EssC, the proteins of T7SS machinery (Jäger et al., 2018), were only present in all strains of S. caprae and some of S. epidermidis. Notably, a secreted serine protease SspA (V8 protease) that degrades fibronectin-binding microbial surface components and recognizes adhesive matrix molecules (MSCRAMMs) to promote invasion (McGavin et al., 1997) was present in all strains of S. caprae and S. epidermidis but not S. capitis. In terms of intercellular aggregation, the ica (intercellular adhesion) locus involved in PNAG/PIA biosynthesis (Otto, 2009), seemed to be present more frequently in S. capitis and S. caprae, while those genes (sdrFGH, ebp, ebh, and atlE) associated with primary attachment appeared to be present almost exclusively in S. epidermidis. Previous studies have reported that most of the S. epidermidis strains carried SdrF, SdrG, and SdrH genes (Bateman et al., 2005). It has been reported that SdrG could necessarily and sufficiently promote in vitro adhesion to fibrinogen of S. epidermidis (Sun et al., 2005; Conrady et al., 2008) and in vivo central venous catheter (CVC) associated infectious disease (Guo et al., 2007). The enrichment of MSCRAMMs in S. epidermidis indicated that S. epidermidis achieved better in vivo colonization than the other two species. Although the ica locus is commonly present in S. caprae and S. capitis, the lack of MSCRAMM-related proteins may limit their ability to form biofilms (Otto, 2009). It is worth mentioning that ica and cap loci which protect the bacteria from the important innate host defense mechanisms are commonly present in S. caprae and might lead to better immune evasion (Otto, 2009; Fournier et al., 2013). T7SS, a secretion pathway for the virulence proteins, protects the bacteria from the host defense system and makes them able to survive in abscesses for a long time (Warne et al., 2016). Identification of T7SS in all S. caprae and some S. epidermidis strains indicated that these strains might possess higher capacity for secretion of virulence factors, as well as higher potential for bacterial pathogenesis. A previous study had shown that T7SS played an essential role in keeping integrity and homeostasis of the staphylococcus aureus membrane. This is crucial when the bacterium faces antimicrobial fatty acids (Lopez et al., 2017). Since T7SS is commonly present in S. caprae, it may suggest that T7SS targeted therapeutics decreases the virulence of S. caprae and makes it more susceptible to fatty acids. Similarly, the distribution of virulence factors involved in “enzyme,” “toxin,” “stress protein,” “metabolic adaptation,” and “regulation and other surface component” in ECG genomes indicates that ECG also has many similarities in terms of pathogenicity. Interestingly, in regard to “metabolic adaptation”-related genes, the isd genes have been only reported to be present in the species S. lugdunensis of CoNS (Heilbronner et al., 2011), while strains of S. caprae and S. capitis analyzed in this research also possess a gene cluster similar to those of S. aureus and S. lugdunensis.
Comparative Genomics Analysis of isd Locus
In order to get iron from the host, pathogens have evolved several mechanisms. S. aureus uses the isd system as a fundamental heme-iron uptake pathway. The S. aureus genome encodes an isd system (isdABCDEFGHI) conferring heme uptake and sortase B (SrtB), which is responsible for anchoring its specific substrates (IsdC) to the bacterial cell wall (Grigg et al., 2010; Liang et al., 2016). The cell walls of S. aureus anchored four Isd proteins (IsdA, IsdB, IsdC, and IsdH). These proteins contain 1 to 3 conserved NEAT domains. Each Isd surface gene at least encodes a secretion signal, a cell wall-anchoring motif and a NEAT domain (Grigg et al., 2010; Heilbronner et al., 2016). IsdB (NEAT1) and IsdH (NEAT1 and NEAT2) can bind to hemoglobin and the haptoglobin-hemoglobin complex but not heme via N-terminal NEAT domains, while IsdH NEAT3 and IsdB NEAT2 contain heme-binding NEAT domains that transfer heme to IsdA, IsdC, and then to IsdEF, which is a membrane-locating transporter (Liu et al., 2008; Muryoi et al., 2008). IsdG and IsdI, the heme oxygenases in the cytoplasm, are responsible to destroy the porphyrin ring, releasing the free iron (Reniere et al., 2007).
To better illustrate the similarities and differences of the isd locus among S. capitis, S. caprae, S. aureus, and S. lugdunensis, we performed a comprehensive comparison of the isd genes within these four species. There are four NEAT proteins in S. caprae (isdA_2, isdC, isdA, and isdL) and S. capitis (isdA_2, isdC, isdA, and isdM), respectively, which contain NEAT domains similar to those of S. aureus and S. lugdunensis (Table 4). Except for isdA_2, the other three NEAT-containing proteins in S. capitis and S. caprae possessed a secretion signal and a putative cell wall-anchoring motif (LPXTG) (Figure 6), which indicated the capacity of cell wall attachment (Cabanes et al., 2002). A comparative genome analysis demonstrated that the isdGEFI and srtB genes were conserved across S. capitis, S. caprae, and S. aureus, while other NEAT domain-containing genes appeared to be less conserved (Figure 6). IsdD, a membrane protein in S. aureus, is absent in S. capitis and S. caprae (Grigg et al., 2010). Phylogenetic analysis of NEAT domains showed that IsdL NEAT2 and NEAT3 and IsdM NEAT were phylogenetically close to IsdB NEAT2, IsdH NEAT3 of S. aureus and IsdB NEAT2 of S. lugdunensis, which were responsible for heme binding. IsdL NEAT1 was clustered together with IsdH NEAT1 and NEAT2, IsdB NEAT1 of S. aureus and IsdB NEAT1 of S. lugdunensis which were responsible for binding to the haptoglobin-hemoglobin complex and to hemoglobin (Figure 7). Multiple sequence alignments of NEAT domains revealed that each putative heme-binding NEAT domain in S. capitis and S. caprae contained an essential YXXXY heme-binding motif (Figure 8A). The first tyrosine of the heme-binding motif (e.g., Y166 of IsdA) non-covalently binds the central iron atom of heme, and the second tyrosine residue (e.g., Y170 of IsdA) provides a stabilizing hydrogen bond to the first tyrosine (Sheldon and Heinrichs, 2015). Except for IsdL NEAT2 of S. capitis, these putative heme-binding NEAT domains also contained a conserved SXXXXY sequence which forms a 310-helix (α-helix) referred to as the “lip” region (Figure 8A). Hemoglobin binding by these NEAT domains in S. aureus is mediated by a conserved five amino acid aromatic motif (YYHFF in IsdH-N1 and FYHYA in IsdH-N2 and IsdB-N1 at positions 164 to 168). Mutation of any one of these key residues severely hinders hemoglobin and/or haptoglobin binding (Sheldon and Heinrichs, 2015). Alignment of the hemoglobin-binding NEAT domains revealed amino acid substitution at position 166 (H166I) and 167 (Y167E) in IsdL NEAT1 which might confer reduction or loss of the function (Figure 8B). These results indicate that these putative heme-binding NEAT domains in S. capitis and S. caprae have the ability to bind to heme, while whether the putative hemoglobin-binding domain (IsdL NEAT1) in S. capitis is functional needs further validation.
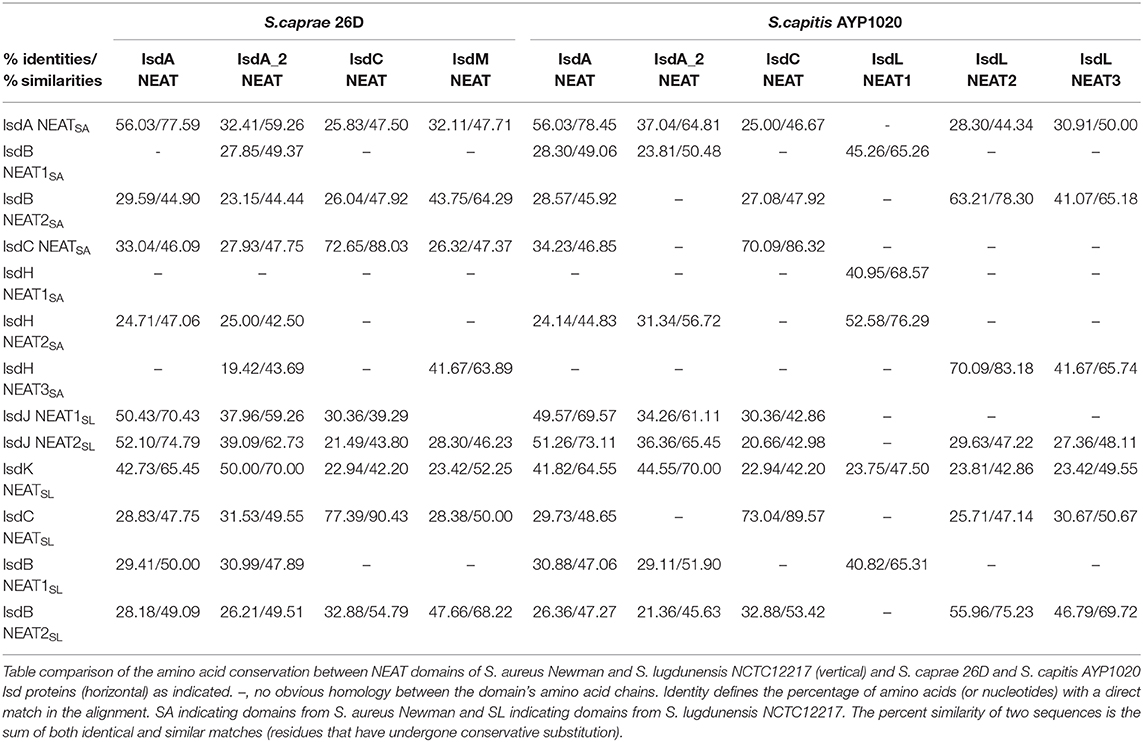
Table 4. Identities and similarities among NEAT domains of S. aureus Newman, S. lugdunensis NCTC12217, S. caprae 26D, and S. capitis AYP1020.
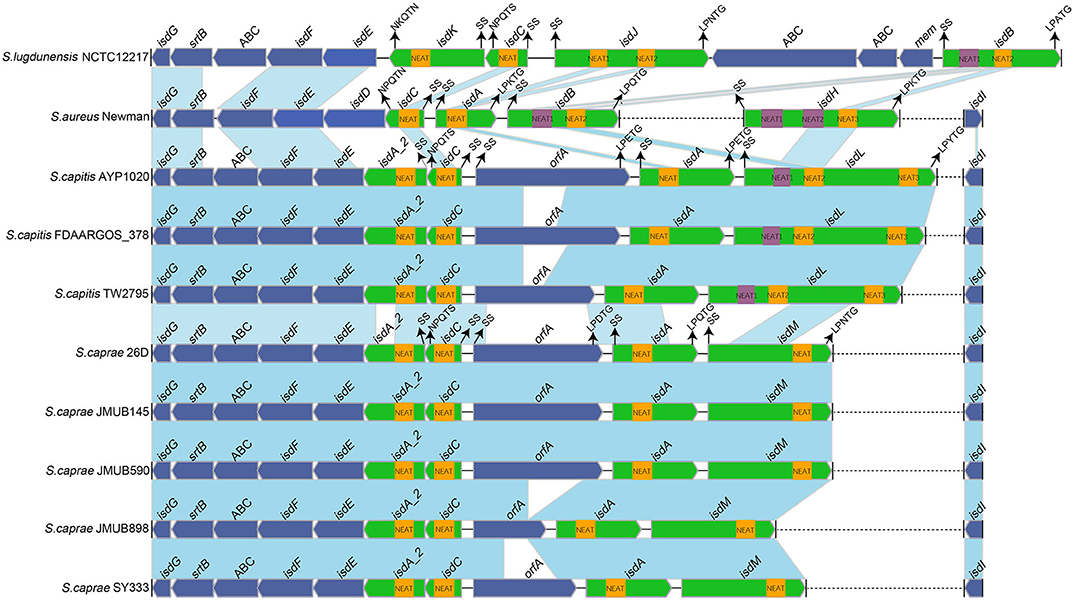
Figure 6. Comparison of the isd loci from completely sequenced genomes of S. caprae and S. capitis and the complete genomes of S. aureus Newman and S. lugdunensis NCTC12217. Blue links connect orthologous genes, and the color depth indicates the degree of identity. Heme-binding NEAT domains are indicated by yellow boxes, while Hb-binding NEAT domains are indicated by purple boxes. Genes are denoted by large arrows, and heme- or Hb-binding genes are colored in green. Dashed lines indicate genes that are not adjacent. The putative cell wall-anchoring motifs, single sequences and the function of NEAT domains in S. aureus Newman are indicated with arrows. SS, secretion signal; orfA, an unknown function putative cell wall-anchoring protein; ABC, ABC transporter.
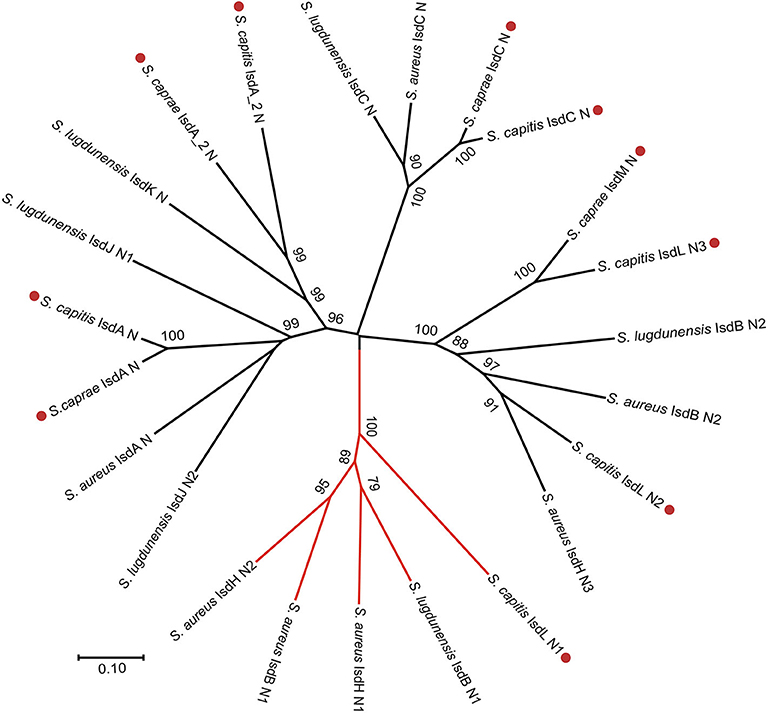
Figure 7. Phylogenetic relationships of NEAT domains in S. capitis AYP1020, S. caprae 26D, S. lugdunensis NCTC12217, and S. aureus Newman. Branches of Hb-binding NEAT domains are in red. Putative NEAT domains from S. capitis or S. caprae are followed by a red dot.

Figure 8. Multiple sequence alignments of the NEAT domains of S. aureus Newman, S. lugdunensis NCTC12217, S. caprae 26D and S. capitis AYP1020. (A) Alignment of the putative heme-binding NEAT domains of S. caprae 26D and S. capitis AYP1020. Conserved motifs are framed in red boxes. (B) Alignment of the putative Hb-binding NEAT domain of S. capitis AYP1020. Conserved motifs are framed in red box.
In this study, we haven't found any NEAT domain that might bind to hemoglobin in S. caprae, and the function of the putative hemoglobin-binding domain (IsdL NEAT1) is still questionable. Indeed, not all heme or hemoglobin-binding Isd proteins contain the LPXTG motif, for example, isdX1 (containing one NEAT domain) and isdX2 (containing five NEAT domains) which are located between isdC and isdE in the isd loci of Bacillus cereus group were secreted without a LPXTG motif (Sheldon and Heinrichs, 2015). Unlike S. aureus, whose NEAT proteins acquire heme from hemoglobin directly at the bacterial surface, Bacillus cereus group secretes IsdX1 to capture heme in the extracellular milieu and relies on NEAT-NEAT interactions to deliver the bound heme to the envelope via IsdC (Maresso et al., 2008). Of note, isdA_2 in S. caprae and S. capitis which is located between isdC and isdE also lacks LPXTG motif and that is similar to isdX1 and isdX2 although no obvious sequence identities were observed between isdA_2 and isdX (isdX1 and isdX2). Based on the characteristics of isdA_2, we hypothesized that IsdA_2 could be secreted out of bacteria cell rather than attached to the cell wall and interact with heme in the extracellular milieu. In addition, a putative cell wall-anchored protein OrfA was found in the isd loci of S. capitis and S. caprae, the function of OrfA remains unknown. In summary, the operation mechanism of Isd systems in S. caprae and S. capitis might be slightly different from that of S. aureus and S. lugdunensis.
S. lugdunensis was once considered as a unique species that harbors an iron-regulated surface determinant locus (isd) among coagulase-negative staphylococci. Discovery of the isd locus in S. capitis and S. caprae would provide evidence for their ability to use heme as an iron source during infection.
Conclusion
In this work, the complete genome sequence of a clinical S. caprae isolate with two resistance plasmids was reported for the first time. S. caprae SY333 showed resistance to several antibiotics, such as erythromycin, clarithromycin, amikacin, azithromycin and penicillin. Two plasmid-encoded resistance genes (blaZ and aadD2) were confirmed to be functional. The pan-genome analysis of the three ECG species showed that their pan-genomes tend to be “open” and functional annotation revealed that core gene category repartition was highly similar across the 3 species. Analysis of the bla region in ECG revealed that the chromosome-encoded and plasmid-encoded bla operons had two distinct evolutionary paths. Virulence factors in ECG differed mostly in adherence, invasion, immune evasion and secretion system. T7SS may play an important role in pathogenesis of S. caprae and S. epidermidis. Genes related to primary attachment are almost exclusively present in S. epidermidis, while intercellular adhesion-related genes are more frequently present in S. caprae and S. capitis. Identification of the isd locus in S. caprae and S. capitis discouraged previous claims that S. lugdunensis was the only coagulase-negative Staphylococcus species with a locus encoding iron-regulated surface determinant (Isd) proteins, as well as indicated that this two species may have the ability to use heme as the nutrient iron source during infection, which could enhance their pathogenic potential.
Data Availability Statement
This article contains previously unpublished data. The name of the repository and accession number(s) are not available.
Ethics Statement
This study was approved by the Ethics Committee of the Central Hospital of Lishui City (China) and informed consent was obtained from the patient.
Author Contributions
ZS, DZ, XZ, QL, HLin, WL, HLiu, and JL collected the strains and performed the experiments. ZS, XL, TX, and HZ analyzed the experimental results and performed the bioinformatics analysis. ZS, TX, QB, and HZ wrote the manuscript. KL, TX, QB, and HZ designed the experiments. All authors read and approved the final manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (81973382), the Science & Technology Project of Wenzhou City, China (Y20170205 and 2019Y0358), and the Natural Science Foundation of Zhejiang Province, China (LQ17H190001 and LY19C060002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank all the colleagues and the reviewers who helped this work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2020.00274/full#supplementary-material
Supplementary Table 1. Gene presence/absence matrix produced by Roary.
Supplementary Figure 1. A network diagram of all virulence factors (VFs) identified in ECG genomes, “Shared” means VFs present in three species, “Accessory” means VFs present in two species and “Species-specific” means VFs present in one species only.
References
Argemi, X., Matelska, D., Ginalski, K., Riegel, P., Hansmann, Y., Bloom, J., et al. (2018). Comparative genomic analysis of Staphylococcus lugdunensis shows a closed pan-genome and multiple barriers to horizontal gene transfer. BMC Genomics 19:621. doi: 10.1186/s12864-018-4978-1
Bagcigil, A. F., Taponen, S., Koort, J., Bengtsson, B., Myllyniemi, A.-L., and Pyörälä, S. (2012). Genetic basis of penicillin resistance of S. aureus isolated in bovine mastitis. Acta Vet. Scand. 54:69. doi: 10.1186/1751-0147-54-69
Bateman, A., Holden, M. T. G., and Yeats, C. (2005). The G5 domain: a potential N-acetylglucosamine recognition domain involved in biofilm formation. Bioinformatics 21, 1301–1303. doi: 10.1093/bioinformatics/bti206
Becker, K., Heilmann, C., and Peters, G. (2014). Coagulase-negative staphylococci. Clin. Microbiol. Rev. 27, 870–926. doi: 10.1128/CMR.00109-13
Benedetti, P., Pellizzer, G., Furlan, F., Nicolin, R., Rassu, M., and Sefton, A. (2008). Staphylococcus caprae meningitis following intraspinal device infection. J. Med. Microbiol. 57, 904–906. doi: 10.1099/jmm.0.2008/000356-0
Bowden, M. G., Chen, W., Singvall, J., Xu, Y., Peacock, S. J., Valtulina, V., et al. (2005). Identification and preliminary characterization of cell-wall-anchored proteins of Staphylococcus epidermidis. Microbiology 151, 1453–1464. doi: 10.1099/mic.0.27534-0
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Cabanes, D., Dehoux, P., Dussurget, O., Frangeul, L., and Cossart, P. (2002). Surface proteins and the pathogenic potential of Listeria monocytogenes. Trends Microbiol. 10, 238–245. doi: 10.1016/S0966-842X(02)02342-9
Casey, A. L., Lambert, P. A., and Elliott, T. S. J. (2007). Staphylococci. Int. J. Antimicrob. Agents 29(Suppl. 3), S23–S32. doi: 10.1016/S0924-8579(07)72175-1
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Chen, Q., Zhou, W., Qian, C., Shen, K., Zhu, X., Zhou, D., et al. (2019). OXA-830, a novel chromosomally encoded extended-spectrum class D β-lactamase in Aeromonas simiae. Front. Microbiol. 10:2732. doi: 10.3389/fmicb.2019.02732
Chen, X., Zhang, Y., Zhang, Z., Zhao, Y., Sun, C., Yang, M., et al. (2018). PGAweb: a web server for bacterial pan-genome analysis. Front. Microbiol. 9:1910. doi: 10.3389/fmicb.2018.01910
CLSI. (2019). Performance Standards for Antimicrobial Susceptibility Testing. 29th Edn. CLSI supplement M100. Wayne, PA: Clinical and Laboratory Standards Institute.
Cock, P. J. A., Antao, T., Chang, J. T., Chapman, B. A., Cox, C. J., Dalke, A., et al. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. doi: 10.1093/bioinformatics/btp163
Comas, I., Moya, A., and González-Candelas, F. (2007). From phylogenetics to phylogenomics: the evolutionary relationships of insect endosymbiotic gamma-Proteobacteria as a test case. Syst. Biol. 56, 1–16. doi: 10.1080/10635150601109759
Conrady, D. G., Brescia, C. C., Horii, K., Weiss, A. A., Hassett, D. J., and Herr, A. B. (2008). A zinc-dependent adhesion module is responsible for intercellular adhesion in staphylococcal biofilms. Proc. Natl. Acad. Sci. U.S.A. 105, 19456–19461. doi: 10.1073/pnas.0807717105
Couvin, D., Bernheim, A., Toffano-Nioche, C., Touchon, M., Michalik, J., Néron, B., et al. (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251. doi: 10.1093/nar/gky425
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1093/bioinformatics/btr088
Diene, S. M., Merhej, V., Henry, M., El Filali, A., Roux, V., Robert, C., et al. (2013). The rhizome of the multidrug-resistant Enterobacter aerogenes genome reveals how new “killer bugs” are created because of a sympatric lifestyle. Mol. Biol. Evol. 30, 369–383. doi: 10.1093/molbev/mss236
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Fournier, P.-E., Gouriet, F., Gimenez, G., Robert, C., and Raoult, D. (2013). Deciphering genomic virulence traits of a Staphylococcus epidermidis strain causing native-valve endocarditis. J. Clin. Microbiol. 51, 1617–1621. doi: 10.1128/JCM.02820-12
Georgiades, K., and Raoult, D. (2010). Defining pathogenic bacterial species in the genomic era. Front. Microbiol. 1:151. doi: 10.3389/fmicb.2010.00151
Grigg, J. C., Ukpabi, G., Gaudin, C. F. M., and Murphy, M. E. P. (2010). Structural biology of heme binding in the Staphylococcus aureus Isd system. J. Inorg. Biochem. 104, 341–348. doi: 10.1016/j.jinorgbio.2009.09.012
Guo, B., Zhao, X., Shi, Y., Zhu, D., and Zhang, Y. (2007). Pathogenic implication of a fibrinogen-binding protein of Staphylococcus epidermidis in a rat model of intravascular-catheter-associated infection. Infect. Immun. 75, 2991–2995. doi: 10.1128/IAI.01741-06
Heilbronner, S., Holden, M. T. G., van Tonder, A., Geoghegan, J. A., Foster, T. J., Parkhill, J., et al. (2011). Genome sequence of Staphylococcus lugdunensis N920143 allows identification of putative colonization and virulence factors. FEMS Microbiol. Lett. 322, 60–67. doi: 10.1111/j.1574-6968.2011.02339.x
Heilbronner, S., Monk, I. R., Brozyna, J. R., Heinrichs, D. E., Skaar, E. P., Peschel, A., et al. (2016). Competing for iron: duplication and amplification of the isd locus in Staphylococcus lugdunensis HKU09-01 provides a competitive advantage to overcome nutritional limitation. PLoS Genet. 12:e1006246. doi: 10.1371/journal.pgen.1006246
Hunter, S., Apweiler, R., Attwood, T. K., Bairoch, A., Bateman, A., Binns, D., et al. (2009). InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215. doi: 10.1093/nar/gkn785
Jackson, R. W., Vinatzer, B., Arnold, D. L., Dorus, S., and Murillo, J. (2011). The influence of the accessory genome on bacterial pathogen evolution. Mob. Genet. Elements 1, 55–65. doi: 10.4161/mge.1.1.16432
Jäger, F., Kneuper, H., and Palmer, T. (2018). EssC is a specificity determinant for Staphylococcus aureus type VII secretion. Microbiology 164, 816–820. doi: 10.1099/mic.0.000650
Kanda, K., Suzuki, E., Hiramatsu, K., Oguri, T., Miura, H., Ezaki, T., et al. (1991). Identification of a methicillin-resistant strain of Staphylococcus caprae from a human clinical specimen. Antimicrob. Agents Chemother. 35, 174–176. doi: 10.1128/AAC.35.1.174
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kwong, S. M., Ramsay, J. P., Jensen, S. O., and Firth, N. (2017). Replication of staphylococcal resistance plasmids. Front. Microbiol. 8:2279. doi: 10.3389/fmicb.2017.02279
Lamers, R. P., Muthukrishnan, G., Castoe, T. A., Tafur, S., Cole, A. M., and Parkinson, C. L. (2012). Phylogenetic relationships among Staphylococcus species and refinement of cluster groups based on multilocus data. BMC Evol. Biol. 12:171. doi: 10.1186/1471-2148-12-171
Liang, X., Liu, B., Zhu, F., Scannapieco, F. A., Haase, E. M., Matthews, S., et al. (2016). A distinct sortase SrtB anchors and processes a streptococcal adhesin AbpA with a novel structural property. Sci. Rep. 6:30966. doi: 10.1038/srep30966
Liu, M., Tanaka, W. N., Zhu, H., Xie, G., Dooley, D. M., and Lei, B. (2008). Direct hemin transfer from IsdA to IsdC in the iron-regulated surface determinant (Isd) heme acquisition system of Staphylococcus aureus. J. Biol. Chem. 283, 6668–6676. doi: 10.1074/jbc.M708372200
Lopez, M. S., Tan, I. S., Yan, D., Kang, J., McCreary, M., Modrusan, Z., et al. (2017). Host-derived fatty acids activate type VII secretion in Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 114, 11223–11228. doi: 10.1073/pnas.1700627114
Ma, X. X., Wang, E. H., Liu, Y., and Luo, E. J. (2011). Antibiotic susceptibility of coagulase-negative staphylococci (CoNS): emergence of teicoplanin-non-susceptible CoNS strains with inducible resistance to vancomycin. J. Med. Microbiol. 60, 1661–1668. doi: 10.1099/jmm.0.034066-0
Maresso, A. W., Garufi, G., and Schneewind, O. (2008). Bacillus anthracis secretes proteins that mediate heme acquisition from hemoglobin. PLoS Pathog. 4:e1000132. doi: 10.1371/journal.ppat.1000132
May, L., Klein, E. Y., Rothman, R. E., and Laxminarayan, R. (2014). Trends in antibiotic resistance in coagulase-negative staphylococci in the United States, 1999 to 2012. Antimicrob. Agents Chemother. 58, 1404–1409. doi: 10.1128/AAC.01908-13
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
McGavin, M. J., Zahradka, C., Rice, K., and Scott, J. E. (1997). Modification of the Staphylococcus aureus fibronectin binding phenotype by V8 protease. Infect. Immun. 65, 2621–2628. doi: 10.1128/IAI.65.7.2621-2628.1997
Medini, D., Donati, C., Tettelin, H., Masignani, V., and Rappuoli, R. (2005). The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594. doi: 10.1016/j.gde.2005.09.006
Muryoi, N., Tiedemann, M. T., Pluym, M., Cheung, J., Heinrichs, D. E., and Stillman, M. J. (2008). Demonstration of the iron-regulated surface determinant (Isd) heme transfer pathway in Staphylococcus aureus. J. Biol. Chem. 283, 28125–28136. doi: 10.1074/jbc.M.802171200
NCBI Resource Coordinators (2016). Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 44, D7–D19. doi: 10.1093/nar/gkv1290
Nourdin-Galindo, G., Sánchez, P., Molina, C. F., Espinoza-Rojas, D. A., Oliver, C., Ruiz, P., et al. (2017). Comparative pan-genome analysis of piscirickettsia salmonis reveals genomic divergences within genogroups. Front. Cell. Infect. Microbiol. 7:459. doi: 10.3389/fcimb.2017.00459
Olsen, J. E., Christensen, H., and Aarestrup, F. M. (2006). Diversity and evolution of blaZ from Staphylococcus aureus and coagulase-negative staphylococci. J. Antimicrob. Chemother. 57, 450–460. doi: 10.1093/jac/dki492
Otto, M. (2009). Staphylococcus epidermidis–the ‘accidental' pathogen. Nat. Rev. Microbiol. 7, 555–567. doi: 10.1038/nrmicro2182
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pereira, L. A., Harnett, G. B., Hodge, M. M., Cattell, J. A., and Speers, D. J. (2014). Real-time PCR assay for detection of blaZ genes in Staphylococcus aureus clinical isolates. J. Clin. Microbiol. 52, 1259–1261. doi: 10.1128/JCM.03413-13
Piette, A., and Verschraegen, G. (2009). Role of coagulase-negative staphylococci in human disease. Vet. Microbiol. 134, 45–54. doi: 10.1016/j.vetmic.2008.09.009
Reniere, M. L., Torres, V. J., and Skaar, E. P. (2007). Intracellular metalloporphyrin metabolism in Staphylococcus aureus. Biometals 20, 333–345. doi: 10.1007/s10534-006-9032-0
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sheldon, J. R., and Heinrichs, D. E. (2015). Recent developments in understanding the iron acquisition strategies of gram positive pathogens. FEMS Microbiol. Rev. 39, 592–630. doi: 10.1093/femsre/fuv009
Shin, J. H., Kim, S. H., Jeong, H. S., Oh, S. H., Kim, H. R., Lee, J. N., et al. (2011). Identification of coagulase-negative staphylococci isolated from continuous ambulatory peritoneal dialysis fluid using 16S ribosomal RNA, tuf, and SodA gene sequencing. Peritoneal Dial. Int. 31, 340–346. doi: 10.3747/pdi.2010.00073
Shuttleworth, R., Behme, R. J., McNabb, A., and Colby, W. D. (1997). Human isolates of Staphylococcus caprae: association with bone and joint infections. J. Clin. Microbiol. 35, 2537–2541. doi: 10.1128/JCM.35.10.2537-2541.1997
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Sun, D., Accavitti, M. A., and Bryers, J. D. (2005). Inhibition of biofilm formation by monoclonal antibodies against Staphylococcus epidermidis RP62A accumulation-associated protein. Clin. Diagn. Lab. Immunol. 12, 93–100. doi: 10.1128/CDLI.12.1.93-100.2005
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. doi: 10.1186/1471-2105-4-41
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477. doi: 10.1016/j.mib.2008.09.006
UniProt Consortium (2015). UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212. doi: 10.1093/nar/gku989
Vandenesch, F., Eykyn, S. J., Bes, M., Meugnier, H., Fleurette, J., and Etienne, J. (1995). Identification and ribotypes of Staphylococcus caprae isolates isolated as human pathogens and from goat milk. J. Clin. Microbiol. 33, 888–892. doi: 10.1128/JCM.33.4.888-892.1995
Warne, B., Harkins, C. P., Harris, S. R., Vatsiou, A., Stanley-Wall, N., Parkhill, J., et al. (2016). The Ess/Type VII secretion system of Staphylococcus aureus shows unexpected genetic diversity. BMC Genomics 17:222. doi: 10.1186/s12864-016-2426-7
Watanabe, S., Aiba, Y., Tan, X.-E., Li, F.-Y., Boonsiri, T., Thitiananpakorn, K., et al. (2018). Complete genome sequencing of three human clinical isolates of Staphylococcus caprae reveals virulence factors similar to those of S. epidermidis and S. capitis. BMC Genomics 19:305. doi: 10.1186/s12864-018-5185-9
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Wu, Y., Zaiden, N., and Cao, B. (2018). The core- and pan-genomic analyses of the genus comamonas: from environmental adaptation to potential virulence. Front. Microbiol. 9:3096. doi: 10.3389/fmicb.2018.03096
Yui Eto, K., Firth, N., Davis, A. M., Kwong, S. M., Krysiak, M., Lee, Y. T., et al. (2019). Evolution of a 72-kilobase cointegrant, conjugative multiresistance plasmid in community-associated methicillin-resistant Staphylococcus aureus isolates from the early 1990s. Antimicrob. Agents Chemother. 63:e01560-19. doi: 10.1128/AAC.01560-19
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Keywords: Epidermidis Cluster Group, bla operon, resistance, isd locus, virulence, pan-genome
Citation: Sun Z, Zhou D, Zhang X, Li Q, Lin H, Lu W, Liu H, Lu J, Lin X, Li K, Xu T, Bao Q and Zhang H (2020) Determining the Genetic Characteristics of Resistance and Virulence of the “Epidermidis Cluster Group” Through Pan-Genome Analysis. Front. Cell. Infect. Microbiol. 10:274. doi: 10.3389/fcimb.2020.00274
Received: 06 March 2020; Accepted: 07 May 2020;
Published: 12 June 2020.
Edited by:
Alexander Horswill, University of Colorado Denver, United StatesReviewed by:
Simon Heilbronner, University of Tübingen, GermanyJens Kreth, Oregon Health and Science University, United States
Copyright © 2020 Sun, Zhou, Zhang, Li, Lin, Lu, Liu, Lu, Lin, Li, Xu, Bao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiyu Bao, YmFvcXlAZ2Vub21pY3MuY24=; Hailin Zhang, emhsd3o5N0Bob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work