 Ben J. Mans1,2,3*
Ben J. Mans1,2,3*- 1Epidemiology, Parasites and Vectors, Agricultural Research Council-Onderstepoort Veterinary Research, Pretoria, South Africa
- 2Department of Veterinary Tropical Diseases, University of Pretoria, Pretoria, South Africa
- 3Department of Life and Consumer Sciences, University of South Africa, Pretoria, South Africa
Species have definitive genomes. Even so, the transcriptional and translational products of the genome are dynamic and subject to change over time. This is especially true for the proteins secreted by ticks at the tick-host feeding interface that represent a complex system known as the sialoverse. The sialoverse represent all of the proteins derived from tick salivary glands for all tick species that may be involved in tick-host interaction and the modulation of the host's defense mechanisms. The current study contemplates the advances made over time to understand and describe the complexity present in the sialoverse. Technological advances at given periods in time allowed detection of functions, genes, and proteins enabling a deeper insight into the complexity of the sialoverse and a concomitant expansion in complexity with as yet, no end in sight. The importance of systematic classification of the sialoverse is highlighted with the realization that our coverage of transcriptome and proteome space remains incomplete, but that complete descriptions may be possible in the future. Even so, analysis and integration of the sialoverse into a comprehensive understanding of tick-host interactions may require further technological advances given the high level of expected complexity that remains to be uncovered.
Introduction
Organisms are finite creatures in space and time with genomes that are definitive for a given species. As such, within the population of organisms that comprise a specific species, their genomes will be similar in identity and synteny over ~95–99% of the total genome. This last 3–5% of intra-species diversity comprise alleles, epigenetic differences, single nucleotide polymorphisms and lost or rearranged genes that forms the basis for phenotypic or strain differences. The contribution of the microbiome that forms with the tick genome the hologenome, adds considerable diversity at individual and population level (Díaz-Sánchez et al., 2019). Horizontal transfer may occur from the microbiome to the core genome, but in this case these transferred genes would then become part of the core genome. However, the hologenome is not considered in the current study, since these are exogenous variable factors that may not be part of the core genetic component of a species. The core genetic component comprises not only the genome, but its translational aspects such as the transcriptome and proteome, which are more dynamic in nature, since the final products such as mRNA, proteins and other metabolites show tissue-specific and temporal expression patterns, different half-life's and concentration levels. The impact of concentration levels of proteins and metabolites at the tick feeding site has recently been explored and shown to be critical for effective functioning (Mans, 2019). An extensive summary of functions for tick proteins involved at the tick-host interface was also presented previously (Mans, 2016, 2019; Mans et al., 2016). The acquisition of new functions by gene duplication has also been explored and shown to play an important part in the expansion of gene families in ticks (Mans et al., 2017). The current review considers the progress on quantitative description of molecules at the feeding site and addresses the question on how our understanding of the number of proteins and functions at the feeding site has changed over time due to technological advances and experimental design (Figure 1).
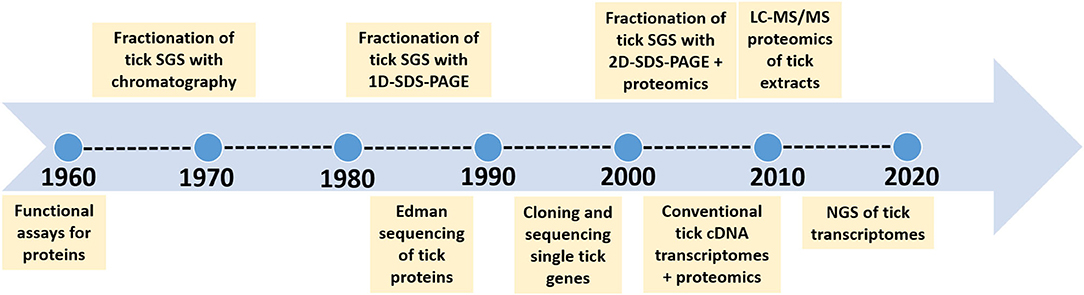
Figure 1. Time line to indicate the major technological advances that improved our estimation of complexity of salivary gland proteins.
Complexity Measured by Morphology and Histochemistry
The recognition that salivary glands have a complex morphology composed of non-secretory and secretory acini (Robinson and Davidson, 1913), as well as multiple cell types within secretory acini, have been well-recognized once cell staining and tissue sectioning could be applied to tick salivary glands (Till, 1959; Chinery, 1965; Balashov, 1972). While soft ticks have a single granular acini, female hard ticks have two types (II and III), while male hard ticks also have a fourth type (IV) (Till, 1959; Roshdy, 1972). Granular acini from both soft and hard ticks all have multiple cell types, with both histochemical staining and immunolocalization indicating that different cells show differential expression of proteins (Roshdy, 1972; Coons and Roshdy, 1973; Mans et al., 2004). Morphological observation was therefore the first indication that salivary glands have higher complexity than may be expected from a simple secretory organ that may be likened to a bag of proteins.
Complexity Measured by Functional Activity
In the absence of fractionation, estimates of complexity may be obtained by measuring unique functions in salivary glands, salivary gland secretions (SGS) or salivary gland extracts (SGE) using empirical methods. As such, it was indicated that tick salivary glands possess numerous functionalities suggesting a complex organ with many functions active at the tick feeding site (Neitz and Vermeulen, 1987). These included enzymatic functions, enzyme inhibitory functions, anticoagulants, cement, toxins and immunogens. Many proteins responsible for these functions were later isolated and identified from numerous tick species, with ~120 experimentally verified tick protein functions to date (reviewed in Mans, 2019). The difficulty in complexity estimation using functional characterization is the sheer immensity of potential known and unknown functions and the amount of effort necessary to characterize these functions (Ribeiro and Arcà, 2009; Mans, 2016).
Complexity Measured by Counting Peaks
Kaire (1966) performed the first chromatographic separation of tick proteins from whole tick extracts to purify the neurotoxin from the Australian paralysis tick, Ixodes holocyclus (Neumann, 1899). However, the first fractionation of tick salivary proteins arguably occurred in the 1960 and 1970s when Neitz purified the first toxins from SGS from the soft tick Ornithodoros savignyi (Audouin, 1826) (sand tampan), now Ornithodoros kalahariensis (Neitz et al., 1969; Howell et al., 1975; Bakkes et al., 2018). The fractionation used combinations of size exclusion, ion exchange and iso-electric focusing methods available at the time. Estimation of the number of proteins that was present in the SGS is dependent on counting protein peaks with corresponding assumptions regarding the complexity of protein numbers within such peaks. Looking at the work of Neitz et al. (1969) in retrospect and doing back of the envelope estimations suggest a low complexity mixture of <100 proteins. For this specific tick species, even with modern HPLC methods, 1-dimensional sodium dodecyl sulfate poly-acrylamide electrophoresis (1D-SDS-PAGE), or 2-dimensional SDS-PAGE (2D-SDS-PAGE), and using SGE rather than SGS (SGE contain both secretory and non-secretory proteins, while SGS should be more abundant in secretory proteins), the complexity estimates did not change much (Gaspar et al., 1995, 1996; Mans et al., 2001, 2002a; Mans and Neitz, 2004a). For other soft tick species the generalization that <200 proteins may be observed in SGE using HPLC, 1D-SDS-PAGE or 2D-SDS-PAGE also holds (Oleaga et al., 2007; Francischetti et al., 2008a,b; Mans et al., 2008).
For the hard tick Amblyomma hebraeum (Koch, 1844) (South African Bont tick), chromatography and microzone electrophoresis suggested a low complexity mixture (Neitz and Vermeulen, 1987). However, it should be noted that the latter study again used SGS from a single feeding stage (partially fed). Whether SGS obtained from chemical induction represents salivary gland complexity has been addressed previously (Mans, 2019), and reservations exist whether such a secretion can be used as accurate measurement of salivary gland protein complexity, since different stimulants results in different proteins secreted (Oliveira et al., 2013).
Complexity Measurement by Chromatographic Fractionation, Edman and Gene Sequencing
Chromatographic fractionation of whole body, SGS or SGE enabled purification of proteins for functional characterization, while development of automated Edman degradation allowed for the first N-terminal amino acid sequences generated for tick salivary gland proteins (Neitz et al., 1983). This approach allowed for the full-length Edman sequencing of a variety of small anticoagulants and platelet aggregation inhibitors (Waxman et al., 1990; Karczewski et al., 1994; Wang et al., 1996). Generation of N-terminal or internal Edman sequences also allowed for the design of degenerate primers or probes that enabled cloning of gene sequences (Keller et al., 1993; Waxman and Connolly, 1993; Gaspar et al., 1996; Joubert et al., 1998; Nienaber et al., 1999; Paesen et al., 1999; Bergman et al., 2000; Valenzuela et al., 2000; Mans et al., 2001, 2002b,c, 2003; Narasimhan et al., 2002; Mulenga et al., 2003). This approach led to generation of ~30 tick salivary gland sequences by 2003. Our insights into salivary gland complexity were heavily skewed at this point in time by the limited number of functions known (Mans and Neitz, 2004b). Even so, we still remain with very few empirically confirmed functions to date (Mans, 2019), although function assignment by homology allowed thousands of potential functions to be assigned to tick proteins (Mans et al., 2016; Pienaar et al., 2018; Ribeiro and Mans, 2020).
Complexity Measured by Counting Bands
The development of high resolution 1D-SDS-PAGE by Laemmli (1970) ushered in a new era of estimating complexity, allowing in theory to differentiate proteins with molecular mass differences of <1,000 Da. Linked with high resolution gel photography, this led to the first glimpses of salivary complexity in the Lone Star tick Amblyomma americanum (Linnaeus, 1758) (McSwain et al., 1982). This study was significant, since it analyzed changes in protein patterns over the course of feeding and showed that differential expression does indeed occur. Counting of the bands indicated at least 100 proteins with different molecular masses. Other studies soon followed with similar estimates in complexity for A. hebraeum, the brown ear tick Rhipicephalus appendiculatus (Neumann, 1901) and the red-legged tick Rhipicephalus evertsi evertsi (Neumann, 1897; Neitz and Gothe, 1986; Viljoen et al., 1986; Wang and Nuttall, 1994; Wang et al., 1999, 2001). At this point in time 1D-SDS-PAGE promised a glimpse at salivary gland protein complexity. However, our ability to identify these proteins with proteomics was not yet at a technological advanced level that would enable this (Rabilloud, 2020). The limitation of 1D-SDS-PAGE to reveal complexity was also limited by the resolution afforded and the fact that members of the same protein family would have very similar molecular weights, even if they have different iso-electric points resulting in underestimation of complexity. As such, 100 bands may very well indicate a 100 protein families rather than a 100 proteins.
Complexity Measured by Counting Spots and Identification by Proteomic Analysis
The advent of 2D-SDS-PAGE that included iso-electric focusing in the first dimension followed by 1D-SDS-PAGE in the second dimension (O'Farrell, 1975), again revolutionized the concept of fractionation of complex mixtures. This produced protein expression maps that correlated with protein iso-electric point and molecular weight represented by spots on a gel. In ticks, the first 2D-SDS-PAGE images were published in 2001 for the deer tick Ixodes scapularis (Say, 1821) and O. kalahariensis (Das et al., 2001; Mans et al., 2001). Analysis of female tick salivary gland extract from O. kalahariensis indicated ~100 proteins on a 2D-electropherogram. The major proteins present in this electropherogram could be confirmed to be soft tick proteins based on protein purification, peptide mass fingerprinting and tick sequences obtained via painstaking single gene sequencing (Mans et al., 2001; Mans and Neitz, 2004a). The 2D-electropherogram for I. scapularis revealed almost 500 spots in SGE (Das et al., 2001), and this complexity was again confirmed (Narasimhan et al., 2007). This latter study also indicated that the proteome changes over the time of feeding using Differential 2D Fluorescence Gel Electrophoresis (DIGE).
The first 2D-SDS-PAGE followed by proteomic analysis was performed for the SGS of A. americanum and the Gulf Coast tick, Amblyomma maculatum (Koch, 1844) (Madden et al., 2002). This revealed ~100–200 protein spots on the 2D-electropherogram. Surprisingly, the majority of spots reacted with anti-sheep serum suggesting that they were host derived. Of 16 prominent spots picked for peptide mass fingerprinting, one spot was identified as a tick protein and five spots as host proteins. It should be considered that at this point only 20 protein sequences were available for A. americanum and only 287 tick proteins were present in Genbank, which may explain the low number of tick proteins identified. This highlighted the impact that the lack of sequence coverage may have on proteomic detection. Other factors that limited the use of 2D-SDS-PAGE as method for saliva analysis is the high salt concentration found in tick saliva that necessitate extra desalting steps (Madden et al., 2002).
Complexity Measured by CDNA Library Sequencing and Proteomics
The advent of complementary DNA (cDNA) library synthesis (Chenchik et al., 1996; Lukyanov et al., 1997) and propagation as unique clones using phage packaging (Kretz et al., 1989), allowed direct sequencing of mRNA derived genes that gave new insight into salivary gland and transcriptome complexity (Table 1). The first cDNA libraries constructed were expression libraries used to identify antigenic proteins recognized by host antiserum and subsequent sequencing of immunogenic clones (Das et al., 2000, 2001; Bishop et al., 2002). This yielded 14 immunogenic sequences for I. scapularis (Das et al., 2001). Larger scale, studies identified 895 immuno-proteins using similar approached in A. americanum (Radulović et al., 2014). Alternative approaches included the use of probes to screen a cDNA library for homologous genes (Sangamnatdej et al., 2002).
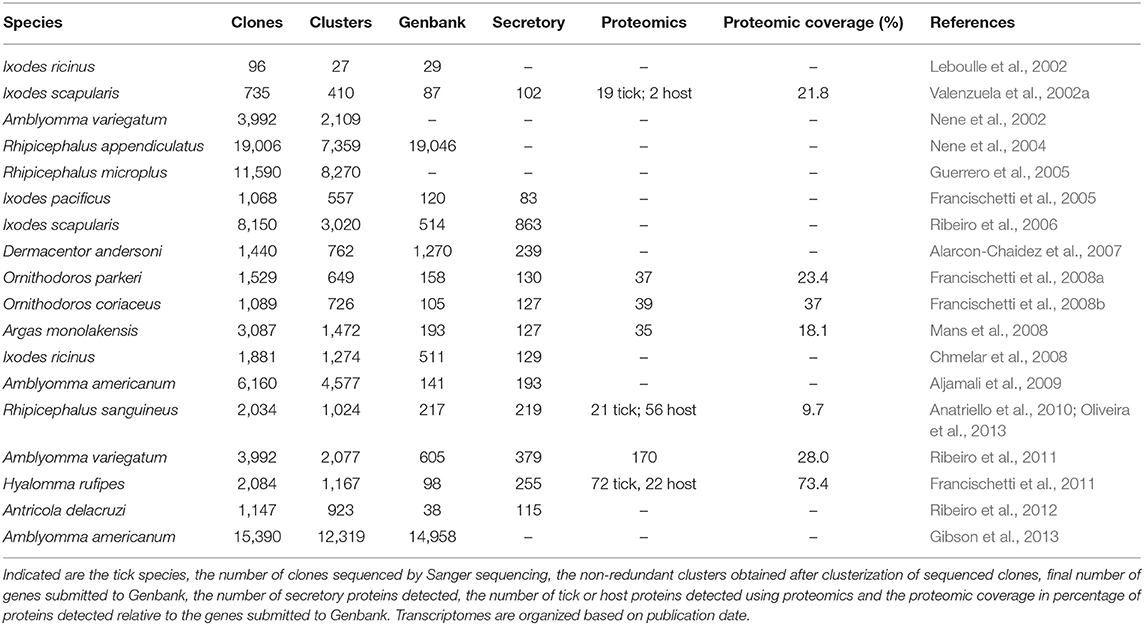
Table 1. Statistics for conventional cDNA library sequencing.
The first tick salivary gland cDNA libraries synthesized and systematically sequenced using Sanger sequencing yielded 36 sequences for the castor bean tick Ixodes ricinus (Linnaeus, 1758) and 87 sequences for I. scapularis, respectively (Leboulle et al., 2002; Valenzuela et al., 2002a). The latter study also identified 19 tick proteins and 2 host proteins using Western blot analysis followed by Edman sequencing and used these sequences to identify corresponding cDNA sequences (Valenzuela et al., 2002a). This already highlighted the importance of having species specific transcriptome sequences available. It also indicated the utility of cDNA library sequencing, when the same cDNA data was used to predict function by homology and subsequent functional characterization of recombinant proteins, leading to the discovery of the anti-coagulant Ixolaris (Francischetti et al., 2002) and a fibrinogenase metalloprotease (Francischetti et al., 2003). The study by Valenzuela et al. (2002a) was further significant in that it represented the first systematic high-throughput randomized sequencing of tick salivary gland cDNA clones (735 clones sequenced that clustered in 410 clusters and yielded 87 unique full-length sequences), that also attempted a systematic classification of salivary transcripts into groups or protein families (7 groups plus various singletons). Secretory proteins accounted for 102 clusters and 310 of the clones sequenced. It also introduced for the first time the term sialome (from the Greek σíελoç = saliva) to describe the set of mRNA and proteins expressed in the salivary glands of ticks. A novel algorithm of deconvoluting Edman sequences obtained from crude SGE fractionated on 1D-SDS-PAGE was also presented, that allowed mixed Edman sequences to be matched to the transcriptome (Valenzuela et al., 2002b). This technique was subsequently successfully used in various transcriptome studies to identify abundant proteins (Francischetti et al., 2008a,b, 2011; Mans et al., 2008; Ribeiro et al., 2011).
Shortly after this groundbreaking study, more studies started to report on systematic conventional cDNA library transcriptome Sanger sequencing. For the Tropical Bont tick, Amblyomma variegatum (Fabricius, 1794), the salivary gland transcriptome was presented as the AvGI (A. variegatum gene index) and sequenced 3,992 clones of which 2,109 was non-redundant and 822 showed similarity to sequences in the database (Nene et al., 2002). No proteomics was performed, and no systematic classification presented, although a high-level gene ontology was assigned. A number of years later the same dataset was re-analyzed with the aim at classifying the genes and resulted in 3,985 EST sequences clustering into 2,077 contigs, of which 605 was submitted to Genbank (Ribeiro et al., 2011). The secretory proteins (379) were classified into 21 groups or families and a 1D-SDS-PAGE fractionation of SGE, from which 24 bands were analyzed by proteomics resulted in the identification of 170 proteins that represented 28% of the EST database submitted to Genbank.
For R. appendiculatus, 9,162 clones from an uninfected cDNA library and 9,844 clones from a Theileria parva infected cDNA library were sequenced (Nene et al., 2004). This resulted in 7,359 non-redundant sequences. Secretory genes were not classified in any systematic manner although gene ontology was presented. No proteomics was performed. This was followed by the Rhipicephalus microplus (Canestrini, 1888) gene index (BmGI) (Asian Blue tick) that was a whole body transcriptome where 11,590 clones were sequenced from a normalized cDNA library resulting in 8,270 unique sequences (Guerrero et al., 2005). A proteomic analysis of larval extract fractionated using 2D-SDS-PAGE, from which 20 spots were selected resulted in 18 proteins being identified (Untalan et al., 2005). This indicated the utility of a representative species specific transcriptome databases.
The salivary gland transcriptome of the Western-Blacklegged tick, Ixodes pacificus (Cooley and Kohls, 1943) was constructed via sequencing of only 1,068 clones that clustered into 557 contigs (Francischetti et al., 2005). Even though few clones were sequenced, classification of secretory proteins still resulted in 15 groups and 83 secretory proteins, creating the notion that the most abundant proteins and therefore those most important for feeding would still be represented in small scale transcriptome sequencing projects. However, a project that sequenced 8,150 clones from I. scapularis from nymphs and adults from various feeding stages resulted in 3,020 contigs and 863 unique secretory proteins (Ribeiro et al., 2006). This study was the first indication that secretory proteins may be present at levels of more than 500 proteins and possibly extending into thousands, suggesting that tick salivary gland diversity may be much more extensive than previously expected. The transcriptome for I. ricinus was also described from four different cDNA libraries (unfed, 24 h after attachment, 4 days—partially fed and 7 days—fully engorged) (Chmelar et al., 2008). A total of 1,881 clones were sequenced that clustered into 1,274 clusters of which 511 was submitted to the nucleotide database. Of these 129 was classified as secretory.
Alarcon-Chaidez et al. (2007) described the salivary gland transcriptome of the Rocky Mountain Wood tick, Dermacentor andersoni (Stiles, 1908). The study sequenced 1,440 clones that clustered into 762 unique sequences. Of these ~75% found homologs in the existing databases. The salivary gland transcriptome for A. americanum was described by Aljamali et al. (2009). A total of 6,160 clones were sequenced from both non-normalized and normalized libraries and clustered in 4,577 contigs. Of these 141 were submitted as proteins to Genbank. Reanalysis of the nucleotide sequences identified 193 secretory proteins in the dataset (Mans et al., 2016). A follow up study sequenced 15,390 clones that yielded 12,319 unique sequences (Gibson et al., 2013). The study observed that 71% of all sequences generated could not be annotated by function assignment by homology. Possible reasons presented were divergence in the tick lineage, the presence of lineage specific genes and limited genomic resources for ticks that would allow homology assignment. The study did not attempt to assign these genes to secretory families and the high level of non-homologous orphan sequences that did not find any hits to other tick sequences remain surprising.
Anatriello et al. (2010) described the salivary gland transcriptome of the brown dog tick, Rhipicephalus sanguineus (Latreille, 1806) and sequenced 2,034 clones that yielded 1,024 non-redundant sequences. Secretory proteins comprised 219 sequences and were classified into 12 protein family classes. A proteomic study was performed of saliva collected by either dopamine or pilocarpine stimulation (Oliveira et al., 2013). Saliva was fractionated on 1D-SDS-PAGE, bands were excised, followed by tryptic digestion and nanoflow reversed-phase liquid chromatography tandem mass spectrometry (nanoRPLC-MS/MS). The transcriptome database previously generated was used for analysis. The protein profiles obtained with dopamine or pilocarpine stimulation differed significantly with pilocarpine stimulation obtaining more bands. For dopamine stimulation only two lipocalins could be identified. For pilocarpine stimulation, 56 rabbit proteins were identified and only 19 tick proteins. Some of the rabbit proteins may have close homologs in ticks, although 16 rabbit proteins were detected that would be mammal specific. Of interest, is that the lipocalins secreted with dopamine were not found in the pilocarpine secretion, which raised again the question regarding representation of secretory proteins obtained with chemical stimulants.
The final ixodid sialome to be analyzed and described in a systematic manner using conventional cDNA sequencing was for the coarse bont-legged tick, Hyalomma rufipes (Koch, 1844) (Francischetti et al., 2011). A total of 2,084 clones were sequenced, clustered into 1,167 contigs of which 255 were classified as secretory and 98 proteins were submitted to Genbank. From 20 1D-SDS-PAGE bands analyzed by MS/MS, 72 tick proteins were identified as well as 22 host proteins. The identified proteins comprised 73.4% of the proteins submitted to Genbank.
By 2006 ~12 soft tick salivary gland proteins have been functionally characterized and single genes cloned and sequenced. No salivary gland transcriptomes was systematically described up to this point for soft ticks. A proteomic analysis of SGE from Ornithodoros erraticus (Lucas, 1849) and Ornithodoros moubata (Murray, 1877), the African hut tampan, based on the proteins then present in the databanks underscored the importance of having species specific transcriptome databases necessary for proteomic analysis (Oleaga et al., 2007). For O. moubata, 48 non-redundant proteins were present in the databank and for 40 2D-SDS-PAGE spots analyzed only two proteins (TSGP1 and moubatin) could be identified. For O. erracticus, no proteins were available in the databanks and from 54 spots analyzed only 7 proteins were identified. In a follow-up study proteomic analysis of saliva from O. moubata was performed using in situ tryptic digest followed by liquid chromatography-MS/MS (LC-MS/MS) (Díaz-Martín et al., 2013). At this point in time 75 sequences were available in the databanks for O. moubata and proteomic analysis identified 193 proteins. Of these, 99.9% of the protein abundance was accounted for by the major lipocalins: TSGP1, TSGP4, and moubatin, while the remaining 0.1% abundance was accounted for by housekeeping proteins similar to sequences from ticks and insects in the database. The presence of housekeeping proteins may be explained by apocrine secretion or perhaps even salivary gland cell degradation given the low abundance found (Mans et al., 2016). As indicated before, the functional provenance of these low abundance proteins at the tick feeding site needs to be confirmed (Mans, 2019).
In 2008, three argasid salivary transcriptomes were reported for the Mono Lake bird tick Argas monolakensis (Schwan et al., 1992), the pajaroello tick Ornithodoros coriaceus (Koch, 1844) and the relapsing fever tick Ornithodoros parkeri (Cooley, 1936) (Francischetti et al., 2008a,b; Mans et al., 2008). For A. monolakensis, 3,087 clones were sequenced and clustered to give 1,472 contigs of which 127 was classified as secretory and 193 proteins were submitted to Genbank (Mans et al., 2008). Analyses of 78 2D-SDS-PAGE spots identified 18 spots and 14 proteins, 14 bands analyzed by Edman sequencing identified 13 proteins, and 116 peaks obtained from HPLC fractionation identified 52 peaks and 27 proteins, resulting in a final number of 35 proteins identified. This comprised 18% of the contigs coding for housekeeping and secretory proteins and 40% of the protein samples analyzed. For O. coriaceus, 1,089 clones were sequenced, resulting in 726 contigs, 127 classified as secretory and 105 proteins submitted to Genbank. Edman sequencing of 1D-SDS-PAGE fractions identified 3 proteins, analysis of 60 2D-SDS-PAGE spots identified 7 proteins and proteomic MS/MS analysis of 61 bands from 1D-SDS-PAGE analysis identified 37 proteins, resulting in a final number of 39 proteins (Francischetti et al., 2008b). This comprised 37% of the contigs submitted to Genbank. For O. parkeri, 1,529 clones were sequenced, resulting in 649 contigs and 158 proteins submitted to Genbank (Francischetti et al., 2008a). Edman sequencing of 1D-SDS-PAGE bands identified 12 proteins, analysis of 60 2D-SDS-PAGE spots identified 11 proteins and proteomic MS/MS analysis of 51 bands from 1D-SDS-PAGE analysis identified 30 proteins, resulting in a final number of 36 proteins (Francischetti et al., 2008a). This comprised 22.7% of the contigs and 42% of the proteomic fractions analyzed. All of the proteins were secretory and the majority belonged to the 5′-nucleotidase (apyrase), basic pancreatic trypsin inhibitor (BPTI), basic tails secretory (BTSP), cystatin, and lipocalin families.
The results obtained for the argasid sialomes indicated that conventional cDNA libraries sequenced at low levels may not represent all proteins present, but that the proteome also do not represent all transcripts found in the sialome. It also indicated that different methodologies may identify different protein sets and are therefore complementary. Even so, it also made the case that specific transcriptomes contribute toward a higher rate of protein identification. It also indicated that for all of these sialomes the major abundant proteins identified were classified as secretory, notably belonging to the BPTI, BTSP and lipocalin families. It also suggested that soft tick sialomes may represent lower complexity than ixodids. Reasons for this may be traced to the short feeding events of soft ticks, where feeding occurs within a few minutes to hours rather than days (Mans and Neitz, 2004b). This result in secretion of a bolus of salivary material and not the differential expression patterns observed for ixodid ticks (McSwain et al., 1982; Wang and Nuttall, 1994).
The salivary transcriptome for female Antricola delacruzi (Estrada-Peña et al., 2004) was also described using conventional cDNA library sequencing (Ribeiro et al., 2012). A total of 1,147 clones were sequenced, resulting in 923 contigs, 115 annotated as secretory proteins with 38 proteins submitted to Genbank. The transcriptome differed completely from those of other soft tick species where the major soft tick protein families are BPTI, BTSP and lipocalin families. Instead, ferritin, mucins with chitin-binding domains and TIL-domain-containing proteins were abundant. A reason for this may lie in the fact that Antricola adults do not feed on blood. The larvae and possibly nymphs do feed on blood (Estrada-Peña et al., 2008), and it may be expected that these life stages may present the more canonical protein families found in blood-feeding argasids.
Conventional Sanger sequencing of cDNA libraries to generate insights into salivary gland transcriptome composition contributed tremendously to our knowledge of salivary gland complexity, even if in the final analysis it became clear that this approach was not exhaustively descriptive or quantitative. This period saw the classification of secretory salivary gland proteins into well-described families (Francischetti et al., 2009), the detection and description of abundant secretory proteins and the observation that protein families are conserved among tick families. It showed that ticks possess their own lineage specific protein repertoires, that gene duplication plays a significant role in creating lineage specific expansions and that the salivary gland protein repertoire may be much more complex than we expected originally (Mans et al., 2008). The data contributed to our ability to detect and identify salivary proteins by proteomic analysis and showed that species-specific sequence databases are crucial for proteomic identification. Where transcriptome data was available, a significant number of genes in the transcriptome could be confirmed by proteomics. This was a golden age in salivary gland discovery and we may have happily continued with this low-level description if the next revolution in sequence technology did not occurred, namely next-generation sequencing.
Complexity Measured by Next-Generation Sequencing of Transcriptomes and Proteomics
The ability to generate automated high-throughput sequence data that was independent of cDNA library construction was made possible by the development of a wide variety of next-generation sequencing technologies that included Roche 454, Ion Torrent, Illumina HiSeq, and MiSeq technologies (Levy and Myers, 2016). These technologies purified mRNA directly, fragmented and directly sequenced all fragments at the same time resulting in large datasets with millions of reads. The reads are then assembled into contigs using a variety of next-generation algorithms (Martin and Wang, 2011). It enables the complete de novo assembly of a whole transcriptomes in the absence of any genome data. It also has the advantage that sequence depth may be converted into sequence coverage, giving an indication of transcript abundance and differential expression.
The first tick salivary gland transcriptome sequenced using these technologies was for A. maculatum using the Roche 454 GS FLX titanium pyrosequencing (Karim et al., 2011). The run generated 1,626,969 reads with an average length of 344 bp. An initial 190,646 contigs were assembled, but were reduced to 72,441 using a size cut-off above 149 bp. A final number of 15,814 were analyzed of which 4,849 were submitted to Genbank. An astounding 3,475 secretory proteins were identified with 304 members of the lipocalin family, the latter comprising almost the number of secretory proteins previously found in conventional cDNA libraries. These numbers were orders of magnitude higher than any transcriptome previously produced and hinted a depth of complexity not previously imagined.
The next transcriptome to be described was for I. ricinus using a combination of Roche 454 and Illumina technology (Schwarz et al., 2013). For Roche 454, 441,381 reads were generated and for Illumina an astounding 67,703,183 reads for the time. Assembly of the combined reads resulted in a total of 272,220 contigs reduced to 82,907 after a size and coverage cutoff. Of these 34,560 were annotated and 8,586 submitted to Genbank. Of the 82,907 contigs, 13% where classified as secretory and of the Genbank sequences, 3,882 was classified as secretory proteins and an astounding 564 as lipocalins. A follow up study expanded the read coverage by 315 million additional reads that were combined with the previous studies reads to generate a new assembly from which 25,808 contigs were extracted (Schwarz et al., 2014). The study also included midgut transcriptomes and focused on the first 24 h of feeding. These contigs were used for proteomic analysis of SGE and identified 1,510 proteins from nymphs and adults at 12, 24, and 36 h post-attachment. This is an impressive improvement in proteome coverage compared to number of proteins identified using conventional cDNA libraries. However, the direct digestion of SGE without fractionation may have influenced the total number of proteins detected since crude SGE presents a very high complexity mixture. The transcriptome and proteome data did not correlate with regards to abundance or relative changes, even though biological and technical replicates showed good correlation. Interestingly, the proteomes for the salivary gland and midgut did not differ significantly and ~60% of all proteins did not show any variation in abundance over all time periods sampled. This may reflect that proteomics detect many housekeeping proteins in addition to secreted proteins. Transcriptome levels varied more suggesting transcriptional changes not reflected in the proteome. A sister study classified the same 25,808 contigs according to function and protein family composition as well as for differential expression (Kotsyfakis et al., 2015). This study indicated significant differences in transcript expression profiles between salivary gland and midgut samples. Transcript expression profiles also change over feeding time for different protein families suggesting that “gene switching” occur. It has been suggested that this is a possible way to evade the immune system of the host. In addition, the study indicated higher levels of non-synonymous substitution in secretory proteins suggesting that this indicate higher rates of positive selection.
In a follow up study for I. ricinus, transcriptomes from single tick salivary glands were generated (Perner et al., 2018). Salivary glands were sampled from ticks fed on rabbits or artificial membranes at 24, 48, and 72 h. Approximately 435 million Illumina paired reads were generated from 18 libraries and assembled with Illumina reads previously generated in other studies and 40,490 coding contigs were extracted and a final 20,773 contigs with RPKM > 10 were annotated. The study identified 1,907 novel protein sequences of which 406 were identified as secretory. The study indicated that individual ticks show differential expression between ticks and over the course of a blood-meal.
The sialotranscriptomes for three different Amblyomma species from Brazil were sequenced using pyrosequencing (Garcia et al., 2014). For Amblyomma parvum (Aragão, 1908) 104,817 reads were generated with a final number of contigs of 3,796 of which 493 was annotated as secreted. For Amblyomma cajennense (Fabricius, 1787) 67,677 reads were generated with a final number of contigs of 4,604 of which 1,015 was annotated as secreted. For Amblyomma triste (Koch, 1844) 442,756 reads were generated with a final number of contigs of 11,240 of which 1,861 was annotated as secreted.
Next-generation sequencing of A. americanum sampled unfed, 12, 18, 24, 36, 48, 72, 120, 144, 168, 192, 216, and 264 h after attachment and generated a total of 344,909,378 paired reads (Karim and Ribeiro, 2015). Assembly resulted in a final number of 5,792 contigs that was analyzed. Of these, 2,153 were classified as secretory. Significant differences in transcript levels were observed over the course of feeding, supporting differential expression and the concept of sialome switching was introduced to suggest that the tick switch between different transcriptomes during the course of feeding.
For the zebra tick, Rhipicephalus pulchellus (Gerstäcker, 1873) 241,229,128 paired Illumina reads were generated from salivary glands of males and females that were unfed or fed for 1, 3, and 7 h as well as 1, 2, 3, 4, 5, 6, and 7 days (Tan et al., 2015). Assembly resulted in 50,460 coding sequences of which 7,134 was secretory. Proteomics was performed using SGE and in situ trypsin digestion followed by LC-MS/MS. This identified 2,231 proteins of which 221 were secretory. Significant differences in transcriptome and proteome expression were observed between males and females.
For Haemaphysalis flava (Neumann, 1897), engorged and semi-engorged female salivary glands were used to generate 162,912,848 paired reads, which after assembly yielded a final number of 54,357 contigs (Xu et al., 2015). Of these, 20,145 had homologs in Genbank and 13,513 could be assigned GO terms. Secretory proteins were not reported.
The salivary gland transcriptomes for brown ear ticks, vectors of T. parva, the causative agent of Corridor disease, East Coast fever and Zimbabwe theileriosis were described (de Castro et al., 2016, 2017). For R. appendiculatus, 430 million paired reads were generated with a final contig number of 21,410 contigs of which 13,996 coded for proteins, of which 2,135 were secretory proteins and 7,414 were annotated as long non-coding RNA. For R. zambeziensis (Walker et al., 1981), ~190 million paired reads were generated, assembled and resulted in 140,703 transcripts. After a FPKM > 1 cut-off a final contig number of 23,631 transcripts were obtained of which 15,737 were coding for proteins of which 2,569 were classified as secretory.
For Amblyomma sculptum (Berlese, 1888), fed and unfed ticks were sampled and ~567 million single reads were generated (Esteves et al., 2017). Assembly resulted in 9,560 contigs of which 2,314 secretory proteins were found in unfed and 2,592 in the fed transcriptomes. Proteomic analysis identified 124 proteins in saliva of which 58 were secretory. Another study on A. sculptum, assembled 195,594,989 Illumina paired reads, 844,529 Ion Torrent reads and 703,210 Roche 454 reads that derived from Rickettsia amblyommii infected and uninfected organs that included ovaries, midguts and salivary glands (Moreira et al., 2017). Assembly resulted in 460,445 contigs of which 27,308 were selected for annotation. Of these 2,177 were annotated as secreted salivary gland proteins.
For Hyalomma excavatum (Koch, 1844), 138,144,530 Illumina paired reads were generated (Ribeiro et al., 2017). After assembly, 53,228 contigs were obtained and 7,875 coding sequences were annotated with 1,796 classified as secretory. For the camel tick, Hyalomma dromedarii (Koch, 1844), male and female ticks at different stages of engorgement were collected from camels in the field (Bensaoud et al., 2018). Illumina sequencing generated 330,285,649 paired-end reads of which 55,819,366 were assembled. After PFAM filtering, 15,342 contigs were annotated of which 1,749 was classified as secretory. The transcriptome study was followed up with proteomic analysis of SGE from ticks collected in the field (Bensaoud et al., 2019a,b). Fractionation of SGE was performed using 1D-SDS-PAGE, after which bands were cut-out, tryptic digested and analyzed using LC-MS/MS. As a target database for the proteomic study, the sialotranscriptome previously generated was used in addition to Acari sequences from Genbank. This identified 1,111 proteins of which 854 were from the Acari database and 257 from the species-specific database. Only 24% of proteins were shared between males and females and 19% of proteins (~211) were classified as secreted.
For I. holocyclus, semi-engorged ticks were collected from animals with confirmed paralysis symptoms, before salivary glands were dissected (Rodriguez-Valle et al., 2018). Illumina sequencing generated 65,035,631 paired reads that produced 134,039 contigs after assembly of which ~3,149 was classified as secretory.
For the cattle tick Rhipicephalus annulatus (Say, 1821), engorged female ticks that were uninfected or infected with Babesia bigemina were used to construct salivary gland transcriptomes (Antunes et al., 2019). Illlumina sequencing generated 40,573,988 paired reads that generated 33,379 contigs in the control and 30,435 contigs in the infected sample of which 16,564 and 15,037 gave significant BLAST hits, respectively. This was reduced to 6,823 and 6,475 unigenes, respectively. Proteomics identified 4,594 proteins in female tick extracts.
For Amblyomma aureolatum (Pallas, 1772), transcriptomes were generated from Rickettsia rickettsii infected and uninfected tissues (Martins et al., 2019). Approximately 242 million reads were generated of which 110 million were from salivary glands. Assembly generated 11,906 contigs of which 11,903 were expressed in salivary glands.
For the Asian longhorned tick, Haemaphysalis longicornis (Neumann, 1901), salivary gland transcriptomes were generated that resulted in 18,313 contigs. The transcriptome was not published, but were used for proteomic analysis with iTRAQ labeling to allow for differential quantification of salivary glands from ~6,000 female ticks that was analyzed as unfed, partially fed, semi-engorged and engorged in three replicates (Ren et al., 2019). Replicate experiments identified 5,059, 5,526, and 5,584 proteins that resulted in 3,667 high-confidence proteins with 2,507 present in all replicates. The majority of proteins identified were housekeeping with significant upregulation of proteins observed during feeding.
A proteomic LC-MS/MS analysis of SGE were performed for the brown ear tick Rhipicephalus bursa (Canestrini and Fanzago, 1878) using the Ixodidae database from Genbank (Couto et al., 2020). SGE were prepared from unfed and fed females, which was also either infected, or not with Babesia ovis. A total of 1,586 proteins were identified even though only 35 proteins exist in Genbank for this tick species. The majority of proteins were housekeeping, which underscore the utility of sequence databases to identify homologous proteins, but also the impact of the absence of databases to identify species specific proteins.
Giachetto et al. (2020) generated salivary gland transcriptomes for engorged female R. microplus that was fed on tick-resistant or tick-susceptible hosts. The transcriptome was assembled from 74,639,552 reads (holstein cattle) and 63,013,658 reads (crossbreed cattle) resulting in 235,451 contigs. Of these 71,757 could be annotated by BLAST analysis but only 1,815 ORFs were extracted of which 20 could be annotated as secretory.
For R. sanguineus, female ticks were collected based on weight rather than just days and included unfed, 1.8 mg (day 2), 3.6 mg (day 6), 7.0 mg (day 6), 10.9 mg (day 8), 24 mg (day 8 and 11), 36 mg (days 6, 10, 13), and a sialotranscriptome was constructed (Tirloni et al., 2020). For 20 different libraries that included replicates, 687 million reads were generated from which 71,643 coding sequences were obtained. These were further reduced to 28,921 transcripts with expression values that would represent a minimum of 0.001% of all transcripts based on TPM value (TPM ≥ 10). Of these 8,178 could be annotated with 4,039 identified as secretory proteins. From a similar time series, proteomic analysis of SGE was performed using LC-MS/MS and the transcriptome database generated. This identified 2,125 proteins of that showed a positive correlation between proteome and transcriptome abundance for those transcripts with TPM > 10 (1,745 transcripts). Of 221 with positive correlation, 30% were secretory. A significant number of host proteins (47) were also identified in the SGE. Variability in expression, both in transcript and protein were less marked than previous studies, most probably since the feeding phases of the ticks were better correlated due to weight rather than time. The study again indicated differential expression and the sialome switching phenomenon, both at transcriptome and proteome level.
To date only one salivary gland transcriptome have been described for soft ticks from next-generation sequencing data, namely for Ornithodoros rostratus (Aragao, 1911) (Araujo et al., 2019). Salivary glands and midguts were collected from fourth instar nymphs that were unfed or 1 and 4 day fed. Sequencing generated 22,395,831 combined Illumina paired reads that were assembled into 40,058 contigs. Filtering based on ORFs generated a final set of 8,031 contigs that were annotated from which 717 proteins were classified as secretory. In contrast to hard ticks, the secretory proteins in the salivary glands were more abundant than housekeeping proteins and represented 67% of all reads. Another study also sequenced the salivary gland transcriptome of the relapsing fever tick, Ornithodoros turicata (Dugès, 1876), but did not describe the transcriptome in a systematic manner (Bourret et al., 2019). The study generated 15,136,406 single-end reads that generated 10,989 contigs after assembly, with 2,138 classified as secretory proteins.
It should be clear from the summary (Table 2) that large variations in data generated, numbers of contigs assembled, final contigs selected for annotation and the final number of contigs submitted to Genbank exists for salivary transcriptomes generated with next-generation sequencing. Given this large variation it may be safe to assume that the transcriptomes for salivary glands may not necessarily be representative of the full complement of proteins expressed in the salivary glands. Additionally, it is not yet clear whether the extremely high number of secretory genes found in the transcriptomes (that increase with each new sampling and sequencing effort) is due to artifacts or a tick-specific mechanism to generate genetic diversity (Mans et al., 2016; Ribeiro and Mans, 2020).
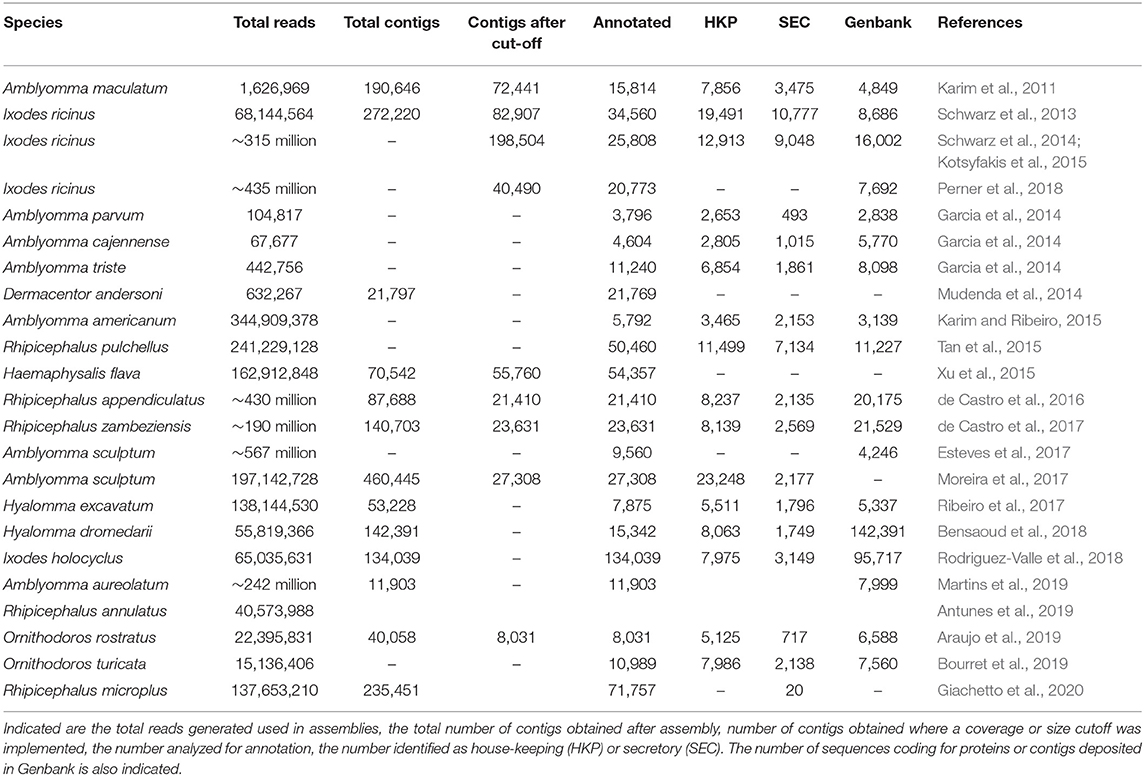
Table 2. Statistics for salivary gland transcriptomes assembled using next-generation sequencing technologies.
Next-Generation Sequencing of Whole Body Transcriptomes
Whole body transcriptomes will by default contain salivary gland derived transcripts. In addition, whole body transcriptomes should at least in theory represent the total protein complexity found in a species at the time of sampling and may as such give a better idea on the upper limits of proteins that may be found in salivary gland transcriptomes.
For the ornate cow tick, Dermacentor reticulatus (Fabricius, 1794), 7 day unfed larvae were used to construct a transcriptome to investigate stress responses in larvae (Villar et al., 2014). A total of 18,946 transcripts were obtained that were reduced to 3,808 unigenes. Proteomics on larval extracts identified 74, 239 and 104 proteins using various approaches, respectively. The low number of proteins identified may be due to sample complexity or the reduction of proteins to unigenes that likely represented loss of paralogous genes. For R. sanguineus, a larval transcriptome was generated for descriptive purposes (De Marco et al., 2017). A total of 5,566,986 short paired-end reads generated 33,396 contigs after assembly and filtering that represented 16,555 unique genes. Dehydration stress in D. variabilis was investigated by sequencing unfed male ticks that was dehydrated (Rosendale et al., 2016). From six libraries, 271,494,907 reads were used for assembly that generated 61,800 contigs that were analyzed, of which ~40,000 found BLAST hits. For engorged female H. longicornis, ~53 million reads generated 65,916 contigs, of which 23,339 could be annotated (Niu et al., 2019). Whole body transcriptomes for larvae (64,474,326) and nymphs (81,612,022) were also generated (Guo et al., 2019). This resulted in 536,336 transcripts and 440,896 unigenes. Of these 22,347 and 15,112 were annotated by KOG and KEGG databases, respectively. For I. ricinus, 15 libraries were constructed for unfed and fed nymphs, unfed males, unfed and fed females (Charrier et al., 2018). From 162,872,698 reads 427,491 contigs were produced that were reduced to a non-redundant dataset of 192,050 contigs. Removal of mammalian and fungal contaminants resulted in 179,316 contigs. Of these 56,809 produced BLAST hits to the Uniref90 or Swissprot databases. Only 12,838 genes were shared with other I. ricinus transcriptome studies, while 36,652 were unique to this study and 23,686 was unique to other transcriptomes bringing the potential number of sequences to 73,176 genes. In a follow-up study, the whole body transcriptomes for a number of additional Ixodes species were generated for use in phylogenomic analysis using combinations of unfed or fed nymphs, males or females (Charrier et al., 2019). This included Ixodes acuminatus (Neumann, 1901) (n = 20,250), Ixodes arboricola (Schulze and Schlottke, 1930) (n = 22,179), Ixodes canisuga Johnston, 1849 (n = 15,238), Ixodes frontalis (Panzer, 1798) (n = 18,187), Ixodes hexagonus (Leach, 1815) (n = 3,215), Ixodes holocyclus (n = 15,520), Ixodes uriae (White, 1852) (49,056), Ixodes ventalloi (Gil Collado, 1936) (n = 16,563) and Ixodes vespertilionis (Koch, 1844) (n = 21,090) with the final number of contigs assembled indicated.
Whole body transcriptomes indicate that expected upper limits for salivary gland transcriptomes may range from ~15,000–30,000 genes that can be annotated with our existing annotated databases. One major problem that may exist is overestimation of existing genes due to miss-assembly, such as insertions or deletion not due to exon-intron splicing, or extension of 5′ or 3′ ends that cause such genes to be identified as unique even though the rest of the gene is 100% identical to the canonical gene. While existing algorithms may identify and remove chimeric transcripts (miss-assembly due to linkage of two canonical genes or fragments), identifying miss-assembled transcripts due to small inserts, deletions or extensions is more difficult with existing algorithms and require extensive manual curation.
Proteomics of Tick Saliva
Apart from the studies mentioned above that specifically focused on salivary gland transcriptome descriptions with added proteomic analysis, some studies focused more specifically on the analysis of tick saliva using proteomics (Table 3). The salivary transcriptome of D. andersoni were sequenced using Roche 454 technology for the purpose of proteomic analysis (Mudenda et al., 2014). A total of 632,267 reads were generated at three time points (Day 0, Day 2, and Day 5). Assembly yielded 21,769 unique sequences coding for proteins. Each time point presented unique transcripts as well as up- or down-regulation of genes during the course of feeding. The study also collected saliva for day 2 and day 5 of feeding and fractionated this with 2D chromatography (in-line cation exchange linked with reversed phase) yielding 30 fractions analyzed by MS/MS. An impressive number of 677 proteins were detected.
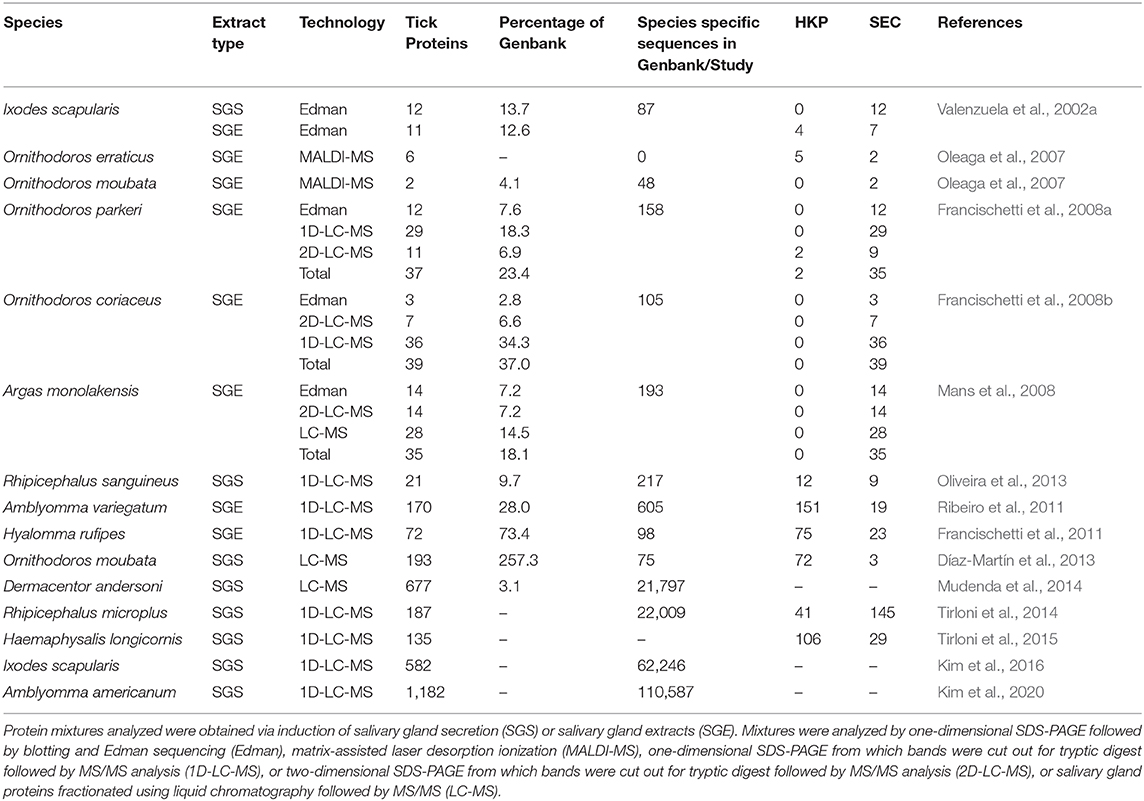
Table 3. Summary of proteomic analysis for tick salivary gland proteins using various technologies.
Tirloni et al. (2014) performed proteomic analysis on saliva collected from partially and fully fed R. microplus. For this, saliva was fractionated on 1D-SDS-PAGE and 42 bands for partially engorged and 15 bands for fully engorged was excised, in-gel trypsin digested and analyzed on LC-MS/MS. Saliva was also directly digested in situ before LC-MS/MS analysis. Spectra were analyzed against an in-house database of 22,009 protein sequences. The study identified 187 tick and 68 host proteins. Another study that also compared saliva collected from partially and fully fed R. microplus by iTRAQ labeling followed by LC-MS/MS analysis, identified 322 unique proteins of which 41 was considered high-confidence and was found in both partially and fully fed samples (Feng et al., 2019).
Using a similar approach to Tirloni et al. (2014), performing in situ trypsin digestion of saliva from nymphs and adults from H. longicornis, followed by LC-MS/MS analysis, 135 tick and 100 rabbit proteins were identified (Tirloni et al., 2015). The authors used the in-house salivary gland transcriptome database of 22,009 sequences from R. microplus and the Ixodidae sequences from Genbank. In a study using a similar methodology that collected saliva from I. scapularis females at 24, 48, 72, 96, and 120 h as well as engorged and detached, a total of 769 tick and 130 rabbit proteins were identified (Kim et al., 2016). The NCBI non-redundant database (62,246 Ixodidae entries) were used for identification, rather than a species-specific database, since this database presumably presents the most up to date collection of I. scapularis sequences derived from both the genome and transcriptomes. The authors indicate that protein profiles change over the course of feeding and suggest that this switching mechanism is used as immune evasion strategy. More recently, the saliva profile of A. americanum was determined using the in situ digestion, LC-MS/MS approach (Kim et al., 2020). Saliva was collected from ticks attached for 24, 48, 72, 96, 120, 144, 168, and 192 h, as well as engorged and detached. The database used was from sequences generated previously (Radulović et al., 2014). This transcriptome was reassembled from the datasets and generated 110,587 contigs used for analysis. This study identified 1,182 tick and 335 rabbit proteins.
From these studies, saliva complexity at any given point seems low and corresponds with roughly 200 proteins. Complexity increase with number of sampling points, since protein expression patterns change over time, but it would seem, also with the size of the species-specific database. With regard to sampling points, the current practice is to sample at 24 h intervals and protein expression patterns may change between sampling events, suggesting that change can occur within hours. At yet, it is not clear how fast this change can occur. Even so, not all proteins identified are secretory, but many are housekeeping and some host-derived (Tables 1, 3). The numbers identified still do not correlate with the almost 10-fold larger number of transcripts identified in the transcriptomes (Mans et al., 2017). This would suggest that transcriptomes generate an over-estimation of complexity or that proteomic analysis are not yet technologically advanced to identify all proteins present in complex salivary mixtures.
The above sections detailed both transcriptome and proteomic studies since these are generally closely linked. It has been indicated that species-specific transcriptomes are essential for detection of lineage specific proteins and to increase proteome coverage. Even so, the use of proteomes to validate transcriptomes is just as valid. In a perfect world, each contig produced by a transcriptome sequencing project should be confirmed and validated by proteomic analysis, since proteins represents functional entities (Mans, 2019). However, the coverage of transcriptomes by proteomes has been dismal to date (Figure 2). In the case of transcriptomes derived from conventional cDNA libraries the transcriptome coverage detected by proteomics was decent, ranging from 10 to 73% (Table 1; Figure 2). This could be explained by the representation of highly abundant transcripts in relatively small transcriptomes, correlating with highly expressed proteins in the proteome. As transcriptome sizes increased, the number of proteins detected in the proteome also increased, but not at the same ratio. As such, while proteins detected by proteomics may range in the thousands, the percentage of transcriptome coverage is lower compared to conventional cDNA derived transcriptomes (Table 3). The potential reasons for this are numerous. Proteomic technology may not yet be sensitive enough to obtain the depths attained by next-generation sequencing of transcriptomes. This may be linked to both hardware limitations of mass spectrometry equipment, but also to sample fractionation strategies. In regard to the latter, the more complex the sample, the lower the expected coverage that may be obtained. Fractionation of samples that decrease sample complexity but retain yields for low abundance proteins should yield higher detection numbers for proteins. The number of time points sampled for proteomics may impact on the number of proteins identified, with more sampling points yielding more proteins. Transcriptomes may present artifacts, i.e., transcripts that do not represent proteins present in a given sample. This may be due to misassemblies or transcription of pseudogenes that are not translated into protein (Mans et al., 2017).
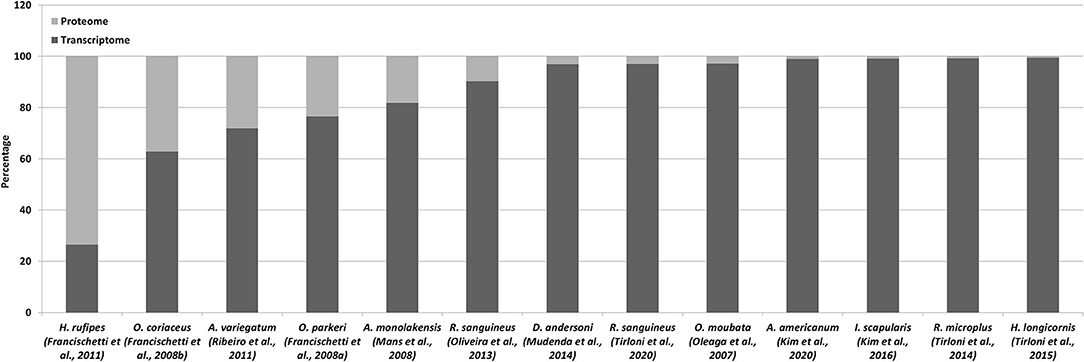
Figure 2. Proteomics of tick transcriptomes. Indicated is the percentage of proteins identified using proteomics relative to the transcriptome database used for identification.
Proteomics of Tick Cement
An integral part of salivary complexity is the cement cone produced by some Ixodes and metastriate ticks (Chinery, 1973; Suppan et al., 2018). These are proteins secreted in soluble form in the saliva that then harden to a matrix that can anchor the tick to the host during feeding. Analysis of the cement cone was historically difficult due to its insolubility. However, the use of chaotropic agents such as urea and SDS to solubilize the cement cone, followed by proteomic analysis have allowed a deeper insight into cement composition. For A. americanum, cement were dissolved in 8M urea followed by SDS-PAGE, before bands were cutout for proteomic analysis. This identified 7 proteins that included a glycine-rich and metalloprotease as well as proteins considered housekeeping using the A. americanum salivary transcriptome and tick database (Karim and Ribeiro, 2015; Bullard et al., 2016). A follow up study solubilized the cement cone in 8M urea, after which alkylation and tryptic digestion were followed with LC-MS/MS identification. This identified 160 proteins using an in-house A. americanum database (Porter et al., 2015; Hollmann et al., 2018). The proteins identified included as major proteins glycine-rich proteins and protease inhibitors as well as a large proportion of house-keeping proteins. A more recent study identified 654 proteins in urea+SDS extracted vs. 388 proteins in SDS vs. 266 proteins in urea alone from cement cones of R. microplus (Villar et al., 2020). The protein profile from cement was also termed the “cementome.” The study also identified 2,264 proteins from salivary gland extract. Of the proteins identified in the cement only 81 seem to be tick-derived while the rest were host-derived. The tick proteins were composed of glycine-rich proteins, protease inhibitors and various enzymes proposed to play a role in cement formation, solidification and maintenance. Complexity of the cement cone therefore seem to increase as extraction methodologies improve. A question that obviously remain is how many proteins may be entrapped during cement formation that do not play a primary role in the cement plug.
The Completeness of Sialotranscriptomes
Next-generation sequencing technologies raised the problem of what would be considered to be true in transcriptomics. In all publications published to date several recurring issues may be observed. Assemblies lead to large numbers of contigs and cut-off values needs to be implemented to reduce the number of contigs analyzed. This is generally based on coverage, either RPKM or TPM values or a subjective selection of the cutoff value. While this result in more trustworthy final assemblies, it raises the question of the origin of those contigs that do not make the selection criteria. Are these assembly artifacts, background transcription artifacts, environmental contamination or contamination from other tick tissues?
To determine how representative a transcriptome is, the presence of expected household genes can be determined. The “completeness” of this set may then be used as a proxy for the quality of a transcriptome. The program “Benchmarking Universal Single-Copy Orthologs or BUSCO” uses the concept of universal genes that are present as singletons in both genomes and transcriptomes (Waterhouse et al., 2018). BUSCO analysis of all published salivary gland transcriptomes generated with next-generation sequence technologies, indicate that the “completeness” of these transcriptomes ranges from 40 to 97% (Figure 3). This would imply that salivary transcriptomes have not been sequenced to depths necessary to recover all genes. However, as indicated above, the BUSCO scores may also have been influenced by the cut-off values used in post-assembly analysis. While universal genes may certainly be indicative of how well housekeeping genes may be represented in transcriptomes, their function as proxy for the completeness of secreted protein families should be considered with caution. Small, abundant, multi-copy, polymorphic families such as the BPTI, BTSP, and lipocalins, may present their own problems during assembly since these genes are likely to be lost during assembly due to non-uniform sequence coverage and assembly artifacts. As such, while salivary gland transcriptomes have been expanded by NGS technologies the results suggest that we may still be missing a third or more of the transcriptome.
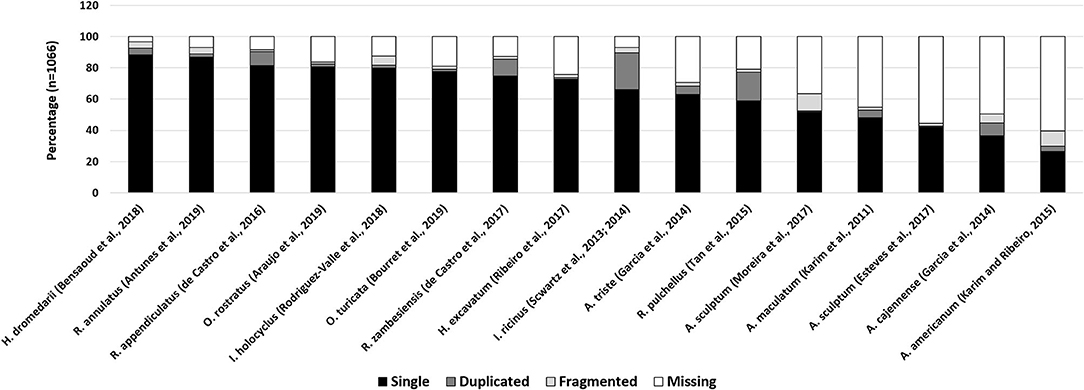
Figure 3. BUSCO analysis of tick salivary gland transcriptomes. Indicated are various published tick transcriptomes and their BUSCO analysis. Complete genes were detected as single copies (single) or multiple copies (duplicated). Genes that are fragmented or missing are also reported.
Complexity Measured by Genome Sequencing
While transcriptome sequencing can certainly give insight into gene diversity, the above discussion indicate that it may not be possible to derive at a final number of genes using transcriptomics. The gold standard for definitively estimating gene numbers remains whole genome sequencing. For ticks, draft genomes of I. scapularis, I. ricinus, H. longicornis, and R. microplus has been published (Cramaro et al., 2015, 2017; Gulia-Nuss et al., 2016; Barrero et al., 2017; Miller et al., 2018; Guerrero et al., 2019).
The first genome for I. scapularis was sequenced using Sanger sequencing (Gulia-Nuss et al., 2016). The estimated genome size was 2.1 Gb with 20,486 predicted genes. BUSCO analysis indicated 69% completeness based on 2,675 reference genes (Mans et al., 2017). The ISE6 cell line from I. scapularis was also sequenced (Miller et al., 2018). This estimated the genome size at 2.8 Gb but if duplications are taken into account the size reduces to 2.2 Gb. BUSCO analysis indicated 95% completeness for 1,066 reference genes. The total number of genes associated with this genome is 37,259 with 24,501 protein coding. However, it has been estimated that as much as 50% of salivary gland proteins may be missing or incomplete in the genome (Ribeiro and Mans, 2020).
The genome for I. ricinus has been sequenced with both Illumina and Pacific Biosciences (PacBio) long read technologies (Cramaro et al., 2015, 2017). The estimated size of the genome is 2.65 Gb from which 516 Mb were sequenced which represent 19.4% of the genome but 67% of the non-repetitive genome. A total of 25,263 proteins were identified based on BLAST similarity searches. However, BUSCO only indicated 55.5% completeness.
The genome for H. longicornis was sequenced using PacBio long read technology (Guerrero et al., 2019). Genome sequencing was facilitated by the parthenogenicity of this species (ability to asexually reproduce with mating) that reduce genome heterogeneity. The total length of the genome was 7.3 Gbp in 34,208 contigs and was 96% complete as assessed with BUSCO. To date a gene count is not available.
The genome of R. microplus was sequenced using an Illumina/PacBio hybrid assembly approach (Barrero et al., 2017). Given the highly repetitive nature of the genome (70%) and the large estimated size (7.1 Gbp), Cot filtration was used to enrich for single, low copy, and moderately repetitive genomic DNA. This yielded an assembly of 2 Gbp with 38,827 genes of which 24,758 is protein coding. BUSCO analysis indicated a completeness of 53.1%. Conversely, a 63,416 non-redundant transcriptome dataset showed 85% completeness, suggesting that the genome assembly still miss 47% proteins encoded in the genome.
While all available tick genomes yield protein coding genes ranging from ~20,000 to 30,000 it is clear that their incomplete nature also do not allow accurate estimations of complexity of the whole genome, nor what may be expected at the tick-host interface.
Complexity Measurements Only as Good as the Existing Databases
It has been indicated that database repositories for sequences has an immense impact on the coverage and complexity detected during proteomic analysis, but also impact on the ability to annotate transcriptomes. In this regard, tick sequences are scattered across many different databases, none which present a complete non-redundant set of sequences. For VectorBase (Giraldo-Calderón et al., 2015), the only datasets available is for I. scapularis, I. ricinus, and R. microplus which is effectively those for which genome datasets exist. For Genbank, the nucleotide core database contain the majority of historical sequences deposited in Genbank, but only links to transcriptome shotgun assembly (TSA) sequences (that include EST and next-generation sequencing assemblies contained in the small read archive database -SRA). For many tick transcriptomes, data is only available in the small read archive database (SRA) as contigs (that may or may not contain full length ORFs) and no protein or nucleotide coding sequences exist. In some instances, no contigs were deposited and only raw sequence read data is available. This generally necessitates either extraction of ORFs or reassembly and annotation to make this data useful for comparative or proteomic analysis, although given the diversity of methods available for transcriptome assembly and analysis, the reproducibility of constructing a published transcriptome is debatable. In many cases as indicated, researchers use in-house sequence databases not deposited anywhere and therefore not available. There is therefore a need for a centralized database that would address all of these shortcomings and would include comprehensive inclusion of all tick transcriptomes sequenced to data as well as accurate annotation. To compound this, the unique lineage specific expansions observed in ticks make extraction of functionally relevant information difficult since proteins may only be annotated as “potential secretory protein” or “unknown function” (Ribeiro and Mans, 2020). In many cases, such proteins cannot even be assigned to existing protein families. In an attempt to partially address this, a database of secretory proteins has recently been constructed that can be searched by reverse position specific BLAST to assign potential secretory proteins to known secretory families (Ribeiro and Mans, 2020). However, this database only attempted classification of those proteins present in the NCBI non-redundant sequence database and excluded TSA and SRA databases. The searchable database will however, allow for better annotation of transcriptomes using a consistent nomenclature.
With regard to proteomic analysis, the data described in publications in tables or supplementary materials is generally useful to document the snapshot provided by a specific study. However, the main usefulness of proteomic data lies in the re-analysis potential of the raw data once sequence databases has been expanded since this may result in higher numbers of proteins identified from the proteomic data. To this end, raw proteomic data can be submitted to the PRoteomics IDEntification database (PRIDE) (Perez-Riverol et al., 2019). To this end, proteomic data for at least 6 tick species is available in PRIDE and include A. americanum (Crispell et al., 2019; Kim et al., 2020), A. sculptum (Esteves et al., 2017), I. scapularis (Villar et al., 2015; Kim et al., 2016), I. ricinus (Cramaro et al., 2015), H. longicornis (Ren et al., 2019), and R. sanguineus (Tirloni et al., 2020).
Other Measures of Complexity: Alternative Splicing, Recombination, Non-Coding RNA and Posttranslational Modification
The current study has thus far largely focused on detection and quantification of genes and the proteins encoded by them, as measures of salivary gland complexity. The impact of exon shuffling and gene duplication has not been considered yet, since these are mechanisms to generate diversity during long-term evolution which is detected retrospectively (Mans et al., 2017). However, alternative splicing may contribute significantly to increase complexity since this may be tissue specific and give rise to protein isoforms (Nilsen and Graveley, 2010). The impact of alternative splicing on complexity has not yet been analyzed in depth in ticks or salivary gland products. For example, the search terms “alternative splicing” and “ticks” retrieve 11 hits in PubMed. Of these, 9 deals with potential alternative splicing in single genes, the majority which is housekeeping (Baxter and Barker, 1998; Guo et al., 1998; Saravanan et al., 2003; Xu et al., 2003; Tabish et al., 2006; Buresova et al., 2009; Olafson et al., 2011; Temeyer et al., 2012; Urbanová et al., 2018). None of the genome, proteome or transcriptome studies described in the current study considered alternative splicing or isoforms extensively, except for the study of Oleaga et al. (2007) that detected numerous isoforms on a two-dimensional gel for the histamine and serotonin-binding protein, TSGP1. It is unclear whether these isoforms were due to alternative splicing or post-translational modification. The absence of extensive detection of alternative splicing in salivary gland proteins may be partially due to the consideration that most secretory tick families are single domain proteins such as the BPTI, BTSP, cystatin or lipocalin families, or composed of multiple domains of these core folds such as for example the various BPTI proteins with double, triple, quadruple and quintuple domains, or composed of multi-domain proteins with well-defined domain structures such as 5'-nucleotidase and the metalloproteases (Francischetti et al., 2009). On this, large scale proteomic studies support the observation that a single main isoform exist for most proteins and that other isoforms may not be functional, or under selective pressure or even expressed (Tress et al., 2017), while splicing of whole domains is also unlikely (Light and Elofsson, 2013). The potential for alternative splicing, exon shuffling and intragenic recombination to generate “new genes on the fly” has been considered to explain the sheer diversity of secretory proteins observed in the salivary glands (Ribeiro and Mans, 2020). Real data is, however, needed to confirm this.
A further level of complexity that may be emerging is that of non-coding RNA. Abundant long non-coding RNA was found in tick transcriptomes (de Castro et al., 2016), while microRNA may perform important housekeeping functions (Barrero et al., 2011; Zhou et al., 2013; Luo et al., 2015, 2019; Shao et al., 2015; Malik et al., 2019; Liu et al., 2020). However, of more interest for the current study is the potential that some of these non-coding RNA may play a role in the regulation of host defense mechanisms. As such, exosomes that carry microRNAs has been described in the saliva of ticks (Hackenberg et al., 2017). It has been proposed that these exomes may deliver microRNAs to immune cells to down-regulate immune modulatory pathways (Hackenberg and Kotsyfakis, 2018; Bensaoud et al., 2019b; Chávez et al., 2019). This will add another layer of complexity to salivary glands of which the magnitude is difficult to estimate. As yet, down-regulation of such immune pathways needs to be proven and it needs to be shown that concentrations of these microRNAs can break the chemical equilibrium barrier to be effective (Mans, 2019).
Post-translational modifications play a large role in regulation of functional networks and in protein function. With regard to protein function involved in tick-host interaction a number of post-translational modifications have been shown to be important for functionality, especially tyrosine sulfation (Shabareesh et al., 2017; Thompson et al., 2017; Vechtova et al., 2018; Franck et al., 2020). Many of the transcriptome studies listed also report many potential modification sites or motifs. However, large scale confirmation of post-translational modifications by proteomic analysis has not yet been performed for ticks (Vechtova et al., 2018). As such, the impact of post-translational modifications on complexity of salivary gland proteins is as yet unknown, but could be quite significant.
Future Perspectives on Salivary Gland Complexity
We are undoubtedly much further along the road to describing and understanding tick salivary gland protein complexity than ever before. It is likely that the next 10–20 years will see the completion of many tick genomes, the validation of many tick transcriptomes by proteomics and final estimates on the complexity of salivary gland protein repertoires for many tick species. This will be facilitated by new technologies such as third generation nanopore sequencing that will hopefully make transcriptome and genome sequencing affordable for the high number of species required to map all orthologs and paralogs. Other challenges are defined by our ignorance of the mechanisms regulating sialome switching, and the mode and possible physiological role of host protein secretion by tick salivary glands. The next challenge will be to analyze all this information to generate a comprehensive, holistic and ultimately understandable synthesis of tick evolution and tick-host interaction. Even so, we may find out that many genes identified in the genomes or transcriptomes may never be validated by proteomics. This may be due to the presence of pseudogenes or expression so transient to remain undetectable by proteomics, or even be functionally irrelevant (Mans et al., 2017; Mans, 2019). In the end, validation by empirical testing will remain crucial.
As indicated, the salivary gland is a complex organ composed of multiple acini with each comprising multiple cell types. While temporal differential expression over the course of feeding has been well-characterized, differential expression also occur within different cell types and acini. To define this expression well is important, since some cells are prone to infection by pathogens, such as T. parva that infect the e-cells of type III acini (Fawcett et al., 1982). In this case the impact of parasite infection on vector expression (Nene et al., 2004), is confounded by non-infected/non-affected members of the same cell type and other cell types since the parasite do not infect all cells of a particular type at the same time (McKeever, 2009). Other cell types are involved in specific biological functions such as water secretion by the e-cells of type III acini (Fawcett et al., 1981). An accurate picture of global differential expression is therefore confounded by the differential expression that occur on smaller scales. The potential to address this issue exist via single cell sequencing approaches (Chattopadhyay et al., 2018). These approaches are hampered by the pyramidal structure of acini and the fragility of salivary gland cell types, but future innovations on non-disruptive separation of acinar cell types may enable single cell sequencing. Alternatives may also be found in laser capture microdissection of cell regions followed by single cell sequencing (Chattopadhyay et al., 2018). It is foreseen that these approaches will certainly enable a deeper dissection of salivary function, localization and parasite-vector interaction.
The majority of studies described for salivary gland transcriptomics are for adult ticks, while whole body transcriptomes are generally generated for larvae or nymphs. Single cell and related technologies that allow sequencing of minute quantities of genetic material may open up the possibility of sequencing larval or nymphal glands made accessible via laser microdissection. This will certainly advance tick salivary gland transcriptomics, since differential expression between life stages is well-recognized (Schwarz et al., 2013; Andreotti et al., 2018).
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work is based on the research supported in part by the National Research Foundation of South Africa (Grant Number: 118540).
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank Jose Ribeiro for critical reading of the manuscript.
References
Alarcon-Chaidez, F. J., Sun, J., and Wikel, S. K. (2007). Transcriptome analysis of the salivary glands of Dermacentor andersoni Stiles (Acari: Ixodidae). Insect Biochem. Mol. Biol. 37, 48–71. doi: 10.1016/j.ibmb.2006.10.002
Aljamali, M. N., Hern, L., Kupfer, D., Downard, S., So, S., Roe, B. A., et al. (2009). Transcriptome analysis of the salivary glands of the female tick Amblyomma americanum (Acari: Ixodidae). Insect Mol. Biol. 18, 129–154. doi: 10.1111/j.1365-2583.2009.00863.x
Anatriello, E., Ribeiro, J. M., de Miranda-Santos, I. K., Brandão, L. G., Anderson, J. M., Valenzuela, J. G., et al. (2010). An insight into the sialotranscriptome of the brown dog tick, Rhipicephalus sanguineus. BMC Genomics 11:450. doi: 10.1186/1471-2164-11-450
Andreotti, R., Giachetto, P. F., and Cunha, R. C. (2018). Advances in tick vaccinology in Brazil: from gene expression to immunoprotection. Front. Biosci. 10, 127–142. doi: 10.2741/s504
Antunes, S., Couto, J., Ferrolho, J., Sanches, G. S., Merino Charrez, J. O., De la Cruz Hernández, N., et al. (2019). Transcriptome and proteome response of Rhipicephalus annulatus tick vector to Babesia bigemina infection. Front. Physiol. 10:318. doi: 10.3389/fphys.2019.00318
Araujo, R. N., Silva, N. C. S., Mendes-Sousa, A., Paim, R., Costa, G. C. A., Dias, L. R., et al. (2019). RNA-seq analysis of the salivary glands and midgut of the argasid tick Ornithodoros rostratus. Sci. Rep. 9:6764. doi: 10.1038/s41598-019-42899-z
Bakkes, D. K., De Klerk, D., Latif, A. A., and Mans, B. J. (2018). Integrative taxonomy of Afrotropical Ornithodoros (Ornithodoros) (Acari: Ixodida: Argasidae). Ticks Tick Borne Dis. 9, 1006–1037. doi: 10.1016/j.ttbdis.2018.03.024
Balashov, Y. S. (1972). Bloodsucking ticks (Ixodideae)—vectors of disease of man and animals. Misc. Pub. Entomol. Soc. Amer. 8, 161–376.
Barrero, R. A., Guerrero, F. D., Black, M., McCooke, J., Chapman, B., Schilkey, F., et al. (2017). Gene-enriched draft genome of the cattle tick Rhipicephalus microplus: assembly by the hybrid Pacific biosciences/illumina approach enabled analysis of the highly repetitive genome. Int. J. Parasitol. 47, 569–583. doi: 10.1016/j.ijpara.2017.03.007
Barrero, R. A., Keeble-Gagnère, G., Zhang, B., Moolhuijzen, P., Ikeo, K., Tateno, Y., et al. (2011). Evolutionary conserved microRNAs are ubiquitously expressed compared to tick-specific miRNAs in the cattle tick Rhipicephalus (Boophilus) microplus. BMC Genomics 12:328. doi: 10.1186/1471-2164-12-328
Baxter, G. D., and Barker, S. C. (1998). Acetylcholinesterase cDNA of the cattle tick, Boophilus microplus: characterisation and role in organophosphate resistance. Insect Biochem. Mol. Biol. 28, 581–589. doi: 10.1016/S0965-1748(98)00034-4
Bensaoud, C., Aounallah, H., Sciani, J. M., Faria, F., Chudzinski-Tavassi, A. M., Bouattour, A., et al. (2019a). Proteomic informed by transcriptomic for salivary glands components of the camel tick Hyalomma dromedarii. BMC Genomics 20:675. doi: 10.1186/s12864-019-6042-1
Bensaoud, C., Hackenberg, M., and Kotsyfakis, M. (2019b). Noncoding RNAs in parasite-vector-host interactions. Trends Parasitol. 35, 715–724. doi: 10.1016/j.pt.2019.06.012
Bensaoud, C., Nishiyama, M. Y. Jr., Ben Hamda, C., Lichtenstein, F., Castro de Oliveira, U., Faria, F., et al. (2018). De novo assembly and annotation of Hyalomma dromedarii tick (Acari: Ixodidae) sialotranscriptome with regard to gender differences in gene expression. Parasit. Vectors 11:314. doi: 10.1186/s13071-018-2874-9
Bergman, D. K., Palmer, M. J., Caimano, M. J., Radolf, J. D., and Wikel, S. K. (2000). Isolation and molecular cloning of a secreted immunosuppresant protein from Dermacentor andersoni salivary gland. J. Parasitol. 86, 516–525. doi: 10.1645/0022-3395(2000)086[0516:IAMCOA]2.0.CO;2
Bishop, R., Lambson, B., Wells, C., Pandit, P., Osaso, J., Nkonge, C., et al. (2002). A cement protein of the tick Rhipicephalus appendiculatus, located in the secretory e cell granules of the type III salivary gland acini, induces strong antibody responses in cattle. Int. J. Parasitol. 32, 833–842. doi: 10.1016/S0020-7519(02)00027-9
Bourret, T. J., Boyle, W. K., Zalud, A. K., Valenzuela, J. G., Oliveira, F., and Lopez, J. E. (2019). The relapsing fever spirochete Borrelia turicatae persists in the highly oxidative environment of its soft-bodied tick vector. Cell. Microbiol. 21:e12987. doi: 10.1111/cmi.12987
Bullard, R., Allen, P., Chao, C. C., Douglas, J., Das, P., Morgan, S. E., et al. (2016). Structural characterization of tick cement cones collected from in vivo and artificial membrane blood-fed lone star ticks (Amblyomma americanum). Ticks Tick Borne Dis. 7, 880–892. doi: 10.1016/j.ttbdis.2016.04.006
Buresova, V., Hajdusek, O., Franta, Z., Sojka, D., and Kopacek, P. (2009). IrAM-An alpha2-macroglobulin from the hard tick Ixodes ricinus: characterization and function in phagocytosis of a potential pathogen Chryseobacterium indologenes. Dev. Comp. Immunol. 33, 489–498. doi: 10.1016/j.dci.2008.09.011
Charrier, N. P., Couton, M., Voordouw, M. J., Rais, O., Durand-Hermouet, A., Hervet, C., et al. (2018). Whole body transcriptomes and new insights into the biology of the tick Ixodes ricinus. Parasit. Vectors 11:364. doi: 10.1186/s13071-018-2932-3
Charrier, N. P., Hermouet, A., Hervet, C., Agoulon, A., Barker, S. C., Heylen, D., et al. (2019). A transcriptome-based phylogenetic study of hard ticks (Ixodidae). Sci. Rep. 9:12923. doi: 10.1038/s41598-019-49641-9
Chattopadhyay, P. K., Roederer, M., and Bolton, D. L. (2018). A deadly dance: the choreography of host-pathogen interactions, as revealed by single-cell technologies. Nat. Commun. 9:4638. doi: 10.1038/s41467-018-06214-0
Chávez, A. S. O., O'Neal, A. J., Santambrogio, L., Kotsyfakis, M., and Pedra, J. H. F. (2019). Message in a vesicle-trans-kingdom intercommunication at the vector-host interface. J. Cell Sci. 132:jcs224212. doi: 10.1242/jcs.224212
Chenchik, A., Diachenko, L., Moqadam, F., Tarabykin, V., Lukyanov, S., and Siebert, P. D. (1996). Full-length cDNA cloning and determination of mRNA 5' and 3' ends by amplification of adaptor-ligated cDNA. Biotechniques 21, 526–534. doi: 10.2144/96213pf02
Chinery, W. A. (1965). Studies on the various glands of the tick Haemaphysalis spinigera Neumann 1897. 3. the salivary glands. Acta Trop. 22, 321–349.
Chinery, W. A. (1973). The nature and origin of the “cement” substance at the site of attachment and feeding of adult Haemaphysalis spinigera (Ixodidae). J. Med. Entomol. 10, 355–362. doi: 10.1093/jmedent/10.4.355
Chmelar, J., Anderson, J. M., Mu, J., Jochim, R. C., Valenzuela, J. G., and Kopecký, J. (2008). Insight into the sialome of the castor bean tick, Ixodes ricinus. BMC Genomics 9:233. doi: 10.1186/1471-2164-9-233
Coons, L. B., and Roshdy, M. A. (1973). Fine structure of the salivary glands of unfed male Dermacentor variabilis (say) (Ixodoidea: Ixodidae). J. Parasitol. 59, 900–912. doi: 10.2307/3278433
Couto, J., Villar, M., Mateos-Hernández, L., Ferrolho, J., Sanches, G. S., Sofia Santos, A., et al. (2020). Quantitative proteomics identifies metabolic pathways affected by Babesia infection and blood feeding in the sialoproteome of the vector Rhipicephalus bursa. Vaccines 8:91. doi: 10.3390/vaccines8010091
Cramaro, W. J., Hunewald, O. E., Bell-Sakyi, L., and Muller, C. P. (2017). Genome scaffolding and annotation for the pathogen vector Ixodes ricinus by ultra-long single molecule sequencing. Parasit. Vectors 10:71. doi: 10.1186/s13071-017-2008-9
Cramaro, W. J., Revets, D., Hunewald, O. E., Sinner, R., Reye, A. L., and Muller, C. P. (2015). Integration of Ixodes ricinus genome sequencing with transcriptome and proteome annotation of the naïve midgut. BMC Genomics 16:871. doi: 10.1186/s12864-015-1981-7
Crispell, G., Commins, S. P., Archer-Hartman, S. A., Choudhary, S., Dharmarajan, G., Azadi, P., et al. (2019). Discovery of alpha-gal-containing antigens in North American tick species believed to induce red meat allergy. Front. Immunol. 10:1056. doi: 10.3389/fimmu.2019.01056
Das, S., Banerjee, G., DePonte, K., Marcantonio, N., Kantor, F. S., and Fikrig, E. (2001). Salp25D, an Ixodes scapularis antioxidant, is 1 of 14 immunodominant antigens in engorged tick salivary glands. J. Infect. Dis. 184, 1056–1064. doi: 10.1086/323351
Das, S., Marcantonio, N., Deponte, K., Telford, S. R., Anderson, J. F., Kantor, F. S., et al. (2000). Salp16, a gene induced in Ixodes scapularis salivary glands during tick feeding. Am. J. Trop. Med. Hyg. 62, 99–105. doi: 10.4269/ajtmh.2000.62.99
de Castro, M. H., de Klerk, D., Pienaar, R., Latif, A. A., Rees, D. J., and Mans, B. J. (2016). De novo assembly and annotation of the salivary gland transcriptome of Rhipicephalus appendiculatus male and female ticks during blood feeding. Ticks Tick Borne Dis. 7, 536–548. doi: 10.1016/j.ttbdis.2016.01.014
de Castro, M. H., de Klerk, D., Pienaar, R., Rees, D. J. G., and Mans, B. J. (2017). Sialotranscriptomics of Rhipicephalus zambeziensis reveals intricate expression profiles of secretory proteins and suggests tight temporal transcriptional regulation during blood-feeding. Parasit. Vectors 10:384. doi: 10.1186/s13071-017-2312-4
De Marco, L., Epis, S., Comandatore, F., Porretta, D., Cafarchia, C., Mastrantonio, V., et al. (2017). Transcriptome of larvae representing the Rhipicephalus sanguineus complex. Mol. Cell. Probes 31, 85–90. doi: 10.1016/j.mcp.2016.02.006
Díaz-Martín, V., Manzano-Román, R., Valero, L., Oleaga, A., Encinas-Grandes, A., and Pérez-Sánchez, R. (2013). An insight into the proteome of the saliva of the argasid tick Ornithodoros moubata reveals important differences in saliva protein composition between the sexes. J. Proteomics 80, 216–235. doi: 10.1016/j.jprot.2013.01.015
Díaz-Sánchez, S., Estrada-Peña, A., Cabezas-Cruz, A., and de la Fuente, J. (2019). Evolutionary insights into the tick hologenome. Trends Parasitol. 35, 725–737. doi: 10.1016/j.pt.2019.06.014
Esteves, E., Maruyama, S. R., Kawahara, R., Fujita, A., Martins, L. A., Righi, A. A., et al. (2017). Analysis of the salivary gland transcriptome of unfed and partially fed Amblyomma sculptum ticks and descriptive proteome of the saliva. Front. Cell Infect. Microbiol. 7:476. doi: 10.3389/fcimb.2017.00476
Estrada-Peña, A., Venzal, J. M., Kocan, K. M., Tramuta, C., Tomassone, L., de la Fuente, J., et al. (2008). Observations on Antricola ticks: small nymphs feed on mammalian hosts and have a salivary gland structure similar to ixodid ticks. J. Parasitol. 94, 953–955. doi: 10.1645/GE-1371.1
Fawcett, D. W., Büscher, G., and Doxsey, S. (1982). Salivary gland of the tick vector of East Coast fever. IV. cell type selectivity and host cell responses to Theileria parva. Tissue Cell 14, 397–414. doi: 10.1016/0040-8166(82)90035-0
Fawcett, D. W., Doxsey, S., and Büscher, G. (1981). Salivary gland of the tick vector (R. appendiculatus) of East Coast fever. II. cellular basis for fluid secretion in the type III acinus. Tissue Cell 13, 231–253. doi: 10.1016/0040-8166(81)90003-3
Feng, L. L., Liu, L., and Cheng, T. Y. (2019). Proteomic analysis of saliva from partially and fully engorged adult female Rhipicephalus microplus (Acari: Ixodidae). Exp. Appl. Acarol. 78, 443–460. doi: 10.1007/s10493-019-00390-4
Francischetti, I. M., Anderson, J. M., Manoukis, N., Pham, V. M., and Ribeiro, J. M. (2011). An insight into the sialotranscriptome and proteome of the coarse bontlegged tick, Hyalomma marginatum rufipes. J. Proteomics 74, 2892–2908. doi: 10.1016/j.jprot.2011.07.015
Francischetti, I. M., Mans, B. J., Meng, Z., Gudderra, N., Veenstra, T. D., Pham, V. M., et al. (2008a). An insight into the sialome of the soft tick, Ornithodorus parkeri. Insect Biochem. Mol. Biol. 38, 1–21. doi: 10.1016/j.ibmb.2007.09.009
Francischetti, I. M., Mather, T. N., and Ribeiro, J. M. (2003). Cloning of a salivary gland metalloprotease and characterization of gelatinase and fibrin(ogen)lytic activities in the saliva of the Lyme disease tick vector Ixodes scapularis. Biochem. Biophys. Res. Commun. 305, 869–875. doi: 10.1016/S0006-291X(03)00857-X
Francischetti, I. M., Meng, Z., Mans, B. J., Gudderra, N., Hall, M., Veenstra, T. D., et al. (2008b). An insight into the salivary transcriptome and proteome of the soft tick and vector of epizootic bovine abortion, Ornithodoros coriaceus. J. Proteomics 71, 493–512. doi: 10.1016/j.jprot.2008.07.006
Francischetti, I. M., My Pham, V., Mans, B. J., Andersen, J. F., Mather, T. N., Lane, R. S., et al. (2005). The transcriptome of the salivary glands of the female western black-legged tick Ixodes pacificus (Acari: Ixodidae). Insect Biochem. Mol. Biol. 35, 1142–1161. doi: 10.1016/j.ibmb.2005.05.007
Francischetti, I. M., Sa-Nunes, A., Mans, B. J., Santos, I. M., and Ribeiro, J. M. (2009). The role of saliva in tick feeding. Front. Biosci. 14, 2051–2088. doi: 10.2741/3363
Francischetti, I. M., Valenzuela, J. G., Andersen, J. F., Mather, T. N., and Ribeiro, J. M. (2002). Ixolaris, a novel recombinant tissue factor pathway inhibitor (TFPI) from the salivary gland of the tick, Ixodes scapularis: identification of factor X and factor Xa as scaffolds for the inhibition of factor VIIa/tissue factor complex. Blood 99, 3602–3612. doi: 10.1182/blood-2001-12-0237
Franck, C., Foster, S. R., Johansen-Leete, J., Chowdhury, S., Cielesh, M., Bhusal, R. P., et al. (2020). Semisynthesis of an evasin from tick saliva reveals a critical role of tyrosine sulfation for chemokine binding and inhibition. Proc. Natl. Acad. Sci. U.S.A. 117, 12657–12664. doi: 10.1073/pnas.2000605117
Garcia, G. R., Gardinassi, L. G., Ribeiro, J. M., Anatriello, E., Ferreira, B. R., Moreira, H. N., et al. (2014). The sialotranscriptome of Amblyomma triste, Amblyomma parvum and Amblyomma cajennense ticks, uncovered by 454-based RNA-seq. Parasit. Vectors 7:430. doi: 10.1186/1756-3305-7-430
Gaspar, A. R., Crause, J. C., and Neitz, A. W. (1995). Identification of anticoagulant activities in the salivary glands of the soft tick, Ornithodoros savignyi. Exp. Appl. Acarol. 19, 117–127. doi: 10.1007/BF00052551
Gaspar, A. R., Joubert, A. M., Crause, J. C., and Neitz, A. W. (1996). Isolation and characterization of an anticoagulant from the salivary glands of the tick, Ornithodoros savignyi (Acari: Argasidae). Exp. Appl. Acarol. 20, 583–598. doi: 10.1007/BF00052809
Giachetto, P. F., Cunha, R. C., Nhani, A. Jr., Garcia, M. V., Ferro, J. A., and Andreotti, R. (2020). Gene expression in the salivary gland of Rhipicephalus (Boophilus) microplus fed on tick-susceptible and tick-resistant hosts. Front. Cell Infect. Microbiol. 9:477. doi: 10.3389/fcimb.2019.00477
Gibson, A. K., Smith, Z., Fuqua, C., Clay, K., and Colbourne, J. K. (2013). Why so many unknown genes? partitioning orphans from a representative transcriptome of the lone star tick Amblyomma americanum. BMC Genomics 14:135. doi: 10.1186/1471-2164-14-135
Giraldo-Calderón, G. I., Emrich, S. J., MacCallum, R. M., Maslen, G., Dialynas, E., Topalis, P., et al. (2015). VectorBase: an updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic Acids Res. 43, D707–D713. doi: 10.1093/nar/gku1117
Guerrero, F. D., Bendele, K. G., Ghaffari, N., Guhlin, J., Gedye, K. R., Lawrence, K. E., et al. (2019). The Pacific biosciences de novo assembled genome dataset from a parthenogenetic New Zealand wild population of the longhorned tick, Haemaphysalis longicornis Neumann, 1901. Data Brief 27:104602. doi: 10.1016/j.dib.2019.104602
Guerrero, F. D., Miller, R. J., Rousseau, M. E., Sunkara, S., Quackenbush, J., Lee, Y., et al. (2005). BmiGI: a database of cDNAs expressed in Boophilus microplus, the tropical/southern cattle tick. Insect Biochem. Mol. Biol. 35, 585–595. doi: 10.1016/j.ibmb.2005.01.020
Gulia-Nuss, M., Nuss, A. B., Meyer, J. M., Sonenshine, D. E., Roe, R. M., Waterhouse, R. M., et al. (2016). Genomic insights into the Ixodes scapularis tick vector of Lyme disease. Nat. Commun. 7:10507. doi: 10.1038/ncomms10507
Guo, J., Sun, Y., Luo, X., Li, M., He, P., He, L., et al. (2019). De novo transcriptome sequencing and comparative analysis of Haemaphysalis flava Neumann, 1897 at larvae and nymph stages. Infect. Genet. Evol. 75:104008. doi: 10.1016/j.meegid.2019.104008
Guo, X., Xu, Q., Harmon, M. A., Jin, X., Laudet, V., Mangelsdorf, D. J., et al. (1998). Isolation of two functional retinoid X receptor subtypes from the ixodid tick, Amblyomma americanum (L.). Mol. Cell. Endocrinol. 139, 45–60. doi: 10.1016/S0303-7207(98)00073-2
Hackenberg, M., and Kotsyfakis, M. (2018). Exosome-mediated pathogen transmission by arthropod vectors. Trends Parasitol. 34, 549–552. doi: 10.1016/j.pt.2018.04.001
Hackenberg, M., Langenberger, D., Schwarz, A., Erhart, J., and Kotsyfakis, M. (2017). In silico target network analysis of de novo-discovered, tick saliva-specific microRNAs reveals important combinatorial effects in their interference with vertebrate host physiology. RNA 23, 1259–1269. doi: 10.1261/rna.061168.117
Hollmann, T., Kim, T. K., Tirloni, L., Radulović, Ž. M., Pinto, A. F. M., Diedrich, J. K., et al. (2018). Identification and characterization of proteins in the Amblyomma americanum tick cement cone. Int. J. Parasitol. 48, 211–224. doi: 10.1016/j.ijpara.2017.08.018
Howell, C. J., Neitz, A. W., and Potgieter, D. J. (1975). Some toxic and chemical properties of the oral secretion of the sand tampan, Ornithodoros savignyi (Audouin) (1827). Onderstepoort J. Vet. Res. 43, 99–102.
Joubert, A. M., Louw, A. I., Joubert, F., and Neitz, A. W. (1998). Cloning, nucleotide sequence and expression of the gene encoding factor Xa inhibitor from the salivary glands of the tick, Ornithodoros savignyi. Exp. Appl. Acarol. 22, 603–619. doi: 10.1023/A:1006198713791
Kaire, G. H. (1966). Isolation of tick paralysis toxin from Ixodes holocyclus. Toxicon 4, 91–97. doi: 10.1016/0041-0101(66)90003-1
Karczewski, J., Endris, R., and Connolly, T. M. (1994). Disagregin is a fibrinogen receptor antagonist lacking the Arg-Gly-Asp sequence from the tick, Ornithodoros moubata. J. Biol. Chem. 269, 6702–6708.
Karim, S., and Ribeiro, J. M. (2015). An insight into the sialome of the Lone Star tick, Amblyomma americanum, with a glimpse on its time dependent gene expression. PLoS ONE 10:e0131292. doi: 10.1371/journal.pone.0131292
Karim, S., Singh, P., and Ribeiro, J. M. C. (2011). A deep insight into the sialotranscriptome of the Gulf Coast tick, Amblyomma maculatum. PLoS ONE 6:e28525. doi: 10.1371/journal.pone.0028525
Keller, P. M., Waxman, L., Arnold, B. A., Schultz, L. D., Condra, C., and Connolly, T. M. (1993). Cloning of the cDNA and expression of moubatin, an inhibitor of platelet aggregation. J. Biol. Chem. 268, 5450–5456.
Kim, T. K., Tirloni, L., Pinto, A. F., Moresco, J., Yates, J. R. 3rd, da Silva Vaz, I. Jr., and Mulenga, A. (2016). Ixodes scapularis tick saliva proteins sequentially secreted every 24 h during blood feeding. PLoS Negl. Trop. Dis. 10:e0004323. doi: 10.1371/journal.pntd.0004323
Kim, T. K., Tirloni, L., Pinto, A. F. M., Diedrich, J. K., Moresco, J. J., Yates, J. R. III., et al. (2020). Time-resolved proteomic profile of Amblyomma americanum tick saliva during feeding. PLoS Negl. Trop. Dis. 14:e0007758. doi: 10.1371/journal.pntd.0007758
Kotsyfakis, M., Schwarz, A., Erhart, J., and Ribeiro, J. M. (2015). Tissue- and time-dependent transcription in Ixodes ricinus salivary glands and midguts when blood feeding on the vertebrate host. Sci. Rep. 5:9103. doi: 10.1038/srep09103
Kretz, P. L., Reid, C. H., Greener, A., and Short, J. M. (1989). Effect of lambda packaging extract mcr restriction activity on DNA cloning. Nucleic Acids Res. 17:5409. doi: 10.1093/nar/17.13.5409
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. doi: 10.1038/227680a0
Leboulle, G., Rochez, C., Louahed, J., Ruti, B., Brossard, M., Bollen, A., et al. (2002). Isolation of Ixodes ricinus salivary gland mRNA encoding factors induced during blood feeding. Am. J. Trop. Med. Hyg. 66, 225–233. doi: 10.4269/ajtmh.2002.66.225
Levy, S. E., and Myers, R. M. (2016). Advancements in next-generation sequencing. Annu. Rev. Genomics Hum. Genet. 17, 95–115. doi: 10.1146/annurev-genom-083115-022413
Light, S., and Elofsson, A. (2013). The impact of splicing on protein domain architecture. Curr. Opin. Struct. Biol. 23, 451–458. doi: 10.1016/j.sbi.2013.02.013
Liu, W., Guo, J., Luo, J., Ren, Q., Chen, Z., Qu, Z., et al. (2020). Analysis of microRNA expression profiles dynamic in different life stages of Haemaphysalis longicornis ticks by deep sequencing of small RNA libraries. Ticks Tick Borne Dis. 11:101427. doi: 10.1016/j.ttbdis.2020.101427
Lukyanov, K., Diatchenko, L., Chenchik, A., Nanisetti, A., Siebert, P., Usman, N., et al. (1997). Construction of cDNA libraries from small amounts of total RNA using the suppression PCR effect. Biochem. Biophys. Res. Commun. 230, 285–288. doi: 10.1006/bbrc.1996.5948
Luo, J., Liu, G. Y., Chen, Z., Ren, Q. Y., Yin, H., Luo, J. X., et al. (2015). Identification and characterization of microRNAs by deep-sequencing in Hyalomma anatolicum anatolicum (Acari: Ixodidae) ticks. Gene 564, 125–133. doi: 10.1016/j.gene.2015.01.019
Luo, J., Ren, Q., Chen, Z., Liu, W., Qu, Z., Xiao, R., et al. (2019). Comparative analysis of microRNA profiles between wild and cultured Haemaphysalis longicornis (Acari, Ixodidae) ticks. Parasite 26:18. doi: 10.1051/parasite/2019018
Madden, R. D., Sauer, J. R., and Dillwith, J. W. (2002). A proteomics approach to characterizing tick salivary secretions. Exp. Appl. Acarol. 28, 77–87. doi: 10.1023/A:1025342015065
Malik, M. I., Nawaz, M., Hassan, I. A., Zhang, H., Gong, H., Cao, J., et al. (2019). A microRNA profile of saliva and role of miR-375 in Haemaphysalis longicornis (Ixodida: Ixodidae). Parasit. Vectors 12:68. doi: 10.1186/s13071-019-3318-x
Mans, B. J. (2016). “Glandular matrices and secretions: blood- feeding arthropods,” in Extracellular Composite Matrices in Arthropods, eds E. Cohen and B. Moussian (Springer International Publishing Switzerland), 625–688.
Mans, B. J. (2019). Chemical equilibrium at the tick-host feeding interface: a critical examination of biological relevance in hematophagous behavior. Front. Physiol. 10:530. doi: 10.3389/fphys.2019.00530
Mans, B. J., Andersen, J. F., Francischetti, I. M., Valenzuela, J. G., Schwan, T. G., Pham, V. M., et al. (2008). Comparative sialomics between hard and soft ticks: implications for the evolution of blood-feeding behavior. Insect Biochem. Mol. Biol. 38, 42–58. doi: 10.1016/j.ibmb.2007.09.003
Mans, B. J., de Castro, M. H., Pienaar, R., de Klerk, D., Gaven, P., Genu, S., et al. (2016). Ancestral reconstruction of tick lineages. Ticks Tick Borne Dis. 7, 509–535. doi: 10.1016/j.ttbdis.2016.02.002
Mans, B. J., Featherston, J., de Castro, M. H., and Pienaar, R. (2017). Gene duplication and protein evolution in tick-host interactions. Front. Cell Infect. Microbiol. 7:413. doi: 10.3389/fcimb.2017.00413
Mans, B. J., Louw, A. I., and Neitz, A. W. (2002b). Amino acid sequence and structure modeling of savignin, a thrombin inhibitor from the tick, Ornithodoros savignyi. Insect Biochem. Mol. Biol. 32, 821–828. doi: 10.1016/S0965-1748(01)00169-2
Mans, B. J., Louw, A. I., and Neitz, A. W. (2002c). Savignygrin, a platelet aggregation inhibitor from the soft tick, Ornithodoros savignyi, presents the RGD integrin recognition motif on the Kunitz-BPTI fold. J. Biol. Chem. 277, 21371–21378. doi: 10.1074/jbc.M112060200
Mans, B. J., Louw, A. I., and Neitz, A. W. (2003). The major tick salivary gland proteins and toxins from the soft tick, Ornithodoros savignyi, are part of the tick Lipocalin family: implications for the origins of tick toxicoses. Mol. Biol. Evol. 20, 1158–1167. doi: 10.1093/molbev/msg126
Mans, B. J., and Neitz, A. W. (2004a). The sand tampan, Ornithodoros savignyi, as a model for tick–host interactions. S. Afr. J. Sci. 100, 283–288.
Mans, B. J., and Neitz, A. W. (2004b). Adaptation of ticks to a blood-feeding environment: evolution from a functional perspective. Insect Biochem. Mol. Biol. 34, 1–17. doi: 10.1016/j.ibmb.2003.09.002
Mans, B. J., Steinmann, C. M., Venter, J. D., Louw, A. I., and Neitz, A. W. (2002a). Pathogenic mechanisms of sand tampan toxicoses induced by the tick, Ornithodoros savignyi. Toxicon 40, 1007–1016. doi: 10.1016/S0041-0101(02)00098-3
Mans, B. J., Venter, J. D., Coons, L. B., Louw, A. I., and Neitz, A. W. (2004). A reassessment of argasid tick salivary gland ultrastructure from an immuno-cytochemical perspective. Exp. Appl. Acarol. 33, 119–129. doi: 10.1023/B:APPA.0000030012.47964.b3
Mans, B. J., Venter, J. D., Vrey, P. J., Louw, A. I., and Neitz, A. W. (2001). Identification of putative proteins involved in granule biogenesis of tick salivary glands. Electrophoresis 22, 1739–1746. doi: 10.1002/1522-2683(200105)22:9<1739::AID-ELPS1739>3.0.CO;2-7
Martin, J. A., and Wang, Z. (2011). Next-generation transcriptome assembly. Nat. Rev. Genet. 12, 671–682. doi: 10.1038/nrg3068
Martins, L. A., Malossi, C. D., Galletti, M. F. B. M., Ribeiro, J. M., Fujita, A., Esteves, E., et al. (2019). The transcriptome of the salivary glands of Amblyomma aureolatum reveals the antimicrobial peptide microplusin as an important factor for the tick protection against Rickettsia rickettsii infection. Front. Physiol. 10:529. doi: 10.3389/fphys.2019.00529
McKeever, D. J. (2009). Bovine immunity - a driver for diversity in Theileria parasites? Trends Parasitol. 25, 269–276. doi: 10.1016/j.pt.2009.03.005
McSwain, J. L., Essenberg, R. C., and Sauer, J. R. (1982). Protein changes in the salivary glands of the female lone star tick, Amblyomma americanum, during feeding. J. Parasitol. 68, 100–106. doi: 10.2307/3281330
Miller, J. R., Koren, S., Dilley, K. A., Harkins, D. M., Stockwell, T. B., Shabman, R. S., et al. (2018). A draft genome sequence for the Ixodes scapularis cell line, ISE6. F1000Res 7:297. doi: 10.12688/f1000research.13635.1
Moreira, H. N. S., Barcelos, R. M., Vidigal, P. M. P., Klein, R. C., Montandon, C. E., Maciel, T. E. F., et al. (2017). A deep insight into the whole transcriptome of midguts, ovaries and salivary glands of the Amblyomma sculptum tick. Parasitol. Int. 66, 64–73. doi: 10.1016/j.parint.2016.10.011
Mudenda, L., Pierlé, S. A., Turse, J. E., Scoles, G. A., Purvine, S. O., Nicora, C. D., et al. (2014). Proteomics informed by transcriptomics identifies novel secreted proteins in Dermacentor andersoni saliva. Int. J. Parasitol. 44, 1029–1037. doi: 10.1016/j.ijpara.2014.07.003
Mulenga, A., Tsuda, A., Onuma, M., and Sugimoto, C. (2003). Four serine proteinase inhibitors (serpin) from the brown ear tick, Rhiphicephalus appendiculatus; cDNA cloning and preliminary characterization. Insect Biochem. Mol. Biol. 33, 267–276. doi: 10.1016/S0965-1748(02)00240-0
Narasimhan, S., Deponte, K., Marcantonio, N., Liang, X., Royce, T. E., Nelson, K. F., et al. (2007). Immunity against Ixodes scapularis salivary proteins expressed within 24 hours of attachment thwarts tick feeding and impairs Borrelia transmission. PLoS ONE 2:e451. doi: 10.1371/journal.pone.0000451
Narasimhan, S., Koski, R. A., Beaulieu, B., Anderson, J. F., Ramamoorthi, N., Kantor, F., et al. (2002). A novel family of anticoagulants from the saliva of Ixodes scapularis. Insect Mol. Biol. 11, 641–650. doi: 10.1046/j.1365-2583.2002.00375.x
Neitz, A. W., Bezuidenhout, J. D., Vermeulen, N. M., Potgieter, D. J., and Howell, C. J. (1983). In search of the causal agents of tick toxicoses. Toxicon S 3, 317–320. doi: 10.1016/0041-0101(83)90219-2
Neitz, A. W., and Gothe, R. (1986). Changes in the protein pattern in the salivary glands of paralysis inducing female Rhipicephalus evertsi evertsi during infestation. Zentralbl. Veterinarmed. B 33, 213–320. doi: 10.1111/j.1439-0450.1986.tb00025.x
Neitz, A. W., Howell, C. J., and Potgieter, D. J. (1969). Purification of the toxic component in the oral secretion of the sand tampan Ornithodoros savignyi (Audouin) (1827). J. S. Afr. Chem. Ind. 22, 142–149.
Neitz, A. W., and Vermeulen, N. M. (1987). Biochemical studies on the salivary glands and haemolymph of Amblyomma hebraeum. Onderstepoort J. Vet. Res. 54, 443–450.
Nene, V., Lee, D., Kanga, S., Skilton, R., Shah, T., de Villiers, E., et al. (2004). Genes transcribed in the salivary glands of female Rhipicephalus appendiculatus ticks infected with Theileria parva. Insect Biochem. Mol. Biol. 34, 1117–1128. doi: 10.1016/j.ibmb.2004.07.002
Nene, V., Lee, D., Quackenbush, J., Skilton, R., Mwaura, S., Gardner, M. J., et al. (2002). AvGI, an index of genes transcribed in the salivary glands of the ixodid tick Amblyomma variegatum. Int. J. Parasitol. 32, 1447–1456. doi: 10.1016/S0020-7519(02)00159-5
Nienaber, J., Gaspar, A. R., and Neitz, A. W. (1999). Savignin, a potent thrombin inhibitor isolated from the salivary glands of the tick, Ornithodoros savignyi (Acari: Argasidae). Exp. Parasitol. 93, 82–91. doi: 10.1006/expr.1999.4448
Nilsen, T. W., and Graveley, B. R. (2010). Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463. doi: 10.1038/nature08909
Niu, D., Zhao, Y., Yang, Y., Yang, R., Gong, X., and Hu, L. (2019). De novo RNA-seq and functional annotation of Haemaphysalis longicornis. Acta Parasitol. 64, 807–820. doi: 10.2478/s11686-019-00103-2
O'Farrell, P. H. (1975). High resolution two-dimensional electrophoresis of proteins. J. Biol. Chem. 250, 4007–4021.
Olafson, P. U., Temeyer, K. B., and Pruett, J. H. (2011). Multiple transcripts encode glucose 6-phosphate dehydrogenase in the southern cattle tick, Rhipicephalus (Boophilus) microplus. Exp. Appl. Acarol. 53, 147–165. doi: 10.1007/s10493-010-9392-6
Oleaga, A., Escudero-Población, A., Camafeita, E., and Pérez-Sánchez, R. (2007). A proteomic approach to the identification of salivary proteins from the argasid ticks Ornithodoros moubata and Ornithodoros erraticus. Insect Biochem. Mol. Biol. 37, 1149–1159. doi: 10.1016/j.ibmb.2007.07.003
Oliveira, C. J., Anatriello, E., de Miranda-Santos, I. K., Francischetti, I. M., Sá-Nunes, A., Ferreira, B. R., et al. (2013). Proteome of Rhipicephalus sanguineus tick saliva induced by the secretagogues pilocarpine and dopamine. Ticks Tick Borne Dis. 4, 469–477. doi: 10.1016/j.ttbdis.2013.05.001
Paesen, G. C., Adams, P. L., Harlos, K., Nuttall, P. A., and Stuart, D. I. (1999). Tick histamine-binding proteins: isolation, cloning, and three dimensional structure. Mol. Cell 3, 661–671. doi: 10.1016/S1097-2765(00)80359-7
Perez-Riverol, Y., Csordas, A., Bai, J., Bernal-Llinares, M., Hewapathirana, S., Kundu, D. J., et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450. doi: 10.1093/nar/gky1106
Perner, J., Kropáčková, S., Kopáček, P., and Ribeiro, J. M. C. (2018). Sialome diversity of ticks revealed by RNAseq of single tick salivary glands. PLoS Negl. Trop. Dis. 12:e0006410. doi: 10.1371/journal.pntd.0006410
Pienaar, R., Neitz, A. W. H., and Mans, B. J. (2018). Tick paralysis: solving an enigma. Vet. Sci. 5:53. doi: 10.3390/vetsci5020053
Porter, L., Radulović, Ž., Kim, T., Braz, G. R., Da Silva Vaz, I. Jr., and Mulenga, A. (2015). Bioinformatic analyses of male and female Amblyomma americanum tick expressed serine protease inhibitors (serpins). Ticks Tick Borne Dis. 6, 16–30. doi: 10.1016/j.ttbdis.2014.08.002
Rabilloud, T. (2020). Paleoproteomics explained to youngsters: how did the wedding of two-dimensional electrophoresis and protein sequencing spark proteomics on: let there be light. J. Proteomics 107, 5–12. doi: 10.1016/j.jprot.2014.03.011
Radulović, Ž. M., Kim, T. K., Porter, L. M., Sze, S. H., Lewis, L., and Mulenga, A. (2014). A 24-48 h fed Amblyomma americanum tick saliva immuno-proteome. BMC Genomics 15:518. doi: 10.1186/1471-2164-15-518
Ren, S., Zhang, B., Xue, X., Wang, X., Zhao, H., Zhang, X., et al. (2019). Salivary gland proteome analysis of developing adult female Haemaphysalis longicornis ticks: molecular motor and TCA cycle-related proteins play an important role throughout development. Parasit. Vectors 12:613. doi: 10.1186/s13071-019-3864-2
Ribeiro, J. M., Alarcon-Chaidez, F., Francischetti, I. M., Mans, B. J., Mather, T. N., Valenzuela, J. G., et al. (2006). An annotated catalog of salivary gland transcripts from Ixodes scapularis ticks. Insect Biochem. Mol. Biol. 36, 111–129. doi: 10.1016/j.ibmb.2005.11.005
Ribeiro, J. M., Anderson, J. M., Manoukis, N. C., Meng, Z., and Francischetti, I. M. (2011). A further insight into the sialome of the tropical bont tick, Amblyomma variegatum. BMC Genomics 12:136. doi: 10.1186/1471-2164-12-136
Ribeiro, J. M., Labruna, M. B., Mans, B. J., Maruyama, S. R., Francischetti, I. M., Barizon, G. C., et al. (2012). The sialotranscriptome of Antricola delacruzi female ticks is compatible with non-hematophagous behavior and an alternative source of food. Insect Biochem. Mol. Biol. 42, 332–342. doi: 10.1016/j.ibmb.2012.01.003
Ribeiro, J. M., Slovák, M., and Francischetti, I. M. (2017). An insight into the sialome of Hyalomma excavatum. Ticks Tick Borne Dis. 8, 201–207. doi: 10.1016/j.ttbdis.2016.08.011
Ribeiro, J. M. C., and Arcà, B. (2009). From sialomes to the sialoverse: an insight into salivary potion of blood-feeding insects. Adv. Insect Physiol. 37, 59–118. doi: 10.1016/S0065-2806(09)37002-2
Ribeiro, J. M. C., and Mans, B. J. (2020). TickSialoFam (TSFam): a database that helps to classify tick salivary proteins, a review on tick salivary protein function and evolution, with considerations on the tick sialome switching phenomenon. Front. Cell. Infect. Microbiol. 10:374. doi: 10.3389/fcimb.2020.00374
Robinson, L. E., and Davidson, J. (1913). The anatomy of Argas persicus (Oken 1818). part II. Parasitology 6, 217–256. doi: 10.1017/S0031182000003073
Rodriguez-Valle, M., Moolhuijzen, P., Barrero, R. A., Ong, C. T., Busch, G., Karbanowicz, T., et al. (2018). Transcriptome and toxin family analysis of the paralysis tick, Ixodes holocyclus. Int. J. Parasitol. 48, 71–82. doi: 10.1016/j.ijpara.2017.07.007
Rosendale, A. J., Romick-Rosendale, L. E., Watanabe, M., Dunlevy, M. E., and Benoit, J. B. (2016). Mechanistic underpinnings of dehydration stress in the American dog tick revealed through RNA-Seq and metabolomics. J. Exp. Biol. 219, 1808–1819. doi: 10.1242/jeb.137315
Roshdy, M. A. (1972). The subgenus Persicargas (Ixodoidea, Argasidae, Argas). 15. histology and histochemistry of the salivary glands of A. (P.) persicus (Oken). J. Med. Entomol. 9, 143–148. doi: 10.1093/jmedent/9.2.143
Sangamnatdej, S., Paesen, G. C., Slovak, M., and Nuttall, P. A. (2002). A high affinity serotonin- and histamine-binding lipocalin from tick saliva. Insect Mol. Biol. 11, 79–86. doi: 10.1046/j.0962-1075.2001.00311.x
Saravanan, T., Weise, C., Sojka, D., and Kopácek, P. (2003). Molecular cloning, structure and bait region splice variants of alpha2-macroglobulin from the soft tick Ornithodoros moubata. Insect Biochem. Mol. Biol. 33, 841–851. doi: 10.1016/S0965-1748(03)00083-3
Schwarz, A., Tenzer, S., Hackenberg, M., Erhart, J., Gerhold-Ay, A., Mazur, J., et al. (2014). A systems level analysis reveals transcriptomic and proteomic complexity in Ixodes ricinus midgut and salivary glands during early attachment and feeding. Mol. Cell. Proteomics 13, 2725–2735. doi: 10.1074/mcp.M114.039289
Schwarz, A., von Reumont, B. M., Erhart, J., Chagas, A. C., Ribeiro, J. M., and Kotsyfakis, M. (2013). De novo Ixodes ricinus salivary gland transcriptome analysis using two next-generation sequencing methodologies. FASEB J. 27, 4745–4756. doi: 10.1096/fj.13-232140
Shabareesh, P. R. V., Kumar, A., Salunke, D. M., and Kaur, K. J. (2017). Structural and functional studies of differentially O-glycosylated analogs of a thrombin inhibitory peptide - variegin. J. Pept. Sci. 23, 880–888. doi: 10.1002/psc.3052
Shao, C. C., Xu, M. J., Chen, Y. Z., Tao, J. P., and Zhu, X. Q. (2015). Comparative profiling of microRNAs in male and female Rhipicephalus sanguineus. Appl. Biochem. Biotechnol. 176, 1928–1936. doi: 10.1007/s12010-015-1688-x
Suppan, J., Engel, B., Marchetti-Deschmann, M., and Nürnberger, S. (2018). Tick attachment cement - reviewing the mysteries of a biological skin plug system. Biol. Rev. Camb. Philos. Soc. 93, 1056–1076. doi: 10.1111/brv.12384
Tabish, M., Clegg, R. A., Turner, P. C., Jonczy, J., Rees, H. H., and Fisher, M. J. (2006). Molecular characterisation of cAMP-dependent protein kinase (PK-A) catalytic subunit isoforms in the male tick, Amblyomma hebraeum. Mol. Biochem. Parasitol. 150, 330–339. doi: 10.1016/j.molbiopara.2006.09.006
Tan, A. W., Francischetti, I. M., Slovak, M., Kini, R. M., and Ribeiro, J. M. (2015). Sexual differences in the sialomes of the zebra tick, Rhipicephalus pulchellus. J. Proteomics 117, 120–144. doi: 10.1016/j.jprot.2014.12.014
Temeyer, K. B., Olafson, P. U., and Pruett, J. H. (2012). Sequence polymorphism in acetylcholinesterase transcripts and genotyping survey of BmAchE1 in laboratory and Mexican strains of Rhipicephalus (Boophilus) microplus. J. Med. Entomol. 49, 555–562. doi: 10.1603/ME11215
Thompson, R. E., Liu, X., Ripoll-Rozada, J., Alonso-García, N., Parker, B. L., Pereira, P. J. B., et al. (2017). Tyrosine sulfation modulates activity of tick-derived thrombin inhibitors. Nat. Chem. 9, 909–917. doi: 10.1038/nchem.2744
Till, W. M. (1959). New cell types in the salivary glands of the brown ear tick, Rhipicephalus appendiculatus Neumann. Nature 184, 1078–1079. doi: 10.1038/1841078a0
Tirloni, L., Islam, M. S., Kim, T. K., Diedrich, J. K., Yates, J. R. III., Pinto, A. F., Mulenga, A., et al. (2015). Saliva from nymph and adult females of Haemaphysalis longicornis: a proteomic study. Parasit. Vectors 8:338. doi: 10.1186/s13071-015-0918-y
Tirloni, L., Lu, S., Calvo, E., Sabadin, G., Di Maggio, L. S., Suzuki, M., et al. (2020). Integrated analysis of sialotranscriptome and sialoproteome of the brown dog tick Rhipicephalus sanguineus (s.l.): insights into gene expression during blood feeding. J. Proteomics. 103899. doi: 10.1016/j.jprot.2020.103899
Tirloni, L., Reck, J., Terra, R. M., Martins, J. R., Mulenga, A., Sherman, N. E., et al. (2014). Proteomic analysis of cattle tick Rhipicephalus (Boophilus) microplus saliva: a comparison between partially and fully engorged females. PLoS ONE 9:e94831. doi: 10.1371/journal.pone.0094831
Tress, M. L., Abasca, F., and Valencia, A. (2017). Alternative splicing may not be the key to proteome complexity. Trends Biochem. Sci. 42, 98–110. doi: 10.1016/j.tibs.2016.08.008
Untalan, P. M., Guerrero, F. D., Haines, L. R., and Pearson, T. W. (2005). Proteome analysis of abundantly expressed proteins from unfed larvae of the cattle tick, Boophilus microplus. Insect Biochem. Mol. Biol. 35, 141–151. doi: 10.1016/j.ibmb.2004.10.009
Urbanová, V., Hajdušek, O., Šíma, R., Franta, Z., Hönig-Mondeková, H., Grunclová, L., et al. (2018). IrC2/Bf - A yeast and Borrelia responsive component of the complement system from the hard tick Ixodes ricinus. Dev. Comp. Immunol. 79, 86–94. doi: 10.1016/j.dci.2017.10.012
Valenzuela, J. G., Charlab, R., Mather, T. N., and Ribeiro, J. M. (2000). Purification, cloning, and expression of a novel salivary anticomplement protein from the tick, Ixodes scapularis. J. Biol. Chem. 275, 18717–18723. doi: 10.1074/jbc.M001486200
Valenzuela, J. G., Francischetti, I. M., Pham, V. M., Garfield, M. K., Mather, T. N., and Ribeiro, J. M. (2002a). Exploring the sialome of the tick Ixodes scapularis. J. Exp. Biol. 205, 2843–2864.
Valenzuela, J. G., Pham, V. M., Garfield, M. K., Francischetti, I. M., and Ribeiro, J. M. (2002b). Toward a description of the sialome of the adult female mosquito Aedes aegypti. Insect Biochem. Mol. Biol. 32, 1101–1122. doi: 10.1016/S0965-1748(02)00047-4
Vechtova, P., Sterbova, J., Sterba, J., Vancova, M., Rego, R. O. M., Selinger, M., et al. (2018). A bite so sweet: the glycobiology interface of tick-host-pathogen interactions. Parasit. Vectors 11:594. doi: 10.1186/s13071-018-3062-7
Viljoen, G. J., Bezuidenhout, J. D., Oberem, P. T., Vermeulen, N. M., Visser, L., Gothe, R., et al. (1986). Isolation of a neurotoxin from the salivary glands of female Rhipicephalus evertsi evertsi. J. Parasitol. 72, 865–874. doi: 10.2307/3281836
Villar, M., Ayllon, N., Alberdi, P., Moreno, A., Moreno, M., Tobes, R., et al. (2015). Integrated metabolomics, transcriptomics and proteomics identifies metabolic pathways affected by Anaplasma phagocytophilum infection in tick cells. Mol. Cell. Proteomics 14, 3154–3372. doi: 10.1074/mcp.M115.051938
Villar, M., Pacheco, I., Merino, O., Contreras, M., Mateos-Hernández, L., Prado, E., et al. (2020). Tick and host-derived compounds detected in the cement complex substance. Biomolecules 10:555. doi: 10.3390/biom10040555
Villar, M., Popara, M., Ayllón, N., Fernández de Mera, I. G., Mateos-Hernández, L., Galindo, R. C., et al. (2014). A systems biology approach to the characterization of stress response in Dermacentor reticulatus tick unfed larvae. PLoS ONE 9:e89564. doi: 10.1371/journal.pone.0089564
Wang, H., Kaufman, W. R., Cui, W. W., and Nuttall, P. A. (2001). Molecular individuality and adaptation of the tick Rhipicephalus appendiculatus in changed feeding environments. Med. Vet. Entomol. 15, 403–412. doi: 10.1046/j.0269-283x.2001.00328.x
Wang, H., Kaufman, W. R., and Nuttall, P. A. (1999). Molecular individuality: polymorphism of salivary gland proteins in three species of ixodid tick. Exp. Appl. Acarol. 23, 969–975. doi: 10.1023/A:1006362929841
Wang, H., and Nuttall, P. A. (1994). Comparison of the proteins in salivary glands, saliva and haemolymph of Rhipicephalus appendiculatus female ticks during feeding. Parasitology 109, 517–523. doi: 10.1017/S003118200008077X
Wang, X., Coons, L. B., Taylor, D. B., Stevens, S. E. Jr., and Gartner, T. K. (1996). Variabilin, a novel RGD-containing antagonist of glycoprotein IIb-IIIa and platelet aggregation inhibitor from the hard tick Dermacentor variabilis. J. Biol. Chem. 271, 17785–17790. doi: 10.1074/jbc.271.30.17785
Waterhouse, R. M., Seppey, M., Simão, F. A., Manni, M., Ioannidis, P., Klioutchnikov, G., et al. (2018). BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548. doi: 10.1093/molbev/msx319
Waxman, L., and Connolly, T. M. (1993). Isolation of an inhibitor selective for collagen-stimulated platelet aggregation from the soft tick Ornithodoros moubata. J. Biol. Chem. 268, 5445–5449.
Waxman, L., Smith, D. E., Arcuri, K. E., and Vlasuk, G. P. (1990). Tick anticoagulant peptide (TAP) is a novel inhibitor of blood coagulation factor Xa. Science 248, 593–596. doi: 10.1126/science.2333510
Xu, G., Fang, Q. Q., Keirans, J. E., and Durden, L. A. (2003). Cloning and sequencing of putative acetylcholinesterase cDNAs from the American dog tick, Dermacentor variabilis, and the brown dog tick, Rhipicephalus sanguineus (Acari: Ixodidae). J. Med. Entomol. 40, 890–896. doi: 10.1603/0022-2585-40.6.890
Xu, X. L., Cheng, T. Y., Yang, H., Yan, F., and Yang, Y. (2015). De novo sequencing, assembly and analysis of salivary gland transcriptome of Haemaphysalis flava and identification of sialoprotein genes. Infect. Genet. Evol. 32, 135–142. doi: 10.1016/j.meegid.2015.03.010
Keywords: tick, sialome, sialoverse, proteome, transcriptome, salivary gland
Citation: Mans BJ (2020) Quantitative Visions of Reality at the Tick-Host Interface: Biochemistry, Genomics, Proteomics, and Transcriptomics as Measures of Complete Inventories of the Tick Sialoverse. Front. Cell. Infect. Microbiol. 10:574405. doi: 10.3389/fcimb.2020.574405
Received: 19 June 2020; Accepted: 13 August 2020;
Published: 11 September 2020.
Edited by:
Lucas Tirloni, National Institute of Allergy and Infectious Diseases (NIH), United StatesReviewed by:
Shahid Karim, University of Southern Mississippi, United StatesElisa Azuara-Liceaga, Universidad Autónoma de la Ciudad de México, Mexico
Copyright © 2020 Mans. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ben J. Mans, bWFuc2JAYXJjLmFncmljLnph