 Austin M. Jeffries
Austin M. Jeffries Ian Marriott*
Ian Marriott*- Department of Biological Sciences, The University of North Carolina at Charlotte, Charlotte, NC, United States
Viral central nervous system (CNS) infections can lead to life threatening encephalitis and long-term neurological deficits in survivors. Resident CNS cell types, such as astrocytes and microglia, are known to produce key inflammatory and antiviral mediators following infection with neurotropic DNA viruses. However, the mechanisms by which glia mediate such responses remain poorly understood. Recently, a class of intracellular pattern recognition receptors (PRRs), collectively known as DNA sensors, have been identified in both leukocytic and non-leukocytic cell types. The ability of such DNA sensors to initiate immune mediator production and contribute to infection resolution in the periphery is increasingly recognized, but our understanding of their role in the CNS remains limited at best. In this review, we describe the evidence for the expression and functionality of DNA sensors in resident brain cells, with a focus on their role in neurotropic virus infections. The available data indicate that glia and neurons can constitutively express, and/or can be induced to express, various disparate DNA sensing molecules previously described in peripheral cell types. Furthermore, multiple lines of investigation suggest that these sensors are functional in resident CNS cells and are required for innate immune responses to viral infections. However, it is less clear whether DNA sensormediated glial responses are beneficial or detrimental, and the answer to this question appears to dependent on the context of the infection with regard to the identity of the pathogen, host cell type, and host species. Defining such parameters will be essential if we are to successfully target these molecules to limit damaging inflammation while allowing beneficial host responses to improve patient outcomes.
Introduction
Infection of the central nervous system (CNS) can result in encephalitis, a condition that is characterized by severe neuroinflammation resulting in fever, headaches, altered consciousness, seizures, and even death (Roos, 1999). Between 2000 and 2010 there were 7.3 encephalitis cases per 100,000, with most identified etiologies (48.2%) being attributable to viral infections (George et al., 2014). Since the mechanisms that lead to CNS inflammation following infection are poorly understood, current treatment strategies include general immune suppression, and/or antiviral therapy (Chaudhuri and Kennedy, 2002; George et al., 2014; Venkatesan and Geocadin, 2014). Traditionally, it was thought that infiltrating peripheral monocytes and leukocytes were the major contributors of pro-inflammatory mediator production in encephalitis as most resident CNS cells were assumed to lack immune functions. However, it is now appreciated that glial cells, most notably microglia and astrocytes, play a critical role in immune surveillance in the CNS and are important contributors to both protective and detrimental host responses to infectious agents (Aloisi, 2000, 2001; Bsibsi et al., 2002, 2006; Bowman et al., 2003; Furr et al., 2008, 2011; Chauhan et al., 2009; Liu et al., 2010; Serramía et al., 2015).
Glial cells can produce an array of proinflammatory and antiviral mediators following infection (Chauhan et al., 2009; Furr and Marriott, 2012) and it is now known that they accomplish this via members of multiple families of pattern recognition receptors (PRRs). These PRRs recognize numerous pathogen-associated molecular patterns (PAMPs) and/or damage associated molecular patterns (DAMPs) and trigger transcription factor activation that, in turn, elicits proinflammatory and antiviral mediator production. Of these, the most widely and best studied glial PRRs are the cell surface and endosomal Toll-like receptors (TLRs) and the cytosolic nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs) (Sterka et al., 2006; Chauhan et al., 2009; Rebsamen et al., 2009; Liu et al., 2010; Dai et al., 2014; Reinert et al., 2016; Su and Zheng, 2017). More recently, multiple classes of cytosolic nucleic acid sensors have been discovered that are likely have an important function during active viral infections due to their intracellular location. These include RNA sensors, such as the retinoic acid-inducible gene-I (RIG-I)-like receptor (RLR) family, and the possible roles and importance of these molecules in glial immune responses have been discussed in depth elsewhere (Furr et al., 2008; Furr and Marriott, 2012; Carty et al., 2014; Nair and Diamond, 2015; Zohaib et al., 2016). However, the importance of DNA sensors, including cyclic guanosine monophosphate-adenosine monophosphate (cGAMP) synthase (cGAS), in viral CNS infections and the initiation of glial immune functions remains more controversial, despite evidence for their antiviral function in peripheral lymphoid and myeloid cells (Unterholzner, 2013; Cai et al., 2014; Dhanwani et al., 2018; Lugrin and Martinon, 2018). In this review article, we will discuss the evidence for the expression and function of DNA sensors in resident CNS cells, their role during viral infections, and their potential as targets for therapeutic intervention.
cGAS/Sting
Perhaps the most well-known and best studied cytosolic DNA sensor is cGAS. This molecule directly binds to double stranded DNA and then catalyzes the production of the secondary messenger, 2′3′cyclic GMP-AMP (cGAMP) (Gao P. et al., 2013; Sun et al., 2013; Zhang X. et al., 2013). This secondary messenger subsequently binds to the downstream adaptor protein stimulator of interferon genes (STING), which initiates the phosphorylation of tank binding kinase 1 (TBK1), and interferon regulatory factor 3 (IRF3), and activates nuclear factor kappalight-chain-enhancer of activated B cells (NF-kB). Such transcription factor activation precipitates the expression of interferon-beta (IFN-β) and other antiviral and/or pro-inflammatory cytokines (Ishikawa et al., 2009; Li et al., 2013; Abe and Barber, 2014; Fang et al., 2017; Sun et al., 2017; Aarreberg et al., 2019). Since its discovery in 2013, cGAS has been demonstrated to play a critical role in recognizing and eliminating a diverse array of pathogens, either through direct recognition of microbial DNA or indirect recognition of retroviral DNA intermediates or damage associate molecular patterns (DAMPS), such as released mitochondrial DNA (Gao D. et al., 2013; Abe and Barber, 2014; Dai et al., 2014; Herzner et al., 2015; Paijo et al., 2016; Fang et al., 2017; Sun et al., 2017; Cheng et al., 2018; Wong et al., 2019). As discussed elsewhere (Cai et al., 2014; Chen et al., 2016; Dhanwani et al., 2018; Ablasser and Chen, 2019), numerous studies have demonstrated the expression and function of cGAS in peripheral leukocytic and non-leukocytic cell types, such as human plasmacytoid dendritic cells, macrophages, monocytes, helper T-lymphocytes, and endothelial cells (as summarized in Table 1). However, less attention has been given to the role of this sensor in the CNS and the immune responses of glial cells (Lahaye et al., 2013; Li et al., 2013; Dai et al., 2014; Ma Z. et al., 2015; West et al., 2015; Bode et al., 2016; Paijo et al., 2016; Vermeire et al., 2016; Luecke et al., 2017; Su and Zheng, 2017; Sun et al., 2017; Swanson et al., 2017).
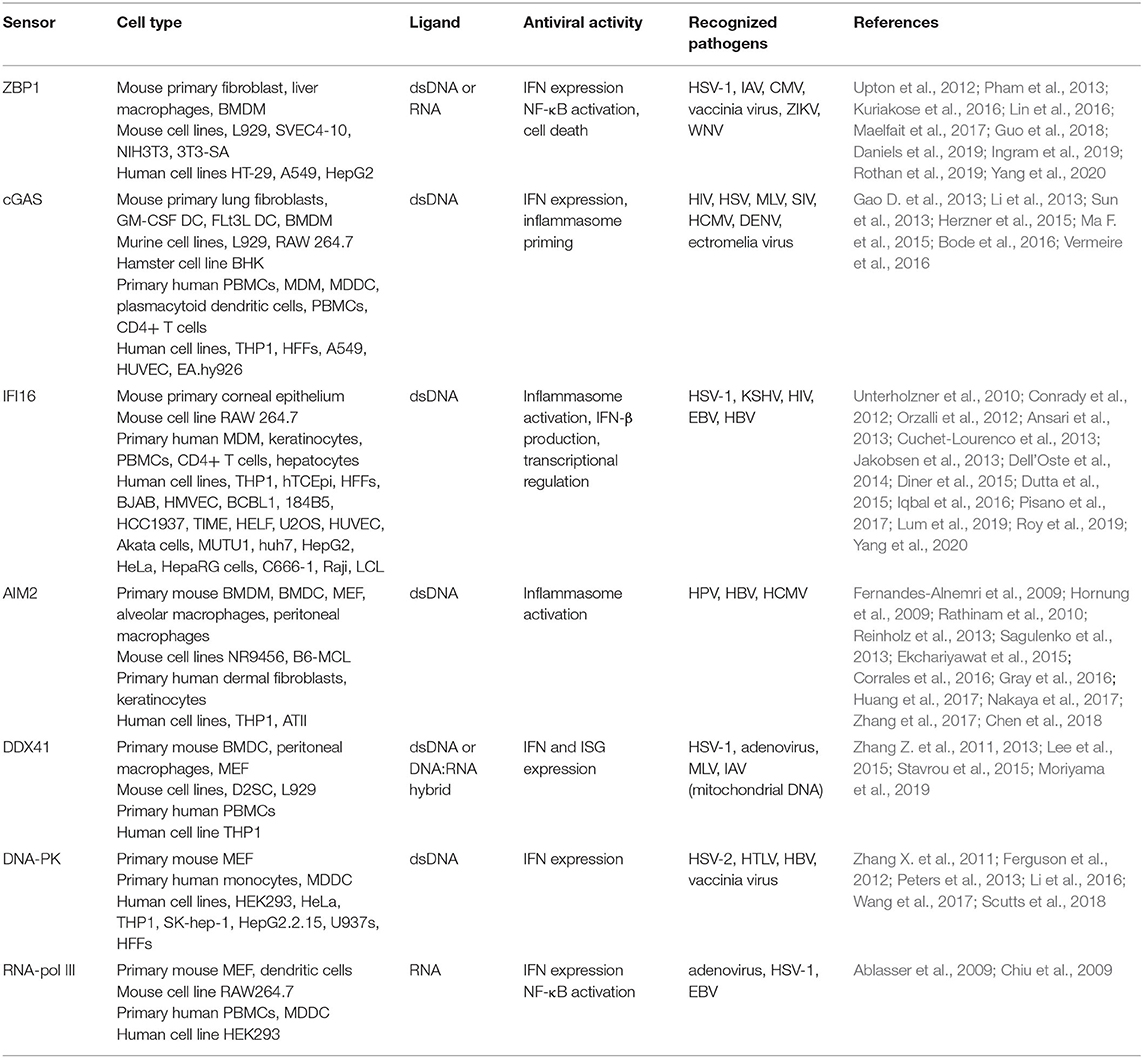
Table 1. Expression and antiviral activity of intracellular DNA sensors in peripheral cell types.
The first description of cGAS expression in resident CNS cell types came from Cox et al. (2015), who established the constitutive and inducible expression of mRNA encoding cGAS in murine microglia. Interestingly, while they observed neither constitutive nor IFN-β-inducible expression of cGAS in murine astrocytes, siRNA-mediated cGAS knockdown decreased IFN-β activity in both microglia and astrocytes following exposure to exogenous dsDNA (Cox et al., 2015). Further support for the presence of cGAS in glia has since been provided by our demonstration that primary human microglia and astrocytes both constitutively express cGAS protein and its downstream adaptor molecule STING (Jeffries and Marriott, 2017).
Circumstantial evidence for the functional importance of cGAS in the brain lays in the recognized ability of many important CNS pathogens to interfere with this sensor and/or it's signaling pathway (as summarized in Table 2). For example, herpes simplex virus 1 (HSV-1), the dsDNA virus that is the most common cause of fatal sporadic encephalitis, has multiple gene products that can interfere with the cGAS-STING signaling pathway (Bradshaw and Venkatesan, 2016). The HSV-1 encoded protein UL37 attenuates the enzymatic activity of cGAS and lowers cGAMP production in human monocytes and fibroblasts, thereby decreasing antiviral gene expression (Zhang et al., 2018). Mutations in UL37 that interfere with its deamidase activity prevent this protein from inhibiting cGAS and lead to lower HSV-1 titers in the brain following infection (Zhang et al., 2018). Interestingly, such an inhibitory activity appears to show species specificity as UL37 molecule does not appear to be important in infections in some non-human primate species (Zhang et al., 2018). Another HSV-1 product, UL41 (virion host shutoff protein), has been shown to decrease IFN-β production in a human epithelial cell line expression system by reducing cGAS protein expression (Su and Zheng, 2017), and its importance in disrupting cGAS-mediated antiviral responses has been illustrated by the ability of cGAS knockdown to increase viral production of a UL41 null mutant HSV-1 strain, but not a wild type strain (Su and Zheng, 2017). Furthermore, additional HSV-1 products have been demonstrated to target down-stream signaling molecules in the cGAS STING pathway. For instance, UL36 can prevent NF-κB activation by cleaving IκBa poly ubiquitin chains while UL46 and ICP27 interact with STING and TBK1 to prevent the activation of IRF3 and interferon stimulated gene (ISG) expression (Christensen et al., 2016; Deschamps and Kalamvoki, 2017; Ye et al., 2017; You et al., 2019). Since all members of the family Herpesviridae appear to target cGAS [Table 2 and as reviewed in Chan and Gack (2016) and Phelan et al. (2020)], and such viruses can cause latent infections, it is tempting to speculate that the inhibition of this sensor may play a critical role in establishing viral latency. Such a mechanism could be of particular importance for HSV encephalitis as the reactivation of a latent infection is thought to be a key contributor to the development of this condition (Menendez and Carr, 2017).
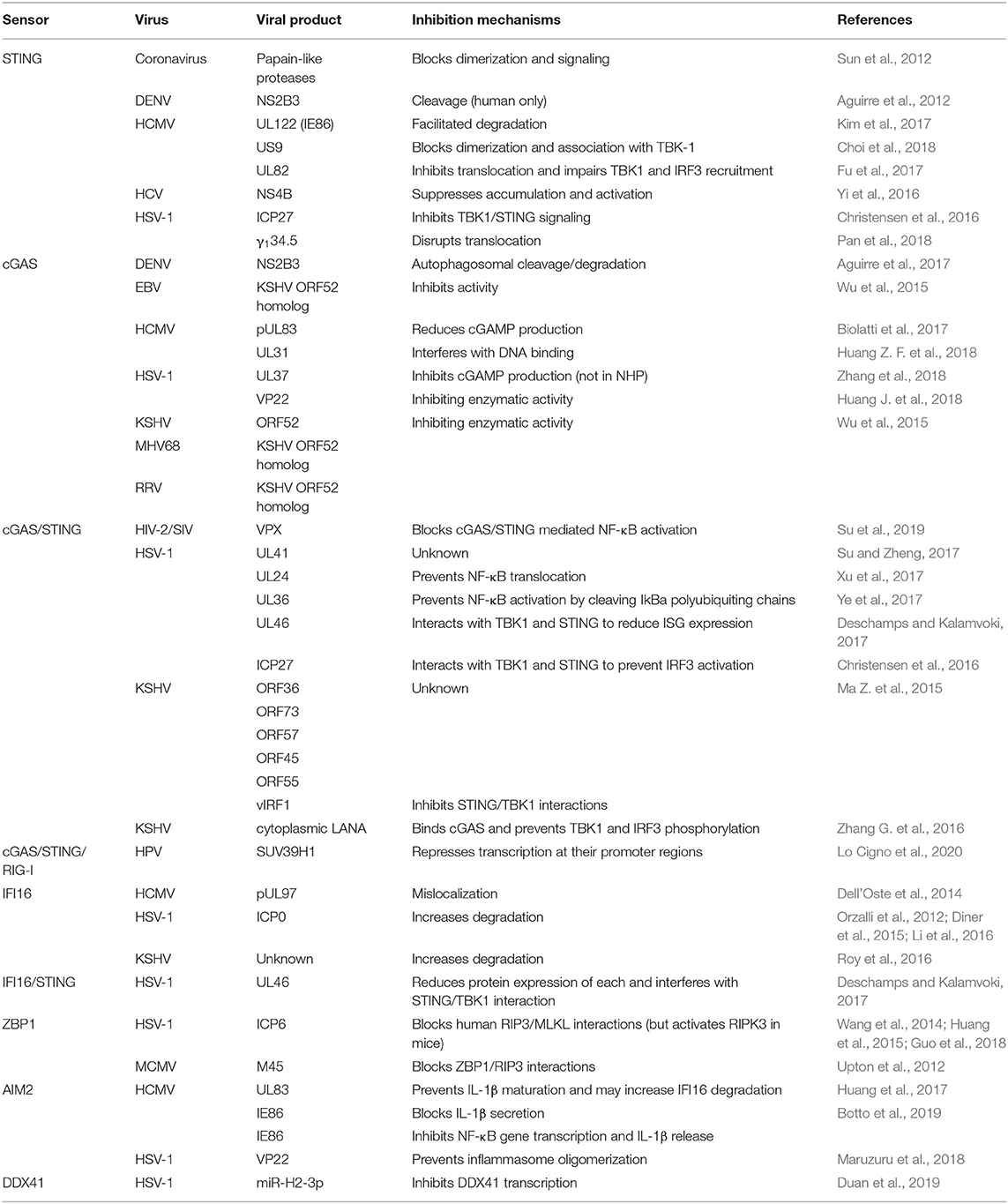
Table 2. Viral inhibitors of DNA sensing pathways.
In addition to DNA viruses, other neurotropic viruses, such as positive stranded RNA viruses from the family Flaviviridae, can also hamper cGAS activity (Table 2). For instance, Dengue virus (DENV) that can cause encephalitis has been shown to target cGAS for degradation and prevent it from detecting released mitochondrial DNA in human monocyte-derived dendritic cells and monocytic and fibroblastic cell lines (Aguirre et al., 2017; Sun et al., 2017).
Specifically, the DENV protein NS2B3 has been shown to directly interact with cGAS and target it for lysosomal degradation (Aguirre et al., 2017). In addition, this viral product can also cleave the downstream signaling molecule STING, further disrupting cGAS-mediated antiviral signaling. Interestingly, this viral product also shows species specificity similar to the HSV-1 product UL37 as NS2B3 targets human STING but does not cleave this molecule in mouse or non-human primate cells (Stabell et al., 2018). Similarly, Zika virus (ZIKV), which came to prominence for its role in CNS and peripheral nerve pathologies including microcephaly and Guillain-Barré syndrome, can cleave STING via its NS2B3 protein and this effect, again, is restricted to human rather than murine cells (Ding et al., 2018). ZIKV can further disrupt cGASSTING signaling by stabilizing caspase-1 protein. This results in cGAS cleavage and reduced expression of type I IFNs and ISGs, and the promotion of inflammasome activation and ZIKV production (Zheng et al., 2018). Importantly, Zheng et al. (2018) demonstrated that cGAS deficiency or caspase-1 inhibition leads to increased cellular levels of ZIKV genetic material. Such findings therefore support the therapeutic potential of augmenting cGAS-STING mediated responses to combat debilitating neurotropic RNA virus infections.
More direct evidence of the importance of cGAS-STING signaling during viral CNS infections comes from the study of HSV-1 infection in STING deficient animals. Parker et al. (2015) demonstrated that STING deficient animals show increased susceptibility to intracerebral HSV-1 infection, with all succumbing within 3–5 days of infection and little mortality in age-matched wildtype animals. Interestingly, it appears that this increased susceptibility is dependent on the route of infection, as STING deficient animals show survival rates comparable to wildtype mice following administration via the cornea, despite the presence of high viral titers in the cornea and trigeminal ganglion (Parker et al., 2015). This phenomenon was subsequently confirmed (Royer and Carr, 2016) and the apparent discrepancy in lethality is likely to be due to difference in the distribution of HSV-1 within the CNS. High viral titers are limited to the trigeminal ganglion following corneal HSV-1 infection, while intracerebral infection results in widely disseminated HSV-1 infection throughout the CNS (Parker et al., 2015). In addition, it should be noted that susceptibility to HSV-1 appears to be strain dependent as STING deficient animals succumb to neuroinvasive strains of HSV-1 following corneal infection more rapidly than wild type animals (Parker et al., 2015). Regardless, it is clear that STING plays a role in HSV-1 neuroinvasion and is critical for protective host responses once the virus has disseminated throughout the CNS (Parker et al., 2015; Royer and Carr, 2016). The increased susceptibility of STING deficient animals to HSV-1 neuroinvasion may be due, at least in part, to a decreased expression of the ISG tetherin [also known as bone marrow stromal antigen 2 (BST2)], as these animals exhibit decreased expression of this ISG (amongst others) during infection (Royer and Carr, 2016), and tetherin depletion has been shown to increase HSV1 titers in the trigeminal ganglion (Royer and Carr, 2016).
Consistent with these studies employing STING deficient animals, treatment with the STING agonist dimethlxanthenone-4-acetic acid (DMXAA) has been demonstrated to increase IFN-β expression by HSV-1 infected mouse fibroblasts and to lower the production of viral particles by these cells (Cerón et al., 2019). Importantly, in vivo DMXAA treatment can lower viral titers in the cornea, trigeminal ganglion, and brainstem, of mice infected with the neuroinvasive McKrae strain of HSV-1, and this is reflected by increased survival and improved neurological outcomes in these animals (Cerón et al., 2019). As such, these studies provide a tantalizing glimpse of the potential of targeting the cGAS-STING pathway to treat CNS infections.
While these studies illustrate the importance of cGAS-STING signaling in HSV-1 infections of the CNS, the specific role of this sensor system in glia was established by Reinert et al. (2016) in a mouse model of HSV-1 encephalitis. They demonstrated that cGAS deficiency resulted in a phenotype that matched that observed in STING deficient mice following ocular HSV-1 infection (Reinert et al., 2016). Furthermore, they established that microglia were the primary producers of IFN-β after HSV-1 challenge and showed that only this glial cell type produced higher viral titers in vitro following the loss of STING (Reinert et al., 2016). In vivo, however, neurons and astrocytes showed greater numbers of HSV-1 viral particles in STING deficient mice (Reinert et al., 2016). This discrepancy was explained by the observation that astrocytes and neurons initiate antiviral programs in vivo in response to IFN-β produced by microglia in a TLR3-dependent manner (Reinert et al., 2016). This suggests that microglia represent the first responders to HSV-1 infection in the CNS.
Consistent with these findings in mice, we have shown that cytosolic administration of a dsDNA ligand can phosphorylate IRF3 and induce IFN-β mRNA expression in primary human microglia and astrocytes (Jeffries and Marriott, 2017), and we have demonstrated that such responses are largely dependent on cGAS expression (Jeffries and Marriott, 2017; Jeffries et al., 2020). Furthermore, we showed that ISG expression is lower in cGAS deficient human microglia both at rest and following infection with HSV-1 (Jeffries et al., 2020). However, while cGAS can contribute to antiviral gene expression, the absence of cGAS had no effect on HSV-1 production in infected human microglia (Jeffries et al., 2020). Given the recognized ability of HSV-1 products to abrogate cGAS-STING signaling in peripheral human but not murine cell types, we assessed cGAS protein levels in human glial cells following HSV-1 challenge. We found that the expression of this sensor was markedly reduced in human microglial and astrocytic cells following infection (Jeffries et al., 2020), highlighting the ability of viruses such as HSV-1 to circumvent PRR-mediated immune responses. As such, improving the stability and/or expression of cGAS/STING signaling components might be a viable approach to combat viral infections of the CNS and/or periphery.
IFI16
Interferon gamma inducible protein 16 (IFI16) is a member of the Pyrin and HIN domain (PHYIN) family of proteins that can serve as an intracellular DNA sensing molecule. PHYIN proteins are characterized by the presence of an N-terminal pyrin domain and one or two Cterminal HIN domains (Unterholzner et al., 2010). The HIN domains bind DNA while the pyrin domain is required for protein-protein interactions (Unterholzner et al., 2010). IFI16, and its mouse ortholog p204, was the first PHYIN family members demonstrated to induce IFN-β in response to transfected DNA (Unterholzner et al., 2010). Additionally, IFI16 has been shown to interact with STING and knockdown of either of these proteins leads to reduced IFN-β production by the human and mouse monocytic cell lines THP-1 and RAW 264.7, respectively (Cridland et al., 2012). Interestingly, similar findings were described in murine astrocytes and microglia where p204 knockdown was shown to reduce IFN-β expression following DNA transfection (Cox et al., 2015), and our own studies indicate that human microglial and astrocytic cells constitutively express robust levels of IFI16 protein (Jeffries et al., 2020).
However, IFI16 does not appear to contribute to IFN-β expression by human foreskin fibroblasts (HFF) stimulated with exogenous DNA (Orzalli et al., 2015). While this finding might be indicative of cell type-specific differences, our studies showing that IFI16 knockdown has no effect on IFN-β protein production by a human microglia cell line following BDNA transfection also indicate that this putative DNA sensor is not required for such responses (Jeffries et al., 2020). Furthermore, Gray et al. (2016) used a mouse model lacking all 13 PHYIN family members to demonstrate that these receptors were dispensable for IFN responses to DNA transfection in bone marrow derived macrophages and mouse embryonic fibroblasts. But despite an apparent lack of involvement in IFN production, this study did identify a requirement for PHYIN family members in inflammasome activation, as characterized by the maturation of the potent pro-inflammatory cytokines IL-1β and IL-18 (Gray et al., 2016). Such a role for IFI16 in linking DNA sensing and inflammasome activation is supported by multiple studies (Ansari et al., 2013, 2015; Johnson et al., 2013; Dutta et al., 2015; Iqbal et al., 2016; Orzalli et al., 2016) and is discussed in depth elsewhere (Dell'Oste et al., 2015), but it is currently unknown whether IFI16-mediated inflammasome activation occurs in CNS cell types.
While there is conflicting evidence for the role of IFI16 in IFN-β responses to foreign DNA challenge, multiple lines of investigation indicate that this DNA sensor contributes to IFN-β and ISG expression following viral infection (Orzalli et al., 2012; Jakobsen et al., 2013; Ansari et al., 2015; Diner et al., 2015; Ma F. et al., 2015; Li et al., 2016; Zhang D. et al., 2016; Jønsson et al., 2017; Lum et al., 2019; Yang et al., 2020). For example, nuclear IRF3 translocation and subsequent IFN production in HSV-1 infected corneal epithelial cells has been shown to be dependent, at least in part, on p204 expression (Conrady et al., 2012), while TBK-1 phosphorylation and subsequent IFN-β expression by HSV-1 infected HFFs was found to require IFI16 and STING (Orzalli et al., 2012).
Interestingly, Orzalli et al. (2012) used attenuated HSV-1 strains to determine that expression of the immediate early viral protein ICP0 leads to IFI16 degradation and reduced nuclear IRF3 translocation, and this degradation was subsequently shown to be dependent on proteasome activity. In addition, another group has shown that HSV-1 can also lead to IFI16 degradation in an ICP0 independent manner, albeit in the U2OS cell line that lacks a functional STING signaling pathway (Cuchet-Lourenco et al., 2013). Moreover, HSV-1 infection markedly lowers IFI16 protein expression in primary human glia and immortalized cells lines, and this observation may explain why knockdown of this DNA sensor fails to alter viral production or IFN-β release by these cells (Jeffries et al., 2020). Such findings are supported in HFFs where IFI16 knockdown does not affect HSV-1 levels in cells infected with wild type HSV-1 but significantly increases viral titers following infection with an ICP0 null mutant virus (Diner et al., 2016). Similarly, IFI16 has been reported to be dispensable for IFN production in mice following HCMV infection (Gray et al., 2016) and this apparent independence may also stem from the reported ability of HCMV to interfere with IFI16 signaling (Dell'Oste et al., 2014). However, it should be noted that the ability of these viruses to reduce IFI16 protein abundance and/or signaling may show cell type specificity as HSV-1 does not elicit such effects in either HeLa cells or U2OS cells, again perhaps due to a lack of a functional STING signaling pathway in the latter (Orzalli et al., 2016; Deschamps and Kalamvoki, 2017).
Since the available evidence indicates that IFI16 has a role in virally-induced IFN signaling and that this is accomplished through via a STING-dependent pathway, it is possible that this molecule could work in concert with cGAS to stimulate antiviral responses. Evidence for this notion comes from the observation that knockdown of either STING, IFI16, or cGAS, in human fibroblasts leads to reduced HSV-1 infection-induced IFN-β expression (Orzalli et al., 2015). Interestingly, in the same study it was noted that cGAS knockdown reduces constitutive IFI16 protein expression and that this effect was dependent on proteasome activity (Orzalli et al., 2015). This suggests that cGAS may stabilize IFI16 protein levels to promote antiviral activity. However, we found no observable difference in IFI16 protein expression in cGAS deficient microglia created with CRISPR/Cas9 approaches (Jeffries et al., 2020) and so it is possible that, like viral ICP0-mediated effects (Orzalli et al., 2016), cGAS-mediated IFI16 stabilization may show cell type specificity.
Additional support for cooperation between IFI16 and cGAS comes from the work of Jønsson et al. (2017) who demonstrated that IFI16 knockdown reduces cGAMP production by THP-1 cells following foreign DNA challenge. Furthermore, they showed that HEK 293T cells stably expressing IFI16 produce higher amounts of cGAMP following intracellular administration of a cGAS expression plasmid than IFI16 deficient cells (Jønsson et al., 2017). Similarly, another study showed that the co-transfection of increasing amounts of an IFI16 expression plasmid with constant levels of STING and cGAS increased IFN-β activity as assessed by IFN promoter driven luciferase activity, and demonstrated the ability of IFI16 and cGAS to interact directly (Almine et al., 2017). It should be noted that these investigators failed to detect significant changes in cGAMP levels in the absence or presence of IFI16 (Almine et al., 2017). Rather, they determined that IFI16 was required for cGAMP to fully activate STING as assessed by CCL5 expression, STING dimerization, and IRF3 nuclear translocation, in response to cGAMP transfection (Almine et al., 2017).
In contrast to such studies that suggest IFI16 acts in concert with cGAS to promote IFN responses, we demonstrated that cGAS knockdown decreased IFN-β production by a human microglial cell line following DNA transfection but IFI16 knockdown did not (Jeffries et al., 2020). Furthermore, we showed that IFI16 knockdown failed to exacerbate the reduction in IFN-β production by DNA stimulated cGAS deficient microglia, and combined cGAS and IFI16 deficiency failed to significantly alter microglial susceptibility to HSV-1 infection over cGAS deficiency alone (Jeffries et al., 2020). However, an explanation for these results may again stem from the ability of HSV-1 to downregulation IFI16 and cGAS expression and/or inhibit their signaling pathways in human microglia following infection (Jeffries et al., 2020). As such, the development of therapeutics that stabilize the expression of either of these sensor proteins could prove to be an attractive approach to combat the devastating consequences of conditions such as HSV-1 encephalitis.
While cGAS and IFI16 might play redundant roles in STING activation, some evidence suggests that they promote similar responses through different mechanisms. For instance, it was found that IFI16 is not required for cGAS/STING/TBK-1 signaling in HFFs following HSV-1 or HCMV infection, but was for the transcription of IFN-β, ISG54, ISG56, and RANTES (Diner et al., 2016). Interestingly, the same investigators found that IFI16 also reduced the transcription of the HSV-1 genes icp27, icp8, and ul30 (Diner et al., 2016). This suggests that the antiviral functions previously attributed to IFI16 may occur through transcriptional regulation, rather than by direct activation of cGAS-STING signaling. However, our own investigations of the role of IFI16 in HSV-1 transcription in infected human microglial cells showed no discernable effect on icp8 expression (Jeffries et al., 2020). This apparent discrepancy is likely due to differences in the HSV-1 strain employed, as the earlier study used an ICP0 mutant strain that prevents IFI16 degradation, while our studies were performed with the neuroinvasive MacIntyre strain (Diner et al., 2016; Jeffries et al., 2020).
The ability of IFI16 to negatively regulate viral transcription has been reported for other herpesviruses, human papillomavirus (HPV), and hepatitis B virus (HBV) (Gariano et al., 2012; Lo Cigno et al., 2015; Roy et al., 2016, 2019; Pisano et al., 2017; Yang et al., 2020), and the modification of heterochromatin and euchromatin appears to be the primary mechanism by which this is accomplished. For example, U2OS cells or an immortalized human keratinocyte cell line (NIKS) overexpressing IFI16 exhibit elevations in heterochromatin markers, such as H3K9m2, and decreases in euchromatin markers, such as H3K4me2, in early and late HPV promoters as determined by chromatin immunoprecipitation (ChiP) analysis (Lo Cigno et al., 2015). In addition, IFI16 has been demonstrated to directly interact with the histone H3-K9 methyltransferases, SUV39h1 and G9a-like protein (GLP), and knockdown of these proteins in a B cell lymphoma latently infected with KSHV (BCBL1 cells) led to increased viral transcription (Roy et al., 2019). Furthermore, in the same study, IFI16 knockdown reduced the recruitment of both methyltransferases to the KSHV genome (Roy et al., 2019).
In a HBV covalently closed circular DNA (cccDNA) model of infection, overexpression of IFI16 has been shown to increase IFN-β and ISG expression along with decreased euchromatin and increased heterochromatin markers on cccDNA (Yang et al., 2020). Interestingly, knockdown of IFI16 in BCBL1 cells increased the transcription of immediate early, early, and late lytic KSHV genes, indicating reactivation of the lytic cycle, while the reintroduction of IFI16 reduced KSHV genome copy numbers (Roy et al., 2016). This was also found to be true for Akata and MUTU1 cell lines latently infected with Epstein-Barr virus (Pisano et al., 2017). As such, it will be important to determine whether IFI16 similarly contributes to HSV-1 latency in CNS cell types, since reactivation of latent infections is a key event in the onset of HSV-1 encephalitis (Menendez and Carr, 2017). If so, IFI16 could be a promising new therapeutic target, either as an intervention during CNS infection or to prevent reactivation of HSV-1 in at-risk populations.
ZBP1
Z-DNA binding protein 1 [ZBP1; also known as DNA-dependent activator of interferon regulatory factors (DAI) and DLM-1] was the first identified cytosolic DNA sensor, and was shown to directly bind dsDNA in murine L929 fibroblast-like cells (Takaoka et al., 2007). Importantly, this study demonstrated ZBP1 can interact with TBK1 and IRF3 and contribute to IFN-β mRNA expression following DNA transfection or infection with HSV-1 (Takaoka et al., 2007). However, it seems that this cytosolic DNA sensor may function in a cell type and ligand specific manner, as ZBP1 knockdown in mouse embryonic fibroblasts (MEFs) has little or no effect on exogenous DNA-induced IFN-β expression (Wang et al., 2008). Similarly, ZBP1 knockdown was found to significantly reduce IFN-β expression in L929 cells in response to BDNA transfection but had no effect in a similarly challenged human lung epithelial cell line (Lippmann et al., 2008).
In addition to the expression of antiviral cytokines, ZBP1 has also been shown to mediate the expression of the pro-inflammatory cytokine IL-6 (Takaoka et al., 2007; Kaiser et al., 2008) subsequently demonstrated the activation of a NF-kB-driven luciferase promoter in HEK 293T cells overexpressing ZBP1. This group identified three RIP homotypic interaction motif (RHIM)-like repeats and hypothesized that such NF-κB activation occurs via a RHIM-dependent interaction with receptor interacting protein 1 (RIP1) in a similar manner to that seen with TLR3 (Kaiser et al., 2008). This hypothesis was confirmed by the demonstration that ZBP1 can interact with both RIP1 and receptor interacting protein 3 (RIP3) through its first RHIM domain, and by the ability of RIP1 knockdown or mutations in the RHIM domain in RIP1 or ZBP1 to decrease NF-kB promoter activation. The ability of ZBP1 to interact with RIP1 and RIP3, and to activate NF-kB-mediated gene transcription was subsequently confirmed in a similar HEK 293 cell expression system (Rebsamen et al., 2009). In is interesting to note, however, that co-expression of ZBP1 and RIP3 was also reported to elicit NF-kB activation in these studies, an observation that is in contrast to TLR3 signaling where RIP3 blocks RIP1-mediated NF-kB activation (Kaiser et al., 2008).
In agreement with these studies in non-CNS cell types, we have determined that murine microglia and astrocytes express ZBP1 in an inducible manner, and found that this sensor contributes to pro-inflammatory cytokine production by glia following HSV-1 infection (Furr et al., 2011). Furthermore, these studies also showed that HSV-1 infection induces the production of soluble neurotoxic mediators by astrocytes and microglia in a ZBP1-dependent manner (Furr et al., 2011). Surprisingly, combined knockdown of ZBP1 and retinoic acid inducible gene 1 (RIG-I) leads to greater reductions in TNF-a and IL-6 production by HSV-1 infected glia than either alone, suggesting that these dissimilar sensors can act in synergy (Crill et al., 2015). Together, these studies suggest a role for ZBP1 in inflammation and/or antiviral immunity both in the periphery and the CNS.
While the available evidence supports a role for ZBP1 as a DNA sensor capable of inducing cytokine production, some studies suggest that ZBP1 plays a broader role in antiviral immunity. For example, ZBP1 has been reported to work in concert with RIP3 in murine fibroblasts and epithelial cells to induce necroptosis following infection with a mutant murine cytomegalovirus (MCMV) strain (Upton et al., 2012) that lacks the expression of m45, a viral product that limits ZBP1/RIP3 interactions due the presence of a RHIM domain (Table 1) (Rebsamen et al., 2009; Upton et al., 2012). Interestingly, a similar immune evasion mechanism has been observed for HSV-1 (Table 1), where the viral protein ICP6 also contains a RHIM domain that is capable of inhibiting necroptosis in peripheral human cells (Guo et al., 2015). Necroptotic cell death initiated by simultaneous treatment with TNF-a and the caspase inhibitor zVAD-FMK was blocked following infection with wild type HSV-1, while an ICP6 deficient HSV-1 strain failed to prevent cell death (Guo et al., 2015; Sawai, 2016). This finding is in sharp contrast to studies in mouse cells, where infection with wild type HSV-1 elicits cell death in a RIP3-dependent manner (Wang et al., 2014; Huang et al., 2015). Surprisingly, expression of ICP6 in MEFs was found to be enough to induce RIP3-dependent cell death, while the presence of ICP6 containing mutations in the RHIM domain did not (Wang et al., 2014; Huang et al., 2015), suggesting that ICP6 may be able to directly induce necroptosis in this cell type (Wang et al., 2014). The reason for these apparently contradictory findings was discovered in more recent studies that show ICP6 has species-dependent effects, inducing necroptosis in cells from mice while inhibiting it in human cells, HSV's primary natural host (Huang et al., 2015; Guo et al., 2018).
Importantly, ZBP1 has been found to be a major contributor to necroptosis in both human and mouse fibroblasts following infection with both an ICP6-deficient and an ICP6 RHIM mutant HSV-1 strain (Guo et al., 2018) and our own observations suggest that this cytosolic DNA sensor functions as a mediator of cell death during HSV-1 infection in glia. Our studies indicate that ZBP1 plays a crucial role in triggering necroptosis in murine glia following infection with a strain of HSV-1 that contains mutations in the ICP6 RHIM domain (unpublished observations). The potential importance of this pathway in antiviral immunity within the CNS is underscored by the decreased survival and increased viral burden in the brain of RIP3 deficient mice following HSV-1 infection (Wang et al., 2014; Huang et al., 2015). However, since necroptosis promotes inflammation, it will be important to determine whether this ZBP-mediated response also contributes to CNS pathology during HSV-1 encephalitis, especially in the human host (Dhuriya and Sharma, 2018).
While it is increasingly clear that ZBP1 is a PRR that is capable of initiating cell death pathways, it is less certain what ligands specifically initiate such as response. ZBP1 was initially shown to directly bind BDNA and it has recently been shown to recognize plasmid DNA (Wang et al., 2008; Semenova et al., 2019). However, other studies have shown that ZBP1 is critical for the induction of necroptosis, pyroptosis, and apoptosis, in cells challenged with influenza virus, a segmented negative strand RNA virus (Kuriakose et al., 2016; Thapa et al., 2016), and this role is discussed in depth elsewhere (Dhuriya and Sharma, 2018). By pharmacologically inhibiting various stages of the MCMV life cycle, Sridharan et al. (2017) were able to determine that active transcription was required for ZBP1-mediated cell death, suggesting that RNA serves as the activating ligand in this response. This notion was subsequently supported by two studies describing the ability of ZBP1 to bind endogenous RNA (Maelfait et al., 2017; Jiao et al., 2020).
An ability to sense both RNA and DNA accounts for the protective role of ZBP1 in influenza virus infection and following exposure to other RNA viruses including West Nile virus (WNV) and ZIKV (Daniels et al., 2019; Rothan et al., 2019). Interestingly, however, ZBP1-mediated protection against these neurotropic flaviviruses appears to be independent of cell death in neurons (Daniels et al., 2019; Rothan et al., 2019). Mice genetically deficient in ZBP1 show worse clinical scores, higher viral burdens in the brain, and increased mortality, following WNV infection than wild type mice (Rothan et al., 2019). Similarly, higher viral burdens and mortality have been observed in WNV challenged RIP3 deficient animals, and this effect was independent of cell death pathways (Daniels et al., 2017). Surprisingly, ZBP1 deficient animals demonstrate higher levels of antiviral and inflammatory cytokines/chemokines following WNV infection (Rothan et al., 2019), and this finding is in contrast to similarly infected RIP3 deficient mice, which demonstrate decreased inflammatory cytokine production (Daniels et al., 2017). As such, it is possible these two molecules have independent roles during neuronal infection, especially since peripheral cells undergo cell death in both a ZBP1 and a RIP3-dependent manner (Daniels et al., 2017; Rothan et al., 2019).
A potential mechanism for the antiviral effects of ZBP1 and RIPK3 during neurotropic RNA virus infections comes from the studies of Daniels et al. (2019) that indicate a neuron-specific function for ZBP1. They found that ZBP1-induced antiviral gene expression in neurons following ZIKV infection occurs in a RIP1 and RIP3-dependent manner, and that loss of any of these signaling molecules results in increased viral burden and mortality (Daniels et al., 2019). Surprisingly, RIP3 deficiency in primary microglial cultures did not result in increased ZIKV replication in these studies suggesting that such protection is intrinsic to neurons. Consistent with this notion, upregulation of the antiviral gene IRG1 was required for protection against both ZIKV and WNV infection in neurons, but this gene was not upregulated in microglia (Daniels et al., 2019), indicating a cell type specific function for ZBP1. Since we have identified a role for this sensor in glia following HSV-1 infection, it will be interesting to see what role, if any, ZBP1 plays in glial responses to neurotropic RNA viruses (Furr et al., 2011; Crill et al., 2015). Regardless, it is apparent that ZBP1 is an important mediator of CNS innate immune responses to both RNA and DNA viruses.
AIM2
Absent in melanoma 2 (AIM2) is another member of the PHYIN family of interferon inducible proteins that has been found to act as a DNA sensor (Bürckstümmer et al., 2009; Fernandes-Alnemri et al., 2009; Hornung et al., 2009; Adamczak et al., 2014). However, unlike IFI16, recognition of dsDNA by AIM2 has been shown to lead exclusively to inflammasome activation and the induction of pyroptosis, an inflammatory form of cell death (Adamczak et al., 2014). Upon binding to dsDNA, AIM2 associates with the downstream signaling molecule apoptosisassociated speck-like protein containing a CARD domain (ASC), which recruits, and activates caspase-1 (Bürckstümmer et al., 2009; Fernandes-Alnemri et al., 2009; Hornung et al., 2009). Caspase-1 then acts as the effector protein to cleave the immature form of IL-1β and IL-18, leading to the maturation and secretion of these potent inflammatory cytokines (Miao et al., 2011). Additionally, caspase-1 can cleave gasdermin D to initiate pyroptotic cell death, characterized by the formation of pores in the plasma membrane and the release of cellular contents into the extracellular environment (Kayagaki et al., 2015; Shi et al., 2015). The AIM2 inflammasome has been shown to form following infection with either DNA or RNA viruses in peripheral myeloid and lymphoid immune cell-types, such as bone marrow derived dendritic cells (BMDCs), bone marrow derived macrophages (BMDM), monocytes, and fibroblasts (Rathinam et al., 2010; Ekchariyawat et al., 2015; Schattgen et al., 2016; Huang et al., 2017; Zhang et al., 2017), and the role of this and other inflammasomes during viral infection is discussed extensively elsewhere (Chen and Ichinohe, 2015; Lupfer et al., 2015; Man et al., 2016; Shrivastava et al., 2016; Lugrin and Martinon, 2018; Zhu et al., 2019).
While the AIM2 inflammasome is recognized to have an antiviral function in peripheral cell types, relatively little is known about its role in CNS infections despite having been shown to be expressed in neurons and glia (Adamczak et al., 2014; Cox et al., 2015). Furthermore, AIM2 has been shown to function as a DNA sensor in neurons as the cytosolic administration of exogenous DNA induces the association of AIM2 with ASC and leads pyroptosis in these cells (Adamczak et al., 2014). Since neuronal cell death is typically detrimental to the host, it appears likely that the proinflammatory nature of the AIM2 inflammasome can be damaging in the context of viral CNS infections. Circumstantial evidence supporting this notion comes from the effect of deleting ataxia-telangiectasia mutated (ATM), a protein known for its role in activating DNA damage responses, in primary murine microglia (Song et al., 2019). Such a deletion results in cytoplasmic DNA accumulation and cellular activation as demonstrated by the retraction of processes (Song et al., 2019). Moreover, co-culture of microglia and neurons with an ATM inhibitor leads to neuronal cell damage, which is reversed with an IL-1 receptor antagonist consistent with a major role for the inflammasome in this effect (Song et al., 2019). Importantly, this study utilized co-immunoprecipitation approaches to demonstrate that inflammasome activation as a result of ASC association with AIM2 rather than other initiator molecules such as NLR family pyrin domain containing 3 (NLRP3) (Song et al., 2019). A detrimental role for AIM2 in CNS pathologies is further supported by the observation that AIM2 deficient mice show less brain atrophy and cognitive defects following stroke than their wild type counterparts (Kim et al., 2020). Furthermore, caspase-1 inhibition resulted in a similar phenotype in these studies indicating that the improved outcome was due to reduced AIM2 inflammasome activation (Kim et al., 2020).
Despite such evidence, some studies suggest that AIM2 can play a protective role in some infections. For example, the neurotropic RNA viruses, WNV and Chikungunya virus (CHIKV), have been shown to activate the AIM2 inflammasome in peripheral dermal fibroblasts and AIM2 knockdown led to increased CHIKV genome copies in these cells (Ekchariyawat et al., 2015). Furthermore, the higher levels of AIM2, caspase 1, IL-1β, and IL-18, found in brain tissue from still births following ZIKV infection provides circumstantial evidence for a role for this sensor (de Sousa et al., 2018). However, it should be noted that the expression of two other inflammasome activators, NLRP3, and NLRP1, were also elevated in this tissue, and it is not clear whether the upregulation of inflammasome components reflect a protective host response or contribute to disease pathology. Similarly, AIM2 expression is upregulated in neurons following infection with enterovirus A71, the causative agent of hand foot and mouth disease and AIM2 knockdown in a neuronal cell line led to decreased IL-1β cleavage and increased viral copy numbers (Yogarajah et al., 2017). Yet, no mechanism has yet been defined for AIM2mediated sensing of RNA viruses.
With regard to neurotropic DNA viruses, AIM2 was initially demonstrated to be dispensable for inflammasome activation following HSV-1 infection in peritoneal macrophages, but was for necessary for such responses to MCMV challenge (Rathinam et al., 2010). Conversely, another study indicated that IFI16 and NLRP3 were the initiators of inflammasome activation in HFFs following HSV-1 infection (Johnson et al., 2013). This apparent discrepancy may be due to the ability of the HSV-1 product VP22 to block AIM2-mediated inflammasome activation by preventing oligomerization (Table 2) (Maruzuru et al., 2018). Intracranial administration of an HSV-1 strain lacking VP22 leads to decreased viral burdens in wildtype mice but not those lacking AIM2, suggesting that this sensor can limit viral replication in the CNS (Maruzuru et al., 2018). Interestingly, the protective functions of AIM2 in the CNS may extend to bacterial pathogens as AIM2 has been shown to contribute to survival following CNS infection with Staphylococcus aureus (Hanamsagar et al., 2014).
It is clear from the available data that out current understanding of the role of AIM2 in the CNS is rudimentary. While some evidence suggests that AIM2 contributes to CNS disease pathology, some indicate protective functions. As such, it may be that AIM2 plays a context-dependent role where this molecule exacerbates sterile inflammation in neurodegenerative diseases when activation tends to be chronic, while acute activation assists in viral or bacterial clearance. Since our understanding of AIM2 in the CNS is based mostly on circumstantial evidence, further study is clearly required to determine the role of this molecule relative to other inflammasome activators, and to determine whether this pathway can be targeted for therapeutic intervention.
DDX41, Ku70/DNA-PK, and, RNA Polymerase III
Several other putative DNA sensors have been identified in peripheral cell types but their role as PRRs in the CNS remains more controversial. DEAD-Box Helicase 41 (DDX41) was first identified as a cytosolic DNA sensor in a murine dendritic cell-like line with the demonstration that this molecule can directly bind dsDNA and interact with the common DNA sensing and antiviral signaling components STING and TBK1 (Zhang Z. et al., 2011). Importantly, DDX41 knockdown was shown to decrease IFN-α production in these cells in response to dsDNA transfection or infection with either HSV-1 or adenovirus (Zhang Z. et al., 2011). Interestingly, DDX41 has also been shown to directly bind cyclic dinucleotides such as cyclic dimeric guanosine monophosphate (c-di-GMP) and cyclic dimeric adenosine monophosphate (c-diAMP), and knockdown of DDX41 prevents STING association with TBK1 or IRF3 and reduces antiviral signaling in response to these molecules (Parvatiyar et al., 2012). As such, it is possible that DDX41 bolsters cGAS-STING signaling by promoting cGAMP- STING interactions. Evidence for such a suggestion comes from the demonstration that DDX41 knockdown further reduces murine leukemia virus (MLV)-induced IFN expression by cGAS deficient macrophages and dendritic cells (Stavrou et al., 2015). Furthermore, IFN-β expression could be rescued in macrophage-like cells following cGAS knockdown with the administration of exogenous cGAMP prior to MLV infection, but this procedure failed to rescue such responses in cells following DDX41 knockdown (Stavrou et al., 2018). Together, these studies suggest that DDX41 can act in a cooperative manner with cGAS to induce STING activation following viral challenge.
To date, it is not known whether DDX41 is expressed in the mammalian CNS. However, the drosophila DDX41 homolog, Abstrakt, has been shown to be involved in visual and CNS system development (Irion and Leptin, 1999; Schmucker et al., 2000). Furthermore, DDX41 is highly expressed in the zebrafish brain and this gene product performs similar antiviral functions to mammalian DDX41 when expressed in a HEK 293 expression system (Ma et al., 2018). Finally, circumstantial evidence of a role for DDX41 in the human CNS lays in the observation that HSV-1 has evolved an evasion mechanism targeting DDX41 (Table 2), suggesting that this molecule can act as a restriction factor for this neurotropic virus (Duan et al., 2019).
DNA protein kinase (DNA-PK) is a protein complex made up of a DNA protein kinase catalytic subunit (DNA-PKcs), ku70, and ku80, and is best known for its role in DNA double stranded break repair. However, several studies have shown that it can bind to transfected DNA and elicit the expression of IFN-β and other ISGs, independent of kinase activity (Ferguson et al., 2012; Harnor et al., 2017; Burleigh et al., 2020). In addition, the Ku70 subunit has also been identified as a possible DNA sensor in studies where plasmid transfected HEK 293 cells produce the type three IFN, IFN-λ1, that can limit HIV replication (Zhang X. et al., 2011). In this work, Ku70 and Ku80 were both found to bind transfected DNA, but only the loss of Ku70 decreased IFN-λ1 expression in these cells (Zhang X. et al., 2011). This finding was confirmed in splenocytes derived from ku70 deficient mice (Zhang X. et al., 2011), and it was later determined that Ku70mediated IFN-λ1 expression requires the expression of STING (Sui et al., 2017). However, it should be noted that these results are in contrast to a more recent report in which signaling through DNA-PK was found to be independent of the presence of STING (Burleigh et al., 2020). While it is presently unclear whether Ku70 functions alone or in concert with Ku80 and the DNA-PKcs to elicit antiviral activity, Ku70/DNA PK has been demonstrated to induce cytokine responses following infection of hepatocyte carcinoma and monocytic cell lines with HBV and human T-cell leukemia virus type 1, respectively (Li et al., 2016; Wang et al., 2017).
Furthermore, vaccinia virus and adenovirus have both been shown to antagonize Ku70/DNA-PK signaling (Peters et al., 2013; Scutts et al., 2018; Burleigh et al., 2020). Together, these studies suggest that ku70/DNA-PK acts as a viral PRR in addition to its DNA repair functions.
Despite evidence for antiviral functions of ku70/DNA-PK in peripheral cell types such as monocytes and fibroblasts, little exists for such a role in the CNS (Li et al., 2016; Burleigh et al., 2020). Expression of DNA-PK in the CNS and its role in DNA repair has been established from the study of mutations in severe combined immunodeficiency (SCID) mice that result in a truncated kinase domain in DNA-PK (Chechlacz et al., 2001; Vemuri et al., 2001). This has been shown to cause increased neuronal cell death in vitro and in vivo, presumably as a consequence of accumulated dsDNA breaks (Chechlacz et al., 2001; Vemuri et al., 2001). Since the DNA repair functions of DNA-PK are found in CNS cell types, the DNA sensing abilities of this molecule seen in peripheral cells may also be retained in the brain and this possibility requires further investigation.
Finally, RNA polymerase III was simultaneously identified as a DNA sensor by two groups as they investigated the mechanisms responsible for RIG-I mediated DNA sensing (Ablasser et al., 2009; Chiu et al., 2009). They demonstrated that poly (dA:dT) was reverse transcribed to RNA that then served as a ligand to activate RIG-I and induce IFN-β production in monocytes, fibroblasts, and dendritic cells (Ablasser et al., 2009; Chiu et al., 2009). Our own work subsequently showed that RNA polymerase III is functionally expressed in murine glia and a human microglial cell line (Crill et al., 2015; Johnson et al., 2020), and that its inhibition reduces HSV-1-induced IRF3 activation and TNF-a production in murine microglia cells and astrocytes (Crill et al., 2015). In addition to HSV-1, evidence suggests that RNA polymerase III also has a role in recognizing varicella-zoster virus (VZV) (Carter-Timofte et al., 2018, 2019). A RNA polymerase III mutation was identified in twins suffering from recurrent VZV CNS vasculitis and PBMCs isolated from them showed reduced antiviral and/or inflammatory cytokine responses to poly(dA:dT) and VZV challenge (Carter-Timofte et al., 2018). Furthermore, additional RNA polymerase III mutations were identified in adult VZV encephalitis patients and PBMCs from these patients similarly showed reduced IFN-β and CXCL10 expression in response to poly(dA:dT). Interestingly, while PBMC cytokine response were unchanged following VZV challenge, patients with these RNA polymerase III mutations showed higher viral gene expression (Carter-Timofte et al., 2019). Together, these studies support a role for RNA polymerase III in combating viral CNS infections.
Concluding Remarks
It is now appreciated that resident CNS cells are important contributors to innate immunity and, due to their location, are likely the first responders to viral CNS infections. Resident CNS cells, especially astrocytes and microglia, are known to express an array of PRRs including TLRs, RLRs, NLRs, and now intracellular DNA sensors. In addition to their expression, multiple studies have demonstrated the functional nature of these sensors in various CNS cell types (as summarized in Figure 1 and Table 3). For example, we have shown that cGAS is required, at least in part, for microglial IFN responses to foreign DNA (Jeffries et al., 2020). However, our understanding of the role of DNA sensors in viral infections is limited and, in some cases, contradictory. It is currently unclear whether DNA sensors are beneficial or detrimental to the host during CNS infections, and it appears likely that outcomes following activation are pathogen, host cell-type, and even species, specific. This is exemplified by the finding that the HSV-1 product ICP6 blocks ZBP1-mediated responses in human cells but activates ZBP1 in murine cells (Guo et al., 2018). Because of this, future research on the role and therapeutic potential of DNA sensors must be cognizant of such variables.
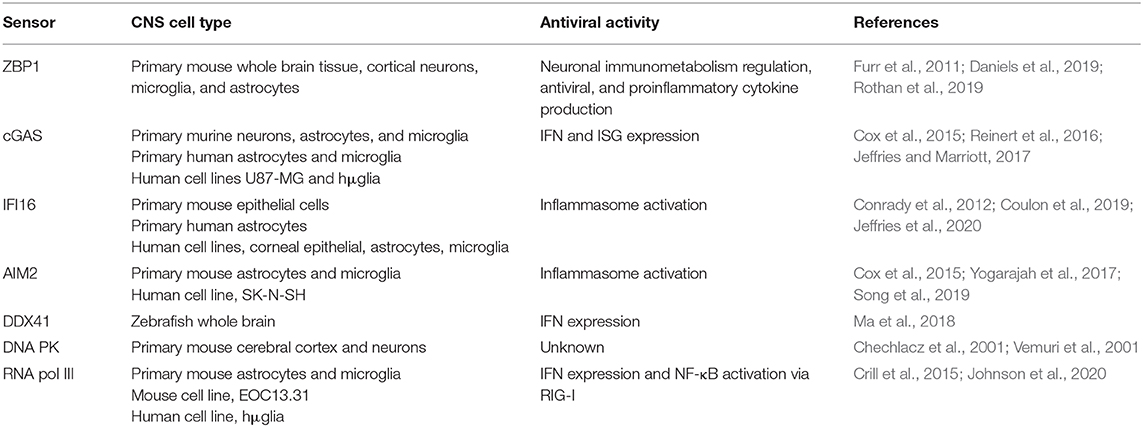
Table 3. Expression and antiviral activity of intracellular DNA sensors in CNS cell types.
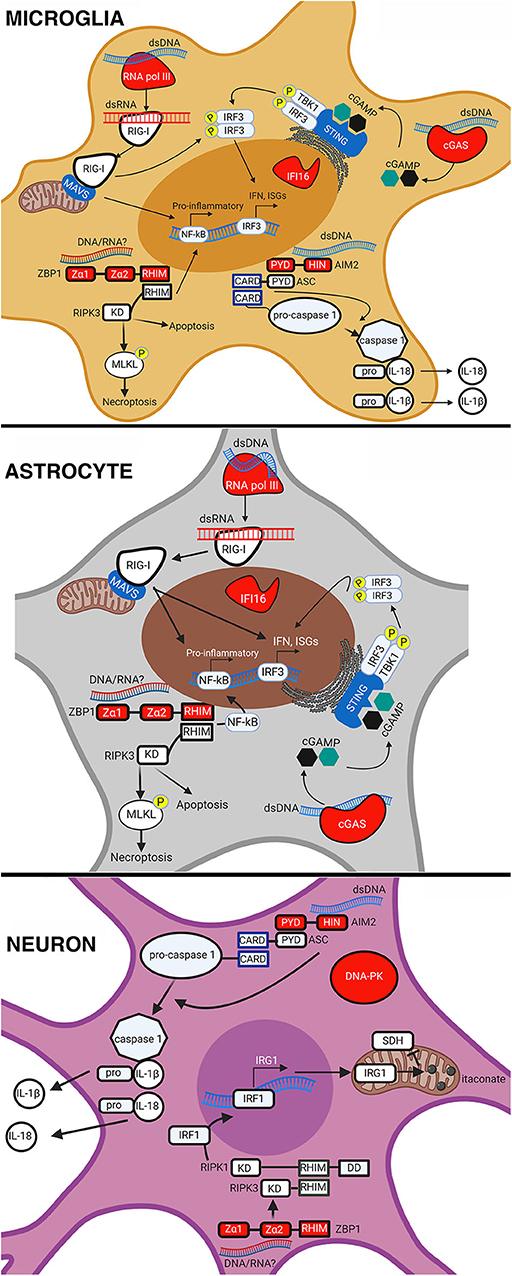
Figure 1. Intracellular DNA sensors in resident CNS cell types. Intracellular nucleic acid sensing by DNA sensors in microglia, astrocytes, and neurons. AIM2 sensing of dsDNA leads to the recruitment of apoptosis-associated speck-like protein containing a CARD (ASC) that then cleaves pro-caspase 1. Active caspase 1 then cleaves the precursor forms of IL18 and IL-1β, causing their maturation and release from the cell. ZBP1 sensing of either dsDNA or RNA causes it to associate with RIPK3, activate the transcription factor NF-κB, and phosphorylate mixed lineage kinase domain-like protein (MLKL) in microglia and astrocytes. This results in pro-inflammatory cytokine expression and execution of necroptosis. In neurons, ZBP1 sensing results in the activation of IRF1, expression of IRG1, production of itaconate, and a reduction in succinate dehydrogenase (SDH) activity. Sensing of dsDNA by cGAS leads to the production of cGAMP, which binds to and activates STING causing the phosphorylation and translocation of interferon regulatory factor 3 (IRF3) to the nucleus. This results in the expression of IFN and ISGs in microglia and astrocytes. RNA pol III senses dsDNA and converts it into dsRNA that can then be sensed by RIG-I in microglia and astrocytes. RIG-I sensing of dsRNA causes it to associate with mitochondrial antiviral-signaling protein (MAVS) leading to activation and translocation of IRF3 and NF-κB, resulting in the expression of IFN, ISGs, and pro-inflammatory cytokines. This figure was created with BioRender.com.
Lastly, it is important to note that DNA sensors may contribute to other CNS pathologies, such as neurodegenerative diseases, which may be initiated or exacerbated by viral infection. For instance, three prime repair exonuclease 1 (TREX1) deficiency can cause accumulation of mislocalized DNA and lead to Aicardi Goutieres syndrome. This condition is characterized by permanent and often severe neurological damage due to IFN overproduction, and the loss of cGAS has been shown to rescue TREX1 deficient mice from disease pathology (Gray et al., 2016). Similarly, neurodegenerative diseases are associated with chronic overproduction of proinflammatory mediators and neuroinflammation in diseases such as Alzheimer's disease could result from the chronic activation of DNA sensors by released DAMPS and/or viral infection. Regardless, it is clear that our understanding of these DNA sensors in the CNS remains rudimentary and further research is needed to define the cell type, species, and pathogen specificity of each. In doing so, it might be possible to target these molecules judiciously to limit damaging inflammation while allowing beneficial host responses to improve patient outcomes.
Author Contributions
AJ and IM co-wrote this literature review article. Both authors contributed to the article and approved the submitted version.
Funding
This work was supported by the NINDS research grant R03 NS097840 awarded to IM.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aarreberg, L. D., Esser-Nobis, K., Driscoll, C., Shuvarikov, A., Roby, J. A., and Gale, M. (2019). Interleukin-1β Induces mtDNA release to activate innate immune signaling via cGAS-STING. Mol. Cell 74, 801–815.e6. doi: 10.1016/j.molcel.2019.02.038
Abe, T., and Barber, G. N. (2014). Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory gene induction necessitates canonical nf- b activation through TBK1. J. Virol. 88, 5328–5341. doi: 10.1128/JVI.00037-14
Ablasser, A., Bauernfeind, F., Hartmann, G., Latz, E., Fitzgerald, K. A., and Hornung, V. (2009). RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III transcribed RNA intermediate. Nat. Immunol. 10, 1065–1072. doi: 10.1038/ni.1779
Ablasser, A., and Chen, Z. J. (2019). CGAS in action: Expanding roles in immunity and inflammation. Science 80:363. doi: 10.1126/science.aat8657
Adamczak, S. E., De Rivero Vaccari, J. P., Dale, G., Brand, F. J., Nonner, D., Bullock, M., et al. (2014). Pyroptotic neuronal cell death mediated by the AIM2 inflammasome. J. Cereb. Blood Flow Metab. 34, 621–629. doi: 10.1038/jcbfm.2013.236
Aguirre, S., Luthra, P., Sanchez-Aparicio, M. T., Maestre, A. M., Patel, J., Lamothe, F., et al. (2017). Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat. Microbiol. 2, 1–11. doi: 10.1038/nmicrobiol.2017.37
Aguirre, S., Maestre, A. M., Pagni, S., Patel, J. R., Savage, T., Gutman, D., et al. (2012). DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 8:e1002934. doi: 10.1371/journal.ppat.1002934
Almine, J. F., O'Hare, C. A. J., Dunphy, G., Haga, I. R., Naik, R. J., Atrih, A., et al. (2017). IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 8:14392. doi: 10.1038/ncomms14392
Aloisi, F. (2000). The role of microglia and astrocytes in CNS immune surveillance and immunopathology. Adv Exp Med Biol, 123–133. doi: 10.1007/978-1-4615-4685-6_10
Ansari, M. A., Dutta, S., Veettil, M. V., Dutta, D., Iqbal, J., Kumar, B., et al. (2015). Herpesvirus genome recognition induced acetylation of nuclear IFI16 Is essential for its cytoplasmic translocation, inflammasome and IFN-β responses. PLoS Pathog. 11:e10050. doi: 10.1371/journal.ppat.1005019
Ansari, M. A., Singh, V. V., Dutta, S., Veettil, M. V., Dutta, D., Chikoti, L., et al. (2013). Constitutive interferon-inducible protein 16-Inflammasome activation during epstein-barr virus latency i, ii, and iii in b and epithelial cells. J. Virol. 87, 8606–8623. doi: 10.1128/JVI.00805-13
Biolatti, M., Dell'Oste, V., Pautasso, S., Gugliesi, F., von Einem, J., Krapp, C., et al. (2017). Human cytomegalovirus tegument protein pp65 (pUL83) dampens type I interferon production by inactivating the DNA sensor cGAS without affecting STING. J. Virol. 92, e01774–17. doi: 10.1128/JVI.01774-17
Bode, C., Fox, M., Tewary, P., Steinhagen, A., Ellerkmann, R. K., Klinman, D., et al. (2016). Human plasmacytoid dentritic cells elicit a Type I Interferon response by sensing DNA via the cGAS-STING signaling pathway. Eur. J. Immunol. 46, 1615–1621. doi: 10.1002/eji.201546113
Botto, S., Abraham, J., Mizuno, N., Pryke, K., Gall, B., Landais, I., et al. (2019). Human cytomegalovirus immediate early 86-kda protein blocks transcription and induces degradation of the immature interleukin-1β protein during virion-mediated activation of the AIM2 inflammasome. MBio 10, e02510–18. doi: 10.1128/mBio.02510-18
Bowman, C. C., Rasley, A., Tranguch, S. L., and Marriott, I. (2003). Cultured astrocytes express toll-like receptors for bacterial products. Glia 43, 281–291. doi: 10.1002/glia.10256
Bradshaw, M. J., and Venkatesan, A. (2016). Herpes simplex virus-1 encephalitis in adults: pathophysiology, diagnosis, and management. Neurotherapeutics 13, 493–508. doi: 10.1007/s13311-016-0433-7
Bsibsi, M., Ravid, R., Gveric, D., and van Noort, J. M. (2002). Broad expression of toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 61, 1013–21. doi: 10.1093/jnen/61.11.1013
Bsibsi, M., Persoon-Deen, C., Verwer, R. W. H., Meeuwsen, S., Ravid, R., and Van Noort, J. M. (2006). Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia 53, 688–695. doi: 10.1002/glia.20328
Bürckstümmer, T., Baumann, C., Blüml, S., Dixit, E., Dürnberger, G., Jahn, H., et al. (2009). An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol. 10, 266–272. doi: 10.1038/ni.1702
Burleigh, K., Maltbaek, J. H., Cambier, S., Green, R., Gale, M., James, R. C., et al. (2020). Human DNA-PK activates a STING-independent DNA sensing pathway. Sci. Immunol. 5:aba4219. doi: 10.1126/sciimmunol.aba4219
Cai, X., Chiu, Y. H., and Chen, Z. J. (2014). The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 54, 289–296. doi: 10.1016/j.molcel.2014.03.040
Carter-Timofte, M. E., Hansen, A. F., Christiansen, M., Paludan, S. R., and Mogensen, T. H. (2019). Mutations in RNA Polymerase III genes and defective DNA sensing in adults with varicella-zoster virus CNS infection. Genes Immun. 20, 214–223. doi: 10.1038/s41435-018-0027-y
Carter-Timofte, M. E., Hansen, A. F., Mardahl, M., Fribourg, S., Rapaport, F., Zhang, S. Y., et al. (2018). Varicella-zoster virus CNS vasculitis and RNA polymerase III gene mutation in identical twins. Neurol. Neuroimmunol. NeuroInflammation 5:e500. doi: 10.1101/244848
Carty, M., Reinert, L., Paludan, S. R., and Bowie, A. G. (2014). Innate antiviral signaling in the central nervous system. Trends Immunol. 35, 79–87. doi: 10.1016/j.it.2013.10.012
Cerón, S., North, B. J., Taylor, S. A., and Leib, D. A. (2019). The STING agonist 5,6-dimethylxanthenone-4-acetic acid (DMXAA) stimulates an antiviral state and protects mice against herpes simplex virus-induced neurological disease. Virology 529, 23–28. doi: 10.1016/j.virol.2019.01.006
Chan, Y. K., and Gack, M. U. (2016). Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Microbiol. 14, 360–373. doi: 10.1038/nrmicro.2016.45
Chaudhuri, A., and Kennedy, P. G. E. (2002). Diagnosis and treatment of viral encephalitis. Postgrad. Med. J. 78, 575–583. doi: 10.1136/pmj.78.924.575
Chauhan, V. S., Sterka, D. G., Furr, S. R., Young, A. B., and Marriott, I. (2009). NOD2 plays an important role in the inflammatory responses of microglia and astrocytes to bacterial CNS pathogens. Glia 57, 414–423. doi: 10.1002/glia.20770
Chechlacz, M., Vemuri, M. C., and Naegele, J. R. (2001). Role of DNA-dependent protein kinase in neuronal survival. J. Neurochem. 78, 141–154. doi: 10.1046/j.1471-4159.2001.00380.x
Chen, H., He, G., Chen, Y., Zhang, X., and Wu, S. (2018). Differential activation of NLRP3, AIM2, and IFI16 inflammasomes in humans with acute and chronic hepatitis B. Viral Immunol. 31, 639–645. doi: 10.1089/vim.2018.0058
Chen, I. Y., and Ichinohe, T. (2015). Response of host inflammasomes to viral infection. Trends Microbiol. 23, 55–63. doi: 10.1016/j.tim.2014.09.007
Chen, Q., Sun, L., and Chen, Z. J. (2016). Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149. doi: 10.1038/ni.3558
Cheng, W. Y., He, X. B., Jia, H. J., Chen, G. H., Jin, Q. W., Long, Z. L., et al. (2018). The cGassting signaling pathway is required for the innate immune response against ectromelia virus. Front. Immunol. 9:1297. doi: 10.3389/fimmu.2018.01297
Chiu, Y. H., MacMillan, J. B., and Chen, Z. J. (2009). RNA Polymerase III detects cytosolic DNA and induces type i interferons through the rig-i pathway. Cell 138, 576–591. doi: 10.1016/j.cell.2009.06.015
Choi, H. J., Park, A., Kang, S., Lee, E., Lee, T. A., Ra, E. A., et al. (2018). Human cytomegalovirus-encoded US9 targets MAVS and STING signaling to evade type i interferon immune responses. Nat. Commun. 9:125. doi: 10.1038/s41467-017-02624-8
Christensen, M. H., Jensen, S. B., Miettinen, J. J., Luecke, S., Prabakaran, T., Reinert, L. S., et al. (2016). HSV−1 ICP 27 targets the TBK 1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. 35, 1385–1399. doi: 10.15252/embj.201593458
Conrady, C. D., Zheng, M., Fitzgerald, K. A., Liu, C., and Carr, D. J. J. (2012). Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal Immunol. 5, 173–183. doi: 10.1038/mi.2011.63
Corrales, L., Woo, S.-R., Williams, J. B., McWhirter, S. M., Dubensky, T. W., and Gajewski, T. F. (2016). Antagonism of the STING pathway via activation of the AIM2 inflammasome by intracellular DNA. J. Immunol. 196, 3191–3198. doi: 10.4049/jimmunol.1502538
Coulon, P. G., Dhanushkodi, N., Prakash, S., Srivastava, R., Roy, S., Alomari, N. I., et al. (2019). NLRP3, NLRP12, and IFI16 inflammasomes induction and caspase-1 activation triggered by virulent HSV-1 strains are associated with severe corneal inflammatory herpetic disease. Front. Immunol. 10:1631. doi: 10.3389/fimmu.2019.01631
Cox, D. J., Field, R. H., Williams, D. G., Baran, M., Bowie, A. G., Cunningham, C., et al. (2015). DNA sensors are expressed in astrocytes and microglia in vitro and are upregulated during gliosis in neurodegenerative disease. Glia 63, 812–825. doi: 10.1002/glia.22786
Cridland, J. A., Curley, E. Z., Wykes, M. N., Schroder, K., Sweet, M. J., Roberts, T. L., et al. (2012). The mammalian PYHIN gene family: Phylogeny, evolution and expression. BMC Evol. Biol. 12:140. doi: 10.1186/1471-2148-12-140
Crill, E. K., Furr-Rogers, S. R., and Marriott, I. (2015). RIG-I is required for VSV-induced cytokine production by murine glia and acts in combination with DAI to initiate responses to HSV-1. Glia 63, 2168–2180. doi: 10.1002/glia.22883
Cuchet-Lourenco, D., Anderson, G., Sloan, E., Orr, A., and Everett, R. D. (2013). The Viral Ubiquitin ligase icp0 is neither sufficient nor necessary for degradation of the cellular dna sensor ifi16 during herpes simplex virus 1 infection. J. Virol. 87, 13422–13432. doi: 10.1128/JVI.02474-13
Dai, P., Wang, W., Cao, H., Avogadri, F., Dai, L., Drexler, I., et al. (2014). Modified Vaccinia Virus ankara triggers type i ifn production in murine conventional dendritic cells via a cGAS/STING-mediated cytosolic DNA-sensing pathway. PLoS Pathog. 10:e1003989. doi: 10.1371/journal.ppat.1003989
Daniels, B. P., Kofman, S. B., Smith, J. R., Norris, G. T., Snyder, A. G., Kolb, J. P., et al. (2019). The nucleotide sensor zbp1 and kinase RIPK3 induce the enzyme irg1 to promote an antiviral metabolic state in neurons. Immunity 50, 64–76.e4. doi: 10.1016/j.immuni.2018.11.017
Daniels, B. P., Snyder, A. G., Olsen, T. M., Orozco, S., Oguin, T. H., Tait, S. W. G., et al. (2017). RIPK3 restricts viral pathogenesis via cell Death-Independent Neuroinflammation. Cell 169, 301–313.e11. doi: 10.1016/j.cell.2017.03.011
de Sousa, J. R., da Silva Azevedo, R. D. S., Martins Filho, A. J., de Araujo, M. T. F., Cruz, E. D. R. M., Vasconcelos, B. C. B., et al. (2018). In situ inflammasome activation results in severe damage to the central nervous system in fatal Zika virus microcephaly cases. Cytokine 111, 255–264. doi: 10.1016/j.cyto.2018.08.008
Dell'Oste, V., Gatti, D., Giorgio, A. G., Gariglio, M., Landolfo, S., and De Andrea, M. (2015). The interferon-inducible DNA-sensor protein IFI16: A key player in the antiviral response. New Microbiol. 38, 5–20.
Dell'Oste, V., Gatti, D., Gugliesi, F., De Andrea, M., Bawadekar, M., Lo Cigno, I., et al. (2014). Innate nuclear sensor ifi16 translocates into the cytoplasm during the early stage of in vitro human cytomegalovirus infection and is entrapped in the egressing virions during the late stage. J. Virol. 88, 6970–6982. doi: 10.1128/JVI.00384-14
Deschamps, T., and Kalamvoki, M. (2017). Evasion of the STING DNA-sensing pathway by VP11/12 of herpes simplex virus 1. J. Virol. 91:e00535–17. doi: 10.1128/JVI.00535-17
Dhanwani, R., Takahashi, M., and Sharma, S. (2018). Cytosolic sensing of immuno-stimulatory DNA, the enemy within. Curr. Opin. Immunol. 50, 82–87. doi: 10.1016/j.coi.2017.11.004
Dhuriya, Y. K., and Sharma, D. (2018). Necroptosis: a regulated inflammatory mode of cell death. J. Neuroinflammation 15:199. doi: 10.1186/s12974-018-1235-0
Diner, B. A., Lum, K. K., Javitt, A., and Cristea, I. M. (2015). Interactions of the antiviral factor interferon gamma-inducible protein 16 (IFI16) mediate immune signaling and herpes simplex virus-1 immunosuppression. Mol. Cell. Proteomics 14, 2341–2356. doi: 10.1074/mcp.M114.047068
Diner, B. A., Lum, K. K., Toettcher, J. E., and Cristea, I. M. (2016). Viral DNA sensors IFI16 and cyclic GMP-AMP synthase possess distinct functions in regulating viral gene expression, immune defenses, and apoptotic responses during herpesvirus infection. MBio 7, e01553–16. doi: 10.1128/mBio.01553-16
Ding, Q., Gaska, J. M., Douam, F., Wei, L., Kim, D., Balev, M., et al. (2018). Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proc. Natl. Acad. Sci. U.S.A. 115, E6310–E6318. doi: 10.1073/pnas.1803406115
Duan, Y., Zeng, J., Fan, S., Liao, Y., Feng, M., Wang, L., et al. (2019). Herpes simplex virus type 1-encoded miR-H2-3p manipulates cytosolic DNA-stimulated antiviral innate immune response by targeting DDX41. Viruses 11:756. doi: 10.3390/v11080756
Dutta, D., Dutta, S., Veettil, M. V., Roy, A., Ansari, M. A., Iqbal, J., et al. (2015). BRCA1 Regulates IFI16 mediated nuclear innate sensing of herpes viral dna and subsequent induction of the innate inflammasome and interferon-β responses. PLOS Pathog. 11:e1005030. doi: 10.1371/journal.ppat.1005030
Ekchariyawat, P., Hamel, R., Bernard, E., Wichit, S., Surasombatpattana, P., Talignani, L., et al. (2015). Inflammasome signaling pathways exert antiviral effect against chikungunya virus in human dermal fibroblasts. Infect. Genet. Evol. 32, 401–408. doi: 10.1016/j.meegid.2015.03.025
Fang, R., Wang, C., Jiang, Q., Lv, M., Gao, P., Yu, X., et al. (2017). NEMO–IKKβ are essential for IRF3 and NF-κB activation in the cGAS–STING pathway. J. Immunol. 199, 3222–3233. doi: 10.4049/jimmunol.1700699
Ferguson, B. J., Mansur, D. S., Peters, N. E., Ren, H., and Smith, G. L. (2012). DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 2012:e00047. doi: 10.7554/eLife.00047.012
Fernandes-Alnemri, T., Yu, J. W., Datta, P., Wu, J., and Alnemri, E. S. (2009). AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513. doi: 10.1038/nature07710
Fu, Y. Z., Su, S., Gao, Y. Q., Wang, P. P., Huang, Z. F., Hu, M. M., et al. (2017). Human cytomegalovirus tegument protein UL82 inhibits STING-mediated signaling to evade antiviral immunity. Cell Host Microbe 21, 231–243. doi: 10.1016/j.chom.2017.01.001
Furr, S. R., Chauhan, V. S., Moerdyk-Schauwecker, M. J., and Marriott, I. (2011). A role for DNA-dependent activator of interferon regulatory factor in the recognition of herpes simplex virus type 1 by glial cells. J. Neuroinflammation 8:99. doi: 10.1186/1742-2094-8-99
Furr, S. R., Chauhan, V. S., Sterka, D., Grdzelishvili, V., and Marriott, I. (2008). Characterization of retinoic acid–inducible gene-I expression in primary murine glia following exposure to vesicular stomatitis virus. J. Neurovirol. 14, 503–513. doi: 10.1080/13550280802337217
Furr, S. R., and Marriott, I. (2012). Viral CNS infection: role of glial pattern recognition receptors in neuroinflammation. Front. Microbiol. 3:201. doi: 10.3389/fmicb.2012.00201
Gao, D., Wu, J., Wu, Y. T., Du, F., Aroh, C., Yan, N., et al. (2013). Cycilc GMP-AMP Synthase is and innate immune sensor of HIV and other retroviruses. Science 341, 903–906. doi: 10.1126/science.1240933
Gao, P., Ascano, M., Wu, Y., Barchet, W., Gaffney, B. L., Zillinger, T., et al. (2013). Cyclic [G(2′,5′)pA(3′,5′)p]is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107. doi: 10.1016/j.cell.2013.04.046
Gariano, G. R., Dell'Oste, V., Bronzini, M., Gatti, D., Luganini, A., de Andrea, M., et al. (2012). The intracellular DNA sensor IFI16 gene acts as restriction factor for human Cytomegalovirus replication. PLoS Pathog. 8:e1002498. doi: 10.1371/journal.ppat.1002498
George, B. P., Schneider, E. B., and Venkatesan, A. (2014). Encephalitis hospitalization rates and inpatient mortality in the United States, 2000-2010. PLoS ONE 9:e0104169. doi: 10.1371/journal.pone.0104169
Gray, E. E., Winship, D., Snyder, J. M., Child, S. J., Geballe, A. P., and Stetson, D. B. (2016). The AIM2-like receptors are dispensable for the interferon response to intracellular DNA. Immunity 45, 255–266. doi: 10.1016/j.immuni.2016.06.015
Guo, H., Gilley, R. P., Fisher, A., Lane, R., Landsteiner, V. J., Ragan, K. B., et al. (2018). Species-independent contribution of ZBP1/DAI/DLM-1-triggered necroptosis in host defense against HSV1. Cell Death Dis. 9:816. doi: 10.1038/s41419-018-0868-3
Guo, H., Omoto, S., Harris, P. A., Finger, J. N., Bertin, J., Gough, P. J., et al. (2015). Herpes simplex virus suppresses necroptosis in human cells. Cell Host Microbe 17, 243–251. doi: 10.1016/j.chom.2015.01.003
Hanamsagar, R., Aldrich, A., and Kielian, T. (2014). Critical role for the AIM2 inflammasome during acute CNS bacterial infection. J. Neurochem. 129, 704–711. doi: 10.1111/jnc.12669
Harnor, S. J., Brennan, A., and Cano, C. (2017). Targeting DNA-dependent protein kinase for cancer therapy. ChemMedChem 12, 895–900. doi: 10.1002/cmdc.201700143
Herzner, A. M., Hagmann, C. A., Goldeck, M., Wolter, S., Kübler, K., Wittmann, S., et al. (2015). Sequence-specific activation of the DNA sensor cGAS by Y-form DNA structures as found in primary HIV-1 cDNA. Nat. Immunol. 16, 1025–1033. doi: 10.1038/ni.3267
Hornung, V., Ablasser, A., Charrel-Dennis, M., Bauernfeind, F., Horvath, G., Caffrey, D. R., et al. (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518. doi: 10.1038/nature07725
Huang, J., You, H., Su, C., Li, Y., Chen, S., and Zheng, C. (2018). Herpes simplex virus 1 tegument protein VP22 abrogates cGAS/STING-mediated antiviral innate immunity. J. Virol. 92, e00841–18. doi: 10.1128/JVI.00841-18
Huang, Y., Liu, L., Ma, D., Liao, Y., Lu, Y., Huang, H., et al. (2017). Human cytomegalovirus triggers the assembly of AIM2 inflammasome in THP-1-derived macrophages. J. Med. Virol. 89, 2188–2195. doi: 10.1002/jmv.24846
Huang, Z., Wu, S. Q., Liang, Y., Zhou, X., Chen, W., Li, L., et al. (2015). RIP1/RIP3 binding to HSV-1 ICP6 initiates necroptosis to restrict virus propagation in mice. Cell Host Microbe. 17, 229–242. doi: 10.1016/j.chom.2015.01.002
Huang, Z. F., Zou, H. M., Liao, B. W., Zhang, H. Y., Yang, Y., Fu, Y. Z., et al. (2018). Human cytomegalovirus protein UL31 inhibits DNA sensing of cGAS to mediate immune evasion. Cell Host Microbe 24, 69–80.e4. doi: 10.1016/j.chom.2018.05.007
Ingram, J. P., Thapa, R. J., Fisher, A., Tummers, B., Zhang, T., Yin, C., et al. (2019). ZBP1/DAI drives RIPK3-mediated cell death induced by IFNs in the absence of RIPK1. J. Immunol. 203, 1348–1355. doi: 10.4049/jimmunol.1900216
Iqbal, J., Ansari, M. A., Kumar, B., Dutta, D., Roy, A., Chikoti, L., et al. (2016). Histone H2BIFI16 recognition of nuclear herpesviral genome induces cytoplasmic interferon-β responses. PLOS Pathog. 12:e1005967. doi: 10.1371/journal.ppat.1005967
Irion, U., and Leptin, M. (1999). Developmental and cell biological functions of the drosophila DEAD-box protein abstrakt. Curr. Biol. 9, 1373–1381. doi: 10.1016/S0960-9822(00)80082-2
Ishikawa, H., Ma, Z., and Barber, G. N. (2009). STING regulates intracellular DNA-mediated, type i interferon-dependent innate immunity. Nature 461, 788–792. doi: 10.1038/nature08476
Jakobsen, M. R., Bak, R. O., Andersen, A., Berg, R. K., Jensen, S. B., Jin, T., et al. (2013). IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. U.S.A. 110:E4571–80. doi: 10.1073/pnas.1311669110
Jeffries, A. M., and Marriott, I. (2017). Human microglia and astrocytes express cGAS-STING viral sensing components. Neurosci. Lett. 658, 53–56. doi: 10.1016/j.neulet.2017.08.039
Jeffries, A. M., Nitika, T. A. W., and Marriott, I. (2020). The intracellular DNA sensors cGAS and IFI16 do not mediate effective antiviral immune responses to HSV-1 in human microglial cells. J. Neurovirol. 26, 544–555. doi: 10.1007/s13365-020-00852-1
Jiao, H., Wachsmuth, L., Kumari, S., Schwarzer, R., Lin, J., Eren, R. O., et al. (2020). Z-nucleicacid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature 580, 391–395. doi: 10.1038/s41586-020-2129-8
Johnson, K. E., Chikoti, L., and Chandran, B. (2013). Herpes simplex virus 1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 Inflammasomes. J. Virol. 87, 5005–5018. doi: 10.1128/JVI.00082-13
Johnson, M. B., Halman, J. R., Burmeister, A. R., Currin, S., Khisamutdinov, E. F., Afonin, K. A., et al. (2020). Retinoic acid inducible gene-I mediated detection of bacterial nucleic acids in human microglial cells. J. Neuroinflammation 17:139. doi: 10.1186/s12974-020-01817-1
Jønsson, K. L., Laustsen, A., Krapp, C., Skipper, K. A., Thavachelvam, K., Hotter, D., et al. (2017). IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat. Commun. 8:14391. doi: 10.1038/ncomms14391
Kaiser, W. J., Upton, J. W., and Mocarski, E. S. (2008). Receptor-interacting protein homotypic interaction motif-dependent control of NF-κB activation via the DNA-dependent activator of IFN regulatory factors. J. Immunol. 181, 6427–6434. doi: 10.4049/jimmunol.181.9.6427
Kayagaki, N., Stowe, I. B., Lee, B. L., O'Rourke, K., Anderson, K., Warming, S., et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. doi: 10.1038/nature15541
Kim, H., Seo, J. S., Lee, S. Y., Ha, K. T., Choi, B. T., Shin, Y., et al. (2020). AIM2 inflammasome contributes to brain injury and chronic post-stroke cognitive impairment in mice. Brain. Behav. Immun. 87, 765–776. doi: 10.1016/j.bbi.2020.03.011
Kim, J. E., Kim, Y. E., Stinski, M. F., Ahn, J. H., and Song, Y. J. (2017). Human cytomegalovirus IE2 86 kDa protein induces STING degradation and inhibits cGAMP-mediated IFN-β induction. Front. Microbiol. 8:1854. doi: 10.3389/fmicb.2017.01854
Kuriakose, T., Man, S. M., Subbarao Malireddi, R. K., Karki, R., Kesavardhana, S., Place, D. E., et al. (2016). ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 1:aag2045. doi: 10.1126/sciimmunol.aag2045
Lahaye, X., Satoh, T., Gentili, M., Cerboni, S., Conrad, C., Hurbain, I., et al. (2013). The Capsids of HIV-1 and HIV-2 determine immune detection of the viral cdna by the innate sensor cgas in dendritic cells. Immunity 39, 1132–1142. doi: 10.1016/j.immuni.2013.11.002
Lee, K. G., Kim, S. S. Y., Kui, L., Voon, D. C. C., Mauduit, M., Bist, P., et al. (2015). Bruton's tyrosine kinase phosphorylates DDX41 and activates its binding of dsDNA and STING to initiate type 1 interferon response. Cell Rep. 10, 1055–1065. doi: 10.1016/j.celrep.2015.01.039
Li, X. D., Wu, J., Gao, D., Wang, H., Sun, L., and Chen, Z. J. (2013). Pivotal roles of cGAScGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394. doi: 10.1126/science.1244040
Li, Y., Wu, Y., Zheng, X., Cong, J., Liu, Y., Li, J., et al. (2016). Cytoplasm-translocated Ku70/80 complex sensing of HBV DNA induces hepatitis-associated chemokine secretion. Front. Immunol. 7:569. doi: 10.3389/fimmu.2016.00569
Lin, J., Kumari, S., Kim, C., Van, T. M., Wachsmuth, L., Polykratis, A., et al. (2016). RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 540, 124–128. doi: 10.1038/nature20558
Lippmann, J., Rothenburg, S., Deigendesch, N., Eitel, J., Meixenberger, K., van Laak, V., et al. (2008). IFNβ responses induced by intracellular bacteria or cytosolic DNA in different human cells do not require ZBP1 (DLM-1/DAI). Cell. Microbiol. 10, 2579–2588. doi: 10.1111/j.1462-5822.2008.01232.x
Liu, X., Chauhan, V. S., Young, A. B., and Marriott, I. (2010). NOD2 mediates inflammatory responses of primary murine glia to Streptococcus pneumoniae. Glia 58, 839–847. doi: 10.1002/glia.20968
Lo Cigno, I., Calati, F., Borgogna, C., Zevini, A., Albertini, S., Martuscelli, L., et al. (2020). Human papillomavirus E7 oncoprotein subverts host innate immunity via SUV39H1-mediated epigenetic silencing of immune sensor genes. J. Virol. 94:19. doi: 10.1128/JVI.01812-19
Lo Cigno, I., De Andrea, M., Borgogna, C., Albertini, S., Landini, M. M., Peretti, A., et al. (2015). The Nuclear DNA Sensor IFI16 acts as a restriction factor for human papillomavirus replication through epigenetic modifications of the viral promoters. J. Virol. 89, 7506–7520. doi: 10.1128/JVI.00013-15
Luecke, S., Holleufer, A., Christensen, M. H., Jønsson, K. L., Boni, G. A., Sørensen, L. K., et al. (2017). cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 18, 1707–1715. doi: 10.15252/embr.201744017
Lugrin, J., and Martinon, F. (2018). The AIM2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol. Rev. 281, 99–114. doi: 10.1111/imr.12618
Lum, K. K., Howard, T. R., Pan, C., and Cristea, I. M. (2019). Charge-mediated pyrin oligomerization nucleates antiviral IFI16 sensing of herpesvirus DNA. MBio 10, e01428–19. doi: 10.1128/mBio.01428-19
Lupfer, C., Malik, A., and Kanneganti, T. D. (2015). Inflammasome control of viral infection. Curr. Opin. Virol. 12, 38–46. doi: 10.1016/j.coviro2015.02.007
Ma, F., Li, B., Liu, S., Iyer, S. S., Yu, Y., Wu, A., et al. (2015). Positive feedback regulation of type I IFN production by the IFN-inducible DNA sensor cGAS. J. Immunol. 194, 1545–1554. doi: 10.4049/jimmunol.1402066
Ma, J. X., Li, J. Y., Fan, D. D., Feng, W., Lin, A. F., Xiang, L. X., et al. (2018). Identification of DEAD-Box RNA helicase DDX41 as a trafficking protein that involves in multiple innate immune signaling pathways in a zebrafish model. Front. Immunol. 9:1327. doi: 10.3389/fimmu.2018.01327
Ma, Z., Jacobs, S. R., West, J. A., Stopford, C., Zhang, Z., Davis, Z., et al. (2015). Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. U.S.A. 112, E4306–E4315. doi: 10.1073/pnas.1503831112
Maelfait, J., Liverpool, L., Bridgeman, A., Ragan, K. B., Upton, J. W., and Rehwinkel, J. (2017). Sensing of viral and endogenous RNA by ZBP 1/ DAI induces necroptosis. EMBO J. 36, 2529–2543. doi: 10.15252/embj.201796476
Man, S. M., Karki, R., and Kanneganti, T. D. (2016). AIM2 inflammasome in infection, cancer, and autoimmunity: role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 46, 269–280. doi: 10.1002/eji.201545839
Maruzuru, Y., Ichinohe, T., Sato, R., Miyake, K., Okano, T., Suzuki, T., et al. (2018). Herpes Simplex Virus 1 VP22 Inhibits AIM2-dependent inflammasome activation to enable efficient viral replication. Cell Host Microbe 23, 254–265.e7. doi: 10.1016/j.chom.2017.12.014
Menendez, C. M., and Carr, D. J. J. (2017). Defining nervous system susceptibility during acute and latent herpes simplex virus-1 infection. J. Neuroimmunol. 308, 43–49. doi: 10.1016/j.jneuroim.2017.02.020
Miao, E. A., Rajan, J. V., and Aderem, A. (2011). Caspase-1-induced pyroptotic cell death. Immunol. Rev. 243, 206–214. doi: 10.1111/j.1600-065X.2011.01044.x
Moriyama, M., Koshiba, T., and Ichinohe, T. (2019). Influenza A virus M2 protein triggers mitochondrial DNA-mediated antiviral immune responses. Nat. Commun. 10:4624. doi: 10.1038/s41467-019-12632-5
Nair, S., and Diamond, M. S. (2015). Innate immune interactions within the central nervous system modulate pathogenesis of viral infections. Curr. Opin. Immunol. 36, 47–53. doi: 10.1016/j.coi.2015.06.011
Nakaya, Y., Lilue, J., Stavrou, S., Moran, E. A., and Ross, S. R. (2017). AIM2-like receptors positively and negatively regulate the interferon response induced by cytosolic DNA. MBio 8:17. doi: 10.1128/mBio.00944-17
Orzalli, M. H., Broekema, N. M., Diner, B. A., Hancks, D. C., Elde, N. C., Cristea, I. M., et al. (2015). CGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc. Natl. Acad. Sci. U.S.A. 112, E1773–E1781. doi: 10.1073/pnas.1424637112
Orzalli, M. H., Broekema, N. M., and Knipe, D. M. (2016). Relative contributions of herpes simplex virus 1 ICP0 and vhs to loss of cellular IFI16 vary in different human cell types. J. Virol. 90, 8351–8359. doi: 10.1128/JVI.00939-16
Orzalli, M. H., DeLuca, N. A., and Knipe, D. M. (2012). Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. U.S.A. 109, E3008–E3017. doi: 10.1073/pnas.1211302109
Paijo, J., Döring, M., Spanier, J., Grabski, E., Nooruzzaman, M., Schmidt, T., et al. (2016). cGAS senses human cytomegalovirus and induces type i interferon responses in human monocyte-derived cells. PLoS Pathog. 12:e1005546. doi: 10.1371/journal.ppat.1005546
Pan, S., Liu, X., Ma, Y., Cao, Y., and He, B. (2018). Herpes simplex virus 1 γ 1 34.5 protein inhibits STING activation that restricts viral replication. J. Virol. 92, e01015–18. doi: 10.1128/JVI.01015-18
Parker, Z. M., Murphy, A. A., and Leib, D. A. (2015). Role of the DNA sensor STING in protection from lethal infection following corneal and intracerebral challenge with herpes simplex virus 1. J. Virol. 89, 11080–11091. doi: 10.1128/JVI.00954-15
Parvatiyar, K., Zhang, Z., Teles, R. M., Ouyang, S., Jiang, Y., Iyer, S. S., et al. (2012). The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type i interferon immune response. Nat. Immunol. 13, 1155–1161. doi: 10.1038/ni.2460
Peters, N. E., Ferguson, B. J., Mazzon, M., Fahy, A. S., Krysztofinska, E., Arribas-Bosacoma, R., et al. (2013). A Mechanism for the inhibition of DNA-PK-mediated DNA sensing by a Virus. PLoS Pathog. 9:1003649. doi: 10.1371/journal.ppat.1003649
Pham, T. H., Kwon, K. M., Kim, Y. E., Kim, K. K., and Ahn, J. H. (2013). DNA sensing-independent inhibition of herpes simplex virus 1 replication by DAI/ZBP1. J. Virol. 87, 3076–3086. doi: 10.1128/JVI.02860-12
Phelan, T., Little, M. A., and Brady, G. (2020). Targeting of the cGAS-STING system by DNA viruses. Biochem. Pharmacol. 174:113831. doi: 10.1016/j.bcp.2020.113831
Pisano, G., Roy, A., Ahmed Ansari, M., Kumar, B., Chikoti, L., and Chandran, B. (2017). Interferon-γ-inducible protein 16 (IFI16) is required for the maintenance of Epstein-Barr virus latency. Virol. J. 14:221. doi: 10.1186/s12985-017-0891-5
Rathinam, V. A. K., Jiang, Z., Waggoner, S. N., Sharma, S., Cole, L. E., Waggoner, L., et al. (2010). The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11, 395–402. doi: 10.1038/ni.1864
Rebsamen, M., Heinz, L. X., Meylan, E., Michallet, M. C., Schroder, K., Hofmann, K., et al. (2009). DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-κB. EMBO Rep. 10, 916–922. doi: 10.1038/embor.2009.109
Reinert, L. S., Lopušná, K., Winther, H., Sun, C., Thomsen, M. K., Nandakumar, R., et al. (2016). Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat. Commun. 7:13348. doi: 10.1038/ncomms13348
Reinholz, M., Kawakami, Y., Salzer, S., Kreuter, A., Dombrowski, Y., Koglin, S., et al. (2013). HPV16 activates the AIM2 inflammasome in keratinocytes. Arch. Dermatol. Res. 305, 723–732. doi: 10.1007/s00403-013-1375-0
Rothan, H. A., Arora, K., Natekar, J. P., Strate, P. G., Brinton, M. A., and Kumar, M. (2019). ZDNA-binding protein 1 is critical for controlling virus replication and survival in west nile virus encephalitis. Front. Microbiol. 10:89. doi: 10.3389/fmicb.2019.02089
Roy, A., Dutta, D., Iqbal, J., Pisano, G., Gjyshi, O., Ansari, M. A., et al. (2016). Nuclear innate immune DNA sensor IFI16 Is degraded during lytic reactivation of kaposi's sarcomaassociated herpesvirus (KSHV): role of ifi16 in maintenance of kshv latency. J. Virol. 90, 8822–8841. doi: 10.1128/JVI.01003-16
Roy, A., Ghosh, A., Kumar, B., and Chandran, B. (2019). IfI16, a nuclear innate immune DNA sensor, mediates epigenetic silencing of herpesvirus genomes by its association with H3K9 methyltransferases SUV39H1 and GLP. Elife 8:e49500. doi: 10.7554/eLife.49500
Royer, D. J., and Carr, D. J. J. (2016). A STING-dependent innate-sensing pathway mediates resistance to corneal HSV-1 infection via upregulation of the antiviral effector tetherin. Mucosal Immunol. 9, 1065–1075. doi: 10.1038/mi.2015.124
Sagulenko, V., Thygesen, S. J., Sester, D. P., Idris, A., Cridland, J. A., Vajjhala, P. R., et al. (2013). AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 20, 1149–1160. doi: 10.1038/cdd.2013.37
Sawai, H. (2016). Induction of apoptosis in TNF-Treated L929 cells in the presence of necrostatin-1. Int. J. Mol. Sci. 17:1678. doi: 10.3390/ijms17101678
Schattgen, S. A., Gao, G., Kurt-Jones, E. A., and Fitzgerald, K. A. (2016). Cutting edge: dna in the lung microenvironment during influenza virus infection tempers inflammation by engaging the DNA Sensor AIM2. J. Immunol. 196, 29–33. doi: 10.4049/jimmunol.1501048
Schmucker, D., Vorbrüggen, G., Yeghiayan, P., Fan, H. Q., Jäckle, H., and Gaul, U. (2000). The Drosophila gene abstrakt, required for visual system development, encodes a putative RNA helicase of the DEAD box protein family. Mech. Dev. 91, 189–196. doi: 10.1016/S0925-4773(99)00298-1
Scutts, S. R., Ember, S. W., Ren, H., Ye, C., Lovejoy, C. A., Mazzon, M., et al. (2018). DNA-PK is targeted by multiple vaccinia virus proteins to inhibit DNA sensing. Cell Rep. 25, 1953–1965.e4. doi: 10.1016/j.celrep.2018.10.034
Semenova, N., Bosnjak, M., Markelc, B., Znidar, K., Cemazar, M., and Heller, L. (2019). Multiple cytosolic DNA sensors bind plasmid DNA after transfection. Nucleic Acids Res. 47, 10235–10246. doi: 10.1093/nar/gkz768
Serramía, M. J., Muñoz-Fernández, M. Á., and Álvarez, S. (2015). HIV-1 increases TLR responses in human primary astrocytes. Sci. Rep. 5:17887. doi: 10.1038/srep17887
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Shrivastava, G., León-Juárez, M., García-Cordero, J., Meza-Sánchez, D. E., and Cedillo-Barrón, L. (2016). Inflammasomes and its importance in viral infections. Immunol. Res. 64, 1101–1117. doi: 10.1007/s12026-016-8873-z
Song, X., Ma, F., and Herrup, K. (2019). Accumulation of cytoplasmic DNA Due to ATM deficiency activates the microglial viral response system with neurotoxic consequences. J. Neurosci. 39, 6378–6394. doi: 10.1523/JNEUROSCI.0774-19.2019
Sridharan, H., Ragan, K. B., Guo, H., Gilley, R. P., Landsteiner, V. J., Kaiser, W. J., et al. (2017). Murine cytomegalovirus IE 3-dependent transcription is required for DAI/ZBP 1mediated necroptosis. EMBO Rep. 18, 1429–1441. doi: 10.15252/embr.201743947
Stabell, A. C., Meyerson, N. R., Gullberg, R. C., Gilchrist, A. R., Webb, K. J., Old, W. M., et al. (2018). Dengue viruses cleave STING in humans but not in nonhuman primates, their presumed natural reservoir. Elife 7:e31919. doi: 10.7554/eLife.31919
Stavrou, S., Aguilera, A. N., Blouch, K., and Ross, S. R. (2018). DDX41 recognizes RNA/DNA retroviral reverse transcripts and is critical for in vivo control of murine leukemia virus infection. MBio 9, e00923–18. doi: 10.1128/mBio.00923-18
Stavrou, S., Blouch, K., Kotla, S., Bass, A., and Ross, S. R. (2015). Nucleic acid recognition orchestrates the anti-viral response to retroviruses. Cell Host Microbe 17, 478–488. doi: 10.1016/j.chom.2015.02.021
Sterka, D., Rati, D. M., and Marriott, I. (2006). Functional expression of NOD2, a novel pattern recognition receptor for bacterial motifs, in primary murine astrocytes. Glia 53, 322–330. doi: 10.1002/glia.20286
Su, C., and Zheng, C. (2017). Herpes simplex virus 1 abrogates the cGAS/STING-mediated cytosolic DNA-sensing pathway via its virion host shutoff protein, UL41. J. Virol. 91, e02414–16. doi: 10.1128/JVI.02414-16
Su, J., Rui, Y., Lou, M., Yin, L., Xiong, H., Zhou, Z., et al. (2019). HIV-2/SIV Vpx targets a novel functional domain of STING to selectively inhibit cGAS–STING-mediated NF-κB signalling. Nat. Microbiol. 4, 2552–2564. doi: 10.1038/s41564-019-0585-4
Sui, H., Zhou, M., Imamichi, H., Jiao, X., Sherman, B. T., Clifford Lane, H., et al. (2017). STING is an essential mediator of the Ku70-mediated production of IFN-γ1 in response to exogenous DNA. Sci. Signal. 10:aah5054. doi: 10.1126/scisignal.aah5054
Sun, B., Sundström, K. B., Chew, J. J., Bist, P., Gan, E. S., Tan, H. C., et al. (2017). Dengue virus activates cGAS through the release of mitochondrial DNA. Sci. Rep. 7:3594. doi: 10.1038/s41598-017-03932-1
Sun, L., Wu, J., Du, F., Chen, X., and Chen, Z. J. (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 39, 786–91. doi: 10.1126/science.1232458
Sun, L., Xing, Y., Chen, X., Zheng, Y., Yang, Y., Nichols, D. B., et al. (2012). Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS ONE 7:30802. doi: 10.1371/journal.pone.0030802
Swanson, K. V., Junkins, R. D., Kurkjian, C. J., Holley-Guthrie, E., Pendse, A. A., Morabiti, R., et al. (2017). A noncanonical function of cGAMP in inflammasome priming and activation. J. Exp. Med. 214, 3611–3262. doi: 10.1084/jem.20171749
Takaoka, A., Wang, Z., Choi, M. K., Yanai, H., Negishi, H., Ban, T., et al. (2007). DAI (DLM1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505. doi: 10.1038/nature06013
Thapa, R. J., Ingram, J. P., Ragan, K. B., Nogusa, S., Boyd, D. F., Benitez, A. A., et al. (2016). DAI senses influenza a virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20, 674–681. doi: 10.1016/j.chom.2016.09.014
Unterholzner, L. (2013). The interferon response to intracellular DNA: why so many receptors? Immunobiology 218, 1312–1321. doi: 10.1016/j.imbio.2013.07.007
Unterholzner, L., Keating, S. E., Baran, M., Horan, K. A., Jensen, S. B., Sharma, S., et al. (2010). IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 11, 997–1004. doi: 10.1038/ni.1932
Upton, J. W., Kaiser, W. J., and Mocarski, E. S. (2012). DAI/ZBP1/DLM-1 Complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297. doi: 10.1016/j.chom.2012.01.016
Vemuri, M. C., Schiller, E., and Naegele, J. R. (2001). Elevated DNA double strand breaks and apoptosis in the CNS of scid mutant mice. Cell Death Differ. 8, 245–255. doi: 10.1038/sj.cdd.4400806
Venkatesan, A., and Geocadin, R. G. (2014). Diagnosis and management of acute encephalitis: a practical approach. Neurol. Clin. Pract. 4, 206–215. doi: 10.1212/CPJ.0000000000000036
Vermeire, J., Roesch, F., Sauter, D., Rua, R., Hotter, D., Van Nuffel, A., et al. (2016). HIV Triggers a cGAS-dependent, Vpu- and Vpr-regulated type i interferon response in CD4+ T Cells. Cell Rep. 17, 413–424. doi: 10.1016/j.celrep.2016.09.023
Wang, J., Kang, L., Song, D., Liu, L., Yang, S., Ma, L., et al. (2017). Ku70 Senses HTLV-1 DNA and modulates HTLV-1 replication. J. Immunol. 199, 2475–2482. doi: 10.4049/jimmunol.1700111
Wang, X., Li, Y., Liu, S., Yu, X., Li, L., Shi, C., et al. (2014). Direct activation of RIP3/MLKLdependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl. Acad. Sci. U.S.A. 111, 15438–15443. doi: 10.1073/pnas.1412767111
Wang, Z. C., Choi, M. K., Ban, T., Yanai, H., Negishi, H., Lu, Y., et al. (2008). Regulation of innate immune responses by DAI (DLM-1/ZBP1) and other DNA-sensing molecules. Proc. Natl. Acad. Sci. U.S.A. 105, 5477–5482. doi: 10.1073/pnas.0801295105
West, A. P., Khoury-Hanold, W., Staron, M., Tal, M. C., Pineda, C. M., Lang, S. M., et al. (2015). Mitochondrial DNA stress primes the antiviral innate immune response. Nature 520, 553–557. doi: 10.1038/nature14156
Wong, E. B., Montoya, B., Ferez, M., Stotesbury, C., and Sigal, L. J. (2019). Resistance to ectromelia virus infection requires cGAS in bone marrow-derived cells which can be bypassed with cGAMP therapy. PLoS Pathog. 15:e1008239. doi: 10.1371/journal.ppat.1008239
Wu, J. J., Li, W., Shao, Y., Avey, D., Fu, B., Gillen, J., et al. (2015). Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe 18, 333–344. doi: 10.1016/j.chom.2015.07.015
Xu, H., Su, C., Pearson, A., Mody, C. H., and Zheng, C. (2017). Herpes simplex virus 1 UL24 abrogates the DNA sensing signal pathway by inhibiting NF-κB activation. J. Virol. 91, e00025–17. doi: 10.1128/JVI.00025-17
Yang, Y., Zhao, X., Wang, Z., Shu, W., Li, L., Li, Y., et al. (2020). Nuclear sensor interferon-inducible protein 16 inhibits the function of hepatitis b virus covalently closed circular DNA by integrating innate immune activation and epigenetic suppression. Hepatology 71, 1154–1169. doi: 10.1002/hep.30897
Ye, R., Su, C., Xu, H., and Zheng, C. (2017). Herpes simplex virus 1 ubiquitin-specific protease ul36 abrogates nf-κb activation in dna sensing signal pathway. J. Virol. 91:e02417–16. doi: 10.1128/JVI.02417-16
Yi, G., Wen, Y., Shu, C., Han, Q., Konan, K. V., Li, P., et al. (2016). Hepatitis C virus NS4B can suppress STING accumulation to evade innate immune responses. J. Virol. 90, 254–265. doi: 10.1128/JVI.01720-15
Yogarajah, T., Ong, K. C., Perera, D., and Wong, K. T. (2017). AIM2 Inflammasome-mediated pyroptosis in enterovirus A71-infected neuronal cells restricts viral replication. Sci. Rep. 7:5845. doi: 10.1038/s41598-017-05589-2
You, H., Zheng, S., Huang, Z., Lin, Y., Shen, Q., and Zheng, C. (2019). Herpes simplex virus 1 tegument protein ul46 inhibits tank-binding kinase 1-mediated signaling. MBio 10, 919–938. doi: 10.1128/mBio.00919-19
Zhang, D., Su, C., and Zheng, C. (2016). Herpes simplex virus 1 serine protease VP24 blocks the dna-sensing signal pathway by abrogating activation of interferon regulatory factor 3. J. Virol. 90, 5824–5829. doi: 10.1128/JVI.00186-16
Zhang, G., Chan, B., Samarina, N., Abere, B., Weidner-Glunde, M., Buch, A., et al. (2016). Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc. Natl. Acad. Sci. U.S.A. 113, E1034–E1043. doi: 10.1073/pnas.1516812113
Zhang, H., Luo, J., Alcorn, J. F., Chen, K., Fan, S., Pilewski, J., et al. (2017). AIM2 inflammasome is critical for influenza-induced lung injury and mortality. J. Immunol. 198, 4383–4393. doi: 10.4049/jimmunol.1600714
Zhang, J., Zhao, J., Xu, S., Li, J., He, S., Zeng, Y., et al. (2018). Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication. Cell Host Microbe 24, 234–248.e5. doi: 10.1016/j.chom.2018.07.004
Zhang, X., Brann, T. W., Zhou, M., Yang, J., Oguariri, R. M., Lidie, K. B., et al. (2011). Cutting Edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol. 186, 4541–4545. doi: 10.4049/jimmunol.1003389
Zhang, X., Shi, H., Wu, J., Zhang, X., Sun, L., Chen, C., et al. (2013). Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 51, 226–235. doi: 10.1016/j.molcel.2013.05.022
Zhang, Z., Bao, M., Lu, N., Weng, L., Yuan, B., and Liu, Y. J. (2013). The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nat. Immunol. 14, 172–178. doi: 10.1038/ni.2492
Zhang, Z., Yuan, B., Bao, M., Lu, N., Kim, T., and Liu, Y. J. (2011). The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol. 12, 959–965. doi: 10.1038/ni.2091
Zheng, Y., Liu, Q., Wu, Y., Ma, L., Zhang, Z., Liu, T., et al. (2018). Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. EMBO J. 37:e99347. doi: 10.15252/embj.201899347
Zhu, W., Zu, X., Liu, S., and Zhang, H. (2019). The absent in melanoma 2 (AIM2) inflammasome in microbial infection. Clin. Chim. Acta 495, 100–108. doi: 10.1016/j.cca.2019.04.052
Keywords: DNA sensors, astrocytes, microglia, neuroinflammation, viral encephalitis
Citation: Jeffries AM and Marriott I (2020) Cytosolic DNA Sensors and CNS Responses to Viral Pathogens. Front. Cell. Infect. Microbiol. 10:576263. doi: 10.3389/fcimb.2020.576263
Received: 25 June 2020; Accepted: 12 August 2020;
Published: 16 September 2020.
Edited by:
Federico Iovino, Karolinska Institutet (KI), SwedenReviewed by:
Maria Kalamvoki, University of Kansas Medical Center, United StatesRaphael Gaudin, UMR9004 Institut de Recherche en Infectiologie de Montpellier (IRIM), France
Copyright © 2020 Jeffries and Marriott. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ian Marriott, aW1hcnJpb3RAdW5jYy5lZHU=