 Md. Aejazur Rahman1
Md. Aejazur Rahman1 Joel N. Glasgow2
Joel N. Glasgow2 Sajid Nadeem2
Sajid Nadeem2 Vineel P. Reddy2
Vineel P. Reddy2 Ritesh R. Sevalkar2
Ritesh R. Sevalkar2 Jack R. Lancaster Jr3
Jack R. Lancaster Jr3 Adrie J. C. Steyn1,2,4*
Adrie J. C. Steyn1,2,4*- 1Africa Health Research Institute, Durban, South Africa
- 2Department of Microbiology, University of Alabama at Birmingham, Birmingham, AL, United States
- 3Department of Pharmacology and Chemical Biology, Vascular Medicine Institute, University of Pittsburgh School of Medicine, Pittsburgh, PA, United States
- 4Centers for AIDS Research and Free Radical Biology, University of Alabama at Birmingham, Birmingham, AL, United States
For centuries, hydrogen sulfide (H2S) was considered primarily as a poisonous gas and environmental hazard. However, with the discovery of prokaryotic and eukaryotic enzymes for H2S production, breakdown, and utilization, H2S has emerged as an important signaling molecule in a wide range of physiological and pathological processes. Hence, H2S is considered a gasotransmitter along with nitric oxide (•NO) and carbon monoxide (CO). Surprisingly, despite having overlapping functions with •NO and CO, the role of host H2S in microbial pathogenesis is understudied and represents a gap in our knowledge. Given the numerous reports that followed the discovery of •NO and CO and their respective roles in microbial pathogenesis, we anticipate a rapid increase in studies that further define the importance of H2S in microbial pathogenesis, which may lead to new virulence paradigms. Therefore, this review provides an overview of sulfide chemistry, enzymatic production of H2S, and the importance of H2S in metabolism and immunity in response to microbial pathogens. We then describe our current understanding of the role of host-derived H2S in tuberculosis (TB) disease, including its influences on host immunity and bioenergetics, and on Mycobacterium tuberculosis (Mtb) growth and survival. Finally, this review discusses the utility of H2S-donor compounds, inhibitors of H2S-producing enzymes, and their potential clinical significance.
Introduction
Although hydrogen sulfide (H2S) was not discovered until 1777 by the Swedish-German chemist Carl Wilhelm Scheele (Mitchell and Davenport, 1924), the description of its biological effects dates to the early 1700s, when Italian physician Bernardino Ramazzini (1633–1714) published his collection of observations regarding workers, their work environments, and occupation-associated illnesses as De Morbis Artificum Diatriba [Treatise on Worker’s Diseases]. Ramazzini is now considered “the father of occupational medicine,” and his compendium contains a chapter entitled, “Diseases of Cleaners of Privies and Cesspits” in which he described a “sewer gas” that caused painful irritation and inflammation in the eyes of sewer workers. Although Ramazzini didn’t know about the chemical nature of the species responsible for this, he noted that this inflammation could lead to bacterial infections and blindness (Felton, 1997; Szabo, 2018).
Until the late 1960s, H2S (we refer to H2S, HS−, and S2− collectively as H2S or sulfide, unless specified otherwise) was still regarded as a foul-smelling toxic and flammable gas (Beauchamp et al., 1984) until it was understood to be produced endogenously in mammals. Indeed, three enzymes are responsible for the majority of H2S production in mammals: cystathionine γ-lyase (CSE), cystathionine β-synthase (CBS), and 3-mercaptopyruvate sulfurtransferase (3-MST). Another source of endogenous H2S is acid labile pools, which function in the presence of endogenous reductants and gut microbiota (Flannigan et al., 2011; Kimura, 2011). Notably, H2S is membrane-permeable and diffuses through cells (Mathai et al., 2009); therefore, it can act as a signaling molecule and/or interact directly with intracellular biomolecules.
H2S has since been increasingly recognized as an important physiological signaling molecule, along with nitric oxide (•NO) and carbon monoxide (CO) (Wang, 2014). H2S is now understood to have pleiotropic effects on human physiology. Indeed, it is an important mediator in cardiovascular (Kanagy et al., 2017; Pan et al., 2017) and respiratory systems (Bazhanov et al., 2017a), the nervous system (Kimura, 2019), and in inflammation and immunity (Bhatia, 2015). Notably, studies on the role of host H2S in bacterial (Benedetti et al., 2017; Saini et al., 2020; Rahman et al., 2020) or viral diseases (Pal et al., 2018), have been sparse. Therefore, it is reasonable to conclude that the role of host H2S in microbial diseases remains understudied.
The objective of this article is to review the physiological role of host-derived H2S in regulating various disease outcomes with an emphasis on its role in tuberculosis (TB). We begin by providing a brief description of H2S biochemistry and the enzymes that produce H2S, followed by the utility of H2S donor compounds and inhibitors of H2S production, which may have therapeutic value for the treatment of TB or other diseases. Next, we highlight the physiological importance of H2S in regulating mammalian metabolism and immunity in response to bacterial and viral infection. We then focus on the role of H2S in modulating immunity and metabolism, and how it contributes to TB pathogenesis. We conclude with brief overview of the role of endogenously produced H2S in bacterial physiology. For additional information, we refer the reader to several excellent review articles on the chemistry of H2S (Szabo, 2007; Mustafa et al., 2009; Wang, 2014; Filipovic et al., 2018; Szabo, 2018) and role of endogenous H2S in bacterial physiology (Shatalin et al., 2011; Mironov et al., 2017; Szabo, 2018; Toliver-Kinsky et al., 2019; Walsh and Giedroc, 2020).
Overview of the Biochemistry and Biophysics of Sulfide
Biochemical Properties
In biological molecules, sulfur can exist in a range of formal oxidation states, from −2 to +6, with H2S in the most reduced (−2) state. The biological chemistry of sulfur covalent compounds can be considered in terms of sulfur’s electronegativity (Sanderson, 1988). This means that its reactivity generally involves products that, relative to reactants, have increased electron density distributed away from the sulfur atom. As a nucleophile, sulfide thus reacts with electrophiles. Depending on the sulfur species and electrophilic reactant, electron redistribution can either be complete, resulting in transfer of an electron(s) from the sulfur-containing molecule to the electrophilic molecule, or partial. In the latter case, within a molecule, partial electron density is rearranged away from the sulfur and toward the electrophile but complete electron transfer does not occur. This sharing of electron density by the sulfur-containing nucleophilic moiety with the electrophilic moiety is a critical determinant of the covalent bond(s) between them, including strength and likelihood of formation.
Historically, oxygen has been the most important atom that, when bound to a nucleophile, results in significant “attraction” of electron density. This is why a change resulting in decreased electron density is referred to as “oxidation,” even if the reaction does not involve oxygen per se. The chemical reactions of sulfide are extensive (Filipovic et al., 2018) and the focus here will be on studies of reactions under physiologically relevant conditions that may be involved in signal transmission.
The protonation equilibria in aqueous solution are of critical importance in understanding the biochemistry of sulfide. Although there are two protonation equilibria for sulfide species,
the pKa of the second reaction is far above physiologically relevant pH (pKa2 = 17 to 19 (Filipovic et al., 2018)) so the sulfide anion (S2−) exists only in trace amounts. Since pKa1 is close to 7 under physiological conditions, both H2S and HS− (which have different chemical profiles) are present in appreciable amounts. In addition, the volatility of H2S is an important parameter experimentally, as described below.
The first reported potential chemical mechanism of protein-mediated signal transmission by sulfide was the modification of protein cysteine to form persulfide (RSSH) (Mustafa et al., 2009). Although the mechanism proposed was S-sulfhydration (a misnomer (Toohey, 2012)), this is highly unlikely since both thiol and sulfide are nucleophiles and a reaction to form persulfide requires an oxidant. A far more likely reaction is H2S with disulfide (RSSR’) or sulfenic acid (RSOH) (Cavallini et al., 1970; Francoleon et al., 2011; Cuevasanta et al., 2015):
Note that reaction (3) is reversible, implying that H2S and persulfide are at least theoretically kinetically interchangeable (Fukuto et al., 2020). Sensitive techniques have revealed the abundant presence of both protein and low molecular weight (cysteine and glutathione) persulfides in cells (Ida et al., 2014; Park et al., 2015; Fu et al., 2019). Akaike and colleagues have recently demonstrated the presence of enzymatic machinery that is capable of both sulfide-independent formation of free cysteine persulfide and direct translational incorporation of cys-SSH into newly synthesized proteins (Akaike et al., 2017). These findings have raised speculation as to whether the direct effector of signaling is sulfide, or whether a product from persulfide, or other polysulfide (Ida et al., 2014; Fukuto et al., 2018), or persulfide derivatives (Doka et al., 2020) is the functional entity (Alvarez et al., 2017; Filipovic et al., 2018; Fukuto et al., 2018).
In its interactions with other small reactive molecules, with the possible exception of hypochlorite (Nagy and Winterbourn, 2010), sulfide is unlikely to be an effective antioxidant under physiologically relevant (low concentration) conditions, although small amounts of oxidized sulfur species from such redox reactions may effect a signaling function as described above (Li and Lancaster, 2013; Nagy et al., 2014). The possibility of crosstalk between sulfide and nitric oxide (•NO) as biological signals (Fukuto et al., 2012; Kevil et al., 2017; Ivanovic-Burmazovic and Filipovic, 2019; Marcolongo et al., 2019) arose upon recognition of a signaling function for sulfide (Hosoki et al., 1997). In addition to overlapping and interactive biological downstream targets of •NO and sulfide (e.g., vasodilation (Kimura, 2015)), several nitrogen oxide species react directly with sulfide and/or oxidized sulfur species to produce a variety of small reactive molecules, including thionitrous acid (HSNO), perthionitrite (ONSS−), polysulfides (HSn−), nitroxyl (HNO), and dinitrososulfite (ON(NO)SO3−; SULFI/NO) which is a diazeniumdiolate •NO/nitroxyl donor. These species could result in multiple effects including cysteine per- and poly-sulfidation (vide supra); however, the likelihood of their formation under biological conditions (with low concentrations of sulfide and •NO or alternative more abundant reactants) is not clear (Kevil et al., 2017). ONSS− in particular has a relatively long lifetime and exhibits potent in vivo hypotensive activity, suggesting potential biological relevance (Cortese-Krott et al., 2015; Bogdandi et al., 2020).
In biological systems transition metal ions are strong electrophiles and interact with nucleophilic sulfur compounds, including sulfide. The focus here is on sulfide reactions with hemoproteins, since this interaction appears to be most important for TB (see section The Role of Host-Derived H2S in Microbial Infections). Prior to the discovery of sulfide signaling activity, its biomedical relevance was dominated by its toxicity, which, as first described by Keilin in 1929 (Keilin, 1929), is primarily due to inhibition of mitochondrial electron transfer at cytochrome c oxidase (CcO) (Petersen, 1977; Olson, 2012a; Nicholls et al., 2013). The mechanism of this inhibition is complex, involving as many as three sulfide molecules, acting both as an electron donor and a ligand to oxidized states of CcO, including ferric heme a3 (Cooper and Brown, 2008). Sulfide also reduces cytochrome c, providing electrons for CcO, and the sulfur oxidation product(s) increases protein persulfidation, thereby possibly potentiating sulfide signaling (Vitvitsky et al., 2018). A similar formation of sulfur oxidation products from endogenously produced sulfide has been reported for intact red blood cells, mediated by hemoglobin (Vitvitsky et al., 2015). Under oxidizing conditions, interaction of sulfide with hemoproteins can result in damaging covalent modification of the heme to form sulfheme, although this occurs only at relatively high sulfide levels (Filipovic et al., 2018).
There are numerous complex factors that determine the nature of the interaction of sulfide with hemoproteins, including the oxidation state of the heme iron, solution protonation state of the sulfide species and resultant solvent (H2O) interactions, dynamic configurations of bound heme and binding to water, access of the sulfide ligand to the distal heme pocket, and interaction of the bound ligand with amino acid residues and water (Pietri et al., 2011; Capece et al., 2013; Boubeta et al., 2020; Fukuto et al., 2020). In terms of ferroheme (not bound to protein) in a non-polar solvent, the hydrosulfide anion (HS−) can bind, but not H2S (Boubeta et al., 2020). The only well-documented ferrous heme sulfide complex in protein is myeloperoxidase (MPO), although no information was provided regarding the protonation state of the bound sulfide ligand (Palinkas et al., 2015). This apparent “exception” may be due to stabilization of bound ligand by an arginine residue in the heme pocket (Boubeta et al., 2020). Sulfide also acts as a reductant for MPO Complexes I and II.
The ferriheme protein that is undoubtedly best characterized for sulfide binding is the hemoglobin I of the bivalve mollusk Lucina pectinata, which delivers environmental sulfide as a respiratory substrate to a bacterial chemoautotrophic symbiont which provides organic carbon to the host (Kraus and Wittenberg, 1990; Kraus et al., 1990; Boubeta et al., 2016). As is true for essentially all ferric hemoproteins (including methemoglobin) it is protonated H2S that initially binds to the ferriheme, with non-existent or very weak binding by hydrosulfide (HS−). Experimental and theoretical evidence (Boubeta et al., 2020) suggests that this discrimination results from the inability of the hydrosulfide anion to access the heme as a result of the protein structure. The rate-limiting step in binding is release of the heme-bound water molecule prior to ligand binding. In some cases, heme-bound H2S deprotonates, leaving hydrosulfide as the final bound species which is stabilized by interaction with distal amino acid residue(s). The proximal heme ligand exerts major influence over the stability of the hydrosulfide complex, as well as the propensity of the sulfide to reduce the heme, forming ferroheme and oxidized sulfur species. Reduction and binding are also accomplished by persulfide interactions with heme proteins (Fukuto et al., 2020).
Biophysical Properties
In the pure state and at standard temperature and pressure (STP) H2S is a gas. Unless exposed to a gaseous interface, this property is irrelevant to its biological actions (Fukuto et al., 2012; Li and Lancaster, 2013); however, it must be taken into account under certain experimental conditions (DeLeon et al., 2012). In a sealed container with headspace, at equilibrium the relative distribution of hydrogen sulfide (H2S) follows Henry’s Law, meaning its concentration in aqueous solution with a pure 1 atm H2S headspace is 110 mM and will decrease proportionately with partial pressure (Filipovic et al., 2018). The volatility of H2S under several laboratory physiological experimental conditions has been reported, with half-life ranging from 0.5 to 5 min (DeLeon et al., 2012).
Sulfide exhibits high turnover rates physiologically, resulting from a balance of production (by enzymes of the transsulfuration pathway, see H2S-Producing Enzymes and Pathways) and consumption (primarily via mitochondrial quinone reductase) with a resultant in vivo concentration in the 4–55 nM range (Kabil and Banerjee, 2014; Benchoam et al., 2019). As a small dissolved non-electrolyte similar to •NO and CO, hydrogen sulfide (H2S) is a highly lipophilic molecule that is freely membrane permeable and does not require facilitated diffusion (Mathai et al., 2009).
H2S-Producing Enzymes and Pathways
The transsulfuration pathway involves the interconversion of cysteine and homocysteine through the intermediate cystathionine, leading to generation of sulfur metabolites and H2S. In mammals, H2S is synthesized within the transsulfuration pathway by two enzymes, namely cystathionine β-synthase (CBS; EC 4.2.1.22) and cystathionine γ-lyase (CSE; EC 4.4.1.1). Another enzyme involved in cysteine catabolism, 3-mercaptopyruvate sulfurtransferase (3-MST or MPST; EC 2.8.1.2) also produces H2S (Figure 1). CBS and CSE require the cofactor pyridoxal-5’-phosphate (PLP, the active form of vitamin B6) and catabolize L-cysteine to produce H2S within the transsulfuration pathway. However, H2S production by the PLP-independent enzyme 3-MST requires another PLP-dependent enzyme, cysteine aminotransferase (CAT, EC 2.6.1.3), to produce H2S from cysteine. Initially, CAT uses cysteine along with α-ketoglutarate to produce 3-mercaptopyruvate and L-glutamate. 3-MST then catalyzes the conversion of 3-mercaptopyruvate to pyruvate and H2S (Shibuya et al., 2009). However, the 3-MST reaction requires an additional reductant, such as thioredoxin (Trx) or dihydrolipoic acid (DHLA), for the release of H2S (Mikami et al., 2011). In contrast, a pathway which does not require PLP for H2S synthesis involves two enzymes, D-amino acid oxidase (DAO) and 3-MST. Here, DAO catalyzes D-cysteine to 3-mercaptopyruvate, the substrate for 3-MST (Figure 1) (Shibuya et al., 2013; Pollegioni et al., 2018).
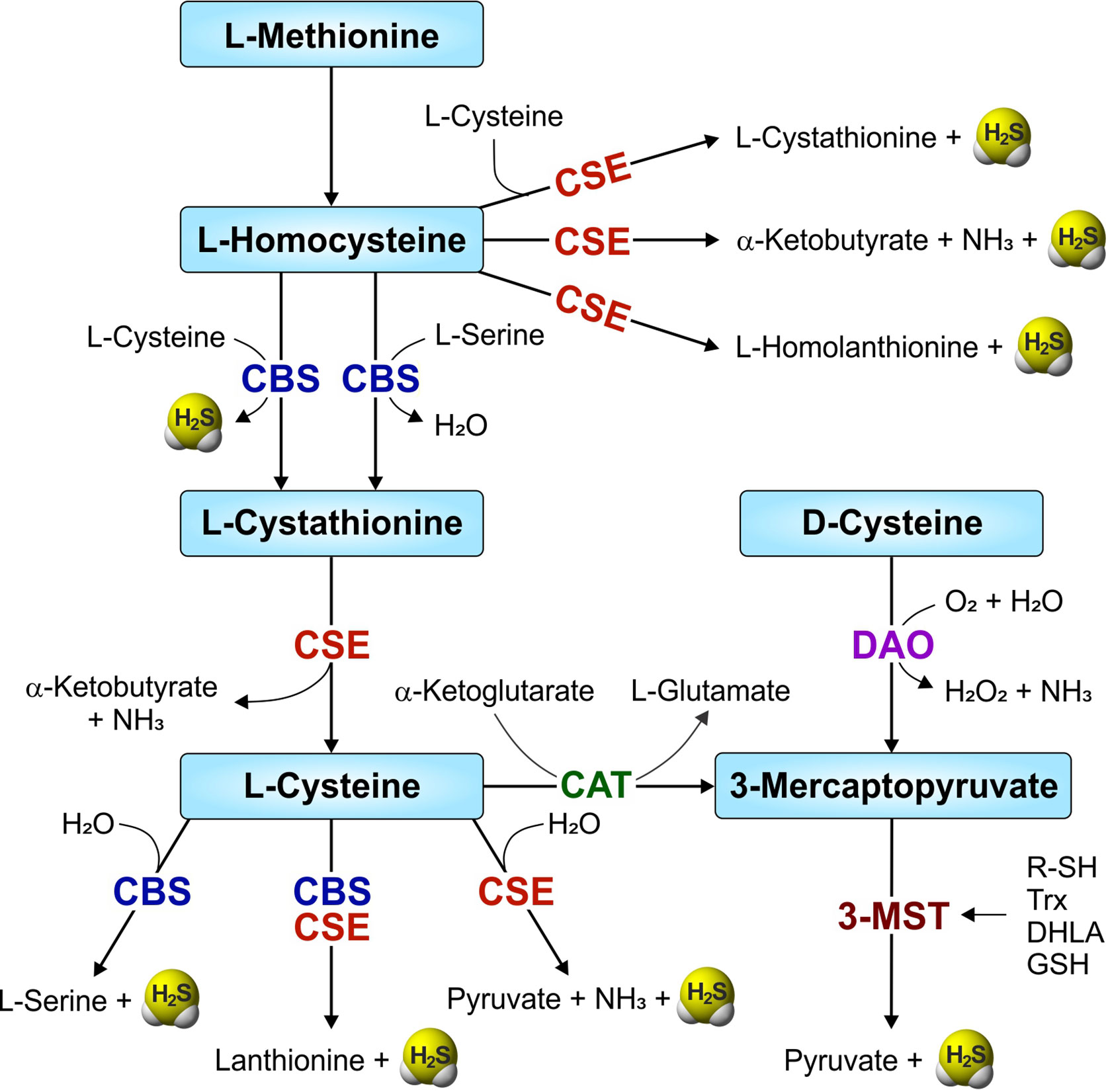
Figure 1 Overview of the mammalian transsulfuration pathway and H2S-producing enzymes. Two enzymes from the transsulfuration pathway—CBS (cystathionine-β-synthase) and CSE (cystathionine-γ-lyase), and another enzyme involved in sulfur metabolism, 3-MST (3-mercaptopyruvate sulfurtransferase) are involved in the production of H2S in mammals. CAT (cysteine aminotransferase) and DAO (D-amino acid oxidase) convert L-cysteine and D-cysteine respectively, to 3-mercaptopyruvate, a substrate for 3-MST. R-SH, thiols; Trx, thioredoxin (in the presence of NADPH/thioredoxin reductase).
Cystathionine β-Synthase
The human CBS enzyme is a homotetramer comprised of ~63 kDa monomers. Each 551-amino acid monomer binds two cofactors, heme and PLP (the active form of vitamin B6), and is composed of three structural domains (Kery et al., 1994; Meier et al., 2001; Ereño-Orbea et al., 2013). The N-terminal domain consisting of residues 1–70 contains a heme-binding pocket, and the heme iron is axially coordinated by Cys52 and His65 (Meier et al., 2001; Taoka et al., 2002). While heme is not required for catalytic activity, it is required for protein folding, subunit assembly and for binding modulatory ligands CO and •NO. The ferric (Fe3+) state of heme iron is relatively inert and more stable, whereas the ferrous (Fe2+) state binds CO and •NO with different affinities (Puranik et al., 2006; Vicente et al., 2014). Both the oxidized (Fe3+) and reduced (Fe2+) state of the human CBS heme iron are low spin, hexa-coordinate species that are axially bound by the ligands His65 and Cys52. In the reduced heme state, binding of CO is hexa-coordinate by replacing the endogenous Cys52 ligand, whereas binding of •NO with heme results in a penta-coordinate species by replacing both ligands. Binding of CO or •NO with heme reduces CBS activity (Taoka and Banerjee, 2001; Vicente et al., 2014). Further, CBS activity is dependent on the oxidation state of the heme iron, as shown by a two-fold reduction in enzyme activity when the heme iron is in the reduced, ferrous (Fe2+) form, compared to the ferric form, which suggests the possibility of redox-associated regulation (Taoka et al., 1998; Taoka et al., 2002; Banerjee and Zou, 2005). The role of heme in regulating CBS catalytic activity is still unclear. However, a CBS deletion mutant lacking the 69 N-terminal residues does not bind heme, and retains only 40% activity (Evande et al., 2004).
The N-terminal domain is followed by a highly conserved catalytic domain that spans residues 71–413 and contains the PLP binding site (Banerjee and Zou, 2005). The C-terminal regulatory domain (residues 414–551) is comprised of tandem “CBS domains” (CBS1 and CBS2), a structural motif known for adenosine nucleotide binding and regulating protein activity via both intrasteric and allosteric effects (Bateman, 1997; Miles and Kraus, 2004; Baykov et al., 2011). CBS catalytic activity is modulated via binding of S-adenosyl-L-methionine (AdoMet or SAM) to two sets of binding sites in the regulatory domain (Finkelstein et al., 1975; Pey et al., 2013); AdoMet binding increases CBS activity 2–3 fold (Taoka et al., 1999) via stabilization of the protein (Prudova et al., 2006). The C-terminal regulatory domain is also critical for maintaining the tetrameric structure of CBS, as tryptic cleavage at Arg413 converts CBS to a dimer of core enzymes (Kery et al., 1998). Notably, C-terminal deletion mutants that lack the regulatory domain lose AdoMet responsiveness, but exhibit increased enzyme activity compared the full-length tetramer form, indicating that the C-terminal regulatory domain serves an autoinhibitory function (Kery et al., 1998; Evande et al., 2004; Banerjee and Zou, 2005).
CBS-mediated H2S production occurs via at least three reactions: 1) converting cysteine to serine and H2S, 2) condensing cysteine and homocysteine to yield cystathionine and H2S, and 3) condensing two cysteine molecules to lanthionine and H2S (Kabil et al., 2011; Giuffre and Vicente, 2018; Majtan et al., 2018). CBS can also catalyze cystine (the oxidized dimer form of cysteine) to form cysteine persulfide, a cysteine with its sulfhydryl group covalently bound to sulfur known as sulfane sulfur (Cys-SSH), pyruvate, and NH3 (Yadav et al., 2016). However, the first and committed step in the mammalian transsulfuration pathway, catalyzed by CBS, is the formation of L-cystathionine and water from the condensation of L-serine and L-homocysteine (Singh et al., 2009; Giuffre and Vicente, 2018; Majtan et al., 2018). Homocysteine is a toxic intermediate in the methionine cycle and the first molecule to enter the transsulfuration pathway for the formation of cysteine (Miles and Kraus, 2004; Singh et al., 2009). AdoMet, an allosteric activator of CBS, is another important intermediate in the methionine cycle that controls the metabolic flux between the transmethylation and transsulfuration routes (Finkelstein et al., 1975; Banerjee and Zou, 2005; Giuffre and Vicente, 2018). CBS is generally considered a cytoplasmic protein, but can be translocated to the nucleus (Kabil et al., 2006) and mitochondria (Bhattacharyya et al., 2013; Teng et al., 2013). While CBS is a major contributor to H2S synthesis throughout the central nervous system (Robert et al., 2003), CBS is also expressed in the liver, kidney, and pancreas (Bao et al., 1998; Kabil et al., 2011; Giuffre and Vicente, 2018).
Cystathionine γ-Lyase
CSE is a homotetrameric enzyme composed of ~44 kDa monomers. Each 405-amino acid monomer consists of two structural domains. The larger N-terminal domain spans residues 9–263, contains the PLP binding pocket and is followed by the smaller C-terminal domain. The PLP cofactor is bound in the active site mainly by Lys212, and Tyr60 and Arg62 from the adjacent subunit (Sun et al., 2009). CSE is localized to the cytoplasm (Ogasawara et al., 1994) and expressed in the cardiovascular system, liver, kidney and lungs and pancreas (Hosoki et al., 1997; Zhao et al., 2001; Yang et al., 2004). Under increased calcium levels or hypoxia, CSE can translocate from the cytosol to the mitochondria of vascular smooth muscle cells, resulting in H2S production within mitochondria (Fu et al., 2012).
CSE is the second enzyme in the transsulfuration pathway and can utilize L-cystathionine to form L-cysteine, α-ketobutyrate and ammonia. In addition, CSE catalyzes other H2S-generating reactions: 1) condensation of L-cysteine and L-homocysteine to produce L-cystathionine and H2S, 2) utilization of two L-cysteine molecules to produce L-lanthionine and H2S, 3) breakdown of L-cysteine into pyruvate, H2S and ammonia, 4) condensation of two molecules of L-homocysteine to generate L-homolanthionine and H2S, and 5) degradation of L-homocysteine to generate α-ketobutyrate, H2S, and ammonia (Chiku et al., 2009; Singh et al., 2009; Giuffre and Vicente, 2018). In addition, CSE can catalyze cystine to form Cys-SSH, pyruvate and NH3, and homocystine (the oxidized dimer of homocysteine) to form homocysteine persulfide (Hcy-SSH), α-ketobutyrate, and NH3 (Yadav et al., 2016). Unlike CBS, CSE can generate H2S solely from homocysteine, an intermediate of the methionine cycle, as well as from cysteine alone. Under Vmax conditions at saturating concentrations (10 mM cysteine or 30 mM homocysteine) CSE forms H2S at a 5-fold faster rate from homocysteine than from cysteine via condensation of two homocysteine molecules (reaction 4 above) (Chiku et al., 2009). However, under physiological conditions (10 μM homocysteine and 100 μM cysteine) over 70% of the H2S produced by CSE is predicted to come from cysteine (reaction 3 above), due to limiting homocysteine concentrations. However, at homocysteine concentrations of 40 μM and above that can occur in hyperhomocysteinemia, CSE is predicted to be the main contributor to H2S production where between 60 and 78% of H2S is derived from homocysteine alone (Chiku et al., 2009; Singh et al., 2009).
3-Mercaptopyruvate Sulfurtransferase
3-MST is the third enzyme in the cysteine catabolism pathway. It is thought to be evolutionarily related to the mitochondrial enzyme thiosulfate sulfurtransferase, known as rhodanese, as 3-MST contains two catalytic active rhodanese-like domains (RLD) (Nagahara et al., 1995). 3-MST is expressed in two isoforms. The full-length 317 amino acid isoform (3-MST-iso1) is comprised of a 20 amino acid N-terminal extension, followed by a 25-residue mitochondrial targeting sequence (MTS), RDL1 (residues 46–162), and RDL2 (residues 167–297) (Frasdorf et al., 2014). 3-MST-iso2 lacks the 20-residue N-terminal extension, exposing the MTS to localize this isoform to the mitochondria and cytoplasm, whereas 3-MST-iso1 is confined to the cytoplasm.
In the cysteine catabolism pathway, CAT converts L-cysteine and α-ketoglutarate into glutamate and 3-mercaptopyruvate (3-MP), as stated above. 3-MP is also generated from D-cysteine by D-amino acid oxidase (DAO) (Shibuya et al., 2013; Pollegioni et al., 2018). 3-MST then transfers the sulfur atom from 3-MP to a nucleophilic Cys248 (the catalytic site in human 3-MST) to generate a 3-MST-bound persulfide and pyruvate. This 3-MST bound persulfide (oxidized form) remains catalytically inactive until after release of H2S which is mediated by a reductant, such as reduced Trx, DHLA, glutathione (GSH), L-cysteine, L-homocysteine or by non-physiological reductants like 2-mercaptoethanol and dithiothreitol (DTT) (Nagahara and Katayama, 2005; Mikami et al., 2011; Yadav et al., 2013).
Unlike CBS and CSE, 3-MST is regulated primarily via its redox sensitivity and exists as a catalytically active monomer or an inactive disulfide-linked homodimer. This inter-subunit disulfide bond can be cleaved by a reducing agent such as reduced Trx, DHLA, GSH, or DTT, acting like a switch to activate 3-MST (Nagahara et al., 2007). Further, 3-MST can be inactivated via oxidation of solvent-exposed cysteines by hydrogen peroxide (H2O2); however, enzymatic activity can be restored in the presence of reducing agents DTT or reduced Trx (Nagahara and Katayama, 2005).
3-MST is involved in a broad range of physiological processes and can generate H2S and thiolate molecules in the cytosol and mitochondria. For example, 3-MST is responsible for detoxifying cyanide (CN−) by converting it to thiocyanate (SCN-), a less toxic molecule that can be safely metabolized and excreted (Nagahara et al., 1999). In the mouse brain, 3-MST has been shown to produce Cys-SSH, as well as glutathione (GSH) persulfide (GSSH), persulfurated cysteine residues on proteins, and H2S2. These sulfur-containing species play a dominant role in signaling and redox homeostasis (Kimura, 2015; Kimura et al., 2017). 3-MST is also referred to as transfer RNA (tRNA) thiouridine modification protein 1 (TUM1) due to its role in thiolation of cytosolic tRNAs. Thiolation of uridine at position 2 stabilizes the tRNA structure and ensures accurate mRNA decoding (Frasdorf et al., 2014). 3-MST is expressed in the perivascular glial cells in the brain, bronchiolar epithelial cells in the lung, myocardial cells in the heart, pericentral hepatic cells in the liver, and proximal renal tubular cells in the kidney (Nagahara et al., 1998).
H2S Research Tools: Inhibitors of H2S-Synthesizing Enzymes and H2S Donor Compounds
Inhibitors of H2S-Synthesizing Enzymes
As mentioned above, there are three H2S-producing enzymes in mammals: CBS, CSE, and 3-MST. The therapeutic potential of reducing endogenous H2S levels by inhibiting the activity of these enzymes has been evaluated in various diseases in small animal models and humans (Szabo, 2007; Vandiver and Snyder, 2012). The most commonly used inhibitors of H2S-producing enzymes are discussed below.
Aminooxyacetic Acid
Aminooxyacetic acid (AOAA) was the first pharmacological inhibitor of CBS to be widely used. AOAA is an inhibitor of CBS (IC50 of ~8.5 μM) and CSE (IC50 of ~1.1 μM) (Asimakopoulou et al., 2013). As described in detail below in section Role of Host H2S in Tuberculosis, data from in vitro and in vivo studies of Mtb infection demonstrate that AOAA reduces H2S production in host cells to reduce H2S-stimulated Mtb growth. Indeed, AOAA treatment of Mtb-infected peritoneal macrophages in vitro reduced Mtb growth compared to untreated controls. Further, intraperitoneal (IP) administration of AOAA to Mtb-infected WT mice reduced lung Mtb burden to the level observed in the lungs of Cbs+/− mice (Saini et al., 2020). Consequently, inhibiting H2S production warrants further evaluation as a potential therapeutic strategy for controlling TB.
AOAA has been employed extensively to study the role of H2S in cancer. While AOAA is not selective for CBS, it remains a useful tool for blockade of CBS-derived H2S in vitro and in animal models. The colon cancer-derived epithelial cell line HCT116 exhibits upregulation of CBS and increased H2S production compared to non-cancerous colon cells. Inhibition of CBS activity with AOAA reduced H2S production, reduced basal cellular respiration, suppressed ATP synthesis, reduced the spare respiratory capacity of HCT116 cells, and inhibited their growth. Further, administration of AOAA to mice bearing colon cancer xenografts slowed tumor growth (Szabo et al., 2013). AOAA treatment inhibited oxygen consumption, reduced ATP levels, and suppressed the proliferation of MDA-MB-231 breast adenocarcinoma cells compared to normal human mammary epithelial cells. In addition, AOAA treatment decreased the growth of pancreatic ductal adenocarcinoma cells (Son et al., 2013), ovarian cancer cell lines in vitro (Bhattacharyya et al., 2013), and MDA-MB-231 breast tumors in nude mice (Thornburg et al., 2008), emphasizing both the role of CBS-derived H2S in cancer pathophysiology and the therapeutic potential of inhibiting CBS enzymatic activity. While AOAA has shown promising results as an anti-cancer treatment in small animal models, it also exhibits dose-dependent toxicity in humans, indicating the need for further design and formulation for future studies.
DL-Propargylglycine
DL-Propargylglycine (PAG) is a selective, irreversible inhibitor of CSE with an IC50 of 40 μM (Sun et al., 2009; Asimakopoulou et al., 2013). In Mtb-infected murine macrophages, PAG treatment reduced H2S production and bacillary burden, indicating that host-derived H2S can modulate Mtb survival in macrophages. Further, PAG increased glycolysis and mitochondrial respiration in Mtb-infected macrophages (Rahman et al., 2020). These observations suggest that excess H2S impedes glycolysis and mitochondrial respiration in host cells during infection and shows H2S to be a key regulator of host central energy metabolism during tuberculosis. The use of PAG in Mtb-infected macrophages in vitro is discussed in more detail in the sections below: H2S Promotes Mtb Growth by Suppressing Pro-Inflammatory Cytokines, Host H2S Suppresses Glycolysis and Oxygen Consumption in Macrophages, and H2S Stimulates Mtb Growth and Metabolism.
In a rat model of hemorrhagic shock, PAG inhibited H2S production which led to increased plasma TNF-α, IL-6, and iNOS levels and accelerated recovery of normal blood pressure (Mok et al., 2004; Mok and Moore, 2008). In a mouse model of endotoxemia, administration of PAG reduced lipopolysaccharide (LPS)-induced elevation of •NO in plasma, and reduced myeloperoxidase, a marker of tissue damage, in the lungs and pancreas (Collin et al., 2005; Li et al., 2005) suggesting an anti-inflammatory role for H2S. The effect of H2S on the production of chemokines has been investigated. In a mouse model of cerulean-induced acute pancreatitis and associated lung injury, the pro-inflammatory effects of H2S were mediated by release of MCP-1, MIP-1α, MIP-2 chemokines, an effect that could be blocked by PAG (Tamizhselvi et al., 2008). Similarly, in the cecal ligation and puncture (CLP) sepsis model, PAG treatment reduced mRNA and protein levels of pro-inflammatory cytokines IL-1β, IL-6, TNF-α in mice, indicating that H2S potentiates systemic inflammation in sepsis (Zhang et al., 2007b). Drawbacks associated with PAG are its relatively low potency and poor cell permeability, requiring the use of high concentrations for CSE inhibition that can also inhibit aspartate and alanine aminotransferases (Szabo, 2007; Asimakopoulou et al., 2013).
D-Penicillamine
D-penicillamine is a penicillin derivative and the D-isomer of dimethylated cysteine that was originally used to treat rheumatoid arthritis, and is now used to treat Wilson Disease due to its ability to chelate accumulated copper, which is characteristic of this genetic disorder (Suarez-Almazor et al., 2000; Litwin et al., 2019). D-penicillamine inhibits both CBS and CSE, but is approximately 30 times more selective against CSE (IC50 of 270 μM) (Brancaleone et al., 2016). In a model of arterial inflammation, administration of D-penicillamine exacerbated the TNF-α inflammatory response by significantly increasing the number of adherent leukocytes as determined by intravital microscopy (Brancaleone et al., 2016). D-Penicillamine can have additional effects unrelated to CSE inhibition. For instance, treatment of murine RAW264.7 macrophage cells with D-penicillamine activates these cells via direct binding of D-penicillamine to cell-surface aldehydes, resulting in increased production of cytokines TNF-α, IL-6, and IL-23 (Li and Uetrecht, 2009). Likewise, D-penicillamine was shown to activate macrophages and T-cells, undergirding the ability of this compound to induce autoimmunity in Brown Norway rats (Masson et al., 2004). Overall, D-penicillamine inhibits CSE and activates immune cells, in addition to other functions. It is possible that this FDA-approved compound can be repurposed for therapeutic use in TB or other diseases where reduction of H2S levels is warranted.
β-Cyanoalanine
β-Cyanoalanine (BCA) is a neurotoxic agent that, unlike PAG, is a reversible inhibitor that acts by transiently modifying the CSE apoenzyme (Whiteman and Winyard, 2011). BCA is a slightly more potent inhibitor of CSE (IC50 of 14 µM) compared to PAG (IC50 of 40 µM) (Asimakopoulou et al., 2013). BCA can inhibit CBS, but only at concentrations exceeding 1 mM (Papapetropoulos et al., 2009; Asimakopoulou et al., 2013). BCA-mediated suppression of endogenous H2S synthesis resulted in enhanced neutrophil adhesion and infiltration, and edema formation in rats. This study suggests an important role for endogenous H2S as a modulator of key components of acute inflammatory responses which might influence the leukocyte-endothelial cell interface (Zanardo et al., 2006). However, BCA also inhibited enzymes such as aspartate β-decarboxylase and alanine aminotransferase, similar to PAG (Alston et al., 1980; Cornell et al., 1984). Undoubtedly, BCA is useful pharmacological tool for studying H2S biology, however further pharmacokinetic analyses are required before more widespread therapeutic use is feasible.
L-Aminoethoxyvinylglycine
L-aminoethoxyvinylglycine (AVG) is a specific inhibitor of CSE with an IC50 of 1.0 μM, although its use is not widespread, likely due to its unclear mechanism (Asimakopoulou et al., 2013). AVG is a natural toxin discovered in fermentation broth that inhibited the growth of Streptomyces cellulosae, and was reported to inhibit enzymes in the transsulfuration pathway (Clausen et al., 1997). Asimakopoulou et al. showed that the IC50 of AVG against CSE is 1 µM, making AVG the most potent inhibitor for this enzyme. Notably, AVG has extremely high selectivity for CSE and does not inhibit CBS activity even at concentrations up to 1 mM (Asimakopoulou et al., 2013). AVG inhibits human CSE via slow, tight, reversible binding to the PLP cofactor in the active by forming a Schiff base bond (Steegborn et al., 1999). Despite having high selectivity for CSE over CBS, AVG has been shown to inhibit other PLP-dependent enzymes, which should be considered when using this compound.
Other Inhibitors
A novel 3-MST inhibitor, HMPSNE, was identified in a drug screen and exhibits an IC50 of ~2–30 μM under various conditions (Hanaoka et al., 2017; Augsburger et al., 2020). HMPSNE suppressed H2S production in a concentration-dependent manner and reduced the proliferation and migration of CT26 murine colon cancer cells. Further, HMPSNE exerted a bell-shaped effect on oxygen consumption rate and extracellular acidification rate in CT26 cells. These observations suggests that 3-MST is the primary enzymatic source of H2S in CT26 cell line (Augsburger et al., 2020). Corvino et al. identified an oxothiazolidine derivative (referred to as compound a2) that efficiently inhibits CSE at concentrations 100-fold lower than PAG in aortic rings ex vivo. Further, compound a2 binding is reversible, offering a further advantage over PAG (Corvino et al., 2016). Use of a novel, highly selective inhibitor of human CBS, CH0004, elevated cellular homocysteine and suppressed H2S production in a dose-dependent manner in cell lines and in liver cancer xenografts. In addition, CH0004 triggered ferroptosis in HepG2 hepatic carcinoma cells and substantially reduced in vivo tumor growth in a xenograft mouse model (Wang et al., 2018).
Inhibitors of H2S-synthesizing enzymes have provided valuable insight into the varied roles of H2S in biological systems. However, like all inhibitors, these compounds can have off-target effects, such as inhibition of protein synthesis and transamination activity, and their use requires care and appropriate controls. Clearly, development of improved inhibitors would greatly benefit current H2S research and future clinical applications.
H2S Donor Compounds
As H2S has emerged as a critical regulator of numerous physiological mechanisms in humans, it has also become clear that a variety of human pathologies are associated with aberrant H2S levels that contribute to disease (Vandiver and Snyder, 2012; Kondo et al., 2013; Bhushan et al., 2014; Polhemus et al., 2014). In biological systems, H2S is produced endogenously by host enzymes or it can be provided through the use of H2S-releasing donor compounds. In this regard, the development of H2S-releasing compounds as potential therapeutic agents has gained considerable interest (Powell et al., 2018). Not surprisingly, several classes of H2S donors have been developed to investigate the role of H2S in disease, and are described in comprehensive reviews (Song Z. J. et al., 2014; Li et al., 2018; Zheng et al., 2018; Levinn et al., 2020). Below, we discuss H2S-donor platforms known to modulate host immunometabolism, which is a key determinant in protection against various diseases including TB.
Inorganic H2S Donors
NaHS and sodium sulfide (Na2S) have been widely used donors of hydrosulfide (HS−, see Overview of the Biochemistry and Biophysics of Sulfide) the deprotonated biologically active form of H2S. In a mouse model of burn- and smoke-induced acute lung injury, subcutaneous administration of NaHS reduced the levels of pro-inflammatory cytokines IL-1 β, IL-6, and IL-8 and increased the anti-inflammatory cytokine IL-10 (Esechie et al., 2008). Similarly, NaHS reduced neutrophil adhesion, attenuated expression of inflammatory mediators such as Tnf, Cox2, and Icam1, and preserved mitochondrial function in various animal models (Fiorucci et al., 2005; Johansen et al., 2006; Elrod et al., 2007; Zhu et al., 2007; Sun et al., 2016). The use of NaHS is widespread in studying the role of H2S in the pathophysiology of various diseases, and is more fully discussed throughout section The Physiological Importance of H2S.
Nambi et al. demonstrated that addition of NaHS restores defective recycling of mycothiol observed in Mtb mutants deleted for components of the membrane-associated oxidoreductase complex (MRC). NaHS also reversed the sensitivity toward oxidative stress and survival of these Mtb mutants in both IFN-γ-activated bone marrow-derived macrophages and in mice, indicating that H2S can play a crucial role in maintaining redox homeostasis in Mtb (Nambi et al., 2015). Of note, release of HS− from NaHS in aqueous solution is nearly instantaneous and can result in locally toxic concentrations for a short duration. This lack of sustained, controlled release has been addressed by the development of organic H2S donor compounds which have shown considerable therapeutic potential (Caliendo et al., 2010; Vandiver and Snyder, 2012; Guo et al., 2013).
Organic H2S Donors
Lawesson's reagent, a sulfurization reagent used in organic synthesis, releases H2S upon hydrolysis, which was shown to reduce TNF-α, IL-1β, reduce myeloperoxidase activity, increase levels of GSH, and protect rats through activation of ATP-sensitive potassium (KATP) channels (Nicolau et al., 2013). Derived from Lawesson’s reagent, GYY4137 is a water soluble slow-releasing H2S donor that more accurately mimics physiological H2S production (Li et al., 2008). Addition of GYY4137 to human cancer cell lines was shown to alter energy metabolism by increasing glycolysis and glucose uptake and inhibiting the excretion of lactate in cancer cells, possibly by suppressing anion exchanger and sodium/proton exchanger activity. This combination of effects led to intracellular acidification and subsequent cell death (Lee et al., 2014). Similarly, GYY4137 released H2S over several days to cause cell cycle arrest and apoptosis in human cancer cell lines and in tumor xenografts in mice, while having no effect on normal lung fibroblasts. These data indicate that prolonged exposure to low levels of H2S can selectively kill cancer cells (Lee et al., 2011).
A recent study showed that exposure of H2S-deficient Cbs+/− macrophages and mice to GYY4137 increased H2S levels that stimulated Mtb growth similar to that observed in WT control cells and mice (Saini et al., 2020). In addition, GYY4137-sourced H2S reduced pro-inflammatory cytokines, altered bioenergetics and increased Mtb growth in Cse−/− macrophages comparable to wild type (Rahman et al., 2020). These studies suggest the utility of GYY4137 for pharmacologically modulating H2S levels in TB. Additional examples of the use of GYY4137 in disease models are presented throughout sections The Physiological Importance of H2S and Role of Host H2S in Tuberculosis.
Mitochondria-Targeted H2S Donors
H2S is known to exert profound effects on cellular bioenergetics and mitochondrial function (Szabo et al., 2014). Two novel mitochondria-targeted slow release H2S donors, anethole dithiolethione (AP39) (Le Trionnaire et al., 2014) and hydroxythiobenzamide (AP123) (Gero et al., 2016) were generated by coupling H2S-donating dithiolethione to a mitochondria-targeting moiety (triphenylphosphonium; TPP+). Hyperglycemia alters mitochondrial membrane potential by significantly increasing the activity of the tricarboxylic acid (TCA) cycle and increasing production of superoxide leading to mitochondrial dysfunction. AP39 provided targeted H2S release within the mitochondria, which reversed hyperglycemia-induced bioenergetic defects, increased cell viability, and minimized the loss of mitochondrial DNA integrity in microvascular endothelial cells undergoing oxidative stress (Gero et al., 2016). In addition, AP39 and AP123 treatment reduced mitochondrial oxidative free radicals (Gero et al., 2016). Similarly, RT01, a novel derivative of AP39, reversed hyperglycemia-induced mitochondrial hyperpolarization, oxidant production, and increased synthesis of ATP, leading to restoration of mitochondrial function in murine brain microvascular endothelial cells (Waters et al., 2017).
Reactive Oxygen Intermediate-Activated H2S Donors
This class of H2S donor limits the release of H2S to situations where reactive oxygen intermediates (ROI) are present and the anti-inflammatory activities of H2S would be particularly beneficial. In this regard, Zhao et al. tested a series of arylboronate-functionalized thiocarbamate (PeroxyTCM) compounds. Upon exposure to H2O2, and to a lesser extent to superoxide and peroxynitrite, PeroxyTCM compounds release carbonyl sulfide (COS) which is quickly converted to H2S by ubiquitous carbonic anhydrase (Zhao and Pluth, 2016; Zhao et al., 2017). In a similar approach, Hu and colleagues developed a ROI-triggered H2S donor, NAB, wherein COS release (and conversion to H2S by carbonic anhydrase) is accompanied by release of a strong fluorophore, allowing for real-time monitoring of H2S release (Hu et al., 2019).
Thiol-Activated H2S Donors
Thiol-activated H2S-releasing agents release H2S upon reacting with thiol-containing molecules such as glutathione and cysteine. Isothiocyanates were first reported as cysteine-activated H2S donors with relatively low release efficiency (Martelli et al., 2014; Martelli et al., 2020). Isothiocyanates have been shown to suppress NF-κB-mediated inflammation (Shehatou and Suddek, 2016), upregulate Nrf2 signaling to protect against oxidation (Huang et al., 2013) and at higher concentrations, induce apoptosis in cancer cell lines by altering mitochondrial function (Sehrawat et al., 2016). Thiol-Activated gem-dithiol-based H2S donors (TAGDD) release H2S in the presence of thiols like glutathione (GSH). TAGDD were able to significantly reduce production of pro-inflammatory cytokines IL-1α, IL-1β, IL-6, TNF-α, GM-CSF, and G-CSF leading to reduced lung inflammation in a mouse model of RSV infection (Bazhanov et al., 2018). Acyl perthiol donors (Zhao et al., 2013), dithioperoxyanhydride (Powell et al., 2018), N-(benzoylthio)benzamides (Zhao et al., 2011), and arylthioamides (Martelli et al., 2013) are H2S donors triggered in the presence of cysteine and/or GSH. A major advantage of the thiol-activated class of H2S donors is that free, naturally occurring thiols such as glutathione are relatively abundant in mammals, which provide a continuous source for H2S release. However, it may prove problematic in clinical settings because some patient groups have reduced levels of glutathione (Wang Y. et al., 2013).
Considerable effort has been focused on designing, testing, and understanding the chemical properties of several classes of H2S donors in a variety of biological systems. A primary consideration is the slow, prolonged release of H2S to better mimic physiological conditions. However, significant gaps in our knowledge remain that hinder clinical use of H2S donors. Continued innovation with a focus on bioavailability and targeted release of H2S will be critical in developing H2S-releasing therapeutic agents for clinical studies.
The Physiological Importance of H2S
Since the discovery of endogenous production of H2S in mammals nearly three decades ago, H2S has gained considerable attention due to its physiological importance and therapeutic potential (Wang, 2012; Zhang Y. et al., 2013; Panthi et al., 2016). Although H2S was long known as a foul smelling and noxious gas, the biological role of H2S was never carefully examined until the end of the last century. Since then, similar to the gasotransmitters •NO and CO for which physiological roles were clearer, the role of H2S has been widely explored in numerous biological systems (Figures 2 and 3).
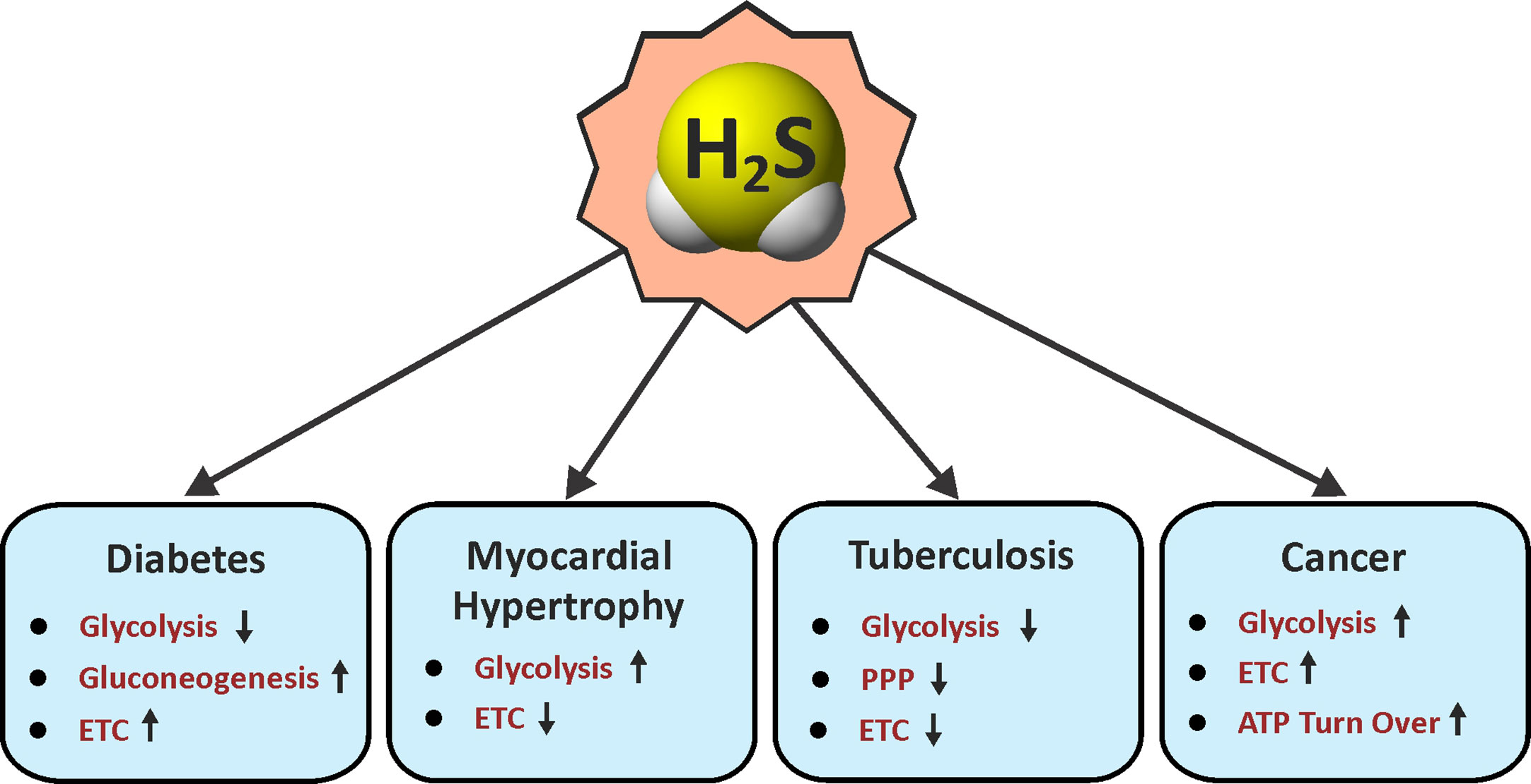
Figure 2 Roles of H2S in energy metabolism. Arrow indicates an H2S-mediated increase (up) or decrease (down) in the pathways associated with metabolism in various diseases. ETC, electron transport chain; PPP, pentose phosphate pathway.
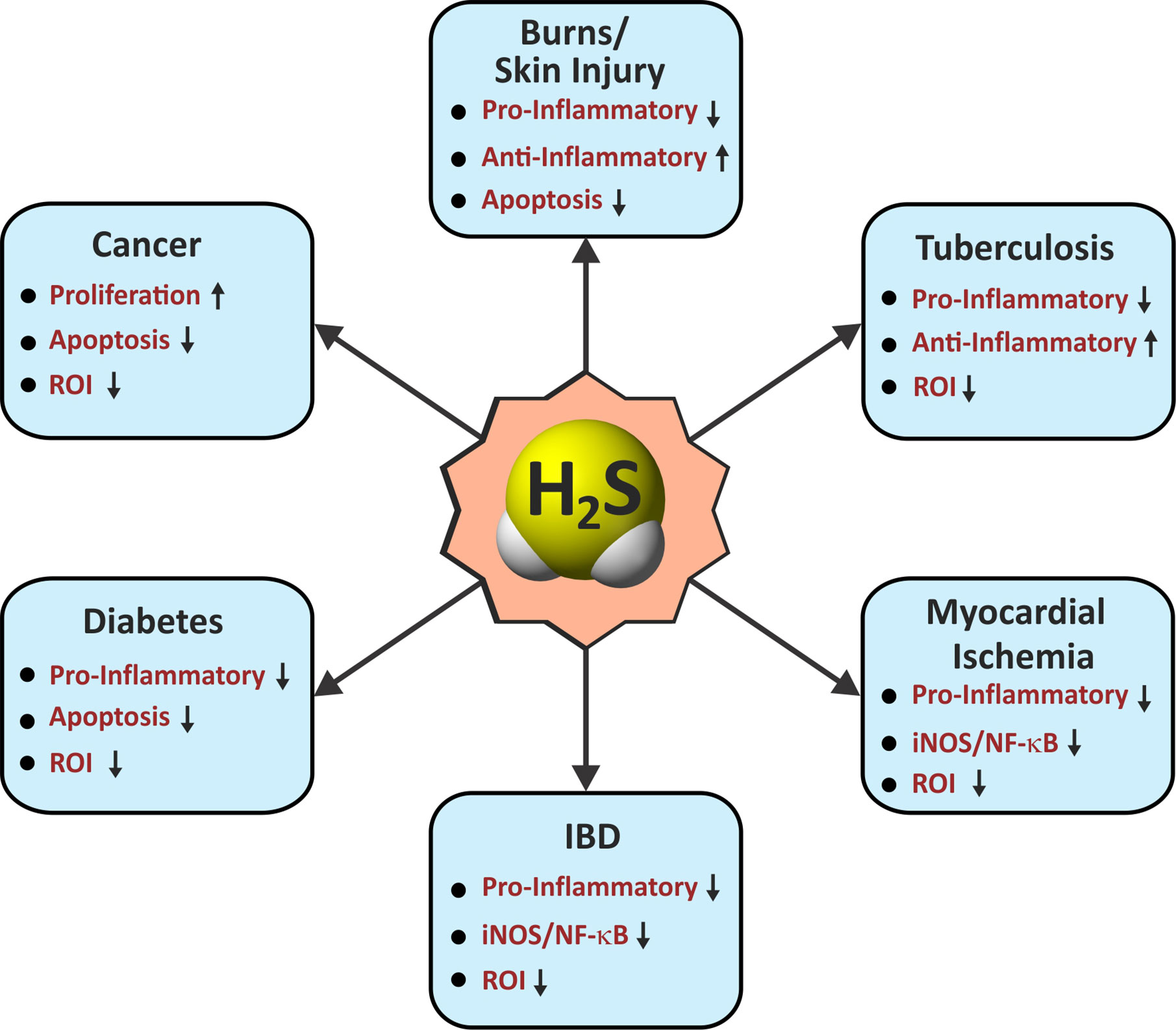
Figure 3 Roles of H2S in immunological pathways. Arrow indicates an H2S-mediated increase (up) or decrease (down) in the activity associated with immunological functions in various diseases. IBD, inflammatory bowel disease; Pro-Inflammatory/Anti-Inflammatory, cytokines; iNOS, inducible nitric oxide synthase; NF-κB, nuclear factor kappa B; ROI, reactive oxygen intermediates.
The first physiological role attributed to H2S was as a neuromodulator (Abe and Kimura, 1996) and neurotransmitter that regulates glutamate receptors, calcium ion concentrations, and cAMP levels (Kimura, 2000; Lu et al., 2008; Kamat et al., 2015). More recently, H2S has emerged as a therapeutic molecule in various central nervous system disorders such as Alzheimer’s disease, ischemia, and injury-related trauma (Zhang and Bian, 2014). Moreover, H2S has been implicated in vasodilation by inducing the relaxation of smooth muscle cells (Hosoki et al., 1997; Zhao et al., 2001; Teague et al., 2002; Zhao and Wang, 2002). Endogenous H2S has also been shown to play a role in regulating erectile dysfunction through relaxing the human corpus cavernosum (d’Emmanuele di Villa Bianca et al., 2009), and has been considered a potential anti-aging molecule by inhibiting free radical formation (Zhang Y. et al., 2013). Overall, a plethora of research has demonstrated a role for H2S in numerous pathophysiological processes such as diabetes (Wu et al., 2009; Szabo, 2012; Ma et al., 2017), hypertension (Wang et al., 2014; Sun et al., 2014; Meng et al., 2015), atherosclerosis (Wang et al., 2009; Zhang et al., 2012; Mani et al., 2014; Wang Z. J. et al., 2017), sepsis (Coletta and Szabo, 2013; Kosir and Podbregar, 2017; Qiu et al., 2018), and respiratory diseases such as asthma (Wang et al., 2011; Chung, 2014). H2S exerts its physiological functions in part through interacting with K+ ion channels (Jiang et al., 2010) and various signaling proteins. These effects are dependent on H2S concentration, which can vary from tissue to tissue or even from cell to cell (Olson, 2012b). Considering the diverse physiological roles of H2S, the following section is limited to a description of how H2S regulates various metabolic and immunological pathways in mammals.
The Role of H2S in Metabolic Regulation and Disorders
Recent evidence suggests that metabolic programming of immune cells is tightly linked to immune cell function and fate (Al-Khami et al., 2017; Patel et al., 2019). Consequently, identifying the molecules, cytokines or microbial products that regulate these metabolic pathways to ultimately affect disease outcomes is the focus of active investigation. One of the first roles identified for H2S was the inhibition of cytochrome c oxidase (Complex IV), a component of the mitochondrial electron transport chain (ETC), which results in reduced ATP production under normoxic conditions (Blackstone et al., 2005). However, at low concentrations H2S increases oxygen consumption, membrane potential and mitochondrial ATP production (Lagoutte et al., 2010). H2S also serves as an oxygen sensor and energy substrate to regulate ATP production under hypoxic conditions (Fu et al., 2012). This function is mediated by a sulfide-quinone reductase whereby electrons derived from H2S are fed into the ETC to stimulate oxidative phosphorylation (OXPHOS) and mitochondrial ATP production (Hildebrandt and Grieshaber, 2008). Moreover, H2S helps maintain mitochondrial integrity by attenuating ROI and reactive nitrogen intermediates (RNI) (Suzuki et al., 2011).
An important mechanism by which H2S functions is through persulfidation of cysteines in target proteins, which can alter protein function (see Overview of the Biochemistry and Biophysics of Sulfide). Unlike nitrosylation, persulfidation accounts for 25–50% of the post-translational modifications of hepatic proteins (Jaffrey and Snyder, 2001; Mustafa et al., 2009). In 2009, Mustafa, et al. reported that 39 proteins in mouse liver lysates were persulfidated following treatment with sodium hydrosulfide (NaHS, a rapid releaser of H2S), several of which are involved in metabolic regulation. One vital enzyme regulated by persulfidation is glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which is an important regulatory enzyme in glycolysis besides its role in gene transcription. This study showed that persulfidation of GAPDH at Cys150, which is critical for catalysis, enhanced its glycolytic activity by ~700%. Furthermore, a 25–30% reduction in GAPDH activity was observed in Cse−/− mice compared to WT controls, suggesting that endogenous H2S can modulate GAPDH activity (Mustafa et al., 2009). Interestingly, the α subunit (ATP5A1) of ATP synthase (Complex V) of the ETC is persulfidated by H2S at two positions, which significantly increases its enzymatic activity in vitro and in vivo (Modis et al., 2016).
Diabetes is a metabolic disorder resulting from impaired insulin secretion and/or insulin resistance that modulates carbohydrate metabolism (Kaneko et al., 2006; Al-Goblan et al., 2014). Several groups have demonstrated that obese participants or those with type 2 diabetes have significantly reduced plasma H2S levels, which is also seen in rodent models of type 1 diabetes (Jain et al., 2010; Whiteman et al., 2010a). However, in streptozotocin (STZ)-diabetic rats, increased Cbs and Cse mRNA and H2S formation were observed in the liver and pancreas (Yusuf et al., 2005), which regulates insulin secretion and resistance. Notably, increased pancreatic CSE expression and H2S production in the Zucker diabetic fatty (ZDF) rat model of diabetes was shown to reduce circulating insulin levels, resulting in hyperglycemia that could be reversed by administration of PAG (see DL-Propargylglycine) (Wu et al., 2009). Inhibition of insulin secretion by H2S is attributed to its ability to activate ATP-sensitive K+ channels (Yang et al., 2005). In another study, H2S was shown to impede insulin secretion by inhibiting Ca2+ channels (Kaneko et al., 2006). Tang et al. further substantiated that exogenous and endogenous H2S inhibits L-type voltage-dependent Ca2+ channels in pancreatic beta cells, and thus regulates insulin secretion in a mouse model (Tang et al., 2013). H2S may inhibit glucose metabolism by reducing intracellular accumulation of ATP, glucose transport, and mitochondrial oxidation in pancreatic beta cells (Kaneko et al., 2006) pointing to several mechanisms whereby H2S can influence insulin secretion in pancreatic beta cells.
H2S can also regulate liver metabolism, which may result in hepatic insulin resistance (Pichette and Gagnon, 2016). In a high fat diet-induced mice model of diabetes, reduced CSE expression led to lower H2S levels and decreased pyruvate carboxylase levels that inhibited gluconeogenesis and prompted glycolysis in the liver (Manna et al., 2014; Peh et al., 2014; Ju et al., 2015). In another study, H2S was reported to impair glucose uptake and increase gluconeogenesis in hepatocytes through increased activity of phosphoenolpyruvate carboxykinase and decreasing glucokinase activity (Zhang L. et al., 2013). The pathogenesis of diabetic complications is also associated with endothelial dysfunction which is linked to enhanced mitochondrial reactive oxygen species (ROI) (Giacco and Brownlee, 2010). H2S replacement therapy blocks the development of endothelial dysfunction by restoring oxidative phosphorylation in addition to improving mitochondrial depolarization, cellular ATP levels and reduced mitochondrial ROI production in hyperglycemic endothelial cells (Suzuki et al., 2011).
H2S has also been implicated in the regulation of cellular bioenergetics in various cancers. In primary ovarian carcinoma cells, a lack of H2S decreases mitochondrial oxygen consumption and enhances ROI production (Bhattacharyya et al., 2013). In colon cancer-derived epithelial cell lines, CBS levels are upregulated with concomitantly increased H2S production. Pharmacological inhibition of CBS in these colon cancer cell lines reduced cell proliferation, invasion, and migration along with suppressed glycolysis and mitochondrial function, suggesting that H2S can facilitate tumor growth (Szabo et al., 2013). In another study, increased levels of H2S in colonocytes activated the sulfide oxidation pathway and inhibited the ETC resulting in reductive stress as indicated by a reduced NAD+/NADH redox ratio (Libiad et al., 2019).
In summary, H2S regulates various facets of metabolism under different pathological conditions (Figure 2). Since metabolic processes are upstream of immune pathways that modulate immunological cell responses, H2S can have multiple roles in dictating disease outcomes.
The Role of H2S in Immune Regulation
The immune system employs various effector mechanisms to protect against microbes and toxic substances. Not surprisingly, perturbations in immune regulation machinery are known to cause human disease. In this regard, numerous studies have shed light on the effects of exogenous or endogenous H2S in regulating immune responses (Wallace and Wang, 2015; Wallace et al., 2015; Fagone et al., 2018).
Considerable attention has been devoted to elucidating the role of H2S as a biological mediator of inflammation. Intriguingly, H2S has been shown to have a dual role in inflammatory processes, and both pro- and anti-inflammatory effects have been reported. The determining factor for a dual role of H2S in inflammation is unclear, but likely depends on the rate of H2S generation and/or H2S concentration (Whiteman et al., 2010b). Increased CSE activity and corresponding plasma H2S levels have been observed in hemorrhagic shock, pancreatitis, edema, and sepsis models of inflammation in mice (Mok et al., 2004; Bhatia et al., 2005; Li et al., 2005). In the cecal ligation and puncture-induced sepsis (CLP) model, administration of the CSE inhibitor PAG reduced leukocyte infiltration into tissues and increased the survival rate in mice. Also, decreased IL-6, TNF-α, and IL-1β levels were observed in the lungs and livers of these mice. Further, NaHS administration caused severe inflammatory damage through increased nuclear factor kappa B (NF-κB) activation (Zhang et al., 2007b; Zhang et al., 2007c). Importantly, LPS injection in mice has been shown to increase the plasma concentration of H2S through increased CSE activity. Inhibition of CSE through PAG administration in these mice reduced LPS-induced myeloperoxidase (MPO) activity in lungs and liver, which was accompanied by less leukocyte infiltration and tissue damage. Further, NaHS treatment led to a significant increase in plasma TNF-α levels, severe tissue damage in lungs, and increased MPO activity in both lungs and liver (Li et al., 2005). Administration of NaHS to the U937 macrophage cell line was shown to activate the p65 subunit of NF-κB and increase mRNA expression and protein levels of NF-κB target genes TNF-α, IL-6, and IL-1β (Zhi et al., 2007). Conversely, H2S can also exhibit anti-inflammatory functions by downregulating pro-inflammatory factors. For example, the H2S donor GYY4137 (Li et al., 2008), a slow releaser of H2S, and NaHS were shown to downregulate expression of the pro-inflammatory mediators TNF-α, ROI, and •NO in LPS-treated neuroblastoma cells and macrophage cells, illustrating the anti-inflammatory and cytoprotective role of H2S in LPS-mediated inflammation (Whiteman et al., 2010b; Yurinskaya et al., 2020). Also, the H2S-releasing compound ATB-429 reduced colitis-induced inflammation in mice by reducing granulocyte infiltration into colon tissue (Fiorucci et al., 2007). H2S also reduced gastric ulcer-related inflammation and promoted healing in a rat model (Wallace et al., 2007a). H2S-treated THP-1 cells incubated with lipid associated membrane proteins (LAMPs) from Mycoplasma pneumonia exhibited reduced production of pro-inflammatory cytokines IL-6 and IL-8 with a concomitant increase in HO-1 expression, suggesting an anti-inflammatory role for H2S (Hu et al., 2020).
Mice subjected to burn injuries had lower H2S plasma levels and increased levels of pro-inflammatory cytokines such as IL-6, IL-8 and TNF-α compared to control mice. Chemical complementation using NaHS reduced these cytokine levels and increased the anti-inflammatory cytokine IL-10 (Zeng et al., 2013). The H2S donor GYY4137 was shown to play a protective role against endotoxin-induced acute lung injury by decreasing iNOS activity and •NO release, and reduced leukocyte infiltration in lung tissues (Zhang et al., 2016). Further, GYY4137 has also been shown to alleviate diabetes-induced atherosclerosis by reducing oxidative stress, decreasing pro-inflammatory cytokines IL-1β, TNF-α, and IL-6, and suppressing the activation of the NLRP3 inflammasome (Zheng et al., 2019). In a rat model of myocardial ischemia-reperfusion injury, intermediate doses of NaHS (1.6 mg/kg) reduced expression of iNOS and NF-κB, and lowered oxidative stress and inflammation in heart tissue (Jeddi et al., 2020).
H2S has also been shown to be protective in various skin-related disorders such as psoriasis and Werner syndrome, an autosomal recessive disorder of premature aging. To model Werner syndrome, Werner fibroblasts were cultured, and it was noted that H2S-generating enzyme levels were lower, with increased oxidative stress and cytosolic aggregates, compared to normal cells. These phenotypic changes were reversed with NaHS and mTOR inhibitor treatment (Talaei et al., 2013). Psoriasis patients were found to have lower levels of serum H2S and higher TNF-α, IL-6, and IL-8 serum levels compared to healthy subjects. Treatment of HaCaT keratinocytes with exogenous H2S inhibited TNF-α-induced upregulation of IL-6, IL-8, and •NO, suggesting that H2S levels negatively correlate with disease severity (Alshorafa et al., 2012). Exogenous H2S supplementation reduced psoriasis symptoms and signs in a skin model of psoriasis (Rodrigues et al., 2015).
H2S has a cytoprotective function upon tissue injury in diabetic cardiomyopathy (DCM). NaHS administration in a rat model of DCM conferred protection from myocardial fibrosis through down-regulation of the JAK-STAT pathway, thereby suppressing inflammation, oxidative stress, and apoptosis (Liu M. et al., 2018). Moreover, in type 2 diabetes patients and in STZ-diabetic rats, lower levels of circulating H2S were reported compared to healthy counterparts, which was associated with increased vascular inflammation (Jain et al., 2010). To model the role of H2S in diabetes-related vascular complications, HUVEC cells were exposed to high glucose (25 mM) and underwent apoptosis associated with an increased Bax/Bcl-2 ratio, caspase-3 activation, and increased ROI, all of which was prevented by pre-treatment with NaHS (Guan et al., 2012). H2S also downregulates miRNA-194 and plays a protective role against fibrotic changes through collagen realignment in diabetic kidneys (John et al., 2017). H2S also improved outcomes in a mouse model of diabetes-associated cognitive decline (DACD), a diabetic complication resulting in cognitive impairment. In one study, administration of NaHS to diabetic mice improved spatial learning, which was associated with modulation of the mitochondrial apoptotic pathway as evidenced by reduced levels of Bax and reduced cleavage of caspase-3 and -9. Further, levels of IL17/IL-23 were decreased in these mice suggesting an overall anti-apoptotic and anti-inflammatory role for H2S (Ma et al., 2017).
Over the past few decades, there has been an upsurge in the prevalence of inflammatory bowel disease (IBD) and other immunologically linked human disorders owing to changes in diet and lifestyle. H2S has been implicated in IBD, however its role remains controversial. H2S has been reported to suppress the expression of pro-inflammatory cytokines including IFN-γ and TNF-α (Li et al., 2007; Wallace et al., 2007b). In a mouse model of dextran sodium sulfate (DSS)-induced colitis, CBS and CSE expression increased in the gut following DSS administration, and inhibition of the CSE with PAG worsened markers of inflammatory disease, suggesting that H2S has anti-inflammatory effects in colitis (Hirata et al., 2011).
H2S has been reported to modulate the expression of genes involved in apoptosis, cell cycle control, and proliferation in a concentration and cell type-dependent manner (Faller et al., 2010; Zhang J. H. et al., 2010; Baskar and Bian, 2011). In human lung fibroblasts, NaHS causes DNA damage and cell cycle arrest (G1) in a concentration-dependent manner. This was coupled with increased expression of Bax, p21, and cytochrome c and stabilization of p53 (Baskar et al., 2007). Similar observations were seen when H2S-treated human and rat aorta smooth muscle cells exhibited increased apoptosis (Yang et al., 2006; Baskar et al., 2008). Another study reported that H2S induced free radical–mediated genomic DNA damage in Chinese hamster ovary cells (Attene-Ramos et al., 2007). Hoffman et al. reported that micromolar concentrations of H2S caused genomic single-strand breaks as a result of ROI generated by the auto-oxidation of H2S (Hoffman et al., 2012). Thus, H2S plays a role in cell growth and proliferation and may be relevant in the development of cancer therapeutics.
In conclusion, it is clear that H2S represents a key signaling molecule that can modulate various processes such as inflammation, oxidative stress, apoptosis, proliferation, and more (Figure 3). Exploiting H2S to regulate these events is likely to alter disease outcome. Moreover, promising results from preclinical studies in diverse pathophysiological conditions including, but not limited to, diabetes, neurodegeneration and ophthalmic disorders, psoriasis, and sepsis highlight the therapeutic potential of H2S.
Role of H2S in Macrophage Polarization
Macrophages are critical effector cells of the innate immune response and can switch between two opposing immunological phenotypes: M1 (pro-inflammatory or classically activated) and M2 (anti-inflammatory or alternatively activated) (Sica and Mantovani, 2012; Sica et al., 2015; Viola et al., 2019). M1 polarization occurs by sensing microbial products (LPS) or stimulation with Th1 cytokines like IFN-γ or GM-CSF. M1 macrophages are characterized by an increased capacity for antigen presentation, a Hif-1α-mediated metabolic shift toward glycolysis and the pentose phosphate pathway (PPP) that ultimately yields ROI for killing pathogens, and secretion of pro-inflammatory mediators like IL-1β, IL-6, TNF-α, IL-12, RNI, and prostaglandin E2 (PGE2) (Sica and Mantovani, 2012; Sica et al., 2015; Khan et al., 2019; Viola et al., 2019). In contrast, M2 polarization can be induced by Th2 cytokines IL-4, IL-13, IL-10, TGF-β, and M-CSF. M2 macrophages are characterized by anti-inflammatory processes, including production of Arg-1, IL-10, TGF-β, IGF-1, and collagen to dampen inflammation and stimulate cell survival and tissue repair (Mantovani et al., 2013; Khan et al., 2019; Viola et al., 2019). M2 macrophages are more reliant on OXPHOS and have reduced rates of glycolysis (Mills and O’Neill, 2016). Numerous studies have reported that H2S modulates inflammatory processes, exerting both pro-inflammatory and anti-inflammatory effects (Figure 4). Specifically, H2S has been implicated in influencing macrophage phenotype, as discussed below.
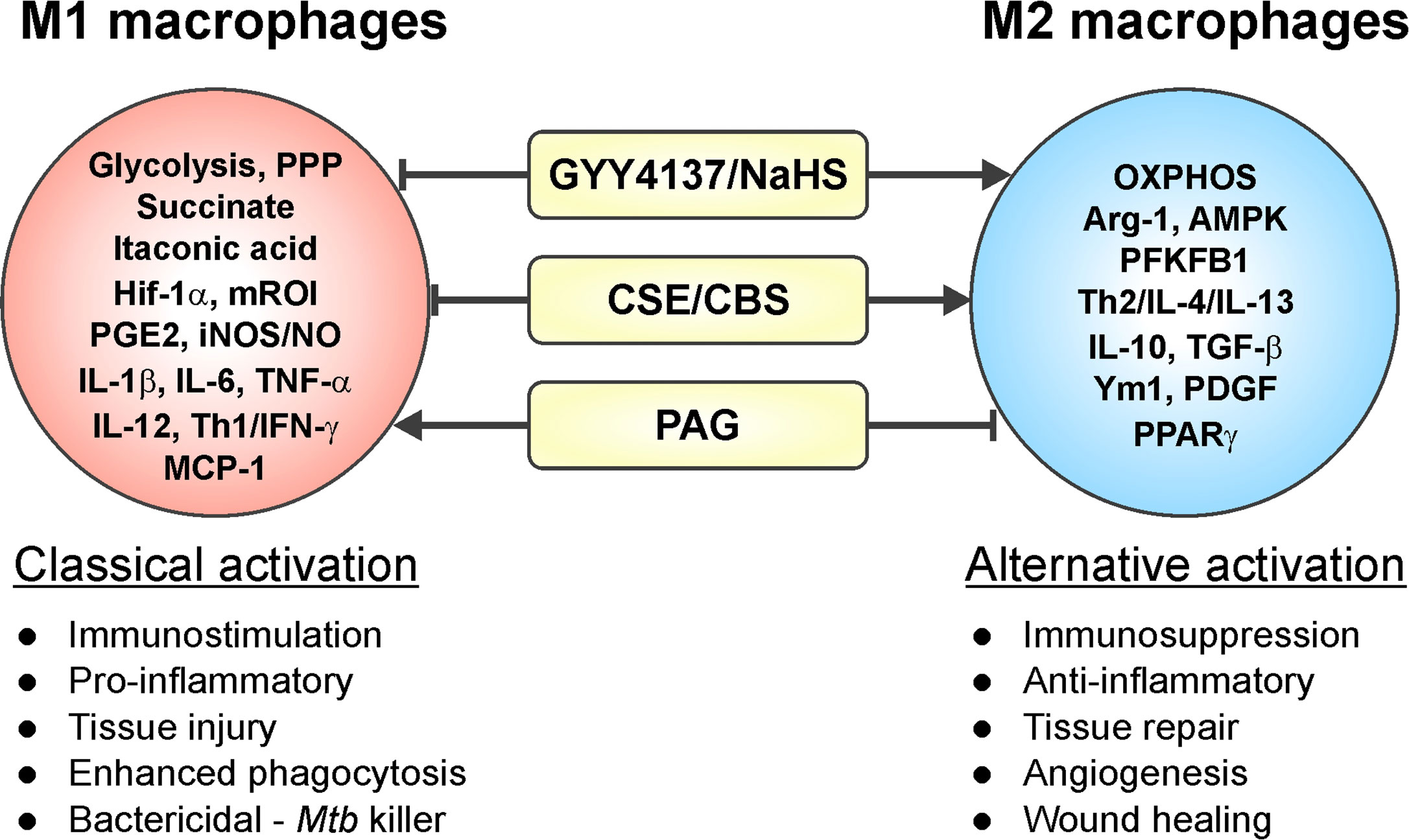
Figure 4 Involvement of H2S in macrophage polarization. Increased levels of H2S in an inflammatory macrophage model (caused by stimuli such as IFN-γ, LPS, or Mtb infection) trigger a phenotypic shift in macrophages leading to an increased M2 phenotype. On the other hand, reduced H2S levels induce a pro-inflammatory response, which drives macrophages toward an M1 phenotype. Hence, H2S can trigger M1 to M2 macrophage polarization with few exceptions.
A study carried out in the RAW264.7 cell line showed that GYY4137 inhibited the LPS-induced release of pro-inflammatory mediators IL-1β, IL-6, TNF-α, •NO, and PGE2, and increased synthesis of the anti-inflammatory cytokine IL-10 in a dose-dependent fashion (Whiteman et al., 2010b). These effects suggest that slow H2S release via GYY4137 can drive LPS-induced M1 macrophages toward an M2 phenotype. In the same study, addition of NaHS, which rapidly releases a bolus of H2S, showed a biphasic effect, with modest reductions in PGE2 at lower NaHS concentrations and increased IL-1β and TNF-α levels higher concentrations (200–1,000 µM) of NaHS (Whiteman et al., 2010b).
In a rat model of renal fibrosis based on unilateral ureteral obstruction, obstructed kidneys exhibited increased CSE expression, loss of CBS expression, and reduced H2S levels compared to controls. Obstructed kidneys also showed considerable macrophage infiltration and fibrosis. Notably, administration of NaHS (5.6 or 56 µg/kg/day, IP) attenuated inflammation by reducing macrophage infiltration and expression of inflammatory cytokines IL-1β, TNF-α, and MCP-1. In contrast, higher doses of NaHS (560 µg/kg/day, IP) increased inflammatory cytokine expression (Song K. et al., 2014).
Oxidized LDL (ox-LDL) is a major risk factor for developing atherosclerosis. Stimulation of macrophages in vitro with ox-LDL reduced CSE expression and H2S levels, and increased production of TNF-α. Conversely, overexpression of CSE or addition of NaHS reduced TNF-α production and endothelial cell adhesion in ox-LDL treated macrophages, showing that suppression of the CSE/H2S axis may be required to initiate and maintain a pro-inflammatory phenotype (Wang X. H. et al., 2013).
Further, exposure of microglia (macrophages in the central nervous system) to rotenone, a common pesticide and inhibitor of mitochondrial complex 1, leads to M1 polarization along with increased production of TNF-α, iNOS, and PGE2. M1 polarization coincided with reduced expression of CBS and lower H2S levels. However, overexpression of CBS in rotenone-treated microglia or exposure to NaHS (50, 100, 500 µM) caused reversion toward the M2 phenotype (Du et al., 2014).
Macrophages are critical for tissue repair following myocardial infarction (MI). In a mouse model of MI, macrophages from Cse−/− mice displayed an M1 phenotype with increased expression of IL-1β, IL-6 and TNF-α compared to WT controls. NaHS treatment of WT and Cse−/− mice reduced pathological cardiac remodeling with a reduction of infarct size. This was associated with an increased number of M2 macrophages that expressed IL-10, Arg-1, and Ym1 (Miao et al., 2016).
The studies mentioned above and numerous others have employed inhibitors of CSE and CBS (PAG and AOAA, respectively), L-cysteine and the H2S donors NaHS and GYY4137, as well as Cse−/− mice within various macrophage inflammatory models, which clearly established H2S as an effector molecule involved in resolution of inflammation by driving macrophages toward an anti-inflammatory M2 phenotype (Castelblanco et al., 2018; Zhou et al., 2019; Sunzini et al., 2020). Similarly, macrophages infected with Mtb exhibited increased CSE expression and H2S levels leading to an anti-inflammatory or M2 macrophage phenotype. This was reflected in increased Mtb growth compared to Cse−/− mice macrophages as discussed further in section H2S and M2 Macrophage Polarization in Tuberculosis (Figure 4). In contrast, a recent study of macrophage polarization during mechanical load-promoted tooth movement reported that load-stimulated periodontal ligament stem cells produce H2S that promotes M1 macrophage polarization and production of pro-inflammatory cytokines (He et al., 2020). Overall, it appears that upregulation of H2S-producing enzymes and/or exogenous H2S supplementation in inflammatory macrophages drives polarization toward an M2 phenotype with few exceptions.
The Role of Host-Derived H2S in Microbial Infections
A large body of literature is available on the chemical biology of H2S and the role of H2S in diverse biological systems. However, studies on the role of H2S in microbial infection are limited. This section focuses on the contribution of H2S to the host response against bacterial and viral infections.
Host-Derived H2S in Response to Bacterial Infections
Apart from studies aimed at elucidating the role of host-derived H2S in Mtb infection (discussed below in Role of Host H2S in Tuberculosis), little is known about the role of host-generated H2S in modulating the course of bacterial infections. However, studies using mouse models of septic shock have examined the role of H2S in response to lipopolysaccharide (LPS), an inflammatory cell wall component of Gram-negative bacteria. Notably, IP injection of LPS in mice increased CSE expression in liver and kidney resulting in increased levels of H2S in tissues and serum (Li et al., 2005). Lung sections of these LPS-treated animals exhibited characteristic signs of inflammatory damage, including interstitial edema, alveolar thickening, and the presence of numerous leukocytes in both the interstitium and alveoli. Inhibition of CSE enzyme activity with PAG triggered an anti-inflammatory effect in LPS-injected animals, thus providing indirect evidence that H2S exerts a pro-inflammatory effect in this model (Li et al., 2005). Another study performed in human macrophages shows that inhibition of NF-κB and ERK prevented LPS-induced increases in H2S, suggesting that H2S acts as an inflammatory mediator via the NF-κB/ERK pathway in macrophages (Badiei et al., 2015). A study by Ahmad et al., using mice deficient in H2S production (Cbs+/−, Cse−/−, and Δ3-Mst mice that exhibit decreased 3-MST expression in the lung and spleen) showed differential cytokine responses compared to WT following LPS injection. Plasma levels of multiple cytokines including TNFα, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12, and IFN-γ were increased in WT mice upon LPS treatment whereas all three H2S-deficient mice showed decreased plasma levels of TNF-α, IL-10, IL-12, and IFN-γ. On the other hand, plasma levels of IL-5 and GM-CSF were increased in H2S-deficient mice while the levels of IL-1β, IL-2, IL-4, and IL-6 were similar to WT mice (Ahmad et al., 2016). However, survival of all three H2S-deficient mice following LPS administration was the same as WT mice.
In the case of Mycoplasma fermentans infection of macrophages, it was shown that H2S inhibits the activation and nuclear translocation of NF-κB, reducing the transcription of pro-inflammatory genes, including MCP-1 (Benedetti et al., 2014). Moreover, M. fermentans infection enhances Nrf2 functions by activating downstream enzymes including HO-1 and SOD1, and by decreasing intracellular ROI levels (Benedetti et al., 2017). H2S also inhibits Keap1 by persulfidation at Cys151, which allows nuclear translocation of Nrf2 and transcriptional activation of cytoprotective genes via binding to an antioxidant/electrophile response element (ARE) in target gene promoters (Hourihan et al., 2013). Persulfidation of Keap1 is important in protecting the host against oxidative stress and cellular senescence, which are generally observed in viral or bacterial infection (Hourihan et al., 2013). Overall, the literature suggests that H2S acts by modulating cytokine responses and by modifying host transcription factors to promote bacterial clearance.
Host H2S in Response to Viral Infections
Several studies reported that H2S has important antiviral and anti-inflammatory activity in respiratory syncytial virus (RSV) infection, since the virus reduced the expression levels of H2S-producing enzymes (Li et al., 2015; Ivanciuc et al., 2016). Intranasal delivery of GYY4137 to RSV-infected mice significantly reduced viral replication and markedly improved clinical disease parameters and pulmonary dysfunction. Similarly, Cse−/− mice showed significantly worse RSV-induced lung disease and increased viral replication compared to WT mice (Ivanciuc et al., 2016). RSV infection of A549 cells leads to activation of two transcription factors, NF-κB and IRF-3. Addition of exogenous H2S to RSV-infected cells leads to a decrease in the levels of these transcription factors, suggesting a role for H2S in regulating NF-κB and IRF-3 (Li et al., 2015). Exogenous H2S using GYY4137 significantly reduced RSV replication in vitro as well as in vivo by targeting viral assembly, replication, and virion release. PAG treatment led to an increase in RSV-induced cytokines IL-6, IL-8, IL-10, and chemokine MIP-1β, and also increased viral replication (Li et al., 2015). A similar effect of H2S was observed during infection with other members of the paramyxoviridae family; Nipah virus (NiV-B) and human metapneumovirus (hMPV) (Ivanciuc et al., 2016). The replication of other pathogenic enveloped RNA viruses from the Orthomyxo-, Filo-, Flavi-, and Bunyavirus families has also been shown to be affected by H2S (Bazhanov et al., 2017b). This same group showed that GYY4137 treatment led to decreased expression of viral proteins and mRNA, suggesting inhibition of an early step of replication. It was reported that the antiviral activity of H2S was due to a decrease of the virus-induced pro-inflammatory mediators NF-κB and IFN-γ (Bazhanov et al., 2017b).
Studies have established a link between H2S and Coxsackie virus B3 (CVB3) infection (Hua et al., 2009; Wu et al., 2015). CVB3 infects cardiomyocytes resulting in immune cell infiltration and cardiac remodeling, which can eventually result in heart failure (Wu et al., 2015). Administration of GYY4137 to CVB3-infected cardiomyocytes suppressed secretion of pro-inflammatory cytokines TNF-α, IL1β, and IL-6 (Wu et al., 2015). Similar to RSV infection, in the case of CVB3, exogenous H2S treatment inhibited NF-κB signaling by reducing degradation of IκBα. CVB3 infection leads to induction of MAPK signaling cascade by activating ERK1/2, p38, and JNK1/2. Exogenous administration of GYY4137 resulted in reduced induction of these pathways, suggesting an anti-inflammatory role for H2S in CVB3 infection (Wu et al., 2015).
The coronavirus disease 2019 (COVID-19) pandemic resulting from severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection is a global health emergency that has led to over 1 million deaths in over two hundred countries as of this writing (WHO, 2020). COVID-19 pathology is variable and patients exhibit a wide range of clinical symptoms (Esakandari et al., 2020; Zhou et al., 2020). COVID-19-related severe respiratory failure is characterized in part by increased numbers of neutrophils and lower numbers of lymphocytes (CD4, CD8, and CD19), as well as increased levels of serum IL-6 and C-reactive protein (CRP) (Giamarellos-Bourboulis et al., 2020; Zhou et al., 2020). It has been speculated that H2S may play a protective role against COVID-19-mediated pathology through several mechanisms: firstly, by altering the function or expression levels of angiotensin converting enzyme 2 (ACE2) and transmembrane protease serine 2 (TMPRSS2) receptors to impede SARS-CoV-2 entry into host cells. Secondly, by inhibiting viral replication through attenuation of virion assembly or release, and thirdly, by suppressing pulmonary immune responses and inflammation (Yang, 2020). A study of 74 COVID-19 patients reported that surviving patients had significantly higher H2S levels than non-survivors, and that serum H2S levels negatively correlated with serum IL-6, CRP, and procalcitonin (markers for bacterial infection, sepsis, and tissue injury) (Renieris et al., 2020). However, these findings should be interpreted with caution since the levels of H2S, which were measured by monobromobimane (MBB) followed by high-performance liquid chromatography, were unphysiologically high (Olson et al., 2014).
Altogether, the available information indicates that during viral infection, H2S modulates NF-κB signaling, which in turn can reduce the virus-induced pro-inflammatory response and improve host survival. Hence, these findings point toward H2S donors as potential therapeutics against viral infections.
Host H2S and Sepsis
Severe sepsis and septic shock are a leading causes of mortality in intensive care units (Medam et al., 2017). Sepsis is caused by a severe systemic infection due to the presence of bacteria or their toxins in blood or tissue, which frequently occurs after hemorrhage, trauma, burn, or abdominal surgery. Studies using mouse models of sepsis [e.g., cecal ligation and puncture (CLP)] show increased expression of CSE in the liver and higher plasma H2S levels upon induction of sepsis (Zhang et al., 2006). Inhibition of H2S production with PAG significantly decreased sepsis-induced systemic inflammation. PAG pre-treatment also considerably reduced the phosphorylation of ERK1/2 in lung and liver 4 h after CLP, coupled with reduced degradation of IκBα and reduced NF-κB activation (Zhang et al., 2008).
H2S also contributes to neurogenic inflammation in the respiratory tract via induction of Substance P (SP) (Zhang et al., 2007a). SP is implicated in inducing the release of pro-inflammatory mediators, enhanced lymphocyte proliferation, and stimulating chemotaxis of lymphocytes, monocytes, and neutrophils via activation of the neurokinin-1 receptor (O’Connor et al., 2004). H2S was also shown to induce systemic inflammation and multiple organ damage characteristic of sepsis via transient-receptor potential-vanilloid-type-1-(TRPV1)-mediated neurogenic inflammation (Ang et al., 2010). The effect of TRPV1 was shown to be mediated through the enhancement of SP production and activation of the ERK/NF-κB pathway (Ang et al., 2011a).
Recent studies have shown that sepsis activates macrophages to produce PGE2, a potent inflammatory mediator, via induction of cyclooxygenase-2 (COX-2), which is important to evoke acute lung injury. H2S upregulates COX-2 and prostaglandin E metabolite (PGEM) in sepsis through the TRPV1 channel. Importantly, inhibition of COX-2 with parecoxib, a potent and selective COX-2 inhibitor, prevents H2S from exacerbating acute lung injury (ALI) and CLP-induced mortality in sepsis. Administration of H2S using NaHS further enhanced the biosynthesis of COX-2 and PGEM, whereas PAG significantly reduced these effects, which improved recovery of the septic injury (Ang et al., 2011b). Delivery of NaHS to sepsis-induced mice leads to a marked increase in lung pro-inflammatory cytokines TNF-α, IL-1β, IL-6, chemokines MIP-1α and MIP-2, and adhesion molecules P-selectin, E-selectin, VCAM-1, and ICAM-1 which affect inflammation and immune cell migration. The levels of cytokine and cell adhesion molecules were reduced upon treatment with parecoxib, which suggests that H2S can act via COX-2 (Ang et al., 2011b).
Other than affecting immune pathways, sepsis is also known to cause mitochondrial dysfunction (Brealey et al., 2002). In the pneumosepsis model, i.e., sepsis induced by Streptococcus pneumonia, it was reported that NaHS infusion reduced local and distant organ injury, which was associated with maintaining mitochondrial function and improved mitochondrial biogenesis (Aslami et al., 2013). In this study, rats were challenged with live S. pneumonia and infused with NaHS to study its effects on metabolism and bioenergetics. Infusion of NaHS reduced heart rate and body temperature, indicative of a hypo–metabolic state. NaHS infusion also reduced sepsis–related lung and kidney injury, increased expression of α–tubulin and protein kinase C-ϵ, which act as regulators of respiration (Rostovtseva et al., 2008; Nowak and Bakajsova-Takacsova, 2018), and help to prevent mitochondrial membrane damage during sepsis (Aslami et al., 2013).
In summary, the evidence presented here indicates that host-derived H2S often plays a pro-inflammatory role in animal models of sepsis, acting via SP, NK1R, or TRPV1. The activation of NK1R or TRPV1 induces systemic inflammatory response via the ERK-NF-κB pathway to induce an exaggerated immune response. In this regard, agents that attenuate H2S production and/or H2S-induced downstream signals improved outcomes. However, in cases where H2S is clearly anti-inflammatory and/or acts to prevent the loss of, or re-establish, host bioenergetic health by upregulating regulators of respiration like α–tubulin and protein kinase C-ϵ, the use of H2S donor compounds should be evaluated further.
Role of Host H2S in Tuberculosis
Mtb is the etiological agent of pulmonary TB. In the lung, Mtb is phagocytosed by alveolar macrophages, where it subverts host killing mechanisms and innate immune responses that promote its survival and dissemination. Importantly, recent studies have established that the metabolic state of innate immune cells and the efficacy of their immune response are interrelated (Gleeson and Sheedy, 2016; O’Neill et al., 2016; Kumar et al., 2019; Van den Bossche et al., 2017). The gasotransmitters •NO and CO, in addition to more recently appreciated H2S, play a vital role in modulating central energy metabolism and effector functions of immune cells. Further, •NO and CO, produced by nitric oxide synthase II (iNOS) and heme oxygenase 1 (HO-1), respectively, are important host factors that can alter Mtb survival and TB disease progression (MacMicking et al., 1997a; MacMicking et al., 1997b; Kumar et al., 2007; Kumar et al., 2008; Chinta et al., 2016; Chinta et al., 2018). Despite functions that overlap with •NO and CO, and its involvement in pathophysiological processes, the role of host-generated H2S in bacterial pathogenesis has been largely overlooked. Inflammatory responses (Zumla et al., 2015; Liu Q.Y. et al., 2018) and metabolic reprogramming of innate and adaptive immune cells are associated with the progression of pulmonary TB (Shi et al., 2016; Kumar et al., 2019). Notably, similar pathophysiological roles are also associated with the effector functions of H2S, depending on concentration and cell type (Bhatia et al., 2005; Zhang et al., 2006; Zhang J. et al., 2010; Miller et al., 2012; Bhatia, 2015; Gaddam et al., 2016). Recent studies have provided insight into the role of host-derived H2S in affecting pathological and immunometabolic aspects of TB (Rahman et al., 2020; Saini et al., 2020). The role of the H2S-producing enzymes CBS and CSE in TB are described in more detail below.
H2S-Producing Enzymes in Human Tuberculous Lungs
Immunohistochemical (IHC) analysis of lesioned and uninvolved human tuberculous lung tissue suggests that CBS is either absent or expressed at undetectable levels. However, variable expression of CSE and 3-MST was observed in TB lungs and depended on the cellular composition around the necrotic and non-necrotic granulomas (Rahman et al., 2020). In short, robust CSE staining was seen in myofibroblasts, epithelioid histiocytes (activated macrophages), giant cells, and other immune cells in the non-necrotic and the granulomatous inflammation layer of the necrotic granuloma. Also, vascular mural smooth muscles cells showed strong CSE staining. Alveolar pneumocytes and bronchiolar epithelial cells exhibited strong 3-MST staining. The bronchiolar epithelium was also weakly positive for CSE. In most CSE-positive cells, CSE was localized to cytoplasmic and nuclear compartments, but unlike CSE, 3-MST was seen mostly in the cytosol. Neither CSE nor 3-MST staining was noted in the adluminal necrotic components of the cavity wall or the central necrotic component of the granuloma. In healthy lung tissue, intense staining of CSE and weaker 3-MST staining was observed in alveolar pneumocytes, respiratory and terminal bronchiolar epithelium, circulating monocytes, scattered desquamated epithelial cells, and in vascular smooth muscle, while no CBS-positive cells were observed. Overall, the number of cells staining positive for CSE and 3-MST around TB lesions was markedly increased compared to healthy lung tissue, suggesting that increased H2S production is a host response to Mtb infection (Rahman et al., 2020).
Role of CSE and CBS in Tuberculosis
Detailed characterization of TB pathology was performed in separate studies employing Cse knock-out (Cse−/−) or Cbs heterozygous knockout (Cbs+/−) mice. Since Cbs−/− mice exhibit early lethality, Cbs+/− mice were used which have a normal life span and ~50% reduction in CBS levels and H2S production (Watanabe et al., 1995; Saini et al., 2020). The course of TB disease was altered in both H2S-deficient mouse models, based on the following observations: i) Mtb-infected Cse−/− and Cbs+/− mice survived significantly longer than WT mice, ii) the Mtb organ burden in the lungs, spleens and livers of Cse−/− and Cbs+/− mice was significantly lower than in WT controls, iii) histopathological analysis of the lungs revealed that Cse−/− and Cbs+/− mice had less consolidated tissue than WT mice over time, and that these mice also had fewer and smaller granulomatous lesions than corresponding WT mice. Moreover, more bacilli were observed in the acid-fast stained lung sections in the WT mice than in the Cse−/−, providing additional evidence for increased CFU in the WT mice. Lastly, iv) Mtb infection led to an increase in CBS and CSE protein levels in the lungs of Cbs+/− and Cse−/− mice as well as WT controls (Rahman et al., 2020; Saini et al., 2020) (Figure 5). Taken together, these data indicate that CSE and CBS exacerbate TB pathology.
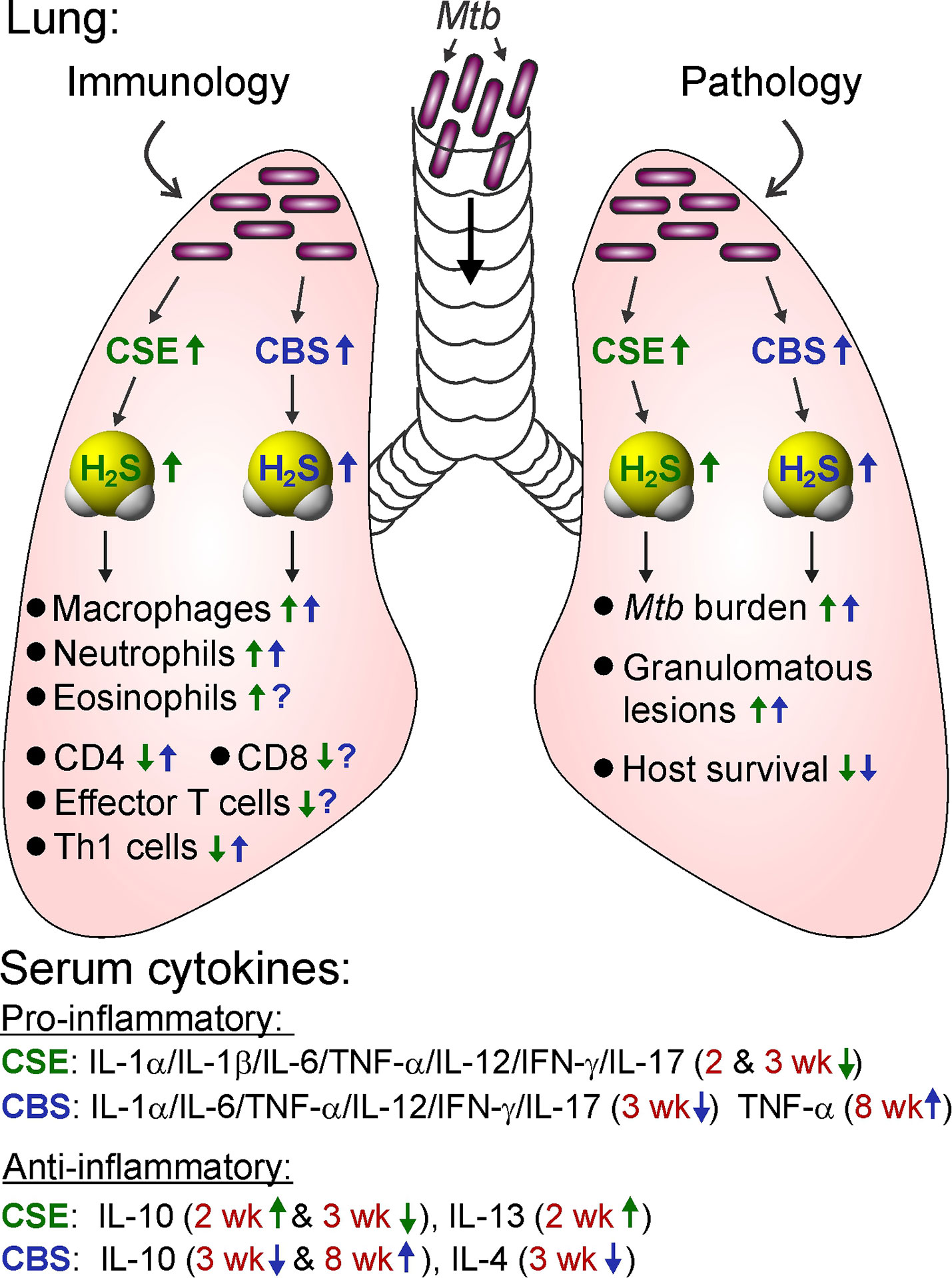
Figure 5 Role of CSE and CBS in the mouse model of TB. Mtb infection of the lung increases CSE and CBS levels, which increases the number of innate immune cells (macrophages, neutrophils, and eosinophils), regulates adaptive immune cells (CD4, CD8, effector T cells—IFNγ+Th1 cells) and serum cytokines. Increased CSE and CBS levels are associated with increased H2S production leading to increased Mtb burden, more granulomatous lesions, and reduced survival. Blue and green text and arrows indicate the regulation caused by CBS and CSE, respectively. Arrows indicate an H2S-mediated increase (up) or decrease (down) in the activity associated with TB disease. Question marks (?) indicate that the role of CBS is unknown. wk, observed time points in weeks.
Immunological Characterization of Cse−/− and Cbs+/− Mice
As discussed above, CSE and CBS contribute to Mtb growth and disease progression. The difference in disease severity between WT and Cse−/− or Cbs+/− mice may result from altered immune responses. Immunological characterization showed no significant differences in the percentages of myeloid or lymphoid cell types in the lungs of Cbs+/− and WT mice prior to, or 3 weeks after infection with Mtb. However, 8 weeks after infection, WT mice exhibited increased percentages of neutrophils, macrophages, CD4+ cells, and CD4+IFN-γ+ cells in the lung compared to Cbs+/− mice (Figure 5). These data suggested that the increase in bacillary burden and decreased survival in WT mice may not result from immunological differences, but rather from the effects of host-derived H2S on Mtb (Saini et al., 2020). Unlike CBS, CSE was shown to contribute to immune dysregulation by promoting an excessive innate immune response and suppressing adaptive immune responses to Mtb infection in the lung (Rahman et al., 2020). The contributions of CSE and CBS to immune regulation in the context of Mtb infection are discussed in more detail below.
H2S and Regulation of Innate Immunity
Significantly increased numbers of alveolar macrophages, neutrophils, and eosinophils were present in the lungs of uninfected Cse−/− mice versus WT (Rahman et al., 2020). However, these differences were reversed after Mtb infection, when significantly more alveolar macrophages, neutrophils, and eosinophils were observed in WT lungs compared to those of Cse−/− mice (Figure 5). These observations suggested that CSE in the WT infected mice promotes an excessive innate immune response, which is consistent with other TB studies that show increased neutrophils can exacerbate TB disease (Nandi and Behar, 2011; Lowe et al., 2012) and that H2S can initiate neutrophil infiltration during Mtb infection (Rahman et al., 2020; Saini et al., 2020) and septic shock (Ang et al., 2011b).
H2S and Regulation of Adaptive Immunity
The lungs of uninfected Cse−/− mice had more CD8+ T cells, but not CD4+ T cells, compared to WT mice (Rahman et al., 2020). At 2- and 6-weeks post-infection, the number of CD4+ and CD8+ T cells and their effector memory populations (CD62LloCD44hi) in the lungs were higher in Cse−/− mice than in WT (Figure 5). Hence, the presence of CSE leads to suppressed adaptive immune response to Mtb infection. The protective role of innate and adaptive immunity to control Mtb infection has been discussed in several reviews (Mogues et al., 2001; Zuniga et al., 2012; O’Garra et al., 2013; Sia and Rengarajan, 2019). Further, examination of regulatory T cells (Tregs, CD4+CD25+) in uninfected mice revealed more cells in WT compared to Cse−/− mice, and no differences in the number of CD8+ Tregs. However, after Mtb infection, the number of CD25+FoxP3− cells was increased in Cse−/− mice compared to WT in both CD4+ and CD8+ T populations. The number of CD4+CD25+FoxP3+ Tregs was lower in Cse−/− mice lungs at 2 weeks post-infection compared to WT, which was reversed after 4 weeks of infection, suggesting that Cse−/− mice can better control pro-inflammatory immune responses at later time points. While a strong immune response resulting from increased innate and adaptive immune cells may be required to control Mtb infection and disease progression, excessive inflammation is detrimental to the host at later stages of infection (Cardona and Cardona, 2019; Okeke and Uzonna, 2019). Tregs are important for controlling pathogen-induced inflammatory responses such as neutrophil activity and T cell proliferation caused by both innate and adaptive immune cells (Cardona and Cardona, 2019; Okeke and Uzonna, 2019). Therefore, a balance between immunosuppressive Tregs and effector T cells, in this case Th1 and Th17 responses against TB, is necessary to simultaneously control Mtb growth and protect host tissue from inflammation-mediated damage (Cardona and Cardona, 2019; Okeke and Uzonna, 2019). Interestingly, in CD4+ and CD8+ T cell populations, the number of IFN-γ+ cells was greater in uninfected WT mice. However, this was reversed upon Mtb infection where Cse−/− mice had significantly more CD4+IFN-γ+ and CD8+IFN-γ+ T cells than the WT (Figure 5). Increased numbers of IFN-γ–producing T (Th1) cells in Cse−/− mice were reflected by the increased number of effector memory T cells, decreased levels of neutrophils (Nandi and Behar, 2011; Rahman et al., 2020), and the subsequent control of Mtb growth in vivo (Rahman et al., 2020).
H2S and Serum Cytokines
Measurement of serum cytokine levels revealed that Cse−/− mice had elevated levels of pro-inflammatory cytokines IL-1α, IL-1β, IL-6, TNF-α, IL-12, IFN-γ, and IL-17, well known for controlling Mtb infection (Zuniga et al., 2012; O’Garra et al., 2013; Domingo-Gonzalez et al., 2016; Sia and Rengarajan, 2019) at 2 or 3 weeks after Mtb infection, compared to WT controls (Rahman et al., 2020). Further, Cse−/− mice had lower levels of anti-inflammatory cytokines IL-10 and IL-13 at 2 weeks post-infection, which was followed by a significant increase in IL-10 levels after 3 weeks of infection. These data suggest precise control of the pro-inflammatory response in Cse−/− mice. Similarly, elevated levels of pro-inflammatory cytokines TNF-α, IL-1α, IL-6, IL-12, IL-17, and IFN-γ were observed in the serum of Mtb-infected Cbs+/− mice compared to WT mice (Saini et al., 2020). The efficient induction of Th1 immunity is critical for defense against Mtb. Understandably, defects in Th1 cytokine (IFN-γ) production are established risk factors for Mtb infection and disease progression in humans and mice (Rossouw et al., 2003; O’Garra et al., 2013; Sia and Rengarajan, 2019). While IFN-γ is pro-inflammatory at the onset of Mtb infection and critical for macrophage activation, it also exerts homeostatic functions that help minimize inflammation-mediated lung damage by limiting neutrophil accumulation (Hu and Ivashkiv, 2009; Nandi and Behar, 2011). Mice that lack IFN-γ are extremely susceptible to Mtb infection due primarily to the inability to activate macrophages, and form large necrotic pulmonary lesions within weeks of Mtb infection (Cooper et al., 1993; Pearl et al., 2001). In agreement with the role of IFN-γ in limiting neutrophil recruitment, increased CSE and CBS levels (which correlate with reduced IFNγ) were associated with neutrophil accumulation and inflammation in infected lungs, leading to progressive Mtb growth and disease progression (Rahman et al., 2020; Saini et al., 2020). However, Cse−/− and Cbs+/− mice are more efficient in controlling Mtb infection due to an increased adaptive response, particularly IFN-γ and IL-17 production (Rahman et al., 2020; Saini et al., 2020). In sum, upregulation of the H2S producing enzymes CSE and CBS in TB is associated with reduced production of pro-inflammatory cytokines and increased levels of anti-inflammatory cytokines, consistent with increased bacterial growth and more severe pathology (Figure 5).
H2S Promotes Mtb Growth by Suppressing Pro-Inflammatory Cytokines
In the context of Mtb infection, H2S was shown to be the CSE- and CBS-generated effector molecule in murine peritoneal macrophages. Mtb infection of murine macrophages led to increased expression of CBS as early as 6 h post-infection (Saini et al., 2020). Similarly, increased CSE and CBS expression was observed 24 h after Mtb infection of macrophages (Rahman et al., 2020), which directly contributed to increased H2S production. Cysteine is used by CSE and CBS to produce H2S. Increasing the cysteine concentration in the culture media of murine macrophages increased H2S production, whereas addition of PAG reduced H2S production. Consistent with this, Mtb growth was reduced in Cse−/− and Cbs+/− macrophages compared to WT controls. Addition of GYY4137 to Cse−/− and Cbs+/− macrophages promoted Mtb growth to the level of WT, whereas addition of PAG or AOAA to WT macrophages reduced Mtb growth to the level of Cse−/− and Cbs+/− macrophages (Figure 6) (Rahman et al., 2020; Saini et al., 2020). These data indicate that H2S is the CBS- and CSE-related effector responsible for Mtb growth. Also, production of pro-inflammatory cytokines IL-1β and IL-6 was higher in Mtb-infected Cse−/− macrophages compared to WT, which was further decreased by treatment with GYY4137. Mtb infection of Cse−/− macrophages significantly increased the percentage of IL-12-positive cells compared to WT. This result was notable since IL-12 links the innate to adaptive immune responses and is involved in the differentiation of Th1 cells for IFN-γ production to control Mtb growth and its dissemination (Cooper et al., 1993; Cooper et al., 1997; Sakai et al., 2016).
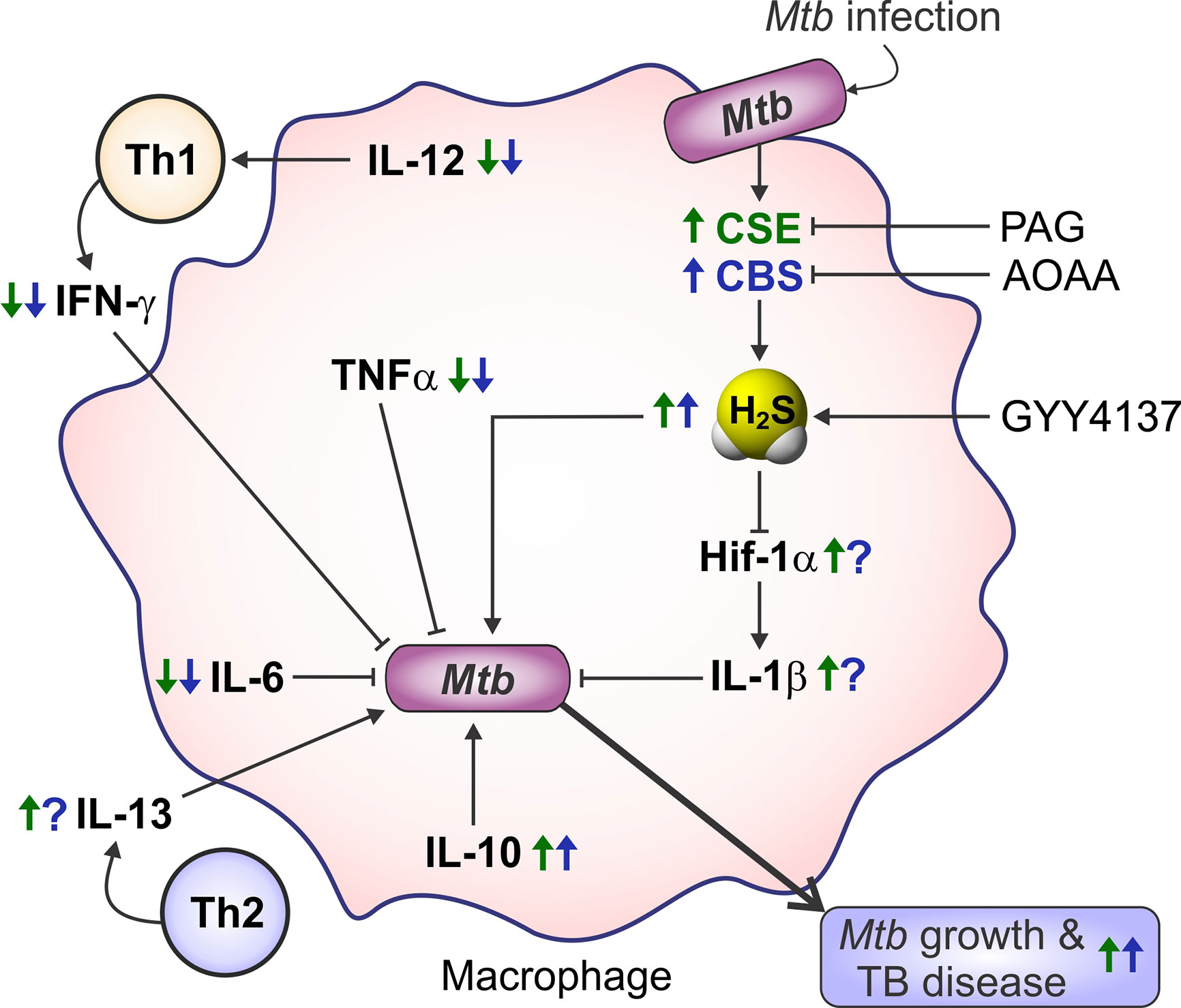
Figure 6 Immune regulation by H2S in Mtb-infected macrophages. Mtb infection increases H2S levels via increased expression of CSE and CBS. Hif-1α promotes the production of pro-inflammatory cytokines IL-1β, TNF-α, IL-6, and IL-12 which are critical for limiting Mtb growth. CSE/H2S reduces Hif-1α levels, thereby reducing the level of pro-inflammatory cytokines, and upregulates the anti-inflammatory Th2 cytokines IL-10 and IL-13, which support intracellular Mtb growth and exacerbate TB pathogenesis. PAG—a specific inhibitor of CSE; AOAA—CBS inhibitor; GYY4137—a slow releaser of H2S. Blue and green arrows indicate the molecular regulation caused by CBS and CSE, respectively. Arrows indicate an H2S-mediated increase (up) or decrease (down) in the activity associated with TB disease. Question marks (?) indicate that the role of CBS is unknown.
Host H2S Suppresses Glycolysis and Oxygen Consumption in Macrophages
H2S is known to modulate glycolysis and mitochondrial respiration (Fu et al., 2012; Modis et al., 2014; Szabo et al., 2014; Liang et al., 2015). The role of host-derived H2S in macrophage energy metabolism was determined using an Agilent Seahorse XF96 flux analyzer to measure the extracellular acidification rate (ECAR) to examine glycolytic functions, and the oxygen consumption rate (OCR) to examine OXPHOS of macrophages (Cumming et al., 2018; Russell et al., 2019). Mtb-infected Cse−/− macrophages exhibited increased glycolysis and a higher glycolytic capacity than WT macrophages (Rahman et al., 2020). These observations are consistent with the notion that H2S production in WT macrophages decelerates glycolysis, making them less capable of controlling Mtb growth and disease progression, which is consistent with recent studies (Cumming et al., 2018; Hackett et al., 2020). Further, OCR, an indicator of cellular respiration, was measured in WT and Cse−/− macrophages. The OCR, basal respiration, and ATP production were increased in uninfected and Mtb-infected Cse−/− macrophages compared to WT controls. Of note, addition of GYY4137 to Cse−/− macrophages reduced the OCR for basal respiration, ATP production, and non-mitochondrial respiration to WT levels. Conversely, addition of the CSE inhibitor PAG to infected WT macrophages increased the basal respiration and non-mitochondrial respiration similar to that of Cse−/− cells. These data clearly show that CSE-generated H2S can modulate energy metabolism in macrophages by suppressing glycolysis and mitochondrial respiration (Figure 7), which can alter immune cell function (Rahman et al., 2020).
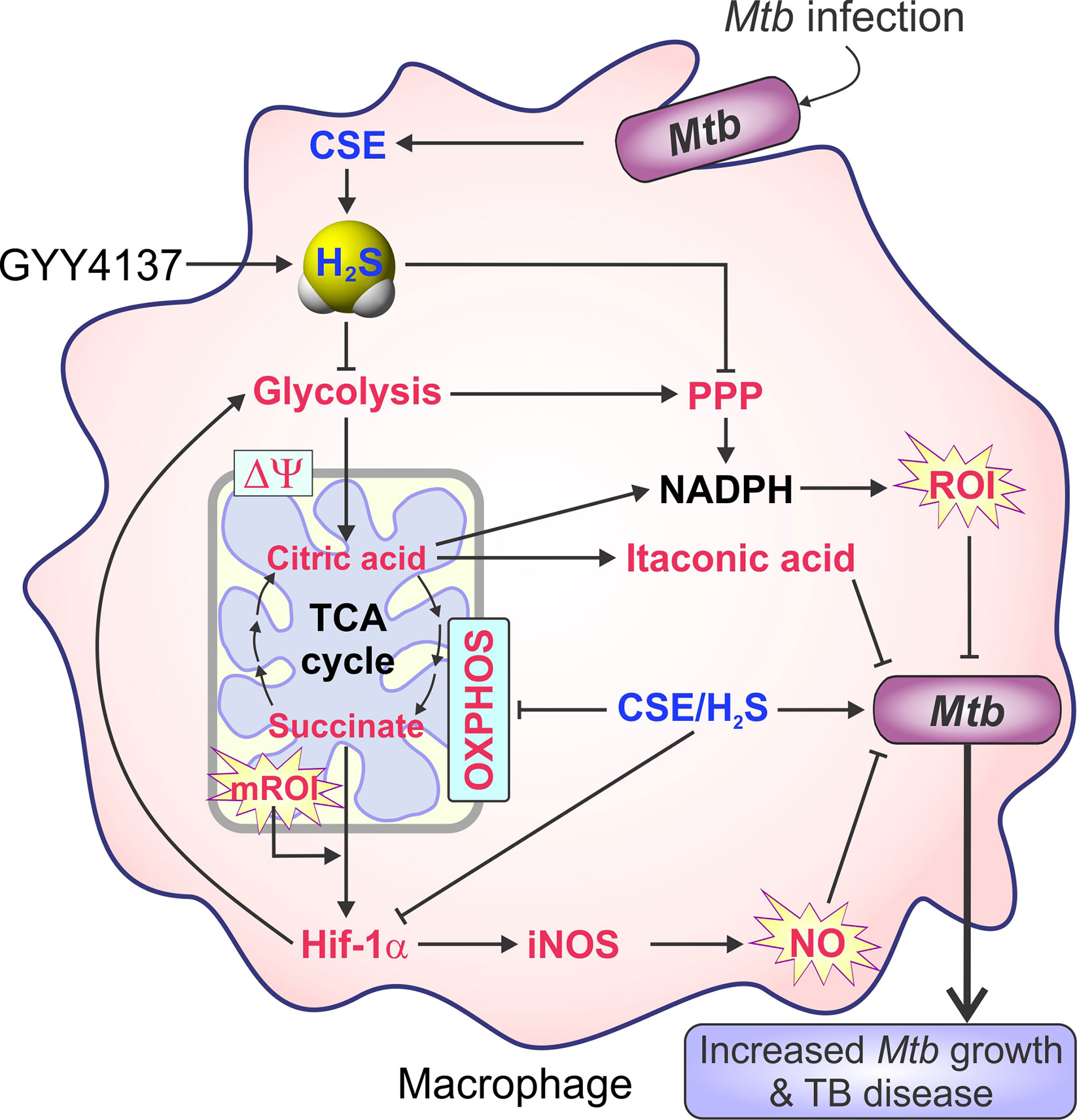
Figure 7 H2S reprograms carbon metabolism in Mtb-infected macrophages. Mtb infection increases H2S production, which suppresses Hif-1α expression followed by reduced glycolytic and PPP, TCA cycle, and oxidative phosphorylation (OXPHOS) activity. Apart from regulating inflammation, Hif-1α also increases the expression of glycolytic enzymes. Hence, Hif-1α links metabolic and immune processes. Molecules in blue text are upregulated, and those in red text are downregulated in Mtb-infected murine macrophages.
H2S Suppresses Central Carbon Metabolism in Macrophages
The bioenergetic differences observed between WT and Cse−/− macrophages suggest that H2S is a regulator of energy metabolism in macrophages. To determine the role of CSE in central carbon metabolism, WT and Cse−/− macrophages were infected with Mtb, with or without GYY4137, and energy-related metabolites were quantified by liquid chromatography–mass spectrometry (LC-MS/MS). This study suggested that excessive H2S produced by upregulation of CSE following Mtb infection suppresses central carbon metabolism (Rahman et al., 2020). For example, while glycolytic metabolites (fructose 6-phosphate, fructose 1,6-bisphosphate, dihydroxyacetone phosphate, phosphoenolpyruvate, pyruvate, and lactate) in uninfected Cse−/− macrophages were moderately increased compared to WT, Mtb infection markedly increased glycolytic metabolites in Cse−/− macrophages. However, addition of GYY4137 to Cse−/− macrophages reduced the levels of these metabolites to that of WT macrophages. Further, a dysregulated TCA cycle with higher levels of citrate and succinate was observed in Cse−/− macrophages, which was restored upon Mtb infection. Lastly, increased levels of α-ketoglutarate and succinate were present in Mtb-infected Cse−/− macrophages, but GYY4137 treatment brought these metabolites down to the level of WT macrophages.
High levels of succinate stabilize HIF-1α, leading to increased expression of IL-1β, a cytokine essential for controlling Mtb growth (Jayaraman et al., 2013; Tannahill et al., 2013). Interestingly, the metabolic phenotype observed in Cse−/− macrophages is characteristic of the metabolic rewiring that occurs in pro-inflammatory macrophages (O’Neill et al., 2016), suggesting that uninfected Cse−/− macrophages are polarized toward a pro-inflammatory phenotype prior to infection. In addition, levels of metabolites in the pentose phosphate pathway (PPP) (ribulose-5-phosphate, sedoheptulose-7-phosphate, and erythrose-4-phosphate) were significantly increased in Mtb-infected Cse−/− macrophages compared to WT. These metabolic differences suggest that increased H2S production in macrophages following Mtb infection suppresses the PPP (Figure 7). Moreover, increased levels of Hif-1α were observed in Mtb-infected Cse−/− macrophages compared to WT controls. Of note, Hif-1α is essential to promote glycolysis and production of pro-inflammatory cytokines like IL-1β, IL-6, IL-12, and IFN-γ (Braverman et al., 2016; Gleeson and Sheedy, 2016; O’Neill et al., 2016; Marks et al., 2017; Wang T. et al., 2017). In different studies, exogenous H2S was shown to increase Hif-1α accumulation under normoxia in Caenorhabditis elegans (Budde and Roth, 2010), or lower Hif-1α levels during hypoxia in cultured cells (Budde and Roth, 2010; Kai et al., 2012; Wu et al., 2012). Overexpression of CSE in HEK293T cells resulted in reduced Hif-1α protein accumulation under hypoxic conditions, whereas under normoxic conditions, addition of 100 μM NaHS to cultures of Hif-1α-overexpressing HEK293T cells also significantly reduced Hif-1α protein levels via inhibition of Hif-1α protein translation (Wu et al., 2012). This indicates that endogenous or exogenous H2S reduces Hif-1α expression irrespective of oxygen tension (Wu et al., 2012). Consistent with this finding, Cse−/− cells showed increased Hif-1α levels after Mtb infection compared to WT cells (Rahman et al., 2020). Overall, these findings suggest that excessive H2S produced by Mtb-infected macrophages suppresses glycolysis and triggers a reduction in Hif-1α levels causing reduced production of pro-inflammatory cytokines and increased bacterial growth.
CSE Suppresses Mitochondrial and Cellular Oxidative Stress
Studies in mice have shown that inhalation of H2S at 80 parts per million reduces inflammation and the formation of reactive oxygen intermediates (ROI) that occurs during ventilator-induced lung injury (Spassov et al., 2017). Since mitochondrial respiration is subject to regulation by H2S, a role for H2S in modulating mitochondrial mass, mitochondrial membrane potential (Δψm), and production of mitochondrial reactive oxygen intermediates (mROI) was investigated during Mtb infection. Mtb infection of WT mouse macrophages significantly decreased mitochondrial mass; however, no change was observed in Cse−/− macrophages (Rahman et al., 2020). Measurement of Δψm revealed that following Mtb infection, overall Δψm was decreased in both Cse−/− and WT macrophages. However, Cse−/− macrophages retained a more polarized Δψm than did WT cells. The observed decrease in Δψm in WT macrophages after Mtb infection is consistent with their lower basal respiration compared to Cse−/− macrophages. Quantitation of mROI revealed that mROI levels in uninfected WT and Cse−/− macrophages were similar; however, Mtb infection lowered mROI levels only in WT cells. Further, cellular reactive nitrogen intermediates (RNI) and cellular ROI were measured. While Mtb infection increased RNI levels in both Cse−/− and WT macrophages, RNI levels in Cse−/− macrophages were consistently higher than in WT macrophages irrespective of infection. The presence of increased RNI/nitric oxide in Cse−/− macrophages is consistent with higher IFN-γ levels seen in Cse−/− macrophages, which can limit Mtb growth. Several studies have demonstrated that at physiological concentrations, H2S can function as an antioxidant by reducing the formation of mROI and RNI (Predmore et al., 2012; Modis et al., 2014; Corsello et al., 2018; Shefa et al., 2018; Xiao et al., 2018). Consistent with this finding, Cse−/− macrophages produce more mROI and cellular RNI following Mtb infection (Figure 7) (Rahman et al., 2020).
H2S and M2 Macrophage Polarization in Tuberculosis
As mentioned above in Role of H2S in Macrophage Polarization, considerable evidence indicates that H2S is involved in macrophage polarization and can drive macrophages toward an anti-inflammatory M2 phenotype (Figure 4). An anti-inflammatory role for H2S was also observed in a recent study that examined the role of H2S in TB using Cse−/− mice and macrophages. For example, infected Cse−/− macrophages, which produce less H2S than infected WT cells, exhibit increased pro-inflammatory cytokine production, increased glycolysis, and flux through the PPP, and higher levels of mROI, iNOS (Nos2), RNI, and Hif-1α expression compared to WT macrophages. This functional profile provided more efficient control of Mtb growth and disease compared to WT macrophages. In contrast, Mtb-infected WT macrophages (which exhibited the highest H2S production) showed increased production of anti-inflammatory cytokines IL-10 and IL-13, and arginase 1/2 (Arg1/2) compared to Cse−/− macrophages. Notably, chemical complementation of Mtb-infected Cse−/− macrophages with GYY4137 produced a functional profile similar to WT cells, characterized by a reduction in pro-inflammatory cytokines IL-1β and IL-6 and reduced levels of glycolytic and PPP intermediates. Overall, these findings indicate that the CSE/H2S axis is a critical determinant of macrophage phenotype in TB disease, and that increased CSE/H2S levels appear to promote M2 type polarization and enhanced TB control (Figure 4) (Rahman et al., 2020).
H2S Stimulates Mtb Growth and Metabolism
H2S is membrane-permeable; therefore, host-derived H2S can influence Mtb residing in host cells. Evidence that H2S directly modulates Mtb growth was provided by addition of GYY4137, a slow releaser of H2S, to mimic host-derived H2S. Addition of low concentrations of GYY4137 (5–25 μM) to Mtb cultures stimulated Mtb growth; however, at higher concentrations growth stimulation was lost, demonstrating a bimodal effect (Saini et al., 2020). Consistent with these observations, Mtb growth was attenuated in Cbs+/− and Cse−/− macrophages, compared to WT, consistent with reduced organ burden and increased survival observed in these mice (Saini et al., 2020; Rahman et al., 2020). Further, addition of GYY4137 to Mtb-infected Cbs+/− and Cse−/− macrophages increased Mtb proliferation versus untreated controls, and addition of the CBS or CSE inhibitors to infected WT macrophages reduced Mtb CFUs to levels seen in Cbs+/− or Cse−/− macrophages respectively (Figure 6). H2S-mediated growth stimulation of Mtb was accompanied by an increased oxygen consumption rate (OCR) and increased intracellular ATP. Further, high-resolution metabolite studies using LC-MS/MS showed that H2S exposure also led to increased levels of glycolytic and TCA cycle metabolic intermediates. The H2S-mediated stimulation of respiration was found to be largely dependent on cytochrome bd-type quinol oxidase (CytBD), as Mtb mutants defective in cytochrome bd oxidase activity showed highly attenuated growth and bioenergetic responses to H2S (Saini et al., 2020). This CytBD-mediated stimulation of respiration in Mtb is consistent with data showing that CytBD in Escherichia coli is resistant to H2S-mediated inhibition of respiration (Forte et al., 2016; Korshunov et al., 2016). Further, H2S was shown to alter gene expression in Mtb. Upon addition of GYY4137, transcriptomic analysis showed upregulation of genes belonging to the DosR/S/T dormancy regulon, CsoR and RicR copper regulons, and several genes involved in sulfur metabolism. In line with these data, H2S supports Mtb entry into in vitro dormancy and recovery from dormancy after reaeration. In addition, Mtb exposed to H2S exhibits enhanced growth under conditions of oxidative stress compared to untreated Mtb, suggesting an antioxidant function for H2S (Saini et al., 2020). Further, a role for H2S in redox balance and antibiotic tolerance in Mtb has been reported (Mishra et al., 2019). Overall, it is reasonable to propose that the presence of H2S at the site of infection can support the growth of Mtb by rewiring central carbon metabolism and improving the bioenergetic health of Mtb (Figure 8).
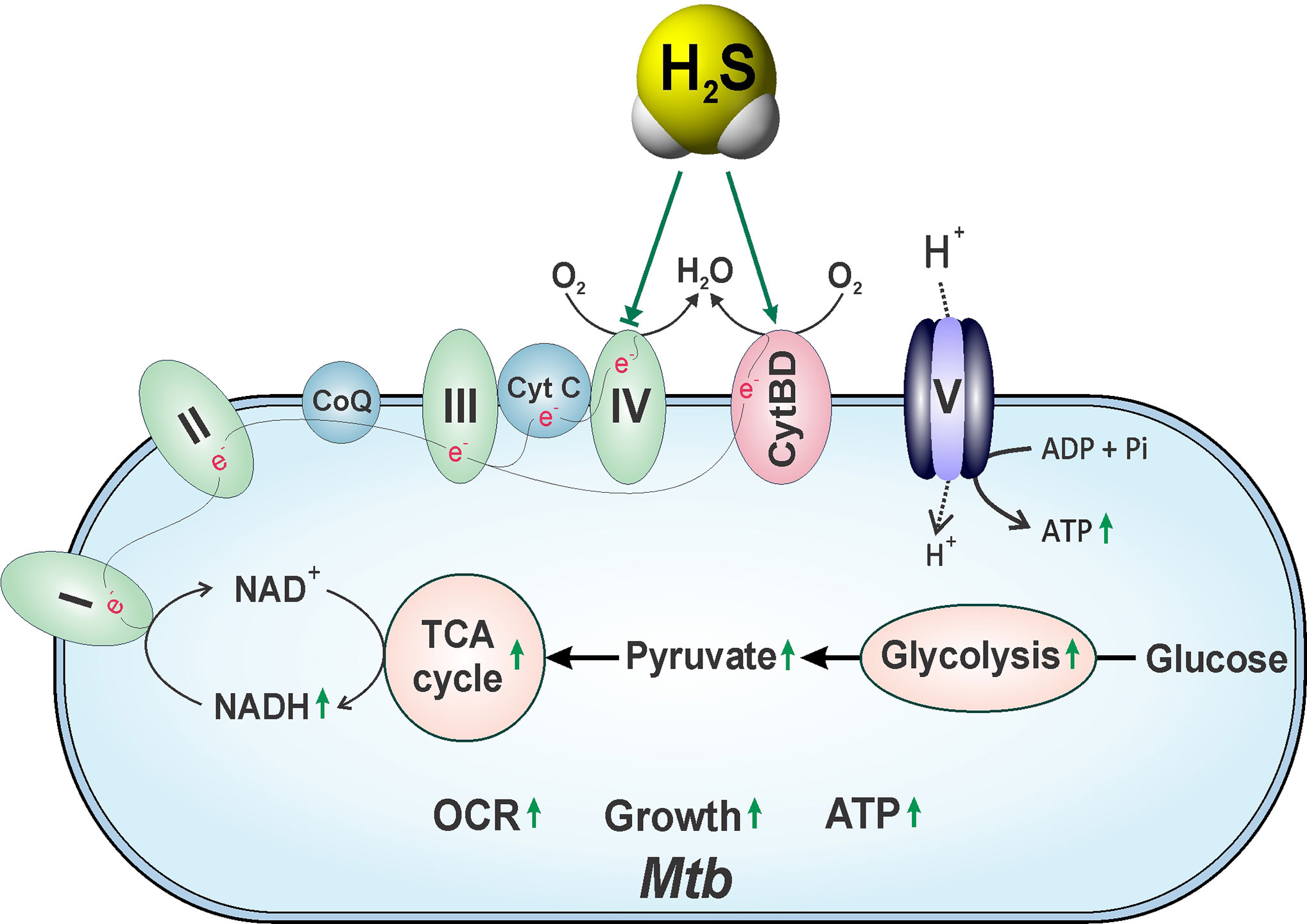
Figure 8 The roles of host-generated H2S in Mtb respiration and growth. Host-generated H2S can stimulate Mtb respiration and bioenergetics predominantly through cytochrome bd oxidase CytBD, thereby increasing bacterial growth and accelerating disease.
Role of Endogenous H2S in Bacterial Physiology
While the focus of this review is to highlight the diverse functions of host-derived H2S in TB and other microbial diseases, a brief overview of the physiological relevance of bacterially-derived H2S is warranted. Unlike H2S in mammals, the production of H2S by bacteria has been known for over 140 years, beginning with the work of French microbiologist Ulysse Gayon in 1877; however, the role of H2S as an important effector molecule in bacterial physiology was not appreciated until the 1960s (Szabo, 2018). Orthologs of mammalian H2S-producing enzymes CBS, CSE, and/or 3-MST are present in most bacterial species, indicating their importance and evolutionary conservation. For example, CBS and CSE activity is the main source of H2S production in Klebsiella pneumoniae, Bacillus anthracis, Pseudomonas aeruginosa, and Staphylococcus aureus, whereas 3-MST is the primary source of H2S production in Escherichia coli (Shatalin et al., 2011; Mironov et al., 2017). Consistent with the presence of bacterial orthologs of CSE and CBS, L-cysteine is also a common substrate for H2S production in many bacteria. Addition of L-cysteine has been shown to stimulate H2S production in B. anthracis, P. aeruginosa, S. aureus, and E. coli (Shatalin et al., 2011; Mironov et al., 2017).
Recent studies have identified specific physiological functions of endogenous H2S in bacteria. A major finding was that H2S can protect bacteria from antibiotics and oxidative stress. Inactivation of CBS and CSE in B. anthracis, P. aeruginosa, and S. aureus or 3-MST in E. coli via genetic deletion or enzymatic inhibitors reduced H2S production by 90% and rendered these pathogens susceptible to gentamycin, ampicillin, and nalidixic acid compared to untreated or WT controls (Shatalin et al., 2011). Further, 3-MST overproduction in E. coli provides increased resistance to spectinomycin. Exogenous H2S (200 μM NaHS) suppressed antibiotic sensitivity in CBS/CSE-double knockout or 3-MST-deficient bacteria. E. coli exhibited increased resistance to gentamycin when cultured in cysteine-rich (0.5 mM) media compared to standard media (<20 μM cysteine) (Shatalin et al., 2011). In contrast, a 3-MST-mutant of E. coli is more sensitive to gentamycin in cysteine-rich media, and was protected by thiol-depleting diamide, suggesting that pro-oxidative cysteine accumulates in this mutant. H2S-deficient bacteria were highly susceptible to peroxide, whereas NaHS protects them from peroxide-generated oxidative stress, indicating that H2S can increase the antioxidant capacity of bacterial cells. In conclusion, H2S provides antibiotic resistance to clinically relevant and evolutionarily distant pathogenic bacteria by alleviating oxidative stress (Shatalin et al., 2011). Another study demonstrated that endogenous H2S produced by 3-MST protects E. coli from oxidative stress and genome damage by lowering cysteine levels and sequestering free iron that drives genotoxic Fenton chemistry that generates hydroxyl radicals (Mironov et al., 2017). Further, 3-MST-derived H2S plays a role in regulating levels of intracellular cysteine, which can be toxic above physiological levels (Mironov et al., 2017). Other reports have shown that exogenous H2S helps E. coli to maintain redox homeostasis and protects bacteria against antibiotic-triggered oxidative stress. Inhibition of H2S biosynthesis reversed antibiotic resistance in multidrug-resistant uropathogenic E. coli, whereas exposure to a bacteria-specific, enzyme-activated H2S donor compound restored drug tolerance (Shukla et al., 2017).
In contrast, a study on Acinetobactor baumannii, a pathogenic bacterium that cannot produce H2S, demonstrated that treatment with exogenous H2S (80–160 μM NaHS) conferred hypersensitivity to various antibiotics (Ng et al., 2020). In fact, exogenous H2S in A. baumannii potentiated the killing effect of antibiotics including gentamycin, colistin, rifampicin, and clarithromycin. Moreover, exposure of a gentamycin-resistant clinical isolate of A. baumannii to NaHS reverted the resistance to gentamycin. H2S exacerbated antibiotic-mediated killing by increasing the Fe2+/Fe3+ ratio (Fe2+ mediates ROI generation via Fenton chemistry), increasing ROI, and by depolarizing the membrane potential which caused a reduction in ATP levels (Ng et al., 2020).
E. coli exposed to 0.5 mM L-cysteine/L-cystine exhibited a transient increased susceptibility to H2O2 resulting in an unusually rapid rate of DNA damage. However, treatment with iron chelators (dipyridyl or desferrioxamine) eliminated this sensitivity, indicating that intracellular free iron mediates the conversion of H2O2 into hydroxyl radicals, the effector of DNA damage (Park and Imlay, 2003). A higher cysteine concentration (10 mM) promoted bacterial respiration and ROI production, and potentiated the killing effect of ampicillin, kanamycin, and ciprofloxacin against bacterial persisters (E. coli, Salmonella enteritidis, A. baumannii, and P. aeruginosa) in the stationary phase, but not in the exponential growth phase, indicating that the synergistic killing effect of cysteine is dependent on the metabolic state of the bacterium (Liu et al., 2020).
Of note, Mtb encodes putative orthologs of CBS (Rv1077), CSE (Rv1079), and 3-MST (Rv2291). Biochemical studies of the Mtb transsulfuration pathway revealed that Rv1079 is a dual-function enzyme with CSE activity (catalyzes cystathionine to cysteine) and cystathionine γ-synthase (CGS) activity (catalyzes O-succinylhomoserine and cysteine to cystathionine) (Wheeler et al., 2005). Interestingly, Rv1079 (CSE/CGS) does not have cysteine desulfhydrase (CDS) activity (CDS converts cysteine to pyruvate, NH3, and H2S) unlike CSE in other organisms including mammals. CDS activity has been observed in WT Mtb as well as a Mtb rv1079 deletion mutant, and this activity was not inhibited by PAG. Hence, the CSD activity in Mtb is catalyzed by an unknown protein(s) using L-cysteine or L-cystine as a substrate (Wheeler et al., 2005). A study showed that the addition of cysteine or other small thiols to either isoniazid (INH) or rifampicin prevents the formation of drug-tolerant and drug-resistant cells in Mtb cultures in a concentration- (8 μM–4 mM) and time-dependent manner (Vilcheze et al., 2017). Moreover, the combination of 4 mM cysteine and INH (7.3 μM, 20 x MIC) sterilized Mtb cultures. The increased killing of INH/cysteine-treated Mtb cultures resulted from increased cellular respiration, ferrous (Fe2+) ion and ROI leading to oxidative stress and DNA damage (Vilcheze et al., 2017). However, this study did not consider a possible role for H2S arising from the CDS activity in Mtb (Wheeler, et al., 2005), which may influence the effects of antibiotics on Mtb.
Finally, to gain insight into the role of bacterial H2S in defense against host immunity, a study using E. coli and S. aureus, common sources of nosocomial infections, showed that endogenous H2S provides resistance against immune-mediated killing (Toliver-Kinsky et al., 2019). This was demonstrated by several observations—1) decreased bacterial killing by mouse leucocytes, when infected with bacteria treated with GYY4137 (0.3 and 1 mM), 2) bacterial clearance from leucocytes or RAW264.7 cells was significantly increased after inhibiting 3-MST in E. coli or CBS/CSE in S. aureus using either an inhibitor or a gene deletion mutant, and 3) Mice infected with H2S-deficient bacteria had a lower burden in the spleen and decreased plasma levels of IL-6 compared to infection with WT bacteria (Toliver-Kinsky et al., 2019). In conclusion, endogenous H2S in bacteria can suppress host immunity and reduce oxidative stress triggered by antibiotics (Figure 9).
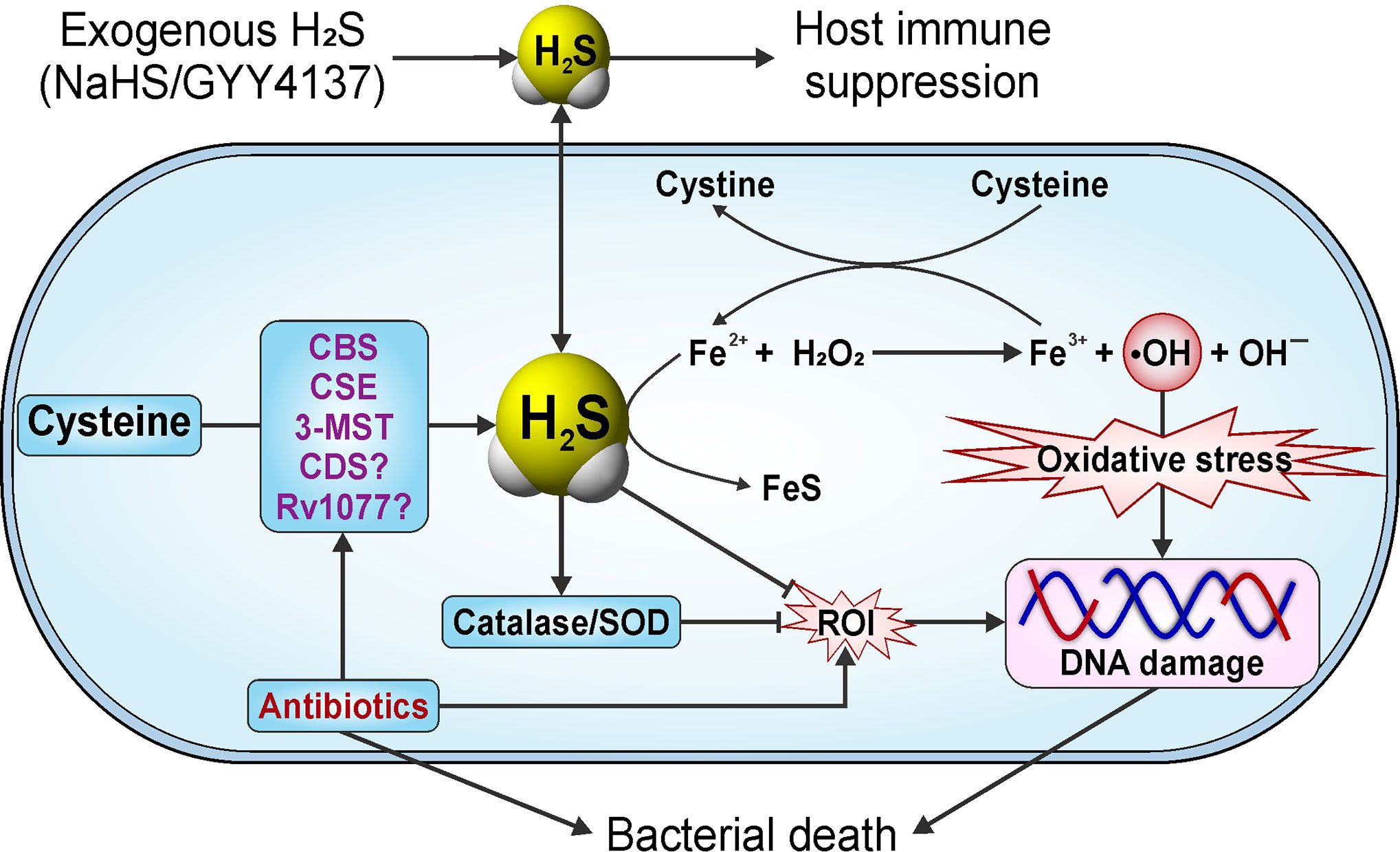
Figure 9 Endogenously produced H2S protects bacteria against oxidative stress. Endogenously produced or exogenous H2S protects bacteria from oxidative stress triggered by antibiotics and/or host against infection. This protection is achieved by multiple mechanisms: i) increased activity of H2S producing enzymes; ii) depletion of excess cysteine (cysteine generates Fe2+ for Fenton chemistry); iii) inhibition of the Fenton reaction, since H2S reacts with H2O2 and free Fe2+); iv) increased activity of catalase and superoxide dismutase (SOD); v) suppression of host immune responses. Question marks (?) indicate that the functions of CDS and Rv1077 in Mtb are unknown.
Conclusion and Future Directions
This review critically summarizes the wide-ranging physiological roles of H2S in mammals and highlights how host-generated H2S impacts Mtb growth, disease progression, and immunometabolism in TB. Despite the documented role of H2S in numerous pathophysiological processes, few studies have attempted to elucidate the role of host-derived H2S in the control of microbial pathogens. The lack of data concerning the intersection of host-derived H2S and infectious disease provides the research community with unique opportunities to make new, innovative contributions to the study of bacterial and viral pathogens. Given the numerous studies that followed the discovery of iNOS (Nos2) and the importance of •NO bioactivity in microbial pathogenesis (MacMicking et al., 1997b; Das et al., 2010), we anticipate a rapid increase in studies that further define the importance of H2S in microbial pathogenesis.
Within the context of TB, several previously unexplored areas of interest have been identified. Firstly, studies have clearly shown that H2S-deficient Cse−/− and Cbs+/- mice have less severe TB disease (Rahman et al., 2020; Saini et al., 2020). This is intriguing, because to the best of our knowledge, the mortality of other knockout mice infected with Mtb has been unchanged or increased compared to wild-type controls. These findings suggest that CBS or CSE are suitable targets for host-directed therapeutic intervention in TB. Since Mtb infection induces excessive production of H2S, which is sensed by Mtb in vivo to promote growth (Saini et al., 2020), and which suppresses host glycolysis (Rahman et al., 2020), it will be important to identify the underlying immunometabolic mechanisms that contribute to disease. For example, how does excessive H2S production following Mtb infection regulate immunity, and how does H2S suppress glycolysis in host cells upon Mtb infection? Secondly, several other lines of investigation may help establish new virulence paradigms. For example, recent reports showing that Mtb suppresses host glycolysis (Cumming et al., 2018; Hackett et al., 2020) are consistent with the demonstration that H2S is the effector molecule that suppresses glycolysis (Rahman et al., 2020) and with studies showing that H2S targets glycolytic enzymes (Guo et al., 2019). Thirdly, while the TB field has benefited greatly from several animal models of TB, no single animal model represents the full histopathological spectrum of human pulmonary TB (Hunter, 2016). Therefore, it is essential that we increase our understanding of the role of H2S-producing enzymes in human pulmonary TB. For example, using flow cytometry, immunohistochemistry, or other novel “-omic” technologies to examine the cellular distribution of H2S-producing enzymes within cavities, necrotic and non-necrotic granulomas of human pulmonary TB (Chinta et al., 2018; Reddy et al., 2018; Rahman et al., 2020) will make important contributions to the field. Although this has been accomplished to a limited extent by a previous study (Rahman et al., 2020), comprehensive characterization of human TB tissues is needed. Also, it is tempting to speculate that exhaled H2S could be used as a volatile biomarker for rapid diagnosis of TB. Fourthly, there is compelling evidence that host-derived H2S is sensed by Mtb to reprogram its metabolism and stimulate growth (Saini et al., 2020). Outstanding questions include: by what mechanism is exogenous H2S sensed by Mtb? How does host-derived H2S reprogram Mtb metabolism to promote virulence and growth? And lastly, how does host-derived H2S modulate Mtb dormancy? Answers to these questions are likely to reveal new insights and establish new paradigms whereby Mtb causes disease. Fifthly, it is difficult to ignore the obvious link between H2S and •NO, as these two gasotransmitters chemically interact with each other (Kolluru et al., 2013) and could possibly regulate the heme prosthetic groups of CBS and iNOS. Of note, CBS- or CSE-generated H2S and iNOS-generated •NO differ substantially in their ability to alter the course of Mtb infection; whereas mice lacking Nos2 are more susceptible to Mtb infection (MacMicking et al., 1997b), Cse−/− and Cbs+/- mice are more resistant to Mtb infection (Rahman et al., 2020; Saini et al., 2020). Hence, it will be important to understand the role of H2S in Nos2-deficient mice, and the role of •NO in Cse−/− and Cbs+/- mice.
In conclusion, a fundamental challenge in the TB field is to improve our understanding of the mechanisms whereby Mtb causes disease. The gasotransmitter H2S plays an unusual role in the control of TB and provides new knowledge which could be exploited for successful TB intervention strategies.
Author Contributions
All authors contributed to the article and approved the submitted version.
Funding
This work was supported by NIH Grants R01AI134810, R01AI137043, R01AI152110, R33AI138280, a Bill and Melinda Gates Foundation Award (OPP1130017), the South African Medical Research Council, and an NRF BRICS Multilateral grant to AJCS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank members of the Steyn lab for critical review of the manuscript.
References
Abe K., Kimura H. (1996). The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 16 (3), 1066–1071. doi: 10.1523/JNEUROSCI.16-03-01066.1996
Ahmad A., Gero D., Olah G., Szabo C. (2016). Effect of endotoxemia in mice genetically deficient in cystathionine-gamma-lyase, cystathionine-beta-synthase or 3-mercaptopyruvate sulfurtransferase. Int. J. Mol. Med. 38 (6), 1683–1692. doi: 10.3892/ijmm.2016.2771
Akaike T., Ida T., Wei F. Y., Nishida M., Kumagai Y., Alam M. M., et al. (2017). Cysteinyl-tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat. Commun. 8 (1), 1177. doi: 10.1038/s41467-017-01311-y
Al-Goblan A. S., Al-Alfi M. A., Khan M. Z. (2014). Mechanism linking diabetes mellitus and obesity. Diabetes Metab. Syndr. Obes. 7, 587–591. doi: 10.2147/DMSO.S67400
Al-Khami A. A., Rodriguez P. C., Ochoa A. C. (2017). Energy metabolic pathways control the fate and function of myeloid immune cells. J. Leukoc. Biol. 102 (2), 369–380. doi: 10.1189/jlb.1VMR1216-535R
Alshorafa A. K., Guo Q., Zeng F., Chen M., Tan G., Tang Z., et al. (2012). Psoriasis is associated with low serum levels of hydrogen sulfide, a potential anti-inflammatory molecule. Tohoku J. Exp. Med. 228 (4), 325–332. doi: 10.1620/tjem.228.325
Alston T. A., Porter D. J., Mela L., Bright H. J. (1980). Inactivation of alanine aminotransferase by the neurotoxin beta-cyano-L-alanine. Biochem. Biophys. Res. Commun. 92 (1), 299–304. doi: 10.1016/0006-291X(80)91552-1
Alvarez L., Bianco C. L., Toscano J. P., Lin J., Akaike T., Fukuto J. M. (2017). Chemical Biology of Hydropersulfides and Related Species: Possible Roles in Cellular Protection and Redox Signaling. Antioxid. Redox Signal 27 (10), 622–633. doi: 10.1089/ars.2017.7081
Ang S. F., Moochhala S. M., Bhatia M. (2010). Hydrogen sulfide promotes transient receptor potential vanilloid 1-mediated neurogenic inflammation in polymicrobial sepsis. Crit. Care Med. 38 (2), 619–628. doi: 10.1097/CCM.0b013e3181c0df00
Ang S. F., Moochhala S. M., MacAry P. A., Bhatia M. (2011a). Hydrogen sulfide and neurogenic inflammation in polymicrobial sepsis: involvement of substance P and ERK-NF-kappaB signaling. PloS One 6 (9), e24535. doi: 10.1371/journal.pone.0024535
Ang S. F., Sio S. W., Moochhala S. M., MacAry P. A., Bhatia M. (2011b). Hydrogen sulfide upregulates cyclooxygenase-2 and prostaglandin E metabolite in sepsis-evoked acute lung injury via transient receptor potential vanilloid type 1 channel activation. J. Immunol. 187 (9), 4778–4787. doi: 10.4049/jimmunol.1101559
Asimakopoulou A., Panopoulos P., Chasapis C. T., Coletta C., Zhou Z., Cirino G., et al. (2013). Selectivity of commonly used pharmacological inhibitors for cystathionine beta synthase (CBS) and cystathionine gamma lyase (CSE). Br. J. Pharmacol. 169 (4), 922–932. doi: 10.1111/bph.12171
Aslami H., Pulskens W. P., Kuipers M. T., Bos A. P., van Kuilenburg A. B., Wanders R. J., et al. (2013). Hydrogen sulfide donor NaHS reduces organ injury in a rat model of pneumococcal pneumosepsis, associated with improved bio-energetic status. PloS One 8 (5), e63497. doi: 10.1371/journal.pone.0063497
Attene-Ramos M. S., Wagner E. D., Gaskins H. R., Plewa M. J. (2007). Hydrogen sulfide induces direct radical-associated DNA damage. Mol. Cancer Res. 5 (5), 455–459. doi: 10.1158/1541-7786.MCR-06-0439
Augsburger F., Randi E. B., Jendly M., Ascencao K., Dilek N., Szabo C. (2020). Role of 3-Mercaptopyruvate Sulfurtransferase in the Regulation of Proliferation, Migration, and Bioenergetics in Murine Colon Cancer Cells. Biomolecules 10 (3), 447. doi: 10.3390/biom10030447
Badiei A., Gieseg S., Davies S., Izani Othman M., Bhatia M. (2015). LPS Up-Regulates Cystathionine gamma -Lyase Gene Expression in Primary Human Macrophages via NF-kappaB/ERK Pathway. Inflammation Allergy Drug Targets 14 (2), 99–104. doi: 10.2174/1871528114666151201201719
Banerjee R., Zou C. G. (2005). Redox regulation and reaction mechanism of human cystathionine-beta-synthase: a PLP-dependent hemesensor protein. Arch. Biochem. Biophys. 433 (1), 144–156. doi: 10.1016/j.abb.2004.08.037
Bao L., Vlcek C., Paces V., Kraus J. P. (1998). Identification and tissue distribution of human cystathionine beta-synthase mRNA isoforms. Arch. Biochem. Biophys. 350 (1), 95–103. doi: 10.1006/abbi.1997.0486
Baskar R., Bian J. (2011). Hydrogen sulfide gas has cell growth regulatory role. Eur. J. Pharmacol. 656 (1-3), 5–9. doi: 10.1016/j.ejphar.2011.01.052
Baskar R., Li L., Moore P. K. (2007). Hydrogen sulfide-induces DNA damage and changes in apoptotic gene expression in human lung fibroblast cells. FASEB J. 21 (1), 247–255. doi: 10.1096/fj.06-6255com
Baskar R., Sparatore A., Del Soldato P., Moore P. K. (2008). Effect of S-diclofenac, a novel hydrogen sulfide releasing derivative inhibit rat vascular smooth muscle cell proliferation. Eur. J. Pharmacol. 594 (1-3), 1–8. doi: 10.1016/j.ejphar.2008.07.029
Bateman A. (1997). The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem. Sci. 22 (1), 12–13. doi: 10.1016/S0968-0004(96)30046-7
Baykov A. A., Tuominen H. K., Lahti R. (2011). The CBS domain: a protein module with an emerging prominent role in regulation. ACS Chem. Biol. 6 (11), 1156–1163. doi: 10.1021/cb200231c
Bazhanov N., Ansar M., Ivanciuc T., Garofalo R. P., Casola A. (2017a). Hydrogen Sulfide: A Novel Player in Airway Development, Pathophysiology of Respiratory Diseases, and Antiviral Defenses. Am. J. Respir. Cell Mol. Biol. 57 (4), 403–410. doi: 10.1165/rcmb.2017-0114TR
Bazhanov N., Escaffre O., Freiberg A. N., Garofalo R. P., Casola A. (2017b). Broad-Range Antiviral Activity of Hydrogen Sulfide Against Highly Pathogenic RNA Viruses. Sci. Rep. 7, 41029. doi: 10.1038/srep41029
Bazhanov N., Ivanciuc T., Wu H., Garofalo M., Kang J., Xian M., et al. (2018). Thiol-Activated Hydrogen Sulfide Donors Antiviral and Anti-Inflammatory Activity in Respiratory Syncytial Virus Infection. Viruses 10 (5), 249. doi: 10.3390/v10050249
Beauchamp R. O. Jr., Bus J. S., Popp J. A., Boreiko C. J., Andjelkovich D. A. (1984). A critical review of the literature on hydrogen sulfide toxicity. Crit. Rev. Toxicol. 13 (1), 25–97. doi: 10.3109/10408448409029321
Benchoam D., Cuevasanta E., Moller M. N., Alvarez B. (2019). Hydrogen Sulfide and Persulfides Oxidation by Biologically Relevant Oxidizing Species. Antioxid. (Basel) 8 (2), 48. doi: 10.3390/antiox8020048
Benedetti F., Davinelli S., Krishnan S., Gallo R. C., Scapagnini G., Zella D., et al. (2014). Sulfur compounds block MCP-1 production by Mycoplasma fermentans-infected macrophages through NF-kappaB inhibition. J. Transl. Med. 12, 145. doi: 10.1186/1479-5876-12-145
Benedetti F., Curreli S., Krishnan S., Davinelli S., Cocchi F., Scapagnini G., et al. (2017). Anti-inflammatory effects of H2S during acute bacterial infection: a review. J. Transl. Med. 15 (1), 100. doi: 10.1186/s12967-017-1206-8
Bhatia M., Wong F. L., Fu D., Lau H. Y., Moochhala S. M., Moore P. K. (2005). Role of hydrogen sulfide in acute pancreatitis and associated lung injury. FASEB J. 19 (6), 623–625. doi: 10.1096/fj.04-3023fje
Bhatia M. (2015). H2S and Inflammation: An Overview. Handb. Exp. Pharmacol. 230, 165–180. doi: 10.1007/978-3-319-18144-8_8
Bhattacharyya S., Saha S., Giri K., Lanza I. R., Nair K. S., Jennings N. B., et al. (2013). Cystathionine beta-synthase (CBS) contributes to advanced ovarian cancer progression and drug resistance. PloS One 8 (11), e79167. doi: 10.1371/journal.pone.0079167
Bhushan S., Kondo K., Polhemus D. J., Otsuka H., Nicholson C. K., Tao Y. X., et al. (2014). Nitrite therapy improves left ventricular function during heart failure via restoration of nitric oxide-mediated cytoprotective signaling. Circ. Res. 114 (8), 1281–1291. doi: 10.1161/CIRCRESAHA.114.301475
Blackstone E., Morrison M., Roth M. B. (2005). H2S induces a suspended animation-like state in mice. Science 308 (5721), 518. doi: 10.1126/science.1108581
Bogdandi V., Ditroi T., Batai I. Z., Sandor Z., Minnion M., Vasas A., et al. (2020). Nitrosopersulfide (SSNO(-)) Is a Unique Cysteine Polysulfidating Agent with Reduction-Resistant Bioactivity. Antioxid. Redox Signal. doi: 10.1089/ars.2020.8049
Boubeta F. M., Bari S. E., Estrin D. A., Boechi L. (2016). Access and Binding of H2S to Hemeproteins: The Case of HbI of Lucina pectinata. J. Phys. Chem. B 120 (36), 9642–9653. doi: 10.1021/acs.jpcb.6b06686
Boubeta F. M., Bieza S. A., Bringas M., Palermo J. C., Boechi L., Estrin D. A., et al. (2020). Hemeproteins as Targets for Sulfide Species. Antioxid. Redox Signal 32 (4), 247–257. doi: 10.1089/ars.2019.7878
Brancaleone V., Esposito I., Gargiulo A., Vellecco V., Asimakopoulou A., Citi V., et al. (2016). D-Penicillamine modulates hydrogen sulfide (H2S) pathway through selective inhibition of cystathionine-gamma-lyase. Br. J. Pharmacol. 173 (9), 1556–1565. doi: 10.1111/bph.13459
Braverman J., Sogi K. M., Benjamin D., Nomura D. K., Stanley S. A. (2016). HIF-1alpha Is an Essential Mediator of IFN-gamma-Dependent Immunity to Mycobacterium tuberculosis. J. Immunol. 197 (4), 1287–1297. doi: 10.4049/jimmunol.1600266
Brealey D., Brand M., Hargreaves I., Heales S., Land J., Smolenski R., et al. (2002). Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 360 (9328), 219–223. doi: 10.1016/S0140-6736(02)09459-X
Budde M. W., Roth M. B. (2010). Hydrogen sulfide increases hypoxia-inducible factor-1 activity independently of von Hippel-Lindau tumor suppressor-1 in C. elegans. Mol. Biol. Cell 21 (1), 212–217. doi: 10.1091/mbc.e09-03-0199
Caliendo G., Cirino G., Santagada V., Wallace J. L. (2010). Synthesis and biological effects of hydrogen sulfide (H2S): development of H2S-releasing drugs as pharmaceuticals. J. Med. Chem. 53 (17), 6275–6286. doi: 10.1021/jm901638j
Capece L., Boechi L., Perissinotti L. L., Arroyo-Manez P., Bikiel D. E., Smulevich G., et al. (2013). Small ligand-globin interactions: reviewing lessons derived from computer simulation. Biochim. Biophys. Acta 1834 (9), 1722–1738. doi: 10.1016/j.bbapap.2013.02.038
Cardona P., Cardona P. J. (2019). Regulatory T Cells in Mycobacterium tuberculosis Infection. Front. Immunol. 10:2139. doi: 10.3389/fimmu.2019.02139
Castelblanco M., Lugrin J., Ehirchiou D., Nasi S., Ishii I., So A., et al. (2018). Hydrogen sulfide inhibits NLRP3 inflammasome activation and reduces cytokine production both in vitro and in a mouse model of inflammation. J. Biol. Chem. 293 (7), 2546–2557. doi: 10.1074/jbc.M117.806869
Cavallini D., Federici G., Barboni E. (1970). Interaction of proteins with sulfide. Eur. J. Biochem. 14 (1), 169–174. doi: 10.1111/j.1432-1033.1970.tb00275.x
Chiku T., Padovani D., Zhu W., Singh S., Vitvitsky V., Banerjee R. (2009). H2S biogenesis by human cystathionine gamma-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 284 (17), 11601–11612. doi: 10.1074/jbc.M808026200
Chinta K. C., Saini V., Glasgow J. N., Mazorodze J. H., Rahman M. A., Reddy D., et al. (2016). The emerging role of gasotransmitters in the pathogenesis of tuberculosis. Nitric. Oxide 59, 28–41. doi: 10.1016/j.niox.2016.06.009
Chinta K. C., Rahman M. A., Saini V., Glasgow J. N., Reddy V. P., Lever J. M., et al. (2018). Microanatomic Distribution of Myeloid Heme Oxygenase-1 Protects against Free Radical-Mediated Immunopathology in Human Tuberculosis. Cell Rep. 25 (7), 1938–1952 e5. doi: 10.1016/j.celrep.2018.10.073
Chung K. F. (2014). Hydrogen sulfide as a potential biomarker of asthma. Expert Rev. Respir. Med. 8 (1), 5–13. doi: 10.1586/17476348.2014.856267
Clausen T., Huber R., Messerschmidt A., Pohlenz H. D., Laber B. (1997). Slow-binding inhibition of Escherichia coli cystathionine beta-lyase by L-aminoethoxyvinylglycine: a kinetic and X-ray study. Biochemistry 36 (41), 12633–12643. doi: 10.1021/bi970630m
Coletta C., Szabo C. (2013). Potential role of hydrogen sulfide in the pathogenesis of vascular dysfunction in septic shock. Curr. Vasc. Pharmacol. 11 (2), 208–221. doi: 10.2174/1570161111311020010
Collin M., Anuar F. B., Murch O., Bhatia M., Moore P. K., Thiemermann C. (2005). Inhibition of endogenous hydrogen sulfide formation reduces the organ injury caused by endotoxemia. Br. J. Pharmacol. 146 (4), . 498–. 505. doi: 10.1038/sj.bjp.0706367
Cooper C. E., Brown G. C. (2008). The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J. Bioenerg. Biomembr. 40 (5), 533–539. doi: 10.1007/s10863-008-9166-6
Cooper A. M., Dalton D. K., Stewart T. A., Griffin J. P., Russell D. G., Orme I. M. (1993). Disseminated tuberculosis in interferon gamma gene-disrupted mice. J. Exp. Med. 178 (6), 2243–2247. doi: 10.1084/jem.178.6.2243
Cooper A. M., Magram J., Ferrante J., Orme I. M. (1997). Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenously infected with mycobacterium tuberculosis. J. Exp. Med. 186 (1), 39–45. doi: 10.1084/jem.186.1.39
Cornell N. W., Zuurendonk P. F., Kerich M. J., Straight C. B. (1984). Selective inhibition of alanine aminotransferase and aspartate aminotransferase in rat hepatocytes. Biochem. J. 220 (3), 707–716. doi: 10.1042/bj2200707
Corsello T., Komaravelli N., Casola A. (2018). Role of Hydrogen Sulfide in NRF2- and Sirtuin-Dependent Maintenance of Cellular Redox Balance. Antioxid. (Basel) 7 (10), 129. doi: 10.3390/antiox7100129
Cortese-Krott M. M., Kuhnle G. G., Dyson A., Fernandez B. O., Grman M., DuMond J. F., et al. (2015). Key bioactive reaction products of the NO/H2S interaction are S/N-hybrid species, polysulfides, and nitroxyl. Proc. Natl. Acad. Sci. U.S.A. 112 (34), E4651–E4660. doi: 10.1073/pnas.1509277112
Corvino A., Severino B., Fiorino F., Frecentese F., Magli E., Perissutti E., et al. (2016). Fragment-based de novo design of a cystathionine gamma-lyase selective inhibitor blocking hydrogen sulfide production. Sci. Rep. 6, 34398. doi: 10.1038/srep34398
Cuevasanta E., Lange M., Bonanata J., Coitino E. L., Ferrer-Sueta G., Filipovic M. R., et al. (2015). Reaction of Hydrogen Sulfide with Disulfide and Sulfenic Acid to Form the Strongly Nucleophilic Persulfide. J. Biol. Chem. 290 (45), 26866–26880. doi: 10.1074/jbc.M115.672816
Cumming B. M., Addicott K. W., Adamson J. H., Steyn A. J. (2018). Mycobacterium tuberculosis induces decelerated bioenergetic metabolism in human macrophages. Elife 7, e39169. doi: 10.7554/eLife.39169
Das P., Lahiri A., Lahiri A., Chakravortty D. (2010). Modulation of the arginase pathway in the context of microbial pathogenesis: a metabolic enzyme moonlighting as an immune modulator. PloS Pathog. 6 (6), e1000899. doi: 10.1371/journal.ppat.1000899
DeLeon E. R., Stoy G. F., Olson K. R. (2012). Passive loss of hydrogen sulfide in biological experiments. Anal. Biochem. 421 (1), 203–207. doi: 10.1016/j.ab.2011.10.016
Doka E., Ida T., Dagnell M., Abiko Y., Luong N. C., Balog N., et al. (2020). Control of protein function through oxidation and reduction of persulfidated states. Sci. Adv. 6 (1), eaax8358. doi: 10.1126/sciadv.aax8358
Domingo-Gonzalez R., Prince O., Cooper A., Khader S. A. (2016). Cytokines and Chemokines in Mycobacterium tuberculosis Infection. Microbiol. Spectr. 4 (5), 10. doi: 10.1128/microbiolspec.TBTB2-0018-2016
d’Emmanuele di Villa Bianca R., Sorrentino R., Maffia P., Mirone V., Imbimbo C., Fusco F., et al. (2009). Hydrogen sulfide as a mediator of human corpus cavernosum smooth-muscle relaxation. Proc. Natl. Acad. Sci. U.S.A. 106 (11), 4513–4518. doi: 10.1073/pnas.0807974105
Du C., Jin M., Hong Y., Li Q., Wang X. H., Xu J. M., et al. (2014). Downregulation of cystathionine beta-synthase/hydrogen sulfide contributes to rotenone-induced microglia polarization toward M1 type. Biochem. Biophys. Res. Commun. 451 (2), 239–245. doi: 10.1016/j.bbrc.2014.07.107
Elrod J. W., Calvert J. W., Morrison J., Doeller J. E., Kraus D. W., Tao L., et al. (2007). Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc. Natl. Acad. Sci. U.S.A. 104 (39), 15560–15565. doi: 10.1073/pnas.0705891104
Ereño-Orbea J., Majtan T., Oyenarte I., Kraus J. P., Martínez-Cruz L. A. (2013). Structural basis of regulation and oligomerization of human cystathionine β-synthase, the central enzyme of transsulfuration. Proc. Natl. Acad. Sci. U. S. A. 110 (40), E3790–E3799. doi: 10.1073/pnas.1313683110
Esakandari H., Nabi-Afjadi M., Fakkari-Afjadi J., Farahmandian N., Miresmaeili S. M., Bahreini E. (2020). A comprehensive review of COVID-19 characteristics. Biol. Proced Online 22, 19. doi: 10.1186/s12575-020-00128-2
Esechie A., Kiss L., Olah G., Horvath E. M., Hawkins H., Szabo C., et al. (2008). Protective effect of hydrogen sulfide in a murine model of acute lung injury induced by combined burn and smoke inhalation. Clin. Sci. (Lond) 115 (3), 91–97. doi: 10.1042/CS20080021
Evande R., Ojha S., Banerjee R. (2004). Visualization of PLP-bound intermediates in hemeless variants of human cystathionine beta-synthase: evidence that lysine 119 is a general base. Arch. Biochem. Biophys. 427 (2), 188–196. doi: 10.1016/j.abb.2004.04.027
Fagone P., Mazzon E., Bramanti P., Bendtzen K., Nicoletti F. (2018). Gasotransmitters and the immune system: Mode of action and novel therapeutic targets. Eur. J. Pharmacol. 834, 92–102. doi: 10.1016/j.ejphar.2018.07.026
Faller S., Ryter S. W., Choi A. M., Loop T., Schmidt R., Hoetzel A. (2010). Inhaled hydrogen sulfide protects against ventilator-induced lung injury. Anesthesiology 113 (1), 104–115. doi: 10.1097/ALN.0b013e3181de7107
Felton J. S. (1997). The heritage of Bernardino Ramazzini. Occup. Med. 47 (3), 167–179. doi: 10.1093/occmed/47.3.167
Filipovic M. R., Zivanovic J., Alvarez B., Banerjee R. (2018). Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 118 (3), 1253–1337. doi: 10.1021/acs.chemrev.7b00205
Finkelstein J. D., Kyle W. E., Martin J. L., Pick A. M. (1975). Activation of cystathionine synthase by adenosylmethionine and adenosylethionine. Biochem. Biophys. Res. Commun. 66 (1), 81–87. doi: 10.1016/S0006-291X(75)80297-X
Fiorucci S., Antonelli E., Distrutti E., Rizzo G., Mencarelli A., Orlandi S., et al. (2005). Inhibition of hydrogen sulfide generation contributes to gastric injury caused by anti-inflammatory nonsteroidal drugs. Gastroenterology 129 (4), 1210–1224. doi: 10.1053/j.gastro.2005.07.060
Fiorucci S., Orlandi S., Mencarelli A., Caliendo G., Santagada V., Distrutti E., et al. (2007). Enhanced activity of a hydrogen sulphide-releasing derivative of mesalamine (ATB-429) in a mouse model of colitis. Br. J. Pharmacol. 150 (8), 996–1002. doi: 10.1038/sj.bjp.0707193
Flannigan K. L., McCoy K. D., Wallace J. L. (2011). Eukaryotic and prokaryotic contributions to colonic hydrogen sulfide synthesis. Am. J. Physiol. Gastrointest. Liver Physiol. 301 (1), G188–G193. doi: 10.1152/ajpgi.00105.2011
Forte E., Borisov V. B., Falabella M., Colaco H. G., Tinajero-Trejo M., Poole R. K., et al. (2016). The Terminal Oxidase Cytochrome bd Promotes Sulfide-resistant Bacterial Respiration and Growth. Sci. Rep. 6, 23788. doi: 10.1038/srep23788
Francoleon N. E., Carrington S. J., Fukuto J. M. (2011). The reaction of H(2)S with oxidized thiols: generation of persulfides and implications to H(2)S biology. Arch. Biochem. Biophys. 516 (2), 146–153. doi: 10.1016/j.abb.2011.09.015
Frasdorf B., Radon C., Leimkuhler S. (2014). Characterization and interaction studies of two isoforms of the dual localized 3-mercaptopyruvate sulfurtransferase TUM1 from humans. J. Biol. Chem. 289 (50), 34543–34556. doi: 10.1074/jbc.M114.605733
Fu M., Zhang W., Wu L., Yang G., Li H., Wang R. (2012). Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc. Natl. Acad. Sci. U.S.A. 109 (8), 2943–2948. doi: 10.1073/pnas.1115634109
Fu L., Liu K., He J., Tian C., Yu X., Yang J. (2019). Direct Proteomic Mapping of Cysteine Persulfidation. Antioxid. Redox Signal 33 (15), 1061–1076. doi: 10.1089/ars.2019.7777
Fukuto J. M., Carrington S. J., Tantillo D. J., Harrison J. G., Ignarro L. J., Freeman B. A., et al. (2012). Small molecule signaling agents: the integrated chemistry and biochemistry of nitrogen oxides, oxides of carbon, dioxygen, hydrogen sulfide, and their derived species. Chem. Res. Toxicol. 25 (4), 769–793. doi: 10.1021/tx2005234
Fukuto J. M., Ignarro L. J., Nagy P., Wink D. A., Kevil C. G., Feelisch M., et al. (2018). Biological hydropersulfides and related polysulfides - a new concept and perspective in redox biology. FEBS Lett. 592 (12), 2140–2152. doi: 10.1002/1873-3468.13090
Fukuto J. M., Vega V. S., Works C., Lin J. (2020). The chemical biology of hydrogen sulfide and related hydropersulfides: interactions with biologically relevant metals and metalloproteins. Curr. Opin. Chem. Biol. 55, 52–58. doi: 10.1016/j.cbpa.2019.11.013
Gaddam R. R., Fraser R., Badiei A., Chambers S., Cogger V. C., Le Couteur D. G., et al. (2016). Cystathionine-Gamma-Lyase Gene Deletion Protects Mice against Inflammation and Liver Sieve Injury following Polymicrobial Sepsis. PloS One 11 (8), e0160521. doi: 10.1371/journal.pone.0160521
Gero D., Torregrossa R., Perry A., Waters A., Le-Trionnaire S., Whatmore J. L., et al. (2016). The novel mitochondria-targeted hydrogen sulfide (H2S) donors AP123 and AP39 protect against hyperglycemic injury in microvascular endothelial cells in vitro. Pharmacol. Res. 113 (Pt A), 186–198. doi: 10.1016/j.phrs.2016.08.019
Giacco F., Brownlee M. (2010). Oxidative stress and diabetic complications. Circ. Res. 107 (9), 1058–1070. doi: 10.1161/CIRCRESAHA.110.223545
Giamarellos-Bourboulis E. J., Netea M. G., Rovina N., Akinosoglou K., 7Antoniadou A., Antonakos N., et al. (2020). Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 27 (6), 992–1000 e3. doi: 10.1016/j.chom.2020.04.009
Giuffre A., Vicente J. B. (2018). Hydrogen Sulfide Biochemistry and Interplay with Other Gaseous Mediators in Mammalian Physiology. Oxid. Med. Cell Longev 2018, 6290931. doi: 10.1155/2018/6290931
Gleeson L. E., Sheedy F. J. (2016). Metabolic reprogramming & inflammation: Fuelling the host response to pathogens. Semin. Immunol. 28 (5), 450–468. doi: 10.1016/j.smim.2016.10.007
Guan Q., Zhang Y., Yu C., Liu Y., Gao L., Zhao J. (2012). Hydrogen sulfide protects against high-glucose-induced apoptosis in endothelial cells. J. Cardiovasc. Pharmacol. 59 (2), 188–193. doi: 10.1097/FJC.0b013e31823b4915
Guo W., Cheng Z. Y., Zhu Y. Z. (2013). Hydrogen sulfide and translational medicine. Acta Pharmacol. Sin. 34 (10), 1284–1291. doi: 10.1038/aps.2013.127
Guo J., Xing H., Chen M., Wang W., Zhang H., Xu S. (2019). H2S inhalation-induced energy metabolism disturbance is involved in LPS mediated hepatocyte apoptosis through mitochondrial pathway. Sci. Total Environ. 663, 380–386. doi: 10.1016/j.scitotenv.2019.01.360
Hackett E. E., Charles-Messance H., O’Leary S. M., Gleeson L. E., Munoz-Wolf N., Case S., et al. (2020). Mycobacterium tuberculosis Limits Host Glycolysis and IL-1beta by Restriction of PFK-M via MicroRNA-21. Cell Rep. 30 (1), 124–136 e4. doi: 10.1016/j.celrep.2019.12.015
Hanaoka K., Sasakura K., Suwanai Y., Toma-Fukai S., Shimamoto K., Takano Y., et al. (2017). Discovery and Mechanistic Characterization of Selective Inhibitors of H2S-producing Enzyme: 3-Mercaptopyruvate Sulfurtransferase (3MST) Targeting Active-site Cysteine Persulfide. Sci. Rep. 7, 40227. doi: 10.1038/srep40227
He D., Liu F., Cui S., Jiang N., Yu H., Zhou Y., et al. (2020). Mechanical load-induced H2S production by periodontal ligament stem cells activates M1 macrophages to promote bone remodeling and tooth movement via STAT1. Stem Cell Res. Ther. 11 (1), 112. doi: 10.1186/s13287-020-01607-9
Hildebrandt T. M., Grieshaber M. K. (2008). Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 275 (13), 3352–3361. doi: 10.1111/j.1742-4658.2008.06482.x
Hirata I., Naito Y., Takagi T., Mizushima K., Suzuki T., Omatsu T., et al. (2011). Endogenous hydrogen sulfide is an anti-inflammatory molecule in dextran sodium sulfate-induced colitis in mice. Dig. Dis. Sci. 56 (5), 1379–1386. doi: 10.1007/s10620-010-1461-5
Hoffman M., Rajapakse A., Shen X., Gates K. S. (2012). Generation of DNA-damaging reactive oxygen species via the autoxidation of hydrogen sulfide under physiologically relevant conditions: chemistry relevant to both the genotoxic and cell signaling properties of H(2)S. Chem. Res. Toxicol. 25 (8), 1609–1615. doi: 10.1021/tx300066z
Hosoki R., Matsuki N., Kimura H. (1997). The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 237 (3), 527–531. doi: 10.1006/bbrc.1997.6878
Hourihan J. M., Kenna J. G., Hayes J. D. (2013). The gasotransmitter hydrogen sulfide induces nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antioxid. Redox Signal 19 (5), 465–481. doi: 10.1089/ars.2012.4944
Hu X., Ivashkiv L. B. (2009). Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity 31 (4), 539–550. doi: 10.1016/j.immuni.2009.09.002
Hu Y., Li X., Fang Y., Shi W., Li X., Chen W., et al. (2019). Reactive oxygen species-triggered off-on fluorescence donor for imaging hydrogen sulfide delivery in living cells. Chem. Sci. 10 (33), 7690–7694. doi: 10.1039/C9SC02323B
Hu X., Zeng W., You X., Ding W., Liu P., Chen L., et al. (2020). Exogenous Hydrogen Sulfide Regulates Mycoplasma pneumoniae Lipid-Associated Membrane Proteins to Induce Expression of Heme Oxygenase-1 and Proinflammatory Cytokines. Inflammation 43 (3), 847–856. doi: 10.1007/s10753-019-01170-3
Hua W., Jiang J., Rong X., Wu R., Qiu H., Zhang Y., et al. (2009). The dual role of the cystathionine gamma-lyase/hydrogen sulfide pathway in CVB3-induced myocarditis in mice. Biochem. Biophys. Res. Commun. 388 (3), 595–600. doi: 10.1016/j.bbrc.2009.08.064
Huang C. S., Lin A. H., Liu C. T., Tsai C. W., Chang I. S., Chen H. W., et al. (2013). Isothiocyanates protect against oxidized LDL-induced endothelial dysfunction by upregulating Nrf2-dependent antioxidation and suppressing NFkappaB activation. Mol. Nutr. Food Res. 57 (11), 1918–1930. doi: 10.1002/mnfr.201300063
Hunter R. L. (2016). Tuberculosis as a three-act play: A new paradigm for the pathogenesis of pulmonary tuberculosis. Tuberculosis (Edinb) 97, 8–17. doi: 10.1016/j.tube.2015.11.010
Ida T., Sawa T., Ihara H., Tsuchiya Y., Watanabe Y., Kumagai Y., et al. (2014). Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. U.S.A. 111 (21), 7606–7611. doi: 10.1073/pnas.1321232111
Ivanciuc T., Sbrana E., Ansar M., Bazhanov N., Szabo C., Casola A., et al. (2016). Hydrogen Sulfide Is an Antiviral and Antiinflammatory End Gasotransmitter in the Airways. Role in Respiratory Syncytial Virus Infection. Am. J. Respir. Cell Mol. Biol. 55 (5), 684–696. doi: 10.1165/rcmb.2015-0385OC
Ivanovic-Burmazovic I., Filipovic M. R. (2019). Saying NO to H2S: A Story of HNO, HSNO, and SSNO(). Inorg. Chem. 58 (7), 4039–4051. doi: 10.1021/acs.inorgchem.8b02592
Jaffrey S. R., Snyder S. H. (2001). The biotin switch method for the detection of S-nitrosylated proteins. Sci. STKE 2001 (86), pl1. doi: 10.1126/stke.2001.86.pl1
Jain S. K., Bull R., Rains J. L., Bass P. F., Levine S. N., Reddy S., et al. (2010). Low levels of hydrogen sulfide in the blood of diabetes patients and streptozotocin-treated rats causes vascular inflammation? Antioxid. Redox Signal 12 (11), 1333–1337. doi: 10.1089/ars.2009.2956
Jayaraman P., Sada-Ovalle I., Nishimura T., Anderson A. C., Kuchroo V. K., Remold H. G., et al. (2013). IL-1beta promotes antimicrobial immunity in macrophages by regulating TNFR signaling and caspase-3 activation. J. Immunol. 190 (8), 4196–4204. doi: 10.4049/jimmunol.1202688
Jeddi S., Gheibi S., Kashfi K., Carlstrom M., Ghasemi A. (2020). Dose-Dependent Effects of Long-Term Administration of Hydrogen Sulfide on Myocardial Ischemia-Reperfusion Injury in Male Wistar Rats: Modulation of RKIP, NF-kappaB, and Oxidative Stress. Int. J. Mol. Sci. 21 (4), 1415. doi: 10.3390/ijms21041415
Jiang B., Tang G., Cao K., Wu L., Wang R. (2010). Molecular mechanism for H2S-induced activation of KATP channels. Antioxid. Redox Signaling 12 (10), 1167–1178. doi: 10.1089/ars.2009.2894
Johansen D., Ytrehus K., Baxter G. F. (2006). Exogenous hydrogen sulfide (H2S) protects against regional myocardial ischemia-reperfusion injury–Evidence for a role of K ATP channels. Basic Res. Cardiol. 101 (1), 53–60. doi: 10.1007/s00395-005-0569-9
John A., Kundu S., Pushpakumar S., Fordham M., Weber G., Mukhopadhyay M., et al. (2017). GYY4137, a Hydrogen Sulfide Donor Modulates miR194-Dependent Collagen Realignment in Diabetic Kidney. Sci. Rep. 7 (1), 10924. doi: 10.1038/s41598-017-11256-3
Ju Y., Untereiner A., Wu L., Yang G. (2015). H2S-induced S-sulfhydration of pyruvate carboxylase contributes to gluconeogenesis in liver cells. Biochim. Biophys. Acta 1850 (11), 2293–2303. doi: 10.1016/j.bbagen.2015.08.003
Kabil O., Banerjee R. (2014). Enzymology of H2S biogenesis, decay and signaling. Antioxid. Redox Signal 20 (5), 770–782. doi: 10.1089/ars.2013.5339
Kabil O., Zhou Y., Banerjee R. (2006). Human cystathionine beta-synthase is a target for sumoylation. Biochemistry 45 (45), 13528–13536. doi: 10.1021/bi0615644
Kabil O., Vitvitsky V., Xie P., Banerjee R. (2011). The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid. Redox Signal 15 (2), 363–372. doi: 10.1089/ars.2010.3781
Kai S., Tanaka T., Daijo H., Harada H., Kishimoto S., Suzuki K., et al. (2012). Hydrogen sulfide inhibits hypoxia- but not anoxia-induced hypoxia-inducible factor 1 activation in a von hippel-lindau- and mitochondria-dependent manner. Antioxid. Redox Signal 16 (3), 203–216. doi: 10.1089/ars.2011.3882
Kamat P. K., Kalani A., Tyagi N. (2015). Role of hydrogen sulfide in brain synaptic remodeling. Methods Enzymol. 555, 207–229. doi: 10.1016/bs.mie.2014.11.025
Kanagy N. L., Szabo C., Papapetropoulos A. (2017). Vascular biology of hydrogen sulfide. Am. J. Physiol. Cell Physiol. 312 (5), C537–C549. doi: 10.1152/ajpcell.00329.2016
Kaneko Y., Kimura Y., Kimura H., Niki I. (2006). L-cysteine inhibits insulin release from the pancreatic beta-cell: possible involvement of metabolic production of hydrogen sulfide, a novel gasotransmitter. Diabetes 55 (5), 1391–1397. doi: 10.2337/db05-1082
Keilin D. (1929). Cytochrome and Respiratory Enzymes. Proc. Royal Soc. Lond. Ser. B 104, 206–252. doi: 10.1098/rspb.1929.0009
Kery V., Bukovska G., Kraus J. P. (1994). Transsulfuration depends on heme in addition to pyridoxal 5’-phosphate. Cystathionine beta-synthase is a heme protein. J. Biol. Chem. 269 (41), 25283–25288.
Kery V., Poneleit L., Kraus J. P. (1998). Trypsin cleavage of human cystathionine beta-synthase into an evolutionarily conserved active core: structural and functional consequences. Arch. Biochem. Biophys. 355 (2), 222–232. doi: 10.1006/abbi.1998.0723
Kevil C., Cortese-Krott M. M., Nagy P., Papapetropoulos A., Feelisch M., Szabo C. (2017). “Cooperative Interactions Between NO and H2S: Chemistry, Biology, Physiology, Pathophysiology,” in Nitric Oxide, Biology and Pathobiology, 3rd ed. Eds. Ignarro L. J., Freeman B. A. (London:Academic Press), 57–83. doi: 10.1016/B978-0-12-804273-1.00005-3
Khan A., Singh V. K., Hunter R. L., Jagannath C. (2019). Macrophage heterogeneity and plasticity in tuberculosis. J. Leukoc. Biol. 106 (2), 275–282. doi: 10.1002/JLB.MR0318-095RR
Kimura Y., Koike S., Shibuya N., Lefer D., Ogasawara Y., Kimura H. (2017). 3-Mercaptopyruvate sulfurtransferase produces potential redox regulators cysteine- and glutathione-persulfide (Cys-SSH and GSSH) together with signaling molecules H2S2, H2S3 and H2S. Sci. Rep. 7 (1), 10459. doi: 10.1038/s41598-017-11004-7
Kimura H. (2000). Hydrogen sulfide induces cyclic AMP and modulates the NMDA receptor. Biochem. Biophys. Res. Commun. 267 (1), 129–133. doi: 10.1006/bbrc.1999.1915
Kimura H. (2011). Hydrogen sulfide: its production, release and functions. Amino Acids 41 (1), 113–121. doi: 10.1007/s00726-010-0510-x
Kimura H. (2015). Physiological Roles of Hydrogen Sulfide and Polysulfides. Handb. Exp. Pharmacol. 230, 61–81. doi: 10.1007/978-3-319-18144-8_3
Kimura H. (2019). Signaling by hydrogen sulfide (H2S) and polysulfides (H2Sn) in the central nervous system. Neurochem. Int. 126, 118–125. doi: 10.1016/j.neuint.2019.01.027
Kolluru G. K., Shen X., Kevil C. G. (2013). A tale of two gases: NO and H2S, foes or friends for life? Redox Biol. 1, 313–318. doi: 10.1016/j.redox.2013.05.001
Kondo K., Bhushan S., King A. L., Prabhu S. D., Hamid T., Koenig S., et al. (2013). H(2)S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 127 (10), 1116–1127. doi: 10.1161/CIRCULATIONAHA.112.000855
Korshunov S., Imlay K. R., Imlay J. A. (2016). The cytochrome bd oxidase of Escherichia coli prevents respiratory inhibition by endogenous and exogenous hydrogen sulfide. Mol. Microbiol. 101 (1), 62–77. doi: 10.1111/mmi.13372
Kosir M., Podbregar M. (2017). Advances in the Diagnosis of Sepsis: Hydrogen Sulfide as a Prognostic Marker of Septic Shock Severity. EJIFCC 28 (2), 134–141.
Kraus D. W., Wittenberg J. B. (1990). Hemoglobins of the Lucina pectinata/bacteria symbiosis. I. Molecular properties, kinetics and equilibria of reactions with ligands. J.Biol.Chem 265 (27), 16043–16053.
Kraus D. W., Wittenberg J. B., Lu J. F., Peisach J. (1990). Hemoglobins of the Lucina pectinata/bacteria symbiosis. II. An electron paramagnetic resonance and optical spectral study of the ferric proteins. J.Biol.Chem 265 (27), 16054–16059.
Kumar A., Toledo J. C., Patel R. P., Lancaster J. R. Jr., Steyn A. J. (2007). Mycobacterium tuberculosis DosS is a redox sensor and DosT is a hypoxia sensor. Proc. Natl. Acad. Sci. U. S. A. 104 (28), 11568–11573. doi: 10.1073/pnas.0705054104
Kumar A., Deshane J. S., Crossman D. K., Bolisetty S., Yan B. S., Kramnik I., et al. (2008). Heme oxygenase-1-derived carbon monoxide induces the Mycobacterium tuberculosis dormancy regulon. J. Biol. Chem. 283 (26), 18032–18039. doi: 10.1074/jbc.M802274200
Kumar R., Singh P., Kolloli A., Shi L., Bushkin Y., Tyagi S., et al. (2019). Immunometabolism of Phagocytes During Mycobacterium tuberculosis Infection. Front. Mol. Biosci. 6:105. doi: 10.3389/fmolb.2019.00105
Lagoutte E., Mimoun S., Andriamihaja M., Chaumontet C., Blachier F., Bouillaud F. (2010). Oxidation of hydrogen sulfide remains a priority in mammalian cells and causes reverse electron transfer in colonocytes. Biochim. Biophys. Acta 1797 (8), 1500–1511. doi: 10.1016/j.bbabio.2010.04.004
Le Trionnaire S., Perry A., Szczesny B., Szabo C., Winyard P. G., Whatmore J. L., et al. (2014). The synthesis and functional evaluation of a mitochondria-targeted hydrogen sulfide donor,(10-oxo-10-(4-(3-thioxo-3 H-1, 2-dithiol-5-yl) phenoxy) decyl) triphenylphosphonium bromide (AP39). MedChemComm 5 (6), 728–736. doi: 10.1039/C3MD00323J
Lee Z. W., Zhou J., Chen C. S., Zhao Y., Tan C. H., Li L., et al. (2011). The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PloS One 6 (6), e21077. doi: 10.1371/journal.pone.0021077
Lee Z. W., Teo X. Y., Tay E. Y., Tan C. H., Hagen T., Moore P. K., et al. (2014). Utilizing hydrogen sulfide as a novel anti-cancer agent by targeting cancer glycolysis and pH imbalance. Br. J. Pharmacol. 171 (18), 4322–4336. doi: 10.1111/bph.12773
Levinn C. M., Cerda M. M., Pluth M. D. (2020). Activatable Small-Molecule Hydrogen Sulfide Donors. Antioxid. Redox Signal 32 (2), 96–109. doi: 10.1089/ars.2019.7841
Li Q., Lancaster J. R. Jr. (2013). Chemical foundations of hydrogen sulfide biology. Nitric. Oxide 35, 21–34. doi: 10.1016/j.niox.2013.07.001
Li J., Uetrecht J. P. (2009). D-penicillamine-induced autoimmunity: relationship to macrophage activation. Chem. Res. Toxicol. 22 (9), 1526–1533. doi: 10.1021/tx900128p
Li L., Bhatia M., Zhu Y. Z., Zhu Y. C., Ramnath R. D., Wang Z. J., et al. (2005). Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 19 (9), 1196–1198. doi: 10.1096/fj.04-3583fje
Li L., Rossoni G., Sparatore A., Lee L. C., Del Soldato P., Moore P. K. (2007). Anti-inflammatory and gastrointestinal effects of a novel diclofenac derivative. Free Radic. Biol. Med. 42 (5), 706–719. doi: 10.1016/j.freeradbiomed.2006.12.011
Li L., Whiteman M., Guan Y. Y., Neo K. L., Cheng Y., Lee S. W., et al. (2008). Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): new insights into the biology of hydrogen sulfide. Circulation 117 (18), 2351–2360. doi: 10.1161/CIRCULATIONAHA.107.753467
Li H., Ma Y., Escaffre O., Ivanciuc T., Komaravelli N., Kelley J. P., et al. (2015). Role of hydrogen sulfide in paramyxovirus infections. J. Virol. 89 (10), 5557–5568. doi: 10.1128/JVI.00264-15
Li Z., Polhemus D. J., Lefer D. J. (2018). Evolution of Hydrogen Sulfide Therapeutics to Treat Cardiovascular Disease. Circ. Res. 123 (5), 590–600. doi: 10.1161/CIRCRESAHA.118.311134
Liang M., Jin S., Wu D. D., Wang M. J., Zhu Y. C. (2015). Hydrogen sulfide improves glucose metabolism and prevents hypertrophy in cardiomyocytes. Nitric. Oxide 46, 114–122. doi: 10.1016/j.niox.2014.12.007
Libiad M., Vitvitsky V., Bostelaar T., Bak D. W., Lee H. J., Sakamoto N., et al. (2019). Hydrogen sulfide perturbs mitochondrial bioenergetics and triggers metabolic reprogramming in colon cells. J. Biol. Chem. 294 (32), 12077–12090. doi: 10.1074/jbc.RA119.009442
Litwin T., Dziezyc K., Czlonkowska A. (2019). Wilson disease-treatment perspectives. Ann. Transl. Med. 7 (Suppl 2), S68. doi: 10.21037/atm.2018.12.09
Liu M., Li Y., Liang B., Li Z., Jiang Z., Chu C., et al. (2018). Hydrogen sulfide attenuates myocardial fibrosis in diabetic rats through the JAK/STAT signaling pathway. Int. J. Mol. Med. 41 (4), 1867–1876. doi: 10.3892/ijmm.2018.3419
Liu Q. Y., Han F., Pan L. P., Jia H. Y., Li Q., Zhang Z. D. (2018). Inflammation responses in patients with pulmonary tuberculosis in an intensive care unit. Exp. Ther. Med. 15 (3), 2719–2726. doi: 10.3892/etm.2018.5775
Liu Y., Yang K., Jia Y., Shi J., Tong Z., Wang Z. (2020). Cysteine Potentiates Bactericidal Antibiotics Activity Against Gram-Negative Bacterial Persisters. Infect. Drug Resist. 13, 2593–2599. doi: 10.2147/IDR.S263225
Lowe D. M., Redford P. S., Wilkinson R. J., O’Garra A., Martineau A. R. (2012). Neutrophils in tuberculosis: friend or foe? Trends Immunol. 33 (1), 14–25. doi: 10.1016/j.it.2011.10.003
Lu M., Hu L. F., Hu G., Bian J. S. (2008). Hydrogen sulfide protects astrocytes against H(2)O(2)-induced neural injury via enhancing glutamate uptake. Free Radic. Biol. Med. 45 (12), 1705–1713. doi: 10.1016/j.freeradbiomed.2008.09.014
Ma S., Zhong D., Ma P., Li G., Hua W., Sun Y., et al. (2017). Exogenous Hydrogen Sulfide Ameliorates Diabetes-Associated Cognitive Decline by Regulating the Mitochondria-Mediated Apoptotic Pathway and IL-23/IL-17 Expression in db/db Mice. Cell Physiol. Biochem. 41 (5), 1838–1850. doi: 10.1159/000471932
MacMicking J. D., Xie Q. W., Nathan C. (1997a). Nitric oxide and macrophage function. Annu. Rev. Immunol. 15, 323–350. doi: 10.1146/annurev.immunol.15.1.323
MacMicking J. D., North R. J., LaCourse R., Mudgett J. S., Shah S. K., Nathan C. F. (1997b). Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 94 (10), 5243–5248. doi: 10.1073/pnas.94.10.5243
Majtan T., Krijt J., Sokolova J., Krizkova M., Ralat M. A., Kent J., et al. (2018). Biogenesis of Hydrogen Sulfide and Thioethers by Cystathionine Beta-Synthase. Antioxid. Redox Signal 28 (4), 311–323. doi: 10.1089/ars.2017.7009
Mani S., Untereiner A., Wu L., Wang R. (2014). Hydrogen sulfide and the pathogenesis of atherosclerosis. Antioxid. Redox Signal 20 (5), 805–817. doi: 10.1089/ars.2013.5324
Manna P., Gungor N., McVie R., Jain S. K. (2014). Decreased cystathionine-gamma-lyase (CSE) activity in livers of type 1 diabetic rats and peripheral blood mononuclear cells (PBMC) of type 1 diabetic patients. J. Biol. Chem. 289 (17), 11767–11778. doi: 10.1074/jbc.M113.524645
Mantovani A., Biswas S. K., Galdiero M. R., Sica A., Locati M. (2013). Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 229 (2), 176–185. doi: 10.1002/path.4133
Marcolongo J. P., Venancio M. F., Rocha W. R., Doctorovich F., Olabe J. A. (2019). NO/H2S “Crosstalk” Reactions. The Role of Thionitrites (SNO(-)) and Perthionitrites (SSNO(-)). Inorg. Chem. 58 (22), 14981–14997. doi: 10.1021/acs.inorgchem.9b01978
Marks E., Naudin C., Nolan G., Goggins B. J., Burns G., Mateer S. W., et al. (2017). Regulation of IL-12p40 by HIF controls Th1/Th17 responses to prevent mucosal inflammation. Mucosal Immunol. 10 (5), 1224–1236. doi: 10.1038/mi.2016.135
Martelli A., Testai L., Citi V., Marino A., Pugliesi I., Barresi E., et al. (2013). Arylthioamides as H2S Donors: l-Cysteine-Activated Releasing Properties and Vascular Effects in Vitro and in Vivo. ACS Med. Chem. Lett. 4 (10), 904–908. doi: 10.1021/ml400239a
Martelli A., Testai L., Citi V., Marino A., Bellagambi F. G., Ghimenti S., et al. (2014). Pharmacological characterization of the vascular effects of aryl isothiocyanates: is hydrogen sulfide the real player? Vascul Pharmacol. 60 (1), 32–41. doi: 10.1016/j.vph.2013.11.003
Martelli A., Citi V., Testai L., Brogi S., Calderone V. (2020). Organic Isothiocyanates as Hydrogen Sulfide Donors. Antioxid. Redox Signal 32 (2), 110–144. doi: 10.1089/ars.2019.7888
Masson M. J., Teranishi M., Shenton J. M., Uetrecht J. P. (2004). Investigation of the involvement of macrophages and T cells in D-penicillamine-induced autoimmunity in the Brown Norway rat. J. Immunotoxicol. 1 (2), 79–93. doi: 10.1080/15476910490496258
Mathai J. C., Missner A., Kugler P., Saparov S. M., Zeidel M. L., Lee J. K., et al. (2009). No facilitator required for membrane transport of hydrogen sulfide. Proc. Natl. Acad. Sci. U.S.A. 106 (39), 16633–16638. doi: 10.1073/pnas.0902952106
Medam S., Zieleskiewicz L., Duclos G., Baumstarck K., Loundou A., Alingrin J., et al. (2017). Risk factors for death in septic shock: A retrospective cohort study comparing trauma and non-trauma patients. Med. (Baltimore) 96 (50), e9241. doi: 10.1097/MD.0000000000009241
Meier M., Janosik M., Kery V., Kraus J. P., Burkhard P. (2001). Structure of human cystathionine beta-synthase: a unique pyridoxal 5’-phosphate-dependent heme protein. EMBO J. 20 (15), 3910–3916. doi: 10.1093/emboj/20.15.3910
Meng G., Ma Y., Xie L., Ferro A., Ji Y. (2015). Emerging role of hydrogen sulfide in hypertension and related cardiovascular diseases. Br. J. Pharmacol. 172 (23), 5501–5511. doi: 10.1111/bph.12900
Miao L., Shen X., Whiteman M., Xin H., Shen Y., Xin X., et al. (2016). Hydrogen Sulfide Mitigates Myocardial Infarction via Promotion of Mitochondrial Biogenesis-Dependent M2 Polarization of Macrophages. Antioxid. Redox Signal 25 (5), 268–281. doi: 10.1089/ars.2015.6577
Mikami Y., Shibuya N., Kimura Y., Nagahara N., Ogasawara Y., Kimura H. (2011). Thioredoxin and dihydrolipoic acid are required for 3-mercaptopyruvate sulfurtransferase to produce hydrogen sulfide. Biochem. J. 439 (3), 479–485. doi: 10.1042/BJ20110841
Miles E. W., Kraus J. P. (2004). Cystathionine beta-synthase: structure, function, regulation, and location of homocystinuria-causing mutations. J. Biol. Chem. 279 (29), 29871–29874. doi: 10.1074/jbc.R400005200
Miller T. W., Wang E. A., Gould S., Stein E. V., Kaur S., Lim L., et al. (2012). Hydrogen sulfide is an endogenous potentiator of T cell activation. J. Biol. Chem. 287 (6), 4211–4221. doi: 10.1074/jbc.M111.307819
Mills E. L., O’Neill L. A. (2016). Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur. J. Immunol. 46 (1), 13–21. doi: 10.1002/eji.201445427
Mironov A., Seregina T., Nagornykh M., Luhachack L. G., Korolkova N., Lopes L. E., et al. (2017). Mechanism of H2S-mediated protection against oxidative stress in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 114 (23), 6022–6027. doi: 10.1073/pnas.1703576114
Mishra R., Kohli S., Malhotra N., Bandyopadhyay P., Mehta M., Munshi M., et al. (2019). Targeting redox heterogeneity to counteract drug tolerance in replicating Mycobacterium tuberculosis. Sci. Transl. Med. 11 (518), eaaw6635. doi: 10.1126/scitranslmed.aaw6635
Mitchell C. W., Davenport S. J. (1924). Hydrogen sulphide literature. Public Health Rep. 39 (1), 1–13. doi: 10.2307/4577000
Modis K., Bos E. M., Calzia E., van Goor H., Coletta C., Papapetropoulos A., et al. (2014). Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part II. Pathophysiological and therapeutic aspects. Br. J. Pharmacol. 171 (8), 2123–2146. doi: 10.1111/bph.12368
Modis K., Ju Y., Ahmad A., Untereiner A. A., Altaany Z., Wu L., et al. (2016). S-Sulfhydration of ATP synthase by hydrogen sulfide stimulates mitochondrial bioenergetics. Pharmacol. Res. 113 (Pt A), 116–124. doi: 10.1016/j.phrs.2016.08.023
Mogues T., Goodrich M. E., Ryan L., LaCourse R., North R. J. (2001). The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J. Exp. Med. 193 (3), 271–280. doi: 10.1084/jem.193.3.271
Mok Y. Y., Moore P. K. (2008). Hydrogen sulphide is pro-inflammatory in haemorrhagic shock. Inflammation Res. 57 (11), 512–518. doi: 10.1007/s00011-008-7231-6
Mok Y. Y., Atan M. S., Yoke Ping C., Zhong Jing W., Bhatia M., Moochhala S., et al. (2004). Role of hydrogen sulphide in haemorrhagic shock in the rat: protective effect of inhibitors of hydrogen sulphide biosynthesis. Br. J. Pharmacol. 143 (7), 881–889. doi: 10.1038/sj.bjp.0706014
Mustafa A. K., Gadalla M. M., Sen N., Kim S., Mu W., Gazi S. K., et al. (2009). H2S signals through protein S-sulfhydration. Sci. Signal 2 (96), ra72. doi: 10.1126/scisignal.2000464
Nagahara N., Katayama A. (2005). Post-translational regulation of mercaptopyruvate sulfurtransferase via a low redox potential cysteine-sulfenate in the maintenance of redox homeostasis. J. Biol. Chem. 280 (41), 34569–34576. doi: 10.1074/jbc.M505643200
Nagahara N., Okazaki T., Nishino T. (1995). Cytosolic mercaptopyruvate sulfurtransferase is evolutionarily related to mitochondrial rhodanese. Striking similarity in active site amino acid sequence and the increase in the mercaptopyruvate sulfurtransferase activity of rhodanese by site-directed mutagenesis. J. Biol. Chem. 270 (27), 16230–16235. doi: 10.1074/jbc.270.27.16230
Nagahara N., Ito T., Kitamura H., Nishino T. (1998). Tissue and subcellular distribution of mercaptopyruvate sulfurtransferase in the rat: confocal laser fluorescence and immunoelectron microscopic studies combined with biochemical analysis. Histochem. Cell Biol. 110 (3), 243–250. doi: 10.1007/s004180050286
Nagahara N., Ito T., Minami M. (1999). Mercaptopyruvate sulfurtransferase as a defense against cyanide toxication: molecular properties and mode of detoxification. Histol Histopathol. 14 (4), 1277–1286. doi: 10.14670/HH-14.1277
Nagahara N., Yoshii T., Abe Y., Matsumura T. (2007). Thioredoxin-dependent enzymatic activation of mercaptopyruvate sulfurtransferase. An intersubunit disulfide bond serves as a redox switch for activation. J. Biol. Chem. 282 (3), 1561–1569. doi: 10.1074/jbc.M605931200
Nagy P., Winterbourn C. C. (2010). Rapid reaction of hydrogen sulfide with the neutrophil oxidant hypochlorous acid to generate polysulfides. Chem. Res. Toxicol. 23 (10), 1541–1543. doi: 10.1021/tx100266a
Nagy P., Palinkas Z., Nagy A., Budai B., Toth I., Vasas A. (2014). Chemical aspects of hydrogen sulfide measurements in physiological samples. Biochim. Biophys. Acta 1840 (2), 876–891. doi: 10.1016/j.bbagen.2013.05.037
Nambi S., Long J. E., Mishra B. B., Baker R., Murphy K. C., Olive A. J., et al. (2015). The Oxidative Stress Network of Mycobacterium tuberculosis Reveals Coordination between Radical Detoxification Systems. Cell Host Microbe 17 (6), 829–837. doi: 10.1016/j.chom.2015.05.008
Nandi B., Behar S. M. (2011). Regulation of neutrophils by interferon-gamma limits lung inflammation during tuberculosis infection. J. Exp. Med. 208 (11), 2251–2262. doi: 10.1084/jem.20110919
Ng S. Y., Ong K. X., Surendran S. T., Sinha A., Lai J. J. H., Chen J., et al. (2020). Hydrogen Sulfide Sensitizes Acinetobacter baumannii to Killing by Antibiotics. Front. Microbiol. 11:1875. doi: 10.3389/fmicb.2020.01875
Nicholls P., Marshall D. C., Cooper C. E., Wilson M. T. (2013). Sulfide inhibition of and metabolism by cytochrome c oxidase. Biochem. Soc. Trans. 41 (5), 1312–1316. doi: 10.1042/BST20130070
Nicolau L. A., Silva R. O., Damasceno S. R., Carvalho N. S., Costa N. R., Aragao K. S., et al. (2013). The hydrogen sulfide donor, Lawesson’s reagent, prevents alendronate-induced gastric damage in rats. Braz. J. Med. Biol. Res. 46 (8), 708–714. doi: 10.1590/1414-431X20133030
Nowak G., Bakajsova-Takacsova D. (2018). Protein kinase Cepsilon targets respiratory chain and mitochondrial membrane potential but not F0 F1 -ATPase in renal cells injured by oxidant. J. Cell Biochem. 119 (11), 9394–9407. doi: 10.1002/jcb.27256
Ogasawara Y., Isoda S., Tanabe S. (1994). Tissue and subcellular distribution of bound and acid-labile sulfur, and the enzymic capacity for sulfide production in the rat. Biol. Pharm. Bull. 17 (12), 1535–1542. doi: 10.1248/bpb.17.1535
Okeke E. B., Uzonna J. E. (2019). The Pivotal Role of Regulatory T Cells in the Regulation of Innate Immune Cells. Front. Immunol. 10:680. doi: 10.3389/fimmu.2019.00680
Olson K. R., DeLeon E. R., Liu F. (2014). Controversies and conundrums in hydrogen sulfide biology. Nitric. Oxide 41, 11–26. doi: 10.1016/j.niox.2014.05.012
Olson K. R. (2012a). Mitochondrial adaptations to utilize hydrogen sulfide for energy and signaling. J. Comp Physiol. B 182 (7), 881–897. doi: 10.1007/s00360-012-0654-y
Olson K. R. (2012b). A practical look at the chemistry and biology of hydrogen sulfide. Antioxid. Redox Signal 17 (1), 32–44. doi: 10.1089/ars.2011.4401
O’Connor T. M., O’Connell J., O’Brien D. II, Goode T., Bredin C. P., Shanahan F. (2004). The role of substance P in inflammatory disease. J. Cell Physiol. 201 (2), 167–180. doi: 10.1002/jcp.20061
O’Garra A., Redford P. S., McNab F. W., Bloom C. II, Wilkinson R. J., Berry M. P. (2013). The immune response in tuberculosis. Annu. Rev. Immunol. 31, 475–527. doi: 10.1146/annurev-immunol-032712-095939
O’Neill L. A., Kishton R. J., Rathmell J. (2016). A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16 (9), 553–565. doi: 10.1038/nri.2016.70
Pal V. K., Bandyopadhyay P., Singh A. (2018). Hydrogen sulfide in physiology and pathogenesis of bacteria and viruses. IUBMB Life 70 (5), 393–410. doi: 10.1002/iub.1740
Palinkas Z., Furtmuller P. G., Nagy A., Jakopitsch C., Pirker K. F., Magierowski M., et al. (2015). Interactions of hydrogen sulfide with myeloperoxidase. Br. J. Pharmacol. 172 (6), 1516–1532. doi: 10.1111/bph.12769
Pan L. L., Qin M., Liu X. H., Zhu Y. Z. (2017). The Role of Hydrogen Sulfide on Cardiovascular Homeostasis: An Overview with Update on Immunomodulation. Front. Pharmacol. 8:686. doi: 10.3389/fphar.2017.00686
Panthi S., Chung H.-J., Jung J., Jeong N. Y. (2016). Physiological importance of hydrogen sulfide: emerging potent neuroprotector and neuromodulator. Oxid. Med. Cell. Longevity 2016, 9049782. doi: 10.1155/2016/9049782
Papapetropoulos A., Pyriochou A., Altaany Z., Yang G., Marazioti A., Zhou Z., et al. (2009). Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 106 (51), 21972–21977. doi: 10.1073/pnas.0908047106
Park S., Imlay J. A. (2003). High levels of intracellular cysteine promote oxidative DNA damage by driving the fenton reaction. J. Bacteriol. 185 (6), 1942–1950. doi: 10.1128/JB.185.6.1942-1950.2003
Park C. M., Macinkovic I., Filipovic M. R., Xian M. (2015). Use of the “tag-switch” method for the detection of protein S-sulfhydration. Methods Enzymol. 555, 39–56. doi: 10.1016/bs.mie.2014.11.033
Patel C. H., Leone R. D., Horton M. R., Powell J. D. (2019). Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat. Rev. Drug Discovery 18 (9), 669–688. doi: 10.1038/s41573-019-0032-5
Pearl J. E., Saunders B., Ehlers S., Orme I. M., Cooper A. M. (2001). Inflammation and lymphocyte activation during mycobacterial infection in the interferon-gamma-deficient mouse. Cell Immunol. 211 (1), 43–50. doi: 10.1006/cimm.2001.1819
Peh M. T., Anwar A. B., Ng D. S., Atan M. S., Kumar S. D., Moore P. K. (2014). Effect of feeding a high fat diet on hydrogen sulfide (H2S) metabolism in the mouse. Nitric. Oxide 41, 138–145. doi: 10.1016/j.niox.2014.03.002
Petersen L. C. (1977). The effect of inhibitors on the oxygen kinetics of cytochrome c oxidase. Biochim. Biophys. Acta 460 (2), 299–307. doi: 10.1016/0005-2728(77)90216-X
Pey A. L., Majtan T., Sanchez-Ruiz J. M., Kraus J. P. (2013). Human cystathionine beta-synthase (CBS) contains two classes of binding sites for S-adenosylmethionine (SAM): complex regulation of CBS activity and stability by SAM. Biochem. J. 449 (1), 109–121. doi: 10.1042/BJ20120731
Pichette J., Gagnon J. (2016). Implications of Hydrogen Sulfide in Glucose Regulation: How H2S Can Alter Glucose Homeostasis through Metabolic Hormones. Oxid. Med. Cell Longev 2016, 3285074. doi: 10.1155/2016/3285074
Pietri R., Roman-Morales E., Lopez-Garriga J. (2011). Hydrogen sulfide and hemeproteins: knowledge and mysteries. Antioxid.Redox.Signal. 15 (2), 393–404. doi: 10.1089/ars.2010.3698
Polhemus D. J., Calvert J. W., Butler J., Lefer D. J. (2014). The cardioprotective actions of hydrogen sulfide in acute myocardial infarction and heart failure. Sci. (Cairo) 2014, 768607. doi: 10.1155/2014/768607
Pollegioni L., Sacchi S., Murtas G. (2018). Human D-Amino Acid Oxidase: Structure, Function, and Regulation. Front. Mol. Biosci. 5:107. doi: 10.3389/fmolb.2018.00107
Powell C. R., Dillon K. M., Matson J. B. (2018). A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 149, 110–123. doi: 10.1016/j.bcp.2017.11.014
Predmore B. L., Lefer D. J., Gojon G. (2012). Hydrogen sulfide in biochemistry and medicine. Antioxid. Redox Signal 17 (1), 119–140. doi: 10.1089/ars.2012.4612
Prudova A., Bauman Z., Braun A., Vitvitsky V., Lu S. C., Banerjee R. (2006). S-adenosylmethionine stabilizes cystathionine beta-synthase and modulates redox capacity. Proc. Natl. Acad. Sci. U.S.A. 103 (17), 6489–6494. doi: 10.1073/pnas.0509531103
Puranik M., Weeks C. L., Lahaye D., Kabil O., Taoka S., Nielsen S. B., et al. (2006). Dynamics of carbon monoxide binding to cystathionine beta-synthase. J. Biol. Chem. 281 (19), 13433–13438. doi: 10.1074/jbc.M600246200
Qiu H., Chen X., Luo Z., Zhao L., Zhang T., Yang N., et al. (2018). Inhibition of endogenous hydrogen sulfide production exacerbates the inflammatory response during urine-derived sepsis-induced kidney injury. Exp. Ther. Med. 16 (4), 2851–2858. doi: 10.3892/etm.2018.6520
Rahman M. A., Cumming B. M., Addicott K. W., Pacl H. T., Russell S. L., Nargan K., et al. (2020). Hydrogen sulfide dysregulates the immune response by suppressing central carbon metabolism to promote tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 117 (12), 6663–6674. doi: 10.1073/pnas.1919211117
Reddy V. P., Chinta K. C., Saini V., Glasgow J. N., Hull T. D., Traylor A., et al. (2018). Ferritin H Deficiency in Myeloid Compartments Dysregulates Host Energy Metabolism and Increases Susceptibility to Mycobacterium tuberculosis Infection. Front. Immunol. 9:860. doi: 10.3389/fimmu.2018.00860
Renieris G., Katrini K., Damoulari C., Akinosoglou K., Psarrakis C., Kyriakopoulou M., et al. (2020). Serum Hydrogen Sulfide and Outcome Association in Pneumonia by the SARS-CoV-2 Corona virus. Shock 54(5), 633–637. doi: 10.1097/SHK.0000000000001562
Robert K., Vialard F., Thiery E., Toyama K., Sinet P. M., Janel N., et al. (2003). Expression of the cystathionine beta synthase (CBS) gene during mouse development and immunolocalization in adult brain. J. Histochem. Cytochem. 51 (3), 363–371. doi: 10.1177/002215540305100311
Rodrigues L., Schmidt T. P., Florenzano J., dos Santos K. T., Cerqueira A. R., Teixeira S. A., et al. (2015). The slow-releasing hydrogen sulphide donor, GYY4137, exhibits novel anti-skin symptoms of psoriasis and related itch. Nitric. Oxide 47), S35. doi: 10.1016/j.niox.2015.02.084
Rossouw M., Nel H. J., Cooke G. S., van Helden P. D., Hoal E. G. (2003). Association between tuberculosis and a polymorphic NFkappaB binding site in the interferon gamma gene. Lancet 361 (9372), 1871–1872. doi: 10.1016/S0140-6736(03)13491-5
Rostovtseva T. K., Sheldon K. L., Hassanzadeh E., Monge C., Saks V., Bezrukov S. M., et al. (2008). Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. U.S.A. 105 (48), 18746–18751. doi: 10.1073/pnas.0806303105
Russell S. L., Lamprecht D. A., Mandizvo T., Jones T. T., Naidoo V., Addicott K. W., et al. (2019). Compromised Metabolic Reprogramming Is an Early Indicator of CD8(+) T Cell Dysfunction during Chronic Mycobacterium tuberculosis Infection. Cell Rep. 29 (11), 3564–3579 e5. doi: 10.1016/j.celrep.2019.11.034
Saini V., Chinta K. C., Reddy V. P., Glasgow J. N., Stein A., Lamprecht D. A., et al. (2020). Hydrogen sulfide stimulates Mycobacterium tuberculosis respiration, growth and pathogenesis. Nat. Commun. 11 (1), 557. doi: 10.1038/s41467-019-14132-y
Sakai S., Kauffman K. D., Sallin M. A., Sharpe A. H., Young H. A., Ganusov V. V., et al. (2016). CD4 T Cell-Derived IFN-gamma Plays a Minimal Role in Control of Pulmonary Mycobacterium tuberculosis Infection and Must Be Actively Repressed by PD-1 to Prevent Lethal Disease. PloS Pathog. 12 (5), e1005667. doi: 10.1371/journal.ppat.1005667
Sanderson R. T. (1988). Principles of electronegativity Part I. General nature. J. Chem. Educ. 65 (2), 112. doi: 10.1021/ed065p112
Sehrawat A., Croix C. S., Baty C. J., Watkins S., Tailor D., Singh R. P., et al. (2016). Inhibition of mitochondrial fusion is an early and critical event in breast cancer cell apoptosis by dietary chemopreventative benzyl isothiocyanate. Mitochondrion 30, 67–77. doi: 10.1016/j.mito.2016.06.006
Shatalin K., Shatalina E., Mironov A., Nudler E. (2011). H2S: a universal defense against antibiotics in bacteria. Science 334 (6058), 986–990. doi: 10.1126/science.1209855
Shefa U., Kim M. S., Jeong N. Y., Jung J. (2018). Antioxidant and Cell-Signaling Functions of Hydrogen Sulfide in the Central Nervous System. Oxid. Med. Cell Longev 2018, 1873962. doi: 10.1155/2018/1873962
Shehatou G. S., Suddek G. M. (2016). Sulforaphane attenuates the development of atherosclerosis and improves endothelial dysfunction in hypercholesterolemic rabbits. Exp. Biol. Med. (Maywood) 241 (4), 426–436. doi: 10.1177/1535370215609695
Shi L., Eugenin E. A., Subbian S. (2016). Immunometabolism in Tuberculosis. Front. Immunol. 7:150. doi: 10.3389/fimmu.2016.00150
Shibuya N., Tanaka M., Yoshida M., Ogasawara Y., Togawa T., Ishii K., et al. (2009). 3-Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid. Redox Signal 11 (4), 703–714. doi: 10.1089/ars.2008.2253
Shibuya N., Koike S., Tanaka M., Ishigami-Yuasa M., Kimura Y., Ogasawara Y., et al. (2013). A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat. Commun. 4, 1366. doi: 10.1038/ncomms2371
Shukla P., Khodade V. S., SharathChandra M., Chauhan P., Mishra S., Siddaramappa S., et al. (2017). “On demand” redox buffering by H2S contributes to antibiotic resistance revealed by a bacteria-specific H2S donor. Chem. Sci. 8 (7), 4967–4972. doi: 10.1039/C7SC00873B
Sia J. K., Rengarajan J. (2019). Immunology of Mycobacterium tuberculosis Infections. Microbiol. Spectr. 7 (4), 10.1128/microbiolspec.GPP3-0022-2018. doi: 10.1128/9781683670131.ch64
Sica A., Mantovani A. (2012). Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122 (3), 787–795. doi: 10.1172/JCI59643
Sica A., Erreni M., Allavena P., Porta C. (2015). Macrophage polarization in pathology. Cell Mol. Life Sci. 72 (21), 4111–4126. doi: 10.1007/s00018-015-1995-y
Singh S., Padovani D., Leslie R. A., Chiku T., Banerjee R. (2009). Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem. 284 (33), 22457–22466. doi: 10.1074/jbc.M109.010868
Son J., Lyssiotis C. A., Ying H., Wang X., Hua S., Ligorio M., et al. (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496 (7443), 101–105. doi: 10.1038/nature12040
Song K., Wang F., Li Q., Shi Y. B., Zheng H. F., Peng H., et al. (2014a). Hydrogen sulfide inhibits the renal fibrosis of obstructive nephropathy. Kidney Int. 85 (6), 1318–1329. doi: 10.1038/ki.2013.449
Song Z. J., Ng M. Y., Lee Z.-W., Dai W., Hagen T., Moore P. K., et al. (2014b). Hydrogen sulfide donors in research and drug development. MedChemComm 5 (5), 557–570. doi: 10.1039/C3MD00362K
Spassov S. G., Donus R., Ihle P. M., Engelstaedter H., Hoetzel A., Faller S. (2017). Hydrogen Sulfide Prevents Formation of Reactive Oxygen Species through PI3K/Akt Signaling and Limits Ventilator-Induced Lung Injury. Oxid. Med. Cell Longev 2017, 3715037. doi: 10.1155/2017/3715037
Steegborn C., Clausen T., Sondermann P., Jacob U., Worbs M., Marinkovic S., et al. (1999). Kinetics and inhibition of recombinant human cystathionine gamma-lyase. Toward the rational control of transsulfuration. J. Biol. Chem. 274 (18), 12675–12684. doi: 10.1074/jbc.274.18.12675
Suarez-Almazor M. E., Spooner C., Belseck E. (2000). Penicillamine for treating rheumatoid arthritis. Cochrane Database Syst. Rev. (4), CD001460. doi: 10.1002/14651858.CD001460
Sun Q., Collins R., Huang S., Holmberg-Schiavone L., Anand G. S., Tan C. H., et al. (2009). Structural basis for the inhibition mechanism of human cystathionine gamma-lyase, an enzyme responsible for the production of H(2)S. J. Biol. Chem. 284 (5), 3076–3085. doi: 10.1074/jbc.M805459200
Sun L., Sun S., Li Y., Pan W., Xie Y., Wang S., et al. (2014). Potential biomarkers predicting risk of pulmonary hypertension in congenital heart disease: the role of homocysteine and hydrogen sulfide, Chin Med J. (Engl) 127 (5), 893–899.
Sun J., Aponte A. M., Menazza S., Gucek M., Steenbergen C., Murphy E. (2016). Additive cardioprotection by pharmacological postconditioning with hydrogen sulfide and nitric oxide donors in mouse heart: S-sulfhydration vs. S-nitrosylation. Cardiovasc. Res. 110 (1), 96–106. doi: 10.1093/cvr/cvw037
Sunzini F., De Stefano S., Chimenti M. S., Melino S. (2020). Hydrogen Sulfide as Potential Regulatory Gasotransmitter in Arthritic Diseases. Int. J. Mol. Sci. 21 (4), 1180. doi: 10.3390/ijms21041180
Suzuki K., Olah G., Modis K., Coletta C., Kulp G., Gero D., et al. (2011). Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc. Natl. Acad. Sci. U.S.A. 108 (33), 13829–13834. doi: 10.1073/pnas.1105121108
Szabo C., Coletta C., Chao C., Modis K., Szczesny B., Papapetropoulos A., et al. (2013). Tumor-derived hydrogen sulfide, produced by cystathionine-beta-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Natl. Acad. Sci. U.S.A. 110 (30), 12474–12479. doi: 10.1073/pnas.1306241110
Szabo C., Ransy C., Modis K., Andriamihaja M., Murghes B., Coletta C., et al. (2014). Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br. J. Pharmacol. 171 (8), 2099–2122. doi: 10.1111/bph.12369
Szabo C. (2007). Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discovery 6 (11), 917–935. doi: 10.1038/nrd2425
Szabo C. (2012). Roles of hydrogen sulfide in the pathogenesis of diabetes mellitus and its complications. Antioxid. Redox Signal 17 (1), 68–80. doi: 10.1089/ars.2011.4451
Szabo C. (2018). A timeline of hydrogen sulfide (H2S) research: From environmental toxin to biological mediator. Biochem. Pharmacol. 149, 5–19. doi: 10.1016/j.bcp.2017.09.010
Talaei F., van Praag V. M., Henning R. H. (2013). Hydrogen sulfide restores a normal morphological phenotype in Werner syndrome fibroblasts, attenuates oxidative damage and modulates mTOR pathway. Pharmacol. Res. 74, 34–44. doi: 10.1016/j.phrs.2013.04.011
Tamizhselvi R., Moore P. K., Bhatia M. (2008). Inhibition of hydrogen sulfide synthesis attenuates chemokine production and protects mice against acute pancreatitis and associated lung injury. Pancreas 36 (4), e24–e31. doi: 10.1097/MPA.0b013e31816857bb
Tang G., Zhang L., Yang G., Wu L., Wang R. (2013). Hydrogen sulfide-induced inhibition of L-type Ca2+ channels and insulin secretion in mouse pancreatic beta cells. Diabetologia 56 (3), 533–541. doi: 10.1007/s00125-012-2806-8
Tannahill G. M., Curtis A. M., Adamik J., Palsson-McDermott E. M., McGettrick A. F., Goel G., et al. (2013). Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496 (7444), 238–242. doi: 10.1038/nature11986
Taoka S., Banerjee R. (2001). Characterization of NO binding to human cystathionine beta-synthase: possible implications of the effects of CO and NO binding to the human enzyme. J. Inorg. Biochem. 87 (4), 245–251. doi: 10.1016/S0162-0134(01)00335-X
Taoka S., Ohja S., Shan X., Kruger W. D., Banerjee R. (1998). Evidence for heme-mediated redox regulation of human cystathionine beta-synthase activity. J. Biol. Chem. 273 (39), 25179–25184. doi: 10.1074/jbc.273.39.25179
Taoka S., Widjaja L., Banerjee R. (1999). Assignment of enzymatic functions to specific regions of the PLP-dependent heme protein cystathionine beta-synthase. Biochemistry 38 (40), 13155–13161. doi: 10.1021/bi990865t
Taoka S., Lepore B. W., Kabil O., Ojha S., Ringe D., Banerjee R. (2002). Human cystathionine beta-synthase is a heme sensor protein. Evidence that the redox sensor is heme and not the vicinal cysteines in the CXXC motif seen in the crystal structure of the truncated enzyme. Biochemistry 41 (33), 10454–10461. doi: 10.1021/bi026052d
Teague B., Asiedu S., Moore P. K. (2002). The smooth muscle relaxant effect of hydrogen sulphide in vitro: evidence for a physiological role to control intestinal contractility. Br. J. Pharmacol. 137 (2), 139–145. doi: 10.1038/sj.bjp.0704858
Teng H., Wu B., Zhao K., Yang G., Wu L., Wang R. (2013). Oxygen-sensitive mitochondrial accumulation of cystathionine β-synthase mediated by Lon protease. Proc. Natl. Acad. Sci. U. S. A. Jul 30;110 (31): 12679–84. doi: 10.1073/pnas.1308487110
Thornburg J. M., Nelson K. K., Clem B. F., Lane A. N., Arumugam S., Simmons A., et al. (2008). Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 10 (5), R84. doi: 10.1186/bcr2154
Toliver-Kinsky T., Cui W., Toro G., Lee S. J., Shatalin K., Nudler E., et al. (2019). H2S, a Bacterial Defense Mechanism against the Host Immune Response. Infect. Immun. 87 (1), e00272–e00218. doi: 10.1128/IAI.00272
Toohey J. II (2012). The conversion of H(2)S to sulfane sulfur. Nat. Rev. Mol. Cell Biol. 13 (12), 803. doi: 10.1038/nrm3391-c1
Van den Bossche J., O’Neill L. A., Menon D. (2017). Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 38 (6), 395–406. doi: 10.1016/j.it.2017.03.001
Vandiver M., Snyder S. H. (2012). Hydrogen sulfide: a gasotransmitter of clinical relevance. J. Mol. Med. (Berl) 90 (3), 255–263. doi: 10.1007/s00109-012-0873-4
Vicente J. B., Colaco H. G., Mendes M. II, Sarti P., Leandro P., Giuffre A. (2014). NO* binds human cystathionine beta-synthase quickly and tightly. J. Biol. Chem. 289 (12), 8579–8587. doi: 10.1074/jbc.M113.507533
Vilcheze C., Hartman T., Weinrick B., Jain P., Weisbrod T. R., Leung L. W., et al. (2017). Enhanced respiration prevents drug tolerance and drug resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 114 (17), 4495–4500. doi: 10.1073/pnas.1704376114
Viola A., Munari F., Sanchez-Rodriguez R., Scolaro T., Castegna A. (2019). The Metabolic Signature of Macrophage Responses. Front. Immunol. 10:1462. doi: 10.3389/fimmu.2019.01462
Vitvitsky V., Yadav P. K., Kurthen A., Banerjee R. (2015). Sulfide oxidation by a noncanonical pathway in red blood cells generates thiosulfate and polysulfides. J. Biol. Chem. 290 (13), 8310–8320. doi: 10.1074/jbc.M115.639831
Vitvitsky V., Miljkovic J. L., Bostelaar T., Adhikari B., Yadav P. K., Steiger A. K., et al. (2018). Cytochrome c Reduction by H2S Potentiates Sulfide Signaling. ACS Chem. Biol. 13 (8), 2300–2307. doi: 10.1021/acschembio.8b00463
Wallace J. L., Wang R. (2015). Hydrogen sulfide-based therapeutics: exploiting a unique but ubiquitous gasotransmitter. Nat. Rev. Drug Discovery 14 (5), 329–345. doi: 10.1038/nrd4433
Wallace J. L., Dicay M., McKnight W., Martin G. R. (2007a). Hydrogen sulfide enhances ulcer healing in rats. FASEB J. 21 (14), 4070–4076. doi: 10.1096/fj.07-8669com
Wallace J. L., Caliendo G., Santagada V., Cirino G., Fiorucci S. (2007b). Gastrointestinal safety and anti-inflammatory effects of a hydrogen sulfide-releasing diclofenac derivative in the rat. Gastroenterology 132 (1), 261–271. doi: 10.1053/j.gastro.2006.11.042
Wallace J. L., Blackler R. W., Chan M. V., Da Silva G. J., Elsheikh W., Flannigan K. L., et al. (2015). Anti-inflammatory and cytoprotective actions of hydrogen sulfide: translation to therapeutics. Antioxid. Redox Signaling 22 (5), 398–410. doi: 10.1089/ars.2014.5901
Walsh B. J. C., Giedroc D. P. (2020). H2S and reactive sulfur signaling at the host-bacterial pathogen interface. J. Biol. Chem. 295 (38), 13150–13168. doi: 10.1074/jbc.REV120.011304
Wang Y., Zhao X., Jin H., Wei H., Li W., Bu D., et al. (2009). Role of hydrogen sulfide in the development of atherosclerotic lesions in apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 29 (2), 173–179. doi: 10.1161/ATVBAHA.108.179333
Wang P., Zhang G., Wondimu T., Ross B., Wang R. (2011). Hydrogen sulfide and asthma. Exp. Physiol. 96 (9), 847–852. doi: 10.1113/expphysiol.2011.057448
Wang C., Han J., Xiao L., Jin C. E., Li D. J., Yang Z. (2014). Role of hydrogen sulfide in portal hypertension and esophagogastric junction vascular disease. World J. Gastroenterol. 20 (4), 1079–1087. doi: 10.3748/wjg.v20.i4.1079
Wang T., Liu H., Lian G., Zhang S. Y., Wang X., Jiang C. (2017). HIF1alpha-Induced Glycolysis Metabolism Is Essential to the Activation of Inflammatory Macrophages. Mediators Inflammation 2017, 9029327. doi: 10.1155/2017/9029327
Wang Z. J., Wu J., Guo W., Zhu Y. Z. (2017). Atherosclerosis and the Hydrogen Sulfide Signaling Pathway - Therapeutic Approaches to Disease Prevention. Cell Physiol. Biochem. 42 (3), 859–875. doi: 10.1159/000478628
Wang L., Cai H., Hu Y., Liu F., Huang S., Zhou Y., et al. (2018). A pharmacological probe identifies cystathionine beta-synthase as a new negative regulator for ferroptosis. Cell Death Dis. 9 (10), 1005. doi: 10.1038/s41419-018-1063-2
Wang X. H., Wang F., You S. J., Cao Y. J., Cao L. D., Han Q., et al. (2013). Dysregulation of cystathionine gamma-lyase (CSE)/hydrogen sulfide pathway contributes to ox-LDL-induced inflammation in macrophage. Cell Signal 25 (11), 2255–2262. doi: 10.1016/j.cellsig.2013.07.010
Wang Y., Chun O. K., Song W. O. (2013). Plasma and dietary antioxidant status as cardiovascular disease risk factors: a review of human studies. Nutrients 5 (8), 2969–3004. doi: 10.3390/nu5082969
Wang R. (2012). Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 92 (2), 791–896. doi: 10.1152/physrev.00017.2011
Wang R. (2014). Gasotransmitters: growing pains and joys. Trends Biochem. Sci. 39 (5), 227–232. doi: 10.1016/j.tibs.2014.03.003
Watanabe M., Osada J., Aratani Y., Kluckman K., Reddick R., Malinow M. R., et al. (1995). Mice deficient in cystathionine beta-synthase: animal models for mild and severe homocyst(e)inemia. Proc. Natl. Acad. Sci. U.S.A. 92 (5), 1585–1589. doi: 10.1073/pnas.92.5.1585
Waters A., Torregrossa R., Gero D., Perry A., Wood M. E., Whiteman M. (2017). RT01, A Novel Derivative of the Mitochondria-targeted Hydrogen Sulfide Donor AP39, Reversed Hyperglycaemia-induced Mitochondrial Dysfunction in Murine Brain Microvascular Endothelial Cells. Free Radical Biol. Med. 112, 157–158. doi: 10.1016/j.freeradbiomed.2017.10.242
Wheeler P. R., Coldham N. G., Keating L., Gordon S. V., Wooff E. E., Parish T., et al. (2005). Functional demonstration of reverse transsulfuration in the Mycobacterium tuberculosis complex reveals that methionine is the preferred sulfur source for pathogenic Mycobacteria. J. Biol. Chem. 280 (9), 8069–8078. doi: 10.1074/jbc.M412540200
Whiteman M., Winyard P. G. (2011). Hydrogen sulfide and inflammation: the good, the bad, the ugly and the promising. Expert Rev. Clin. Pharmacol. 4 (1), 13–32. doi: 10.1586/ecp.10.134
WHO. (2020). Available at: https://www.who.int/emergencies/diseases/novel-coronavirus-2019
Whiteman M., Gooding K. M., Whatmore J. L., Ball C. II, Mawson D., Skinner K., et al. (2010a). Adiposity is a major determinant of plasma levels of the novel vasodilator hydrogen sulphide. Diabetologia 53 (8), 1722–1726. doi: 10.1007/s00125-010-1761-5
Whiteman M., Li L., Rose P., Tan C. H., Parkinson D. B., Moore P. K. (2010b). The effect of hydrogen sulfide donors on lipopolysaccharide-induced formation of inflammatory mediators in macrophages. Antioxid. Redox Signal 12 (10), 1147–1154. doi: 10.1089/ars.2009.2899
Wu L., Yang W., Jia X., Yang G., Duridanova D., Cao K., et al. (2009). Pancreatic islet overproduction of H2S and suppressed insulin release in Zucker diabetic rats. Lab. Invest. 89 (1), 59–67. doi: 10.1038/labinvest.2008.109
Wu B., Teng H., Yang G., Wu L., Wang R. (2012). Hydrogen sulfide inhibits the translational expression of hypoxia-inducible factor-1alpha. Br. J. Pharmacol. 167 (7), 1492–1505. doi: 10.1111/j.1476-5381.2012.02113.x
Wu Z., Peng H., Du Q., Lin W., Liu Y. (2015). GYY4137, a hydrogen sulfidereleasing molecule, inhibits the inflammatory response by suppressing the activation of nuclear factorkappa B and mitogenactivated protein kinases in Coxsackie virus B3infected rat cardiomyocytes. Mol. Med. Rep. 11 (3), 1837–1844. doi: 10.3892/mmr.2014.2901
Xiao Q., Ying J., Xiang L., Zhang C. (2018). The biologic effect of hydrogen sulfide and its function in various diseases. Med. (Baltimore) 97 (44), e13065. doi: 10.1097/MD.0000000000013065
Yadav P. K., Yamada K., Chiku T., Koutmos M., Banerjee R. (2013). Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J. Biol. Chem. 288 (27), 20002–20013. doi: 10.1074/jbc.M113.466177
Yadav P. K., Martinov M., Vitvitsky V., Seravalli J., Wedmann R., Filipovic M. R., et al. (2016). Biosynthesis and Reactivity of Cysteine Persulfides in Signaling. J. Am. Chem. Soc. 138 (1), 289–299. doi: 10.1021/jacs.5b10494
Yang G., Sun X., Wang R. (2004). Hydrogen sulfide-induced apoptosis of human aorta smooth muscle cells via the activation of mitogen-activated protein kinases and caspase-3. FASEB J. 18 (14), 1782–1784. doi: 10.1096/fj.04-2279fje
Yang W., Yang G., Jia X., Wu L., Wang R. (2005). Activation of KATP channels by H2S in rat insulin-secreting cells and the underlying mechanisms. J. Physiol. 569 (Pt 2), 519–531. doi: 10.1113/jphysiol.2005.097642
Yang G., Wu L., Wang R. (2006). Pro-apoptotic effect of endogenous H2S on human aorta smooth muscle cells. FASEB J. 20 (3), 553–555. doi: 10.1096/fj.05-4712fje
Yang G. (2020). H2S as a potential defense against COVID-19? Am. J. Physiol. Cell Physiol. 319 (2), C244–C249. doi: 10.1152/ajpcell.00187.2020
Yurinskaya M. M., Krasnov G. S., Kulikova D. A., Zatsepina O. G., Vinokurov M. G., Chuvakova L. N., et al. (2020). H2S counteracts proinflammatory effects of LPS through modulation of multiple pathways in human cells. Inflammation Res. 69 (5), 481–495. doi: 10.1007/s00011-020-01329-x
Yusuf M., Kwong Huat B. T., Hsu A., Whiteman M., Bhatia M., Moore P. K. (2005). Streptozotocin-induced diabetes in the rat is associated with enhanced tissue hydrogen sulfide biosynthesis. Biochem. Biophys. Res. Commun. 333 (4), 1146–1152. doi: 10.1016/j.bbrc.2005.06.021
Zanardo R. C., Brancaleone V., Distrutti E., Fiorucci S., Cirino G., Wallace J. L. (2006). Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 20 (12), 2118–2120. doi: 10.1096/fj.06-6270fje
Zeng J., Lin X., Fan H., Li C. (2013). Hydrogen sulfide attenuates the inflammatory response in a mouse burn injury model. Mol. Med. Rep. 8 (4), 1204–1208. doi: 10.3892/mmr.2013.1610
Zhang X., Bian J. S. (2014). Hydrogen sulfide: a neuromodulator and neuroprotectant in the central nervous system. ACS Chem. Neurosci. 5 (10), 876–883. doi: 10.1021/cn500185g
Zhang H., Zhi L., Moore P. K., Bhatia M. (2006). Role of hydrogen sulfide in cecal ligation and puncture-induced sepsis in the mouse. Am. J. Physiol. Lung Cell Mol. Physiol. 290 (6), L1193–L1201. doi: 10.1152/ajplung.00489.2005
Zhang H., Hegde A., Ng S. W., Adhikari S., Moochhala S. M., Bhatia M. (2007a). Hydrogen sulfide up-regulates substance P in polymicrobial sepsis-associated lung injury. J. Immunol. 179 (6), 4153–4160. doi: 10.4049/jimmunol.179.6.4153
Zhang H., Zhi L., Moochhala S., Moore P. K., Bhatia M. (2007b). Hydrogen sulfide acts as an inflammatory mediator in cecal ligation and puncture-induced sepsis in mice by upregulating the production of cytokines and chemokines via NF-κB. Am. J. Physiol. - Lung Cell. Mol. Physiol. 292 (4), 960–971. doi: 10.1152/ajplung.00388.2006
Zhang H., Zhi L., Moochhala S. M., Moore P. K., Bhatia M. (2007c). Endogenous hydrogen sulfide regulates leukocyte trafficking in cecal ligation and puncture-induced sepsis. J. Leukoc. Biol. 82 (4), 894–905. doi: 10.1189/jlb.0407237
Zhang H., Moochhala S. M., Bhatia M. (2008). Endogenous hydrogen sulfide regulates inflammatory response by activating the ERK pathway in polymicrobial sepsis. J. Immunol. 181 (6), 4320–4331. doi: 10.4049/jimmunol.181.6.4320
Zhang H., Guo C., Wu D., Zhang A., Gu T., Wang L., et al. (2012). Hydrogen sulfide inhibits the development of atherosclerosis with suppressing CX3CR1 and CX3CL1 expression. PloS One 7 (7), e41147. doi: 10.1371/journal.pone.0041147
Zhang L., Yang G., Untereiner A., Ju Y., Wu L., Wang R. (2013). Hydrogen sulfide impairs glucose utilization and increases gluconeogenesis in hepatocytes. Endocrinology 154 (1), 114–126. doi: 10.1210/en.2012-1658
Zhang Y., Tang Z. H., Ren Z., Qu S. L., Liu M. H., Liu L. S., et al. (2013). Hydrogen sulfide, the next potent preventive and therapeutic agent in aging and age-associated diseases. Mol. Cell Biol. 33 (6), 1104–1113. doi: 10.1128/MCB.01215-12
Zhang H. X., Liu S. J., Tang X. L., Duan G. L., Ni X., Zhu X. Y., et al. (2016). H2S Attenuates LPS-Induced Acute Lung Injury by Reducing Oxidative/Nitrative Stress and Inflammation. Cell Physiol. Biochem. 40 (6), 1603–1612. doi: 10.1159/000453210
Zhang J., Sio S. W., Moochhala S., Bhatia M. (2010). Role of hydrogen sulfide in severe burn injury-induced inflammation in mice. Mol. Med. 16 (9-10), 417–424. doi: 10.2119/molmed.2010.00027
Zhang J. H., Dong Z., Chu L. (2010). Hydrogen sulfide induces apoptosis in human periodontium cells. J. Periodontal Res. 45 (1), 71–78. doi: 10.1111/j.1600-0765.2009.01202.x
Zhao Y., Pluth M. D. (2016). Hydrogen Sulfide Donors Activated by Reactive Oxygen Species. Angew Chem. Int. Ed Engl. 55 (47), 14638–14642. doi: 10.1002/anie.201608052
Zhao W., Wang R. (2002). H(2)S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am. J. Physiol. Heart Circ. Physiol. 283 (2), H474–H480. doi: 10.1152/ajpheart.00013.2002
Zhao W., Zhang J., Lu Y., Wang R. (2001). The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 20 (21), 6008–6016. doi: 10.1093/emboj/20.21.6008
Zhao Y., Wang H., Xian M. (2011). Cysteine-activated hydrogen sulfide (H2S) donors. J. Am. Chem. Soc. 133 (1), 15–17. doi: 10.1021/ja1085723
Zhao Y., Bhushan S., Yang C., Otsuka H., Stein J. D., Pacheco A., et al. (2013). Controllable hydrogen sulfide donors and their activity against myocardial ischemia-reperfusion injury. ACS Chem. Biol. 8 (6), 1283–1290. doi: 10.1021/cb400090d
Zhao Y., Henthorn H. A., Pluth M. D. (2017). Kinetic insights into hydrogen sulfide delivery from caged-carbonyl sulfide isomeric donor platforms. J. Am. Chem. Soc. 139 (45), 16365–16376. doi: 10.1021/jacs.7b09527
Zheng Y., Yu B., De La Cruz L. K., Roy Choudhury M., Anifowose A., Wang B. (2018). Toward Hydrogen Sulfide Based Therapeutics: Critical Drug Delivery and Developability Issues. Med. Res. Rev. 38 (1), 57–100. doi: 10.1002/med.21433
Zheng Q., Pan L., Ji Y. (2019). H2S protects against diabetes-accelerated atherosclerosis by preventing the activation of NLRP3 inflammasome. J. BioMed. Res. 34 (2), 94–102. doi: 10.7555/JBR.33.20190071
Zhi L., Ang A. D., Zhang H., Moore P. K., Bhatia M. (2007). Hydrogen sulfide induces the synthesis of proinflammatory cytokines in human monocyte cell line U937 via the ERK-NF-kappaB pathway. J. Leukoc. Biol. 81 (5), 1322–1332. doi: 10.1189/jlb.1006599
Zhou X., Chu X., Xin D., Li T., Bai X., Qiu J., et al. (2019). L-Cysteine-Derived H2S Promotes Microglia M2 Polarization via Activation of the AMPK Pathway in Hypoxia-Ischemic Neonatal Mice. Front. Mol. Neurosci. 12:58. doi: 10.3389/fnmol.2019.00058
Zhou F., Yu T., Du R., Fan G., Liu Y., Liu Z., et al. (2020). Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395 (10229), 1054–1062. doi: 10.1016/S0140-6736(20)30566-3
Zhu Y. Z., Wang Z. J., Ho P., Loke Y. Y., Zhu Y. C., Huang S. H., et al. (2007). Hydrogen sulfide and its possible roles in myocardial ischemia in experimental rats. J. Appl. Physiol. (1985) 102 (1), 261–268. doi: 10.1152/japplphysiol.00096.2006
Zumla A., Rao M., Parida S. K., Keshavjee S., Cassell G., Wallis R., et al. (2015). Inflammation and tuberculosis: host-directed therapies. J. Intern Med. 277 (4), 373–387. doi: 10.1111/joim.12256
Keywords: H2S, hydrogen sulfide, Mycobacterium tuberculosis, immunometabolism, inflammation, CSE, CBS, 3-MST
Citation: Rahman MA, Glasgow JN, Nadeem S, Reddy VP, Sevalkar RR, Lancaster JR Jr and Steyn AJC (2020) The Role of Host-Generated H2S in Microbial Pathogenesis: New Perspectives on Tuberculosis. Front. Cell. Infect. Microbiol. 10:586923. doi: 10.3389/fcimb.2020.586923
Received: 24 July 2020; Accepted: 13 October 2020;
Published: 10 November 2020.
Edited by:
Patricia Ann Champion, University of Notre Dame, United StatesReviewed by:
Alessandro Giuffre, Institute of Molecular Biology and Pathology (CNR), ItalyRajagopal Kammara, Central Food Technological Research Institute (CSIR), India
Copyright © 2020 Rahman, Glasgow, Nadeem, Reddy, Sevalkar, Lancaster and Steyn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adrie J. C. Steyn, YXN0ZXluQHVhYi5lZHU=