 Liqi Zhu
Liqi Zhu Chen Zhu1,2
Chen Zhu1,2 Quan Zhang
Quan Zhang- 1Institute of Comparative Medicine, College of Veterinary Medicine, Yangzhou University, Yangzhou, China
- 2Jiangsu Co-Innovation Center for Prevention and Control of Important Animal Infectious Diseases and Zoonoses, Yangzhou University, Yangzhou, China
- 3Affiliated Hospital of Yangzhou University, Yangzhou University, Yangzhou, China
It has been well documented that cytolethal distending toxin (CDT) from Helicobacter hepaticus (H. hepaticus), Campylobacter jejuni (C. jejuni) and other Gram-negative intestinal pathogens is linked to the inflammatory bowel disease (IBD). However, the mechanisms underlying the progression of H. hepaticus induced colitis remains unclear. In this study, male B6.129P2-IL10tm1Cgn/J mice were infected by H. hepaticus and ΔCdtB H. hepaticus for 6, 12, 18, and 24 weeks. Histopathology, H. hepaticus colonization levels, expression of inflammatory cytokines, signaling pathways, and content of NO in proximal colon were examined. We found that Cytolethal distending toxin subunit B (CdtB) deletion had no influence on colonization ability of H. hepaticus in colon of B6.129P2-IL10tm1cgn/J mice, and there was no significant difference in abundance of colonic H. hepaticus over infection duration. H. hepaticus aggravated rectocele and proximal colonic inflammation, especially at 24 WPI, while ΔCdtB H. hepaticus could not cause significant symptom. Furthermore, mRNA levels of Il-6, Tnf-α, Il-1β, and iNOS significantly increased in the proximal colon of H. hepaticus-infected mice compared to ΔCdtB H. hepaticus infected group from 12 WPI to 24 WPI. In addition, the elevated content of NO and activated Stat3 and Jak2 in colon were observed in H. hepaticus infected mice. These data demonstrated that CdtB promote colitis development in male B6.129P2-IL10tm1Cgn/J mice by induction of inflammatory response and activation of Jak-Stat signaling pathway.
Introduction
Helicobacter hepaticus (H. hepaticus) belongs to Proteobacteria, ϵ-proteobacteria, Heliobacterium. H. hepaticus, the prototype of human enterohepatic helicobacters, infection is characterized by colitis (Xu et al., 2018), and induce colon cancer (Falsafi and Mahboubi, 2013) in susceptible mice strains. Immunodeficient mice infected by H. hepaticus are often used as animal models for inflammatory bowel disease (IBD) studies (Cahill et al., 1997). In general, H. hepaticus can cause IBD in IL-10 deficient mice, but is an enteric symbiotic bacterium in wild-type C57/BL6 mice. These results may be caused by the secretion of a high level anti-inflammatory cytokine by macrophages, which were activated by large polysaccharide secreted from H. hepaticus (Danne et al., 2017). But there are also contrary studies pointing out that H. hepaticus does not induce or potentiate colitis in IL-10 deficient Mice (Dieleman et al., 2000). Moreover, Mark et al. found that H. hepaticus do not induce typical colonic inflammation symptoms unless it co-infected with the potential probiotic Lactobacillus reuteri in Germ-free mice (Whary et al., 2011). Therefore, the mechanism of H. hepaticus inducing to inflammatory bowel disease is complicated, which needs to be further explored.
Cytolethal distending toxin (CDT), a known major virulence of H. hepaticus, is a type of AB2 toxin found in Gram-negative bacteria, including Heliobacterium, Salmonella, Shigella, Campylobacter jejuni, and Escherichia coli. The CdtA and CdtB subunits produce non-spherical amino acid extensions that interact with the CdtC subunit (Avenaud et al., 2004). Usually, the subunits CdtA and CdtC play the role of connecting transportation, transferring the active subunit CdtB into cells (Smith and Bayles, 2006). It has been reported that the biological functions of CdtB include phosphatase activity, blocking cell cycle, and eventually leading to cell death (Ge et al., 2008). CdtB is similar to members of the DNaseI family, which can bind to cations and contain DNA-catalyzed residues, but its hydrolysis activity to DNA is about 100 times lower than that of DNaseI (Dassanayake et al., 2005). Meanwhile, it activates the ATM-dependent DNA damage reaction in a myc-dependent pathway (Guerra et al., 2010). Many studies have found that CDT plays an important role in induction of IBD and carcinogenesis (Young et al., 2004; Ge et al., 2017). In previous studies, H. hepaticus infection has been shown significantly induced ileum and cecum Ifn-γ production and mucosal IgA secretion as well as down-regulated Il-10 (Kullberg et al., 1998; Ge et al., 2005). In a recent study, Ge et al. demonstrated that CdtB could upregulate Tnf-α and Il-6 expression and cause DNA double-strand break in cecal epithelial cells, as well as Stat3 activation, leading to precancerous lesions (Ge et al., 2017). However, the roles of CdtB of H. hepaticus induced colitis are still to be delineated.
Therefore, the purpose of this study was to investigate the effects of CdtB of H. hepaticus infection on inflammatory bowel disease by using a CdtB deleted H. hepaticus strain, and further clarify the mechanism of H. hepaticus infection induced colitis.
Materials and Methods
Mice
Female and male B6.129P2-IL10tm1Cgn/J mice (Il10−/− mice) were purchased from Jackson Laboratory (Stock No: 002251), all mice were bred and maintained in an accredited specific pathogen-free facility, and in vivo experiments were conducted in accordance with the China laboratory Act (2017) under a Project License [SYXK (su) 2007-0044] authorized by Jiangsu Provincial Science and Technology Department and approved by Institutional Animal Care and Use Committee (IACUC) of Yangzhou University. The mice were free of Helicobacter species as assessed by PCR as previously described (Qin et al., 2016).
Bacterial Culture and Collection
CdtB mutant strain (ΔCdtB H. hepaticus) was constructed by Zhu et al. (2020), and conserved in our lab. In brief, the CdtB gene was replaced to Chloramphenicol resistant gene (Cm) by homologous recombination through electro-transforming the CdtA-Cm-CdtC fragment to H. hepaticus competent cell; then, the positive transformants were continuously subcultured on Brucella agar plates with chloramphenicol for five times. H. hepaticus 3B1 (ATCC 51449) and ΔCdtB H. hepaticus were cultured on Brucella agar plates (BD, USA) supplemented with 5% defibrinated sheep blood and antibiotics for 4–5 days under microaerobic conditions (85% N2, 10% CO2, 5% O2) at 37°C. Bacteria were harvested in PBS and used for oral infection when OD600 reading was 1 (Xu et al., 2018).
Experimental Details
Approximately 60 5-week-old male mice were randomly divided into three groups. In order to promote the colonization of H. hepaticus in colon and rectum, the mice drank free water with gentamicin for 1 week (Kennedy et al., 2018), and then were infected with H. hepaticus and ΔCdtB H. hepaticus at the age of 6 weeks. The control group was inoculated with PBS. The mice were inoculated intragastrically with 200 μl of PBS containing 2 × 108 H. hepaticus (Xu et al., 2018) every 2 days for a week, and the infection was confirmed by feces sampling. Five mice from each group were euthanized at 6, 12, 18, 24 WPI, respectively. Colon tissues were collected and stored at −80°C for analysis.
DNA Extraction and H. hepaticus Quantitative PCR Analysis
The mice were fasted overnight before sacrificed. The proximal colon segments in the same position were selected, then the tissue and bacterial DNA was extracted according to manufacturer’s instructions using the TIANamp Bacteria DNA kit (Tiangen, Beijing). Abundance of H. hepaticus in colon was determined according to HH1450 gene primers by qPCR according to the method established by Ge et al. (2005). In brief, to quantitative detect the copy number of H. hepaticus, the tissue DNA was diluted to 10 ng/μl and used as a template for quantitation PCR assay in the Applied Biosystems StepOne Real Time PCR System (ABI) using Universal SYBR Green master (ABI). Serial dilutions of H. hepaticus DNA, including 2 × 107, 2 × 106, 2 × 104, 2 × 102, 2 × 101, and 2 fg, were used to generate a standard curve.
Histopathology
During necropsy, colon tissues of each animal were fixed in 4% paraformaldehyde for 24 h, and proximal, middle, and distal part embedded in paraffin for microscopy examination respectively. For histopathological analysis, tissue samples were cut into 4 μm sections, deparaffinized, and stained with hematoxylin and eosin (H&E). Histopathologic parameters were graded as described previously (Berg et al., 1996; Erdman et al., 2009). In brief, score from 0 to 4 was based on the following criteria: (grade 0) normal colonic mucosa without lymphocyte infiltration and disorganization; (grade 1) mild inflammation with mononuclear cell infiltrates in the lamina propria accompanied by minimal epithelial structure change; (grade 2) lesions tended to involve more of the colon than grade 1 lesions, or were more frequent; (grade 3) lesions involved a large area of the mucosa or were more frequent than grade 2 lesions; (grade 4) lesions usually involved most of the colonic section and were more severe than grade 3 lesions. Each sample was randomly selected for five slices, and given a score based on the criteria described above and the summation of these scores provided a total colonic disease score per mouse. The detailed pathological characteristics was referred to Table 1. The disease scores could range from 0 (no change in any segment) to a maximum of 20 (grade 4 lesions in all five segments).

Table 1 The criteria of Histologic analysis (Berg et al., 1996).
Cytokines Quantification by Real-Time RT-PCR
Total RNA was extracted from proximal colon tissue trituration in liquid nitrogen using Trizol (Roche, Germany). cDNA was synthesized from 1 μg total RNA using Prime Script RT reagent Kit with gDNA Eraser (Takara, Dalian). Transcript levels of colonic were amplified by qPCR using the Universal SYBR Green master and mRNA level of the GAPDH gene in each cDNA sample was measured and used for normalization. qPCR primers (Il-1β: TGATGAGAATGACCTCTTCT,CTTCTTCAAAGATGAAGGAAA; Il-6: TAGTCCTTCCTACCCCAATTTCC,TTGGTCCTTAGCCACTCCTTC; Tnf-α: AACTAGTGGTGCCAGCCGAT,CTTCACAGAGCAATGACTCC; iNOS: GTTGAAGACTGAGACTCTGG,GACTAGGCTACTCCGTGGA; MMP-9: TGGGGGGCAACTCGGC,GGAATGATCTAAGCCCAG; ZO-1: GGGGCCTACACTGATCAAGA,TGGAGATGAGGCTTCTGCTT; GAPDH: CCATCACCATCTTCCAGGAG,CCTGCTTCACCACCTTCTTG) were synthesized by Sangon Biotech (Shanghai) Co., Ltd. All samples were run in triplicate in the Applied Biosystems StepOne Real-Time PCR System. Relative expression levels were calculated using 2-ΔΔCT method, as previously described (Livak and Schmittgen, 2001).
ELISA
Total proteins were extracted from proximal colon tissue trituration in liquid nitrogen using IP Lysis Buffer (Thermo, USA) containing 1% PMSF and 1% phosphatase inhibitor cocktail. The samples were performed ultrasonic cracking at a power of 200 W for 5 s, then centrifuge at 12,000 rpm for 30 min at 4°C, and the Il-6 content from supernatant was detected with the Mouse IL-6 ELISA kit (Boster, Wuhan).
Western Blot Analysis
Total proteins were extracted from proximal colon tissue trituration in liquid nitrogen using RIPA lysis buffer (Thermo, USA) containing 1% PMSF and 1% phosphatase inhibitor cocktail. The samples were performed ultrasonic cracking at a power of 200 W for 5 s, then centrifuge at 12,000 rpm for 15 min at 4°C. The amount of protein was determined by BCA kit, and 40 μg of total proteins was loaded into 10% SDS-PAGE. After electrophoresis, proteins were transferred to a 0.22 μm PVDF membrane at 300 mA for 1.5 h, 5% skim milk was used for blocking at room temperature for 2 h. The membranes were incubated with primary antibody overnight at 4°C, including p-Stat3, Stat3, p-Jak2, Jak2, GAPDH (1:1,000, Cell Signaling Technology), and then incubated with secondary antibody conjugated to horseradish peroxidase (1:3,000, Cell Signaling Technology) at room temperature for 1 h. The proteins were visualized by Amersham ECL Select Western Blotting Detection Reagent.
Statistical Analysis
Statistical analysis was performed using SPSS 18.0 software. Pathological scores were analyzed using a Mann-Whitney nonparametric U test. H. hepaticus colonization levels, cytokines, ELISA, and protein expression were analyzed using unpaired two-tailed Student’s t test. Figures were drawn using GraphPad 8.0 software, * refers to the P value when compared with the control group, * for P < 0.05, ** for P < 0.01, and *** for P < 0.001.
Results
Lack of CdtB Did Not Affect the Colonization Efficiency of ΔCdtB H. hepaticus
It was reported that the ΔCdtB H. hepaticus lose their colonization efficiency over time in outbred Swiss Webster mice, inbred A/JCr mice and Il-10−/− mice (C57BL/6 background) (Ge et al., 2005; Pratt et al., 2006; Ge et al., 2007). We also examined the abundance of wild type H. hepaticus and CdtB mutant H. hepaticus in B6.129P2-IL10tm1Cgn/J mice of this study. Quantitative PCR was used to obtain the standard curve of H. hepaticus, and the abundance of two strains in the colon of Il10−/− mice was detected at necropsy. The results revealed that H. hepaticus and ΔCdtB H. hepaticus could be able to colonize in the colon of Il10−/− mice over the infection time (Figure 1), and there was no significant difference between H. hepaticus and ΔCdtB H. hepaticus colonization levels.
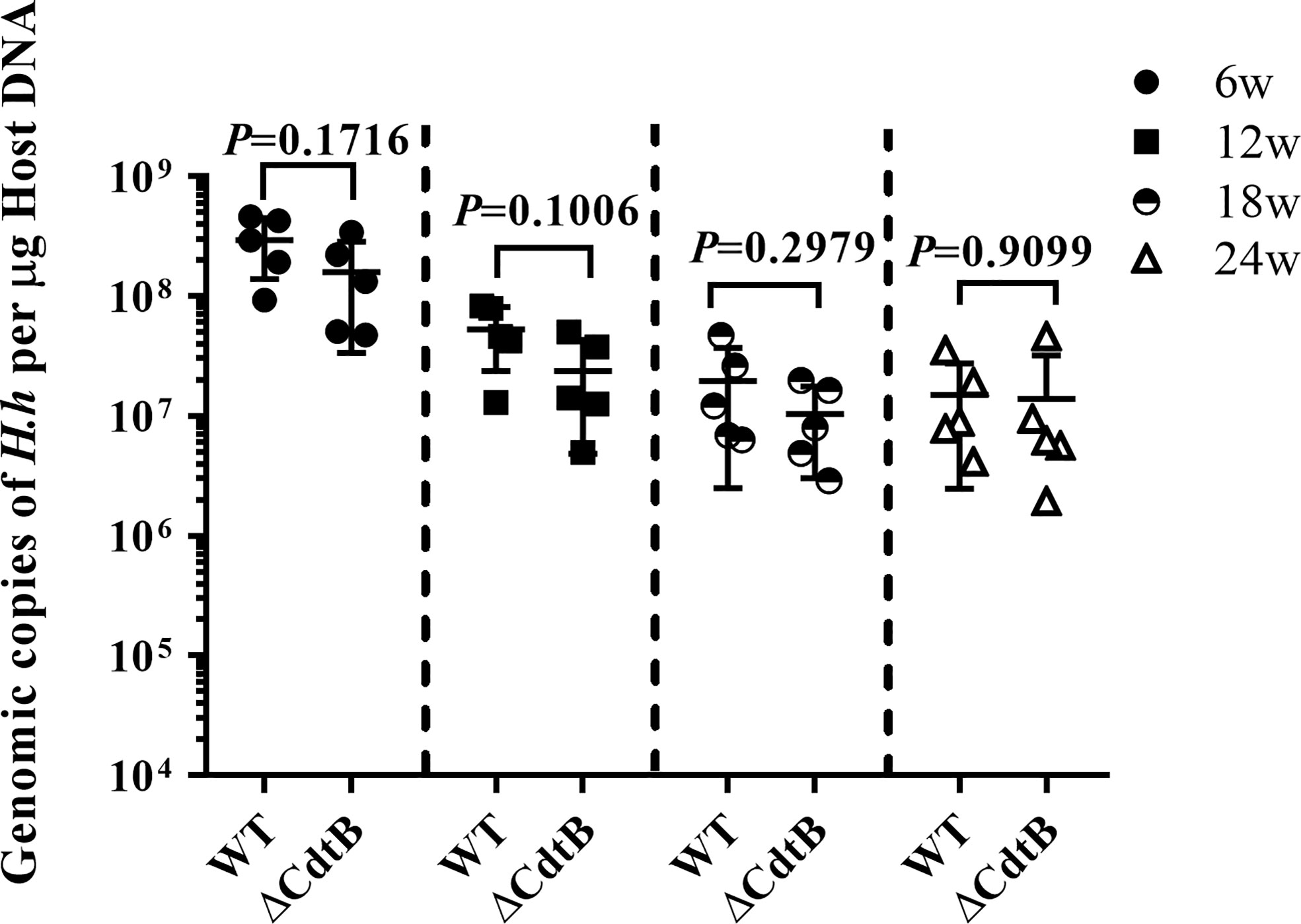
Figure 1 The absence of CdtB did not affect the colonic colonization of H. hepaticus in mice. The levels of H. hepaticus in the respective samples were expressed as its genomic copies per μg of mouse colon DNA. Data are expressed as the means ± SEM (n = 5/group). Statistics were analyzed using unpaired two-tailed Student’s t test.
H. hepaticus Infection Induced Colitis in Il-10−/− Mice
The weight/length ratio of the colon is an important parameter in the evaluation of intestinal inflammation. The results showed that there was no difference between the groups at 6 WPI, while anorectal prolapse was found in WT H. hepaticus infected mice (Figure 2A), and the weight/length ratio of mice infected by H. hepaticus was of significant difference compared with ΔCdtB H. hepaticus group at 24 WPI (P = 0.022) (Figures 2B, C). In addition, the symptoms of rectocele and watery stools in H. hepaticus group gradually worsened compared with ΔCdtB H. hepaticus group, suggesting that CdtB is an important virulence factor of H. hepaticus induced colitis in Il-10−/− mice.
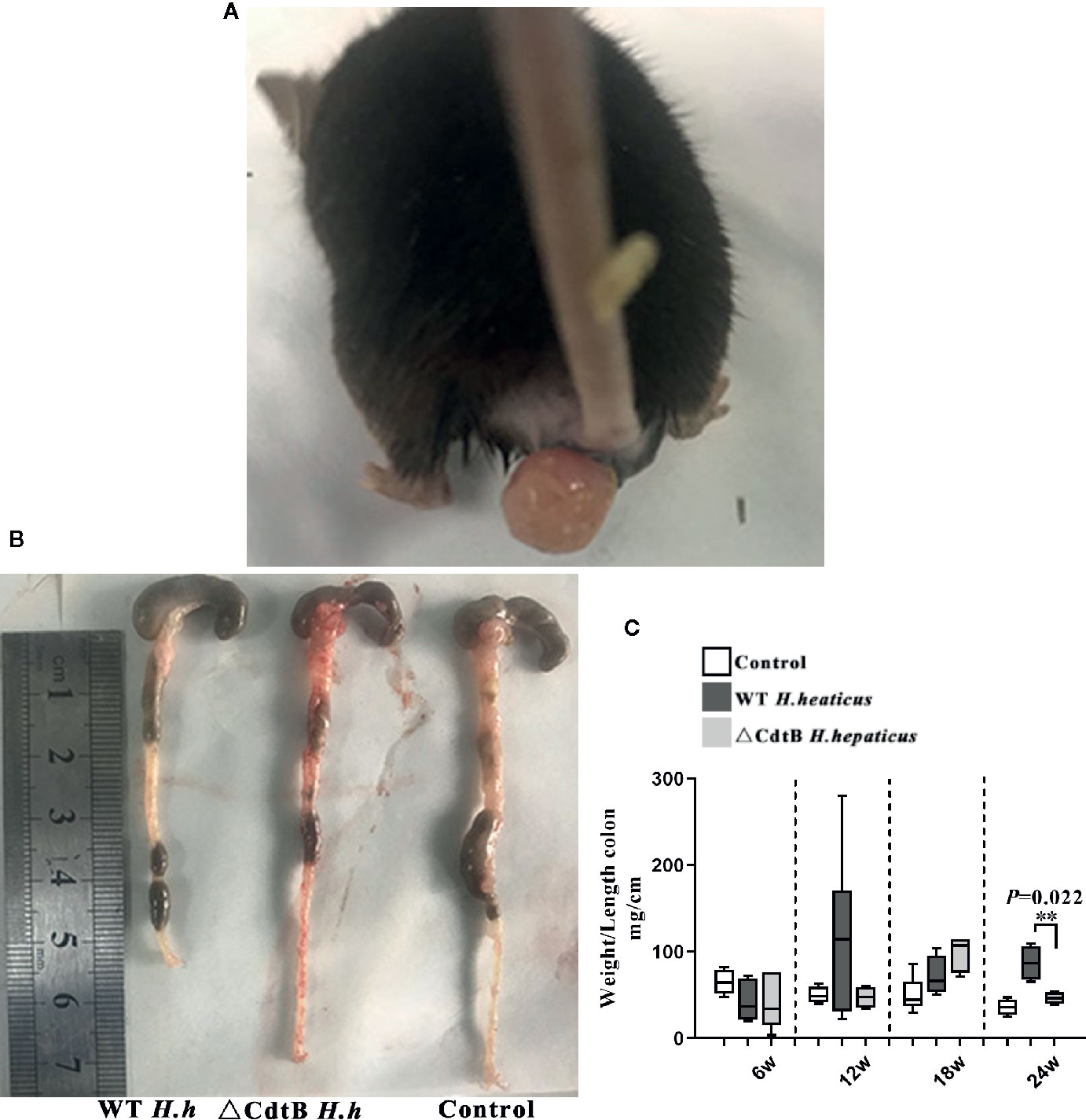
Figure 2 Deletion of CdtB alleviated colitis symptoms induced by (H) hepaticus infection. (A) Rectocele of mice in H. hepaticus group; (B) colonic segment of the experimental groups at 24 w; (C) weight/length ratio of the colon; Data are expressed as the mean ± SEM (n = 5/group). **represents P values <0.01 compared with the control group.
CdtB Promoted the Severity of Colitis Over the Infection
Histopathological examination was performed to characterize the role of CdtB in H. hepaticus infection induced colitis in Il-10−/− mice. Pathological changes included inflammation, edema, epithelial defect, atrophy, hyperplasia, and dysplasia. As shown in Figure 3, there was no inflammation observed in H. hepaticus infected mice at 6 WPI, but H. hepaticus infection promoted inflammation and edema, with mild epithelial defect, atrophy, hyperplasia, and dysplasia at 12 WPI. While at 18 WPI, proximal bowel inflammation score of H. hepaticus infected mice was significantly higher than that of ΔCdtB H. hepaticus infected group and severe inflammation were presented with inflammatory cells infiltrating the lamina propria and submucosa (P < 0.05). Furthermore, fibrin exudation in the intestinal glands and mucous epithelial exfoliation were also found in H. hepaticus infected mice. However, no fibrosis was observed (data not shown). By contrast, ΔCdtB H. hepaticus infected group developed into mild inflammation without typical pathological changes. Taken together, these data suggest that CdtB has a significant effect on H. hepaticus-induced inflammation. Interestingly, the proximal colon lesion was more obvious in the H. hepaticus infected group. Therefore, subsequent studies focused on the proximal colon.
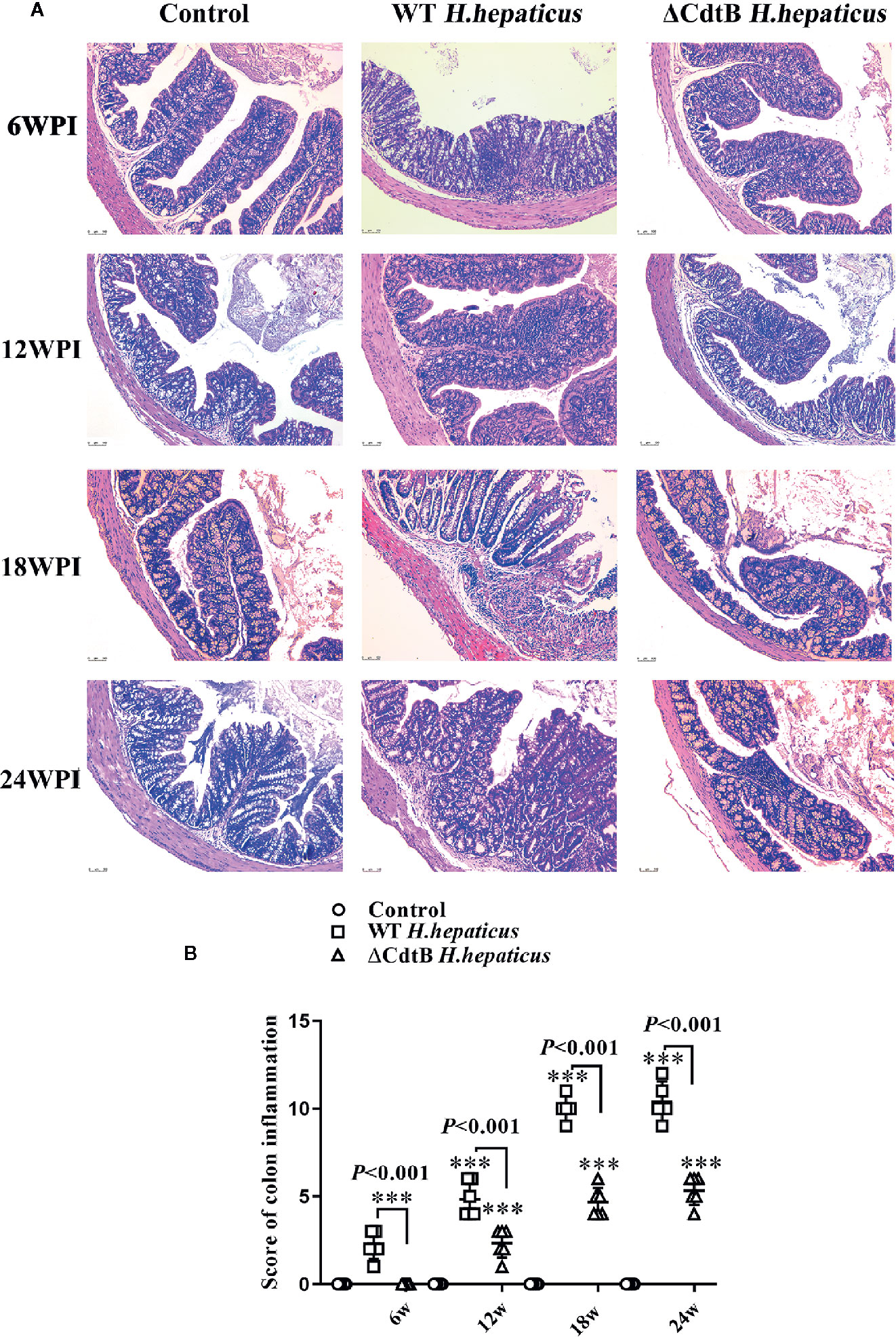
Figure 3 The colonic mucosa of colitic mice was attenuated by CdtB mutant. (A) Sections of the colonic mucosa were stained with hematoxylin and eosin (×100). At 6 WPI, the control colonic mucosa was integrated, crypts and goblet cells were evenly distributed, and a few scattered lymphocytes were found in submucosa; but WT H. hepaticus induced mild mononuclear cell infiltrates in lamina propria accompanied by slight depletion of mucus from goblet cells. At 12 WPI, ΔCdtB H. hepaticus induced a few multifocal mononuclear cell infiltrates in the lamina propria accompanied by slight to no depletion of mucus from goblet cells; while, WT H. hepaticus induced mild inflammatory cell infiltrates in the lamina propria accompanied by depletion of mucus from goblet cells. During the 18–24 WPI, the change of colonic symptoms caused by ΔCdtB H. hepaticus was not obvious; but WT H. hepaticus induced progressive colitic symptoms, including moderate lamina propria or submucosa inflammation, mucin depletion, crypts abscesses or hyperplasia and enlargement, etc. (B) Microscopic scores were assigned to the different groups according to the criteria described (Erdman et al., 2009). Data are expressed as the means ± SEM (n = 5/group). ***P < 0.001 when compared with the control group.
CdtB Induce Proinflammatory Response in Proximal Colon
Tnf-α, Il-1β, Il-6, and iNOS are crucial in the pathogenesis of colitis (Luo and Zhang, 2017). In addition, iNOS has also been shown to be associated with the occurrence of intestinal inflammation because the increase of iNOS expression in inflammatory regions is related to histological inflammatory parameters (Palatka et al., 2005). As shown in Figure 4, the results showed that only iNOS have differences between infected groups at 6 WPI. While H. hepaticus significantly increased the Il-1β (P = 0.041), Il-6 (P = 0.0095), Tnf-α (P = 0.0133), iNOS mRNA transcription level (P = 0.0049) at 12 WPI. Similarly, at 18 WPI and 24 WPI, CdtB promoted cytokine production detected by qPCR (excluding the Tnf-α at 24 WPI).
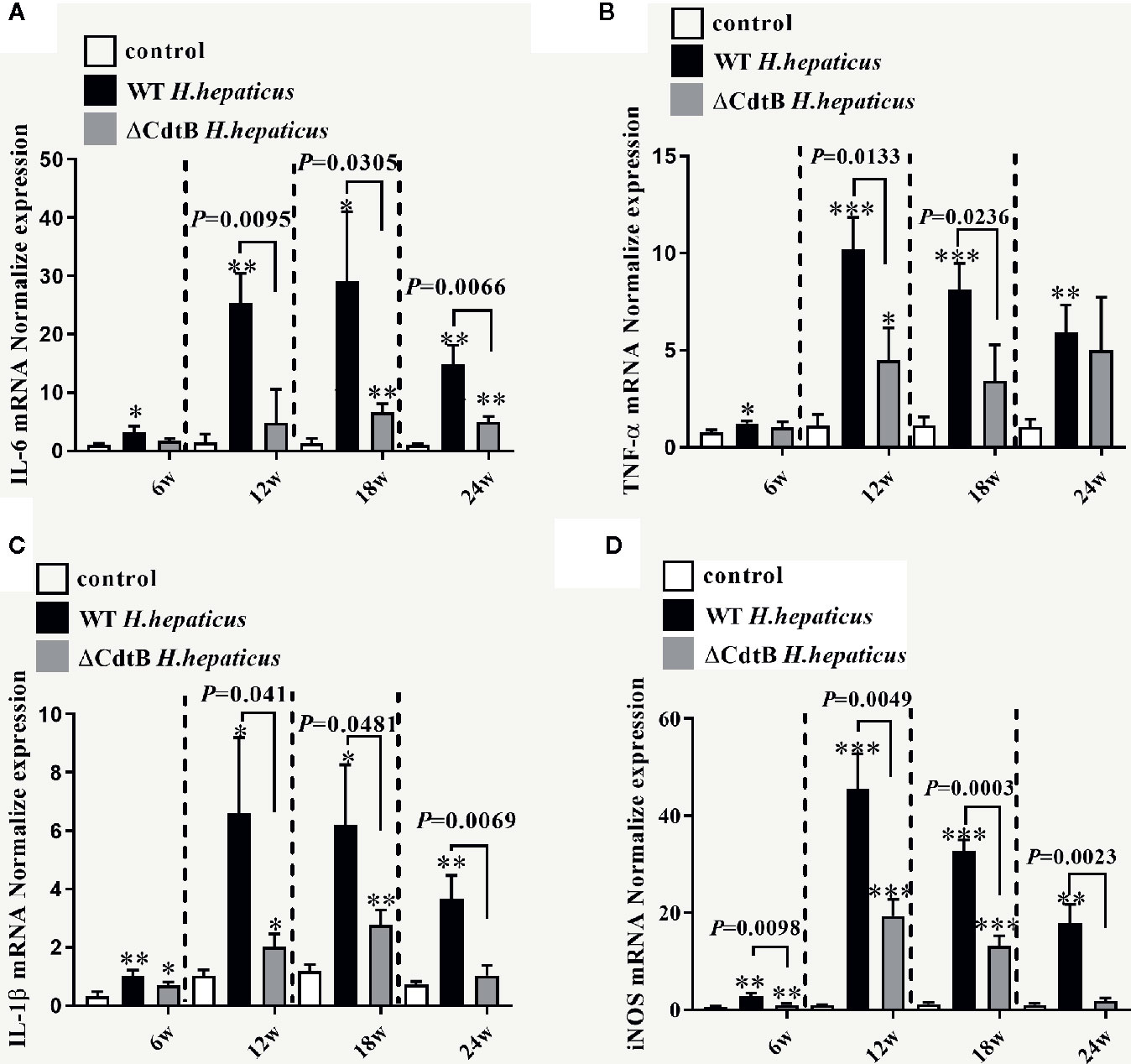
Figure 4 H. hepaticus infection induced the expression of pro-inflammatory cytokines. Colonic gene expression of pro-inflammatory cytokines (A) Il-6, (B) Tnf-α, (C) Il-1β, and (D) iNOS was analyzed by real-time qPCR and normalized with the housekeeping gene GAPDH. Data are expressed as the means ± SEM (n = 5/group). * refers to the P value when compared with the control group, *P < 0.05, **P < 0.01, and ***P < 0.001.
To better determine Il-6 protein levels, the expression of Il-6 in the proximal colon was detected by ELISA. Consistent with the results obtained by qPCR, there was no significant difference between the infected groups at 6 WPI (Figure 5). However, H. hepaticus infection significantly increased the Il-6 protein expression levels compared to ΔCdtB H. hepaticus at 12 WPI (P = 0.0357). Semblable results appeared at 18 WPI (P = 0.0349) and 24 WPI (P = 0.0076), but Il-6 protein levels of H. hepaticus group decline from 12 WPI to 24 WPI. This may due to the immune tolerance of the colon. Taken together, the data showed that CdtB up-regulated Il-6 protein expression during infection.
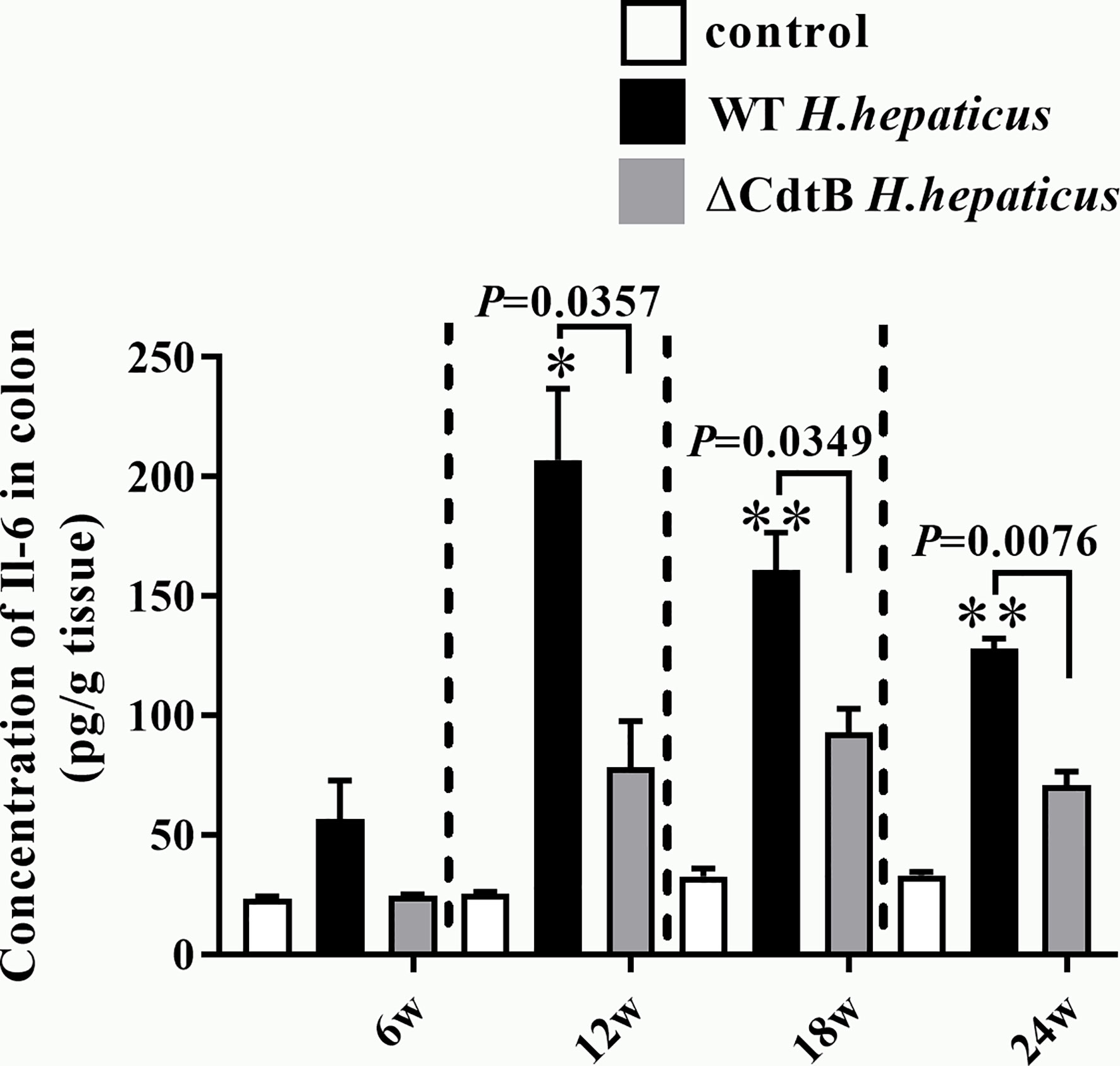
Figure 5 H. hepaticus infection induced the protein expression of Il-6 in proximal colon. The cytokine levels of proximal colon tissue were expressed as the concentration in pg/g. Data are expressed as the means ± SEM (n = 5/group). *refers to the P value when compared with the control group, *for P < 0.05, **for P < 0.01.
Tight Junctions and extracellular matrix are important for intestinal barrier stability (Bollyky et al., 2011; Parikh et al., 2019). Zo-1 can prevent bacterial translocation, improve intestinal permeability (Arrieta et al., 2006), and maintain epithelial integrity to reduce colitis (Macdonald and Monteleone, 2005). Mmp-9 is a family of proteolytic zinc enzyme and calcium-dependent structural proteins that degrade the extracellular matrix and are involved in the pathogenesis of IBD and experimental colitis (Koelink et al., 2014). However, there was no significant change in Mmp-9 and Zo-1 associated with colitis during the experiment probably for immune protection of the colon (data not shown). These data suggest that CdtB enhances the production of proinflammatory cytokines of Tnf-α, Il-1β, Il-6, and iNOS during infection.
CdtB Increase NO Content in Proximal Colon
The content of NO was detected due to the differences in iNOS between H. hepatius infected and ΔCdtB H. hepaticus infected mice. It has been proposed that the increase of NO produced by iNOS will react with superoxide to form peroxynitrite, resulting in harmful changes in protein structure and function (Rachmilewitz et al., 1995). In the current study, from 12 WPI to 24 WPI, NO content in the gut was both significantly increased in ΔCdtB and WT H. hepaticus infection groups, especially raised much higher in WT H. hepaticus infection group (Figure 6). While, there was no difference between H. hepaticus groups at each time point (Figure 6). The data showed that CdtB caused the body to produce more NO during infection, which may link to CdtB ability of DNA damage and proto-oncogene expression.
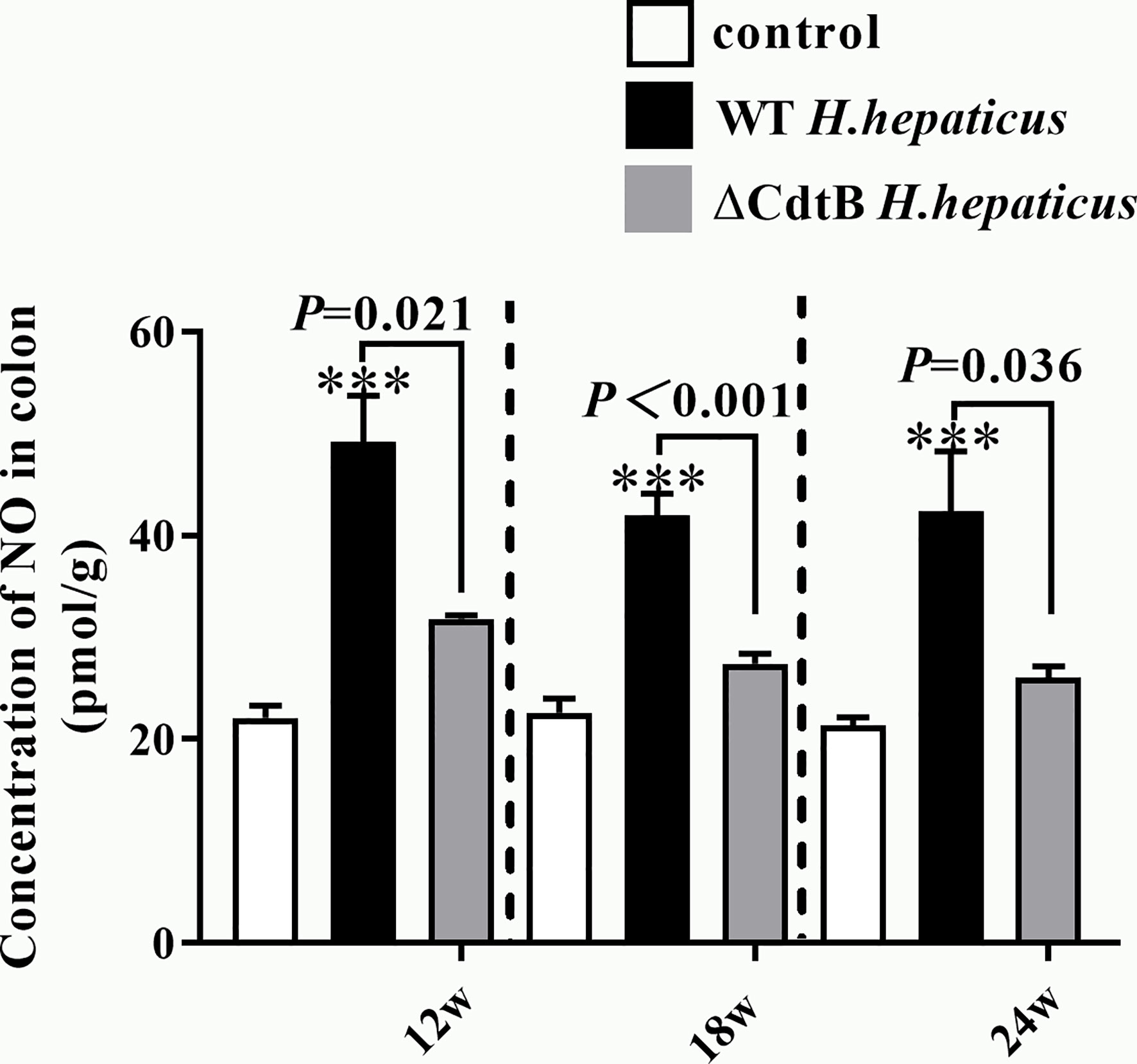
Figure 6 H. hepaticus infection induced the content of NO. The content of NO in colon tissue was expressed as the concentration in pmol/g. Data are expressed as the means ± SEM (n = 5/group). ***means P < 0.001 as compared to Control.
CdtB Promoted the Activation of Jak-Stat Signaling Pathways
The Jak-Stat signaling pathway plays a pivotal role in the pathogenesis of IBD, such as controlling and maintaining the development and differentiation of mucosal immune system and T help cells (Coskun et al., 2013). In order to understand whether IBD caused by H. hepaticus infection is related to this pathway, the protein extracted from the colonic epithelium was detected by immunoblotting (Figure 7A). The statistical result (Figure 7B) showed that only the phosphorylation of Jak2 (p-Jak2) was significantly increased at 6 WPI in H. hepaticus groups compared to ΔCdtB H. hepaticus; while, at 18 and 24 WPI, both p-Jak2 and p-Stat3 was significantly increased in H. hepaticus groups compared to ΔCdtB H. hepaticus. These data suggest that CdtB promote the JAK-STAT signaling pathway activation during H. hepaticus infection, and plays an important role in H. hepaticus-induced colitis.
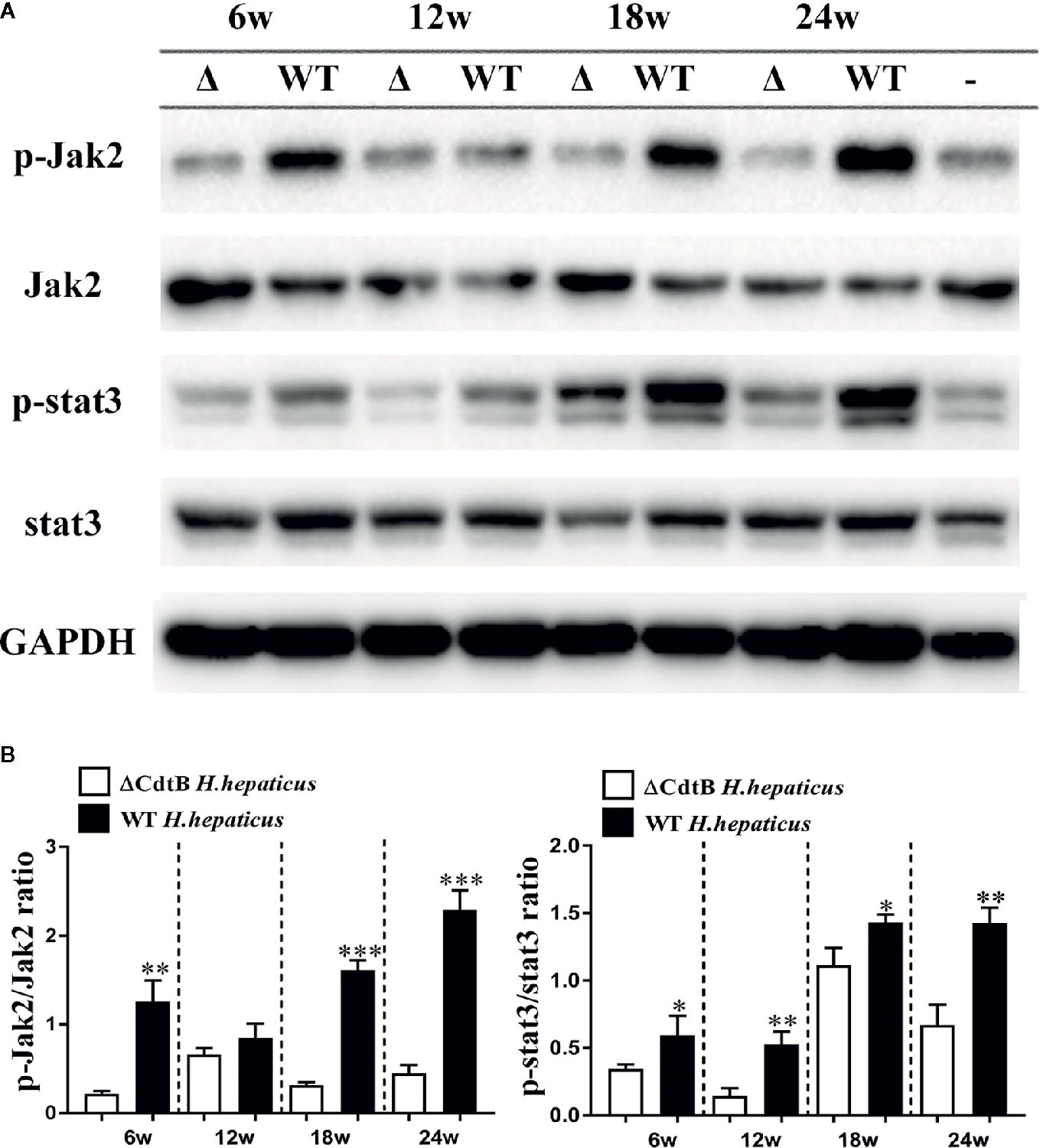
Figure 7 H. hepaticus promoted activation of Jak2 and Stat3. (A) Western blotting and densitometric analysis of p-Jak2, Jak2, p-Stat3, Stat3, and GAPDH; WT represents wild type H. hepaticus, Δ represents ΔCdtB H. hepaticus, and – represents control sample of 24 WPI. (B) The Statistical data are expressed as the mean SD (n = 5). *P < 0.05 as compared to ΔCdtB H. hepaticus, **P < 0.01 as compared to ΔCdtB H. hepaticus, ***P < 0.001 as compared to ΔCdtB H. hepaticus.
Discussion
It has been reported that CdtB is essential for the long-term colonization of H. hepaticus in the gut of Swiss Webster and A/JCr mice with complete immune system (Ge et al., 2005; Ge et al., 2007). In contrast, the ΔCdtB H. hepaticus used in this study and H. hepaticus 3B1 had similar levels of colonization in B6.129P2-IL10tm1Cgn/J mice, which are consistent with the results obtained by Ge (Ge et al., 2017).This difference may be due to mouse strains that have different immune tolerance to H. hepaticus infection. In our study, H. hepaticus did not induce the expected degree of colitis. In the initial experiment, the common infection procedure failed to infect Il-10−/− mice from the Jackson Laboratory. Interestingly, when we hybridized male Il-10−/− mice from Jackson Laboratory with female wild type B6 mice from Model Animal Research Center of Nanjing University, then hybridized female Il10+/− heterozygote with male Il-10−/− mice, the homozygote of offspring obtained were highly susceptible to H. hepaticus, which suggested the important role of gut flora in the process of H. hepaticus infection. Pretreatment of mice with gentamicin drinking water can eliminate part of the normal microflora of the gut, and increase the colonization of H. hepaticus in the cecum and colon. Compared with other reports, the continuous infection at 24 WPI of mice in this study did not induce severe colitis, which may be due to the fact that some bacterial which was not completely eliminated by gentamicin protected the gut from H. hepaticus colonization. To explore possible reasons, we performed fecal microbiota transplantation experiment in the next stage (data not shown), and high-throughput sequencing related to gut flora also requires further research.
In this study, CdtB up-regulated the transcription of Il-6 and induced Jak2/Stat3 signaling pathways. Colitis in Jak3 knockout mice is thought to be caused by a defect in the intestinal barrier. Jak3 knockout mouse model showing Il-2, Il-4, Il-7, Il-9, and Il-15 conduction defects, elevated levels of Il-6 and Il-17a and severe immunodeficiency, defects in barrier function lead to Ulcerative colitis (UC) (Baird et al., 1998; Mishra et al., 2013). It is currently believed that the Il-6/Stat3 pathway promotes the apoptosis resistance of intestinal T lymphocytes by inducing the activation of the downstream anti-apoptotic genes Bcl-2 and Bcl-xl, resulting in the massive release of inflammatory factors and the continued destruction of the organization (Carey et al., 2008). Suzuki et al. (2001) knocked down the Il-6 gene in mice induced by dextran sodium sulfate (DSS), the severity of colitis and the level of activated Stat3 were significantly reduced, and Il-22 activated Stat3 to promote intestinal epithelium cellular (IEC) homeostasis and tissue healing indicate that the activation of Stat3 is an important factor in the persistence of colon inflammation. In UC, the differentiation of lamina propria T cells into Th2 cells is mediated by Il-4, which produces Th2 type cytokines Il-4, Il-5, and Il-13, and naive T cells are sent out through Jak1–Jak3 Signal and activate Stat6. Barrett et al. (2008) found that the rs10758669 risk site associated with UC is located in the Jak2 region. In the pattern-recognition receptor (PRR)–induced UC, its anti-inflammatory cytokines gradually decreased with the decrease of Jak2 expression. PRR-induced reduction of inflammatory factors such as Il-10, Il-4, Il-22 depends on Jak to regulate inflammation and induce UC (Hedl et al., 2016).
Colitis can cause massive infiltration of immune cells in the inflamed tissues of the colon, release a variety of pro-inflammatory cytokines, and produce high levels of NO and lipid peroxidation (Araujo et al., 2017). Proinflammatory cytokines are the basic regulators of colitis development (Araujo et al., 2017), and imbalance between intestinal mucosal immunity can damage the colonic mucosa and affect intestinal homeostasis (Zhang et al., 2017). CdtB can significantly stimulate cells to produce NO, which may be related to the induction of DNA damage and the upregulation of proto-oncogene expression. In this study, it was observed that CdtB can up-regulate Jak2/Stat3 phosphorylation, suggesting that the promotion of Jak/Stat pathway may be involved in the effect of H. hepaticus CdtB on colitis, which is related to the expression of proinflammatory cytokine-related genes. The key role of Stat3 signal transduction in inflammation has been demonstrated in colitis-related mouse models (Bollrath et al., 2009; Grivennikov et al., 2009). Infection of mice with H. hepaticus can cause colitis, including rectocele, watery stool, intestinal epithelial damage, inflammatory cell infiltration, and reduction in colon length. Studies have shown that CdtB can induce mammalian cells cytopathy through a series of cellular reactions (Ge et al., 2008). Although there are many reports on clarifying CdtB-induced cytopathy, the pathogenic mechanism of CdtB in bacterial infected animal has not been fully clarified. This study demonstrated that CdtB promoted the progression of inflammation within 24 weeks in B6.129P2-IL10tm1Cgn/J mice infected with H. hepaticus, and up-regulated the transcription of Il-6 and Tnf-α in the proximal colon during infection. In addition, the mutant of CdtB reduced the ability of H. hepaticus to activate Jak2 and Stat3 signaling pathways, but did not affect the colonization efficiency of H. hepaticus. Taken together, these data suggest that CdtB may enhance H. hepaticus’ inflammatory ability by activating the Il-6/Jak2/Stat3 signaling pathway. In view of the role of CdtB in promoting the occurrence of gut inflammation, the role of bacterial CDT on the pathogenicity of intestinal is worthy of further study.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Yangzhou University.
Author Contributions
LZ processed data and wrote manuscript. CZ constructed the CdtB deficient H. hepaticus strain and carried out animal experiments. SC carried out the histological and immunoblotting experiments. QZ designed the experimental scheme and provided funds. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Key Research and Development Program of China (2017YFD0502303, 2017YFD0501605), the Key Research and Development Program of Jiangsu Province (BE2020674), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), High-end Talent Support Program of Yangzhou University and the Young and Middle-Aged Academic Leaders Plan of Yangzhou University.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Zhongming Ge for kind advice for constructing the CdtB mutant Helicobacter hepaticus for this study.
References
Araujo, D. F. S., Guerra, G. C. B., Pintado, M. M. E., Sousa, Y. R. F., Algieri, F., Rodriguez-Nogales, A., et al. (2017). Intestinal anti-inflammatory effects of goat whey on DNBS-induced colitis in mice. PloS One 12, e0185382. doi: 10.1371/journal.pone.0185382
Arrieta, M. C., Bistritz, L., Meddings, J. B. (2006). Alterations in intestinal permeability. Gut 55, 1512–1520. doi: 10.1136/gut.2005.085373
Avenaud, P., Castroviejo, M., Claret, S., Rosenbaum, J., Megraud, F., Menard, A. (2004). Expression and activity of the cytolethal distending toxin of Helicobacter hepaticus. Biochem. Biophys. Res. Commun. 318, 739–745. doi: 10.1016/j.bbrc.2004.04.089
Baird, A. M., Thomis, D. C., Berg, L. J. (1998). T cell development and activation in Jak3-deficient mice. J. Leukoc. Biol. 63, 669–677. doi: 10.1002/jlb.63.6.669
Barrett, J. C., Hansoul, S., Nicolae, D. L., Cho, J. H., Duerr, R. H., Rioux, J. D., et al. (2008). Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 40, 955–962. doi: 10.1038/ng.175
Berg, D. J., Davidson, N., Kühn, R., Müller, W., Menon, S., Holland, G., et al. (1996). Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J. Clin. Invest. 98, 1010–1020. doi: 10.1172/JCI118861
Bollrath, J., Phesse, T. J., Von Burstin, V. A., Putoczki, T., Bennecke, M., Bateman, T., et al. (2009). gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 15, 91–102. doi: 10.1016/j.ccr.2009.01.002
Bollyky, P. L., Wu, R. P., Falk, B. A., Lord, J. D., Long, S. A., Preisinger, A., et al. (2011). ECM components guide IL-10 producing regulatory T-cell (TR1) induction from effector memory T-cell precursors. Proc. Natl. Acad. Sci. U.S.A. 108, 7938–7943. doi: 10.1073/pnas.1017360108
Cahill, R. J., Foltz, C. J., Fox, J. G., Dangler, C. A., Powrie, F., Schauer, D. B. (1997). Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect. Immun. 65, 3126–3131. doi: 10.1128/IAI.65.8.3126-3131.1997
Carey, R., Jurickova, I., Ballard, E., Bonkowski, E., Han, X., Xu, H., et al. (2008). Activation of an IL-6:STAT3-dependent transcriptome in pediatric-onset inflammatory bowel disease. Inflammation Bowel Dis. 14, 446–457. doi: 10.1002/ibd.20342
Coskun, M., Salem, M., Pedersen, J., Nielsen, O. H. (2013). Involvement of JAK/STAT signaling in the pathogenesis of inflammatory bowel disease. Pharmacol. Res. 76, 1–8. doi: 10.1016/j.phrs.2013.06.007
Danne, C., Ryzhakov, G., Martínez-López, M., Ilott, N. E., Franchini, F., Cuskin, F., et al. (2017). A Large Polysaccharide Produced by Helicobacter hepaticus Induces an Anti-inflammatory Gene Signature in Macrophages. Cell Host Microbe 22, 733–745.e735. doi: 10.1016/j.chom.2017.11.002
Dassanayake, R. P., Griep, M. A., Duhamel, G. E. (2005). The cytolethal distending toxin B sub-unit of Helicobacter hepaticus is a Ca2+- and Mg2+-dependent neutral nuclease. FEMS Microbiol. Lett. 251, 219–225. doi: 10.1016/j.femsle.2005.08.005
Dieleman, L. A., Arends, A., Tonkonogy, S. L., Goerres, M. S., Craft, D. W., Grenther, W., et al. (2000). Helicobacter hepaticus does not induce or potentiate colitis in interleukin-10-deficient mice. Infect. Immun. 68, 5107–5113. doi: 10.1128/IAI.68.9.5107-5113.2000
Erdman, S. E., Rao, V. P., Poutahidis, T., Rogers, A. B., Taylor, C. L., Jackson, E. A., et al. (2009). Nitric oxide and TNF-alpha trigger colonic inflammation and carcinogenesis in Helicobacter hepaticus-infected, Rag2-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 106, 1027–1032. doi: 10.1073/pnas.0812347106
Falsafi, T., Mahboubi, M. (2013). Helicobacter hepaticus, a new pathogenic species of the Helicobacter genus: Similarities and differences with H. pylori. Iran J. Microbiol. 5, 185–194.
Ge, Z., Feng, Y., Whary, M. T., Nambiar, P. R., Xu, S., Ng, V., et al. (2005). Cytolethal distending toxin is essential for Helicobacter hepaticus colonization in outbred Swiss Webster mice. Infect. Immun. 73, 3559–3567. doi: 10.1128/IAI.73.6.3559-3567.2005
Ge, Z., Rogers, A. B., Feng, Y., Lee, A., Xu, S., Taylor, N. S., et al. (2007). Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell Microbiol. 9, 2070–2080. doi: 10.1111/j.1462-5822.2007.00939.x
Ge, Z., Schauer, D. B., Fox, J. G. (2008). In vivo virulence properties of bacterial cytolethal-distending toxin. Cell Microbiol. 10, 1599–1607. doi: 10.1111/j.1462-5822.2008.01173.x
Ge, Z., Feng, Y., Ge, L., Parry, N., Muthupalani, S., Fox, J. G. (2017). Helicobacter hepaticus cytolethal distending toxin promotes intestinal carcinogenesis in 129Rag2-deficient mice. Cell Microbiol. 19, e12728 doi: 10.1111/cmi.12728
Grivennikov, S., Karin, E., Terzic, J., Mucida, D., Yu, G. Y., Vallabhapurapu, S., et al. (2009). IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15, 103–113. doi: 10.1016/j.ccr.2009.01.001
Guerra, L., Albihn, A., Tronnersjo, S., Yan, Q., Guidi, R., Stenerlow, B., et al. (2010). Myc is required for activation of the ATM-dependent checkpoints in response to DNA damage. PloS One 5, e8924. doi: 10.1371/journal.pone.0008924
Hedl, M., Proctor, D. D., Abraham, C. (2016). JAK2 Disease-Risk Variants Are Gain of Function and JAK Signaling Threshold Determines Innate Receptor-Induced Proinflammatory Cytokine Secretion in Macrophages. J. Immunol. 197, 3695–3704. doi: 10.4049/jimmunol.1600845
Kennedy, E. A., King, K. Y., Baldridge, M. T. (2018). Mouse Microbiota Models: Comparing Germ-Free Mice and Antibiotics Treatment as Tools for Modifying Gut Bacteria. Front. Physiol. 9, 1534. doi: 10.3389/fphys.2018.01534
Koelink, P. J., Overbeek, S. A., Braber, S., Morgan, M. E., Henricks, P. A., Abdul Roda, M., et al. (2014). Collagen degradation and neutrophilic infiltration: a vicious circle in inflammatory bowel disease. Gut 63, 578–587. doi: 10.1136/gutjnl-2012-303252
Kullberg, M. C., Ward, J. M., Gorelick, P. L., Caspar, P., Hieny, S., Cheever, A., et al. (1998). Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect. Immun. 66, 5157–5166. doi: 10.1128/IAI.66.11.5157-5166.1998
Livak, K. J., Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Luo, C., Zhang, H. (2017). The Role of Proinflammatory Pathways in the Pathogenesis of Colitis-Associated Colorectal Cancer. Mediators Inflammation 2017, 5126048. doi: 10.1155/2017/5126048
Macdonald, T. T., Monteleone, G. (2005). Immunity, inflammation, and allergy in the gut. Science 307, 1920–1925. doi: 10.1126/science.1106442
Mishra, J., Verma, R. K., Alpini, G., Meng, F., Kumar, N. (2013). Role of Janus kinase 3 in mucosal differentiation and predisposition to colitis. J. Biol. Chem. 288, 31795–31806. doi: 10.1074/jbc.M113.504126
Palatka, K., Serfozo, Z., Vereb, Z., Hargitay, Z., Lontay, B., Erdodi, F., et al. (2005). Changes in the expression and distribution of the inducible and endothelial nitric oxide synthase in mucosal biopsy specimens of inflammatory bowel disease. Scand. J. Gastroenterol. 40, 670–680. doi: 10.1080/00365520510015539
Parikh, K., Antanaviciute, A., Fawkner-Corbett, D., Jagielowicz, M., Aulicino, A., Lagerholm, C., et al. (2019). Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature 567, 49–55. doi: 10.1038/s41586-019-0992-y
Pratt, J. S., Sachen, K. L., Wood, H. D., Eaton, K. A., Young, V. B. (2006). Modulation of host immune responses by the cytolethal distending toxin of Helicobacter hepaticus. Infect. Immun. 74, 4496–4504. doi: 10.1128/IAI.00503-06
Qin, H., Tang, G., Yi, P., Pan, X., Huang, H., Chang, R., et al. (2016). Diagnosis of Genus Helicobacter through a hemi-nested PCR assay of 16S rRNA. Saudi Pharm. J. 24, 265–272. doi: 10.1016/j.jsps.2016.04.015
Rachmilewitz, D., Karmeli, F., Okon, E., Bursztyn, M. (1995). Experimental colitis is ameliorated by inhibition of nitric oxide synthase activity. Gut 37, 247–255. doi: 10.1136/gut.37.2.247
Smith, J. L., Bayles, D. O. (2006). The contribution of cytolethal distending toxin to bacterial pathogenesis. Crit. Rev. Microbiol. 32, 227–248. doi: 10.1080/10408410601023557
Suzuki, A., Hanada, T., Mitsuyama, K., Yoshida, T., Kamizono, S., Hoshino, T., et al. (2001). CIS3/SOCS3/SSI3 plays a negative regulatory role in STAT3 activation and intestinal inflammation. J. Exp. Med. 193, 471–481. doi: 10.1084/jem.193.4.471
Whary, M. T., Taylor, N. S., Feng, Y., Ge, Z., Muthupalani, S., Versalovic, J., et al. (2011). Lactobacillus reuteri promotes Helicobacter hepaticus-associated typhlocolitis in gnotobiotic B6.129P2-IL-10(tm1Cgn) (IL-10(-/-) ) mice. Immunology 133, 165–178. doi: 10.1111/j.1365-2567.2011.03423.x
Xu, M., Pokrovskii, M., Ding, Y., Yi, R., Au, C., Harrison, O. J., et al. (2018). c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 554, 373–377. doi: 10.1038/nature25500
Young, V. B., Knox, K. A., Pratt, J. S., Cortez, J. S., Mansfield, L. S., Rogers, A. B., et al. (2004). In vitro and in vivo characterization of Helicobacter hepaticus cytolethal distending toxin mutants. Infect. Immun. 72, 2521–2527. doi: 10.1128/IAI.72.5.2521-2527.2004
Zhang, M., Sun, K., Wu, Y., Yang, Y., Tso, P., Wu, Z. (2017). Interactions between Intestinal Microbiota and Host Immune Response in Inflammatory Bowel Disease. Front. Immunol. 8, 942. doi: 10.3389/fimmu.2017.00942
Keywords: Helicobacter hepaticus, CdtB, colitis, Jak-Stat, IL-10−/− mice
Citation: Zhu L, Zhu C, Cao S and Zhang Q (2021) Helicobacter hepaticus Induce Colitis in Male IL-10−/− Mice Dependent by Cytolethal Distending Toxin B and via the Activation of Jak/Stat Signaling Pathway. Front. Cell. Infect. Microbiol. 11:616218. doi: 10.3389/fcimb.2021.616218
Received: 11 October 2020; Accepted: 09 February 2021;
Published: 12 March 2021.
Edited by:
Teresa Thurston, Imperial College London, United KingdomReviewed by:
Xingmin Sun, University of South Florida, United StatesGeorge Liechti, Uniformed Services University of the Health Sciences, United States
Copyright © 2021 Zhu, Zhu, Cao and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quan Zhang, enF1YW5AeXp1LmVkdS5jbg==; orcid.org/0000-0001-6389-6500