 Patricia M. Sikorski
Patricia M. Sikorski Alessandra G. Commodaro
Alessandra G. Commodaro Michael E. Grigg
Michael E. Grigg- 1Molecular Parasitology Section, Laboratory of Parasitic Diseases, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, United States
- 2Department of Microbiology and Immunology, Georgetown University Medical Center, Georgetown University, Washington, DC, United States
The infection competence of the protozoan pathogen Toxoplasma gondii is critically dependent on the parasite’s ability to inactivate the host complement system. Toxoplasma actively resists complement-mediated killing in non-immune serum by recruiting host-derived complement regulatory proteins C4BP and Factor H (FH) to the parasite surface to inactivate surface-bound C3 and limit formation of the C5b-9 membrane attack complex (MAC). While decreased complement activation on the parasite surface certainly protects Toxoplasma from immediate lysis, the biological effector functions of C3 split products C3b and C3a are maintained, which includes opsonization of the parasite for phagocytosis and potent immunomodulatory effects that promote pro-inflammatory responses and alters mucosal defenses during infection, respectively. In this review, we discuss how complement regulation by Toxoplasma controls parasite burden systemically but drives exacerbated immune responses locally in the gut of genetically susceptible C57BL/6J mice. In effect, Toxoplasma has evolved to strike a balance with the complement system, by inactivating complement to protect the parasite from immediate serum killing, it generates sufficient C3 catabolites that signal through their cognate receptors to stimulate protective immunity. This regulation ultimately controls tachyzoite proliferation and promotes host survival, parasite persistence, and transmissibility to new hosts.
Introduction
Insect and vertebrate complement systems play critical roles in the defense against invading microbial pathogens and the regulation of inflammatory responses. Apicomplexan parasites, which comprise a diverse group of obligatory intracellular parasites, have evolved sophisticated strategies to regulate or inactivate this humoral first line of defense to promote their infection competency in both insects and mammalian hosts (Belachew, 2018; Shao et al., 2019). These parasites possess complex lifecycles consisting of both sexual and asexual stages that typically infect multiple hosts, and so, must overcome a myriad of species-specific immune defenses in order to initiate infection. The most studied among these parasites include Toxoplasma, Cryptosporidium, Eimeria, and Plasmodium, which cause various infectious diseases of human and veterinary importance (Votýpka et al., 2017). This paper reviews the strategies employed by Toxoplasma gondii to both inactivate and regulate the complement cascade and highlights similar strategies employed by other Apicomplexan parasites to facilitate the establishment of a persistent, transmissible infection.
The Complement System
The complement system is an evolutionarily conserved first line of defense that rapidly activates against invading pathogens. This defense system consists of a set of circulating liver-derived soluble proteins, membrane bound receptors, and regulators that function in a highly coordinated proteolytic cascade to opsonize and lyse invading microbes in addition to mobilizing the cellular arm of the immune response (Figure 1—schematic of complement system pathway activation and regulation). Activation of this cascade of more than 50 molecules and their complement receptors occur via three different pathways: the classical (CP), lectin (LP), and alternative (AP) pathways. Activation of these pathways leads to the formation of pathway-specific complexes known as C3 convertases, in which all three pathways converge to facilitate the cleavage of the central molecule C3 into effector proteins C3a and C3b. Successful complement activation culminates in the assembly of a pore-forming protein on the pathogen surface (referred to as the membrane attack complex, or MAC) that mediates pathogen lysis.
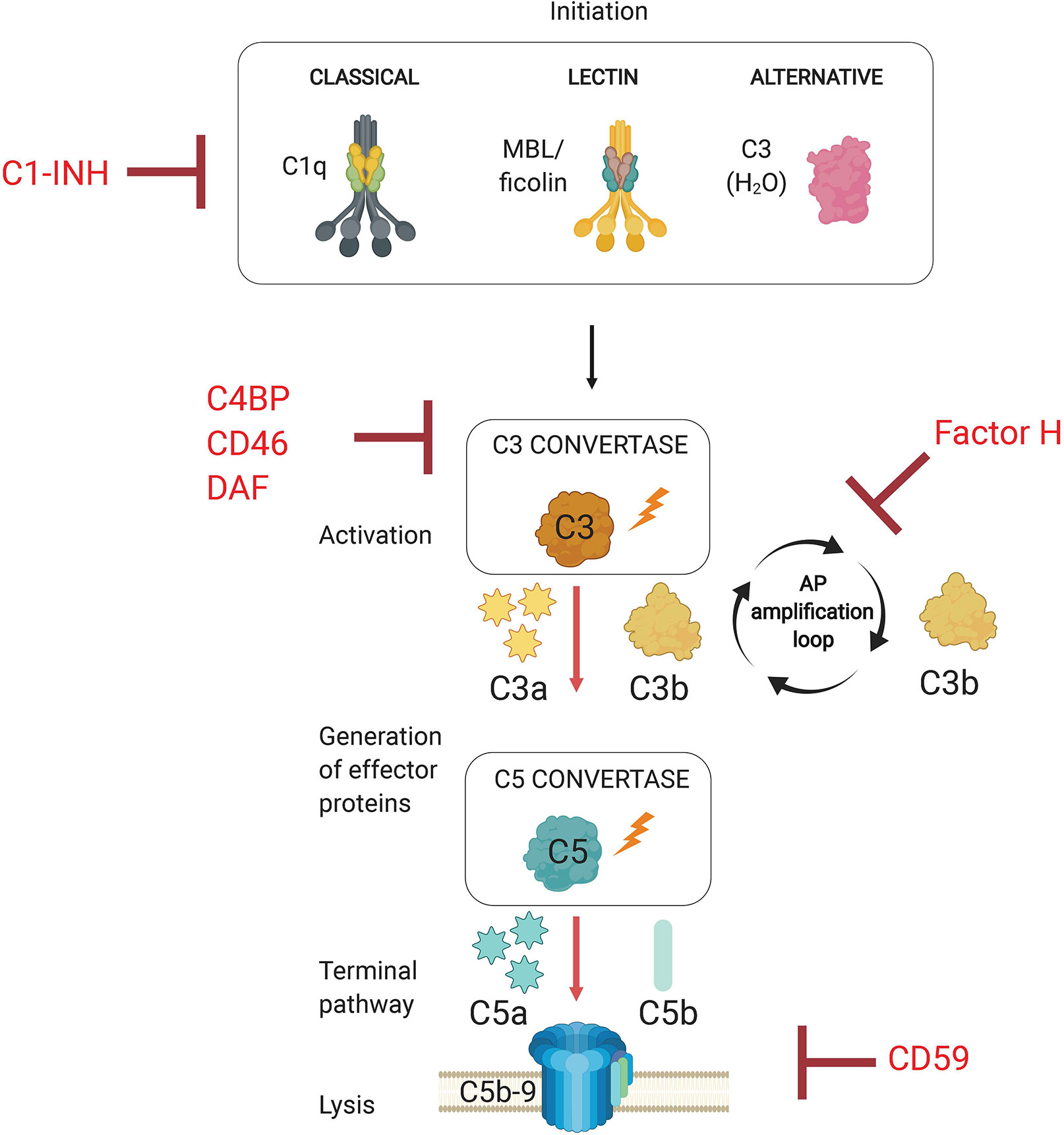
Figure 1 Overview of the Complement System. The complement system is activated via three separate pathways (classical, lectin, and alternative) that have distinct recognition mechanisms (C1q, MBL/ficolins, and C3b(H2O), respectively). Complement activation results in the formation of C3 convertases. All three pathways converge at C3 activation via C3 convertases, which generates effector molecules C3a and C3b. C3b allows for progression to the terminal pathway by forming C5 convertases, which cleave C5 into C5a and C5b. The terminal pathway requires C5 activation product C5b to initiate the assembly of the membrane attack complex (MAC, C5b-9). The pore inserts into the cell membrane and promotes pathogen lysis. Complement regulators target complement pathway initiation (C1-INH, C1 esterase inhibitor), convertase formation (C4BP, C4b-binding protein; CD46; DAF, decay accelerating factor), and the alternative pathway amplification loop (Factor H).
The complement system is activated upon detection of invading pathogens through specialized and pathway-specific recognition molecules that recognize pathogen-associated molecular patterns (PAMPs). Initiation of the classical pathway requires the recognition of pathogen bound IgM or IgG by pattern recognition molecule C1q. The lectin pathway is activated when microbial sugars are bound by the pattern recognition molecule mannose-binding lectin (MBL). The alternative pathway, however, does not rely on a recognition molecule for activation, but rather on the failure to regulate the continuous low-level spontaneous hydrolysis of C3 into C3(H2O), which indiscriminately probes foreign and self surfaces alike. The ability of the AP to discriminate between self and non-self surfaces relies on the presence of membrane bound and soluble complement regulatory proteins. Importantly, the AP is a critical component of host defense because it amplifies the complement response independent of the pathway that initiates the response, so it represents a critical target for regulation by host and pathogens alike. Specifically, the AP utilizes CP and LP activated C3b as a platform for rapidly generating new AP C3 convertases which establishes a positive feedback loop to amplify C3 cleavage (Thurman and Holers, 2006). Due to its non-discriminatory nature and potential for rapid amplification, this process must necessarily be tightly controlled by host regulator proteins to limit inflammation and damage to host cells. Complement regulators target various points within the proteolytic cascade, which includes inactivation of proteases associated with C1q and mannose binding lectin (MBL), cleavage of active C3b into inactive iC3b and C3dg, accelerating the decay of C3 and C5 convertases, and preventing insertion of the membrane attack complex into the plasma membrane (Schmidt et al., 2016).
Protection against pathogens occurs when the complement system produces several biologically active effector molecules, which include opsonins (C3b and its catabolites iC3b, C3d and C3dg), anaphylatoxins (C3a and C5a), and the membrane attack complex (MAC). The most direct effector function is the lysis of pathogens through MAC. Because many pathogens have evolved mechanisms to limit complement activation and prevent the progression of the cascade, generation of C3b and anaphylatoxins ensure additional layers of immune defense. These effector functions are exerted by the interaction between complement split product effectors and their host receptors to facilitate biological processes such as phagocytosis, chemotaxis, and inflammation (Figure 2A). In the absence of antibodies, C3b and its catabolites (iC3b, C3d, and C3dg) function as major opsonins recognized by complement receptors 1, 3, and 4 (CR1, CR3, CR4) that are expressed on myeloid cells and aid in phagocytosis and pathogen clearance. Anaphylatoxins C3a and C5a are critical danger signals that exert their function through interactions with cognate receptors C3aR and C5aR expressed on both immune and non-immune cells. Signaling through these receptors induces several critical pro-inflammatory immunological responses, including chemotaxis, oxidative burst, immune cell activation, vasodilation and induction of cytokines (Klos et al., 2009). Complement split products also play a critical role in bridging innate immunity and adaptive immunity (Figure 2B). For example, C3b inactivation products C3dg and C3d covalently linked to antigen are recognized by complement receptor 2 (CR2, CD21) expressed on B cells. Co-ligation of CR2 with the B cell receptor (BCR) via C3d-antigen complexes amplifies B cell signaling, lowers the threshold for B cell activation, and thus this interaction is critical for enhancing B cell responses (Carroll, 2008). Recent work has also implicated local intracellular complement activation and the subsequent production of anaphylatoxins (C3a, C5a) during cognate interactions between antigen presenting cells (APCs) and CD4+ T cells (Heeger et al., 2005; Strainic et al., 2008). Upregulation of and signaling through cognate receptors C3aR and C5aR have been shown to regulate T cell activation, lineage commitment and proinflammatory Th1 cytokine production (Strainic et al., 2008; Liszewski et al., 2013).
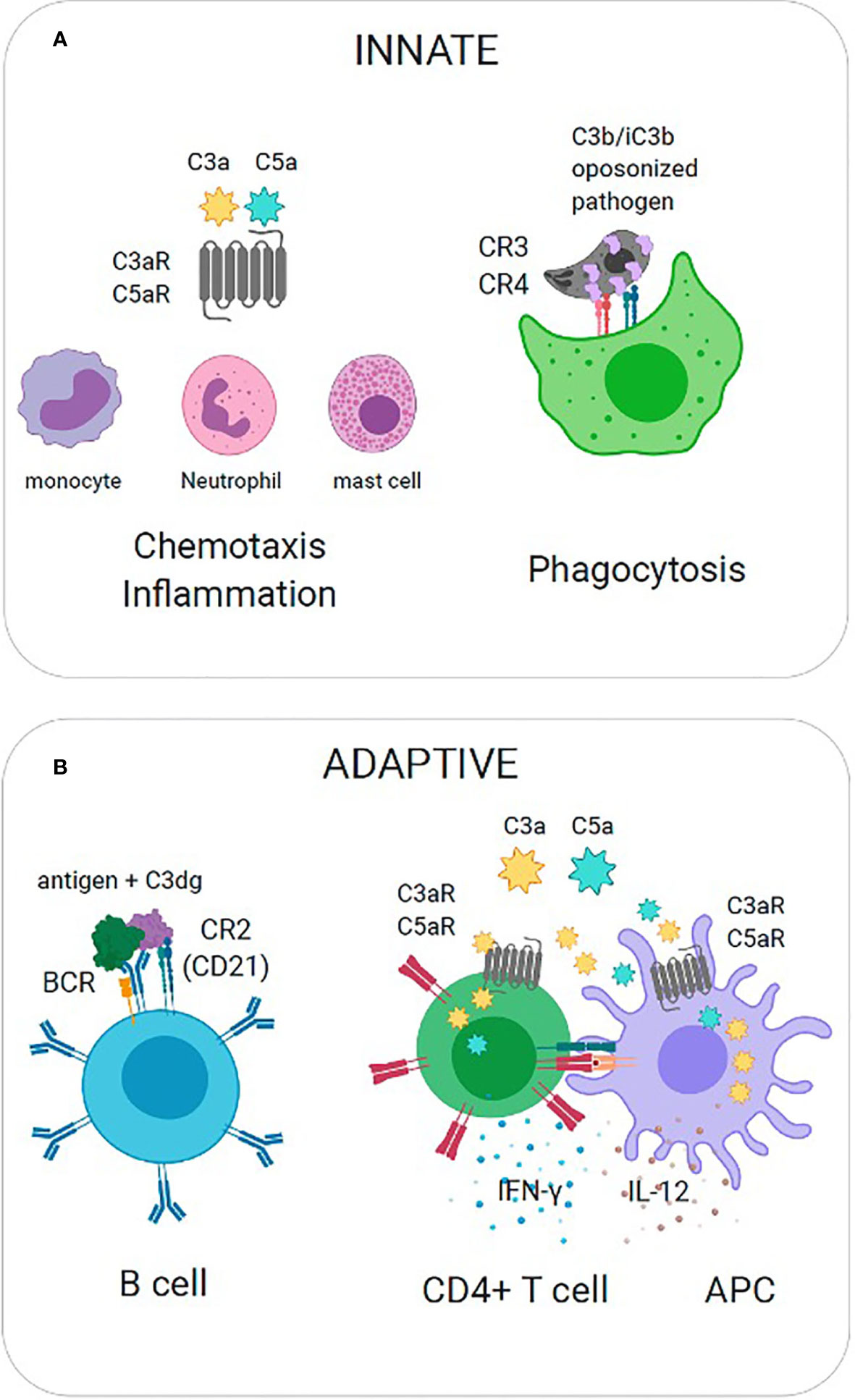
Figure 2 Effector Functions of Complement Split Products. (A) Innate immunity. (Left) C3a is recognized by cognate receptor C3aR to mediate recruitment of innate immune cells (mast cells, monocytes, neutrophils) and promote inflammation. (Right) C3b is covalently coupled to pathogen surfaces and promotes opsonization. C3b is recognized by complement receptors (CR3, CR4) to promote phagocytosis. (B) Adaptive immunity. (Left) Co-ligation of complement receptor 2 with the B cell receptor (BCR) on B cells via C3dg-antigen complexes amplifies B cell signaling and lowers the threshold for B cell activation. (Right) The local activation and secretion of complement anaphylatoxins (C3a, C5a) and upregulation of cognate receptors (C3aR, C5aR) is triggered during T cell-APC interactions. Autocrine and paracrine signaling through C3aR and C5aR promote Th1 cytokine production (IL-12, IFN).
Complement Activation and Resistance: The Apicomplexa
Apicomplexan parasites, not unlike bacteria, viruses, and fungi, activate the complement system. Complement evasion is a critical step in the establishment of infection, hence parasites have evolved multiple sophisticated strategies to overcome serum killing. Mechanisms of parasite complement evasion include the recruitment of host regulators by parasite surface molecules, expression of complement regulator protein orthologs, and expression of parasite-encoded proteins that target and/or inactivate complement function (recently reviewed by Shao et al., 2019).
While parasites share common evasion strategies, these mechanisms are achieved by unique parasite-specific factors. Plasmodium spp. evades both mosquito and human complement systems to facilitate the survival and transmission of the parasite from vector to host. Genetic studies have identified the 6-CYS protein Pfs47 as a critical factor facilitating parasite transmission in mosquitos by its ability to regulate the insect complement-like immune system (Molina-Cruz et al., 2013). Additional recently identified parasite factors that recruit human regulator Factor H to facilitate C3b inactivation include pfGAP50 expressed by gametes in the mosquito midgut and Pf92 expressed during the blood-stage (Simon et al., 2013; Kennedy et al., 2016). In addition, parasites bind plasminogen during the intraerythrocytic stage and mediate its conversion to plasmin in order to inactivate C3b, however the parasite factor(s) that facilitate this interaction have not been determined (Reiss et al., 2021).
Complement evasion by Cryptosporidium and Toxoplasma, on the contrary, is less extensively studied. While studies have established that Cryptosporidium parvum binds C3 and classical (C1q) and lectin (mannose-binding lectin, MBL) pathway molecules (Petry et al., 2008), little is known about resistance mechanisms. Our group has confirmed earlier work demonstrating that Toxoplasma inactivates C3 (Fuhrman and Joiner, 1989; Sikorski et al., 2020) but the precise molecular details have only recently been elucidated. We utilized improved strategies to study complement interactions with T. gondii using flow cytometry, leading to a significant advancement in our understanding of complement activation and regulation by T. gondii. Our work has for the first time revealed important contributions of the lectin pathway as well as parasite genotype to complement activation. Our studies showed strain-specific differences in C3b deposition between Type I and Type II strains that was partially attributed to the differences in surface glycans and lectin pathway activation, indicating that increased C3b deposition on Type II strains correlated with greater lectin binding and MBL recognition. However, despite these differences, both strains were equally capable of inactivating C3b and were resistant to serum killing. Our data showed that resistance to serum killing was due to the ability of T. gondii to recruit the alternative pathway regulator Factor H (FH) and classical and lectin pathway regulator C4b-binding protein (C4BP) to the parasite cell surface (Fuhrman and Joiner, 1989; Sikorski et al., 2020). The importance of the alternative pathway was highlighted by its critical role mediating serum resistance, irrespective of its limited contribution in the initiation of the complement system. Our studies demonstrated that blocking Factor H, but not C4BP, resulted in greater C5b-9 deposition and greater parasite susceptibility to serum killing, suggesting that the AP functions principally as a potent mediator that amplifies complement activation and is thus an important target for T. gondii regulation. Although the T. gondii parasite factor(s) that bind C3b, FH, and C4BP have not been identified, their discovery would directly address the role of individual and cumulative parasite factors in the molecular basis of protection against serum killing, and would further provide an opportunity to unearth potential targets for therapeutic intervention to render parasites more susceptible to complement attack. Collectively, these studies demonstrate how apicomplexan parasites employ multiple strategies to target C3 in order to successfully resist serum killing.
Complement: Protective and Pathological Role During T. gondii Infection
Toxoplasma gondii is a highly prevalent and successful protozoan parasite that can infect any nucleated cell in all mammals. Non felid, intermediate hosts acquire T. gondii by ingesting oocysts from contaminated water or food, or by eating infected meat containing tissue cysts. Upon ingestion, parasites excyst and invade the intestinal epithelium where they differentiate and rapidly replicate asexually as tachyzoites. The parasites then disseminate systemically to distal organs to mediate acute infection before establishing a chronic infection by the formation of tissue cysts (Dubey et al., 1997). The pathogenic tachyzoite form can be easily grown in vitro and chronic stages can be maintained in animal models, thus mice represent an experimentally tractable model system optimal for studying this host-pathogen interaction.
Until recently, T. gondii complement resistance mechanisms and the biological significance of the complement system in vivo during acute infection were unknown. The role of complement during systemic T. gondii intraperitoneal infection was recently evaluated using complement deficient C57BL/6 mice. In the absence of C3, mice died acutely and exhibited higher parasite loads whereas control mice survived. This study demonstrated that both the presence of C3 as well as the parasite’s ability to inactivate C3 are important factors regulating parasite proliferation and contributing to host survival in vivo (Sikorski et al., 2020). One limitation of using C3 deficient animals is that the protective effect observed could not be specifically assigned to either C3a or C3b split product effector function (Sikorski et al., 2020). The observed reduction in T. gondii-specific antibodies in this model was, however, consistent with the known function of C3dg tagged antigen to enhance humoral responses (Sikorski et al., 2020), suggesting that C3b opsonization of T. gondii antigen is critical for priming humoral immunity. Importantly, this study suggested that T. gondii strikes a critical balance in fine tuning complement system activation, by evading serum killing to promote parasite persistence, while preserving the ability of complement effectors C3a and C3b to activate sufficient host immunity to regulate parasite proliferation and promote both host survival and parasite transmissibility.
Other studies have attempted to elucidate the role of anaphylatoxin signaling in acute T. gondii infection. Anaphylatoxin signaling has been implicated in regulating Th1 responses, which are critical in host resistance to T. gondii infection. The first studies demonstrated increased susceptibility of C5ar1−/− C3ar1−/− double knockout (DKO) mice to T. gondii infection. Intraperitoneal injection of 20 Me49 cysts resulted in acute death within 12 days in the DKO mice whereas all WT mice survived for greater than 50 days, the longest time point studied. The DKO mice had significant reductions in IL-12 and IFN-γ secretion in splenic cultures stimulated with STAg (soluble Toxoplasma antigen) and supported a protective role for anaphylatoxin receptor signaling (Strainic et al., 2008). These findings are consistent with an established role for locally produced C3a and C5a in regulating Th1 responses through autocrine and paracrine C3aR/C5aR receptor signaling (Strainic et al., 2008; Liszewski et al., 2013). More recently, infection studies in C5ar1−/− mice showed that these mice also die acutely, but with a much less dramatic phenotype or kinetic than the DKO mice (Briukhovetska et al., 2020). In this study, 50 cysts of the Type II strain Me49 were injected intraperitoneally. At this dose, both WT and C5ar1−/− mice were susceptible to intraperitoneal infection, with 60% of WT mice dead within 30 days compared to 80% of C5ar1−/− infected mice, which exhibited a statistically significant higher parasite load during acute disease (Briukhovetska et al., 2020). The authors argued that C3b and its degradation products likely promoted the release of the anaphylatoxin C5a that bound the C5aR1 expressed on CD8α+ dendritic cells (DCs) which were activated by T. gondii-induced TLR signaling to amplify IL-12 and IFN-γ production (Briukhovetska et al., 2020). Evidence for decreased levels of serum proinflammatory cytokines and increased IL-10 serum levels in C5ar1−/− mice, and the reduction of IL-12 secretion from C5ar1−/− splenic DCs in response to STAg in vitro suggested that complement signaling was important for priming appropriate Th1 responses to control parasite replication. Collectively, these studies suggest that loss of systemic complement and anaphylatoxin receptor signaling play a significant role in the failure to sense or initiate proper humoral and cytokine responses against acute T. gondii infection, respectively, which is necessary to control parasite proliferation during acute disease.
To better understand the relative and contributing roles of complement effectors C3, C3b, and associated anaphylatoxin receptor signaling on immunological priming and control of parasite proliferation, we infected C57BL/6J mice perorally with 40 cysts of the Me49 strain. C57BL/6 mice develop an acute, Th1 CD4+ T cell-mediated lethal ileitis within 8 days of Toxoplasma peroral infection that results in tissue destruction and necrosis of the intestinal mucosa. This immunopathology is associated with exacerbated Th1 immune responses, which include increased production of the inflammatory mediators IFNγ, TNF-α, nitric oxide (Liesenfeld et al., 1996; Khan et al., 1997; Liesenfeld et al., 1999), Th1 and Th17 cytokines (Rachinel et al., 2004; Vossenkämper et al., 2004), and shifts in the intestinal microbiota (Heimesaat et al., 2006; Molloy et al., 2013; Wang et al., 2019). Systemic complement and anaphylatoxin receptor signaling is emerging as an important component of the intestinal immune response by its ability to regulate epithelial barrier integrity, the microbiota, and oral tolerance (Pekkarinen et al., 2013; Nissilä et al., 2017; Zhang et al., 2018; Benis et al., 2019; Qi et al., 2020), however the contribution of complement in the protection of mucosal barriers during acute T. gondii infection has not been directly addressed.
To investigate this question, we infected C57BL/6J WT and C3 deficient animals with 40 Me49 tissue cysts perorally and assessed survival. As a control, we also infected WT and C3 deficient animals with 25 tachyzoites of the Type I RH strain intraperitoneally to demonstrate that C3 is protective during infection regardless of parasite genotype (Figure 3A). In stark contrast to the i.p. model in which all mice die acutely, 100% of C3 deficient mice survived T. gondii infection whereas all WT mice died acutely, within 10 days (Figure 3B). Histologic examination of the spleen and intestines at days 4, 6, and 8 (Figures 3C, D) post infection showed striking differences in pathology, highlighted by areas of necrosis in the white pulp of the spleen of infected WT mice compared to C3−/− mice, and a severe necrosis of the ilea, predominantly within the villi, with significant inflammation, a loss of columnar epithelial cells and a large accumulation of granulocytes that was largely absent in the C3−/− infected mice. Parasite load was determined by plaquing a portion of the spleen and small intestine of WT versus C3 deficient mice at day 6 post infection and showed a similar level of parasites present (with no significant difference detected in the tissues examined; data not shown).
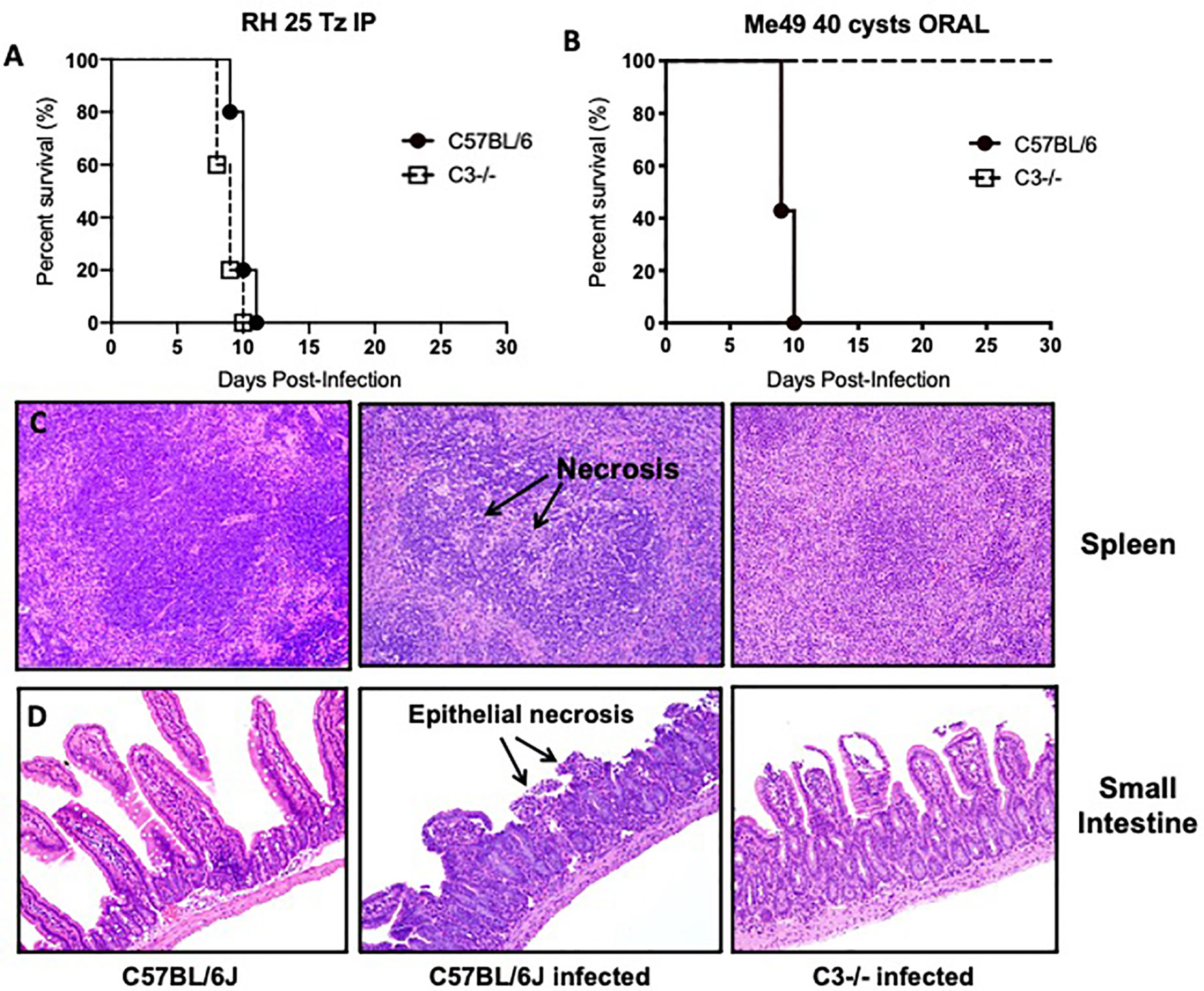
Figure 3 Lethal ileitis in Toxoplasma gondii perorally infected C57BL/6J mice is dependent on C3. (A) Survival of 6- to 8-week-old C57BL6/J (n = 5, closed circle) and C3−/− (n=5, open square) female mice infected with 25 tachyzoites injected intraperitoneally of the RH strain of Toxoplasma gondii. Data shown are for one of two independently performed experiments. (B) Survival of 6- to 8-week-old C57BL6/J (n=5, closed circle) and C3−/− (n=5, open square) female mice infected by oral gavage with 40 cysts of the Me49 strain of Toxoplasma gondii. Data shown are for one of two independently performed experiments. (C) Histological examination of the spleen of C57BL6/J or C3−/− mice stained with hematoxylin-eosin (HE) at day 8 post-infection with 40 cysts of Me49 by oral gavage. (D) Histological examination of the small intestine of C57BL6/J or C3−/− mice stained with hematoxylin-eosin (HE) at day 8 post-infection with 40 cysts of Me49 by oral gavage.
Consistent with phenotypes observed in other murine models of inflammatory-driven acute colitis, complement deficient mice were protected from T. gondii-induced lethal ileitis and survived. We hypothesize that dysregulation of the local complement response contributed detrimental inflammation and led to the pathogenesis of acute infection. Similar phenotypes have previously been observed in perorally infected mice deficient in the inducible NO synthetase enzyme (iNOS−/−) and may suggest that complement likewise plays an analogous role by exacerbating the immunopathology observed in this oral model of acute ileitis. Nitric oxide is an important mediator restricting intracellular pathogen growth. iNOS−/− mice exhibited greater dissemination and parasite burden but survived significantly longer than control mice infected perorally (Khan et al., 1997; Scharton-Kersten et al., 1997). Whereas control mice exhibited exacerbated cytokine production and necrosis, the prolonged survival of iNOS−/− mice was attributed to control of the dysregulated inflammatory response that occurs in B6 mice. Unlike the C3 deficient mice, iNOS−/− mice eventually succumb to infection, largely the result of their inability to control parasite proliferation (Khan et al., 1997). These studies indicated that iNOS and NO production is critical for parasite killing. Further, these findings suggest that acute ileitis is critically dependent on the level of IL-10 present, a critical meditator of immune homeostasis during proinflammatory Th1 responses, both in genetically susceptible C57BL/6 mice and resistant BALB/c. Indeed, mice deficient in IL-10 show an increased susceptibility to T. gondii infection compared to control animals (Gazzinelli et al., 1996; Suzuki et al., 2000). The increased susceptibility was not attributed to increases in parasite burden, but rather to the inability to control the pro-inflammatory response. Together, the studies highlighted illustrate that several immunological factors contribute toward tipping the balance towards immunopathology in T. gondii-induced ileitis, and further investigations are required to determine how protective versus pathological roles of complement contribute to the immunopathology associated with oral infection of genetically susceptible C57BL/6 mice. In the next section, we discuss how studies addressing the role of complement in other intestinal inflammatory disorders may provide insight into the observed dichotomous role for complement in the pathogenesis of acute T. gondii peroral infection.
Additional Insights and Outstanding Questions
The intestinal pathology induced by oral T. gondii infection shares similarities with human inflammatory bowel disease (IBD) (Liesenfeld, 2002). Recent studies in the IBD field support a model in which complement has both a pathogenic and a protective role and specifically, that local complement production plays a central role in the pathophysiology of these inflammatory diseases, including the established experimental mouse model of acute dextran-sulfate induced (DSS) colitis, a model for human inflammatory bowel disease. Complement is also produced extra-hepatically by immune cells and epithelial cells, including intestinal enterocytes, and may have specific functions at local sites (Morgan and Gasque, 1997; Lubbers et al., 2017). Mice deficient in C3 or Factor B were protected from acute colitis induction 5 days post DSS treatment and exhibited improved clinical outcome (Elvington et al., 2015). In contrast to previous studies (Lu et al., 2010), complement deficient mice unexpectedly died within 5 days of the DSS recovery period, indicating a protective role for complement after induction of colitis (Schepp-Berglind et al., 2012). Mortality was attributed to impaired epithelial barrier function in the absence of complement, leading to translocation of commensals and increased endotoxins, and reduction of mucosal tissue proliferation during the repair process (Schepp-Berglind et al., 2012; Elvington et al., 2015). C5 deficiency in an earlier study was also shown to make mice more susceptible to colitis after 10 days, supporting this protective role (Deguchi et al., 2005). Reported discrepancies in phenotypes across various studies are likely due to differences in disease severity attributed to dose of DSS and mouse genotype. Thus, understanding the contributions of complement to dual protective and pathogenic functions in T. gondii infection may not only rely on host genotypes but also parasite genotype and dose and thus requires further study.
Anaphylatoxin receptor signaling has also been shown to play a role in regulating pro-inflammatory responses at the intestinal barrier. Reports of a pathogenic role for C5a in colitis models are supported by the amelioration of disease pathology in mice and rats using C5aR deficient animals (Johswich et al., 2009), C5a blockade (Chen et al., 2011) or treatment with C5a agonists (Woodruff et al., 2003). Complement activation also contributed to the development of colitis-associated colorectal cancer (CAC), supported by tumor repression in complement deficient mice (C3, C5, or C5aR) (Ning et al., 2015). Mechanistic studies revealed that complement deficiency reduced proinflammatory cytokine production produced by neutrophils (IL-1β and IL-17) in the colonic tissues, indicating that C5a is a potent inducer of this response. While C3aR deficiency in BALB/c was partially protective in DDS-induced colitis, there was no significant effect observed in C57BL/6 mice (Wende et al., 2013), indicating that differences between these two mice strains are important factors in these phenotypes. Given this evidence and the recently demonstrated role of C5aR signaling in systemic T. gondii infection (Briukhovetska et al., 2020), it is highly likely that anaphylatoxins play an important role in promoting inflammation in the gut during T. gondii infection.
It is tempting to speculate that complement at mucosal barriers may need greater regulation. IBD in humans and mice is associated with increased complement activation (Ahrenstedt et al., 1990; Elvington et al., 2015; Preisker et al., 2019) and reduced expression of complement regulatory proteins in the gut epithelium (Berstad and Brandtzaeg, 1998; Scheinin et al., 1999). Accordingly, mice deficient in DAF (CD59) are more susceptible to DSS-induced colitis (Lin et al., 2004). In the previously discussed studies, while complement deficient mice died by day 5 after DSS recovery, the wild type mice treated with complement inhibitors survived the DSS treatment, suggesting complement regulation or inhibition over deficiency is more protective and may have important therapeutic implications for inflammatory conditions (Schepp-Berglind et al., 2012; Elvington et al., 2015).
Lastly, the interaction between intestinal complement, commensal bacteria, and Toxoplasma during acute infection remains to be fully elucidated. T. gondii infection is associated with microbial dysbiosis (Molloy et al., 2013; Wang et al., 2019) and emerging evidence for the role complement plays in maintaining gut homeostasis and regulating commensal microbiota is a factor that must be carefully evaluated (Benis et al., 2019; Qi et al., 2020). Equally important is to consider whether the interaction of T. gondii with complement and its regulators may contribute adversely to the outcome of this complex environment. Our previous work has shown that parasite genotype impacts complement deposition. Additional studies are required to test if the capacity of Type II strains, that show greater C3b deposition (Sikorski et al., 2020) could potentially induce greater local levels of C3a and C5a, which play a role in pro-inflammatory signaling by their cognate anaphylatoxin receptors. It is also possible that greater opsonization of Type II strains may contribute to the increased internalization of Type II strains via phagocytosis. Recent studies have shown that avirulent Type II strains preferentially enter macrophages through phagocytosis but avoid elimination by escaping the phagolysosome in murine macrophages (Zhao et al., 2014), in which the authors hypothesized this mode of entry promotes an enhanced immune stimulation and greater control of acute infection.
The identification of parasite factors that activate and regulate complement are critical for determining whether direct parasite activation and regulation of the complement system is occurring in vivo. Studies from the Plasmodium field have identified several developmentally regulated 6-CYS surface proteins that regulate complement in both the mosquito and human host (Molina-Cruz et al., 2013; Kennedy et al., 2016). Comparative modeling studies and the crystal structure of Pf12 has determined that the 6-CYS proteins share tertiary structural homology with T. gondii SRS (SAG-1 related sequences) surface proteins (He et al., 2002; Gerloff et al., 2005; Arredondo et al., 2012; Tonkin et al., 2013). This evidence points to a potential role for the structurally homologous T. gondii SRS surface proteins (Gerloff et al., 2005; Tonkin et al., 2013) to possess an analogous role. Though the mechanism for Factor H recruitment by the Plasmodium 6-CYS protein Pf92 remains elusive, it is well established that FH is recruited to host cell surfaces through its ability to bind host ligands such as sialic acid and sulfated proteoglycans (SPGs) (Blaum, 2017). Toxoplasma is known to interact with both sialic acid and SPGs via microneme proteins 1 and 4 (MIC1, MIC4) and SRS57, and thus these proteins are prime candidates for future study to determine their capacity in recruiting FH (Ortega-Barria and Boothroyd, 1999; Dzierszinski et al., 2000; Jacquet et al., 2001; Sardinha-Silva et al., 2019). Interestingly, recent analyses done in our lab have determined that the developmentally regulated SRS superfamily of surface proteins are significantly expanded in T. gondii (Jung et al., 2004; Wasmuth et al., 2012). While the biological significance of this expansion remains elusive, it is tempting to speculate that the SRS superfamily plays a role in overcoming immunological barriers to establish successful infection in a wide host range. Specifically, are there stage-specific surface or secreted proteins that activate or regulate complement in a species-specific manner, i.e. intermediate versus definitive host, or does a universally expressed SRS protein facilitate this process across several species? All of these intriguing questions require additional studies.
Concluding Remarks
The data presented in this review highlight a protective and pathogenic role for the complement system during acute T. gondii infection, depending on the route of infection. We are just beginning to understand how parasite modulation of complement activation may impact complement effector functions required to strike the optimal balance in the intermediate host. Evading serum killing ensures parasite survival, persistence and transmission to new hosts, while maintaining the generation of critical effector proteins (iC3b, C3a, C5a) that are required for stimulating sufficient humoral and Th1 immunity to regulate tachyzoite proliferation. We argue that future investigations will further unravel the immunomodulatory role of complement activation and regulation during T. gondii infection in the gut. Additional studies into the parasite factors that interact with and regulate complement are required to better understand the dynamics that occur between parasite and host systemically and locally in the gut.
Author Contributions
Review topic was solicited by MG, PS, and AC. PS wrote the first draft with editing from MG and AC. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID) at the National Institutes of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ahrenstedt Ö, Knutson L., Nilsson B., Nilsson-Ekdahl K., Odlind B., Hällgren R. (1990). Enhanced Local Production of Complement Components in the Small Intestines of Patients with Crohn’s Disease. New Engl. J. Med. 322 (19), 1345–1349. doi: 10.1056/NEJM199005103221903
Arredondo S. A., Cai M., Takayama Y., MacDonald N. J., Anderson D. E., Aravind L., et al. (2012). Structure of the Plasmodium 6-cysteine s48/45 domain. Proc. Natl. Acad. Sci. 109 (17), 6692–6697. doi: 10.1073/pnas.1204363109
Belachew E. B. (2018). Immune Response and Evasion Mechanisms of Plasmodium falciparum Parasites. J. Immunol. Res. 2018:6529681. doi: 10.1155/2018/6529681
Benis N., Wells J. M., Smits M. A., Kar S. K., van der Hee B., dos Santos V. A. P. M., et al. (2019). High-level integration of murine intestinal transcriptomics data highlights the importance of the complement system in mucosal homeostasis. BMC Genomics 20 (1), 1028. doi: 10.1186/s12864-019-6390-x
Berstad A. E., Brandtzaeg P. (1998). Expression of cell membrane complement regulatory glycoproteins along the normal and diseased human gastrointestinal tract. Gut 42 (4), 522–529. doi: 10.1136/gut.42.4.522
Blaum B. S. (2017). The lectin self of complement factor H. Curr. Opin. Struct. Biol. 44, 111–118. doi: 10.1016/j.sbi.2017.01.005
Briukhovetska D., Ohm B., Mey F. T., Aliberti J., Kleingarn M., Huber-Lang M., et al. (2020). C5aR1 Activation Drives Early IFN-γ Production to Control Experimental Toxoplasma gondii Infection. Front. Immunol. 11:1397 (1397), 1–15. doi: 10.3389/fimmu.2020.01397
Carroll M. C. (2008). Complement and humoral immunity. Vaccine 26 Suppl 8 (0 8), I28–I33. doi: 10.1016/j.vaccine.2008.11.022
Chen G., Yang Y., Gao X., Dou Y., Wang H., Han G., et al. (2011). Blockade of complement activation product C5a activity using specific antibody attenuates intestinal damage in trinitrobenzene sulfonic acid induced model of colitis. Lab. Invest. 91 (3), 472–483. doi: 10.1038/labinvest.2010.183
Deguchi Y., Andoh A., Inatomi O., Araki Y., Hata K., Tsujikawa T., et al. (2005). Development of dextran sulfate sodium-induced colitis is aggravated in mice genetically deficient for complement C5. Int. J. Mol. Med. 16 (4), 605–608. doi: 10.3892/ijmm.16.4.605
Dubey J. P., Speer C. A., Shen S. K., Kwok O. C., Blixt J. A. (1997). Oocyst-induced murine toxoplasmosis: life cycle, pathogenicity, and stage conversion in mice fed Toxoplasma gondii oocysts. J. Parasitol. 83 (5), 870–882. doi: 10.2307/3284282
Dzierszinski F., Mortuaire M., Cesbron-Delauw M. F., Tomavo S. (2000). Targeted disruption of the glycosylphosphatidylinositol-anchored surface antigen SAG3 gene in Toxoplasma gondii decreases host cell adhesion and drastically reduces virulence in mice. Mol. Microbiol. 37 (3), 574–582. doi: 10.1046/j.1365-2958.2000.02014.x
Elvington M., Schepp-Berglind J., Tomlinson S. (2015). Regulation of the alternative pathway of complement modulates injury and immunity in a chronic model of dextran sulphate sodium-induced colitis. Clin. Exp. Immunol. 179 (3), 500–508. doi: 10.1111/cei.12464
Fuhrman S. A., Joiner K. A. (1989). Toxoplasma gondii: mechanism of resistance to complement-mediated killing. J. Immunol. 142 (3), 940–947.
Gazzinelli R. T., Wysocka M., Hieny S., Scharton-Kersten T., Cheever A., Kühn R., et al. (1996). In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J. Immunol. 157 (2), 798–805.
Gerloff D. L., Creasey A., Maslau S., Carter R. (2005). Structural models for the protein family characterized by gamete surface protein Pfs230 of & lt;em<Plasmodium falciparum</em>. Proc. Natl. Acad. Sci. U. States America 102 (38), 13598–13603. doi: 10.1073/pnas.0502378102
He X.-l., Grigg M. E., Boothroyd J. C., Garcia K. C. (2002). Structure of the immunodominant surface antigen from the Toxoplasma gondii SRS superfamily. Nat. Struct. Biol. 9 (8), 606–611. doi: 10.1038/nsb819
Heeger P. S., Lalli P. N., Lin F., Valujskikh A., Liu J., Muqim N., et al. (2005). Decay-accelerating factor modulates induction of T cell immunity. J. Exp. Med. 201 (10), 1523–1530. doi: 10.1084/jem.20041967
Heimesaat M. M., Bereswill S., Fischer A., Fuchs D., Struck D., Niebergall J., et al. (2006). Gram-negative bacteria aggravate murine small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii. J. Immunol. 177 (12), 8785–8795. doi: 10.4049/jimmunol.177.12.8785
Jacquet A., Coulon L., De Nève J., Daminet V., Haumont M., Garcia L., et al. (2001). The surface antigen SAG3 mediates the attachment of Toxoplasma gondii to cell-surface proteoglycans. Mol. Biochem. Parasitol. 116 (1), 35–44. doi: 10.1016/s0166-6851(01)00297-3
Johswich K., Martin M., Bleich A., Kracht M., Dittrich-Breiholz O., Gessner J. E., et al. (2009). Role of the C5a receptor (C5aR) in acute and chronic dextran sulfate-induced models of inflammatory bowel disease. Inflammation Bowel Dis. 15 (12), 1812–1823. doi: 10.1002/ibd.21012
Jung C., Lee C. Y., Grigg M. E. (2004). The SRS superfamily of Toxoplasma surface proteins. Int. J. Parasitol. 34 (3), 285–296. doi: 10.1016/j.ijpara.2003.12.004
Kennedy A. T., Schmidt C. Q., Thompson J. K., Weiss G. E., Taechalertpaisarn T., Gilson P. R., et al. (2016). Recruitment of Factor H as a Novel Complement Evasion Strategy for Blood-Stage Plasmodium falciparum Infection. J. Immunol. 196 (3), 1239–1248. doi: 10.4049/jimmunol.1501581
Khan I. A., Schwartzman J. D., Matsuura T., Kasper L. H. (1997). A dichotomous role for nitric oxide during acute Toxoplasma gondii infection in mice. Proc. Natl. Acad. Sci. U. S. A. 94 (25), 13955–13960. doi: 10.1073/pnas.94.25.13955
Klos A., Tenner A. J., Johswich K.-O., Ager R. R., Reis E. S., Köhl J. (2009). The role of the anaphylatoxins in health and disease. Mol. Immunol. 46 (14), 2753–2766. doi: 10.1016/j.molimm.2009.04.027
Liesenfeld O., Kosek J., Remington J. S., Suzuki Y. (1996). Association of CD4+ T cell-dependent, interferon-gamma-mediated necrosis of the small intestine with genetic susceptibility of mice to peroral infection with Toxoplasma gondii. J. Exp. Med. 184 (2), 597–607. doi: 10.1084/jem.184.2.597
Liesenfeld O., Kang H., Park D., Nguyen T. A., Parkhe C. V., Watanabe H., et al. (1999). TNF-alpha, nitric oxide and IFN-gamma are all critical for development of necrosis in the small intestine and early mortality in genetically susceptible mice infected perorally with Toxoplasma gondii. Parasite Immunol. 21 (7), 365–376. doi: 10.1046/j.1365-3024.1999.00237.x
Liesenfeld O. (2002). Oral Infection of C57BL/6 Mice with Toxoplasma gondii: A New Model of Inflammatory Bowel Disease? J. Infect. Dis. 185 (Supplement_1), S96–S101. doi: 10.1086/338006
Lin F., Spencer D., Hatala D. A., Levine A. D., Medof M. E. (2004). Decay-accelerating factor deficiency increases susceptibility to dextran sulfate sodium-induced colitis: role for complement in inflammatory bowel disease. J. Immunol. 172 (6), 3836–3841. doi: 10.4049/jimmunol.172.6.3836
Liszewski M. K., Kolev M., Le Friec G., Leung M., Bertram P. G., Fara A. F., et al. (2013). Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 39 (6), 1143–1157. doi: 10.1016/j.immuni.2013.10.018
Lu F., Fernandes S. M., Davis A. E. ,. 3. (2010). The role of the complement and contact systems in the dextran sulfate sodium-induced colitis model: the effect of C1 inhibitor in inflammatory bowel disease. Am. J. Physiol. Gastrointest. Liver Physiol. 298 (6), G878–G883. doi: 10.1152/ajpgi.00400.2009
Lubbers R., van Essen M. F., van Kooten C., Trouw L. A. (2017). Production of complement components by cells of the immune system. Clin. Exp. Immunol. 188 (2), 183–194. doi: 10.1111/cei.12952
Molina-Cruz A., Garver L. S., Alabaster A., Bangiolo L., Haile A., Winikor J., et al. (2013). The human malaria parasite Pfs47 gene mediates evasion of the mosquito immune system. Sci. (New York N.Y.) 340 (6135), 984–987. doi: 10.1126/science.1235264
Molloy M. J., Grainger J. R., Bouladoux N., Hand T. W., Koo L. Y., Naik S., et al. (2013). Intraluminal containment of commensal outgrowth in the gut during infection-induced dysbiosis. Cell Host Microbe 14 (3), 318–328. doi: 10.1016/j.chom.2013.08.003
Morgan B. P., Gasque P. (1997). Extrahepatic complement biosynthesis: where, when and why? Clin. Exp. Immunol. 107 (1), 1–7. doi: 10.1046/j.1365-2249.1997.d01-890.x
Ning C., Li Y. Y., Wang Y., Han G. C., Wang R. X., Xiao H., et al. (2015). Complement activation promotes colitis-associated carcinogenesis through activating intestinal IL-1β/IL-17A axis. Mucosal Immunol. 8 (6), 1275–1284. doi: 10.1038/mi.2015.18
Nissilä E., Korpela K., Lokki A. I., Paakkanen R., Jokiranta S., de Vos W. M., et al. (2017). C4B gene influences intestinal microbiota through complement activation in patients with paediatric-onset inflammatory bowel disease. Clin. Exp. Immunol. 190 (3), 394–405. doi: 10.1111/cei.13040
Ortega-Barria E., Boothroyd J. C. (1999). A Toxoplasma lectin-like activity specific for sulfated polysaccharides is involved in host cell infection. J. Biol. Chem. 274 (3), 1267–1276. doi: 10.1074/jbc.274.3.1267
Pekkarinen P. T., Vaali K., Jarva H., Kekäläinen E., Hetemäki I., Junnikkala S., et al. (2013). Impaired intestinal tolerance in the absence of a functional complement system. J. Allergy Clin. Immunol. 131 (4), 1167–1175. doi: 10.1016/j.jaci.2012.09.004
Petry F., Jakobi V., Wagner S., Tessema T. S., Thiel S., Loos M. (2008). Binding and activation of human and mouse complement by Cryptosporidium parvum (Apicomplexa) and susceptibility of C1q- and MBL-deficient mice to infection. Mol. Immunol. 45 (12), 3392–3400. doi: 10.1016/j.molimm.2008.04.010
Preisker S., Brethack A. K., Bokemeyer A., Bettenworth D., Sina C., Derer S. (2019). Crohn’s Disease Patients in Remission Display an Enhanced Intestinal IgM⁺ B Cell Count in Concert with a Strong Activation of the Intestinal Complement System. Cells 8 (1), 1–13. doi: 10.3390/cells8010078
Qi H., Wei J., Gao Y., Yang Y., Li Y., Zhu H., et al. (2020). Reg4 and complement factor D prevent the overgrowth of E. coli in the mouse gut. Commun. Biol. 3 (483), 1–15. doi: 10.1038/s42003-020-01219-2
Rachinel N., Buzoni-Gatel D., Dutta C., Mennechet F. J., Luangsay S., Minns L. A., et al. (2004). The induction of acute ileitis by a single microbial antigen of Toxoplasma gondii. J. Immunol. 173 (4), 2725–2735. doi: 10.4049/jimmunol.173.4.2725
Reiss T., Theis H. I., Gonzalez-Delgado A., Vega-Rodriguez J., Zipfel P. F., Skerka C., et al. (2021). Acquisition of human plasminogen facilitates complement evasion by the malaria parasite Plasmodium falciparum. Eur. J. Immunol. 51, 490–493. doi: 10.1002/eji.202048718
Sardinha-Silva A., Mendonça-Natividade F. C., Pinzan C. F., Lopes C. D., Costa D. L., Jacot D., et al. (2019). The lectin-specific activity of Toxoplasma gondii microneme proteins 1 and 4 binds Toll-like receptor 2 and 4 N-glycans to regulate innate immune priming. PloS Pathog. 15 (6), e1007871–e1007871. doi: 10.1371/journal.ppat.1007871
Scharton-Kersten T. M., Yap G., Magram J., Sher A. (1997). Inducible Nitric Oxide Is Essential for Host Control of Persistent but Not Acute Infection with the Intracellular Pathogen Toxoplasma gondii. J. Exp. Med. 185 (7), 1261–1274. doi: 10.1084/jem.185.7.1261
Scheinin T., Böhling T., Halme L., Kontiainen S., Bjørge L., Meri S. (1999). Decreased expression of protectin (CD59) in gut epithelium in ulcerative colitis and Crohn’s disease. Hum. Pathol. 30 (12), 1427–1430. doi: 10.1016/S0046-8177(99)90163-6
Schepp-Berglind J., Atkinson C., Elvington M., Qiao F., Mannon P., Tomlinson S. (2012). Complement-dependent injury and protection in a murine model of acute dextran sulfate sodium-induced colitis. J. Immunol. (Baltimore Md. 1950) 188 (12), 6309–6318. doi: 10.4049/jimmunol.1200553
Schmidt C. Q., Lambris J. D., Ricklin D. (2016). Protection of host cells by complement regulators. Immunol. Rev. 274 (1), 152–171. doi: 10.1111/imr.12475
Shao S., Sun X., Chen Y., Zhan B., Zhu X. (2019). Complement Evasion: An Effective Strategy That Parasites Utilize to Survive in the Host. Front. Microbiol. 10:532. doi: 10.3389/fmicb.2019.00532
Sikorski P. M., Commodaro A. G., Grigg M. E. (2020). Toxoplasma gondii Recruits Factor H and C4b-Binding Protein to Mediate Resistance to Serum Killing and Promote Parasite Persistence in vivo. Front. Immunol. 10:3105. doi: 10.3389/fimmu.2019.03105
Simon N., Lasonder E., Scheuermayer M., Kuehn A., Tews S., Fischer R., et al. (2013). Malaria parasites co-opt human factor H to prevent complement-mediated lysis in the mosquito midgut. Cell Host Microbe 13 (1), 29–41. doi: 10.1016/j.chom.2012.11.013
Strainic M. G., Liu J., Huang D., An F., Lalli P. N., Muqim N., et al. (2008). Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity 28 (3), 425–435. doi: 10.1016/j.immuni.2008.02.001
Suzuki Y., Sher A., Yap G., Park D., Neyer L. E., Liesenfeld O., et al. (2000). IL-10 is required for prevention of necrosis in the small intestine and mortality in both genetically resistant BALB/c and susceptible C57BL/6 mice following peroral infection with Toxoplasma gondii. J. Immunol. 164 (10), 5375–5382. doi: 10.4049/jimmunol.164.10.5375
Thurman J. M., Holers V. M. (2006). The Central Role of the Alternative Complement Pathway in Human Disease. J. Immunol. 176 (3), 1305–1310. doi: 10.4049/jimmunol.176.3.1305
Tonkin M. L., Arredondo S. A., Loveless B. C., Serpa J. J., Makepeace K. A., Sundar N., et al. (2013). Structural and biochemical characterization of Plasmodium falciparum 12 (Pf12) reveals a unique interdomain organization and the potential for an antiparallel arrangement with Pf41. J. Biol. Chem. 288 (18), 12805–12817. doi: 10.1074/jbc.M113.455667
Vossenkämper A., Struck D., Alvarado-Esquivel C., Went T., Takeda K., Akira S., et al. (2004). Both IL-12 and IL-18 contribute to small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii, but IL-12 is dominant over IL-18 in parasite control. Eur. J. Immunol. 34 (11), 3197–3207. doi: 10.1002/eji.200424993
Votýpka J., Modrý D., Oborník M., Šlapeta J., Lukeš J., Chapman D. J. (2017). “Apicomplexa,” in Handbook of the Protists. Eds. Archibald J. M., Simpson A. G. B., Slamovits C. H., Margulis L., Melkonian M., Corliss J. O. (Cham: Springer International Publishing), 1–58.
Wang S., El-Fahmawi A., Christian D. A., Fang Q., Radaelli E., Chen L., et al. (2019). Infection-Induced Intestinal Dysbiosis Is Mediated by Macrophage Activation and Nitrate Production. mBio 10 (3):e00935-19, 1–13. doi: 10.1128/mBio.00935-19
Wasmuth J. D., Pszenny V., Haile S., Jansen E. M., Gast A. T., Sher A., et al. (2012). Integrated Bioinformatic and Targeted Deletion Analyses of the SRS Gene Superfamily Identify SRS29C as a Negative Regulator of Toxoplasma Virulence. mBio 3 (6), e00321–e00312. doi: 10.1128/mBio.00321-12
Wende E., Laudeley R., Bleich A., Bleich E., Wetsel R. A., Glage S., et al. (2013). The complement anaphylatoxin C3a receptor (C3aR) contributes to the inflammatory response in dextran sulfate sodium (DSS)-induced colitis in mice. PloS One 8 (4), e62257. doi: 10.1371/journal.pone.0062257
Woodruff T. M., Arumugam T. V., Shiels I. A., Reid R. C., Fairlie D. P., Taylor S. M. (2003). A potent human C5a receptor antagonist protects against disease pathology in a rat model of inflammatory bowel disease. J. Immunol. 171 (10), 5514–5520. doi: 10.4049/jimmunol.171.10.5514
Zhang J., Ye J., Ren Y., Zuo J., Dai W., He Y., et al. (2018). Intracellular activation of complement C3 in Paneth cells improves repair of intestinal epithelia during acute injury. Immunotherapy 10 (15), 1325–1336. doi: 10.2217/imt-2018-0122
Keywords: complement, Toxoplasma gondii, C4BP, factor H, regulation, immune evasion
Citation: Sikorski PM, Commodaro AG and Grigg ME (2021) A Protective and Pathogenic Role for Complement During Acute Toxoplasma gondii Infection. Front. Cell. Infect. Microbiol. 11:634610. doi: 10.3389/fcimb.2021.634610
Received: 28 November 2020; Accepted: 06 January 2021;
Published: 22 February 2021.
Edited by:
Chandra Ramakrishnan, University of Zurich, SwitzerlandReviewed by:
Eric Y. Denkers, University of New Mexico, United StatesCarsten Lüder, Universitätsmedizin Göttingen, Germany
Copyright © 2021 Sikorski, Commodaro and Grigg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael E. Grigg, Z3JpZ2dtQG5pYWlkLm5paC5nb3Y=
†Present address: Patricia M. Sikorski, Department of Neurology, George Washington University, Washington, DC, United States