 Chong Chen1†‡
Chong Chen1†‡ Min Wang1,2‡Tao Huang1Dong-liang Huang3Shuai Yu4Hui-min Zhao1
Min Wang1,2‡Tao Huang1Dong-liang Huang3Shuai Yu4Hui-min Zhao1 Xiang-xiang Fu2Xin-xin Li2
Xiang-xiang Fu2Xin-xin Li2 Hua Wu1*
Hua Wu1*- 1Department of Clinical Laboratory, Hainan General Hospital, Haikou, Hainan, China
- 2Department of Pharmacy, Hainan General Hospital (Hainan Medical University Hainan Hospital), Haikou, Hainan, China
- 3Emergency Department, Hainan Armed Police Force Hospital, Haikou, Hainan, China
- 4Department of Tropical Medicine, Hainan Hospital of People’s Liberation Army (PLA) General Hospital, Sanya, Hainan, China
Introduction: Rarely does Pandoraea occur in bloodstream infections (BSI), although it’s typically found in cystic fibrosis. This study aims to decipher the genetic map and obtain insights of clinical symptoms into Pandoraea from BSI patients.
Methods: 30 suspected BSI patients' diagnostic records and medical histories were recorded. Pandoraea spp. isolates were collected and subjected to antimicrobial susceptibility testing, Sanger sequencing and Whole-genome sequencing (WGS).
Results: Of the 30 clinical cases, five (16.67%) ultimately died, whereas 25 (83.33%) are alive. 30 purified Pandoraea isolates showed high degree of MIC values to Meropenem, Amoxicillin and Potassium Clavulanate, Gentamicin, and Ceftazidime. Then, all isolates were identified as P. pneumonica based on the 16S rRNA-based phylogenetic analysis. Among 28 genomes of them, the average genome size and average GC contents were 5,397,568 bp, and 62.43%, respectively. However, WP1 displayed high similarity (90.6%) to reference Pandoraea sp. LMG 31114. Genetic differences between the tested isolates and LMG 31114 suggested that the outbreak’s causative pathogen could be a novel cluster of P. pneumonica. The genomes accumulated mutations at an estimated rate of 1.3 × 10-7 mutations/year/site. Moreover, 26 clinical isolates within the P. pneumonica cluster were formed in July 2014, revealing a tendency to develop regional endemic patterns.
Conclusion: BSI caused by this novel cluster of P. pneumonica is linked to significant morbidity and mortality. Such cluster remains a critical public health challenge due to their regional epidemiological patterns and antibiotic treatment risk. This study contributed to the basis on pathogen identification, disease diagnosis, and BSI treatment.
Introduction
Bloodstream infections (BSI), a growing public health burden and concern worldwide, are life-threatening human inflammation and dysfunction caused by a dysregulated host response to bacterial, fungal, and viral infections. Approximately 575,000–677,000 clinical cases/year and 79,000–94,000 deaths/year due to BSI have been reported in North America, whereas Europe experiences approximately 1,200,000 episodes and 157,000 deaths per year (Goto and Al-Hasan, 2013). The underlying cause of the infection may be used to distinguish or classify BSI cases. For instance, diagnostic codes for pneumonia, peritonitis, or urinary tract infections may be assigned to patients with BSI resulting from respiratory, gastrointestinal, or urinary tract sources of infection, respectively. Furthermore, BSI can go unnoticed as a cause of mortality, especially in individuals with end-stage renal or hepatic diseases, advanced cancer, or other terminal illnesses.
Pandoraea spp. serves as opportunistic pathogens in patients with cystic fibrosis (CF); however, their pathogenic mechanisms have not yet been systematically explored or understood. Epithelial cell invasion and increased levels of pro-inflammatory cytokines (interleukin [IL]-6 and IL-8) usually occur in these cases (Caraher et al., 2008; Green and Jones, 2018). Additionally, a CF individual with Pandoraea spp. was associated with BSI (Johnson et al., 2004) whereas six cases without CF were reported (Stryjewski et al., 2003; Falces-Romero et al., 2016; Xiao et al., 2019; Bodendoerfer et al., 2021; Gawalkar et al., 2021; Singh et al., 2021). Itoh et al. reported the first case of obstructive cholangitis related to bacteremia caused by P. apista which was identified using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and BLAST algorithms based on 16s ribosomal RNA sequence (Itoh et al., 2022). Another case of Pandoraea pnomenusa infection was recorded locally in Portugal revealed that the increased usage of carbapenems result in increased morbidity (Ramos Oliveira et al., 2023). Given the recent increase in the incidence rate of Pandoraea spp., it is imperative to comprehensively demonstrate its prevalence.
Most cases of CF caused by Pandoraea have been observed in Europe, America, and Australia, whereas in Asia, this type of case has rarely been reported (Lin et al., 2019; Singh et al., 2021). Patients with BSI caused by Pandoraea are even rarer. In addition to epidemiological data, the paucity of clinical reports indicate that the diagnosis of Pandoraea spp. is underestimated because of the challenges associated with standard diagnostic testing. For example, laboratory workers probably misidentify this microorganism as Ralstonia spp., Stenotrophomonas spp., or Burkholderia spp. when using only widely used assays, including the VITEK 2 automated microbiological system and standard phenotypic approaches (McMenamin et al., 2000; Bosshard et al., 2006). Furthermore, Sanger sequencing based on 16S ribosomal RNA and gyrB gene has low resolution for the classification of closely related Pandoraea spp. and poor traceability (Coenye and LiPuma, 2002; Pimentel and MacLeod, 2008). In contrast, high-throughput sequencing allows for classification and genetic evolutionary analysis based on bacterial whole genome. Besides, a whole map of core genes, antimicrobial resistance genes and virulence genes of infectious agents would be drawn (Shelenkov et al., 2020). It greatly benefits BSI patients in terms of an accurate diagnosis, preventing illness from worsening, shortening their hospital stay, and even improving survival rates.
In this study, the clinical presentations of 30 patients with suspected bacteremia or BSI were recorded. Pandoraea spp. were obtained from patient samples and subjected to whole-genome sequencing (WGS) and bioinformatic analyses to systematically analyze their epidemiology. This research offers a basic understanding of the relationship between P. pneumonica and clinical symptoms in patients as well as a theoretical direction for molecular diagnosis of this emerging pathogen.
Materials and methods
Clinical specimens
Based on hospital records, past patient data were retrieved. Sex, age, address, occupation, length of hospital stay, time required to isolate Pandoraea species, clinical symptoms at different times of the day, diagnostic evidence, use of antibiotics, use of antimicrobial drugs, underlying medical disorders, and patient outcomes were included in the data. The isolation of Pandoraea spp. from the blood was considered an infection based on the combined judgment of two infectious disease consultants.
Initial enriched cultures from positive blood culture mixtures were transferred to the enrichment culture plates using cell separation gradient solution on sheep blood agar and incubated aerobically at 37°C for 24–48 h. For morphological observations (the presence and kind of hemolysis, morphology, color, shape, size, and consistency) and primary identification, gram-negative bacteria were carefully sub-cultured on blood agar and MacConkey agar plates (Oxoid Ltd., Basingstoke, United Kingdom).
Testing for antimicrobial susceptibility of Pandoraea isolates
The Pandoraea isolates were subjected for antimicrobial susceptibility testing, according to the detailed guidelines of the Clinical and Laboratory Standards Institute (CLSI). Mueller-Hinton Broth was applied for bacterial growth. The following 12 antibiotics were used in this study: Ceftazidime (CAZ), Piperacillin/Tazobactam (TZP), Amoxicillin/Clavulanate (AMC), Imipenem (IPM), Meropenem (MEM), Doxycycline (DOX), Chloramphenicol (CHL), Cefoperazone/sulbactam (SCF), Tigecycline (TGC), Ciprofloxacin (CIP), Trimethoprim-sulfamethoxazole (SXT) and Gentamicin (GEN). Since there were no criteria for antibiotic resistance of Pandoraea in CLSI at current, the Burkholderia cepacian was used as quality control organisms. Finally, the MIC values were determined visually after incubation for 18 hours.
DNA extraction and genome sequencing
The whole genome of Pandoraea was extracted and purified using a 12224-250 DNeasy UltraClean Microbial Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. DNA quality was determined by 1.0% agarose gel electrophoresis, and the 260/280 absorbance ratios were determined using a NanoDrop2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
For short-read sequencing, paired-end libraries were constructed following DNA fragmentation, end repair, and adaptor joining using Nextera XT library preparation kits (Illumina, San Diego, CA, USA). The libraries were cleaned using AMPure beads (Beckman Coulter Inc., Brea, CA, USA). Genomic libraries were assayed using a Flex Fluorometer Qubit4.0 and sequenced using a MiSeq sequencer (Illumina, San Diego, CA, USA).
For long-read sequencing, individual DNA libraries were prepared following DNA fragmentation, end repair, A-tailing, and adaptor joining using sequencing kit chemistries SQK-LSK109 (Oxford Nanopore Technologies, Oxford, United Kingdom). After library quantification using a Flex Fluorometer Qubit4.0, approximately 700 ng library was loaded onto an Oxford Nanopore MinION platform (Oxford Nanopore Technologies [ONT], Oxford, United Kingdom) for sequencing.
Genome assembly and annotation
Quality control of Illumina sequencing reads and adapter trimming were performed using fastp (v0.23.2) with default parameters (Chen et al., 2018). De novo genome assembly based on clean reads from high-throughput sequencing was performed using SPAdes v.3.11 with default parameters (Prjibelski et al., 2020). To construct the complete genome, a hybrid assembly based on Illumina short reads and ONT long reads of WP1 was constructed using Unicycler 0.4.8 (Wick et al., 2017). Genome size and GC content were simultaneously measured and recorded. Subsequently, Prokka v1.1.3 was used to predict putative protein-coding genes with default parameters (Seemann, 2014).
Comparative genomic analysis
Phylogenetic analysis based on 16S rDNA gene sequences was performed, and a neighbor-joining tree was visualized using MEGA X (Kumar et al., 2018). The average nucleotide identity (ANI) among all pairwise combinations of the whole genome of Pandoraea was computed using the BLASTN alignment tool in the Pyani package and visualized via an interactive heatmap (Pritchard et al., 2016). Core-genome single nucleotide polymorphism (SNP) analysis was performed using Parsnp software (V1.2) with the parameters “-c -x” (Kille et al., 2024). Core SNP positions were extracted using Harvesttools (Treangen et al., 2014) and then analyzed for recombination using the Gubbins software (Croucher et al., 2015). Tandem SNP loci after the removal of recombination were used to construct maximum likelihood trees using RaxML (Stamatakis et al., 2005), with the model set to GTRGAMMA and bootstrap of 1000 times.
Pan-genome analysis and identification of virulence and antibiotic resistance factors
The pan-genome of Pandoraea spp. was performed using Prokka v1.1.3 tool generated feature files, and the families of homologous genes for Pandoraea spp. were assessed using Roary v3.11 with similarity cutoff value of 90.0% (Page et al., 2015). The virulence signatures and antimicrobial resistance genes were predicted from the representative sequences of pan-genome using the ABRicate software (v. 0.8.10) when both similarity and coverage were set to 60%.
Inference of the evolutionary history of the novel Pandoraea pneumonica group
The best nucleotide substitution model general time-reversible (GTR) calculated using jModelTest 2.1.10 was selected for Bayesian inference using the core SNP alignment (Darriba et al., 2012). Molecular clock models (Strict and uncorrelated relaxed) and tree models (Constant Size, Exponential Growth, Expansion Growth, Bayesian SkyGrid, and Bayesian Skyline) were nested to test for convergence in the data set, and each model was run for 100,000,000 states at a 10,000 sampling frequency using BEAST software (v1.8.4). The Tracer program in BEAST was used to verify the convergence of every model combination (ESS of the tree model over 200). The maximum clade credibility (MCC) tree was generated using TreeAnnotator program in BEAST based on the best model (strict molecular clock and Bayesian Skyline tree model) and visualized using FigTree software (v1.4.3). The posterior mean values and 95% highest posterior density intervals of divergence time were estimated using 10 million Markov chain Monte Carlo generations, by sampling every 5000, and discarding the first 10% as burn-ins.
Results and discussion
Clinical symptoms of the infected patients
Pandoraeae spp. are usually isolated from environmental samples (soil, water, etc.) (Coenye et al., 2000). However, reports of BSI associated with comorbidities or complications are extremely rare. Herein, we described a large P. pneumonica outbreak in 30 patients. Between October 2021 and April 2024, 22 men and eight women were enrolled in this study (Table 1). One patient was 19 years old, seven were 35–49 years old, 15 were 50–69 years old, and seven were > 69 years old. In Hainan, 22 cases were found between May 15 and November 15 of the flood season, whereas eight cases were found at other times. During the flood season in extremely vulnerable communities in flooded areas, outbreaks of pathogenic infectious diseases, such as E. coli (Shah et al., 2016) and Vibrio spp (Esteves et al., 2015; Brumfield et al., 2021). increased. This might indicate a pathway of spread from the environment to humans. Itoh et al. have reported a similar clinical case (Itoh et al., 2022). The potential source of P. apista resulting in bacteremia was attributed to contamination of this pathogen from the environment during endoscopic ultrasound-guided hepaticogastrostomy surgery. Among the underlying medical disorders, 17 patients had gastrointestinal tract inflammation or peritoneal dialysis-associated peritonitis, 15 had high blood pressure, 10 were divided into a low-protein group, six had renal insufficiency, and four had anemia. The hypothesis that food-sourced E. coli cause BSI has been previously reported (Day et al., 2019). Further evidence is required to support the idea that eating food contaminated with pathogens can cause gastrointestinal tract inflammation and increase the risk of BSI caused by Pandoraeae spp. Additionally, four patients developed infections as a result of physical trauma, seven patients contracted infections after exposure to cold, and 17 patients had triggers that were unclear. In cases of severe trauma, 14.1% of patients with BSI had an initial serum procalcitonin level > 2 ng/mL (Rajkumari et al., 2013).
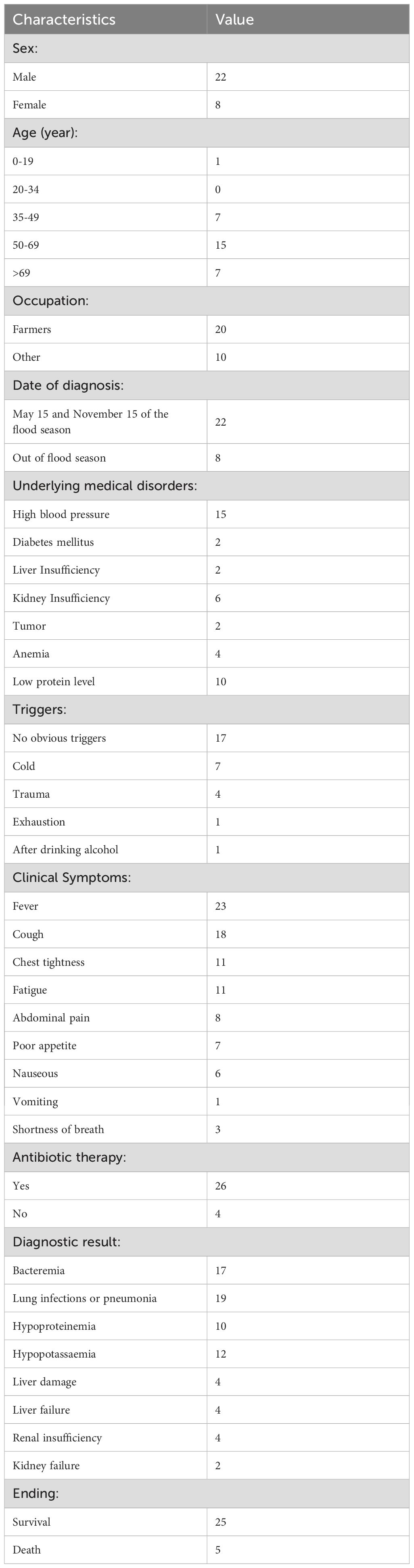
Table 1. Patient characteristics in an outbreak of P. pneumonica among BSI patients in Hainan during October 2021–April 2024.
The assessment of clinical symptoms in patients with suspected bacteremia or BSI during this outbreak is not always straightforward. Twenty-three patients had fever, 11 had chest tightness, 11 had exhaustion, eight had abdominal pain, seven had poor appetite, six had nausea, and 3 had decreased consciousness during the infection period (Table 1). BSI is caused by a wide variety of pathogens but carries clinical syndromes with considerable overlap, including fever, chills, and changes in mental status (Martinez and Wolk, 2016). In addition to Pandoraeae spp., Ralstonia pickettii, Escherichia coli, Burkholderia cepacian, and Leifsonia aquatica was detected in the blood cultures of four, one, one and one patients, respectively. Additionally, 19 patients had potential pulmonary infection, among which five were validated to have co-infections (two with severe acute respiratory syndrome coronavirus 2, one with Klebsiella pneumoniae, and two with P. aeruginosa). Notably, one patient with acute diffuse peritonitis, one patient who had co-infection with B. cepacian, and three patients with pulmonary co-infections died (Supplementary Table S1). Even while this study was unable to provide direct evidence for the higher death rate among individuals with co-infections or co-morbidities, we must focus on the distinct variations among pathogens, co-infection complexity, surveillance data, and several factors linked to unfavorable consequences (such as disability and death) and therapeutic challenges. Quick and precise molecular testing will contribute to rapid precision therapy. This necessitates a methodological understanding of the entire pathogen genome sequence.
Antibiotic susceptibility of Pandoraea isolates
Most of the patients survived with antibiotic treatments, but there were still four deaths who were treated with antibiotics. Therefore, we then tested for the antibiotic susceptibility profiles of these Pandoraea isolates. The MIC50, MIC90, MIC range and percent susceptibility for tested antibiotics are listed in Table 2. Referenced to the criteria of Burkholderia cepacian and the quality control organisms (Clinical and Laboratory Standards Institute (CLSI), 2025), the tested Pandoraea isolates showed high degree of MIC values to MEM (MIC50 and MIC90: ≧16 μg/mL), AMC (MIC50 and MIC90: ≧128/64 μg/mL), GEN (MIC50 and MIC90: ≧16 μg/mL), and CAZ (MIC50 and MIC90: ≧128 μg/mL). It was also reported that high frequency (76.7%, 23/30) of Pandoraea isolates exhibited resistance to MEM (Lin et al., 2019). In contrast, MIC50 and MIC90 values of DOX and TGC were at least 8-fold lower than MEM, AMC, GEN and CAZ. Collectively, these findings suggested that the Pandoraea isolates had potential resistance to various antibiotics. However, there is currently no criteria for the evaluation of MIC values in Pandoraea, we were unable to directly determine the resistance level of the isolates in this study. Further investigations are needed to understand the susceptibility patterns of Pandoraea, which would help clinicians choose effective antibiotics and inform public health strategies to combat resistance.
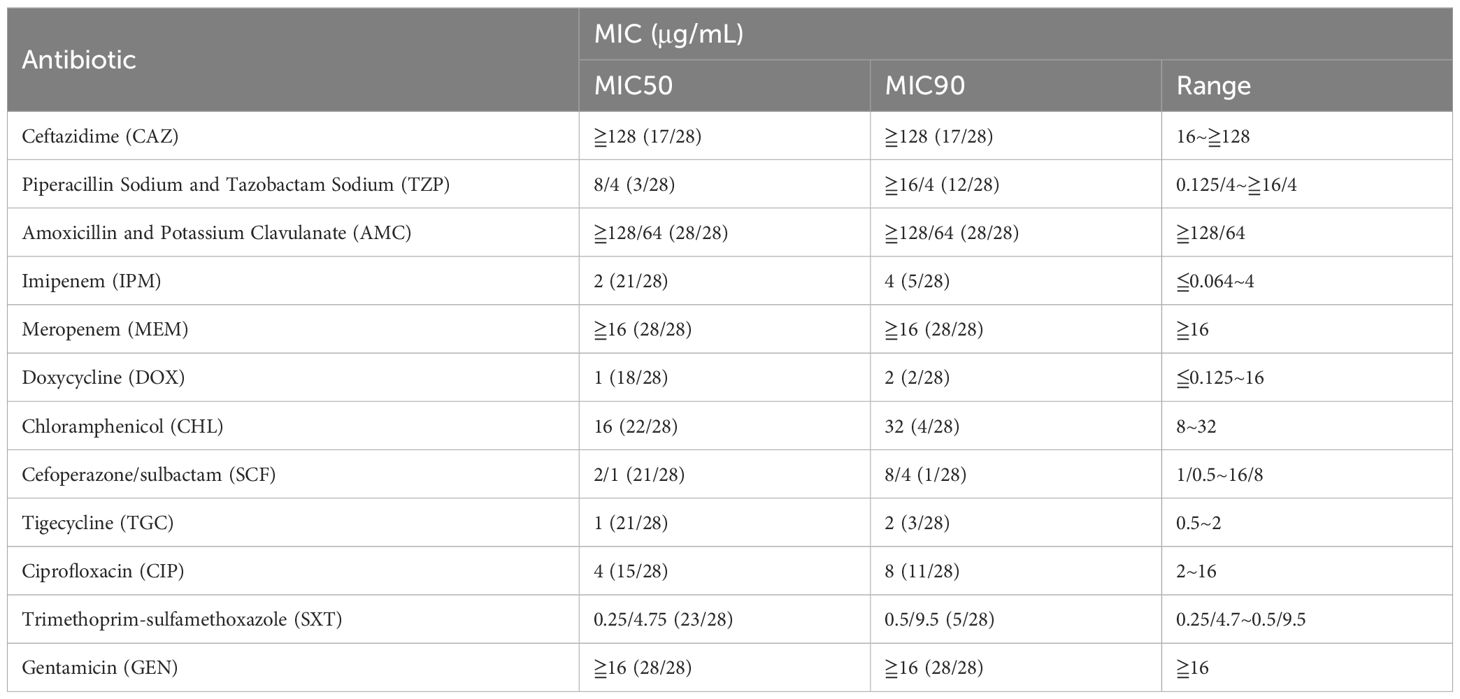
Table 2. MIC value and activity spectrum of 12 antibiotics against P. pneumonica clinical isolates.
Determination of the novel Pandoraea pneumonica group based on genome sequencing
Whole blood samples were collected for the isolation of Pandoraea spp. and 30 isolates were used for further analysis. A phylogenetic tree based on 16S rDNA gene sequences was constructed using representative sequences of the genus Pandoraea and our isolates. All 28 isolates and Pandoraea sp. LMG 31114 were divided into the same large clusters (Figure 1). Thus, it has been suggested that they originated from a common, distant ancestor. Pandoraea sp. LMG 31114 was firstly isolated from CF patient in the United States in 2009 (Peeters et al., 2019).
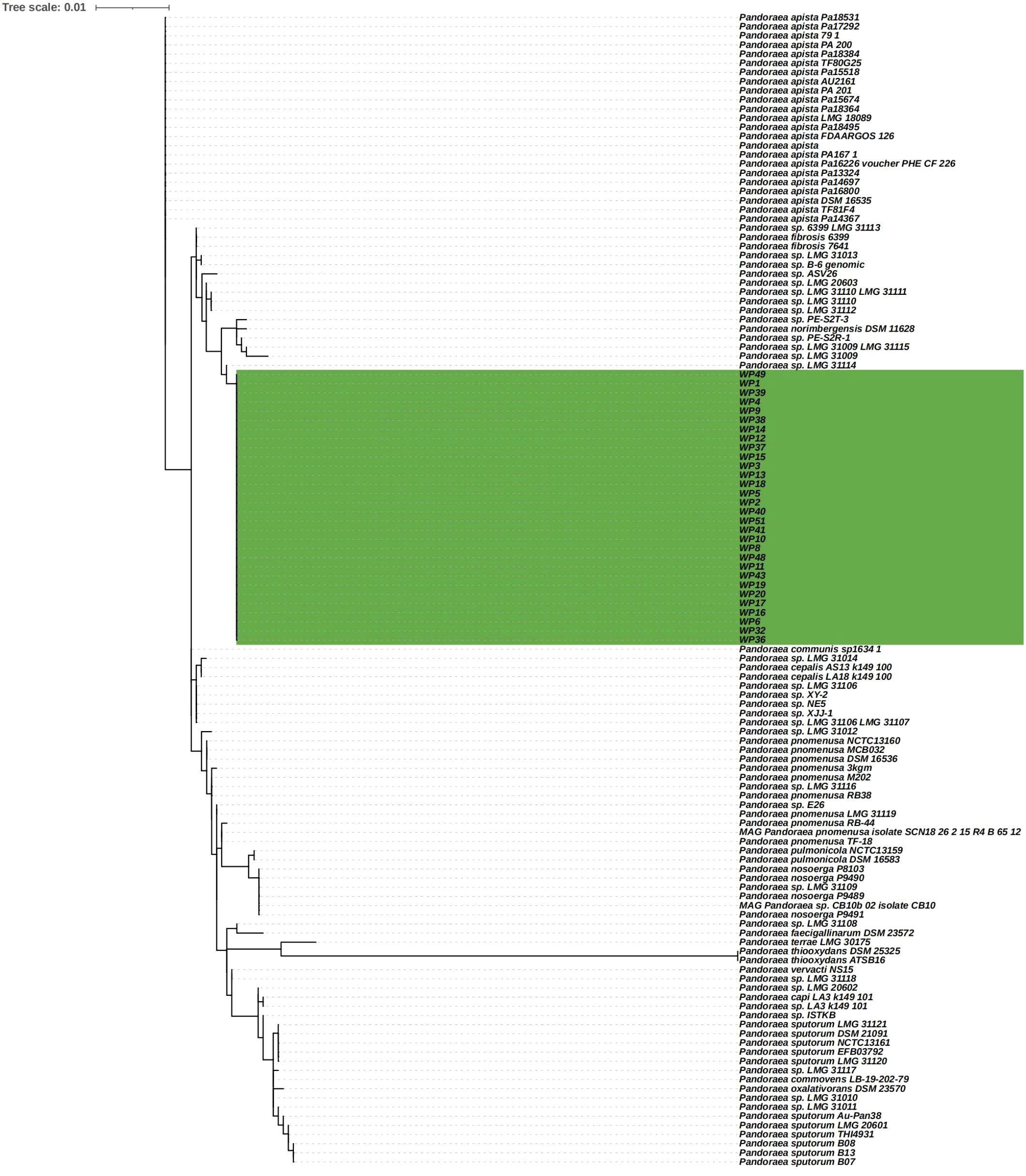
Figure 1. Phylogenetic tree based on 16S rDNA gene sequencing showing P. pneumonica isolates and other related reference strain from the NCBI database.
The complete genome of pathogenic microorganisms are plastic on an evolutionary time scale. But high-throughput sequencing can be applied for research on overall structure, organization and function of them. The complete genome sequence of WP1 was obtained by Nanopore sequencing. The WP1 genome has one chromosome of 5,773,419 bp, containing 5,100 coding sequences, 12 rRNA genes, 68 tRNA genes, and one tmRNA. Subsequently, draft genomes of the remaining 27 isolates were sequenced (Supplementary Table S2). Among the genomes of the 28 Pandoraea isolates, the minimum, maximum, and average genome size was 4,442,224 bp, 5,826,184 bp, and 5,397,568 bp, respectively. The minimum, maximum, and, average GC contents were 62.11%, 62.51%, and 62.43%, respectively. To the best of our knowledge, only a few whole genome sequences of P. pneumonica have been reported. Peeters et al. demonstrated that P. pneumonica sp. nov. (type strain LMG 31114T = CCUG 73388T) from a patient with CF had a 5,845,078 bp nucleotide sequence with 62.5% GC content (Peeters et al., 2019). Their genome size and GC content were consistent with those of the 28 isolates in this study.
Furthermore, the homology of the WP1 whole genome was analyzed via ANI analysis. As shown in Figure 2, WP1 displayed high similarity (90.6%) to Pandoraea sp. LMG 31114. The genus is a cohesive group that shares a high degree of nucleotide sequence similarity (> 85.0%), with two species belonging to the same species differing by < 5% from each other at the nucleotide level, according to ANI analysis (Goris et al., 2007; Richter and Rossello-Mora, 2009). Thus, we hypothesized that WP1 is a novel subspecies or even a novel member of a species similar to P. pneumonica.
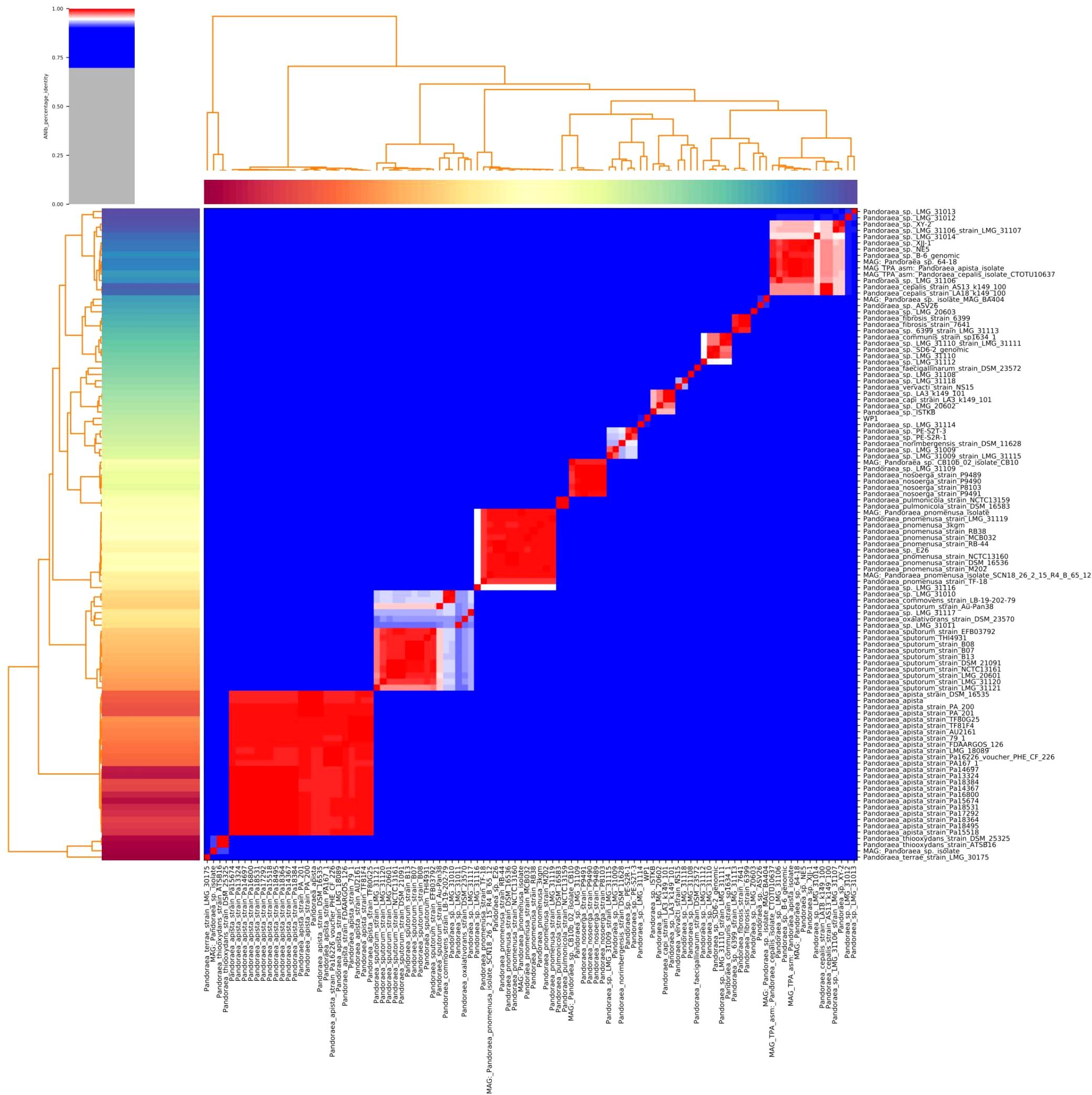
Figure 2. Heatmap of the average nucleotide identity (ANI) values for whole genomes, sequenced from related Pandoraea spp. in the NCBI database, including WP1 from this study. Values range from 0 (0% ANI) to 1 (100% ANI): gray represents 0% ANI.
The SNP classification strategy on the core genome (defined as orthologous sequences conserved in all aligned genomes) allows phylogenetic reconstruction, clade discrimination, and genetic distance calculation by SNP alignment, base-by-base comparison, and consideration of evolutionary models with masked recombination locations as well as mobile elements (Abdel-Glil et al., 2022). As shown in Supplementary Figure S1, 28 isolates clustered together, but diverged in LMG 31114. The inner genetic distance within the 28 tested isolates was < 100 bases, whereas the genetic distance within LMG 31114 was > 100,000 bases. This result further suggests that these isolates can be defined as a novel cluster of P. pneumonica.
Potential virulence factors and antibiotic resistance genes
The total number of pan genes in this new P. pneumonica cluster was 5425, of which the core and soft core genes were 1919 and 1095, respectively, accounting for 55% of the total number of genes (Figures 3A, B). In total, 46 potential virulence factor genes were predicted, of which 38 were core genes, including cheY with 99.24% coverage, tufA 2 with 98.65% coverage, tufA 1 with 98.65% coverage, adeG with 97.61% coverage, and flgG with 97.59% coverage (Supplementary Table S3). However, the similarity between these genes was < 85% compared to those reported in the Virulence Factor Database. adeG, which encodes the cation/multidrug efflux pump AdeG, serves as a biofilm-controlling response regulator related to biofilm formation (the major virulence factor of Acinetobacter baumannii) (Mea et al., 2021; Shan et al., 2022).
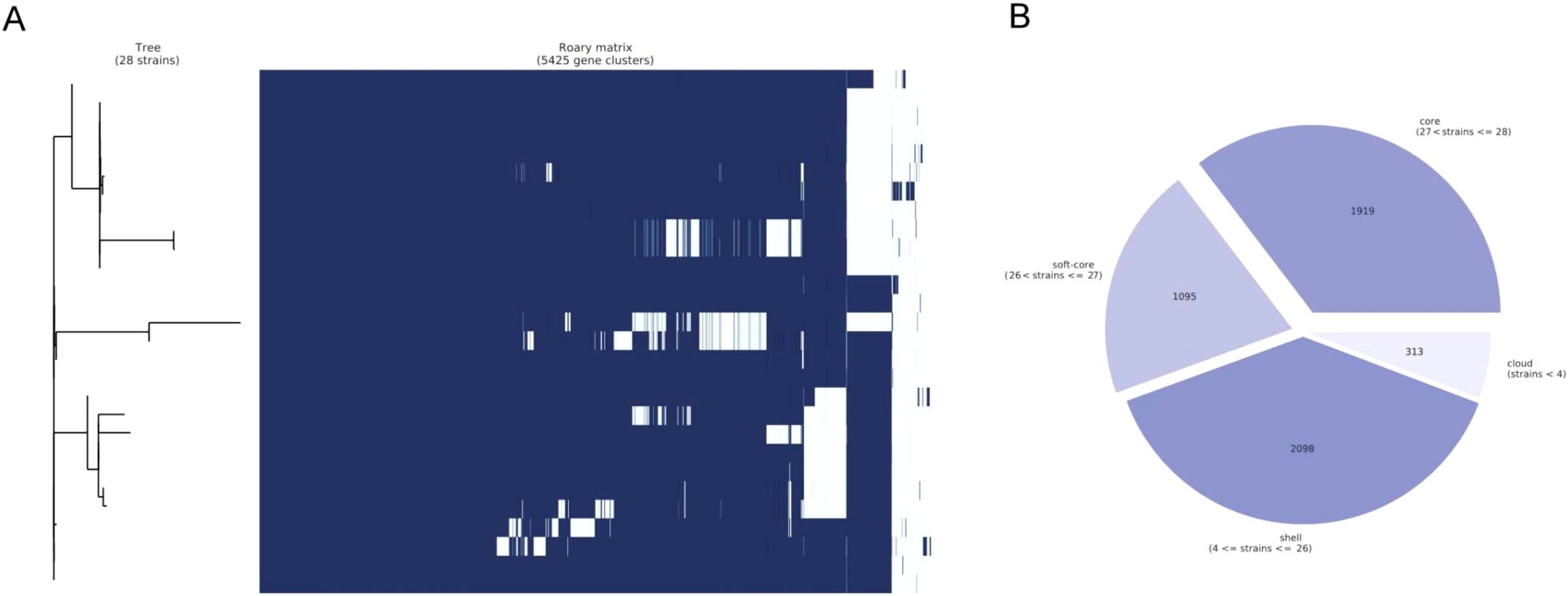
Figure 3. Pan-genome analysis. (A) Clustered heatmap based on the shared genes. Multiple sequence alignments were performed for each core gene in each isolate, and combined together to build the phylogenetic tree. (B) Statistical information on core and soft-core genes. During the statistical analysis, core genes were defined as genes present in 28 isolates, soft genes were defined as genes present in 27 isolates, shell genes were present in at least 4 isolates and no greater than 26 isolates, and cloud genes were present in less than 4 isolates.
Seventeen potential resistance genes were identified, of which 10 were core genes, including ceoB with 99.42% coverage, MexB with 97.33% coverage, MuxB with 96.04% coverage, MexA with 84.72% coverage, and OrpM with 85.6% coverage (Supplementary Table S4). Except for OXA-157 (85.31% coverage and 86.61% identity), the similarity between the remaining genes and drug resistance genes in the Comprehensive Antibiotic Resistance Database was < 85%. Combined with phenotype results of this study, we highlighted the potential relation between MEM and OXA-157. Previously, the influence of OXA-151 to OXA-159 on the MEM-resistant phenotype of Pandoraea spp. was observed by Schneider et al (Schneider and Bauernfeind, 2015). Thus, OXA-157, coding oxacillinases (β-lactamases of the molecular class D), may serve as a critical factor on MEM resistance. MuxB contributes to muxABC-opmB synthesis, a putative resistance nodulation cell division (RND)-type multidrug efflux pump system, in Pseudomonas aeruginosa (Lorusso et al., 2022). Three membrane-bound subunits (MexA, MexB, and OprM) anchor the inner and outer membranes associated with another RND system MexAB-OprM (Paulsen et al., 1996; Tseng et al., 1999; Akama et al., 2004). Based on the outcomes of gene disruption, such a mechanism might be considered to be essential for the resistance to CAZ and CIP in P. pneumonica (Li and Plésiat, 2016). Further research is required on additional resistance phenotypes and pathogenic pathways.
Evolution of the novel Pandoraea pneumonica group
The ML phylogeny was inputted into TempEST to examine the temporal signal of the dataset. A linear association between evolutionary distance and divergence time was observed. The genomes accumulated mutations at a rate of 1.3 × 10-7 substitutions per site annually, or roughly 0.27 SNPs (Figure 4). Based on root-to-tip divergence analysis, the most recent common ancestor of the P. pneumonica lineage was estimated to have emerged in 1993. The WP9 and WP2 clusters were formed between December 2021 and January 2022, whereas other distinct clinical isolates within the P. pneumonica cluster were formed at the earliest in July 2014. We also hypothesized that isolates with comparable geographic origins might carry a clonal link and develop regional endemic patterns. Overall, this P. pneumonica cluster contained few SNPs and was genetically homogeneous. These results indicate that all strains exhibited a high degree of relatedness and a close phylogenetic link. To the best of our knowledge, this is the first study to demonstrate the rate of mutations in the core genome of P. pneumonica based on the whole genome sequences acquired through high-throughput sequencing. Usually, a genome-wide mutation rate of approximately 10-7 mutations/year/site occur in E. coli (White et al., 2024), Shigella (Holt et al., 2013), and Chlamydia psittaci (White et al., 2022; Kasimov et al., 2023). Genetic epidemiology in this study revealed clusters of isolates, which may indicate that patients contracted the infection from a common source. This finding highlights the advantages of integrating epidemiological data from WGS into public health surveillance and research.
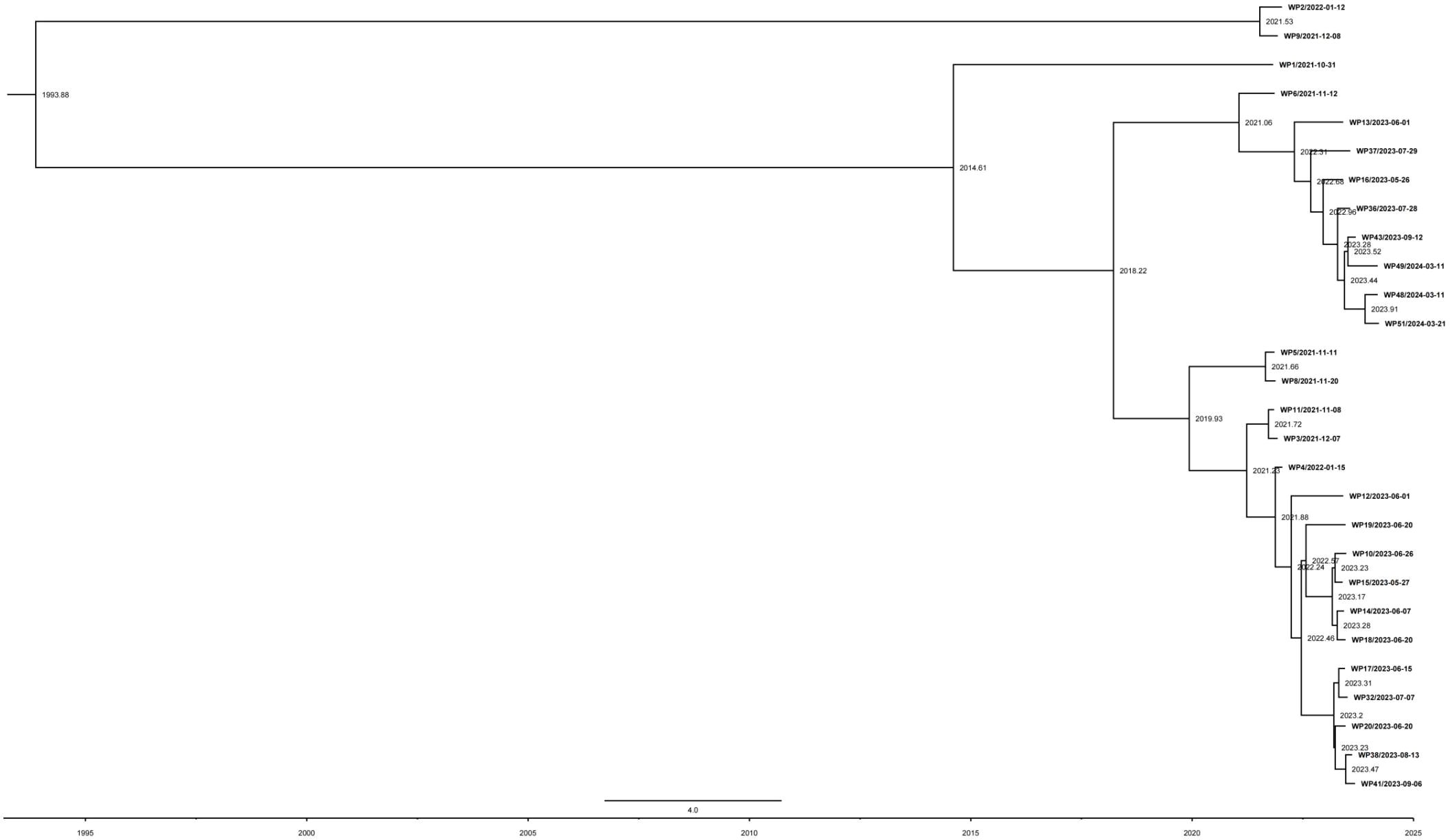
Figure 4. Evolutionary reconstruction of P. pneumonica isolates. A time-calibrated maximum clade credibility tree was inferred from core-genome single-nucleotide polymorphisms (SNPs) from P. pneumonica genomes. The X-axis represents the emergence time estimates.
Conclusion
Our study highlighted the benefits of bacterial WGS for precise isolate identification and epidemic classification. Based on these findings, including clinical symptoms and whole-genome bioinformatics analysis, we concluded that a novel cluster of P. pneumonica serves as a critical emerging pathogen in people with BSI. Comorbidities or complications make the patient’s condition more difficult. This outbreak report demonstrates the importance of the vigilance of microbiologists and doctors when novel members of pathogenic microorganisms are detected. Medical testing laboratories must develop and update cutting-edge molecular sequencing technologies. Additionally, a more comprehensive genomic epidemiology study is necessary to promote future diagnosis and therapy of this pathogen.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found here: [https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1250782, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1250798, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1250801, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1250804, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1250810, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251137, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251149, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251200, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251207, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251212, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251214, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251218, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251221 https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251234, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251238 https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251241, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251244 https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251246, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251249, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251253, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251255, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251270, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251274, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251280 https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251282, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251283, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251288, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1251289].
Ethics statement
The studies involving humans were approved by Medical Ethics Committee of Hainan General Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from gifted from another research group. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements.
Author contributions
CC: Data curation, Writing – original draft, Formal Analysis, Project administration, Writing – review & editing. MW: Formal Analysis, Methodology, Writing – original draft. TH: Supervision, Writing – review & editing. D-LH: Formal Analysis, Writing – review & editing. SY: Methodology, Writing – original draft. H-MZ: Writing – original draft. XF: Formal Analysis, Methodology, Writing – original draft. X-XL: Methodology, Software, Writing – original draft. HW: Conceptualization, Funding acquisition, Supervision, Writing – review & editing, Project administration, Resources, Validation.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the Key Research and Development Program of Hainan Province (ZDYF2022SHFZ050) and National Natural Science Fund Cultivating 530 Project of Hainan General Hospital and Hainan Clinical Research Center.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2025.1560634/full#supplementary-material
Supplementary Figure 1 | Maximum likelihood phylogeny inferred from the core-genome single-nucleotide polymorphisms (SNPs) in 28 genomes identified by Parsnp among the isolates. Branch lengths represent the nucleotide substitutions per site, as indicated by the scale bar.
Supplementary Table 1 | Summary of patient conditions.
Supplementary Table 2 | Basic genome information on P. pneumonica isolates, including genome length and GC content.
Supplementary Table 3 | Basic information on potential virulence factor genes, including gene name, genomic location, coverage, similarity, and expression product.
Supplementary Table 4 | Basic information on potential resistance genes, including gene name, genomic location, coverage, similarity, and expression product.
References
Abdel-Glil, M. Y., Thomas, P., Brandt, C., Melzer, F., Subbaiyan, A., Chaudhuri, P., et al. (2022). Core genome multilocus sequence typing scheme for improved characterization and epidemiological surveillance of pathogenic brucella. J. Clin. Microbiol. 60, e0031122. doi: 10.1128/jcm.00311-22
Akama, H., Matsuura, T., Kashiwagi, S., Yoneyama, H., Narita, S., Tsukihara, T., et al. (2004). Crystal structure of the membrane fusion protein, MexA, of the multidrug transporter in Pseudomonas aeruginosa. J. Biol. Chem. 279, 25939–25942. doi: 10.1074/jbc.C400164200
Bodendoerfer, E., Personnic, N., Mestres, C. A., Wilhelm, M. J., Meyer, L., Hasse, B. (2021). Possible prosthetic valve endocarditis by pandoraea pnomenusa and specific virulence mechanisms. Infection Drug resistance. 14, 1319–1324. doi: 10.2147/IDR.S301138
Bosshard, P. P., Zbinden, R., Abels, S., Boddinghaus, B., Altwegg, M., Bottger, E. C. (2006). 16S rRNA gene sequencing versus the API 20 NE system and the VITEK 2 ID-GNB card for identification of nonfermenting Gram-negative bacteria in the clinical laboratory. J. Clin. Microbiol. 44, 1359–1366. doi: 10.1128/JCM.44.4.1359-1366.2006
Brumfield, K. D., Usmani, M., Chen, K. M., Gangwar, M., Jutla, A. S., Huq, A., et al. (2021). Environmental parameters associated with incidence and transmission of pathogenic Vibrio spp. Environ. Microbiol. 23, 7314–7340. doi: 10.1111/1462-2920.15716
Caraher, E., Collins, J., Herbert, G., Murphy, P. G., Gallagher, C. G., Crowe, M. J., et al. (2008). Evaluation of in vitro virulence characteristics of the genus Pandoraea in lung epithelial cells. J. Med. Microbiol. 57, 15–20. doi: 10.1099/jmm.0.47544-0
Chen, S., Zhou, Y., Chen, Y., Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–ii90. doi: 10.1093/bioinformatics/bty560
Clinical and Laboratory Standards Institute (CLSI) (2025). Performance Standards for Antimicrobial Susceptibility Testing. 35th edition (USA: Clinical and Laboratory Standards Institute).
Coenye, T., Falsen, E., Hoste, B., Ohlen, M., Goris, J., Govan, J. R., et al. (2000). Description of Pandoraea gen. nov. with Pandoraea apista sp. nov., Pandoraea pulmonicola sp. nov., Pandoraea pnomenusa sp. nov., Pandoraea sputorum sp. nov. and Pandoraea norimbergensis comb. nov. Int. J. Syst. Evol. Microbiol. 50 Pt 2, 887–899. doi: 10.1099/00207713-50-2-887
Coenye, T., LiPuma, J. J. (2002). Use of the gyrB gene for the identification of Pandoraea species. FEMS Microbiol. letters. 208, 15–19. doi: 10.1111/j.1574-6968.2002.tb11053.x
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43, e15. doi: 10.1093/nar/gku1196
Darriba, D., Taboada, G. L., Doallo, R., Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772. doi: 10.1038/nmeth.2109
Day, M. J., Hopkins, K. L., Wareham, D. W., Toleman, M. A., Elviss, N., Randall, L., et al. (2019). Extended-spectrum beta-lactamase-producing Escherichia coli in human-derived and foodchain-derived samples from England, Wales, and Scotland: an epidemiological surveillance and typing study. Lancet Infect. diseases. 19, 1325–1335. doi: 10.1016/S1473-3099(19)30273-7
Esteves, K., Hervio-Heath, D., Mosser, T., Rodier, C., Tournoud, M. G., Jumas-Bilak, E., et al. (2015). Rapid proliferation of Vibrio parahaemolyticus, Vibrio vulnificus, and Vibrio cholerae during freshwater flash floods in French Mediterranean coastal lagoons. Appl. Environ. Microbiol. 81, 7600–7609. doi: 10.1128/AEM.01848-15
Falces-Romero, I., Gutierrez-Arroyo, A., Romero-Gomez, M. P. (2016). Catheter-associated bacteremia by Pandoraea pnomenusa in an infant with acute lymphoblastic leukemia. Medicina clinica. 147, 132. doi: 10.1016/j.medcli.2016.04.009
Gawalkar, A. A., Kasinadhuni, G., Kanaujia, R., Rajan, P., Vijay, J., Revaiah, P. C., et al. (2021). Prosthetic aortic valve dehiscence following infective endocarditis by a rare bacterium - Pandoraea pnomenusa. J. Cardiol. cases. 24, 27–29. doi: 10.1016/j.jccase.2020.12.003
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Goto, M., Al-Hasan, M. N. (2013). Overall burden of bloodstream infection and nosocomial bloodstream infection in North America and Europe. Clin. Microbiol. Infect. 19, 501–509. doi: 10.1111/1469-0691.12195
Green, H., Jones, A. M. (2018). Emerging Gram-negative bacteria: pathogenic or innocent bystanders. Curr. Opin. pulmonary Med. 24, 592–598. doi: 10.1097/MCP.0000000000000517
Holt, K. E., Thieu Nga, T. V., Thanh, D. P., Vinh, H., Kim, D. W., Vu Tra, M. P., et al. (2013). Tracking the establishment of local endemic populations of an emergent enteric pathogen. Proc. Natl. Acad. Sci. United States America. 110, 17522–17527. doi: 10.1073/pnas.1308632110
Itoh, N., Akazawa, N., Ishibana, Y., Masuishi, T., Nakata, A., Murakami, H. (2022). Clinical and microbiological features of obstructive cholangitis with bloodstream infection caused by Pandoraea apista identified by MALDI-TOF mass spectrometry and ribosomal RNA sequencing in a cancer patient. BMC Infect. diseases. 22, 529. doi: 10.1186/s12879-022-07514-z
Johnson, L. N., Han, J. Y., Moskowitz, S. M., Burns, J. L., Qin, X., Englund, J. A. (2004). Pandoraea bacteremia in a cystic fibrosis patient with associated systemic illness. Pediatr. Infect. Dis. J. 23, 881–882. doi: 10.1097/01.inf.0000136857.74561.3c
Kasimov, V., White, R. T., Foxwell, J., Jenkins, C., Gedye, K., Pannekoek, Y., et al. (2023). Whole-genome sequencing of Chlamydia psittaci from Australasian avian hosts: A genomics approach to a pathogen that still ruffles feathers. Microbial Genomics 9 (7), 1–13. doi: 10.1099/mgen.0.001072
Kille, B., Nute, M. G., Huang, V., Kim, E., Phillippy, A. M., Treangen, T. J. (2024). Parsnp 2.0: scalable core-genome alignment for massive microbial datasets. Bioinformatics 40 (5), 1–5. doi: 10.1093/bioinformatics/btae311
Kumar, S., Stecher, G., Li, M., Knyaz, C., Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. evolution. 35, 1547–1549. doi: 10.1093/molbev/msy096
Li, X.-Z., Plésiat, P. (2016). Antimicrobial drug efflux pumps in pseudomonas aeruginosa. In: Efflux-Mediated Antimicrobial Resistance in Bacteria. 14, 359–400. doi: 10.1007/978-3-319-39658-3
Lin, C., Luo, N., Xu, Q., Zhang, J., Cai, M., Zheng, G., et al. (2019). Pneumonia due to Pandoraea Apista after evacuation of traumatic intracranial hematomas:a case report and literature review. BMC Infect. diseases. 19, 869. doi: 10.1186/s12879-019-4420-6
Lorusso, A. B., Carrara, J. A., Barroso, C. D. N., Tuon, F. F., Faoro, H. (2022). Role of efflux pumps on antimicrobial resistance in pseudomonas aeruginosa. Int. J. Mol. Sci. 23 (24), 1–21. doi: 10.3390/ijms232415779
Martinez, R. M., Wolk, D. M. (2016). Bloodstream infections. Microbiol. Spectr. 4 (4), 653–89. doi: 10.1128/microbiolspec.DMIH2-0031-2016
McMenamin, J. D., Zaccone, T. M., Coenye, T., Vandamme, P., LiPuma, J. J. (2000). Misidentification of Burkholderia cepacia in US cystic fibrosis treatment centers: an analysis of 1,051 recent sputum isolates. Chest. 117, 1661–1665. doi: 10.1378/chest.117.6.1661
Mea, H. J., Yong, P. V. C., Wong, E. H. (2021). An overview of Acinetobacter baumannii pathogenesis: Motility, adherence and biofilm formation. Microbiological Res. 247, 126722. doi: 10.1016/j.micres.2021.126722
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Paulsen, I. T., Brown, M. H., Skurray, R. A. (1996). Proton-dependent multidrug efflux systems. Microbiological Rev. 60, 575–608. doi: 10.1128/mr.60.4.575-608.1996
Peeters, C., Canck, E. D., Cnockaert, M., De Brandt, E., Snauwaert, C., Verheyde, B., et al. (2019). Comparative genomics of pandoraea, a genus enriched in xenobiotic biodegradation and metabolism. Front. Microbiol. 10, 2556. doi: 10.3389/fmicb.2019.02556
Pimentel, J. D., MacLeod, C. (2008). Misidentification of Pandoraea sputorum isolated from sputum of a patient with cystic fibrosis and review of Pandoraea species infections in transplant patients. J. Clin. Microbiol. 46, 3165–3168. doi: 10.1128/JCM.00855-08
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A., Korobeynikov, A. (2020). Using SPAdes de novo assembler. Curr. Protoc. Bioinf. 70, e102. doi: 10.1002/cpbi.v70.1
Rajkumari, N., Mathur, P., Sharma, S., Gupta, B., Bhoi, S., Misra, M. C. (2013). Procalcitonin as a predictor of sepsis and outcome in severe trauma patients: a prospective study. J. Lab. physicians. 5, 100–108. doi: 10.4103/0974-2727.119852
Ramos Oliveira, S., Costa Monteiro, I., Rodrigues, C., Soares, M. J. (2023). Fever in a patient with a central venous catheter colonized by pandoraea pnomenusa. Acta Med. portuguesa. 36, 127–130. doi: 10.20344/amp.16176
Richter, M., Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. United States America. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Schneider, I., Bauernfeind, A. (2015). Intrinsic carbapenem-hydrolyzing oxacillinases from members of the genus Pandoraea. Antimicrobial Agents chemotherapy. 59, 7136–7141. doi: 10.1128/AAC.01112-15
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics. 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shah, M. S., Eppinger, M., Ahmed, S., Shah, A. A., Hameed, A., Hasan, F. (2016). Flooding adds pathogenic Escherichia coli strains to the water sources in southern Khyber Pakhtunkhwa, Pakistan. Indian J. Med. Microbiol. 34, 483–488. doi: 10.4103/0255-0857.195350
Shan, W., Kan, J., Cai, X., Yin, M. (2022). Insights into mucoid Acinetobacter baumannii: A review of microbiological characteristics, virulence, and pathogenic mechanisms in a threatening nosocomial pathogen. Microbiological Res. 261, 127057. doi: 10.1016/j.micres.2022.127057
Shelenkov, A., Mikhaylova, Y., Yanushevich, Y., Samoilov, A., Petrova, L., Fomina, V., et al. (2020). Molecular typing, characterization of antimicrobial resistance, virulence profiling and analysis of whole-genome sequence of clinical klebsiella pneumoniae isolates. Antibiotics 9 (5), 1–15. doi: 10.3390/antibiotics9050261
Singh, S., Sahu, C., Patel, S. S., Garg, A., Ghoshal, U. (2021). Pandoraea apista bacteremia in a COVID-positive man: A rare coinfection case report from north India. J. Lab. physicians. 13, 192–194. doi: 10.1055/s-0041-1730847
Stamatakis, A., Ludwig, T., Meier, H. (2005). RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics. 21, 456–463. doi: 10.1093/bioinformatics/bti191
Stryjewski, M. E., LiPuma, J. J., Messier, R. H., Jr., Reller, L. B., Alexander, B. D. (2003). Sepsis, multiple organ failure, and death due to Pandoraea pnomenusa infection after lung transplantation. J. Clin. Microbiol. 41, 2255–2257. doi: 10.1128/JCM.41.5.2255-2257.2003
Treangen, T. J., Ondov, B. D., Koren, S., Phillippy, A. M. (2014). The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15, 524. doi: 10.1186/s13059-014-0524-x
Tseng, T. T., Gratwick, K. S., Kollman, J., Park, D., Nies, D. H., Goffeau, A., et al. (1999). The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development proteins. J. Mol. Microbiol. Biotechnol. 1, 107–125.
White, R. T., Anstey, S. I., Kasimov, V., Jenkins, C., Devlin, J., El-Hage, C., et al. (2022). One clone to rule them all: Culture-independent genomics of Chlamydia psittaci from equine and avian hosts in Australia. Microbial Genomics 8 (10), 1–12. doi: 10.1099/mgen.0.000888
White, R. T., Bull, M. J., Barker, C. R., Arnott, J. M., Wootton, M., Jones, L. S., et al. (2024). Genomic epidemiology reveals geographical clustering of multidrug-resistant Escherichia coli ST131 associated with bacteraemia in Wales. Nat. Commun. 15, 1371. doi: 10.1038/s41467-024-45608-1
Wick, R. R., Judd, L. M., Gorrie, C. L., Holt, K. E. (2017). Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PloS Comput. Biol. 13, e1005595. doi: 10.1371/journal.pcbi.1005595
Keywords: Pandoraea pneumonica, bloodstream infection, genomic epidemiology, whole-genome sequencing, pan-genome analysis
Citation: Chen C, Wang M, Huang T, Huang D-l, Yu S, Zhao H-m, Fu X-x, Li X-x and Wu H (2025) Genomic epidemiology of a novel Pandoraea pneumonica group caused severe bloodstream infection in Hainan, China, 2021-2024. Front. Cell. Infect. Microbiol. 15:1560634. doi: 10.3389/fcimb.2025.1560634
Received: 14 January 2025; Accepted: 24 March 2025;
Published: 28 April 2025.
Edited by:
Costas C. Papagiannitsis, University of Thessaly, GreeceReviewed by:
Rui Pang, South China Agricultural University, ChinaPandora Jim Tsolakidou, General Hospital of Volos, Greece
Copyright © 2025 Chen, Wang, Huang, Huang, Yu, Zhao, Fu, Li and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hua Wu, c3lqeWt3dWh1YUAxNjMuY29t
†Present address: Chong Chen, Department of Clinical Laboratory, Hainan Second People's Hospital, Wuzhishan, Hainan, China
‡These authors have contributed equally to this work