 Daniel Turner
Daniel Turner Chen Kang
Chen Kang Pietro Mesirca3
Pietro Mesirca3 Juan Hong
Juan Hong Alexey V. Glukhov
Alexey V. Glukhov Rajan Sah
Rajan Sah- 1Department of Medicine, University of Wisconsin-Madison School of Medicine and Public Health, Madison, WI, United States
- 2Cardiovascular Division, Department of Internal Medicine, Washington University School of Medicine, St. Louis, MO, United States
- 3Institut de Génomique Fonctionnelle, Université de Montpellier, CNRS, INSERM, Montpellier, France
The understanding of the electrophysiological mechanisms that underlie mechanosensitivity of the sinoatrial node (SAN), the primary pacemaker of the heart, has been evolving over the past century. The heart is constantly exposed to a dynamic mechanical environment; as such, the SAN has numerous canonical and emerging mechanosensitive ion channels and signaling pathways that govern its ability to respond to both fast (within second or on beat-to-beat manner) and slow (minutes) timescales. This review summarizes the effects of mechanical loading on the SAN activity and reviews putative candidates, including fast mechanoactivated channels (Piezo, TREK, and BK) and slow mechanoresponsive ion channels [including volume-regulated chloride channels and transient receptor potential (TRP)], as well as the components of mechanochemical signal transduction, which may contribute to SAN mechanosensitivity. Furthermore, we examine the structural foundation for both mechano-electrical and mechanochemical signal transduction and discuss the role of specialized membrane nanodomains, namely, caveolae, in mechanical regulation of both membrane and calcium clock components of the so-called coupled-clock pacemaker system responsible for SAN automaticity. Finally, we emphasize how these mechanically activated changes contribute to the pathophysiology of SAN dysfunction and discuss controversial areas necessitating future investigations. Though the exact mechanisms of SAN mechanosensitivity are currently unknown, identification of such components, their impact into SAN pacemaking, and pathological remodeling may provide new therapeutic targets for the treatment of SAN dysfunction and associated rhythm abnormalities.
Introduction
The heart is continuously experiencing a dynamic mechanical environment, both on a beat-to-beat basis (e.g., fluctuating blood pressure and exercise) and chronically (e.g., elevated venous return and high blood pressure). Alterations in intra-cardiac pressure and/or volume preload/afterload may influence cardiac performance to coordinate cardiac output with venous return and arterial blood supply, in a cardiac autonomous fashion. This process involves activation of complex mechano-electrical [i.e., mechanically induced changes in cardiac action potential (AP) morphology, frequency, and propagation] and mechanochemical (i.e., changes in various second messenger signaling that are ultimately translated into regulation of calcium handling) signal transduction feedback mechanisms that autoregulate the frequency and the force of cardiac muscle contraction (Figure 1). An important component of such autoregulation includes changes in heart rate controlled by the heart's primary pacemaker, the sinoatrial node (SAN). SAN response to altered hemodynamic load is described via the Bainbridge response: an increase in heart rate upon right atrial pressure/volume increase, which may help in matching cardiac output to venous return (1). SAN mechanosensitivity and associated changes in pacemaker activity have been demonstrated at multiple levels, including isolated heart (2, 3) as well as in isolated SAN cells (4), and have been linked to mechanosensitive mechanisms (5, 6) intrinsic to pacemaker cells (7). In this review, we summarize the emerging understanding of cellular and molecular mechanisms that could be involved in SAN mechanosensing and pacemaker rate attenuation. Though the exact components of mechano-electro-chemical signal transduction, specifically involved in SAN mechanosensitivity, are not currently identified, here, we overview possible candidates that might be responsible for both fast (i.e., within seconds or on beat-to-beat manner) and slow (minutes) changes in SAN automaticity in response to mechanical stress. Specifically, we focus on canonical mechanoactivated channels (Piezo, TREK, and BK), slow mechanoresponsive ion channels (including volume-regulated chloride channels (ClC), and transient receptor potential (TRP) channels), and the components of mechanochemical signal transduction, including reactive oxygen species (ROS), cyclic adenosine monophosphate (cAMP), and inositol trisphosphate (IP3). Mechanosensitivity of these pathways has been described in either non-pacemaker cardiomyocytes (atrial or ventricular) or non-cardiac cells. Here, we show the expression profile of mechanosensitive ion channels in murine SAN (Figure 2) and discuss how these ion channels, as well as various mechano-chemical signaling pathways, could potentially modulate membrane and calcium clock components of the so-called coupled-clock pacemaker system (8), contributing to SAN mechanosensitivity and changing in heart rate upon alterations in intra-cardiac mechanics.
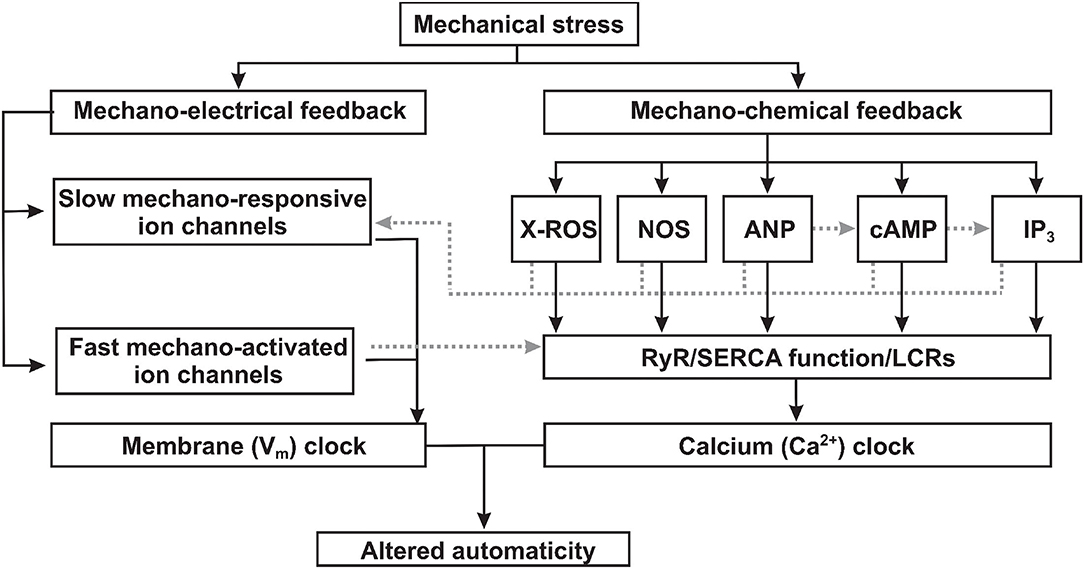
Figure 1. Proposed mechanisms of mechano-electrical and mechano-chemical feedback contributions to sinoatrial node mechanosensitivity. Mechanical stress (1) triggers mechano-electrical signal transduction pathways via both slow mechano-responsive and fast mechano-activated ion channels directly changing the membrane (Vm) clock component of the coupled-clock pacemaker system; and (2) activates mechano-chemical feedback via various signaling factors which alters the function of the calcium (Ca2+) clock component of the coupled-clock pacemaker system. ROS, reactive oxygen species; NOS, nitric oxide synthase; ANP, atrial natriuretic peptide; cAMP, cyclic adenoside monophosphate; IP3, inositol triphosphate; RyR, ryanodine receptor; SERCA, sarcoplasmic reticulum Ca2+-ATPase; LCRs, local calcium releases.
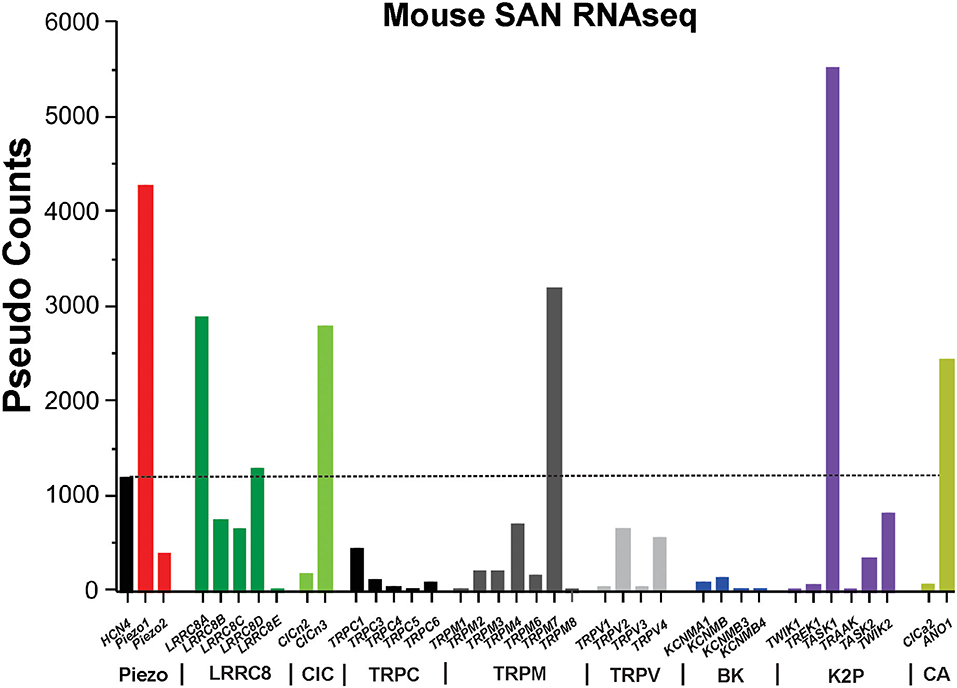
Figure 2. RNAseq of mouse sinoatrial node. The graph shows the absolute values (in pseudo-counts) for mRNA expression level. Horizontal dotted line indicates HCN4 level. LRRC8, leucine-rich repeat containing 8 family chloride channels; ClC, chloride channel; TRPC/M/V, transient receptor potential cation/melastatin/vanilloid subtype ion channels; BK, Ca2+-activated “big” potassium ion channels; K2P, two-pore domain potassium ion channels; CA, calcium-activated chloride ion channels; HCN4, hyperpolarization activated cyclic nucleotide gated cation channel 4.
Sinoatrial Node Anatomy
The SAN is a small body of specialized cardiac tissues located within the wall of the right atrium of the heart, laterally to the entrance of the superior vena cava, anatomically described by Silverman and Hollman (9). The SAN has a crescent-shaped structure positioned along the crista terminalis and running between the superior and inferior venae cavae, usually being arranged around a prominent nodal artery. The SAN is functionally insulated from the surrounding atrial myocardium, except for several critical conduction pathways (10–13). Indeed, the SAN requires anatomical (fibroblasts, adipose tissue, and blood vessels) and/or functional barriers (paucity of connexins) (13–16) to protect it from the hyperpolarizing influence of the surrounding atrium in order to function as a leading pacemaker. The presence of conduction barriers and pathways may explain how a small cluster of pacemaker cells in the SAN pacemaker complex manages to depolarize separate, widely distributed areas of the right atrium as evidenced functionally by exit points and breakthroughs (17–21). The autonomic nervous system and humoral factors can further regulate conduction through these pathways, contributing to pacemaker automaticity and ultimately determining heart rate (22–24).
Mechanosensitivity of the Sinoatrial Node
The SAN is well-positioned anatomically to sense both coronary and atrial blood pressure changes, providing a structural basis for hemodynamic regulation of heart rate via SAN mechanosensitivity. Changes in venous blood flow to the heart not only affect the volume available for atrial contraction and subsequent ventricular filling but also has an impact on the diastolic atrial dimension. Increased right atrial filling distends the atrial wall, including the SAN myocytes, which may consequently influence the pacemaker function and heart rate. This mechano-modulation of pacemaker activity was first described in 1915, when Bainbridge observed an increase in heart rate associated with right atrial distension from intravenous fluid injection in anesthetized dogs (1). While Bainbridge originally attributed this phenomenon to altering autonomic inputs, a study performed in dogs by Brooks et al. (25) determined that this positive chronotropic response was insensitive to adrenergic and cholinergic receptor blockade, and also to denervation, suggesting SAN intrinsic regulation of SAN automaticity. Conversely, in 1963, James and Nadeau demonstrated a bradycardic SAN response in dogs upon injection of fluid into the right atrium, while controlling for temperature, pH, osmolarity, oxygen, and ionic content (3). It was not until 1978, when Donald and Shepherd (2) performed controlled observations of human atrial and SAN mechanosensitivity by developing an experimental method that did not increase arterial blood pressure in humans (baroreceptor “depressor reflex”), that SAN mechanosensitivity was observed in humans. By placing subjects into a “supine” position, these researchers were able to observe an increase in heart rate concurrent with an increase in venous return to the heart. Lastly, Cooper et al. (4) determined that direct moderate stretch on isolated SAN cells, a possible consequence of increased venous pressure in vivo, induced elevated beating rate, confirming “for the first time, that the positive chronotropic response of the heart to stretch is, at least in part, encoded on the level of individual sinoatrial node pacemaker cells.” Please refer to the review by Quinn and Kohl for a deeper examination on the history of SAN mechanosensitivity and canonical mediators (6).
While these cornerstone studies demonstrate the immediate or “fast” response of stretch on SAN automaticity, there is also growing evidence of “slow” activating channels (>1 min), ClC-2, for example (26), which can be activated by long-term pressure increases normally associated with the slow force response in the working myocardium (27). Prior to its naming, the slow force response was observed in feline and canine models. Gertrude et al. observed in isolated cat nodal tissue that sustained stretch accelerated beating rate and even induced spontaneous beating from quiescent nodal cells (28). From a similar group of researchers, Brooks et al. observed a similar response in anesthetized dogs (25). Using in situ SAN stretch, they observed a biphasic response to SAN stretch with an immediate acceleration of beating rate followed by a decrease to a rate still above pre-stretch levels (25). These gradual (over the course of minutes) and reversible changes in beating rate and cardiac contractility inherent of the slow force response may play a role in more delayed changes in SAN automaticity via slowly activating mechanosensitive channels (26, 29, 30) and various mechano-chemical signaling pathways (31–35).
Electrophysiological Mechanisms of Sinoatrial Node Mechanosensitivity
Overview of Sinoatrial Node Pacemaker Activity
Spontaneous beating of SAN myocytes is initiated, sustained, and regulated by a complex coupled system of cellular “clocks” that integrates ion channels and transporters on the cell membrane surface or “voltage clock,” with subcellular Ca2+ handling machinery, also referred to as an intracellular “Ca2+ clock” (8, 36, 37) (Figure 3). The firing of SAN cells is due to diastolic depolarization, a slow depolarizing phase of the membrane potential (Vm), mediated by the concomitant action of both membrane and Ca2+ clocks. Since SAN cells lack IK1 current expression (41), following the minimum, or most hyperpolarized diastolic potential, potassium IK current (IKs, and IKr) conductance decreases, allowing the inward hyperpolarization-activated current (If) (42, 43) and a low-threshold, voltage-gated T-type Ca2+ current (ICa,T), which contribute to the early fraction of diastolic depolarization (Figure 3) (44). In addition, L-type Cav1.3 Ca2+ channels open during diastolic depolarization to generate an inward Ca2+ current (45, 46) and enabling the sustained inward Na+ current Ist (47). Local Ca2+ release (LCR) from the sarcoplasmic reticulum (SR) via subsarcolemmal ryanodine receptors (RyRs) generates small increments in intracellular Ca2+ concentration. These LCRs activate Na+/Ca2+ exchange (NCX) to pump Ca2+ out of the cell in exchange for Na+ ions at a ratio of 1 Ca2+:3 Na+, to generate an inward NCX current (INCX), and this contributes to both early and late phases of diastolic depolarization (48) and subsequent depolarization of the Vm to the threshold of the next beat (Figure 3). The exact molecular mechanisms responsible for SR Ca2+ release during late diastole are not completely understood. While some studies show that such local Ca2+ release events are spontaneous, independent of transmembrane potential, and likely include stochastic opening of hyperphosphorylated RyRs (49–51), other evidence suggest that these events might be triggered by Ca2+ entry via low-voltage activated T-type Ca2+ channels (52) or Cav1.3 L-type Ca2+ channels (46). Particularly, recent studies indicate that Cav1.3 channel activity contributes to generation and synchronization of diastolic LCRs (46) and that Cav1.3 is necessary for the Ca2+ clock function during SAN firing (53). Overall, the sum of If, ICa,T, INCX and Cav1.3-mediated L-type Ca2+ current (ICa,L) contributes to diastolic depolarization required to ultimately trigger activation of cardiac Cav1.2-mediated ICa,L that initiates the AP, and global Ca2+-induced Ca2+ release. In nature, neither clock functions in the absence of the other. Abundant evidence indicates that functional interactions between the two clock components are critical for normal SAN automaticity (8, 36, 37, 46).
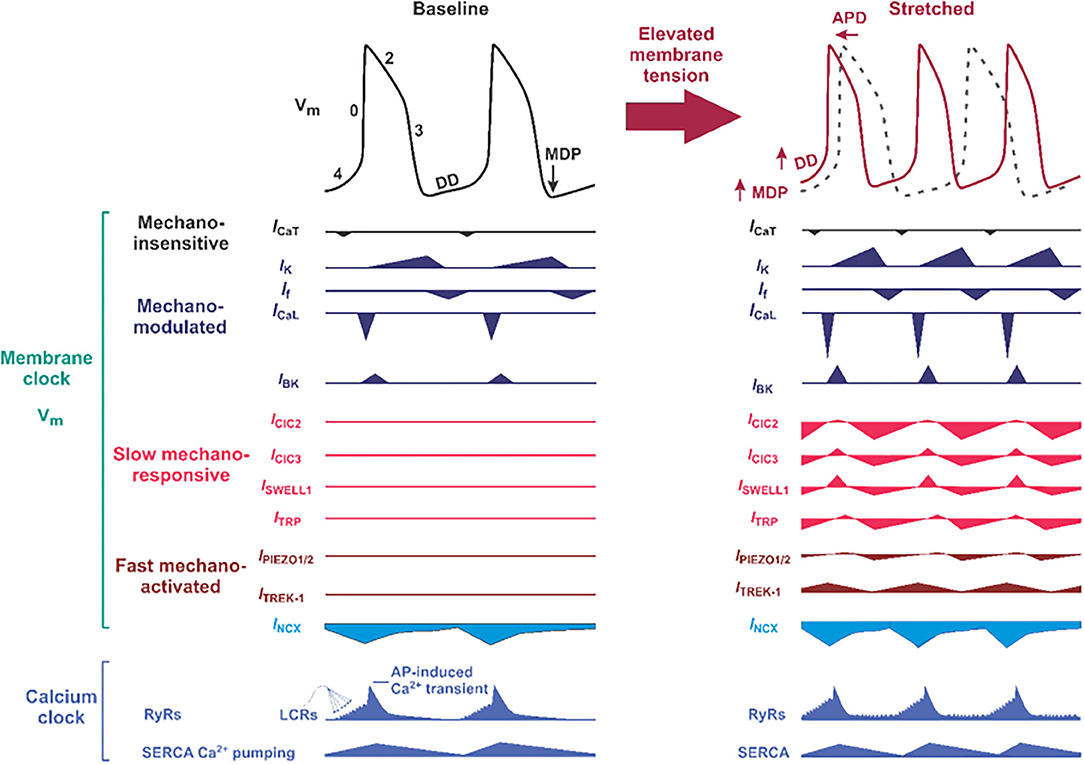
Figure 3. Proposed molecular composition of the mechano-electrical signal transduction in the sinoatrial node (SAN) cell. (Left) Typical SAN membrane action potential (black trace) and the timing of membrane (Vm) clock and calcium (Ca2+) clock components of the coupled-clock pacemaker system are shown. The phases of the action potentials are labeled including phase 4, in which diastolic depolarization (DD) that is key to automatic pacemaker activity takes place. APD, action potential duration; MDP, maximum diastolic potential; DD, diastolic depolarization; ICa,T and ICa,L, T- and L-type voltage-dependent Ca2+ currents; INCX, sodium-calcium exchange current; IK, rapid (IKr) and slow (IKs) delayed rectifier potassium currents; If, HCN4 “funny” current; SERCA, sarco-endoplasmic reticulum ATPase; LCRs, local Ca2+ releases. Below “classical” ion channels defined as mechano-modulated as indicated by various authors (38–40) which can have their normal activity altered by mechanical stress, proposed slow mechano-responsive, and fast mechano-activated ion channels are shown. ClC, chloride channels; SWELL1, swelling-activated leucine-rich repeat containing 8 (LRRC8) family chloride channels; TRP, transient receptor potential ion channels. (Right) Proposed changes in SAN action potential morphology (solod red trace on top of the black dotted trace for baseline condition) under mechanical stress. Below, proposed contribution of slow mechano-responsive and fast mechano-activated ion channels is shown for each ion channel.
There has been significant interest in determining the underlying cellular and molecular mechanisms responsible for intrinsic SAN mechanosensitivity. Mechanical modulation of SAN pacemaking adds another level of complexity to SAN automaticity that has been proposed by Quinn and Kohl as the additional “mechanics-clock” (6, 7) or, more accurately, as a third coupled oscillator. The authors specifically highlighted that in case of fast, beat-to-beat changes in heart rate, the voltage and Ca2+ clocks do not inherently account for the rapid response of the SAN to changes in hemodynamic load and that another set of mechanisms must contribute to spontaneous diastolic depolarization of the SAN. The importance of “mechanics-clock” could be further supported by the fact that stretching of quiescent tissue frequently induces spontaneous activity. In particular, arrhythmic isolated hearts of Prosobranch gastropod become rhythmic when the pacemaker tissue is stretched by with internal perfusion and improve in form as pressure is increased (25). It may be more accurate to describe mechanical modulation of SAN pacemaking as an additional coupled oscillator since it could be applied to both fast and slow changes in heart rate through coupling with fast and slow mechanical oscillators, respectively.
Since ion channels are both central for the regulation of SAN automaticity and can sense mechanical stimuli via various mechanisms, they provide plausible molecular candidates for SAN mechanosensitivity. Ion channels may be grouped into two categories with respect to mechanosensitivity: (1) directly mechanoactivated (fast) and (2) indirectly mechanoresponsive (slow) (Figure 1). Although both categories of channels can change their open probability and other biophysical characteristics in response to mechanical stimulation, they differ in how mechanical forces transduce these effects. Fast directly mechanoactivated ion channels (Piezo1-2, TREK-1, TRAAK, and BK channels) are intrinsically sensitive to mechanical forces applied to the protein or to the lipid bilayer in which the channel resides and do not require any other associated proteins or protein complexes to confer mechano-responsiveness (54, 55). Slow indirectly mechanoresponsive ion channels are polymodal ion channels (TRP channels, SWELL1/LRRC8, and ClC) that respond to mechanical forces in cell-type specific contexts but may not themselves be intrinsically mechanosensitive, for example, when reconstituted in a minimal lipid membrane, devoid of other cellular proteins, with some channels displaying faster activation kinetics and some slower.
RNA Sequencing Identifies Highly Expressed and Enriched Mechanosensitive Ion Channels in Mouse Sinoatrial Node
There are a multitude of mechanosensitive and mechanoresponsive ion channels expressed in mammalian cells. However, without knowledge of the expression level of these channels in SAN, their relevance to SAN physiology is entirely speculative. To guide our discussion of the molecular mechanisms of SAN mechanosensitivity, we examined the expression levels of Piezo1-2, LRRC8a-e, TRPCs, TRPVs, TRPMs, K2P, and ClCs in a genome-wide RNA sequencing data set derived from murine SAN (Figure 2). Hyperpolarization-activated cyclic nucleotide-gated potassium channel 4 (HCN4) is robustly expressed and enriched in murine SAN. Remarkably, the most highly expressed mechanosensitive and mechanoresponsive ion channels (or essential components) in SAN are Piezo1 (3.6-fold greater than HCN4), LRRC8a [SWELL1/volume-regulated anion channel (VRAC), 2.4-fold greater than HCN4], ANO1 (2.04-fold greater than HCN4), and TASK-1 (4.6-fold greater than HCN4). Among TRP channels, the cells expressing TRPM7 were the most present (2.2-folds more than cells expressing HCN4), followed by cells expressing TRPM4, TRPV2, TRPV4, and TRPC1. Regarding mechanosensitive K2P channels, the number of counts for TREK-1 was lower than the number of HCN4. RNA sequencing data did not detect any cells expressing TRAAK. On the contrary, there were more counts for TASK1 than HCN4. Among ClC channels, the number of CICn3 counts was more than that for HCN4, the opposite for CICn2. Furthermore, RNA sequencing showed that the BK channel alpha subunit (KNCMA1) is lowly represented in SAN. Based on these data, we will discuss primarily those mechanosensitive/responsive ion channels that are highly expressed in SAN relative to HCN4. However, one limitation of mouse SAN is that they are the only known species to have a negative chronotropic response to sustained SAN stretch (56). Furthermore, please refer to Table 1 as a resource for where the following ion channels have been confirmed, how they were analyzed, and their literature sources.
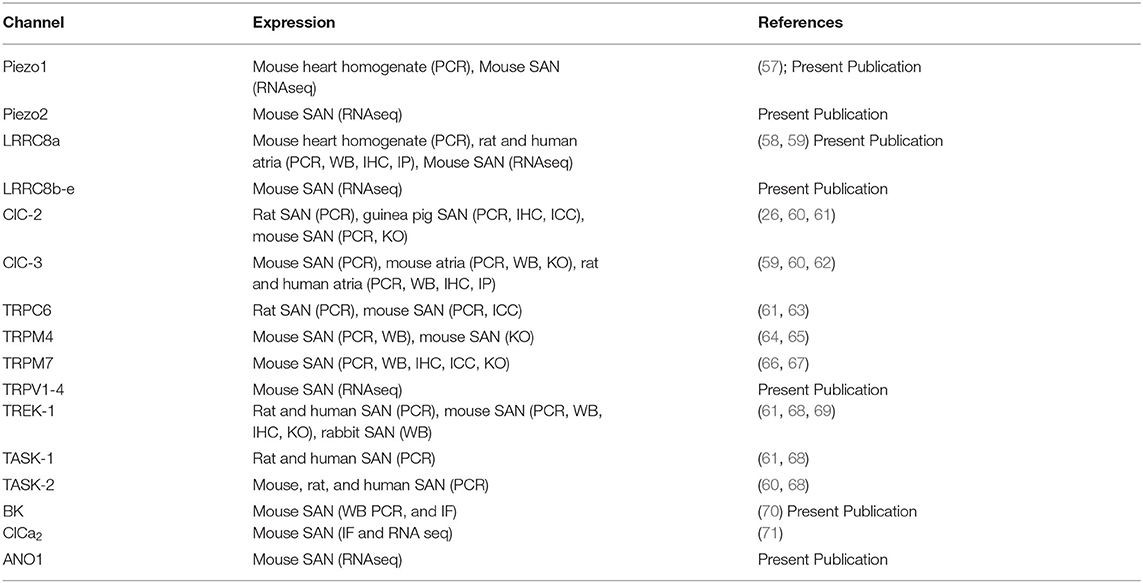
Table 1. Compiled mechanosensitive ion channels discussed in the review, their known expression and detection method, and respective references.
Directly Mechanoactivated Ion Channels
Piezo Channels
Piezo1 and Piezo2 ion channels are bona fide mechanoactivated cation channels with established roles in the cardiovascular system (57, 72) and provide another potential mediator of SAN mechanosensitivity. These non-selective cationic channels may be activated by shear stress from nearby blood flow (laminar or turbulent) as well as membrane stretch induced by increased blood pressure, and their activation is highly sensitive to mechanical stimulus variation in frequency and duration (73). In addition, they are non-selective and are therefore permeable to Ca2+ and Na+, in addition to K+, and have a relatively low threshold for mechanoactivation (57, 74). Although there have been no studies directly examining Piezo1 in the SAN, it is expressed in cardiac tissue (54) and appears highly expressed in the murine SAN (Figure 2). Moreover, Piezo1 plays an important role in the regulation of vascular tone (54) and baroreceptor pressure sensing (55). Since Piezo1 channels provide a depolarizing current in response to mechanoactivation, they are good candidates for mechanically activated increases in SAN automaticity and heart rate acceleration (72). Moreover, Piezo1 channels activate rapidly (within milliseconds) and are responsive to phasic, high-frequency mechanical inputs, such as systolic contractions, but may also be modulated by more gradual mechanical inputs (75), such as increases in atrial filling pressures and could therefore govern SAN mechanosensitivity on a beat-to-beat basis and during periods of chronic stretch. Future studies with targeted genetic deletion of Piezo1 from SAN may directly test these hypotheses.
TREK-1 Channels
Cardiac cells have two-pore domain potassium currents with little time- or voltage-dependency, also known as background currents, that regulate resting membrane potential and cell excitability (76). The family of cloned mammalian background K+ channels includes 14 members encoded by different genes. The members were divided into six subfamilies, TWIK, TREK, TASK, TALK, THIK, and TRESK, on the basis of sequence homology and functional similarities (77). Two-pore domain potassium channels are typically insensitive to conventional K+ channel blockers such as 4-AP, TEA, Ba2+, Cs+, and glibenclamide (76), but they are sensitive to membrane stretch, changes in extracellular or intracellular pH, fatty acids, and inhalation anesthetic agents (e.g., isoflurane) and are regulated by second messenger phosphorylation (76). Structurally, these channels possess four transmembrane domains and two pore domains; each subunit contains two pore-forming domains, so two subunits can form a complete pore of the channels. In murine SAN (Figure 2), two subtypes of two-pore domain potassium channels are mainly expressed: stretch-activated K+ channel TREK-1 (78–80) and the acid-sensitive K+ channel TASK1 (81–83). Under basal conditions, the activity of the TREK channels is low; however, applying negative pressure to the cell membrane reversibly activates TREK-1 (84). In addition, laminar shear stress stimulates TREK-1, whereas the cell shrinkage induced by extracellular hyperosmolarity reduces the amplitude of TREK-1 (84). Indeed, it has been shown that TREK-1 mechanosensitivity is mediated directly by the lipid membrane perturbations and changes in plasma membrane tension (85). Given its expression in the SAN (Figure 2), it is a candidate contributor to SAN mechanosensitivity on a fast (<1 s) basis.
A number of studies on zebrafish and mice that inhibited plasma membrane trafficking of TREK-1 by inactivating the interacting proteins POPDC1 and POPDC2 revealed exercise- and age-dependent sick sinus syndrome and atrioventricular block (78, 86), suggesting a role for TREK-1 in cardiac automaticity. Similarly, transgenic overexpression of a C-terminal truncation of beta IV spectrin, which also disrupts TREK-1 plasma membrane trafficking, results in sick sinus syndrome (87). These studies provide indirect evidence of TREK-1-mediated effects on SAN automaticity. However, more direct evidence was provided by Unudurthi et al. (69). The authors determined that TREK-1 protein is indeed expressed in both murine and rabbit SAN, and TREK-1-like background currents were reduced in patch-clamped SAN cells isolated from cardiac-specific TREK-1 KO mice (αMHC-Kcnk2f/f). Also, freely moving, telemetered αMHC-Kcnk2f/f mice exhibited sinus bradycardia at rest, consistent with studies by Hund et al. (87) where disrupted plasma membrane TREK-1 trafficking induced sick sinus syndrome. Paradoxically, isolated TREK-1 KO SAN cells exhibited increased rather than decreased firing rates as compared with wild-type (WT) SAN. Furthermore, exercise and treatment with epinephrine uncovered stress-induced sinus pauses in αMHC-Kcnk2f/f mice via unclear mechanisms, or possibly via variation in sympathetic and parasympathetic activity. Finally, intrinsic heart rates measured in telemetered αMHC-Kcnk2f/f mice with atropine and propranolol treatment exhibited no significant differences, suggesting that neurohumoral inputs are important for TREK-1-dependent regulation of SAN automaticity. These studies illustrate the incomplete understanding of TREK-1 and its contribution to SAN mechanosensitivity, which requires further investigation.
BK Channels
BK (large-conductance Ca2+- and voltage-activated K+) channels are another promising contributor to SAN mechanosensitivity and automaticity [reviewed in (88)]. These channels are characterized as having large single-channel conductance and selective inhibitors and are regulated by voltage and Ca2+ (70). Imlach et al. (89) determined that BK channel inhibition via paxilline caused a reduction in heart rate in isolated mouse and rat hearts but not in hearts from Kcnma1 KO mice. This finding was confirmed at a cellular level when Lai et al. (70) demonstrated that paxilline applied directly to isolated mouse SAN cells reduced AP firing rate in WT mice but not in Kcnma1 KO mice. Lastly, Zhao et al. (90) found that BK channels are mechanosensitive to a small degree, showing an increased activity in chick ventricular myocytes plated on stretched extracellular matrix. Given this and their expression in murine SAN (Figure 2), BK channels are a putative contributor to SAN mechanosensitivity. In this case, SAN membrane depolarization, increases in cytosolic Ca2+, and mechanical stimulation from SAN/atrial systole all coincide to activate BK channels after the peak of the SAN AP to augment AP repolarization, re-initiation of diastolic depolarization, and heart rate acceleration (Figure 3). Based on this model, mechanoactivation of BK channels must be relatively rapid to contribute to SAN AP repolarization, as published data suggest; however, it remains unclear if these channels are rapidly or slowly mechanoactivated in SAN.
Mechanoresponsive Transient Receptor Channels
Putative candidates for stretch-responsive non-selective cation channels include TRP channels expressed in murine SAN: TRPC1, TRPC3, TRPM4, TRPM7, TRPV2, and TRPV4 (Figure 2). A number of TRP channels that we found expressed in murine SAN have been described as mechanoresponsive either directly or indirectly (29, 91). However, thus far, only TRPM4 and TRPM7 have been studied in the context of SAN function. TRPM4 is an intracellular Ca2+-activated, non-selective cation channel, which is possibly indirectly mechanoresponsive (29). At negative membrane potentials, TRPM4 activation allows Na+ influx, leading to the membrane depolarization, whereas, at the positive membrane potentials, TRPM4 allows K+ efflux, leading to membrane repolarization (92, 93) (Figure 3). In rodent SAN, TRPM4 is thought to contribute to diastolic depolarization and a positive chronotropic response in response to stretch (64, 65, 94). TRPM7, an ion channel and protein kinase (chanzyme), permeable to both divalent cations, including Zn2+, Mg2+, and Ca2+, as well as monovalent cations such as Na+ and K+ (95, 96), is broadly expressed. TRPM7 is highly expressed in murine SAN at the mRNA level (Figure 2) and generates a robust current in both SAN and atrioventricular node cells (66). Both cardiac- and SAN-targeted TRPM7 deletion impaired cardiac automaticity (67); however, the mechanism was proposed to be via regulation of HCN4 and If current rather than a direct effect on diastolic depolarization via channel activity (66, 67). While it is clear that none of these TRP channels are intrinsically mechanoactivated (97, 98), it is possible that some of these channels form part of a mechanosensory system (29) and therefore may be mechanoresponsive within specific cellular contexts (99). Testing these hypotheses would require directly measuring these mechanoresponsive currents in isolated SAN, as performed by Kohl et al. (4, 100), in genetic knock-outs of each of these putative mechanoresponsive channels or using specific pharmacologic inhibitors.
Volume-Regulated Anion Channels
Another mechanoresponsive ionic current that has been implicated in the regulation of SAN automaticity is ICl,SWELL or the swell-activated chloride current. This ionic current may be carried by VRAC or ClC ion channels, both of which are most commonly activated by cell swelling, which is typically achieved by applying hypotonic or hypo-osmolar solution to cells. However, in a few studies, anion or chloride conductances were demonstrated by application of mechanical forces, as described in further details below. VRACs are activated by cell swelling, ubiquitously expressed in various mammalian cell types and thought to be implicated in many physiological and pathophysiological processes, including fluid secretion, glutamate release, membrane potential regulation, and apoptosis [summarized in the review article (101)]. Although the biophysical properties of VRACs have been well-characterized in multiple cell types over the course of decades, the molecular identity of VRAC remained a mystery until the Patapoutian (58) and Jentsch (102) groups identified leucine-rich repeat containing 8a (LRRC8a, also known as SWELL1) as a required component of a heterohexameric channel complex consisting of various stoichiometries of LRRC8a, and/or LRRC8b,c,d,e. Although the function of the VRAC current has been attributed to cell volume regulation in response to relatively non-physiological osmotic gradients, the broad tissue expression pattern of LRRC8 proteins and presence of VRAC/ICl,SWELL current in a multitude of cell types (103–108), including cardiac myocytes (109–113) that are rarely exposed to hypotonic swelling, suggests that the actual physiological role of VRAC and LRRC8 channels remains unknown. Indeed, experiments using magnetic dynabeads bound to monoclonal antibodies for beta1-integrins demonstrated activation of ICl,SWELL in cardiac myocytes in response to mechanical force applied via magnetic fields (109, 110), supporting the notion that ICl,SWELL is mechanoresponsive in cardiac cells. Therefore, given the high mRNA counts of LRRC8a (SWELL1) and associated subunits LRRC8b,c,d in murine SAN relative to HCN4 (Figure 2), the contribution of SWELL1-mediated ICl,SWELL to pacemaker activity and response to stretch warrants further investigation.
Elegant studies by Hagiwara et al. (114) demonstrated that mechanical inflation of isolated rabbit SAN cells using positive pressure via the patch-pipette in whole-cell configuration induces an outwardly rectifying, stretch-activated anion current that is inhibited by chloride channel blockers, 4,4′-diisothiocyano-2,2′-stilbenedisulfonic acid (DIDs) and 9-anthracenecarboxylic acid (9-AC). Also, this current exhibits a shift in reversal potential consistent with a chloride conductance (115) and has a sequence of anion permeability (I− > > Br− > Cl− > F−) similar to VRAC or LRRC8 channels. ICl,SWELL activates over the course of minutes (~2 min), which implies responsiveness to tonic changes in membrane tension, as may be expected from gradual atrial stretch-associated increased venous return, but relatively unaffected by phasic changes associated with beat-to-beat changes. Based on the outwardly rectifying current–voltage relationship, and reversal potential around the Cl− reversal potential (ECl = −30 mV), we speculate that inward chloride current at voltages below −30 mV may contribute to diastolic depolarization and SAN automaticity, while outward current at voltages above −30 mV may contribute to SAN AP shortening (116) (Figure 3). The integrated effects on automaticity and the response to stretch, however, are likely to be complex.
Similarly, Decher et al. found in guinea-pig atrial myocytes that ICl,SWELL induced by osmotic swelling leads to a shortening of AP duration that was inhibited by DCPIB (a relatively selective ICl,SWELL inhibitor) (117). Furthermore, Seol et al. found the ICl,SWELL can be activated by axial stretch in cardiomyocytes isolated from the pulmonary vein (30, 59); and Egorov et al. determined that ICl,SWELL activation in response to mechanical stretch can depolarize resting membrane potential, generate arrhythmic substrates, and confirm that it can also shorten APs (59). In isolated rabbit SAN tissue, Arai et al. also showed that application of various non-specific anion channel blockers that can block VRACs, such as DIDs, caused a reduction in the stretch-induced increase in firing rate at a high level of distension (118). On the other hand, Cooper et al. (56) reported that the application of 9-AC at 1 mM concentration had no effect on the stretch-induced increase in heart rate when a significant stretch-stimulus was applied, suggesting that ICl,SWELL may not underlie the SAN response to mechanical stretch. However, application of such high concentrations of 9-AC is highly non-specific and therefore complicates the interpretation of this result. Furthermore, the use of different stretching techniques between the two studies may account for the differences observed. These studies, albeit contradictory, indicate the potential role of ICl,SWELL in modulating SAN function on a slow, non-beat-to-beat basis, which may be present during periods of chronic stretch, and demonstrate the need for additional experiments. Since SWELL1 (LRRC8a) and LRRC8 subunit proteins are now known to encode ICl,SWELL in numerous other cell types (103–108), future studies examining cardiac specific and SAN targeted Swell1 KO mice, transient knockdown in isolated cells, and/or more specific small molecules such as DCPIB will provide important new insights into the contribution of ICl,SWELL and VRAC in cardiac automaticity and the response to SAN stretch.
ClC Anion Channels
Other candidates for the molecular identity for ICl,SWELL in SAN are the ClC ion channels. While both ClC-2 and ClC-3 have been studied in cardiac myocytes (26, 62), and ClC-3 has a high mRNA count in murine SAN (Figure 2), only ClC-2 has been directly studied with respect to regulating SAN automaticity. Huang et al. (26) showed that inwardly rectifying chloride current induced by osmotic swelling in isolated guinea-pig SAN pacemaker myocytes could be blocked though intracellular dialysis of anti-ClC-2 antibody, which did not affect other pacemaker currents, including If, ICa,L, and IKs and the volume-regulated outwardly rectifying Cl− current (ICl,vol). Anti-ClC-2 antibody reversed the changes in SAN APs induced by osmotic swelling. The authors also showed that ClC-2 KO (ClCN2−/−) mice demonstrate a decreased chronotropic response to acute exercise stress when compared with their age-matched ClCN2+/+ and ClCN2+/− littermates. It was then concluded that targeted inactivation of ClC-2 does not alter intrinsic heart rate but prevented the positive chronotropic effect of acute exercise stress through sympathetic regulation of ClC-2 channels. While ClC-2 channels may contribute in part to cardiac ICl,SWELL, there have been no studies examining the signaling mechanisms underlying ClC-2 mechanoresponsivity.
ClC-3, on the other hand, has been proposed to be mechanoresponsive in osteoblasts (119) and has been shown to be expressed in cardiac myocytes, to underlie ICl,SWELL, and to be involved in numerous pathophysiological processes, including ischemic preconditioning, myocardial hypertrophy, and heart failure (120). However, there have yet to be any studies directly examining ClC-3 in SAN cells, and neither global nor cardiac specific ClC-3 KO mice were noted to show differences in heart rates (62).
Other Chloride Channels
Other possible contributors to SAN mechanosensitivity are calcium-activated chloride channels (CaCCs) such as anoctamin1 (ANO1) and chloride channel accessory 2 (ClCA2). Ye et al. (121) determined that ANO1 is expressed in mouse ventricular myocytes and facilitates accelerated AP repolarization. Given that ANO1 is implicated to be mechanoresponsive (121) and expressed in murine SAN (Figure 2), it is plausible that ANO1 may contribute similarly to shorten pacemaker potentials, as Sung et al. speculated (122). In addition, Mao et al. found that ClCA2 is highly expressed in SAN tissue and, when mutated, induces mild conduction block and ectopic pacemaker activity (71). While no study has examined ClCA2 mechanosensitivity, given its calcium-activated properties, it is likely to be affect by pressure-induced calcium transients (123). Given these findings, it is feasible that calcium-activated chloride channels could play a partial role in the response of SAN beating rate to stretch.
Caveolae-Mediated Ion Channel Mechanosensitivity
Interestingly, besides ANO1 and ClCA2, all the aforementioned ion channels affected by mechanical stress have been found to associate with caveolae, which are abundantly expressed in SAN cells (5, 124, 125) and are known to mediate cellular response to mechanical stress by reserving “extra” cell membrane to buffer mechanical forces and contribute to cell volume regulation (126–128). Caveolae are small, 50- to 100-nm omega-shaped membrane invaginations of the plasma membrane enriched by sphingolipids, cholesterol, cavin proteins, and caveolin proteins. Caveolin-3 (129) is the dominant isoform in muscle cell caveolae; however, caveolin-1 has also been found in atrial myocytes (129, 130). It has been shown that caveolae compartmentalize multiple ion channels involved in SAN pacemaker activity, such as canonical contributors to the SAN AP such as HCN channels (131), L-type calcium channels, and Kv1.5 channels (132), as well as anion channels such as SWELL1 (LRRC8a), ClC-2, and ClC-3 (59, 103, 105, 108). Stretch-induced disruption of caveolae may participate directly or indirectly via localization of other signaling factors (133) in the activation of mechanoresponsive ion channels, including VRAC and ClC ion channels responsible for ICl,SWELL (134). Specific surface membrane proteins may not only affect changes in membrane potential but also directly or indirectly regulate intracellular Ca2+ cycling; on the other hand, intracellular Ca2+ cycling proteins may also regulate Vm via Ca2+ modulation of surface membrane electrogenic molecules (135).
Mechanochemical Signal Transduction
While changes in cardiac morphology are attributed to mechano-electrical signal transduction via regulating the activity of mechanosensitive ion channels, mechanochemical signal transduction could be described as a mechano-induced regulation of various second messenger signaling pathways that are ultimately translated into changes of calcium handling and ion channel activity. Here, we focus on mechano-dependent regulation of ROS, cAMP, and IP3 signaling pathways (Figure 4). It should be noted that mechano-electrical and mechanochemical feedbacks are not mutually independent but rather interact in a complex and dynamic manner as described below. While mechanochemical signal transduction pathways could be involved in the regulation of various ion channels via different post-translational modifications (such as phosphorylation, nitrosylation, and oxidation), activation of mechano-electrical feedback can significantly modify intracellular ion composition affecting intracellular Ca2+ signaling. Below, we briefly summarize several key mechano-chemical signaling pathways that could be involved in SAN mechanosensitivity. Though the role of these pathways has not been demonstrated in SAN mechanosensitivity, it is feasible that they have an impact on mechanical heart rate modulation.

Figure 4. Proposed effect of mechanical loading on caveolae-associated signaling pathways and their effect on sinoatrial node calcium clock function. Mechanical stress: (A) Activates X-ROS signaling via NOX2 (NAPDH oxidase 2) which stimulates CaMKII activity and upregulates RyR (ryanodine receptor) function leading to an accelerated calcium clock, as well as sensitizes SAC (stretch-activated channels) for increased activation. (B) Displaces the GPCR alpha subunit, activating AC (adenylyl cyclase)-mediated conversion of ATP to cAMP, which activates PKA leading to an accelerated calcium clock via RyR activation and PLB (phospholamban) inhibition of SERCA (sarcoplasmic reticulum Ca2+-ATPase). (C) Induces PLC (phospholipase C)-mediated conversion of PIP2 to IP3 (inositol triphosphate), which activates IP3 receptors (IP3R) and an accelerated calcium clock and also activates VRACs (volume regulated anion channels). All three pathways lead to accelerated cardiomyocyte calcium cycling, LCR (local calcium releases) rate, and increased activity of mechanosensitive channels.
Petroff et al. (33) were the first to use confocal microscopy to monitor subcellular Ca2+ events in cardiomyocytes during stretch and to provide direct evidence that stretch modulates the elementary Ca2+ release process, the Ca2+ spark. Stretch-induced increases in Ca2+ spark frequency are a phenomenon consistently observed in myocytes (136, 137), also in response to other mechanical stimuli, such as shear stress and afterload (123). A single myocyte stretch event immediately—within milliseconds—triggers a burst of Ca2+ sparks, which is reversible and declines within seconds (137, 138). While in ventricular myocytes these sparks are restricted in time and space, unique patterns of RyR expression and the presence of bridging RyR groups between large clusters demonstrated in the SAN cells (139–141) could lead to the generation of propagating LCR events as demonstrated in modeling studies by Stern and colleagues (141). This mechanism of stretch-induced increase in Ca2+ spark activity might be also present in the SAN and potentially contribute to mechanical regulation of Ca2+-clock activity (Figure 4).
NOX2–Reactive Oxygen Species
Mechanical modulation of Ca2+ spark activity was linked to stretch-induced activation of ROS signaling that is also graded in a stretch-dependent manner (142). The stretch-induced NOX2-dependent ROS response sensitizes RyR to Ca2+ possibly through direct oxidation but may also do so indirectly via oxidation of calmodulin, displacing it from the RyR and promoting activation (143) or via RyR phosphorylation by oxidized CaMKII (144). These pathways of mechano-transduction are termed X-ROS signaling and require an intact microtubule network and functions independently of stretch-activated channels (SACs) and transsarcolemmal Ca2+ influx (33, 137). Furthermore, X-ROS signaling is confined to dyads (the cytosolic space between the sarcolemmal and SR membranes) (145) and has been proposed to be an important regulator of beat-to-beat adaptation to hemodynamic load in working cardiomyocytes (142). However, these pathways have not been confirmed specifically in SAN myocytes, but they are known to contain the necessary components (146).
In addition to regulation of Ca2+ signaling, the X-ROS pathway has also been found to be involved in the modulation of mechanoresponsive ion channels as well (147). Patch-clamp studies on stretched ventricular myocytes revealed NOX-dependent modulation of SACs (148), and this modulation may be facilitated by co-localization of NOX2 and SAC in caveolae (149, 150). Although SACs are not involved in X-ROS signaling per se, these channels may contribute to stretch-induced modulation of AP as discussed above for mechano-electrical signal transduction. Additionally, ICl,SWELL activated by osmotic swelling has been found to be controlled by an angiotensin II-dependent ROS cascade that is previously implicated by integrin stretch (113). This is consistent with persistent activation of ICl,SWELL and ROS present in several models of cardiac disease. Furthermore, Gradogna et al. demonstrated that LRRC8 channel subunits and their currents are differentially modulated by oxidation depending on LRRC8 channel subunit composition (151). Given that inflammation and oxidation are present in the setting of hypertension, it is possible that SAN mechanosensitivity could differ from other cardiac regions due to varying SWELL1 subunit expression (151).
Nitric Oxide Synthase
Nitric oxide synthase (NOS) also plays a discrete role facilitating cardiac stretch as Petroff et al. demonstrated that stretch increases nitric oxide (NO) production with concurrent increases in Ca2+ spark frequency and transient amplitudes (33). Pharmacological inhibition or genetic deletion of both neuronal NOS (nNOS) and endothelial NOS (eNOS) demonstrates that subtypes contribute to the increase of systolic Ca2+, but only nNOS participated in the afterload induced Ca2+ sparks (152). Due to the short lifetime of NO, its effective signaling range is limited and dependent on the diffusion distance, amount produced, and the buffer capacity of the cell (33). Therefore, one possible explanation for the distinct effects of nNOS vs. eNOS-derived NO on Ca2+ sparks is their different subcellular localizations. While eNOS is localized at the caveolae (153, 154), nNOS is preferentially localized at the SR membrane in the vicinity of RyR, and nNOS increases RyR Ca2+ leak, directly by S-nitrosylation or indirectly via CaMKII (155). In addition, nNOS facilitates SERCA Ca2+ reuptake (155), which may compensate for the increased SR Ca2+ leak and reduced basal ICa,L (156). In regard to the SAN, in a study by Vila-Petroff et al. using exogenous NO donors, high levels of NO induced a large increase in cGMP and a negative inotropic effect, while low levels of NO increased cAMP and caused positive inotropy via cGMP-independent activation of adenylyl cyclase (157). Furthermore, it has been shown that inhibition of NOS has a negative chronotropic effect on SAN activity and that NOS activation has an opposite effect, indicating that SAN function is somewhat dependent on NOS activity (158, 159). However, unlike X-ROS signaling, NO mechanosensitivity operates on a slower time scale of minutes rather than seconds (33), suggesting that it may play a more significant role in conditions where chronic stretch is a factor (i.e., hypertension and chamber filling pressures).
Atrial Natriuretic Peptide
Another important factor in myocyte stretch signaling is atrial natriuretic peptide (ANP) (160). Similar to X-ROS, ANP is a mechanosensitive signaling factor that is activated by a caveolae and angiotensin II-dependent pathway (161). ANP has been found to enhance reflex bradycardias (162); therefore, it is likely that ANP has a compensatory mechanosensitive role on the SAN, acting to restore it to normal function in response to elevated stretch. ANP has been found to shift midpoint activation of pacemaking If current toward less negative potentials (163) and thus accelerate SAN rhythm. ANP is also able to increase intracellular cGMP and cAMP levels (163), which play a crucial role in SAN automaticity via phosphorylation of the calcium clock proteins (50, 164). Indeed, ANP has been identified as a critical regulator of SAN automaticity (38, 165).
Inositol Trisphosphate
IP3Rs are another type of SR Ca2+ release channels, which are activated by IP3 through the hydrolysis of phosphatidylinositol-(4, 5)-bisphosphate by phospholipase C and thus may also contribute to the LCR generation via hypersensitization of RyRs. They are highly abundant in atrial and SAN myocytes (166–168), and recent studies demonstrated that this signaling pathway might be confined within specific microdomains, including lipid rafts and dorsal ruffles (169). Stimulation of IP3Rs was found to accelerate spontaneous beating rate of the mouse SAN likely through regulation of Ca2+ spark activity and RyR function (170). In rabbit ventricular myocytes, upregulation of IP3R-induced Ca2+ releases was detected and linked to enhanced spontaneous SR Ca2+ releases (170). It has been shown that mechanical stretch can directly activate phospholipase C with production of IP3 (171), which may subsequently modulate SAN automaticity (Figure 4).
Mechanochemical Signal Transduction and Caveolae
While there are numerous mechanochemical signaling factors that may affect SAN automaticity, they are united as facilitators of cardiac mechano-transduction through their association with caveolae membrane structures (Figure 4) (123, 172–174). For example, NOS (173), NOX2-mediated ROS (150), and calcium dynamics (123) are all affected by the presence of caveolae, which are suspected to play an inhibitory role on these factors, which are disrupted by shear stress. Digging deeper, angiotensin II mediates activation of cAMP production (175) and X-ROS through caveolae membrane structures (176), further linking the discussed factors to these structures. The suspected regulation of these signaling factors of SAN automaticity by caveolae may explain the connection between caveolae loss and cardiac pathology (177) as chronic shear stress depletes caveolae (178), allowing these factors to activate constitutively and/or enter unusual feedback loops. For example, as shear stress increases, caveolae flatten and release NOS, which should reduce the initial mechanical stimuli and allow caveolae to reform. However, if the mechanical stimulus is prolonged, membrane caveolae density will decrease, eliminating a crucial regulator of nNOS activity. Without this negative regulation, these signaling factors can enter positive feedback loops, inducing the generation of excess ROS from sarcolemmal and mitochondrial sources that can ultimately lead to changes in myocyte electrophysiology as calcium kinetics are subsequently altered. For these reasons, it is highly plausible that SAN caveolae may regulate downstream signaling factors that are known to alter SAN automaticity and consequently heart rate.
Pathophysiology
While physiological stretch provides a critical autoregulatory feedback loop to adjust SAN pacemaker rate upon hemodynamic changes, pathophysiological stretch (and physiological stretch applied to diseased myocardium) can lead to SAN dysfunction and trigger cardiac arrhythmias (6, 7, 179). It has been shown that conditions associated with atrial pressure and/or volume preload/afterload, including heart failure, atrial fibrillation, hypertension, and valvular disease, are common comorbidities linked to SAN dysfunction or sick sinus syndrome (23, 180–183). Sick sinus syndrome is manifested clinically as arrhythmias that can include sinus bradycardia, sinus pauses or arrest, sinoatrial exit block, or alternating brady- and tachyarrhythmias (184). These manifestations can lead to chronotropic incompetence, which is an inadequate heart rate response to exercise or stress (184). Electrophysiological mechanisms that underlie SAN dysfunction in the setting of pathologically elevated atrial stretch are not completely understood and may vary for different conditions. The mechanisms could include abnormal functioning, expression, and/or regulation of the components of mechano-electrical and mechanochemical signal transduction and may be also conditioned by structural remodeling of the SAN.
Importantly, in the setting of sinus node dysfunction when SAN is not able to maintain physiologically robust rhythm, mechanical stretch can enhance automaticity in latent atrial pacemakers or provoke arrhythmogenic activity in the working myocardium to form ectopic foci and trigger atrial fibrillation (30, 185, 186). Though distribution patterns of stretch-induced atrial ectopic foci are not currently known, pulmonary veins represent the most common source of atrial fibrillation ectopy (187, 188). Mechanical stretch of pulmonary vein myocardium has been shown to promote arrhythmogenic activity from this region and may initiate atrial fibrillation (186, 189). Recently, we have demonstrated that stretch-induced activation of ICl,SWELL in rat pulmonary veins leads to membrane depolarization and decreased AP amplitude, which trigger conduction discontinuities and both ectopic and reentrant activities (30, 59). We also found that downregulation of caveolin-3 protein expression and disruption of caveolae structures during chronic hypertension in spontaneously hypertensive rats significantly facilitates activation of ICl,SWELL and increase the sensitivity of pulmonary vein in response to stretch to 10- to 50-fold (59). The increased sensitivity to stretch could be linked to the presence of constitutively active ICl,SWELL that has been previously reported in failing (a pacing-induced congestive heart failure model) canine ventricular myocytes (111) and in human atrial myocytes obtained from patients with right atrial enlargement and/or elevated left ventricular end-diastolic pressure (112). Similar results of constitutively (i.e., without hypotonic stress) active, DIDS-sensitive ICl,SWELL current was shown in cultured neonatal rat ventricular hypertrophic myocytes induced by cyclic mechanical stretch (190) and in mouse ventricular myocytes isolated from hearts subjected to 4 weeks' transverse aortic constriction (TAC) (191).
Pressure overload for 8 weeks using the TAC mouse model demonstrated a smaller basally active ICl,SWELL as well as a significantly reduced hypotonic solution-induced ICl,SWELL (191). Similar decreases in hypotonic ICl,SWELL current without basal activation are observed in rabbit hypertrophied ventricular cells after treatment of volume and pressure overload (192) and spontaneous hypertrophic ventricular cells from caveolin-3-deficient mice (193). These may indicate that in an early adaptive stage of cardiac pressure/volume overload, ICl,SWELL is basally activated by persistent mechanical stretch of the cell membrane and thus contributes to SAN tachycardia as well as facilitate atrial ectopy, as discussed earlier. However, attenuation of ICl,SWELL mechanical sensitivity by long-term mechanical stretch of the plasma membrane may contribute to depressed SAN function and contribute to transformation to a non-adaptive stage. Indeed, our preliminary findings indicate that in 8-week post-myocardial infarction mouse model of heart failure, mRNA protein expression levels of ClC-2 and ClC-3 mechanosensitive chloride channels are significantly downregulated within the intercaval region of the right atrium, which correlates with a significantly enhanced cardiomyocyte membrane tension and downregulation of caveolae structures (194).
Pathological stretch can also affect mechanochemical signal transduction and contribute to stretch-mediated ectopic foci and atrial arrhythmogenesis. Stretch-induced activation of ROS systems via activation and upregulation of NADPH oxidases NOX2 and NOX4 have been linked to an increase in oxidation of RyRs and concomitant rise in spontaneous Ca2+ release event frequency, elevated Ca2+ leak, and significant increase in atrial fibrillation susceptibility (195). It should be also noted that chronic mechanical stretch may dramatically attenuate the protein expression profile of various ion channels and Ca2+-handling proteins, including those involved in mechano-electrical and mechanochemical signal transductions, further contributing to SAN dysfunction and atrial arrhythmogenesis.
Summary
Emerging evidence demonstrates that mechano-electrical and mechanochemical signal transduction pathways could be implicated in mechanical modulation of SAN function and thus represent an important mechanism for intrinsic regulation of cardiac rhythm. This adds another level of complexity to SAN automaticity and could be described as a “mechanics-clock” component of the pacemaker system (6, 7). Though the exact components of mechano-electro-chemical signal transduction involved in SAN mechanosensitivity are currently unknown, as summarized in the current review, these may involve a number of complex signaling feedback mechanisms that alter the function of both the voltage and calcium pacemaker clocks. As discussed, these mechanisms may interplay with each other, providing a precise attenuation of the SAN beating rate in response to various mechanical stimuli. Disruption of SAN function and regulation has been observed with multiple pathological conditions that are associated with atrial pressure/volume overload and thus may involve the remodeling of the components of the mechano-electro-chemical feedback loops in the SAN. Identification of such components, their impact into SAN pacemaking, and pathological remodeling may provide new therapeutic targets for the treatment of SAN dysfunction and associated rhythm abnormalities. Moreover, linking molecular components of mechano-electro-chemical signaling to certain cellular nanodomains and nanostructures may introduce a novel framework for therapeutic approaches for pacemaker dysfunction treatment targeted at preventing the degradation of cardiac cytoarchitecture.
Author Contributions
RS and AG conceived the topic of the review and steered the general direction on what they wanted the review to cover. DT, CK, and JH examined the details of SAN mechanosensitivity and organized the literature into a concise review. PM and MM produced, analyzed, and wrote significant portions of the review related to RNAseq data. All authors allocated time and effort into writing/editing the manuscript and creating figures, small differences in effort are reflected in the author order.
Funding
This work was supported by NIH R01HL141214, American Heart Association 16SDG29120011, the Wisconsin Partnership Program 4140 (to AG), VA Merit Award I01BX005072, NIH R01DK106009 (RS), NIH predoctoral training grant T32GM008688 (to DT), and the Fondation Leducq TNE 19CV03 FANTASY (MM).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The figures were generated using CorelDRAW 2018 software and BioRender.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.662410/full#supplementary-material
References
1. Bainbridge FA. The influence of venous filling upon the rate of the heart. J Physiol. (1915) 50:65–84. doi: 10.1113/jphysiol.1915.sp001736
2. Donald DE, Shepherd JT. Reflexes from the heart and lungs: physiological curiosities or important regulatory mechanisms. Cardiovasc Res. (1978) 12:446–69. doi: 10.1093/cvr/12.8.449
3. James TN, Nadeau RA. Sinus bradycardia during injections directly into the sinus node artery. Am J Physiol. (1963) 204:9–15. doi: 10.1152/ajplegacy.1963.204.1.9
4. Cooper PJ, Lei M, Cheng LX, Kohl P. Selected contribution: axial stretch increases spontaneous pacemaker activity in rabbit isolated sinoatrial node cells. J Appl Physiol. (2000) 89:2099–104. doi: 10.1152/jappl.2000.89.5.2099
5. MacDonald EA, Madl J, Greiner J, Ramadan AF, Wells SM, Torrente AG, et al. Sinoatrial node structure, mechanics, electrophysiology and the chronotropic response to stretch in rabbit and mouse. Front Physiol. (2020) 11:809. doi: 10.3389/fphys.2020.00809
6. Quinn TA, Kohl P. Cardiac mechano-electric coupling: acute effects of mechanical stimulation on heart rate and rhythm. Physiol Rev. (2021) 101:37–92. doi: 10.1152/physrev.00036.2019
7. Quinn TA, Kohl P. Mechano-sensitivity of cardiac pacemaker function: pathophysiological relevance, experimental implications, and conceptual integration with other mechanisms of rhythmicity. Prog Biophys Mol Biol. (2012) 110:257–68. doi: 10.1016/j.pbiomolbio.2012.08.008
8. Lakatta EG, Maltsev VA, Vinogradova TM. A coupled SYSTEM of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart's pacemaker. Circ Res. (2010) 106:659–73. doi: 10.1161/CIRCRESAHA.109.206078
9. Silverman ME, Hollman A. Discovery of the sinus node by Keith and Flack: on the centennial of their 1907 publication. Heart. (2007) 93:1184–7. doi: 10.1136/hrt.2006.105049
10. Boyett MR, Honjo H, Kodama I. The sinoatrial node, a heterogeneous pacemaker structure. Cardiovasc Res. (2000) 47:658–87. doi: 10.1016/S0008-6363(00)00135-8
11. Fedorov VV, Glukhov AV, Chang R. Conduction barriers and pathways of the sinoatrial pacemaker complex: their role in normal rhythm and atrial arrhythmias. Am J Physiol Heart Circ Physiol. (2012) 302:H1773–83. doi: 10.1152/ajpheart.00892.2011
12. Fedorov VV, Glukhov AV, Chang R, Kostecki G, Aferol H, Hucker WJ, et al. Optical mapping of the isolated coronary-perfused human sinus node. J Am Coll Cardiol. (2010) 56:1386–94. doi: 10.1016/j.jacc.2010.03.098
13. Fedorov VV, Schuessler RB, Hemphill M, Ambrosi CM, Chang R, Voloshina AS, et al. Structural and functional evidence for discrete exit pathways that connect the canine sinoatrial node and atria. Circ Res. (2009) 104:915–23. doi: 10.1161/CIRCRESAHA.108.193193
14. Camelliti P, Green CR, LeGrice I, Kohl P. Fibroblast network in rabbit sinoatrial node: structural and functional identification of homogeneous and heterogeneous cell coupling. Circ Res. (2004) 94:828–35. doi: 10.1161/01.RES.0000122382.19400.14
15. Liu J, Dobrzynski H, Yanni J, Boyett MR, Lei M. Organisation of the mouse sinoatrial node: structure and expression of HCN channels. Cardiovasc Res. (2007) 73:729–38. doi: 10.1016/j.cardiores.2006.11.016
16. Zicha S, Fernandez-Velasco M, Lonardo G, L'Heureux N, Nattel S. Sinus node dysfunction and hyperpolarization-activated (HCN) channel subunit remodeling in a canine heart failure model. Cardiovasc Res. (2005) 66:472–81. doi: 10.1016/j.cardiores.2005.02.011
17. Boineau JP, Canavan TE, Schuessler RB, Cain ME, Corr PB, Cox JL. Demonstration of a widely distributed atrial pacemaker complex in the human heart. Circulation. (1988) 77:1221–37. doi: 10.1161/01.CIR.77.6.1221
18. Kirchhof CJ, Bonke FI, Allessie MA, Lammers WJ. The influence of the atrial myocardium on impulse formation in the rabbit sinus node. Pflugers Arch. (1987) 410:198–203. doi: 10.1007/BF00581916
19. Li N, Hansen BJ, Csepe TA, Zhao J, Ignozzi AJ, Sul LV, et al. Redundant and diverse intranodal pacemakers and conduction pathways protect the human sinoatrial node from failure. Sci Transl Med. (2017) 9:eaam5607. doi: 10.1126/scitranslmed.aam5607
20. Li N, Kalyanasundaram A, Hansen BJ, Artiga EJ, Sharma R, Abudulwahed SH, et al. Impaired neuronal sodium channels cause intranodal conduction failure and reentrant arrhythmias in human sinoatrial node. Nat Commun. (2020) 11:512. doi: 10.1038/s41467-019-14039-8
21. Schuessler RB. Abnormal sinus node function in clinical arrhythmias. J Cardiovasc Electrophysiol. (2003) 14:215–7. doi: 10.1046/j.1540-8167.2003.02229.x
22. Glukhov AV, Fedorov VV, Anderson ME, Mohler PJ, Efimov IR. Functional anatomy of the murine sinus node: high-resolution optical mapping of ankyrin-B heterozygous mice. Am J Physiol Heart Circ Physiol. (2010) 299:H482–91. doi: 10.1152/ajpheart.00756.2009
23. Glukhov AV, Hage LT, Hansen BJ, Pedraza-Toscano A, Vargas-Pinto P, Hamlin RL, et al. Sinoatrial node reentry in a canine chronic left ventricular infarct model: role of intranodal fibrosis and heterogeneity of refractoriness. Circ Arrhythm Electrophysiol. (2013) 6:984–94. doi: 10.1161/CIRCEP.113.000404
24. Shibata N, Inada S, Mitsui K, Honjo H, Yamamoto M, Niwa R, et al. Pacemaker shift in the rabbit sinoatrial node in response to vagal nerve stimulation. Exp Physiol. (2001) 86:177–84. doi: 10.1113/eph8602100
25. Brooks CM, Lu HH, Lange G, Mangi R, Shaw RB, Geoly K. Effects of localized stretch of the sinoatrial node region of the dog heart. Am J Physiol. (1966) 211:1197–202. doi: 10.1152/ajplegacy.1966.211.5.1197
26. Huang ZM, Prasad C, Britton FC, Ye LL, Hatton WJ, Duan D. Functional role of CLC-2 chloride inward rectifier channels in cardiac sinoatrial nodal pacemaker cells. J Mol Cell Cardiol. (2009) 47:121–32. doi: 10.1016/j.yjmcc.2009.04.008
27. Parmley WW, Chuck L. Length-dependent changes in myocardial contractile state. Am J Physiol. (1973) 224:1195–9. doi: 10.1152/ajplegacy.1973.224.5.1195
28. Lange G, Lu HH, Chang A, Brooks CM. Effect of stretch on the isolated cat sinoatrial node. Am J Physiol. (1966) 211:1192–6. doi: 10.1152/ajplegacy.1966.211.5.1192
29. Hill-Eubanks DC, Gonzales AL, Sonkusare SK, Nelson MT. Vascular TRP channels: performing under pressure and going with the flow. Physiology. (2014) 29:343–60. doi: 10.1152/physiol.00009.2014
30. Seol CA, Kim WT, Ha JM, Choe H, Jang YJ, Youm JB, et al. Stretch-activated currents in cardiomyocytes isolated from rabbit pulmonary veins. Prog Biophys Mol Biol. (2008) 97:217–31. doi: 10.1016/j.pbiomolbio.2008.02.008
31. Caldiz CI, Garciarena CD, Dulce RA, Novaretto LP, Yeves AM, Ennis IL, et al. Mitochondrial reactive oxygen species activate the slow force response to stretch in feline myocardium. J Physiol. (2007) 584:895–905. doi: 10.1113/jphysiol.2007.141689
32. Neves JS, Castro-Ferreira R, Ladeiras-Lopes R, Neiva-Sousa M, Leite-Moreira AM, Almeida-Coelho J, et al. The effects of angiotensin II signaling pathway in the systolic response to acute stretch in the normal and ischemic myocardium. Peptides. (2013) 47:77–84. doi: 10.1016/j.peptides.2013.07.004
33. Petroff MG, Kim SH, Pepe S, Dessy C, Marban E, Balligand JL, et al. Endogenous nitric oxide mechanisms mediate the stretch dependence of Ca2+ release in cardiomyocytes. Nat Cell Biol. (2001) 3:867–73. doi: 10.1038/ncb1001-867
34. Reil JC, Reil GH, Kovacs A, Sequeira V, Waddingham MT, Lodi M, et al. CaMKII activity contributes to homeometric autoregulation of the heart: a novel mechanism for the Anrep effect. J Physiol. (2020) 598:3129–53. doi: 10.1113/JP279607
35. Todaka K, Ogino K, Gu A, Burkhoff D. Effect of ventricular stretch on contractile strength, calcium transient, and cAMP in intact canine hearts. Am J Physiol. (1998) 274:H990–1000. doi: 10.1152/ajpheart.1998.274.3.H990
36. Lakatta EG, DiFrancesco D. What keeps us ticking: a funny current, a calcium clock, or both? J Mol Cell Cardiol. (2009) 47:157–70. doi: 10.1016/j.yjmcc.2009.03.022
37. Torrente AG, Zhang R, Zaini A, Giani JF, Kang J, Lamp ST, et al. Burst pacemaker activity of the sinoatrial node in sodium-calcium exchanger knockout mice. Proc Natl Acad Sci USA. (2015) 112:9769–74. doi: 10.1073/pnas.1505670112
38. Fenske S, Hennis K, Rotzer RD, Brox VF, Becirovic E, Scharr A, et al. cAMP-dependent regulation of HCN4 controls the tonic entrainment process in sinoatrial node pacemaker cells. Nat Commun. (2020) 11:5555. doi: 10.1038/s41467-020-19304-9
39. Kraichely RE, Strege PR, Sarr MG, Kendrick ML, Farrugia G. Lysophosphatidyl choline modulates mechanosensitive L-type Ca2+ current in circular smooth muscle cells from human jejunum. Am J Physiol Gastrointest Liver Physiol. (2009) 296:G833–9. doi: 10.1152/ajpgi.90610.2008
40. Laitko U, Morris CE. Membrane tension accelerates rate-limiting voltage-dependent activation and slow inactivation steps in a Shaker channel. J Gen Physiol. (2004) 123:135–54. doi: 10.1085/jgp.200308965
41. Irisawa H, Brown HF, Giles W. Cardiac pacemaking in the sinoatrial node. Physiol Rev. (1993) 73:197–227. doi: 10.1152/physrev.1993.73.1.197
42. DiFrancesco D. The role of the funny current in pacemaker activity. Circ Res. (2010) 106:434–46. doi: 10.1161/CIRCRESAHA.109.208041
43. Severi S, Fantini M, Charawi LA, DiFrancesco D. An updated computational model of rabbit sinoatrial action potential to investigate the mechanisms of heart rate modulation. J Physiol. (2012) 590:4483–99. doi: 10.1113/jphysiol.2012.229435
44. Mangoni ME, Nargeot J. Genesis and regulation of the heart automaticity. Physiol Rev. (2008) 88:919–82. doi: 10.1152/physrev.00018.2007
45. Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, et al. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci USA. (2003) 100:5543–8. doi: 10.1073/pnas.0935295100
46. Torrente AG, Mesirca P, Neco P, Rizzetto R, Dubel S, Barrere C, et al. L-type Cav1.3 channels regulate ryanodine receptor-dependent Ca2+ release during sino-atrial node pacemaker activity. Cardiovasc Res. (2016) 109:451–61. doi: 10.1093/cvr/cvw006
47. Toyoda F, Mesirca P, Dubel S, Ding WG, Striessnig J, Mangoni ME, et al. CaV1.3 L-type Ca(2+) channel contributes to the heartbeat by generating a dihydropyridine-sensitive persistent Na(+) current. Sci Rep. (2017) 7:7869. doi: 10.1038/s41598-017-08191-8
48. Monfredi O, Maltseva LA, Spurgeon HA, Boyett MR, Lakatta EG, Maltsev VA. Beat-to-beat variation in periodicity of local calcium releases contributes to intrinsic variations of spontaneous cycle length in isolated single sinoatrial node cells. PLoS ONE. (2013) 8:e67247. doi: 10.1371/journal.pone.0067247
49. Vinogradova TM, Bogdanov KY, Lakatta EG. beta-Adrenergic stimulation modulates ryanodine receptor Ca(2+) release during diastolic depolarization to accelerate pacemaker activity in rabbit sinoatrial nodal cells. Circ Res. (2002) 90:73–9. doi: 10.1161/hh0102.102271
50. Vinogradova TM, Lyashkov AE, Zhu W, Ruknudin AM, Sirenko S, Yang D, et al. High basal protein kinase A-dependent phosphorylation drives rhythmic internal Ca2+ store oscillations and spontaneous beating of cardiac pacemaker cells. Circ Res. (2006) 98:505–14. doi: 10.1161/01.RES.0000204575.94040.d1
51. Vinogradova TM, Zhou YY, Maltsev V, Lyashkov A, Stern M, Lakatta EG. Rhythmic ryanodine receptor Ca2+ releases during diastolic depolarization of sinoatrial pacemaker cells do not require membrane depolarization. Circ Res. (2004) 94:802–9. doi: 10.1161/01.RES.0000122045.55331.0F
52. Huser J, Blatter LA, Lipsius SL. Intracellular Ca2+ release contributes to automaticity in cat atrial pacemaker cells. J Physiol. (2000) 524:415–22. doi: 10.1111/j.1469-7793.2000.00415.x
53. Baudot M, Torre E, Bidaud I, Louradour J, Torrente AG, Fossier L, et al. Concomitant genetic ablation of L-type Cav1.3 (alpha1D) and T-type Cav3.1 (alpha1G) Ca(2+) channels disrupts heart automaticity. Sci Rep. (2020) 10:18906. doi: 10.1038/s41598-020-76049-7
54. Bagriantsev SN, Gracheva EO, Gallagher PG. Piezo proteins: regulators of mechanosensation and other cellular processes. J Biol Chem. (2014) 289:31673–81. doi: 10.1074/jbc.R114.612697
55. Duchemin AL, Vignes H, Vermot J. Mechanically activated piezo channels modulate outflow tract valve development through the Yap1 and Klf2-Notch signaling axis. Elife. (2019) 8:e44706. doi: 10.7554/eLife.44706
56. Cooper PJ, Kohl P. Species- and preparation-dependence of stretch effects on sino-atrial node pacemaking. Ann N Y Acad Sci. (2005) 1047:324–35. doi: 10.1196/annals.1341.029
57. Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science. (2010) 330:55–60. doi: 10.1126/science.1193270
58. Qiu Z, Dubin AE, Mathur J, Tu B, Reddy K, Miraglia LJ, et al. SWELL1, a plasma membrane protein, is an essential component of volume-regulated anion channel. Cell. (2014) 157:447–58. doi: 10.1016/j.cell.2014.03.024
59. Egorov YV, Lang D, Tyan L, Turner D, Lim E, Piro ZD, et al. Caveolae-mediated activation of mechanosensitive chloride channels in pulmonary veins triggers atrial arrhythmogenesis. J Am Heart Assoc. (2019) 8:e012748. doi: 10.1161/JAHA.119.012748
60. Marionneau C, Couette B, Liu J, Li H, Mangoni ME, Nargeot J, et al. Specific pattern of ionic channel gene expression associated with pacemaker activity in the mouse heart. J Physiol. (2005) 562(Pt 1):223–34. doi: 10.1113/jphysiol.2004.074047
61. Yanni J, Tellez JO, Maczewski M, Mackiewicz U, Beresewicz A., Billeter R., et al. Changes in ion channel gene expression underlying heart failure-induced sinoatrial node dysfunction. Circ. Heart Fail. (2011) 4:496–508. doi: 10.1161/CIRCHEARTFAILURE.110.957647
62. Xiong D, Heyman NS, Airey J, Zhang M, Singer CA, Rawat S, et al. Cardiac-specific, inducible ClC-3 gene deletion eliminates native volume-sensitive chloride channels and produces myocardial hypertrophy in adult mice. J Mol Cell Cardiol. (2010) 48:211–9. doi: 10.1016/j.yjmcc.2009.07.003
63. Ju YK, Chu Y, Chaulet H, Lai D, Gervasio OL, Graham RM, et al. Store-operated Ca2+ influx and expression of TRPC genes in mouse sinoatrial node. Circ Res. (2007) 100:1605–14. doi: 10.1161/CIRCRESAHA.107.152181
64. Demion M, Bois P, Launay P, Guinamard R. TRPM4, a Ca2+-activated nonselective cation channel in mouse sino-atrial node cells. Cardiovasc Res. (2007) 73:531–8. doi: 10.1016/j.cardiores.2006.11.023
65. Hof T, Simard C, Rouet R, Salle L, Guinamard R. Implication of the TRPM4 nonselective cation channel in mammalian sinus rhythm. Heart Rhythm. (2013) 10:1683–9. doi: 10.1016/j.hrthm.2013.08.014
66. Sah R, Mesirca P, Mason X, Gibson W, Bates-Withers C, Van den Boogert M, et al. Timing of myocardial trpm7 deletion during cardiogenesis variably disrupts adult ventricular function, conduction, and repolarization. Circulation. (2013) 128:101–14. doi: 10.1161/CIRCULATIONAHA.112.000768
67. Sah R, Mesirca P, Van den Boogert M, Rosen J, Mably J, Mangoni ME, et al. Ion channel-kinase TRPM7 is required for maintaining cardiac automaticity. Proc Natl Acad Sci USA. (2013) 110:E3037–46. doi: 10.1073/pnas.1311865110
68. Brennan JA, Chen Q, Gams A, Dyavanapalli J, Mendelowitz D, Peng W, et al. Evidence of superior and inferior sinoatrial nodes in the mammalian heart. JACC Clin Electrophysiol. (2020) 6:1827–40. doi: 10.1016/j.jacep.2020.09.012
69. Unudurthi SD, Wu X, Qian L, Amari F, Onal B, Li N, et al. Two-Pore K+ channel TREK-1 regulates sinoatrial node membrane excitability. J Am Heart Assoc. (2016) 5:e002865. doi: 10.1161/JAHA.115.002865
70. Lai MH, Wu Y, Gao Z, Anderson ME, Dalziel JE, Meredith AL. BK channels regulate sinoatrial node firing rate and cardiac pacing in vivo. Am J Physiol Heart Circ Physiol. (2014) 307:H1327–38. doi: 10.1152/ajpheart.00354.2014
71. Mao Z, Wang Y, Peng H, He F, Zhu L, Huang H, et al. A newly identified missense mutation in CLCA2 is associated with autosomal dominant cardiac conduction block. Gene. (2019) 714:143990. doi: 10.1016/j.gene.2019.143990
72. Beech DJ, Kalli AC. Force sensing by piezo channels in cardiovascular health and disease. Arterioscler Thromb Vasc Biol. (2019) 39:2228–39. doi: 10.1161/ATVBAHA.119.313348
73. Lewis AH, Cui AF, McDonald MF, Grandl J. Transduction of repetitive mechanical stimuli by piezo1 and piezo2 ion channels. Cell Rep. (2017) 19:2572–85. doi: 10.1016/j.celrep.2017.05.079
74. Douguet D, Patel A, Xu A, Vanhoutte PM, Honore E. Piezo ion channels in cardiovascular mechanobiology. Trends Pharmacol Sci. (2019) 40:956–70. doi: 10.1016/j.tips.2019.10.002
75. Miyamoto T, Mochizuki T, Nakagomi H, Kira S, Watanabe M, Takayama Y, et al. Functional role for Piezo1 in stretch-evoked Ca(2+) influx and ATP release in urothelial cell cultures. J Biol Chem. (2014) 289:16565–75. doi: 10.1074/jbc.M113.528638
76. O'Connell AD, Morton MJ, Hunter M. Two-pore domain K+ channels-molecular sensors. Biochim Biophys Acta. (2002) 1566:152–61. doi: 10.1016/S0005-2736(02)00597-7
77. Enyedi P, Czirjak G. Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev. (2010) 90:559–605. doi: 10.1152/physrev.00029.2009
78. Froese A, Breher SS, Waldeyer C, Schindler RF, Nikolaev VO, Rinne S, et al. Popeye domain containing proteins are essential for stress-mediated modulation of cardiac pacemaking in mice. J Clin Invest. (2012) 122:1119–30. doi: 10.1172/JCI59410
79. Tan JH, Liu W, Saint DA. Trek-like potassium channels in rat cardiac ventricular myocytes are activated by intracellular ATP. J Membr Biol. (2002) 185:201–7. doi: 10.1007/s00232-001-0123-0
80. Xian Tao L, Dyachenko V, Zuzarte M, Putzke C, Preisig-Muller R, Isenberg G, et al. The stretch-activated potassium channel TREK-1 in rat cardiac ventricular muscle. Cardiovasc Res. (2006) 69:86–97. doi: 10.1016/j.cardiores.2005.08.018
81. Decher N, Wemhoner K, Rinne S, Netter MF, Zuzarte M, Aller MI, et al. Knock-out of the potassium channel TASK-1 leads to a prolonged QT interval and a disturbed QRS complex. Cell Physiol Biochem. (2011) 28:77–86. doi: 10.1159/000331715
82. Limberg SH, Netter MF, Rolfes C, Rinne S, Schlichthorl G, Zuzarte M, et al. TASK-1 channels may modulate action potential duration of human atrial cardiomyocytes. Cell Physiol Biochem. (2011) 28:613–24. doi: 10.1159/000335757
83. Putzke C, Wemhoner K, Sachse FB, Rinne S, Schlichthorl G, Li XT, et al. The acid-sensitive potassium channel TASK-1 in rat cardiac muscle. Cardiovasc Res. (2007) 75:59–68. doi: 10.1016/j.cardiores.2007.02.025
84. Patel AJ, Honore E, Maingret F, Lesage F, Fink M, Duprat F, et al. A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J. (1998) 17:4283–90. doi: 10.1093/emboj/17.15.4283
85. Brohawn SG, Su Z, MacKinnon R. Mechanosensitivity is mediated directly by the lipid membrane in TRAAK and TREK1 K+ channels. Proc Natl Acad Sci USA. (2014) 111:3614–9. doi: 10.1073/pnas.1320768111
86. Kirchmaier BC, Poon KL, Schwerte T, Huisken J, Winkler C, Jungblut B, et al. The Popeye domain containing 2 (popdc2) gene in zebrafish is required for heart and skeletal muscle development. Dev Biol. (2012) 363:438–50. doi: 10.1016/j.ydbio.2012.01.015
87. Hund TJ, Snyder JS, Wu X, Glynn P, Koval OM, Onal B, et al. beta(IV)-Spectrin regulates TREK-1 membrane targeting in the heart. Cardiovasc Res. (2014) 102:166–75. doi: 10.1093/cvr/cvu008
88. Takahashi K, Naruse K. Stretch-activated BK channel and heart function. Prog Biophys Mol Biol. (2012) 110:239–44. doi: 10.1016/j.pbiomolbio.2012.08.001
89. Imlach WL, Finch SC, Miller JH, Meredith AL, Dalziel JE. A role for BK channels in heart rate regulation in rodents. PLoS ONE. (2010) 5:e8698. doi: 10.1371/journal.pone.0008698
90. Zhao H, Yu Y, Wu X, Liu S, Liu B, Du J, et al. A role of BK channel in regulation of Ca(2+) channel in ventricular myocytes by substrate stiffness. Biophys J. (2017) 112:1406–16. doi: 10.1016/j.bpj.2017.01.036
91. Inoue R, Jian Z, Kawarabayashi Y. Mechanosensitive TRP channels in cardiovascular pathophysiology. Pharmacol Ther. (2009) 123:371–85. doi: 10.1016/j.pharmthera.2009.05.009
92. Launay P, Fleig A, Perraud AL, Scharenberg AM, Penner R, Kinet JP. TRPM4 is a Ca2+-activated nonselective cation channel mediating cell membrane depolarization. Cell. (2002) 109:397–407. doi: 10.1016/S0092-8674(02)00719-5
93. Ramsey IS, Delling M, Clapham DE. An introduction to TRP channels. Annu Rev Physiol. (2006) 68:619–47. doi: 10.1146/annurev.physiol.68.040204.100431
94. Little SC, Mohler PJ. TRPM4 modulates sinus node diastolic depolarization. Heart Rhythm. (2013) 10:1690–1. doi: 10.1016/j.hrthm.2013.08.026
95. Li M, Jiang J, Yue L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J Gen Physiol. (2006) 127:525–37. doi: 10.1085/jgp.200609502
96. Runnels LW, Yue L, Clapham DE. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. (2001) 291:1043–7. doi: 10.1126/science.1058519
97. Gottlieb P, Folgering J, Maroto R, Raso A, Wood TG, Kurosky A, et al. Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflugers Arch. (2008) 455:1097–103. doi: 10.1007/s00424-007-0359-3
98. Nikolaev YA, Cox CD, Ridone P, Rohde PR, Cordero-Morales JF, Vasquez V, et al. Mammalian TRP ion channels are insensitive to membrane stretch. J Cell Sci. (2019) 132:jcs238360. doi: 10.1242/jcs.238360
99. Oancea E, Wolfe JT, Clapham DE. Functional TRPM7 channels accumulate at the plasma membrane in response to fluid flow. Circ Res. (2006) 98:245–53. doi: 10.1161/01.RES.0000200179.29375.cc
100. Kohl P, Hunter P, Noble D. Stretch-induced changes in heart rate and rhythm: clinical observations, experiments and mathematical models. Prog Biophys Mol Biol. (1999) 71:91–138. doi: 10.1016/S0079-6107(98)00038-8
101. Osei-Owusu J, Yang J, Vitery MDC, Qiu Z. Molecular biology and physiology of volume-regulated anion channel (VRAC). Curr Top Membr. (2018) 81:177–203. doi: 10.1016/bs.ctm.2018.07.005
102. Voss FK, Ullrich F, Munch J, Lazarow K, Lutter D, Mah N, et al. Identification of LRRC8 heteromers as an essential component of the volume-regulated anion channel VRAC. Science. (2014) 344:634–8. doi: 10.1126/science.1252826
103. Alghanem AF, Abello J, Maurer JM, Kumar A, Ta CM, Gunasekar SK, et al. The SWELL1-LRRC8 complex regulates endothelial AKT-eNOS signaling and vascular function. Elife. (2021) 10:e61313. doi: 10.7554/eLife.61313
104. Kang C, Xie L, Gunasekar SK, Mishra A, Zhang Y, Pai S, et al. SWELL1 is a glucose sensor regulating beta-cell excitability and systemic glycaemia. Nat Commun. (2018) 9:367. doi: 10.1038/s41467-017-02664-0
105. Kumar A, Xie L, Ta CM, Hinton AO, Gunasekar SK, Minerath RA, et al. SWELL1 regulates skeletal muscle cell size, intracellular signaling, adiposity and glucose metabolism. Elife. (2020) 9:e58941. doi: 10.7554/eLife.58941
106. Stuhlmann T, Planells-Cases R, Jentsch TJ. LRRC8/VRAC anion channels enhance beta-cell glucose sensing and insulin secretion. Nat Commun. (2018) 9:1974. doi: 10.1038/s41467-018-04353-y
107. Yang J, Vitery MDC, Chen J, Osei-Owusu J, Chu J, Qiu Z. Glutamate-Releasing SWELL1 channel in astrocytes modulates synaptic transmission and promotes brain damage in stroke. Neuron. (2019) 102:813–27.e816. doi: 10.1016/j.neuron.2019.03.029
108. Zhang Y, Xie L, Gunasekar SK, Tong D, Mishra A, Gibson WJ, et al. SWELL1 is a regulator of adipocyte size, insulin signalling and glucose homeostasis. Nat Cell Biol. (2017) 19:504–17. doi: 10.1038/ncb3514
109. Browe DM, Baumgarten CM. Stretch of beta 1 integrin activates an outwardly rectifying chloride current via FAK and Src in rabbit ventricular myocytes. J Gen Physiol. (2003) 122:689–702. doi: 10.1085/jgp.200308899
110. Brower KJ. Insomnia, alcoholism and relapse. Sleep Med Rev. (2003) 7:523–39. doi: 10.1016/S1087-0792(03)90005-0
111. Clemo HF, Stambler BS, Baumgarten CM. Swelling-activated chloride current is persistently activated in ventricular myocytes from dogs with tachycardia-induced congestive heart failure. Circ Res. (1999) 84:157–65. doi: 10.1161/01.RES.84.2.157
112. Patel DG, Higgins RS, Baumgarten CM. Swelling-activated Cl current, ICl,SWELL, is chronically activated in diseased human atrial myocytes. Biophys J. (2003) 84:233a.
113. Ren Z, Raucci FJ Jr, Browe DM, Baumgarten CM. Regulation of swelling-activated Cl(-) current by angiotensin II signalling and NADPH oxidase in rabbit ventricle. Cardiovasc Res. (2008) 77:73–80. doi: 10.1093/cvr/cvm031
114. Hagiwara N, Masuda H, Shoda M, Irisawa H. Stretch-activated anion currents of rabbit cardiac myocytes. J Physiol. (1992) 456:285–302. doi: 10.1113/jphysiol.1992.sp019337
115. Hagiwara N, Irisawa H, Kasanuki H, Hosoda S. Background current in sino-atrial node cells of the rabbit heart. J Physiol. (1992) 448:53–72. doi: 10.1113/jphysiol.1992.sp019029
116. Kopton RA, Baillie JS, Rafferty SA, Moss R, Zgierski-Johnston CM, Prykhozhij SV, et al. Cardiac electrophysiological effects of light-activated chloride channels. Front Physiol. (2018) 9:1806. doi: 10.3389/fphys.2018.01806
117. Decher N, Lang HJ, Nilius B, Bruggemann A, Busch AE, Steinmeyer K. DCPIB is a novel selective blocker of I(Cl,swell) and prevents swelling-induced shortening of guinea-pig atrial action potential duration. Br J Pharmacol. (2001) 134:1467–79. doi: 10.1038/sj.bjp.0704413
118. Arai A, Kodama I, Toyama J. Roles of Cl- channels and Ca2+ mobilization in stretch-induced increase of SA node pacemaker activity. Am J Physiol. (1996) 270:H1726–35. doi: 10.1152/ajpheart.1996.270.5.H1726
119. Wang D, Wang H, Gao F, Wang K, Dong F. ClC-3 promotes osteogenic differentiation in MC3T3-E1 cell after dynamic compression. J Cell Biochem. (2017) 118:1606–13. doi: 10.1002/jcb.25823
120. Liang W, Huang L, Zhao D, He JZ, Sharma P, Liu J, et al. Swelling-activated Cl- currents and intracellular CLC-3 are involved in proliferation of human pulmonary artery smooth muscle cells. J Hypertens. (2014) 32:318–30. doi: 10.1097/HJH.0000000000000013
121. Ye Z, Wu MM, Wang CY, Li YC, Yu CJ, Gong YF, et al. Characterization of cardiac anoctamin1 Ca(2)(+)-activated chloride channels and functional role in ischemia-induced arrhythmias. J Cell Physiol. (2015) 230:337–46. doi: 10.1002/jcp.24709
122. Sung TS, Hwang SJ, Koh SD, Bayguinov Y, Peri LE, Blair PJ, et al. The cells and conductance mediating cholinergic neurotransmission in the murine proximal stomach. J Physiol. (2018) 596:1549–74. doi: 10.1113/JP275478
123. Tanaka S, Fujio Y, Nakayama H. Caveolae-specific CaMKII signaling in the regulation of voltage-dependent calcium channel and cardiac hypertrophy. Front Physiol. (2018) 9:1081. doi: 10.3389/fphys.2018.01081
124. Barbuti A, Terragni B, Brioschi C, DiFrancesco D. Localization of f-channels to caveolae mediates specific beta2-adrenergic receptor modulation of rate in sinoatrial myocytes. J Mol Cell Cardiol. (2007) 42:71–78. doi: 10.1016/j.yjmcc.2006.09.018
125. Masson-Pevet M, Gros D, Besselsen E. The caveolae in rabbit sinus node and atrium. Cell Tissue Res. (1980) 208:183–96. doi: 10.1007/BF00234869
126. Echarri A, Del Pozo MA. Caveolae - mechanosensitive membrane invaginations linked to actin filaments. J Cell Sci. (2015) 128:2747–58. doi: 10.1242/jcs.153940
127. Gilbert G, Ducret T, Savineau JP, Marthan R, Quignard JF. Caveolae are involved in mechanotransduction during pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. (2016) 310:L1078–87. doi: 10.1152/ajplung.00198.2015
128. Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. (2007) 8:185–94. doi: 10.1038/nrm2122
129. Pfleger C, Ebeling G, Blasche R, Patton M, Patel HH, Kasper M, et al. Detection of caveolin-3/caveolin-1/P2X7R complexes in mice atrial cardiomyocytes in vivo and in vitro. Histochem Cell Biol. (2012) 138:231–41. doi: 10.1007/s00418-012-0961-0
130. Volonte D, McTiernan CF, Drab M, Kasper M, Galbiati F. Caveolin-1 and caveolin-3 form heterooligomeric complexes in atrial cardiac myocytes that are required for doxorubicin-induced apoptosis. Am J Physiol Heart Circ Physiol. (2008) 294:H392–401. doi: 10.1152/ajpheart.01039.2007
131. Barbuti A, Scavone A, Mazzocchi N, Terragni B, Baruscotti M, Difrancesco D. A caveolin-binding domain in the HCN4 channels mediates functional interaction with caveolin proteins. J Mol Cell Cardiol. (2012) 53:187–95. doi: 10.1016/j.yjmcc.2012.05.013
132. Shibata EF, Brown TL, Washburn ZW, Bai J, Revak TJ, Butters CA. Autonomic regulation of voltage-gated cardiac ion channels. J Cardiovasc Electrophysiol. (2006) 17 (Suppl. 1):S34–42. doi: 10.1111/j.1540-8167.2006.00387.x
133. Ichishima K, Yamamoto S, Iwamoto T, Ehara T. alpha-Adrenoceptor-mediated depletion of phosphatidylinositol 4, 5-bisphosphate inhibits activation of volume-regulated anion channels in mouse ventricular myocytes. Br J Pharmacol. (2010) 161:193–206. doi: 10.1111/j.1476-5381.2010.00896.x
134. Gunasekar SK, Xie L, Sah R. SWELL signalling in adipocytes: can fat ‘feel' fat? Adipocyte. (2019) 8:223–8. doi: 10.1080/21623945.2019.1612223
135. Kohl P, Sachs F, Franz MR. Cardiac Mechano-Electric Coupling and Arrhythmias. Oxford: OUP Oxford (2011).
136. Iribe G, Ward CW, Camelliti P, Bollensdorff C, Mason F, Burton RA, et al. Axial stretch of rat single ventricular cardiomyocytes causes an acute and transient increase in Ca2+ spark rate. Circ Res. (2009) 104:787–95. doi: 10.1161/CIRCRESAHA.108.193334
137. Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. (2011) 333:1440–5. doi: 10.1126/science.1202768
138. Iribe G, Kohl P. Axial stretch enhances sarcoplasmic reticulum Ca2+ leak and cellular Ca2+ reuptake in guinea pig ventricular myocytes: experiments and models. Prog Biophys Mol Biol. (2008) 97:298–311. doi: 10.1016/j.pbiomolbio.2008.02.012
139. Lyashkov AE, Juhaszova M, Dobrzynski H, Vinogradova TM, Maltsev VA, Juhasz O, et al. Calcium cycling protein density and functional importance to automaticity of isolated sinoatrial nodal cells are independent of cell size. Circ Res. (2007) 100:1723–31. doi: 10.1161/CIRCRESAHA.107.153676
140. Musa H, Lei M, Honjo H, Jones SA, Dobrzynski H, Lancaster MK, et al. Heterogeneous expression of Ca(2+) handling proteins in rabbit sinoatrial node. J Histochem Cytochem. (2002) 50:311–24. doi: 10.1177/002215540205000303
141. Stern MD, Maltseva LA, Juhaszova M, Sollott SJ, Lakatta EG, Maltsev VA. Hierarchical clustering of ryanodine receptors enables emergence of a calcium clock in sinoatrial node cells. J Gen Physiol. (2014) 143:577–604. doi: 10.1085/jgp.201311123
142. Prosser BL, Khairallah RJ, Ziman AP, Ward CW, Lederer WJ. X-ROS signaling in the heart and skeletal muscle: stretch-dependent local ROS regulates [Ca(2)(+)]i. J Mol Cell Cardiol. (2013) 58:172–81. doi: 10.1016/j.yjmcc.2012.11.011
143. Balog EM, Norton LE, Thomas DD, Fruen BR. Role of calmodulin methionine residues in mediating productive association with cardiac ryanodine receptors. Am J Physiol Heart Circ Physiol. (2006) 290:H794–9. doi: 10.1152/ajpheart.00706.2005
144. Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. (2008) 133:462–74. doi: 10.1016/j.cell.2008.02.048
145. Dries E, Bito V, Lenaerts I, Antoons G, Sipido KR, Macquaide N. Selective modulation of coupled ryanodine receptors during microdomain activation of calcium/calmodulin-dependent kinase II in the dyadic cleft. Circ Res. (2013) 113:1242–52. doi: 10.1161/CIRCRESAHA.113.301896
146. Wu Y, Anderson ME. CaMKII in sinoatrial node physiology and dysfunction. Front Pharmacol. (2014) 5:48. doi: 10.3389/fphar.2014.00048
147. Dyachenko V, Husse B, Rueckschloss U, Isenberg G. Mechanical deformation of ventricular myocytes modulates both TRPC6 and Kir2.3 channels. Cell Calcium. (2009) 45:38–54. doi: 10.1016/j.ceca.2008.06.003
148. Dyachenko V, Rueckschloss U, Isenberg G. Modulation of cardiac mechanosensitive ion channels involves superoxide, nitric oxide and peroxynitrite. Cell Calcium. (2009) 45:55–64. doi: 10.1016/j.ceca.2008.06.002
149. Gervasio OL, Whitehead NP, Yeung EW, Phillips WD, Allen DG. TRPC1 binds to caveolin-3 and is regulated by Src kinase - role in Duchenne muscular dystrophy. J Cell Sci. (2008) 121:2246–55. doi: 10.1242/jcs.032003
150. Noel J, Wang H, Hong N, Tao JQ, Yu K, Sorokina EM, et al. PECAM-1 and caveolae form the mechanosensing complex necessary for NOX2 activation and angiogenic signaling with stopped flow in pulmonary endothelium. Am J Physiol Lung Cell Mol Physiol. (2013) 305:L805–18. doi: 10.1152/ajplung.00123.2013
151. Gradogna A, Gavazzo P, Boccaccio A, Pusch M. Subunit-dependent oxidative stress sensitivity of LRRC8 volume-regulated anion channels. J Physiol. (2017) 595:6719–33. doi: 10.1113/JP274795
152. Jian Z, Han H, Zhang T, Puglisi J, Izu LT, Shaw JA, et al. Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Sci Signal. (2014) 7:ra27. doi: 10.1126/scisignal.2005046
153. Massion PB, Dessy C, Desjardins F, Pelat M, Havaux X, Belge C, et al. Cardiomyocyte-restricted overexpression of endothelial nitric oxide synthase (NOS3) attenuates beta-adrenergic stimulation and reinforces vagal inhibition of cardiac contraction. Circulation. (2004) 110:2666–72. doi: 10.1161/01.CIR.0000145608.80855.BC
154. Michel JB, Feron O, Sacks D, Michel T. Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+-calmodulin and caveolin. J Biol Chem. (1997) 272:15583–86. doi: 10.1074/jbc.272.25.15583
155. Vielma AZ, Leon L, Fernandez IC, Gonzalez DR, Boric MP. Nitric oxide synthase 1 modulates basal and beta-adrenergic-stimulated contractility by rapid and reversible redox-dependent s-nitrosylation of the heart. PLoS ONE. (2016) 11:e0160813. doi: 10.1371/journal.pone.0160813
156. Burkard N, Rokita AG, Kaufmann SG, Hallhuber M, Wu R, Hu K, et al. Conditional neuronal nitric oxide synthase overexpression impairs myocardial contractility. Circ Res. (2007) 100:e32–44. doi: 10.1161/01.RES.0000259042.04576.6a
157. Vila-Petroff MG, Younes A, Egan J, Lakatta EG, Sollott SJ. Activation of distinct cAMP-dependent and cGMP-dependent pathways by nitric oxide in cardiac myocytes. Circ Res. (1999) 84:1020–31. doi: 10.1161/01.RES.84.9.1020
158. Chowdhary S, Harrington D, Bonser RS, Coote JH, Townend JN. Chronotropic effects of nitric oxide in the denervated human heart. J Physiol. (2002) 541:645–51. doi: 10.1113/jphysiol.2001.015107
159. Musialek P. Nitric oxide stimulation of cardiac pacemaking in the sino-atrial node through the activation of a novel signalling pathway: overview of in vitro and in vivo evidence for a new basic mechanism in the control of heart rate. Przegl Lek. (2002) 59:691–4.
160. Zhang YH, Youm JB, Earm YE. Stretch-activated non-selective cation channel: a causal link between mechanical stretch and atrial natriuretic peptide secretion. Prog Biophys Mol Biol. (2008) 98:1–9. doi: 10.1016/j.pbiomolbio.2008.05.005
161. Oh YB, Gao S, Lim JM, Kim HT, Park BH, Kim SH. Caveolae are essential for angiotensin II type 1 receptor-mediated ANP secretion. Peptides. (2011) 32:1422–30. doi: 10.1016/j.peptides.2011.06.002
162. Thomas CJ, Allen AM, McAllen RM, Woods RL. ANP potentiates nonarterial baroreflex bradycardia: evidence from sinoaortic denervation in rats. Auton Neurosci. (2002) 97:89–98. doi: 10.1016/S1566-0702(02)00049-8
163. Lonardo G, Cerbai E, Casini S, Giunti G, Bonacchi M, Battaglia F, et al. Atrial natriuretic peptide modulates the hyperpolarization-activated current (If) in human atrial myocytes. Cardiovasc Res. (2004) 63:528–36. doi: 10.1016/j.cardiores.2004.03.004
164. Li Y, Sirenko S, Riordon DR, Yang D, Spurgeon H, Lakatta EG, et al. CaMKII-dependent phosphorylation regulates basal cardiac pacemaker function via modulation of local Ca2+ releases. Am J Physiol Heart Circ Physiol. (2016) 311:H532–44. doi: 10.1152/ajpheart.00765.2015
165. Rose RA, Kabir MG, Backx PH. Altered heart rate and sinoatrial node function in mice lacking the cAMP regulator phosphoinositide 3-kinase-gamma. Circ Res. (2007) 101:1274–82. doi: 10.1161/CIRCRESAHA.107.158428
166. Ju YK, Liu J, Lee BH, Lai D, Woodcock EA, Lei M, et al. Distribution and functional role of inositol 1,4,5-trisphosphate receptors in mouse sinoatrial node. Circ Res. (2011) 109:848–57. doi: 10.1161/CIRCRESAHA.111.243824
167. Kapoor N, Tran A, Kang J, Zhang R, Philipson KD, Goldhaber JI. Regulation of calcium clock-mediated pacemaking by inositol-1,4,5-trisphosphate receptors in mouse sinoatrial nodal cells. J Physiol. (2015) 593:2649–63. doi: 10.1113/JP270082
168. Mery A, Aimond F, Menard C, Mikoshiba K, Michalak M, Puceat M. Initiation of embryonic cardiac pacemaker activity by inositol 1,4,5-trisphosphate-dependent calcium signaling. Mol Biol Cell. (2005) 16:2414–23. doi: 10.1091/mbc.e04-10-0883
169. Levental I, Grzybek M, Simons K. Greasing their way: lipid modifications determine protein association with membrane rafts. Biochemistry. (2010) 49:6305–16. doi: 10.1021/bi100882y
170. Domeier TL, Zima AV, Maxwell JT, Huke S, Mignery GA, Blatter LA. IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol. (2008) 294:H596–604. doi: 10.1152/ajpheart.01155.2007
171. Dassouli A, Sulpice JC, Roux S, Crozatier B. Stretch-induced inositol trisphosphate and tetrakisphosphate production in rat cardiomyocytes. J Mol Cell Cardiol. (1993) 25:973–82. doi: 10.1006/jmcc.1993.1109
172. Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, et al. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev. (2018) 98:1627–738. doi: 10.1152/physrev.00038.2017
173. Mineo C, Shaul PW. Regulation of eNOS in caveolae. Adv Exp Med Biol. (2012) 729:51–62. doi: 10.1007/978-1-4614-1222-9_4
174. Zhang Y, Peng F, Gao B, Ingram AJ, Krepinsky JC. Mechanical strain-induced RhoA activation requires NADPH oxidase-mediated ROS generation in caveolae. Antioxid Redox Signal. (2010) 13:959–73. doi: 10.1089/ars.2009.2908
175. Simo-Cheyou ER, Youreva V, Srivastava AK. cAMP attenuates angiotensin-II-induced Egr-1 expression via PKA-dependent signaling pathway in vascular smooth muscle cells. Can J Physiol Pharmacol. (2017) 95:928–37. doi: 10.1139/cjpp-2017-0035
176. Forrester SJ, Elliott KJ, Kawai T, Obama T, Boyer MJ, Preston KJ, et al. Caveolin-1 deletion prevents hypertensive vascular remodeling induced by angiotensin II. Hypertension. (2017) 69:79–86. doi: 10.1161/HYPERTENSIONAHA.116.08278
177. Fridolfsson HN, Patel HH. Caveolin and caveolae in age associated cardiovascular disease. J Geriatr Cardiol. (2013) 10:66–74. doi: 10.3969/j.issn.1671-5411.2013.01.011
178. Sinha B, Koster D, Ruez R, Gonnord P, Bastiani M, Abankwa D, et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell. (2011) 144:402–13. doi: 10.1016/j.cell.2010.12.031
179. Peyronnet R, Nerbonne JM, Kohl P. Cardiac mechano-gated ion channels and arrhythmias. Circ Res. (2016) 118:311–29. doi: 10.1161/CIRCRESAHA.115.305043
180. Jensen PN, Gronroos NN, Chen LY, Folsom AR, deFilippi C, Heckbert SR, et al. Incidence of and risk factors for sick sinus syndrome in the general population. J Am Coll Cardiol. (2014) 64:531–8. doi: 10.1016/j.jacc.2014.03.056
181. Lou Q, Hansen BJ, Fedorenko O, Csepe TA, Kalyanasundaram A, Li N, et al. Upregulation of adenosine A1 receptors facilitates sinoatrial node dysfunction in chronic canine heart failure by exacerbating nodal conduction abnormalities revealed by novel dual-sided intramural optical mapping. Circulation. (2014) 130:315–24. doi: 10.1161/CIRCULATIONAHA.113.007086
182. Nakao S, Hirakawa A, Fukushima R, Kobayashi M, Machida N. The anatomical basis of bradycardia-tachycardia syndrome in elderly dogs with chronic degenerative valvular disease. J Comp Pathol. (2012) 146:175–82. doi: 10.1016/j.jcpa.2011.03.016
183. Sanders P, Kistler PM, Morton JB, Spence SJ, Kalman JM. Remodeling of sinus node function in patients with congestive heart failure: reduction in sinus node reserve. Circulation. (2004) 110:897–903. doi: 10.1161/01.CIR.0000139336.69955.AB
184. Mesirca P, Fedorov VV, Hund TJ, Torrente AG, Bidaud I, Mohler PJ, et al. Pharmacologic approach to sinoatrial node dysfunction. Annu Rev Pharmacol Toxicol. (2021) 61:757–78. doi: 10.1146/annurev-pharmtox-031120-115815
185. Bode F, Katchman A, Woosley RL, Franz MR. Gadolinium decreases stretch-induced vulnerability to atrial fibrillation. Circulation. (2000) 101:2200–5. doi: 10.1161/01.CIR.101.18.2200
186. Chang SL, Chen YC, Chen YJ, Wangcharoen W, Lee SH, Lin CI, et al. Mechanoelectrical feedback regulates the arrhythmogenic activity of pulmonary veins. Heart. (2007) 93:82–8. doi: 10.1136/hrt.2006.089359
187. Haissaguerre M, Jais P, Shah DC, Takahashi A, Hocini M, Quiniou G, et al. Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N Engl J Med. (1998) 339:659–66. doi: 10.1056/NEJM199809033391003
188. Tabatabaei N, Asirvatham SJ. Supravalvular arrhythmia: identifying and ablating the substrate. Circ Arrhythm Electrophysiol. (2009) 2:316–26. doi: 10.1161/CIRCEP.108.847962
189. Walters TE, Lee G, Spence S, Larobina M, Atkinson V, Antippa P, et al. Acute atrial stretch results in conduction slowing and complex signals at the pulmonary vein to left atrial junction: insights into the mechanism of pulmonary vein arrhythmogenesis. Circ Arrhythm Electrophysiol. (2014) 7:1189–97. doi: 10.1161/CIRCEP.114.001894
190. de Jonge HW, Dekkers DH, Tilly BC, Lamers JM. Cyclic stretch and endothelin-1 mediated activation of chloride channels in cultured neonatal rat ventricular myocytes. Clin Sci. (2002) 103 (Suppl. 48):148S−51S. doi: 10.1042/CS103S148S
191. Yamamoto S, Kita S, Iyoda T, Yamada T, Iwamoto T. New molecular mechanisms for cardiovascular disease: cardiac hypertrophy and cell-volume regulation. J Pharmacol Sci. (2011) 116:343–9. doi: 10.1254/jphs.10R31FM
192. van Borren MM, Verkerk AO, Vanharanta SK, Baartscheer A, Coronel R, Ravesloot JH. Reduced swelling-activated Cl(-) current densities in hypertrophied ventricular myocytes of rabbits with heart failure. Cardiovasc Res. (2002) 53:869–78. doi: 10.1016/S0008-6363(01)00507-7
193. Yamamoto S, Kita S, Iyoda T, Yamada T, Ehara T, Iwamoto T. Caveolin-3 modulates the activity of the volume-regulated anion channel in mouse ventricular cells. Biophys J. (2011) 100:S170. doi: 10.1016/j.bpj.2010.12.1668
194. Piro ZD, Lodin R, Tyan L, Lim E, Lang D, Glukhov A. Region-specific stretch-induced disruption of caveolae decreases expression of mechanosensitive chloride channels and stimulates fibrogenesis promoting arrhythmogenic atrial ectopy in failing mice. Biophys J. (2019) 116:375a. doi: 10.1016/j.bpj.2018.11.2040
Keywords: automaticity, ion channel, cardiac, stretch activated, calcium, heart rate
Citation: Turner D, Kang C, Mesirca P, Hong J, Mangoni ME, Glukhov AV and Sah R (2021) Electrophysiological and Molecular Mechanisms of Sinoatrial Node Mechanosensitivity. Front. Cardiovasc. Med. 8:662410. doi: 10.3389/fcvm.2021.662410
Received: 01 February 2021; Accepted: 24 June 2021;
Published: 09 August 2021.
Edited by:
Futoshi Toyoda, Shiga University of Medical Science, JapanReviewed by:
Eilidh A. MacDonald, University of Glasgow, United KingdomT. Alexander Quinn, Dalhousie University, Canada
Copyright © 2021 Turner, Kang, Mesirca, Hong, Mangoni, Glukhov and Sah. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajan Sah, cmFqYW4uc2FoQHd1c3RsLmVkdQ==; Alexey V. Glukhov, YWdsdWtob3ZAbWVkaWNpbmUud2lzYy5lZHU=