
Autism and autism spectrum disorders (ASDs) are thought to be caused mostly by genetic factors with an additional contribution from unidentified environmental risk factors. Identifying these environmental risk factors could improve both prevention and development of therapeutics, but the nature of these factors is not well understood.
Clues to the identification of one environmental risk factor come from studies of a specific set of genetic diseases in which the frequency of autism or ASD is significantly increased. These diseases include tuberous sclerosis (40–50% of afflicted individuals have autism), type 1 Neurofibromatosis (a several-fold increase in frequency of autism in afflicted individuals), and autism with macrocephaly (Mansheim, 1979; Gillberg and Forsell, 1984; Goffin et al., 2001). As has been noted previously by several others (for example, see Kelleher and Bear, 2008), the genes affected in these disorders, Tsc1/Tsc2, Nf1, and PTEN, respectively, each encode inhibitors of a specific signaling pathway called the PI3K/Tor pathway (Hay and Sonenberg, 2004). Nf1 encodes a Ras GTPase activator protein, which reduces the ability of Ras to activate the lipid kinase PI3K (Xu et al., 1990; Rodriguez-Viciana et al., 1994). PTEN encodes a lipid phosphatase that opposes the effects of PI3K. Tsc1/Tsc2 encode pathway inhibitors that are negatively regulated by PI3K activity (Manning et al., 2002). One effect of PI3K/Tor activity is increased mRNA translation, which is mediated by activation of the translation initiation activator eIF-4E. This observation is of interest because a fourth disease gene that increases the frequency of autism, Fragile X, encodes a translational inhibitor that might oppose the effects of eIF-4E (Brown et al., 1982; see Figure 1). Thus, mutations in any of these four genes both increase mRNA translation and increase the incidence of autism.
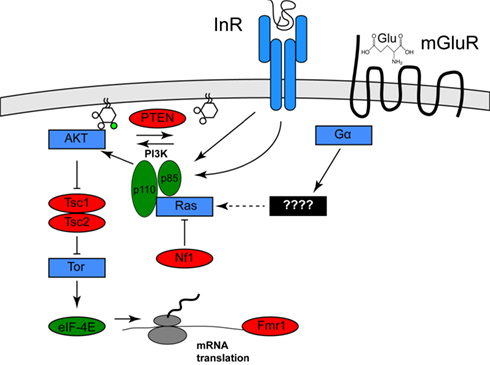
Figure 1. Activation of the Pi3K/Tor pathway by insulin and glutamate. The relationships among the genes described in the text are shown. Proteins shown in red are PI3K/Tor pathway inhibitors, and autism is associated with loss of function mutation in these genes, whereas the proteins shown in green are PI3K/Tor pathway activators and autism is associated with gain of function alterations in these genes. Proteins shown in blue represent other pathway activators. For clarity, several molecular intermediates are omitted. See Hay and Sonenberg (2004) for a more complete molecular description of this pathway.
Other evidence supports the idea that hyperactivation of the PI3K/Tor pathway causes autism and ASD. Recently, Neves-Pereira et al. (2009) suggested that translocations or point mutations that hyperactivate eIF-4E are causal for autism. In addition, studies of copy-number variants associated with autism revealed that hyperactivated PI3K variants were found in numbers far in excess of those expected by chance (Serajee et al., 2003; Cuscó et al., 2009).
In neurons, the PI3K/Tor signaling pathway affects a form of synaptic plasticity that has been implicated in autism (Bear et al., 2004). This form of synaptic plasticity is called long term depression (LTD) mediated by metabotropic glutamate receptors (mGluR), which are G protein coupled receptors for which glutamate is ligand. The best characterized example of mGluR-LTD occurs at the synapse between the Schaffer collaterals and CA1 pyramidal cells of the hippocampus. At this synapse, mGluR-LTD induction requires dendritic protein synthesis (Huber et al., 2001), activation of both PI3K and Tor (Hou and Klann, 2004), and ultimately internalization of AMPA-type glutamate receptors (Snyder et al., 2001). In addition, loss of Fmr1, the protein affected in Fragile X, increases the magnitude of mGluR-LTD (Huber et al., 2002; Waung and Huber, 2009) suggesting that Fmr1 normally functions to inhibit LTD by inhibiting translation of specific dendritic messages. These observations and others have led to the proposal that at least some of the deficits in autism might reflect increased sensitivity to mGluR-LTD induction (Bear et al., 2004; Kelleher and Bear, 2008) as a consequence of hyperactivation of the PI3K/Tor pathway.
The PI3K/Tor pathway is the major intracellular effector of insulin signals, and insulin signaling is predicted to activate the PI3K/Tor pathway in a manner similar to the genetic changes described above (Scott et al., 1998). Insulin can cross the blood–brain barrier (Schwartz and Porte, 2005) and insulin receptors are present and can regulate synaptic activity in relevant portions of the brain, including the hippocampus, cerebellum, and prefrontal cortex (Zhao et al., 2004, 2006; Dou et al., 2005). Furthermore, Huang et al. (2004) reported that insulin application evokes a LTD of synaptic activity (insulin-LTD) in the hippocampal CA1 region that is very similar mechanistically to mGluR-LTD: insulin-LTD, like mGluR-LTD, requires PI3K and Tor activity, dendritic protein synthesis, and removal of AMPA receptors from the cell surface. Thus, it appears that insulin is capable of activating the precise cellular pathway implicated in autism. For these reasons I hypothesize that insulin signaling contributes to development of autism in genetically susceptible individuals by contributing to PI3K/Tor pathway activation in neurons.
In further support of this hypothesis, Gardener et al. (2009) recently reported that in a large meta-analysis of maternal factors linked to autism, gestational diabetes was associated with the greatest increase (twofold) in incidence of autism. Although insulin does not cross the placenta, the elevation of fetal blood glucose levels as a consequence of maternal diabetes is predicted to increase fetal insulin secretion, and thus hyperactivate the fetal PI3K/Tor pathway. Another strong risk factor was advanced parental age, which may be a consequence of an age-dependent impairment of glucose tolerance short of diabetes. Furthermore, preliminary studies indicated that administration of a “ketogenic diet,” in which calories from carbohydrates are replaced with calories from fat, was therapeutic in individuals with autism and ASD (Evangeliou et al., 2003). This effect might result from the strong suppression of insulin secretion that accompanies this diet (Volek et al., 2008), which is predicted to attenuate activation of the PI3K/Tor pathway.
The incidence of gestational diabetes has recently increased (Ferrara et al., 2004), an increase that might be associated with the increased incidence in impaired glucose tolerance and hyperinsulinemia in the general population. Although the reported incidence of autism and ASD has also increased, it is not yet clear if this represents a true increase or rather a consequence of greater awareness and broader diagnostic criteria. However, if a true increase in the incidence of autism is occurring, then this increase could be a consequence of the increased incidence in hyperinsulinemia in the general population.
Thus this hypothesis provides a mechanism for previously unexplained observations concerning the occurrence and treatment of autism as well as for any true increase in the incidence of autism and ASD that might be occurring. This hypothesis also raises new possibilities for prevention and therapeutic intervention for autism.
Acknowledgments
I am grateful to Frank Masiarz and James McNew for comments on the manuscript, and James McNew for assistance with preparation of Figure 1.
References
Bear, M. F., Huber, K. M., and Warren, S. T. (2004). The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377.
Brown, W. T., Friedman, E., Jenkins, E. C., Brooks, J., Wisniewski, K., Raguthu, S., and French, J. H. (1982). Association of fragile X syndrome with autism. Lancet 319, 100.
Cuscó, I., Medrano, A., Gener, B., Vilardell, M., Gallastegui, F., Villa, O., González, E., Rodríguez-Santiago, B., Vilella, E., Del Campo, M., and Pérez-Jurado, L. A. (2009). Autism-specific copy number variants further implicate the phosphatidylinositol signaling pathway and the glutamatergic synapse in the etiology of the disorder. Hum. Mol. Genet. 18, 1795–1804.
Dou, J.-T., Chen, M., Dufour, F., Alkon, D. L., and Zhao, W.-Q. (2005). Insulin receptor signaling in long-term memory consolidation following spatial learning. Learn. Mem. 12, 646–655.
Evangeliou, A., Vlachonikolis, I., Mihailidou, H., Spilioti, M., Skarpalezou, A., Makaronas, N., Prokopiou, A., Christodoulou, P., Liapi-Adamidou, G., Helidonis, E., Sbyrakis, S., and Smeitink, J. (2003). Application of a ketogenic diet in children with autistic behavior: pilot study. J. Child Neurol. 18, 113–118.
Ferrara, A., Kahn, H. S., Quesenberry, C. P., Riley, C., and Hedderson, M. M. (2004). An increase in the incidence of gestational diabetes mellitus: Northern California, 1991-2000. Obstet. Gynecol. 103, 526–533.
Gardener, H., Spiegelman, D., and Buka, S. L. (2009). Prenatal risk factors for autism: comprehensive meta-analysis. Br. J. Psychiatry 195, 7–14.
Gillberg, C., and Forsell, C. (1984). Childhood psychosis and neurofibromatosis – more than a coincidence?. J. Autism Dev. Disord. 14, 1–8.
Goffin, A., Hoefsloot, L. H., Bosgoed, E., Swillen, A., and Fryns, J. P. (2001). PTEN mutation in a family with Cowden syndrome and autism. Am. J. Med. Genet. 105, 521–524.
Hou, L., and Klann, E. (2004). Activation of the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway is required for metabotropic glutamate receptor-dependent long-term depression. J. Neurosci. 24, 6352–6361.
Huang, C.-C., Lee, C.-C., and Hsu, K.-S. (2004). An investigation into signal transduction mechanisms involved in insulin-induced long-term depression in the CA1 region of the hippocampus. J. Neurochem. 89, 217–231.
Huber, K. M., Gallagher, S. M., Warren, S. T., and Bear, M. F. (2002). Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc. Natl. Acad. Sci. U.S.A. 99, 7746–7750.
Huber, K. M., Roder, J. C., and Bear, M. F. (2001). Chemical induction of mGluR5- and protein synthesis – dependent long-term depression in hippocampal area CA1. J. Neurophysiol. 86, 321–325.
Kelleher, R. J. III, and Bear, M. F. (2008). The autistic neuron: troubled translation? Cell 135, 401–406.
Manning, B. D., Tee, A. R., Logsdon, M. N., Blenis, J., and Cantley, L. C. (2002). Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 10, 151–162.
Neves-Pereira, M., Müller, B., Massie, D., Williams, J. H., O’Brien, P. C., Hughes, A., Shen, S. B., Clair, D. S., and Miedzybrodzka, Z. (2009). Deregulation of EIF4E: a novel mechanism for autism. J. Med. Genet. 46, 759–765.
Rodriguez-Viciana, P., Warne, P. H., Dhand, R., Vanhaesebroeck, B., Gout, I., Fry, M. J., Waterfield, M. D., and Downward, J. (1994). Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 370, 527–532.
Scott, P. H., Brunn, G. J., Kohn, A. D., Roth, R. A., and Lawrence, J. C. Jr. (1998). Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 95, 7772–7777.
Serajee, F. J., Nabi, R., Zhong, H., and Mahbubul Huq, A. H. (2003). Association of INPP1, PIK3CG, and TSC2 gene variants with autistic disorder: implications for phosphatidylinositol signalling in autism. J. Med. Genet. 40, e119.
Snyder, E. M., Philpot, B. D., Huber, K. M., Dong, X., Fallon, J. R., and Bear, M. F. (2001). Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat. Neurosci. 4, 1079–1085.
Volek, J. S., Fernandez, M. L., Feinman, R. D., and Phinney, S. D. (2008). Dietary carbohydrate restriction induces a unique metabolic state positively affecting atherogenic dyslipidemia, fatty acid partitioning, and metabolic syndrome. Prog. Lipid Res. 47, 307–318.
Waung, M. W., and Huber, K. M. (2009). Protein translation in synaptic plasticity: mGluR-LTD, Fragile X. Curr. Opin. Neurobiol. 19, 319–326.
Xu, G. F., Lin, B., Tanaka, K., Dunn, D., Wood, D., Gesteland, R., White, R., Weiss, R., and Tamanoi, F. (1990). The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 63, 835–841.
Zhao, W.-Q., Chen, H., Quon, M. J., and Alkon, D. L. (2004). Insulin and the insulin receptor in experimental models of learning and memory. Eur. J. Pharmacol. 490, 71–81.
Citation: Stern M (2011) Insulin signaling and autism. Front. Endocrin. 2:54. doi: 10.3389/fendo.2011.00054
Received: 18 August 2011;
Accepted: 29 September 2011;
Published online: 14 October 2011.
Copyright: © 2011 Stern. This is an open-access article subject to a non-exclusive license between the authors and Frontiers Media SA, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and other Frontiers conditions are complied with.
*Correspondence:c3Rlcm5AcmljZS5lZHU=