Abstract
The proliferative pool and regenerative potential of neural stem cells diminishes with age, a phenomenon that may be exacerbated in prodromal and mild Alzheimer’s disease (AD) brains. In parallel, the neuroactive progesterone metabolite, allopregnanolone (APα), along with a host of other factors, is decreased in the AD brain. Results of preclinical analyses demonstrate that APα is a potent inducer of neural progenitor proliferation of both rodent and human derived neural progenitor cells in vitro. In vivo, APα significantly increased neurogenesis within the subgranular zone of the dentate gyrus and subventricular zone of the 3xTgAD mouse model. Functionally, APα reversed the learning and memory deficits of 3xTgAD mice prior to and following the onset of AD pathology and was comparably efficacious in aged normal mice. In addition to inducing regenerative responses in mouse models of AD, APα significantly reduced beta-amyloid burden, beta-amyloid binding alcohol dehydrogenase load, and microglial activation. In parallel, APα increased markers of white matter generation and cholesterol homeostasis. Analyses to determine the optimal treatment regimen in the 3xTgAD mouse brain indicated that a treatment regimen of APα once per week was optimal for both inducing neurogenesis and reducing AD pathology. Pharmacokinetic analyses indicated that APα is rapidly increased in both plasma and brain following a single dose. APα is most efficacious when administered once per week which will contribute to its margin of safety. Further, analyses in both animals and humans have provided parameters for safe APα dosage exposure in humans. From a translational perspective, APα is a small molecular weight, blood brain barrier penetrant molecule with substantial preclinical efficacy data as a potential Alzheimer’s therapeutic with existing safety data in animals and humans. To our knowledge, APα is the only small molecule that both promotes neural progenitor regeneration in brain and simultaneously reduces AD pathology burden.
Introduction
Dynamic neural stem-cell proliferation zones apparent in the developing central nervous system are also present in the adult brain, primarily restricted to the dentate gyrus subgranular zone (SGZ; Altman and Das, 1965; Cameron et al., 1993; reviewed in Liu and Brinton, 2010) of the hippocampus and the subventricular zone (SVZ) of the lateral ventricle (Altman, 1969; Luskin, 1993; reviewed in Liu and Brinton, 2010). These regions retain regenerative potential throughout the life span with marked declines in aged brain (Kuhn et al., 1996; Cameron and McKay, 1999; reviewed in Liu and Brinton, 2010). Adult mammalian neurogenesis has been confirmed in humans (Eriksson et al., 1998). Quantitatively a remarkable degree of neurogenesis occurs in the rat dentate gyrus on a daily basis with 9,400 cells proliferating with a cell cycle time of 25 h (Cameron and McKay, 2001). By this estimate, ∼9,000 new cells would be generated in the dentate gyrus each day or more than 250,000 per month. Within 1–2 weeks of 5-bromo-2′-deoxyuridine (BrdU) injection, 50–60% of all BrdU-labeled (BrdU+) cells in the dentate gyrus express neuron-specific markers. The number of new granule neurons generated each month is 6% of the total size of the granule cell population and 30–60% of the size of the afferent and efferent populations. Although most newborn granule cells do not survive, 3% of these cells are replaced every month which corresponds to ∼0.1% per day in young adult mice and rats (Kempermann et al., 1997; Cameron and McKay, 1999; Liu and Brinton, 2010). In the rat dentate gyrus, 85% of the granule cells are formed postnatally (Bayer and Altman, 1974). Immature granule cells reach their highest level within the second postnatal week, while mature granule cells reach asymptotic levels at 2 months of age (Bayer and Altman, 1974). Proliferation of granule cell precursors is age-associated and proliferation of these cells declines with increased age (Kuhn et al., 1996). A decline in granule cell proliferation correlated with high levels of corticosteroids that decreased neurogenic potential (Cameron and McKay, 2001). The rat SVZ is estimated to produce 30,000 neuroblasts per day (Alvarez-Buylla et al., 2000). Together with the 9,000 new granule cells, roughly 40,000 new cells are available to regenerate the adult rat brain every day. Relative to juvenile rats, year-old rats displayed a 94% reduction in neurogenesis due to a 92% drop in cell production (McDonald and Wojtowicz, 2005). Aging alone does not overtly weaken the intrinsic neuronal properties of newborn granule cells. In the aged rodent, the relatively few new neurons that survive in the dentate gyrus develop typical granule cell morphology and spine density (Morgenstern et al., 2008). Surviving neurons possess a synaptic density of afferent glutamatergic connections comparable to that of neurons born in young adults (Morgenstern et al., 2008). Decreased neurogenesis has been proposed as a factor in the age-related decline of cognitive ability (Bizon and Gallagher, 2003; Drapeau et al., 2003; Kempermann et al., 2004).
Newborn granule cells arise from the hippocampal SGZ within the dentate gyrus and functionally integrate into the preexisting neural circuitry (van Praag et al., 2002) to influence hippocampal-dependent processes including spatial pattern recognition (Clelland et al., 2009). Experimental methods that influence neurogenesis include exercise (van Praag et al., 1999, 2002; Pereira et al., 2007; Rodriguez et al., 2011), environmental enrichment (Thuret et al., 2009; Rodriguez et al., 2011), deep brain stimulation (Toda et al., 2008; Encinas et al., 2011; Stone et al., 2011), stem-cell transplantation (Blurton-Jones et al., 2009), and pharmacologic intervention by peptide growth factor delivery (Massa et al., 2010) or small molecules (Malberg et al., 2000; Mayo et al., 2005; Wang et al., 2005; Taupin, 2009; Pieper et al., 2010). Glutamatergic and cholinergic synaptic input from the surrounding hippocampal circuitry penetrate the SGZ of the adult dentate gyrus. However, the local microenvironment of the adult dentate gyrus SGZ retains an embryonic-like state wherein nearly exclusive gamma-aminobutyric acid type A receptor (GABAAR) chloride channel depolarizing inputs surround progenitors in the neurogenic niche (Tozuka et al., 2005). While GABA is typically an inhibitory neurotransmitter, GABA is excitatory to neural progenitor cells and triggers an efflux of chloride to modulate a cascade of molecular events that regulate cell proliferation (Figure 1). Adult-generated granule cells initially receive GABAergic input from local interneurons, isolated from extrinsic excitatory input (Overstreet Wadiche et al., 2005). Adult-generated granule cells mature at a slow rate in comparison to the perinatal granule cells which are exposed to the hyperproliferative environment required for brain development. Nevertheless, GABAAR excitation initiates a cascade of events leading to calcium influx in adult neuroprogenitor cells subsequently inducing the accumulation of a neurogenic transcription factor, NeuroD (Tozuka et al., 2005). Work by Hisatsune’s group found significant inward currents in Type-2 cells but not Type-1 cells following GABA exposure (Tozuka et al., 2005). In response to depolarization events, transcription factor-activated processes promote mitosis, and subsequent granule cell maturation (Overstreet-Wadiche et al., 2006) that occurs in the weeks following cell division.
Figure 1

Allopregnanolone (APα) mechanism of action promotes neurogenesis. (1) APα activates the GABA type A receptor to initiate the efflux of chloride ions (Cl−) from neural progenitor and neural stem cells. (2) Extrusion of Cl− from the intracellular compartment leads to membrane depolarization and activation of the voltage dependent L-type calcium channel. (3) The intracellular calcium (Ca++) rise activates Ca++dependent kinases that ultimately lead to regulation of gene expression and protein synthesis of cell cycle proteins. (4) Involving the transcription factor cyclic AMP response element binding protein (CREB) signaling pathway, APα up-regulates the expression of cell cycle genes that promote neural progenitor mitosis while simultaneously down regulating genes that repress cell division. (5) The mechanism of APα-induced neurogenesis takes advantage of the developmentally regulated direction of Cl− flux to induce neurogenesis in those cells that are phenotypically competent to divide while not activating those mechanisms in mature neurons (Wang et al., 2005; Brinton and Wang, 2006). Adult hippocampal neural progenitor cells (BrdU+ cells immunolabeled green; NeuN+ cells are colored red; coronal section of mouse hippocampal dentate gyrus; scale bar = 50 μm) in the image above are shown as an illustrative example of neurogenesis within the mouse dentate gyrus subgranular zone (SGZ). Newly born granule cells proliferate and develop into neurons and glia along the border between the hilus and the granule cell layer. Migration (through the vertical space of the granule cell layer (GCL); arrows indicate sequence of temporal development) and integration of these cells occurs within the days and weeks following proliferation. Newly born neurons that survive will continue to mature and send axonal projections to form mossy fiber synapses in the CA3 subfield and dendrites to extend into the molecular cell layer (MCL) to receive glutamatergic afferents from the perforant pathway of the entorhinal cortex.
Neurosteroids bind to GABAAR at sites that differ from GABA, benzodiazepines, ethanol, and barbiturate binding sites and can act as positive or negative modulators of GABAAR function (Gee et al., 1987, 1988; reviewed in Liu and Brinton, 2010). Allopregnanolone (APα; 3α-hydroxy-5α-pregnan-20-one; also known as AP, Allo, or THP) is a potent positive allosteric activator of the GABAAR channels which at nanomolar concentrations enhances the action of GABA at GABAAR and at higher concentrations directly activates GABAAR. APα binds to two transmembrane sites of the heteropentameric GABAAR (Hosie et al., 2006). GABA and neurosteroid transmembrane binding sites each occur twice per channel complex. The binding sites are conserved throughout all GABAAR subtypes with the general subunit stoichiometry 2α:2β:1γ with the γ subunit replaced by the neurosteroid-sensitive δ subunit in some extrasynaptic channel complexes. The αβγ and αβδ receptor composites are pharmacologically distinct. GABA plays a key role in generation of the spontaneous network activity of immature dentate granule cells (Owens and Kriegstein, 2002; Sipila et al., 2004). In neuroprogenitor cells, GABAergic depolarization of the uniquely reversed membrane potential underlies the trophic actions of APα (Figure 1). Adult dentate granule cells, devoid of the α1 subunit, are also subjected to tonic GABAergic signaling via δ subunit containing GABAARs mediated by surrounding synaptic boutons of local interneurons (Overstreet Wadiche et al., 2005). GABAARs have been observed by electron microscopy at membrane sites on BrdU co-labeled adult hippocampal SGZ progenitor cells (Mayo et al., 2005). APα-potentiated GABAergic stimulation of neural progenitor cells elicits an efflux of chloride and a concomitant influx of calcium that contributes to the induction of cell signaling events leading to gene transcription of mitotic genes and downregulation of anti-mitotic genes (Figure 1). GABAergic signaling in the SVZ also controls proliferation of adult progenitor cells within the neurogenic niche (Liu et al., 2005). The survival and maturation potential of newborn neural cells as evidenced through enhanced learning and memory (Figure 2; Wang et al., 2010; Singh et al., 2011) is achieved through modulation of GABAergic signaling with optimal pulses of APα (Figure 3; Chen et al., 2011).
Figure 2
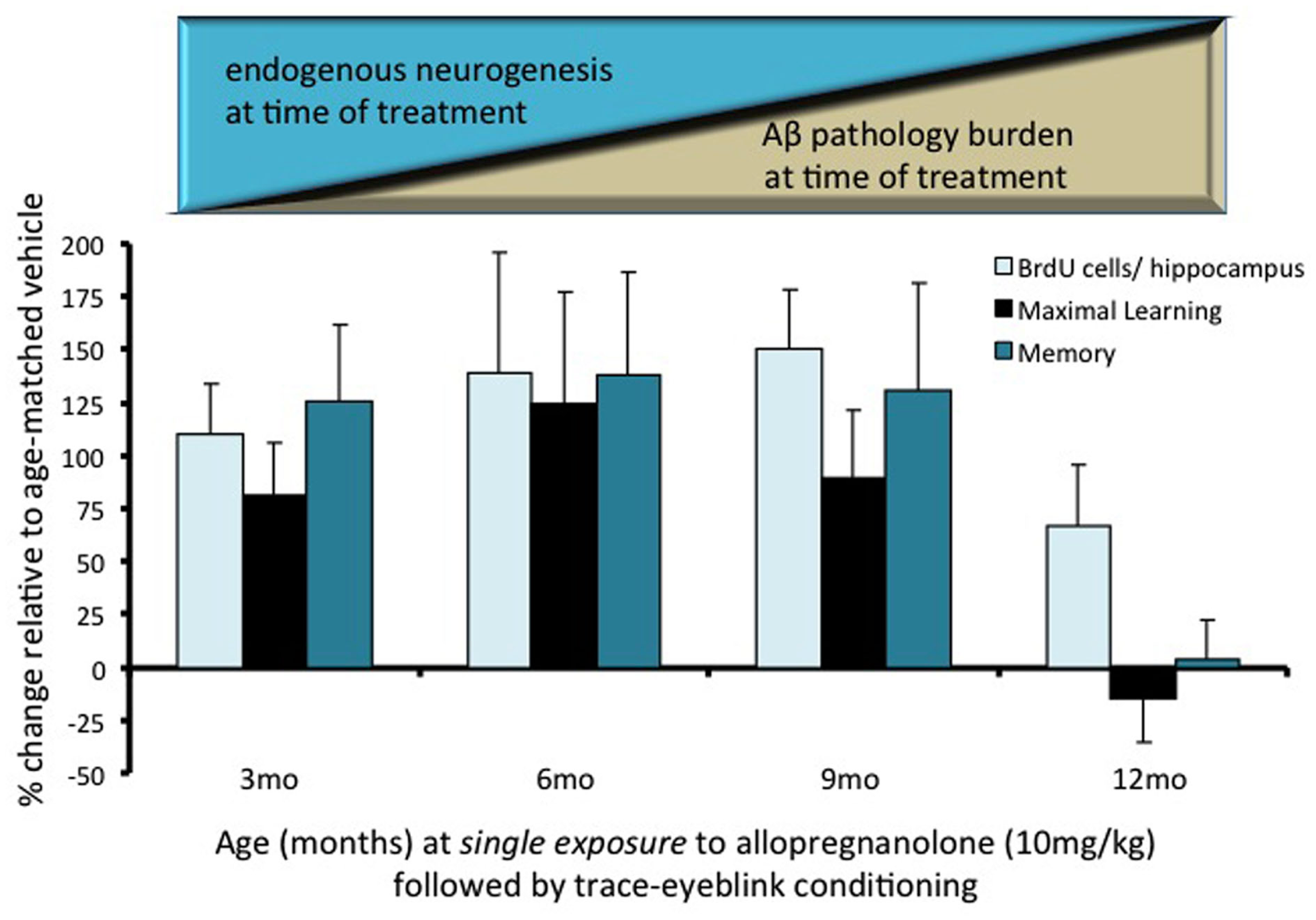
Cognitive efficacy of allopregnanolone (APα) prior to extraneuronal beta-amyloid plaque. Triple-transgenic Alzheimer’s disease (3xTgAD) mice were exposed to a single dose of APα. Data were plotted as percent change relative to age-matched vehicle control to assess the age-related differences in response to APα administered at 3-, 6-, 9-, or 12-months of age. Mice were treated with either APα (subcutaneous, 10 mg/kg) or vehicle and 1 h later with bromodeoxyuridine (BrdU) (intraperitoneal, 100 mg/kg). Learning and memory performances were measured by trace eyeblink conditioning, a hippocampal-dependent task. Mice were trained by pairing delivery of a tone (conditioned stimulus; CS, 250 ms, 2 kHz, 85 dB) as the conditioned stimulus followed by a 250-ms period of no stimuli, followed by a mild periorbital shock (unconditioned stimulus; US, 100 ms) to elicit an eyeblink response. Mice received two blocks of 30 trials per day (30–60 s intertrial intervals, 3–4 h interblock intervals). This behavioral paradigm is subthreshold for inducing neurogenesis (Wang et al., 2010; Singh et al., 2011). One week following a single dose of APα, mice were subjected to trace eyeblink conditioning, with 1 day of habituation and 5 days of paired training. Following paired training, mice were left undisturbed in their home cages for 9 days and subsequently were tested for memory. Following the final learning trial, BrdU+ cell survival/hippocampus was measured at the end of the study, 3-weeks following a single dose of APα and the thymidine analog DNA-synthesis marker BrdU. Bars represent percent change ± sem in response to a single exposure to APα compared to age-matched vehicle at 3-, 6-, 9-, 12-months of age in 3xTgAD mice (n ≥ 7; Wang et al., 2010; Singh et al., 2011). Within 3 weeks following a single exposure to APα, neurogenesis, maximal learning, and memory indicators were increased ∼100% relative to age-matched vehicle control in adult male 3xTgAD mice when administered at ages prior to overt AD pathology. The 3xTgAD mouse model displays age-associated decrements in endogenous neurogenic cell survival in the subgranular zone (SGZ) as compared with the non-transgenic mice in addition to age-associated increments in Aβ pathology burden (depicted supra to bar graph). At 12-months of age, intra- and extraneuronal Aβ 6E10 antibody staining is apparent and plaque structures are developed in subiculum (Wang et al., 2010; Singh et al., 2011). The therapeutic response to APα was specific to the transgenic AD phenotype, as the age-matched non-transgenic mice did not benefit from a single exposure to APα. Remarkably, a single exposure to APα increased neurogenesis and subsequent cell survival in aged non-transgenic mice when administered at 15-months of age (non-Tg data not in figure; Singh et al., 2011). At 12-months of age, the point when extraneuronal plaques are known to be present in this AD mouse model, a single exposure to APα was ineffective. At ages prior to extraneuronal Aβ plaques, APα significantly (P < 0.05) increased BrdU+ cell survival, maximal learning, and memory function relative to age-matched vehicle control.
Figure 3
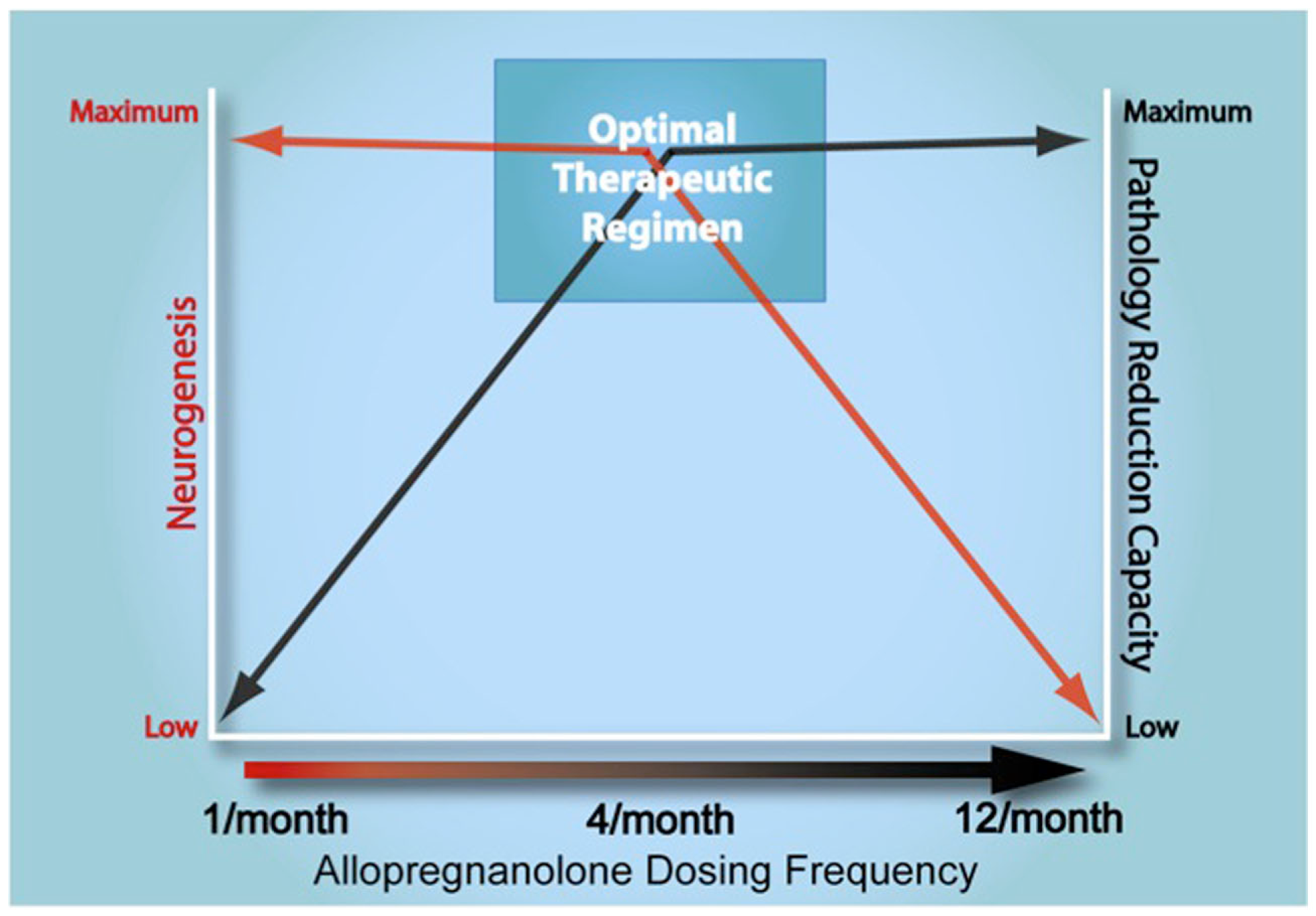
Optimal allopregnanolone therapeutic regimen. Triple-transgenic Alzheimer’s disease (3xTgAD) mice were treated subcutaneously with allopregnanolone (APα; 10 mg/kg) once per month (1/month), once per week (4/month), or every other day (12/month) to determine extent of neurogenesis (depicted by red colored arrow) and pathology reduction capacity (depicted by black colored arrow) on double y-axes (Chen et al., 2011). The dosing frequency determined the therapeutic efficacy for both neurogenic and pathological endpoints. All three APα treatments were initiated when 3xTgAD mice reached 3 months of age. Upon completion of each treatment paradigm, BrdU+ labeled nuclei were counted to assess neurogenesis. Both the 1/month APα treatment and the 4/month APα treatment induced a significant increase in neurogenesis, with the latter regimen yielding the greater increase in neurogenesis. However, the 3/week/3 months (12/month) treatment induced a significant decrease in neurogenesis. Brain sections from 3xTgAD mice treated with APα or vehicle were immunostained. Aβ immunoreactivity was detected and indicated that the 1/week/6 months (4/month) APα treatment significantly decreased Aβ immunoreactivity. Efficacy of Aβ reduction in 4/month was comparable to the 12/month APα treatment whereas the 1/month APα treatment was ineffective at reducing Aβ immunoreactivity (Chen et al., 2011). From these dosing frequency studies, we conclude that the optimal treatment regimen for AD is to intervene as early as possible with once per week administration of APα to simultaneously promote neurogenesis and subsequent cell survival.
Enzymatic conversion of progesterone to its metabolites occurs in the brain and periphery. Neurosteroids are synthesized in the central and peripheral nervous system, particularly in myelinating glial cells, astrocytes, and several neuronal cell types. A region-specific expression pattern of progesterone converting enzymes is evident in both hippocampus and cortex. In steroidogenic cells, biosynthesis of neuroactive steroids is controlled by the 18-kDa outer mitochondrial membrane translocator protein (TSPO; Rupprecht et al., 2010) and TSPO-associated proteins including the steroidogenic acute regulatory protein (StAR) to form a macromolecular signaling complex which transports cholesterol through the mitochondrial inner membrane to the mitochondrial matrix to be converted into pregnenolone by the cytochrome P450 side-chain cleavage (CYP450scc) enzyme (Liu et al., 2006). Pregnenolone subsequently diffuses back into the cytoplasm whereby its conversion to progesterone is facilitated by the 3β-hydroxysteroid dehydrogenase (3β-HSD) enzyme. APα is synthesized in the brain from progesterone by the sequential action of 5α-reductase (5α-R) type-I and 3α-hydroxysteroid dehydrogenase (3α-HSD; Mellon et al., 2001; Mellon, 2007). The rate-limiting step is the unidirectional reduction of progesterone to 5α-dihydroprogesterone by 5α-R. Subsequently, 3α-HSD catalyzes conversion of 5α-dihydroprogesterone into APα. Interestingly, 5α-R and 3α-HSD are functionally expressed in pluripotent progenitor cells (Melcangi et al., 1996).
APα and other trophic factors are diminished in the brains of Alzheimer’s disease (AD) patients compared to age-matched controls (Weill-Engerer et al., 2002; Marx et al., 2006; Naylor et al., 2010). An early feature of AD is the loss of episodic and semantic memory (Perry et al., 2000). AD diagnostic imaging studies using volumetric MRI reveal a decreased hippocampal volume due to atrophy of gray matter, i.e., neurodegeneration in amnestic subtype mild cognitive impairment in the progression to AD (Whitwell et al., 2007). Neuropathological hallmarks of AD include extracellular and intracellular deposition of β-amyloid (Aβ) protein, neurofibrillary tangles and neurodegeneration. AD causes extensive neurodegeneration to the cholinergic neurons of the nucleus basalis and to a greater extent to the noradrenergic neurons of the locus coeruleus (Zarow et al., 2003). In addition to the usual culprits that lead to neurodegeneration, it was discovered that a subpopulation of dysfunctional neurons in AD display aberrant entry into the cell cycle and replicate their genomes but fail to divide leading to a fatal apoptotic mechanism (Busser et al., 1998; Herrup, 2010). There have been no neurogenic therapies developed to prevent neurodegenerative mechanisms of aberrant cell cycle entry. The fundamental role of neurogenesis in AD etiology is not without controversy. Several studies have reported decreased neurogenesis in AD mouse models (Lazarov and Marr, 2010). However, there are few human studies to confirm or reject the importance of neurogenesis in AD. Controversial findings by Jin et al. (2004) showed increased expression of doublecortin (DCX), a marker for new neurons, in a cohort of senile patients and suggested that neurogenesis is increased as a compensatory mechanism of neurodegeneration. The report by Boekhoorn et al. (2006) challenged the findings reported by Jin et al. and demonstrated that in human presenile AD brain, most of the proliferation could be accounted for by glial and vasculature-associated changes and found no evidence for altered neurogenesis in the dentate gyrus. Thus, in AD brain, the extent of neural proliferation and its potential for regenerative therapeutic responsiveness remains an active area of investigation.
Preclinical Discovery of Allopregnanolone as a Pro-Neurogenic Agent in vitro
Our discovery investigations of APα effects in immature hippocampal cells began nearly two decades ago beginning in vitro then advancing to in vivo studies, all reviewed herein (Brinton, 1994). It was known that seizure activity was associated with aberrant hippocampal nerve cell growth and that APα protected against seizure activity (Brinton, 1994). What was not known at the time was whether APα prevented against aberrant circuit development. Our initial attempts to address this question were in vitro analyses using videomicroscopy of hippocampal neurons in culture to determine the impact of APα on neurite outgrowth in real time (Brinton, 1994). Results of these analyses revealed that APα induced a significant decrease in the area and length of neurites within 40 min of exposure. A concomitant decrement in the number and length of filopodia decorating neuritic extensions also occurred within the same time frame. An unforeseen outcome was the observation that within APα-treated neurons, retrograde transport of intracellular organelles occurred. APα-induced regression of neuronal morphology and retrograde transport of organelles was only observed in hippocampal cells that had not yet established contact with other neuronal or glial cells in culture. Established structural connections between neurons or glia did not regress during APα exposure. The cellular selectivity for the morphological effect of APα was an early indicator of a more significant effect of APα which we later discovered. Neither the inactive stereoisomer 3β-hydroxy-5β-pregnan-20-one nor progesterone had a significant effect upon any of the morphological parameters assessed (Brinton, 1994). In more mature hippocampal cultures in which structural synaptic connections had been established, APα protected neurons against picrotoxin-induced cell death. APα activated chloride ion channels, proved to be an initial step in the biochemical mechanism underlying both the retraction and later neurogenesis. Interestingly, 17β-estradiol a known neurotrophic factor (Brinton, 2009) rapidly reversed the filopodial regression induced by APα indicating that neurosteroids acting singly and in combination play complex roles in generation and differentiation of developing neural cells.
Subsequent to the observation of APα-induced neurite regression of hippocampal neurons in culture, we made the serendipitous discovery that APα was increasing mitosis of hippocampal neurons in culture. We then understood that the APα-induced regression of neurites was a prelude to APα-induced mitosis. At the time this discovery was made, understanding of neurogenesis in brain was limited to embryonic development in the mammalian brain and regeneration of neurons required for song in the bird brain. From the vantage point of our current understanding, the hippocampal neurons undergoing mitosis in the primary hippocampal cell cultures were likely neural progenitor cells. This early discovery of APα-induced mitosis was supported by biochemical analyses demonstrating that APα induced a significant increase in 3H-thymidine uptake indicating increased DNA synthesis during the S phase of the cell cycle (Wang et al., 2005). These early foundational studies were followed up with the advent of an understanding of the neural progenitor pool within the adult mammalian brain. We determined APα proliferative efficacy in rodent embryonic and adult neural progenitor cells and embryonic derived human neural progenitor cells. In each of these cell types, APα induced a significant increase in markers of DNA synthesis, BrdU and 3H-thymidine incorporation, MuLV-GFP-labeled mitotic neural progenitor cells by FACS analysis and unbiased quantitative stereology of BrdU-positive cells in both the subgranular and subventricular proliferative zones (Wang et al., 2005).
In vitro, the magnitude of APα-induced neurogenesis ranged from 20 to 30% in the rat hippocampal neural progenitor cells to 37–49% in human cortical neural progenitors (Wang et al., 2005). The efficacy of APα as a neurogenic factor is comparable to that induced by bFGF + heparin. As was the case for cultured embryonic hippocampal neurons, APα-induced progenitor cell proliferation was stereoisomer specific, as 3β-hydroxy-5β-pregnan-20-one did not increase 3H-thymidine uptake (Wang et al., 2005). NPC markers nestin and Tuj1 were identified by immunofluorescence and indicated that the newly formed cells were indeed neuronal. To determine the mechanism of action induced by APα in progenitor cells, microarray analysis of cell cycle genes demonstrated that APα increased the expression of mitotic genes and inhibited the expression of anti-mitotic genes. APα-induced proliferation of neural progenitors was antagonized by the both voltage-gated L-type calcium channel blocker nifedipine indicating a calcium-dependent mechanism for neuroproliferation (Figure 1; Wang et al., 2005).
Consistent with a calcium-dependent mechanism for neuroproliferation, APα induced a rapid increase in intracellular calcium in hippocampal neurons via a GABAAR-activated L-type calcium channel (Wang and Brinton, 2008). Following APα treatment, regulation of intracellular calcium concentration was measured in E18 rat hippocampal neurons using ratiometric Fura2-AM imaging. Results indicated that APα rapidly increased the intracellular calcium concentration in a dose-dependent and developmentally regulated manner, with an EC50 of 110 ± 15 nM and a maximal response occurring at 3 days in vitro. The stereoisomers 3β-hydroxy-5α-hydroxy-pregnan-20-one and 3β-hydroxy-5β-hydroxy-pregnan-20-one, as well as progesterone, were without significant effect. APα-induced intracellular calcium concentration increase was not observed in calcium depleted medium and was blocked in the presence of the broad spectrum calcium channel blocker lanthanum ion, or the L-type calcium channel blocker nifedipine. Furthermore, the GABAAR blockers bicuculline and picrotoxin abolished the APα-induced intracellular calcium concentration rise (Wang and Brinton, 2008). The in vitro neurogenic properties of APα coupled with a low molecular weight, easy penetration of the blood brain barrier and lack of toxicity, were key elements that guided our efforts toward in vivo studies of APα as a neurogenic regenerative therapeutic with the ambitious aim of restoring synaptic connections in victims of AD.
Allopregnanolone as a Multifaceted Pro-Neurogenic Agent in the Triple-Transgenic AD Mouse Model
To evaluate APα as a potential therapy for AD it is essential to obtain preclinical in vivo evidence of APα safety and efficacy. The triple-transgenic mouse model of Alzheimer’s (3xTgAD), an AD model is characterized by overexpression of Swedish mutant APP, mutant P301L Tau in the homozygous mutant of presenilin 1 (M146V) knock-in mouse (Oddo et al., 2003a,b). These mice have been used extensively as an AD research model as they age-dependently develop hallmarks that include hippocampal tangle-like pathology, neurological deficits, and intraneuronal and extraneuronal Aβ deposition. Several completed in vivo studies have demonstrated the pro-neurogenic and promnesic effects of APα. We review here three studies from our laboratory in which a correlation was made between APα-induced neural progenitor cell survival and improved memory function in 3xTgAD mice (Wang et al., 2010; Chen et al., 2011; Singh et al., 2011). The basal concentration of APα in blood plasma (0.47 ng/ml ± 0.88) was significantly lower (P < 0.05) than in cortex (10.36 ng/g ± 1.43) in young adult male non-transgenic (non-Tg) mice, indicating a higher brain accumulation, consistent with locally synthesized APα in hippocampus and cortex. In contrast, 3xTgAD mice exhibited a lower basal level of APα in the cerebral cortex (3xTgAD, 6.49 ng/g ± 2.02 versus non-Tg, 10.36 ng/g ± 1.43), suggesting either impairment of APα synthesis or accelerated APα metabolism in 3xTgAD mice brain (Wang et al., 2010). A decline in neurogenesis occurs within the SGZ and SVZ in male and female 3xTgAD mice in correlation with AD pathology (Brinton and Wang, 2006; Rodriguez et al., 2008, 2009; Wang et al., 2010). APα promoted neurogenesis in the hippocampal SGZ to reverse learning and memory deficits. Neural progenitor cell proliferation and subsequent cell survival was determined by analysis of BrdU incorporation. We verified the phenotype of in vivo newly formed BrdU+ cells by triple-immunolabeling coronal sections of mouse hippocampi adjacent to those that were stereologically analyzed and derived from 3-month-old 3xTgAD mice treated with a single subcutaneous injection of 10 mg/kg APα (Wang et al., 2010). The phenotypic markers assessed were doublecortin DCX, to label young immature neurons; NeuN, to label mature neurons; and glial fibrillary acidic protein (GFAP), to label astrocytes. Confocal microscopy identified BrdU-positive cells colocalized with DCX alone or together with NeuN. Newly born cell survival was further confirmed by immunolabeling coronal sections derived from APα (subcutaneous, 10 mg/kg)-treated 3xTgAD mouse dentate gyrus 22 days post-APα treatment and post-behavioral analyses. BrdU+ cells were located deep within the granular cell layer, indicating the migration of newly formed cells from the SGZ to the granule cell layer (Wang et al., 2010). Collectively, these data indicated that newly formed cells, generated following APα treatment, express a neuronal phenotype.
Learning and memory function was assessed using the hippocampal-dependent trace eyeblink conditioning paradigm. At 3 months of age, the basal level of BrdU+ cells in the SGZ of 3xTgAD mice was significantly lower relative to non-transgenic (non-Tg) mice, despite the lack of evident AD pathology (Wang et al., 2010). APα significantly increased, in a dose-dependent manner, BrdU+ cells in the SGZ of 3xTgAD mice and restored SGZ proliferation to normal magnitude of non-Tg mice (Wang et al., 2010). Coinciding with deficits in adult neural progenitor proliferation, 3xTgAD mice exhibited deficits in learning and memory. APα reversed the cognitive deficits to restore learning and memory performance to the level of normal non-Tg mice (Wang et al., 2010). In 3-month-old 3xTgAD mice, APα increased proliferation and promoted the survival of newly born hippocampal dentate granule cells (Wang et al., 2010). Neural progenitor cell numbers significantly correlated with APα-induced memory performance. Interestingly, the early deficits in neurogenesis detected in this genetic model of AD were evident prior to immunodetectable β-amyloid (Aβ).
To broaden this preclinical discovery work, studies were extended to 3xTgAD mice aged 6-, 9-, 12-months, therapeutically relevant to early and mid-stage AD. Comparable to the 3-month mice, APα increased proliferative activity, promoted the survival of newly born hippocampal dentate granule cells, and restored cognition affected by AD pathology in 6-, 9-month-old 3xTgAD mice while having no significant impact on their age-matched normal non-Tg counterparts (Singh et al., 2011). BrdU+ neural progenitor cell survival was assessed after 3 weeks, following a single exposure to APα (10 mg/kg). During the 3-week period, a hippocampal-dependent associative learning and memory task was performed which evidently allowed granule cells sufficient time to proliferate and migrate at the border between the hilus and the granule cell layer subsequent to their incorporation into the existing neural network. APα significantly increased survival of BrdU+ cells and recovered hippocampal-dependent cognition in 6-, and 9-month-old 3xTgAD mice, in the presence of intraneuronal Aβ, whereas APα was ineffective subsequent to development of extraneuronal Aβ plaques in 12-month-old mice. Surprisingly, cognition was restored to maximum by the first day of trace eyeblink conditioning, only 1 week following a single exposure to APα. Hippocampal-dependent associative learning accomplished by repeated trials of an auditory tone followed by a mildly aversive shock stimulus, was sustained throughout behavioral training. Learning and memory function in APα-treated 3xTgAD mice was 100% greater in magnitude compared to the age-matched vehicle-treated group (Figure 2) and was remarkably comparable to the maximal normal non-Tg mouse performance. Furthermore, we observed an upward trend albeit not statistically significant, toward efficacy in non-Tg at 12 months of age. The behavioral experiment was then extended to include 15-month-old non-Tg mice to determine whether or not APα presented a therapeutic benefit to older non-AD mice that may have a greater degree of age-related diminution of neurogenic growth factors. Our results indicated that hippocampal BrdU+ cell survival was significantly increased by a single exposure to APα and cognitive performance was significantly enhanced in 15-month-old male non-Tg mice (Singh et al., 2011). In normal non-AD men as early as age 40, a decline in APα has been reported (Genazzani et al., 1998). The therapeutic benefit found in aged non-Tg mice suggests that at least in males, APα therapy could supplement an age- and gender-related decline in this neurogenic factor. These findings provided preclinical evidence of APα-promoted survival of newly generated cells and paralleled restoration of cognitive performance in the pre-plaque phase of AD pathology and in late-stage normal aging.
To further advance efforts to assess the preclinical efficacy of APα for AD, our group designed long-term studies. The studies were designed to test APα using the same age of enrollment (3xTgAD male 3 months of age; prior to overt intraneuronal Aβ), neurogenically efficacious dose of 10 mg/kg via subcutaneous route of administration, matching our previous studies. In addition to neurogenic efficacy, the long-term studies were extended to determine the disease modifying effects afforded by the therapeutic regimen. Specifically, Chen et al. (2011) tested three treatment regimens – once per month, once per week, and every other day (Figure 3). Based on measured endpoints of Aβ oligomers by immunostain and immunoblot approaches, the every other day treatment regimen was maximally efficacious but did not increase neurogenesis. The once per month APα treatment was efficacious for proliferation of neural progenitor cells in the SGZ (and SVZ, unpublished) but not for decreased Aβ pathology. The once per month treatment regimen (Chen et al., 2011) was similar to the single exposure treatment paradigm that improved learning and memory performance after a single exposure (Singh et al., 2011). Analyses to determine the optimal treatment paradigm indicated that APα administered once per week for 6 months was maximally efficacious for both neurogenic and anti-amyloidogenic endpoints (Figure 3). In parallel to the 3-month-old mice administered once per week APα, we simultaneously began treatment of a 6-month-old male 3xTgAD group. When APα was administered beginning at 6 months of age, the age at which intraneuronal plaques are apparent in this mouse model, the appearance of Aβ pathology paralleled cessation of APα efficacy. Once intraneuronal Aβ is extracellularly localized, the efficacy of APα is largely diminished. This suggested to us that intraneuronal Aβ accumulation is a determining factor that focuses the window of APα therapeutic efficacy on the early stages of AD.
There is a well-established relationship between cholesterol homeostasis and Aβ generation. Increasing evidence indicates that altered cholesterol metabolism is linked to AD pathology (Schumacher et al., 2004; Mellon et al., 2008). In addition to the mechanism of action whereby APα induces neurogenesis through excitation of GABAAR chloride channels in neural progenitor cells (Figure 1; Wang et al., 2005, 2008, 2010; Brinton and Wang, 2006; Wang and Brinton, 2008), APα regulates cholesterol homeostasis via the Liver-X-receptor (LXR) and pregnane-X-receptor (PXR) system (Chen et al., 2011). LXR, a nuclear hormone receptor abundant in the brain (Whitney et al., 2002), acts as a molecular sensor of cholesterol levels and initiates cholesterol clearance (Whitney et al., 2002). LXR activation increases cholesterol efflux through up-regulating ABCA1 and ApoE expression, and prevents the hyper-activation of γ-secretase and over-production of Aβ (Whitney et al., 2002; Shenoy et al., 2004; Jiang et al., 2008). LXR activation has been demonstrated to improve cognitive function in multiple mouse models of amyloidogenesis (Schultz et al., 2000; Whitney et al., 2002; Yang et al., 2006; Xiong et al., 2008; Donkin et al., 2010; Leduc et al., 2010). Importantly, LXR ligands have been shown to activate pregnane-X-receptor (PXR; Riddell et al., 2007). Results from our analyses indicated that in parallel with an APα-induced increase in LXR expression in the pre-pathology condition, APα also increased PXR expression in the pre-pathology 3xTgAD mouse brain (Chen et al., 2011). PXR activation induces cytochrome P450 3A (CYP3A) enzymes including CYP3A4 and CYP3A13 and leads to cholesterol hydroxylation and activation of organic anion transporters for cholesterol extrusion (Sun et al., 2003). In addition to increased LXR and PXR expression, APα treatment initiated in pre-Aβ pathology 3-month-old 3xTgAD mice treated once per week for 6 months displayed increased expression of 3-hydroxy-3-methyl-glutaryl-CoA-reductase (HMG-CoA-R; Chen et al., 2011). The increase in HMG-CoA-R is at first paradoxical as it is the rate-limiting enzyme in cholesterol synthesis. HMG-CoA reductase is also required for oxysterol generation which activate LXR and PXR-mediated gene transcription for cholesterol and lipid transport proteins (Leduc et al., 2010). Thus, the APα-induced increase in brain LXR and PXR leads to increased cholesterol efflux, thereby reducing γ-secretase activation by cholesterol-laden lipid rafts. Increased cholesterol efflux provides a plausible mechanism to explain how APα decreased the generation of both 27 and 56 kDa intraneuronal Aβ oligomers after 6 months of once per week treatment (Chen et al., 2011).
Our findings suggest that in vivo, brain cholesterol homeostasis and intraneuronal Aβ are tightly coupled with APα efficacy (Chen et al., 2011). Deposition of Aβ in the extracellular compartment disconnected this coupled pathway and led to a loss of APα efficacy in advanced stages of AD-like pathology in the 3xTgAD model. APα significantly reduced Aβ generation in hippocampus, cortex, and amygdala, which was paralleled by decreased mitochondrial Aβ-binding alcohol dehydrogenase (ABAD) and reduced microglia activation assessed as reduced expression of OX42 (Chen et al., 2011). APα has also been shown to increase myelin basic protein in organotypic slice cultures of rat cerebellum (Schumacher et al., 2004) and delay demyelination in Niemann–Pick C mice (Mellon et al., 2008). APα may stimulate oligodendrocyte progenitor cells in addition to neural progenitor cells. In oligodendrocytes, the myelin marker CNPase was increased by once per week APα, indicating pro-myelinating capabilities in this mouse model (Chen et al., 2011). Collectively, APα is a multifaceted neurosteroid that promotes neurogenesis while simultaneously reducing AD pathology in the 3xTgAD mouse model.
Translation of Preclinical Allopregnanolone Studies to Clinical Alzheimer’s Disease Therapy
Currently there are no effective treatments to delay progression of Alzheimer’s, halt the degenerative process or effectively treat the disease. In fact, since 2003, there have been no new drug approvals for AD by the United States Food and Drug Administration (FDA). Of the five FDA-approved AD drugs, four are cholinesterase inhibitors and memantine is an N-methyl-d-aspartate (NMDA) receptor antagonist. As part of the recent AD Neuroimaging Initiative or “ADNI”, mild cognitive impairment patients who received cholinesterase inhibitors with or without memantine were more functionally impaired, showed greater decline in scores, and progressed to dementia sooner than patients who did not receive treatment (Schneider et al., 2011). Phase 3 clinical trials have failed to therapeutically modify AD via the inhibition of amyloid cascade targets including a gamma-secretase modulator (tarenflurbil), gamma-secretase inhibitor (semagacestat), and beta-secretase inhibitor (tramiprosate; Karran et al., 2011). Most AD researchers agree that it is ineffective to evaluate therapeutic efficacy in people that have reached the later stages of AD, when irreversible damage has occurred. To modify disease progression, therapies need to be instituted early to target specific physiological stages of AD progression. One strategy to detect dysfunction of the neuroendocrine system would be to access blood levels of APα in persons at risk for AD. Preclinical biomarkers to predict clinical therapeutic potential and clinical biomarkers that confirm target engagement and dose selection are needed as prognostic and diagnostic tools for AD disease progression to select and track treatment strategies relevant to each pathological stage of dementia (Buckholtz, 2011). Currently, large-scale multicenter controlled studies are being conducted in US, Europe, Japan, and Australia (ADNI) to systematically develop and validate candidate biomarkers such as cerebrospinal fluid proteins and structural and functional imaging signatures (Hampel et al., 2008). Ideally, complementary brain imaging and fluid biomarkers will be developed for clinical evaluation of AD.
As an endogenous metabolite of progesterone, men and women are exposed to APα throughout their lifetime. During reproductive years, women are chronically exposed to APα concentrations ranging from less than 1 nmol/l (0.32 ng/ml) to over 4 nmol/l (1.27 ng/ml) during the luteal phase (Genazzani et al., 1998). During pregnancy, blood production rate of APα can reach 100 mg/24 h (Dombroski et al., 1997) and remain high throughout the third trimester of pregnancy at levels up to 157 nmol/l (50 ng/ml), which while associated with drowsiness, is not associated with adverse effects for either mother or fetus (Luisi et al., 2000). In response to acute stress, the fetal brain can increase synthesis of APα de novo independent of maternal supply and of the hypothalamic–pituitary–adrenal axis (Nguyen et al., 2003). Women receiving progesterone therapy can generate between 0.9 nmol/l (0.3 ng/ml) and 4.2 nmol/l (1.3 ng/ml) of APα, consistent with levels generated during the menstrual cycle. Analyses following a single intramuscular injection of 200 mg progesterone (APα precursor) indicated similar profiles in men and women with peak levels of APα of 35 nmol/l (11 ng/ml) for men and 41 nmol/l (13 ng/ml) for women in the first hours after administration (Soderpalm et al., 2004). Post-mortem human brain tissue from women revealed APα levels in the range of 14–21 ng/g (Bixo et al., 1997). Oral delivery of APα would be convenient and readily tolerated however this route presents a challenge because of the low solubility properties of APα and first-pass metabolism in the digestive tract and liver. Results of our preclinical analyses predict that the optimal therapeutic APα regimen will be a once per week transdermal or subcutaneous administration (Figure 3). Initial pharmacokinetic analyses of a topically applied formulation of APα in rabbit, indicated that APα is absorbed transdermally to reach the blood circulation with accumulation in the brain that is relatively slowly eliminated when compared to intravenous dosing (Table 1). Safety is most critical for translational studies. In the past decade, several clinical studies by Bäckström’s group at Umeå University, Sweden have demonstrated that APα is safe with short-lived, mild self-reported sedation (drowsiness). Following dosing of APα 0.09 mg/kg (three cumulative doses of 0.015, 0.03, 0.045 mg/kg within 1 h) fully bioavailable intravenous administration, van Broekhoven et al. (2007) measured maximum blood levels of 150 nmol/l (48 ng/ml) in men and 100 nmol/l (32 ng/ml) in women (Table 1). Interestingly, mean levels of APα were found to be higher in men compared to women at baseline (men, 2.4 nmol/l or 0.76 ng/ml versus women, 0.4 nmol/l or 0.13 ng/ml) and after each of three doses of intravenous APα. Volume of distribution, elimination half-life, and the area under the curve (AUC) adjusted for body weight did not differ between men and women in the clinical study (van Broekhoven et al., 2007). To date, APα has not been studied in clinical trials for the treatment of AD.
Table 1
| Purpose | Species | Route | APα (mg/kg) | Vehicle | Frequency of exposure | Endpoint | Blood (APα) | Brain (APα) | Results | Safety | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Allopregnanolone (APα) preclinical efficacy and route of administration development in mouse model of Alzheimer’s disease | Mouse (male) | SC | 10 | PBS/5% EtOH | Single dose | 24 h | 4 ng/ml (12.5 nmol/l) C24 h | 3.5 ng/g (∼11 nmol/l) C24 h | Increased neurogenesis | No adverse effects | Wang et al. (2010) |
| TD | 10–50 | Proprietary formulations | Single dose | 24 h | – | – | Increased neurogenesis | No adverse effects | Brinton et al. unpublished | ||
| IN | 3–10 | ||||||||||
| SC | 10 | PBS/5% EtOH | Single dose | 1 month | – | – | Increased neurogenesis and cell survival; hippocampal-dependent learning and memory | No adverse effects | Singh et al. (2011), Chen et al. (2011) | ||
| SC | 10 | 20% HBCD | Once/week | 6 month | – | – | Increased neurogenesis and cell survival; decreased AD pathology | No serious adverse effects; 20 min hyperactivity followed by brief sedation | Chen et al. (2011) | ||
| Bioavailability/pharmacokinetic preclinical development | Rabbit (female, n = 9) | IV | 3 | 20% HBCD | Single dose | 24 h time course | 1,176 ng/ml (3,692 nmol/l) Cmax; 0.08 h Tmax | 1,181 ng/g (∼3,706 nmol/l) Cmax; 0.5 h Tmax | Rapid uptake, rapid elimination from blood and brain | IV – unresponsive 20–30 min, one death indicated upper dose limit for acute toxicity | Brinton et al.1 |
| Rabbit (female, n = 9) | TD | 5 | 100% DMSO | Single dose | 24 h time course | 9.6 ng/ml (30 nmol/l) Cmax; 0.25 h Tmax | 151 ng/g (∼474 nmol/l) Cmax; 4 h Tmax | Low peak uptake, rapid elimination from blood, slow elimination from brain | TD – no adverse effects other than local skin erythema/edema from topical DMSO | ||
| Pharmacokinetics of GABAA function in induced-seizure model | Rat (male, n = 5) | ICV | 0.00125–0.075 | 45% HBCD | Single dose | 1 h time course | 33 ng/ml (104 nmol/l) Cmax; 0.08 h Tmax | 278 ng/g (∼873 nmol/l) Cmax; 0.08 h Tmax | Dose- and time-dependent positive modulation of GABAAR chloride channel function in brain, anti-convulsant activity | No adverse effects reported | Concas et al. (1996) |
| Preclinical model of Neimann–Pick C demyelination disorder | Mouse postnatal day 7 | SC | 25 | 20% HBCD | Once/2 weeks | 4.3 month (end of lifespan) | – | – | Extended lifespan; corpus callosum white matter hyperintensity by diffusion tensor imaging; delayed demyelination | Sedation | Ahmad et al. (2005) |
| Preclinical ethanol substitution | Non-human primate (male, n = 9) | SC | 0.17–10 (ED50 0.40–1.78) | 4% HBCD | Twice weekly testing | 30–60 min post-dosing | – | – | No sex differences in APα substitution for ethanol; stereoisomer specific | Ethanol-like discriminative stimulus effects | Grant et al. (2008) |
| Non-human primate (female, n = 8) | 0.17–10 (ED50 0.89–1.07) | – | – | ||||||||
| Clinical trial | Human (healthy male, n = 9) | IV | 0.09 | Albumin solution | Acute 1 h cumulative doses | During 1 h cumulative dosing | 48 ng/ml (150 nmol/l) Cmax | – | Male – decreased saccadic eye movement; decrease in contentedness | No adverse effects other than self-reported sedation | van Broekhoven et al. (2007) |
| Human (healthy female on oral contraceptive, n = 9) | 32 ng/ml (100 nmol/l) Cmax | – | Female – decreased saccadic eye movement; increase in contentedness | ||||||||
| Clinical trial | Human (healthy female, n = 10) | IV | 0.09 | Albumin solution | Acute 1 h cumulative doses | During 1 h cumulative dosing | 22 ng/ml (70 nmol/l) Cmax | – | Decreased saccadic eye movement | No adverse effects other than fatigue, mild nausea | Timby et al. (2006) |
| Clinical trial | Human (healthy female, n = 28 crossover) | IV | 0.07 | Albumin solution | Single dose | 10–55 min post-dosing | 22–45 ng/ml (70–140 nmol/l) Cmax | – | Acutely impaired free verbal recall episodic memory due to mild sedation with high variability; no acute effect on semantic or working memory | No adverse effects other than self-reported sedation | Kask et al. (2008) |
| Clinical trial | Human (healthy female, n = 12; PMDD, n = 16) | IV | 0.05 | Albumin solution | Single dose | 0–20 min post-dosing | 16–22 ng/ml (50–70 nmol/l) Cmax | – | No effect on startle response or prepulse inhibition to startle | No adverse effects other than self-reported sedation | Kask et al. (2009) |
Allopregnanolone (APα) preclinical and clinical studies (previous human experience).
APα blood plasma and brain concentrations and reported safety by multiple routes of administration. Pharmacokinetic properties are best studied by an intravenous (IV) dose as this route of administration has the greatest quantitative potential, assumed to be 100% bioavailable, and is useful for comparison to alternative routes of administration. Preclinical APα dosing studies with subcutaneous (SC), transdermal (TD), intranasal (IN), intracerebroventricular (ICV), and IV routes of administration were tabulated with human IV dosing. Although the differences in the tabulated doses appear large, when bioavailability and species-specific allometric conversions are taken into account the preclinical studies that demonstrate neuroregenerative efficacy in an AD mouse model are within the range of the safe dosage for humans. In the rabbit pharmacokinetic study, the bioavailability of TD APα was ∼1.5% (APα TD area under curve of 17.7 h*ng/ml versus IV 732.1 h*ng/ml plasma), Brinton, SRI, Intl. subcontracted pharmacokinetic study unpublished. The conversion factor for APα is 3.1398 (APα formula weight 318.49 g/mol) to simplify mathematical conversion from nanograms per milliliter to nanomoles per liter. For example, APα 50 ng/ml × 3.1398 = APα 157 nmol/l corresponding to the upper level of plasma APα measured during the third trimester of human pregnancy (Luisi et al., 2000) and safely achieved with human IV dosing (van Broekhoven et al., 2007; Kask et al., 2008). Neurosteroid levels from post-mortem brain tissue were measured in nanograms per gram and for comparison purposes are estimated to equal nanograms per milliliter and then converted to nanomoles per liter. 1Brinton, SRI, Intl. subcontracted pharmacokinetic study unpublished.
Human, as well as rodent, brains exhibit a significant and profound reduction in the proliferative pool and regenerative potential of neural stem cells as they age, a phenomenon that may be exacerbated in prodromal and mild AD brains. Both the pool of neural stem cells and their proliferative potential are compromised in the course of AD (Lazarov et al., 2010; Winner et al., 2011). In parallel, APα content, along with a host of other factors, is diminished in the brains of AD patients compared to age-matched controls (Weill-Engerer et al., 2002; Marx et al., 2006; Naylor et al., 2010). While de novo synthesis of APα in brain is diminished, peripheral delivery could restore levels to normal. A deficient milieu of endogenous neurosteroids and depleted neurogenesis demonstrate the therapeutic need for APα in the human brain. Promnesic effects are not achievable with every APα dosing regimen or intervention point. Decrements in learning and memory have been reported in animals and humans although these studies are either acute measures of memory performance within minutes of a sedative dose (Kask et al., 2008) or chronic treatment paradigms that mimic stress conditions (Turkmen et al., 2006). When high doses or chronic treatment regimens are employed, APα acts similar to benzodiazepines to reduce learning and impair memory. APα administered twice daily at high doses to male rats for several consecutive days decreased performance on the Morris water maze, escape latency, path length, and thigmotaxis and it was determined that pretreatment induced a partial tolerance against acute APα effects (Turkmen et al., 2006). It is important to consider that GABAergic synapses are not universally influenced by APα and that many factors including GABAAR subunit composition and receptor density influence the local cellular response. Thus for therapeutic use of APα, it is imperative to determine the proper dosing regimen which may be specific to each indication. Aside from AD, the proper APα dosing regimen has shown therapeutic potential in research models of several brain disorders or injuries including catamenial epilepsy (Reddy and Rogawski, 2001; Rogawski, 2003), spontaneous seizures (Concas et al., 1996), diabetic neuropathy (Leonelli et al., 2007), Niemann–Pick type C neurodegenerative disorder (Griffin et al., 2004; Ahmad et al., 2005), and traumatic brain injury (He et al., 2004a,b; Djebaili et al., 2005). APα is absorbed by multiple routes of administration, penetrates the blood brain barrier well, exhibits a wide margin of safety particularly when given transdermally or subcutaneously and for the 3xTgAD mouse model is most efficacious when administered only once per week which also contributes to its margin of safety (Table 1). Importantly, APα is a small molecular weight, blood brain barrier penetrant molecule with safety data in animals and humans (Timby et al., 2006; van Broekhoven et al., 2007; Grant et al., 2008; Kask et al., 2008, 2009; Wang et al., 2010). To our knowledge, APα is the only small molecule agent that both promotes regenerative function in the brain and simultaneously reduces AD pathology burden.
Conclusion
Herein we reviewed preclinical evidence for APα-induced promotion of neurogenesis (Wang et al., 2005, 2010), recovery of learning and memory function (Wang et al., 2010; Singh et al., 2011), and reduction of AD pathology burden (Chen et al., 2011) in the 3xTgAD mouse model. Moreover, APα induction of cell cycle gene expression (Wang et al., 2005) and key regulators of cholesterol homeostasis (Chen et al., 2011) provide mechanistic plausibility for its therapeutic efficacy to promote neurogenesis and cognitive function while reducing AD pathology. Our data show that regeneration is achieved with either once per month or once per week regimen of APα. Reduction of AD pathology can be achieved with once per week or every other day regimens. The combination of regeneration and reduction of pathology was achievable with the once per week APα treatment regimen. Together with the dosing frequency, the magnitude of pathology at the start of treatment intervention is critical to therapeutic efficacy. Administration of APα prior to and during the early stages of AD pathology significantly increased the regenerative response in brain while also reducing burden of pathology in an AD mouse model. APα treatment initiated at the point of Aβ plaque generation was not efficacious indicating that APα targets regenerative and pathology-reducing mechanisms present during the early to mid-stages of the disease. Based on the therapeutic efficacy of APα in a preclinical AD mouse model and in normal aged mice, we predict that APα has potential therapeutic benefit in humans to delay progression in persons with familial early onset AD and to prevent and delay disease in late onset AD. In these populations, APα could be an effective therapy to promote the regenerative potential and myelination capacity of the brain to prevent or delay progression of mild cognitive impairment to clinically diagnosed AD. In summary, targeting a unique mechanism of action, APα promotes the innate regenerative capability of the brain by increasing the number and survival of newly generated neurons.
Statements
Acknowledgments
This research was supported by grants from National Institute on Aging U01 AG031115, Alzheimer’s Drug Discovery Foundation, and the Kenneth T and Eileen L. Norris Foundation to RDB.
Conflict of interest
Patents pending on allopregnanolone as a therapeutic for mild cognitive impairment and Alzheimer’s disease.
References
1
AhmadI.Lope-PiedrafitaS.BiX.HicksC.YaoY.YuC.ChaitkinE.HowisonC. M.WebergL.TrouardT. P.EricksonR. P. (2005). Allopregnanolone treatment, both as a single injection or repetitively, delays demyelination and enhances survival of Niemann-Pick C mice. J. Neurosci. Res.82, 811–821.10.1002/jnr.20685
2
AltmanJ. (1969). Autoradiographic and histological studies of postnatal neurogenesis. IV. Cell proliferation and migration in the anterior forebrain, with special reference to persisting neurogenesis in the olfactory bulb. J. Comp. Neurol.137, 433–457.10.1002/cne.901370404
3
AltmanJ.DasG. D. (1965). Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J. Comp. Neurol.124, 319–335.10.1002/cne.901240303
4
Alvarez-BuyllaA.HerreraD. G.WichterleH. (2000). The subventricular zone: source of neuronal precursors for brain repair. Prog. Brain Res.127, 1–11.10.1016/S0079-6123(00)27002-7
5
BayerS. A.AltmanJ. (1974). Hippocampal development in the rat: cytogenesis and morphogenesis examined with autoradiography and low-level X-irradiation. J. Comp. Neurol.158, 55–79.10.1002/cne.901580105
6
BixoM.AnderssonA.WinbladB.PurdyR. H.BackstromT. (1997). Progesterone, 5alpha-pregnane-3,20-dione and 3alpha-hydroxy-5alpha-pregnane-20-one in specific regions of the human female brain in different endocrine states. Brain Res.764, 173–178.10.1016/S0006-8993(97)00455-1
7
BizonJ. L.GallagherM. (2003). Production of new cells in the rat dentate gyrus over the lifespan: relation to cognitive decline. Eur. J. Neurosci.18, 215–219.10.1046/j.1460-9568.2003.02733.x
8
Blurton-JonesM.KitazawaM.Martinez-CoriaH.CastelloN. A.MullerF. J.LoringJ. F.YamasakiT. R.PoonW. W.GreenK. N.LaferlaF. M. (2009). Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A.106, 13594–13599.10.1073/pnas.0901402106
9
BoekhoornK.JoelsM.LucassenP. J. (2006). Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol. Dis.24, 1–14.10.1016/j.nbd.2006.04.017
10
BrintonR. D. (1994). The neurosteroid 3 alpha-hydroxy-5 alpha-pregnan-20-one induces cytoarchitectural regression in cultured fetal hippocampal neurons. J. Neurosci.14, 2763–2774.
11
BrintonR. D. (2009). Estrogen-induced plasticity from cells to circuits: predictions for cognitive function. Trends Pharmacol. Sci.30, 212–222.10.1016/j.tips.2008.12.006
12
BrintonR. D.WangJ. M. (2006). Preclinical analyses of the therapeutic potential of allopregnanolone to promote neurogenesis in vitro and in vivo in transgenic mouse model of Alzheimer’s disease. Curr. Alzheimer Res.3, 11–17.10.2174/156720506775697160
13
BuckholtzN. S. (2011). Perspective: in search of biomarkers. Nature475, S8.10.1038/475008a
14
BusserJ.GeldmacherD. S.HerrupK. (1998). Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brain. J. Neurosci.18, 2801–2807.
15
CameronH. A.McKayR. D. (1999). Restoring production of hippocampal neurons in old age. Nat. Neurosci.2, 894–897.10.1038/13197
16
CameronH. A.McKayR. D. (2001). Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J. Comp. Neurol.435, 406–417.10.1002/cne.1040
17
CameronH. A.WoolleyC. S.McEwenB. S.GouldE. (1993). Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience56, 337–344.10.1016/0306-4522(93)90335-D
18
ChenS.WangJ. M.IrwinR. W.YaoJ.LiuL.BrintonR. D. (2011). Allopregnanolone promotes regeneration and reduces beta-amyloid burden in a preclinical model of Alzheimer’s disease. PLoS ONE6, e24293.10.1371/journal.pone.0024293
19
ClellandC. D.ChoiM.RombergC.ClemensonG. D.Jr.FragniereA.TyersP.JessbergerS.SaksidaL. M.BarkerR. A.GageF. H.BusseyT. J. (2009). A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science325, 210–213.10.1126/science.1173215
20
ConcasA.MostallinoM. C.PerraC.LenerR.RoscettiG.BarbacciaM. L.PurdyR. H.BiggioG. (1996). Functional correlation between allopregnanolone and [35S]-TBPS binding in the brain of rats exposed to isoniazid, pentylenetetrazol or stress. Br. J. Pharmacol.118, 839–846.
21
DjebailiM.GuoQ.PettusE. H.HoffmanS. W.SteinD. G. (2005). The neurosteroids progesterone and allopregnanolone reduce cell death, gliosis, and functional deficits after traumatic brain injury in rats. J. Neurotrauma22, 106–118.10.1089/neu.2005.22.106
22
DombroskiR. A.CaseyM. L.MacdonaldP. C. (1997). 5-Alpha-dihydroprogesterone formation in human placenta from 5alpha-pregnan-3beta/alpha-ol-20-ones and 5-pregnan-3beta-yl-20-one sulfate. J. Steroid Biochem. Mol. Biol.63, 155–163.10.1016/S0960-0760(97)00058-7
23
DonkinJ. J.StukasS.Hirsch-ReinshagenV.NamjoshiD.WilkinsonA.MayS.ChanJ.FanJ.CollinsJ.WellingtonC. L. (2010). ATP-binding cassette transporter A1 mediates the beneficial effects of the liver-X-receptor agonist GW3965 on object recognition memory and amyloid burden in APP/PS1 mice. J. Biol. Chem.285, 34144–34154.10.1074/jbc.M110.108100
24
DrapeauE.MayoW.AurousseauC.Le MoalM.PiazzaP. V.AbrousD. N. (2003). Spatial memory performances of aged rats in the water maze predict levels of hippocampal neurogenesis. Proc. Natl. Acad. Sci. U.S.A.100, 14385–14390.10.1073/pnas.2334169100
25
EncinasJ. M.HamaniC.LozanoA. M.EnikolopovG. (2011). Neurogenic hippocampal targets of deep brain stimulation. J. Comp. Neurol.519, 6–20.10.1002/cne.22564
26
ErikssonP. S.PerfilievaE.Bjork-ErikssonT.AlbornA. M.NordborgC.PetersonD. A.GageF. H. (1998). Neurogenesis in the adult human hippocampus. Nat. Med.4, 1313–1317.10.1038/3305
27
GeeK. W.BolgerM. B.BrintonR. E.CoiriniH.McewenB. S. (1988). Steroid modulation of the chloride ionophore in rat brain: structure-activity requirements, regional dependence and mechanism of action. J. Pharmacol. Exp. Ther.246, 803–812.
28
GeeK. W.ChangW. C.BrintonR. E.McewenB. S. (1987). GABA-dependent modulation of the Cl− ionophore by steroids in rat brain. Eur. J. Pharmacol.136, 419–423.10.1016/0014-2999(87)90317-7
29
GenazzaniA. R.PetragliaF.BernardiF.CasarosaE.SalvestroniC.TonettiA.NappiR. E.LuisiS.PalumboM.PurdyR. H.LuisiM. (1998). Circulating levels of allopregnanolone in humans: gender, age, and endocrine influences. J. Clin. Endocrinol. Metab.83, 2099–2103.10.1210/jc.83.6.2099
30
GrantK. A.HelmsC. M.RogersL. S.PurdyR. H. (2008). Neuroactive steroid stereospecificity of ethanol-like discriminative stimulus effects in monkeys. J. Pharmacol. Exp. Ther.326, 354–361.10.1124/jpet.108.137315
31
GriffinL. D.GongW.VerotL.MellonS. H. (2004). Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat. Med.10, 704–711.10.1038/nm1073
32
HampelH.BurgerK.TeipelS. J.BokdeA. L.ZetterbergH.BlennowK. (2008). Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimer’s Dement.4, 38–48.10.1016/j.jalz.2008.05.068
33
HeJ.EvansC. O.HoffmanS. W.OyesikuN. M.SteinD. G. (2004a). Progesterone and allopregnanolone reduce inflammatory cytokines after traumatic brain injury. Exp. Neurol.189, 404–412.10.1016/j.expneurol.2004.06.008
34
HeJ.HoffmanS. W.SteinD. G. (2004b). Allopregnanolone, a progesterone metabolite, enhances behavioral recovery and decreases neuronal loss after traumatic brain injury. Restor. Neurol. Neurosci.22, 19–31.
35
HerrupK. (2010). The involvement of cell cycle events in the pathogenesis of Alzheimer’s disease. Alzheimer’s Res. Ther.2, 13.10.1186/alzrt37
36
HosieA. M.WilkinsM. E.Da SilvaH. M.SmartT. G. (2006). Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature444, 486–489.10.1038/nature05324
37
JiangQ.LeeC. Y.MandrekarS.WilkinsonB.CramerP.ZelcerN.MannK.LambB.WillsonT. M.CollinsJ. L.RichardsonJ. C.SmithJ. D.ComeryT. A.RiddellD.HoltzmanD. M.TontonozP.LandrethG. E. (2008). ApoE promotes the proteolytic degradation of Abeta. Neuron58, 681–693.
38
JinK.PeelA. L.MaoX. O.XieL.CottrellB. A.HenshallD. C.GreenbergD. A. (2004). Increased hippocampal neurogenesis in Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A.101, 343–347.10.1073/pnas.2634794100
39
KarranE.MerckenM.De StrooperB. (2011). The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discov.10, 698–712.10.1038/nrd3505
40
KaskK.BackstromT.LundgrenP.Sundstrom PoromaaI. (2009). Allopregnanolone has no effect on startle response and prepulse inhibition of startle response in patients with premenstrual dysphoric disorder or healthy controls. Pharmacol. Biochem. Behav.92, 608–613.10.1016/j.pbb.2009.02.014
41
KaskK.BackstromT.NilssonL. G.Sundstrom-PoromaaI. (2008). Allopregnanolone impairs episodic memory in healthy women. Psychopharmacology (Berl.)199, 161–168.10.1007/s00213-008-1150-7
42
KempermannG.KuhnH. G.GageF. H. (1997). Genetic influence on neurogenesis in the dentate gyrus of adult mice. Proc. Natl. Acad. Sci. U.S.A.94, 10409–10414.10.1073/pnas.94.19.10409
43
KempermannG.WiskottL.GageF. H. (2004). Functional significance of adult neurogenesis. Curr. Opin. Neurobiol.14, 186–191.10.1016/j.conb.2004.03.001
44
KuhnH. G.Dickinson-AnsonH.GageF. H. (1996). Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J. Neurosci.16, 2027–2033.
45
LazarovO.MarrR. A. (2010). Neurogenesis and Alzheimer’s disease: at the crossroads. Exp. Neurol.223, 267–281.10.1016/j.expneurol.2009.08.009
46
LazarovO.MattsonM. P.PetersonD. A.PimplikarS. W.Van PraagH. (2010). When neurogenesis encounters aging and disease. Trends Neurosci.33, 569–579.10.1016/j.tins.2010.09.003
47
LeducV.Jasmin-BelangerS.PoirierJ. (2010). APOE and cholesterol homeostasis in Alzheimer’s disease. Trends. Mol. Med.16, 469–477.10.1016/j.molmed.2010.07.008
48
LeonelliE.BianchiR.CavalettiG.CarusoD.CrippaD.Garcia-SeguraL. M.LauriaG.MagnaghiV.RoglioI.MelcangiR. C. (2007). Progesterone and its derivatives are neuroprotective agents in experimental diabetic neuropathy: a multimodal analysis. Neuroscience144, 1293–1304.10.1016/j.neuroscience.2006.11.014
49
LiuJ.RoneM. B.PapadopoulosV. (2006). Protein-protein interactions mediate mitochondrial cholesterol transport and steroid biosynthesis. J. Biol. Chem.281, 38879–38893.10.1074/jbc.M513490200
50
LiuL.BrintonR. D. (2010). “Gonadal hormones, neurosteroids and clinical progestins as neurogenic regenerative agents: therapeutic implications,” in Hormones in Neurodegeneration, Neuroprotection, and Neurogenesis, eds GravanisA. G.MellonS. H. (Hoboken, NJ: Wiley-Blackwell), 281–303.
51
LiuX.WangQ.HaydarT. F.BordeyA. (2005). Nonsynaptic GABA signaling in postnatal subventricular zone controls proliferation of GFAP-expressing progenitors. Nat. Neurosci.8, 1179–1187.10.1038/nn1522
52
LuisiS.PetragliaF.BenedettoC.NappiR. E.BernardiF.FadaltiM.ReisF. M.LuisiM.GenazzaniA. R. (2000). Serum allopregnanolone levels in pregnant women: changes during pregnancy, at delivery, and in hypertensive patients. J. Clin. Endocrinol. Metab.85, 2429–2433.10.1210/jc.85.7.2429
53
LuskinM. B. (1993). Restricted proliferation and migration of postnatally generated neurons derived from the forebrain subventricular zone. Neuron11, 173–189.10.1016/0896-6273(93)90281-U
54
MalbergJ. E.EischA. J.NestlerE. J.DumanR. S. (2000). Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci.20, 9104–9110.
55
MarxC. E.TrostW. T.ShampineL. J.StevensR. D.HuletteC. M.SteffensD. C.ErvinJ. F.ButterfieldM. I.BlazerD. G.MassingM. W.LiebermanJ. A. (2006). The neurosteroid allopregnanolone is reduced in prefrontal cortex in Alzheimer’s disease. Biol. Psychiatry60, 1287–1294.10.1016/j.biopsych.2006.06.017
56
MassaS. M.YangT.XieY.ShiJ.BilgenM.JoyceJ. N.NehamaD.RajadasJ.LongoF. M. (2010). Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J. Clin. Invest.120, 1774–1785.10.1172/JCI41356
57
MayoW.LemaireV.MalaterreJ.RodriguezJ. J.CayreM.StewartM. G.KharoubyM.RougonG.Le MoalM.PiazzaP. V.AbrousD. N. (2005). Pregnenolone sulfate enhances neurogenesis and PSA-NCAM in young and aged hippocampus. Neurobiol. Aging26, 103–114.10.1016/j.neurobiolaging.2005.09.004
58
McDonaldH. Y.WojtowiczJ. M. (2005). Dynamics of neurogenesis in the dentate gyrus of adult rats. Neurosci. Lett.385, 70–75.10.1016/j.neulet.2005.05.041
59
MelcangiR. C.FroelichsthalP.MartiniL.VescoviA. L. (1996). Steroid metabolizing enzymes in pluripotential progenitor central nervous system cells: effect of differentiation and maturation. Neuroscience72, 467–475.10.1016/0306-4522(95)00522-6
60
MellonS. H. (2007). Neurosteroid regulation of central nervous system development. Pharmacol. Ther.116, 107–124.10.1016/j.pharmthera.2007.04.011
61
MellonS. H.GongW.SchonemannM. D. (2008). Endogenous and synthetic neurosteroids in treatment of Niemann-Pick type C disease. Brain Res. Rev.57, 410–420.10.1016/j.brainresrev.2007.05.012
62
MellonS. H.GriffinL. D.CompagnoneN. A. (2001). Biosynthesis and action of neurosteroids. Brain Res. Brain Res. Rev.37, 3–12.10.1016/S0165-0173(01)00109-6
63
MorgensternN. A.LombardiG.SchinderA. F. (2008). Newborn granule cells in the ageing dentate gyrus. J. Physiol. (Lond.)586, 3751–3757.10.1113/jphysiol.2008.154807
64
NaylorJ. C.KiltsJ. D.HuletteC. M.SteffensD. C.BlazerD. G.ErvinJ. F.StraussJ. L.AllenT. B.MassingM. W.PayneV. M.YoussefN. A.ShampineL. J.MarxC. E. (2010). Allopregnanolone levels are reduced in temporal cortex in patients with Alzheimer’s disease compared to cognitively intact control subjects. Biochim. Biophys. Acta1801, 951–959.10.1016/j.bbalip.2010.05.006
65
NguyenP. N.BilliardsS. S.WalkerD. W.HirstJ. J. (2003). Changes in 5alpha-pregnane steroids and neurosteroidogenic enzyme expression in the perinatal sheep. Pediatr. Res.53, 956–964.10.1203/01.PDR.0000064905.64688.10
66
OddoS.CaccamoA.KitazawaM.TsengB. P.LaferlaF. M. (2003a). Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol. Aging24, 1063–1070.10.1016/j.neurobiolaging.2003.08.012
67
OddoS.CaccamoA.ShepherdJ. D.MurphyM. P.GoldeT. E.KayedR.MetherateR.MattsonM. P.AkbariY.LaferlaF. M. (2003b). Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron39, 409–421.10.1016/S0896-6273(03)00434-3
68
Overstreet WadicheL.BrombergD. A.BensenA. L.WestbrookG. L. (2005). GABAergic signaling to newborn neurons in dentate gyrus. J. Neurophysiol.94, 4528–4532.10.1152/jn.00633.2005
69
Overstreet-WadicheL. S.BensenA. L.WestbrookG. L. (2006). Delayed development of adult-generated granule cells in dentate gyrus. J. Neurosci.26, 2326–2334.10.1523/JNEUROSCI.5508-05.2006
70
OwensD. F.KriegsteinA. R. (2002). Is there more to GABA than synaptic inhibition? Nature reviews. Neuroscience3, 715–727.
71
PereiraA. C.HuddlestonD. E.BrickmanA. M.SosunovA. A.HenR.McKhannG. M.SloanR.GageF. H.BrownT. R.SmallS. A. (2007). An in vivo correlate of exercise-induced neurogenesis in the adult dentate gyrus. Proc. Natl. Acad. Sci. U.S.A.104, 5638–5643.10.1073/pnas.0708611104
72
PerryR. J.WatsonP.HodgesJ. R. (2000). The nature and staging of attention dysfunction in early (minimal and mild) Alzheimer’s disease: relationship to episodic and semantic memory impairment. Neuropsychologia38, 252–271.10.1016/S0028-3932(99)00079-2
73
PieperA. A.XieS.CapotaE.EstillS. J.ZhongJ.LongJ. M.BeckerG. L.HuntingtonP.GoldmanS. E.ShenC. H.CapotaM.BrittJ. K.KottiT.UreK.BratD. J.WilliamsN. S.MacmillanK. S.NaidooJ.MelitoL.HsiehJ.De BrabanderJ.ReadyJ. M.McKnightS. L. (2010). Discovery of a proneurogenic, neuroprotective chemical. Cell142, 39–51.10.1016/j.cell.2010.06.018
74
ReddyD. S.RogawskiM. A. (2001). Enhanced anticonvulsant activity of neuroactive steroids in a rat model of catamenial epilepsy. Epilepsia42, 337–344.10.1046/j.1528-1157.2001.10200.x
75
RiddellD. R.ZhouH.ComeryT. A.KouranovaE.LoC. F.WarwickH. K.RingR. H.KirkseyY.AschmiesS.XuJ.KubekK.HirstW. D.GonzalesC.ChenY.MurphyE.LeonardS.VasylyevD.OganesianA.MartoneR. L.PangalosM. N.ReinhartP. H.JacobsenJ. S. (2007). The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer’s disease. Mol. Cell. Neurosci.34, 621–628.10.1016/j.mcn.2007.01.011
76
RodriguezJ. J.JonesV. C.TabuchiM.AllanS. M.KnightE. M.LaferlaF. M.OddoS.VerkhratskyA. (2008). Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS ONE3, e2935.10.1371/journal.pone.0002935
77
RodriguezJ. J.JonesV. C.VerkhratskyA. (2009). Impaired cell proliferation in the subventricular zone in an Alzheimer’s disease model. Neuroreport20, 907–912.10.1097/WNR.0b013e32832be77d
78
RodriguezJ. J.NoristaniH. N.OlabarriaM.FletcherJ.SomervilleT. D.YehC. Y.VerkhratskyA. (2011). Voluntary running and environmental enrichment restores impaired hippocampal neurogenesis in a triple transgenic mouse model of Alzheimer’s disease. Curr. Alzheimer Res.8, 707–717.10.2174/156720511797633214
79
RogawskiM. A. (2003). Progesterone, neurosteroids, and the hormonal basis of catamenial epilepsy. Ann. Neurol.53, 288–291.10.1002/ana.10534
80
RupprechtR.PapadopoulosV.RammesG.BaghaiT. C.FanJ.AkulaN.GroyerG.AdamsD.SchumacherM. (2010). Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug Discov.9, 971–988.10.1038/nrd3295
81
SchneiderL. S.InselP. S.WeinerM. W. (2011). Treatment with cholinesterase inhibitors and memantine of patients in the Alzheimer’s disease neuroimaging initiative. Arch. Neurol.68, 58–66.10.1001/archneurol.2011.69
82
SchultzJ. R.TuH.LukA.RepaJ. J.MedinaJ. C.LiL.SchwendnerS.WangS.ThoolenM.MangelsdorfD. J.LustigK. D.ShanB. (2000). Role of LXRs in control of lipogenesis. Genes Dev.14, 2831–2838.10.1101/gad.850400
83
SchumacherM.GuennounR.RobertF.CarelliC.GagoN.GhoumariA.Gonzalez DeniselleM. C.GonzalezS. L.IbanezC.LabombardaF.CoiriniH.BaulieuE. E.De NicolaA. F. (2004). Local synthesis and dual actions of progesterone in the nervous system: neuroprotection and myelination. Growth Horm. IGF Res.14(Suppl. A), S18–S33.10.1016/j.ghir.2004.03.007
84
ShenoyS. D.SpencerT. A.Mercer-HainesN. A.AlipourM.GarganoM. D.Runge-MorrisM.KocarekT. A. (2004). CYP3A induction by liver x receptor ligands in primary cultured rat and mouse hepatocytes is mediated by the pregnane X receptor. Drug Metab. Dispos.32, 66–71.10.1124/dmd.32.1.66
85
SinghC.LiuL.WangJ. M.IrwinR. W.YaoJ.ChenS.HenryS.ThompsonR. F.BrintonR. D. (2011). Allopregnanolone restores hippocampal-dependent learning and memory and neural progenitor survival in aging 3xTgAD and nonTg mice. Neurobiol. Aging. [Epub ahead of print].10.1016/j.neurobiolaging.2011.06.008
86
SipilaS.HuttuK.VoipioJ.KailaK. (2004). GABA uptake via GABA transporter-1 modulates GABAergic transmission in the immature hippocampus. J. Neurosci.24, 5877–5880.10.1523/JNEUROSCI.1287-04.2004
87
SoderpalmA. H.LindseyS.PurdyR. H.HaugerR.Wit DeH. (2004). Administration of progesterone produces mild sedative-like effects in men and women. Psychoneuroendocrinology29, 339–354.10.1016/S0306-4530(03)00033-7
88
StoneS. S.TeixeiraC. M.DevitoL. M.ZaslavskyK.JosselynS. A.LozanoA. M.FranklandP. W. (2011). Stimulation of entorhinal cortex promotes adult neurogenesis and facilitates spatial memory. J. Neurosci.31, 13469–13484.10.1523/JNEUROSCI.3100-11.2011
89
SunY.YaoJ.KimT. W.TallA. R. (2003). Expression of liver X receptor target genes decreases cellular amyloid beta peptide secretion. J. Biol. Chem.278, 27688–27694.10.1074/jbc.M212801200
90
TaupinP. (2009). Nootropic agents stimulate neurogenesis. Brain Cells, Inc.: WO2007104035. Expert Opin. Ther. Pat.19, 727–730.10.1517/13543770802665725
91
ThuretS.ToniN.AignerS.YeoG. W.GageF. H. (2009). Hippocampus-dependent learning is associated with adult neurogenesis in MRL/MpJ mice. Hippocampus19, 658–669.10.1002/hipo.20550
92
TimbyE.BalgardM.NybergS.SpigsetO.AnderssonA.Porankiewicz-AsplundJ.PurdyR. H.ZhuD.BackstromT.PoromaaI. S. (2006). Pharmacokinetic and behavioral effects of allopregnanolone in healthy women. Psychopharmacology (Berl.)186, 414–424.10.1007/s00213-005-0148-7
93
TodaH.HamaniC.FawcettA. P.HutchisonW. D.LozanoA. M. (2008). The regulation of adult rodent hippocampal neurogenesis by deep brain stimulation. J. Neurosurg.108, 132–138.10.3171/JNS/2008/108/01/0132
94
TozukaY.FukudaS.NambaT.SekiT.HisatsuneT. (2005). GABAergic excitation promotes neuronal differentiation in adult hippocampal progenitor cells. Neuron47, 803–815.10.1016/j.neuron.2005.08.023
95
TurkmenS.LofgrenM.BirznieceV.BackstromT.JohanssonI. M. (2006). Tolerance development to Morris water maze test impairments induced by acute allopregnanolone. Neuroscience139, 651–659.10.1016/j.neuroscience.2005.12.031
96
van BroekhovenF.BackstromT.Van LuijtelaarG.BuitelaarJ. K.SmitsP.VerkesR. J. (2007). Effects of allopregnanolone on sedation in men, and in women on oral contraceptives. Psychoneuroendocrinology32, 555–564.10.1016/j.psyneuen.2007.03.009
97
van PraagH.KempermannG.GageF. H. (1999). Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat. Neurosci.2, 266–270.10.1038/6368
98
van PraagH.SchinderA. F.ChristieB. R.ToniN.PalmerT. D.GageF. H. (2002). Functional neurogenesis in the adult hippocampus. Nature415, 1030–1034.10.1038/4151030a
99
WangJ. M.BrintonR. D. (2008). Allopregnanolone-induced rise in intracellular calcium in embryonic hippocampal neurons parallels their proliferative potential. BMC Neurosci.9(Suppl. 2), S11.10.1186/1471-2202-9-S2-S11
100
WangJ. M.JohnstonP. B.BallB. G.BrintonR. D. (2005). The neurosteroid allopregnanolone promotes proliferation of rodent and human neural progenitor cells and regulates cell-cycle gene and protein expression. J. Neurosci.25, 4706–4718.10.1523/JNEUROSCI.4127-04.2005
101
WangJ. M.LiuL.IrwinR. W.ChenS.BrintonR. D. (2008). Regenerative potential of allopregnanolone. Brain Res. Rev.57, 398–409.10.1016/j.brainresrev.2007.08.010
102
WangJ. M.SinghC.LiuL.IrwinR. W.ChenS.ChungE. J.ThompsonR. F.BrintonR. D. (2010). Allopregnanolone reverses neurogenic and cognitive deficits in mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A.107, 6498–6503.10.1073/pnas.0909422107
103
Weill-EngererS.DavidJ. P.SazdovitchV.LiereP.EychenneB.PianosA.SchumacherM.DelacourteA.BaulieuE. E.AkwaY. (2002). Neurosteroid quantification in human brain regions: comparison between Alzheimer’s and nondemented patients. J. Clin. Endocrinol. Metab.87, 5138–5143.10.1210/jc.2002-020878
104
WhitneyK. D.WatsonM. A.CollinsJ. L.BensonW. G.StoneT. M.NumerickM. J.TippinT. K.WilsonJ. G.WinegarD. A.KliewerS. A. (2002). Regulation of cholesterol homeostasis by the liver X receptors in the central nervous system. Mol. Endocrinol.16, 1378–1385.10.1210/me.16.6.1378
105
WhitwellJ. L.PrzybelskiS. A.WeigandS. D.KnopmanD. S.BoeveB. F.PetersenR. C.JackC. R.Jr. (2007). 3D maps from multiple MRI illustrate changing atrophy patterns as subjects progress from mild cognitive impairment to Alzheimer’s disease. Brain130, 1777–1786.10.1093/brain/awl388
106
WinnerB.KohlZ.GageF. H. (2011). Neurodegenerative disease and adult neurogenesis. Eur. J. Neurosci.33, 1139–1151.10.1111/j.1460-9568.2011.07613.x
107
XiongH.CallaghanD.JonesA.WalkerD. G.LueL. F.BeachT. G.SueL. I.WoulfeJ.XuH.StanimirovicD. B.ZhangW. (2008). Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol. Dis.29, 422–437.10.1016/j.nbd.2007.10.005
108
YangY.VarvelN. H.LambB. T.HerrupK. (2006). Ectopic cell cycle events link human Alzheimer’s disease and amyloid precursor protein transgenic mouse models. J. Neurosci.26, 775–784.10.1523/JNEUROSCI.4247-05.2006
109
ZarowC.LynessS. A.MortimerJ. A.ChuiH. C. (2003). Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch. Neurol.60, 337–341.10.1001/archneur.60.3.337
Summary
Keywords
allopregnanolone, Alzheimer’s disease, β-amyloid, neurogenesis, regeneration, cholesterol homeostasis, myelin, treatment regimen
Citation
Irwin RW, Wang JM, Chen S and Brinton RD (2012) Neuroregenerative Mechanisms of Allopregnanolone in Alzheimer’s Disease. Front. Endocrin. 2:117. doi: 10.3389/fendo.2011.00117
Received
26 October 2011
Accepted
27 December 2011
Published
12 January 2012
Volume
2 - 2011
Edited by
Hubert Vaudry, University of Rouen, France
Reviewed by
Synthia H. Mellon, University of California San Francisco, USA; Michael Schumacher, INSERM, France; Roberto Cosimo Melcangi, Università degli Studi di MIlano, Italy
Copyright
© 2012 Irwin, Wang, Chen and Brinton.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Roberta Diaz Brinton, Department of Pharmacology and Pharmaceutical Sciences, Pharmaceutical Sciences Center, University of Southern California, 1985 Zonal Avenue, Los Angeles, CA 90089-9121, USA. e-mail: rbrinton@usc.edu
This article was submitted to Frontiers in Neuroendocrine Science, a specialty of Frontiers in Endocrinology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.