

- 1INSERM U1016, Institut Cochin, Université Paris-Descartes, Paris, France
- 2CNRS UMR5203, INSERM U661, Institut de Génomique Fonctionnelle, Université Montpellier 1, 2, Montpellier, France
Kallmann syndrome (KS) is a developmental disease that associates hypogonadism and a deficiency of the sense of smell. The reproductive phenotype of KS results from the primary interruption of the olfactory, vomeronasal, and terminal nerve fibers in the frontonasal region, which in turn disrupts the embryonic migration of neuroendocrine gonadotropin-releasing hormone (GnRH) synthesizing cells from the nose to the brain. This is a highly heterogeneous genetic disease, and mutations in any of the nine genes identified so far have been found in approximately 30% of the KS patients. PROKR2 and PROK2, which encode the G protein-coupled prokineticin receptor-2 and its ligand prokineticin-2, respectively, are two of these genes. Homozygous knockout mice for the orthologous genes exhibit a phenotype reminiscent of the KS features, but biallelic mutations in PROKR2 or PROK2 (autosomal recessive mode of disease transmission) have been found only in a minority of the patients, whereas most patients carrying mutations in these genes are heterozygotes. The mutations, mainly missense mutations, have deleterious effects on PROKR2 signaling in transfected cells, ranging from defective cell surface-targeting of the receptor to defective coupling to G proteins or impaired receptor-ligand interaction, but the same mutations have also been found in apparently unaffected individuals, which suggests a digenic/oligogenic mode of inheritance of the disease in heterozygous patients. This non-Mendelian mode of inheritance has so far been confirmed only in a few patients. However, it may account for the unusually high proportion of KS sporadic cases compared to familial cases.
Introduction
Kallmann syndrome (KS) is a developmental disease that associates hypogonadotropic hypogonadism, due to gonadotropin-releasing hormone (GnRH) deficiency, and anosmia, related to the absence or hypoplasia of the olfactory bulbs (Kallmann, 1944; deMorsier, 1954; Naftolin et al., 1971). The degree of the hypogonadism and that of the smell deficiency can vary significantly, not only between unrelated KS patients, but also between patients from the same family. The prevalence of KS has been estimated at one out of 8000 in boys. In girls, the prevalence is thought to be five times lower, but it is probably underestimated because some affected females only have mild hypogonadism, and also because primary amenorrhea in females often remains unexplored (Jones and Kemmann, 1976).
Pathohistological studies of fetuses with olfactory bulb agenesis have shown that the reproductive phenotype of KS results from a pathological sequence in embryonic life, whereby premature interruption of the olfactory, vomeronasal, and terminal nerve fibers in the frontonasal region disrupts the migration of neuroendocrine GnRH cells, which normally migrate from the nose to the brain along these nerve fibers (Schwanzel-Fukuda and Pfaff, 1989; Teixeira et al., 2010). What causes the primary failure of these fibers to establish proper contact with the forebrain is, however, still unknown.
Kallmann syndrome is genetically heterogeneous and involves various modes of transmission, specifically, X-chromosome linked, autosomal recessive, autosomal dominant with incomplete penetrance, and also digenic/oligogenic inheritance (Dodé and Hardelin, 2009; Sykiotis et al., 2010). Because the common infertility in affected individuals and, most importantly, the incomplete penetrance of the disease impede genetic linkage analysis, researchers have used various strategies to identify genes involved in KS, including mutation screening in genes that are disrupted by deletion or translocation breakpoints in chromosomal rearrangements associated with the disease phenotype, and candidate gene approaches. Nine causal genes have been reported to date, namely, by chronological order of discovery, KAL1 (Franco et al., 1991; Legouis et al., 1991; Hardelin et al., 1992), FGFR1 (Dodé et al., 2003), PROKR2 and PROK2 (Dodé et al., 2006), FGF8 (Falardeau et al., 2008), CHD7 (Kim et al., 2008; Jongmans et al., 2009), WDR11 (Kim et al., 2010), HS6ST1 (Tornberg et al., 2011), and SEMA3A (Hanchate et al., 2012; Young et al., 2012) (Table 1). Various loss-of-function mutations in KAL1, encoding the extracellular matrix glycoprotein anosmin-1, and in FGFR1 or FGF8, encoding fibroblast growth factor receptor-1 and fibroblast growth factor-8, underlie the X-chromosome linked form and an autosomal dominant form of KS, respectively. Mutations in KAL1 and FGFR1/FGF8 account for roughly 8 and 10% of all KS cases, respectively. The KAL1 gene product, anosmin-1, binds to heparan-sulfate glycosaminoglycans, and may act as a co-receptor for FGF signaling through FGFR1, which also requires interaction with heparan-sulfate glycosaminoglycans for receptor activation. Mutations in the genes encoding heparan-sulfate 6-O-sulfotransferase 1, an enzyme involved in glycosaminoglycan modifications, WDR11, an intracellular protein that interacts with the transcription factor EMX1, and semaphorin 3A, a secreted protein involved in axonal pathfinding, have also been found in some KS patients. Mutations in the chromodomain helicase DNA-binding protein 7 gene (CHD7) are present in approximately 70% of the patients affected by the CHARGE syndrome, which in most patients includes KS (Pinto et al., 2005), and mutations in this gene have been found in some patients who initially presented with KS. Finally, mutations in PROKR2 and PROK2, encoding prokineticin receptor-2 and prokineticin-2, respectively, have been identified in approximately 9% of the KS patients, both in heterozygous and in homozygous or compound heterozygous states.
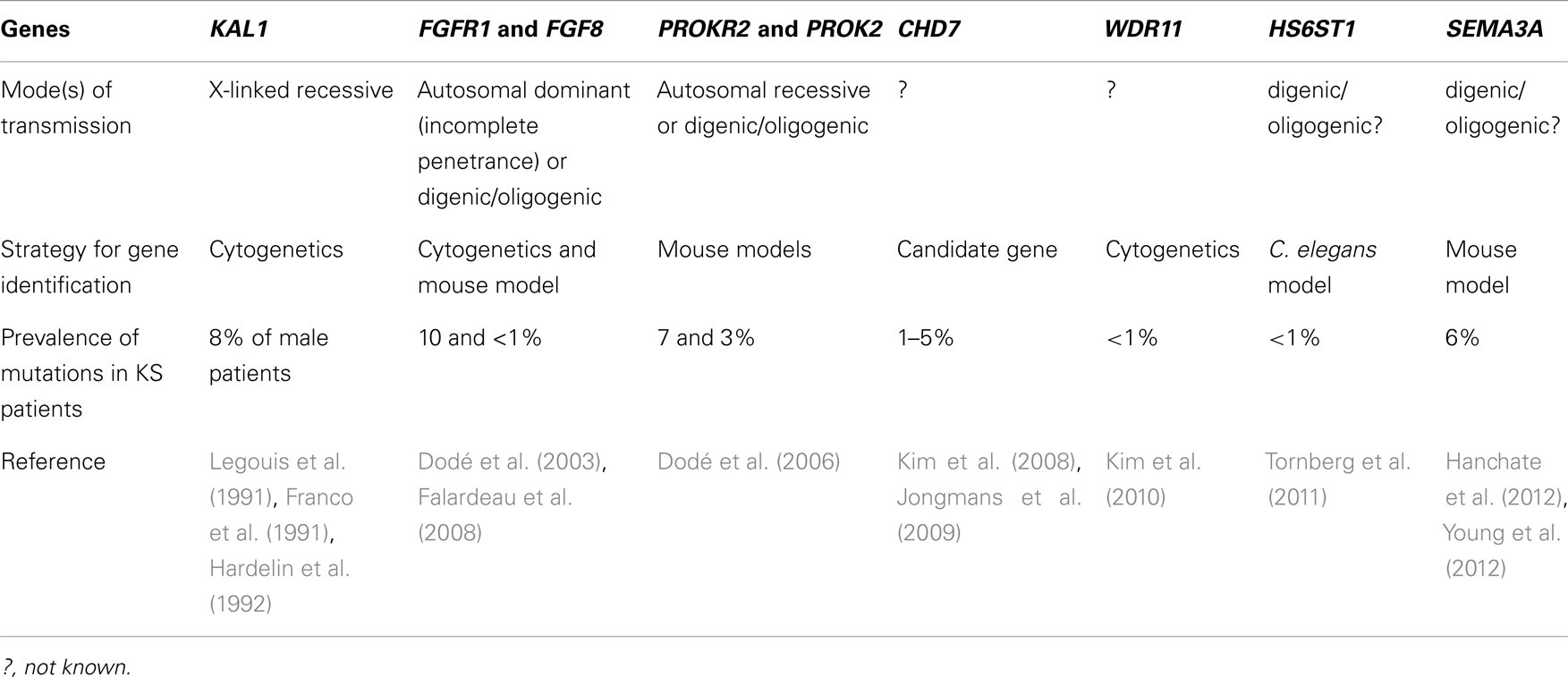
Table 1. KS genes.
Prokineticins and Their Receptors
Prokineticins are secreted cysteine-rich proteins that possess diverse biological activities. The first identified member of this family was isolated from the venom of the black mamba snake (Joubert and Strydom, 1980), and was named mamba intestinal toxin 1 (MIT1) owing to its ability to induce intestinal contraction (Schweitz et al., 1999). Then, a small protein of similar size (77 amino acid residues, 8 kDa), with 58% sequence identity with MIT1, was isolated from skin secretions in the amphibian Bombina variegata, and called Bv8 (Mollay et al., 1999). Soon after, two mammalian proteins of this family were identified and named prokineticin-1 and -2 (PROK1 and PROK2) (Li et al., 2001; Kaser et al., 2003). PROK1 has 80% sequence identity with MIT1 and 58% identity with PROK2 (Li et al., 2001). The amino-terminal domain of prokineticins contains a sequence of six amino acid residues (AVITGA), which is conserved in all mammalian and non-mammalian orthologs. Substitutions, deletions, or insertions to this hexapeptide result in the loss of agonist activity on prokineticin receptors (Kaser et al., 2003; Bullock et al., 2004). The carboxy-terminal region of prokineticins contains 10 cysteine residues forming five disulfide bonds. Apart from its potent effect on gastrointestinal smooth muscle contraction, PROK1 was also characterized as an angiogenic factor with specific effects on steroidogenic glands, thus earning its initial name EG-VEGF (endocrine gland vascular endothelial growth factor) (LeCouter et al., 2003). Mouse and human orthologs of Bv8, also known as PROK2, have been involved in a variety of biological activities, including effects on neuronal survival, gastrointestinal smooth muscle contraction (Li et al., 2001), circadian locomotor rhythm (Cheng et al., 2002), survival and migration of adrenal cortical capillary endothelial cells (LeCouter et al., 2003). PROK2 also has a role in appetite regulation and its anorectic effect is mediated partly by the melanocortin system (Gardiner et al., 2006).
Prokineticins can bind to two different G protein-coupled receptors, prokineticin receptor-1 and -2 (PROKR1 and PROKR2), which have about 85% sequence identity. These receptors were characterized simultaneously by three different groups (Lin et al., 2002; Masuda et al., 2002; Soga et al., 2002). Both are able to bind to PROK1 and PROK2 with similar nanomolar range affinities. They have a central core formed by seven transmembrane α-helical segments (TM1–TM7) connected by intracellular (i1–i3) and extracellular (e1–e3) loops, an extracellular amino-terminal end, and an intracellular carboxy-terminal end. These plasma membrane receptors operate as molecular switches to relay extracellular ligand-activation to intracellular heterotrimeric G proteins. PROKR1 is mainly expressed in peripheral tissues, including endocrine glands and organs of the reproductive system, the gastrointestinal tract, lungs, and the circulatory system (Soga et al., 2002; Battersby et al., 2004), whereas PROKR2 shows relatively localized distribution in the central nervous system (Cheng et al., 2002; Lin et al., 2002).
The olfactory bulb is one of the few areas in the mammalian brain that produce neurons throughout life. New interneurons originating from progenitors in the subventricular zone (SVZ) are continually added to the olfactory bulbs. The mRNAs of both receptors (PROKR1 and PROKR2) are expressed in the SVZ and the olfactory bulbs. The expression of PROKR1 have been detected in these areas although less abundantly than PROKR2. Whereas PROK1 mRNAs were not detected in any of these brain region (Ng et al., 2005). It was first shown that PROK2 functions as a chemoattractant for these neuronal progenitors, which follow a rostral migratory stream. Accordingly, PROK2 deficiency in mice leads to a loss of normal olfactory bulb architecture, and accumulation of neuronal progenitors in the rostral migratory stream (Ng et al., 2005). Soon after, it was found that Prokr2−/− knockout mice exhibit early hypoplasia of the olfactory bulbs and severe atrophy of the reproductive organs in both sexes, a phenotype reminiscent of the KS features. In addition, immunohistochemical analysis of these mice revealed that the neuroendocrine GnRH cells were absent from the hypothalamus (Matsumoto et al., 2006).
PROK2 is a clock-controlled gene: the level of its messenger RNA shows a circadian oscillation profile in the suprachiasmatic nuclei (Cheng et al., 2002; Li et al., 2006). It has been postulated that PROK2 signaling through PROKR2 is a suprachiasmatic nuclei clock output signal that regulates circadian rhythms (Prosser et al., 2007; Li et al., 2012). PROK2-null mice show accelerated acquisition of food anticipatory activity during a daytime food restriction (Li et al., 2006), exhibit reduced total sleep time predominantly during the light period, and also have an impaired response to sleep disturbance (Hu et al., 2007).
PROK2 is a functional target gene of proneural basic helix-loop-helix (bHLH) factors. Neurogenin-1 (NGN1) and MASH1 activate PROK2 transcription by binding to E-box motifs on the PROK2 promoter with the same set of E-boxes critical for another pair of bHLH factors, CLOCK and BMAL1, in the regulation of circadian clock (Cheng et al., 2002; Zhang et al., 2007).
Complex Genetics of Kallmann Syndrome Caused by Mutations in PROKR2 or PROK2
We first considered that PROKR2 was a relevant KS candidate because of the KS-like phenotype of PROKR2-null mice (see above). We thus sequenced the two coding exons of PROKR2 in a cohort of patients affected by KS, and identified 10 different mutations (one frame-shifting and nine missense mutations) in 14 patients, either in heterozygous state (10 cases) or in homozygous or compound heterozygous state (4 cases) (Dodé et al., 2006) (Table 2). Notably, most of these mutations were missense mutations, and many were also found in apparently unaffected individuals, thus initially raising some questions regarding their pathogenic role. A deleterious effect on the signaling activity of PROKR2 was, however, confirmed in transfected HEK-293 cells for most of the mutations (Cole et al., 2008; Monnier et al., 2009).
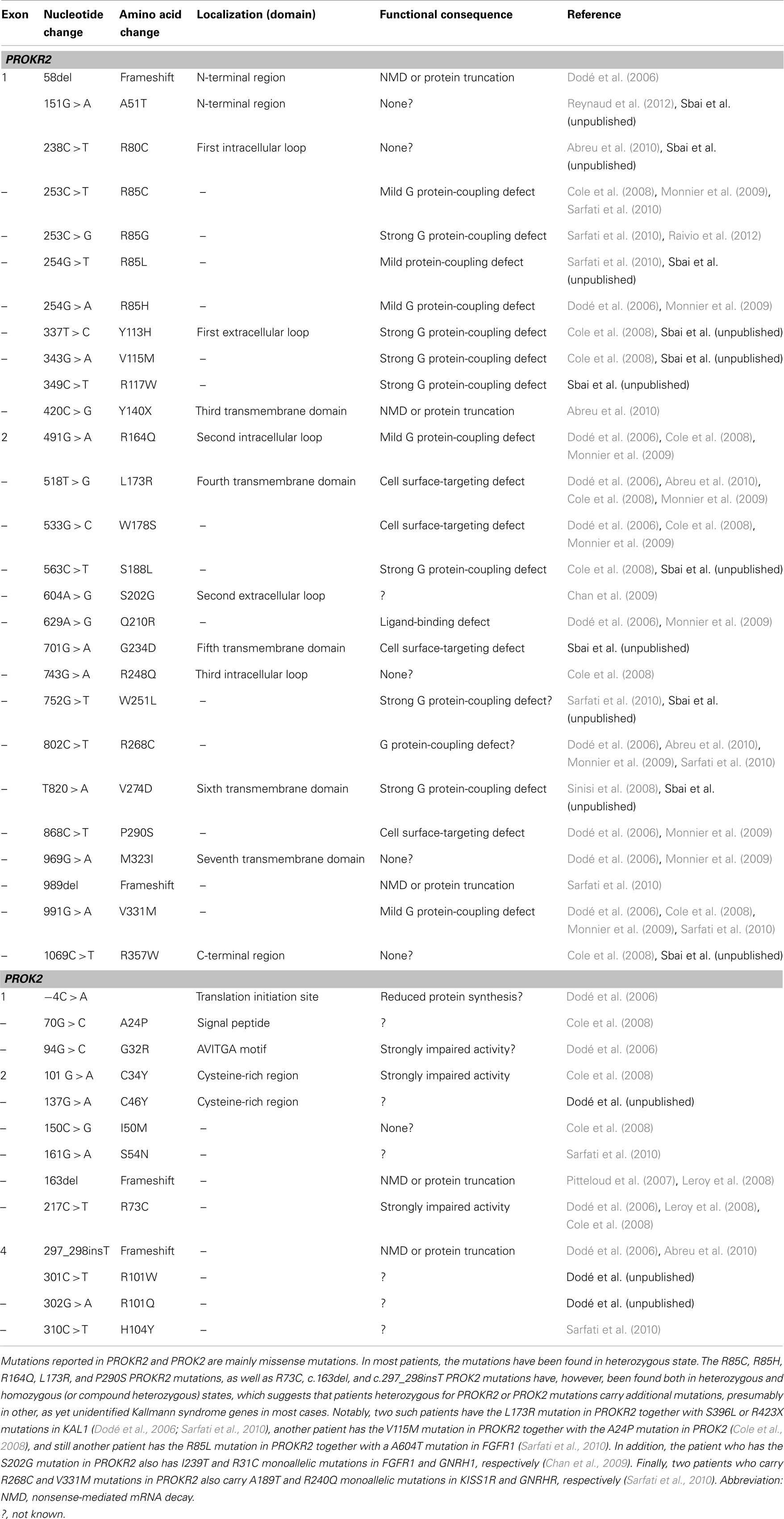
Table 2. PROKR2 and PROK2 mutations in Kallmann syndrome.
Then, we considered the possibility that mutations in PROK2 also account for some KS cases, especially since mutant mice defective in PROK2 showed a marked reduction in the size of their olfactory bulbs. PROK2 contains four coding exons (Bullock et al., 2004), of which exon 3, encoding an arginine- and lysine-rich peptide of 21 amino acid residues, may or may not be included in the mature transcript due to alternative splicing (Wechselberger et al., 1999) (Figure 1). We sequenced the entire coding sequence of PROK2 in the patients, and identified four different point mutations (two missense mutations, one frame-shifting mutation, and one single nucleotide substitution in the translation initiation sequence), all in the heterozygous state (Dodé et al., 2006). PROK2 mutations in homozygous state were subsequently found in a few patients, and a KS-like phenotype was concomitantly reported in PROK2-null mutant mice (Pitteloud et al., 2007; Leroy et al., 2008). Since then, additional mutations in PROKR2 and PROK2 have been reported in KS patients. A list of the mutations, together with the corresponding references, is provided in Table 2.
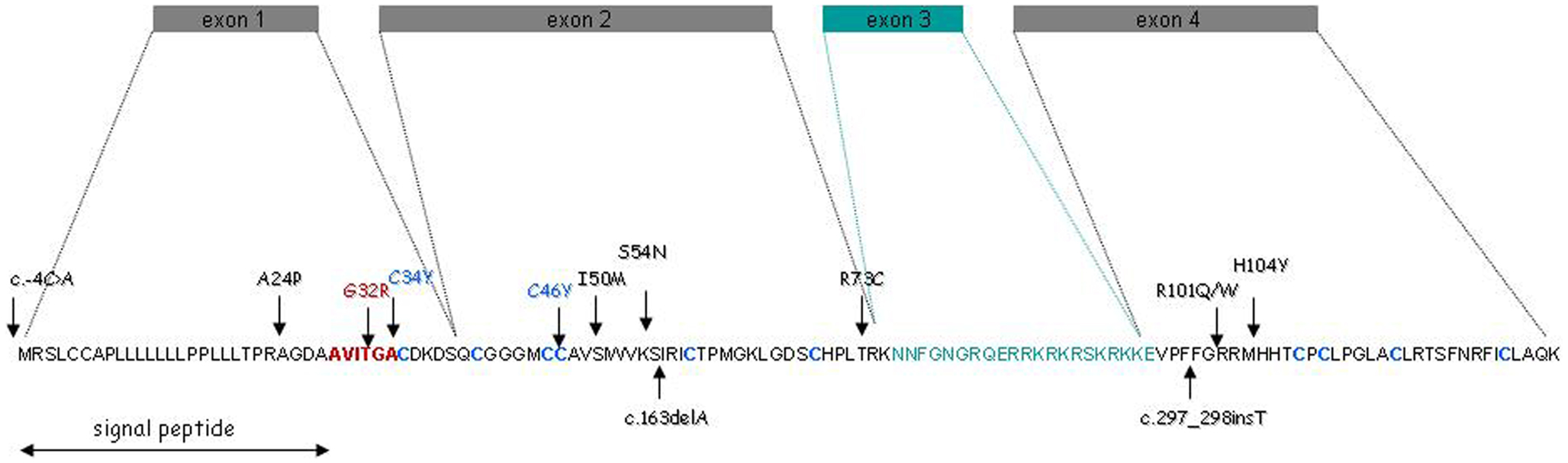
Figure 1. PROK2 gene structure and amino acid sequence. The green box and green amino acid sequence denote the alternative exon and encoded peptide, respectively. The AVITGA motif is shown in red, and the 10 cysteinyl residues (forming five disulfide bonds) are in blue. Vertical arrows indicate the positions of the mutations identified in the patients.
The finding, for given PROKR2 and PROK2 mutations, of both heterozygous and homozygous (or compound heterozygous) unrelated patients is quite remarkable, and argues in favor of a digenic or oligogenic mode of inheritance in heterozygous patients. To date, digenic inheritance of KS has been shown in few patients who had monoallelic missense mutations both in PROKR2 or PROK2, and in other KS genes (KAL1, FGFR1) or genes underlying normosmic congenital hypogonadotropic hypogonadism (GNRHR, KISS1R) (Dodé et al., 2006; Cole et al., 2008; Raivio et al., 2009; Martin et al., 2010; Sarfati et al., 2010). The other patients harboring monoallelic mutations in PROKR2 or PROK2 are expected to carry at least one additional pathogenic mutation in as yet uncharacterized genes. Indeed, mutations in any of the currently known KS genes have been identified in only 30% of all KS patients, thus indicating that other disease genes remain to be discovered. Notably, it has been found that patients carrying biallelic mutations in PROKR2 or PROK2 consistently have a severe, complete KS phenotype, whereas the phenotype of patients carrying monoallelic mutations in these genes is more variable, and likely depends on the additional genetic hits in these patients (Martin et al., 2010; Sarfati et al., 2010).
Kallmann syndrome patients who carry biallelic mutations in PROK2 or PROKR2 do not seem to have any of the non-olfactory, non-reproductive occasional anomalies that have been reported in the previously characterized genetic forms of the disease (i.e., X-linked KAL1 form and autosomal dominant KAL2 form), specifically, bimanual synkinesis, renal agenesis, dental agenesis, and cleft lip or palate, and even though sleep disorders or increased body mass index have been reported in some of these patients, there is so far no evidence that these clinical features can be ascribed to the PROKR2 and PROK2 mutations, despite potential roles of these genes in sleep-wake regulation and ingestive behavior (see above). In addition, plasma cortisol levels were measured during 24 h in five patients mutated in PROK2 or PROKR2, including one patient with biallelic PROKR2 mutations, and normal circadian variation was observed in all cases (Sarfati et al., 2010), which argues against a major contribution of PROK2/PROKR2 signaling to physiological circadian variation of plasma cortisol levels in humans. Of course, this result does not exclude the presence of more subtle defects in the patients.
Consequences of the PROKR2 and PROK2 Missense Mutations on Receptor Signaling Activity
A total of 24 PROKR2 missense mutations have been identified (Dodé et al., 2006; Cole et al., 2008; Sinisi et al., 2008; Chan et al., 2009; Abreu et al., 2010; Sarfati et al., 2010) in KS patients (Table 2; Figure 2). Most of the mutant receptors have been characterized regarding their ability to induce intracellular release of calcium upon PROK2-stimulation, their cell surface expression, and their PROK2-binding, in mammalian cell lines (Cole et al., 2008; Monnier et al., 2009; Abreu et al., 2012; Raivio et al., 2012; Sbai et al., in preparation). Although PROKR2 can activate different G protein pathways (Lin et al., 2002; Soga et al., 2002; Chen et al., 2005), its coupling to Gq, leading to intracellular release of calcium, represents the best characterized transduction mechanism. Only five mutants (A51T, R80C, R248Q, M321I, R357W) have properties similar to the wild-type PROKR2, thus calling into question the pathogenic effect of these missense variants. Notably, the R268C mutation has also been found in heterozygous state in a relatively large proportion (174 out of 2203, i.e., 7.9%) of individuals from the African American general population, and in homozygous state in six individuals from the same population (0.3%; see Exome Variant Server website URL: http://evs.gs.washington.edu/EVS/). Moreover, a clear deleterious effect of the R268C mutation on PROKR2 signaling through Gq protein activation could not be found in transfected HEK-293 cells, thus calling into question the pathogenic effect of this missense variant too. Most of the PROKR2 mutations, however, impair cell surface expression, ligand-binding, or G protein-binding of the receptor (Figure 2). Three mutations affecting conserved residues located in the middle of the transmembrane helices, W178S, G234D, and P290S, impede targeting of the receptor to the cell surface. The Q210R mutant receptor is present at the cell surface, but is not able to bind to the ligand. The other mutants impair, either mildly or strongly, intracellular release of calcium in the Gq signaling pathway, and for some of them this effect might be due to the low expression of the mutant at the cell surface. Interestingly, the mutations that mostly impair the signaling are located nearby the extracellular side or the intracellular side of the receptor, and for these mutants, the loss-of-function is associated to a loss of cell surface expression. The mutations that result in a mildly impaired signaling activity are located in the intracellular loops, in agreement with the important role played by these loops in G protein-coupling. In addition, when wild-type and mutant receptors were coexpressed in HEK-293 cells, none of the mutant receptors that were retained within the cells did affect cell surface-targeting of the wild-type receptor, and none of the mutant receptors properly addressed at the plasma membrane did affect wild-type receptor signaling activity. This argues against a dominant negative effect of the mutations in vivo.
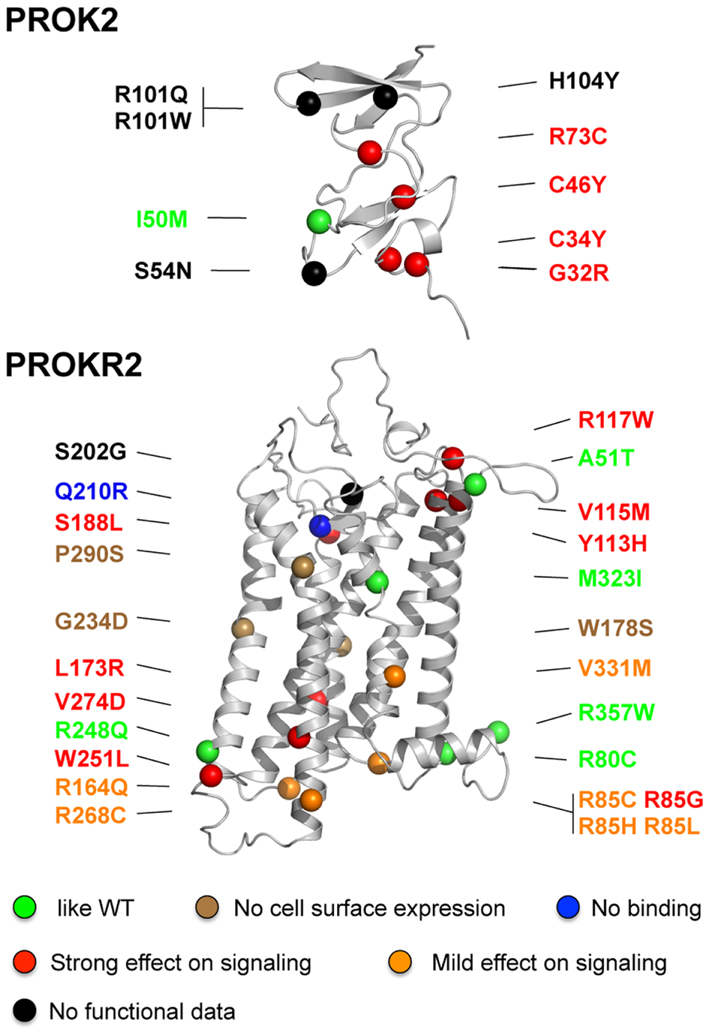
Figure 2. Positions of missense mutations in PROK2 and PROKR2 in the structural models of the ligand and the receptor. The mutations are classified in different categories according to their effects on PROKR2 signaling activity: similar to wild-type (green), absence of the receptor at the cell surface (brown), absence of ligand-binding (blue), and strong or mild effect on signaling (red and orange, respectively). The mutations for which functional data are not available are denoted in black. The colored balls indicate the atom of alpha carbon of the polypeptidic chain of the mutated residue. Note that residue R357 is located in the proximal part of the C-terminal region of the receptor.
A total of 10 missense mutations have been identified in PROK2 (Table 2, Figure 2) (Dodé et al., 2006; Cole et al., 2008; Leroy et al., 2008; Sarfati et al., 2010). A mutation located at the translation initiation site (−4C > A) likely reduced the protein synthesis. Mutations of conserved residues in prokineticins are expected to strongly impair PROK2 activity such as those in the N-terminal conserved region AVITGA important for prokineticin activity (G32R), and those of the conserved cysteines (C34Y and C46Y). In addition, mutations that introduce an additional cysteine (R73C) in the cysteine-rich region of PROK2 may affect folding of the hormone then leading to a loss-of-function. This is consistent with the functional analysis of Cole et al. (2008) who have examined the signaling properties of three PROK2 mutants: the C34Y and R73C mutations totally abolished and strongly impaired the intracellular calcium release activity of PROKR2, respectively, whereas the I50M mutation had an activity similar to that of the wild-type PROK2. The functional consequence of the other mutations (A24P, S54N, R101Q, R101W, and H104Y) are more difficult to predict in the absence of model of the complex between PROK2 and PROKR2. Further studies will be necessary to better analyze whether the mutations in PROK2 affect the process of binding and/or activation of PROKR2 by the hormone.
Conclusion
The characterization of new genes involved in KS is a difficult goal. Because of the hypogonadotropic hypogonadism, sizes of the KS families are small and the mode of inheritance is very often difficult to establish. Animal models in which a gene has been inactivated and involving a KS phenotype are an alternative approach for the identification of new KS genes. The inactivation of PROK2 or PROKR2 lead to defective olfactory morphogenesis and hypogonadism in mice and humans. Curiously, PROK2 and PROKR2 mutations in homozygous state were found in a few patients and the main part of the KS patients carried only heterozygous mutations. In all of the functional studies done so far, only Gq signaling pathways have been investigated to characterize the PROK2 and PROKR2 mutants and further studies will be necessary to analyze other signaling pathways. Interestingly, few patients who had monoallelic missense mutations both in PROK2 or PROKR2, and in other KS or normosmic congenital hypogonadotropic hypogonadism genes, raising the idea of oligogenism. So far, this hypothesis has been validated only in a few patients suggesting that the second mutation in the other heterozygous patients resides in unexplored regions of the genes or as yet undiscovered KS genes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jean-Pierre Hardelin for critical reading of the manuscript, Oualid Sbai and Carine Monnier for sharing unpublished data, and Guillaume Lebon for his help in preparing the 3D models of PROKR2 and PROK2. This work was supported by ANR-09-GENO-017-01 and GIS maladies rares (Project A09051KS).
References
Abreu, A. P., Kaiser, U. B., and Latronico, A. C. (2010). The role of prokineticins in the pathogenesis of hypogonadotropic hypogonadism. Neuroendocrinology 91, 283–290.
Abreu, A. P., Noel, S. D., Xu, S., Carroll, R. S., Latronico, A. C., and Kaiser, U. B. (2012). Evidence of the importance of the first intracellular loop of prokineticin receptor 2 in receptor function. Mol. Endocrinol. 26, 1417–1427.
Battersby, S., Critchley, H. O., Morgan, K., Millar, R. P., and Jabbour, H. N. (2004). Expression and regulation of the prokineticins (endocrine gland-derived vascular endothelial growth factor and Bv8) and their receptors in the human endometrium across the menstrual cycle. J. Clin. Endocrinol. Metab. 89, 2463–2469.
Bullock, C. M., Li, J.-D., and Zhou, Q.-Y. (2004). Structural determinants required for the bioactivities of prokineticins and identification of prokineticin receptor antagonists. Mol. Pharmacol. 65, 582–588.
Chan, Y. M., de Guillebon, A., Lang-Muritano, M., Plummer, L., Cerrato, F., Tsiaras, S., et al. (2009). GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. U.S.A. 106, 11703–11708.
Chen, J., Kuei, C., Sutton, S., Wilson, S., Yu, J., Kamme, F., et al. (2005). Identification and pharmacological characterization of prokineticin 2 β as a selective ligand for prokineticin receptor 1. Mol. Pharmacol. 67, 2070–2076.
Cheng, M., Bullock, C. M., Li, C., Lee, A. G., Bermak, J. C., Belluzzi, J., et al. (2002). Prokineticin 2 transmits the behavioural circadian rhythm of the suprachiasmatic nucleus. Nature 417, 405–410.
Cole, L. W., Sidis, Y., Zhang, C., Quinton, R., Plummer, L., Pignatelli, D., et al. (2008). Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin-releasing hormone deficiency: molecular genetics and clinical spectrum. J. Clin. Endocrinol. Metab. 93, 3551–3559.
deMorsier, G. (1954). Etudes sur les dysraphies crânio-encéphaliques. I. Agénésie des lobes olfactifs (telencéphaloschizis latéral) et des commissures calleuse et antérieure (telencéphaloschizis médian). La dysplasie olfacto-génitale. Schweiz. Arch. Neurol. Psychiatr. 74, 309–361.
Dodé, C., Levilliers, J., Dupont, J.-M., De Paepe, A., Le Dû, N., Soussi-Yanicostas, N., et al. (2003). Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 33, 463–465.
Dodé, C., Teixeira, L., Levilliers, J., Fouveaut, C., Bouchard, P., Kottler, M.-L., et al. (2006). Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2:e175. doi:10.1371/journal.pgen.0020175
Falardeau, J., Chung, W. C., Beenken, A., Raivio, T., Plummer, L., Sidis, Y., et al. (2008). Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J. Clin. Invest. 118, 2822–2831.
Franco, B., Guioli, S., Pragliola, A., Incerti, B., Bardoni, B., Tonlorenzi, R., et al. (1991). A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature 353, 529–536.
Gardiner, J. V., Bataveljic, A., Patel, N. A., Bewick, G. A., Roy, D., Campbell, D., et al. (2006). Prokineticin 2 is a hypothalamic neuropeptide that potently inhibits food intake. Diabetes 59, 397–406.
Hanchate, N. K., Giacobini, P., Lhuillier, P., Parkash, J., Espy, C., Fouveaut, C., et al. (2012). SEMA3A, a gene involved in axonal pathfinding, is mutated in some patients with Kallmann syndrome. PLoS Genet. 8:e1002896. doi:10.1371/journal.pgen.1002896
Hardelin, J.-P., Levilliers, J., del Castillo, I., Cohen-Salmon, M., Legouis, R., Blanchard, S., et al. (1992). X chromosome-linked Kallmann syndrome: stop mutations validate the candidate gene. Proc. Natl. Acad. Sci. U.S.A. 89, 8190–8194.
Hu, W. P., Li, J. D., Zhang, C., Boehmer, L., Siegel, J. M., and Zhou, Q. Y. (2007). Altered circadian and homeostatic sleep regulation in prokineticin 2-deficient mice. Sleep 30, 247–256.
Jones, J., and Kemmann, E. (1976). Olfacto-genital dysplasia in the female. Obstet. Gynecol. Annu. 5, 443.
Jongmans, M. C., van Ravenswaaij-Arts, C. M., Pitteloud, N., Ogata, T., Sato, N., Claahsen-van der Grinten, H. L., et al. (2009). CHD7 mutations in patients initially diagnosed with Kallmann syndrome – the clinical overlap with CHARGE syndrome. Clin. Genet. 75, 65–71.
Joubert, F. J., and Strydom, D. J. (1980). Snake venom: the amino acid sequence of protein A from Dendroaspis polylepis (black mamba) venom. Hoppe-Seyler’s Z. Physiol. Chem. 361, 1787–1794.
Kallmann, F. J. (1944). The genetic aspects of primary eunuchoidism. Am. J. Ment. Defic. 48, 203–236.
Kaser, A., Winklmayr, M., Lepperdinger, G., and Kreil, G. (2003). The AVIT protein family. EMBO Rep. 4, 469–473.
Kim, H. G., Ahn, J.-W., Kurth, I., Ullmann, R., Kim, H.-T., Kulharya, A., et al. (2010). WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 87, 465–479.
Kim, H. G., Kurth, I., Lan, F., Meliciani, I., Wenzel, W., Eom, S. H., et al. (2008). Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 83, 511–519.
LeCouter, J., Lin, R., Tejada, M., Frantz, G., Peale, F., Hillan, K. J., et al. (2003). The endocrine-gland-derived VEGF homologue Bv8 promotes angiogenesis in the testis: localization of Bv8 receptors to endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 100, 2685–2690.
Legouis, R., Hardelin, J.-P., Levilliers, J., Claverie, J.-M., Compain, S., Wunderle, V., et al. (1991). The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell 67, 423–435.
Leroy, C., Fouveaut, C., Leclercq, S., Jacquemont, S., Boullay, H. D., Lespinasse, J., et al. (2008). Biallelic mutations in the prokineticin-2 gene in two sporadic cases of Kallmann syndrome. Eur. J. Hum. Genet. 16, 865–868.
Li, J. D., Hu, W. P., Boehmer, L., Cheng, M. Y., Lee, A. G., Siegel, J. M., et al. (2006). Attenuated circadian rhythms in mice lacking the prokineticin 2 gene. J. Neurosci. 26, 11615–11623.
Li, J. D., Hu, W. P., and Zhou, Q. Y. (2012). The circadian output signals from the suprachiasmatic nuclei. Prog. Brain Res. 199, 119–127.
Li, M., Bullock, C. M., Knauer, D. J., Ehlert, F. J., and Zhou, Q. Y. (2001). Identification of two prokineticin cDNAs: recombinant proteins potently contract gastrointestinal smooth muscle. Mol. Pharmacol. 59, 692–698.
Lin, D. C., Bullock, C. M., Ehlert, F. J., Tian, H., and Zhou, Q. Y. (2002). Identification and molecular characterization of two closely related G protein-coupled receptors activated by prokineticins/endocrine gland vascular endothelial growth factor. J. Biol. Chem. 277, 19276–19280.
Martin, C., Balasubramanian, R., Dwyer, A. A., Au, M. G., Sidis, Y., Kaiser, U. B., et al. (2010). The role of the prokineticin 2 pathway in human reproduction: evidence from the study of human and murine gene mutations. Endocr. Rev. 32, 225–246.
Masuda, Y., Takatsu, Y., Terao, Y., Kumano, S., Ishibashi, Y., Suenaga, M., et al. (2002). Isolation and identification of EG-VEGF/prokineticins as cognate ligands for two orphan G-protein-coupled-receptors. Biochem. Biophys. Res. Commun. 293, 396–402.
Matsumoto, S.-I., Yamazaki, C., Masumoto, K.-H., Nagano, M., Naito, M., Soga, T., et al. (2006). Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc. Natl. Acad. Sci. U.S.A. 103, 4140–4145.
Mollay, C., Wechselberger, C., Mignogna, G., Negri, L., Melchiorri, P., Barra, D., et al. (1999). Bv8, a small protein from frog skin and its homologue from snake venom induce hyperalgesia in rats. Eur. J. Pharmacol. 374, 189–196.
Monnier, C., Dodé, C., Fabre, L., Teixeira, L., Labesse, G., Pin, J.-P., et al. (2009). PROKR2 missense mutations associated with Kallmann syndrome impair receptor signalling activity. Hum. Mol. Genet. 18, 75–81.
Naftolin, F., Harris, G. W., and Bobrow, M. (1971). Effect of purified luteinizing hormone releasing factor on normal and hypogonadotropic anosmic men. Nature 232, 496–497.
Ng, K. L., Li, J.-D., Cheng, M. Y., Leslie, F. M., Lee, A. G., and Zhou, Q.-Y. (2005). Dependence of olfactory bulb morphogenesis on prokineticin 2 signaling. Nature 308, 1923–1927.
Pinto, G., Abadie, V., Mesnage, R., Blustajn, J., Cabrol, S., et al. (2005). CHARGE syndrome includes hypogonadotropic hypogonadism and abnormal olfactory bulb development. J. Clin. Endocrinol. Metab. 90, 5621–5626.
Pitteloud, N., Zhang, C., Pignatelli, D., Li, J.-D., Raivio, T., Cole, L. W., et al. (2007). Loss-of-function mutation in the prokineticin 2 gene causes Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. U.S.A. 104, 17447–17452.
Prosser, H. M., Bradley, A., Chesham, J. E., Ebing, F. J. P., Hastings, M. H., and Maywood, E. S. (2007). Prokineticin receptor 2 (Prokr2) is essential for the regulation of circadian behaviour by suprachiasmatic nuclei. Proc. Natl. Acad. Sci. U.S.A. 104, 648–653.
Raivio, T., Avbelj, M., McCabe, M. J., Romero, C. J., Dwyer, A. A., Tommiska, J., et al. (2012). Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J. Clin. Endocrinol. Metab. 97, E694–E699.
Raivio, T., Sidis, Y., Plummer, L., Chen, H., Ma, J., Mukherjee, A., et al. (2009). Impaired fibroblast growth factor receptor 1 signaling as a cause of normosmic idiopathic hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 94, 4380–4390.
Reynaud, R., Jayakody, S. A., Monnier, C., Saveanu, A., Bouligand, J., Guedj, A. M., et al. (2012). PROKR2 variants in multiple hypopituitarism with pituitary stalk interruption. J. Clin. Endocrinol. Metab. 97, 1068–1073.
Sarfati, J., Guiochon-Mantel, A., Rondard, P., Arnulf, I., Garcia-Pineiro, A., Wolczynski, S., et al. (2010). A comparative phenotypic study of Kallmann syndrome patients carrying monoallelic and biallelic mutations in the prokineticin 2 or prokineticin receptor 2 genes. J. Clin. Endocrinol. Metab. 95, 659–969.
Schwanzel-Fukuda, M., and Pfaff, D. W. (1989). Origin of luteinizing hormone-releasing hormone neurons. Nature 338, 161–164.
Schweitz, H., Pacaud, P., Diochot, S., Moinier, D., and Lazdunski, M. (1999). MIT1, a black mamba toxin with a new and highly potent activity on intestinal contraction. FEBS Lett. 461, 183–188.
Sinisi, A. A., Asci, R., Bellastella, G., Maione, L., Esposito, D., Elefante, A., et al. (2008). Homozygous mutation in the prokineticin-receptor 2 gene (Val274Asp) presenting as reversible Kallmann syndrome and persistent oligozoospermia: case report. Hum. Reprod. 23, 2380–2384.
Soga, T., Matsumoto, S., Oda, T., Saito, T., Hiyama, H., Takasaki, J., et al. (2002). Molecular cloning and characterization of prokineticin receptors. Biochim. Biophys. Acta 1579, 173–179.
Sykiotis, G. P., Plummer, L., Hughes, V. A., Au, M., Durrani, S., Nayak-Young, S., et al. (2010). Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc. Natl. Acad. Sci. U.S.A. 107, 15140–15144.
Teixeira, L., Guimiot, F., Dodé, C., Fallet-Bianco, C., Millar, R. P., Delezoide, A.-L., et al. (2010). Defective migration of neuroendocrine GnRH cells in human arrhinencephalic conditions. J. Clin. Invest. 120, 3668–3672.
Tornberg, J., Sykiotis, G. P., Keefe, K., Plummer, L., Hoang, X., Hall, J. E., et al. (2011). Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. U.S.A. 108, 11524–11529.
Wechselberger, C., Puglisi, R., Engel, E., Lepperdinger, G., Boitani, C., and Kreil, G. (1999). The mammalian homologues of frog Bv8 are mainly expressed in spermatocytes. FEBS Lett. 462, 177–181.
Young, J., Metay, C., Bouligand, J., Tou, B., Francou, B., Maione, L., et al. (2012). SEMA3A deletion in a family with Kallmann syndrome validates the role of semaphorin 3A in human puberty and olfactory system development. Hum. Reprod. 27, 1460–1465.
Keywords: PROK2, PROKR2, Kallmann syndrome, hypogonadotropic hypogonadism, anosmia, digenic/oligogenic mode of inheritance
Citation: Dodé C and Rondard P (2013) PROK2/PROKR2 signaling and Kallmann syndrome. Front. Endocrinol. 4:19. doi: 10.3389/fendo.2013.00019
Received: 17 September 2012; Paper pending published: 25 October 2012;
Accepted: 14 February 2013; Published online: 12 April 2013.
Edited by:
Hubert Vaudry, University of Rouen, FranceCopyright: © 2013 Dodé and Rondard. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Catherine Dodé, INSERM U1016, Institut Cochin, Département de génétique et développement, 24 rue du Faubourg Saint-Jacques, 75014 Paris, France. e-mail:Y2F0aGVyaW5lLmRvZGVAaW5zZXJtLmZy