Abstract
Dynamin-2 is a ubiquitously expressed mechano-GTPase involved in different stages of the secretory pathway. Its most well-known function relates to the scission of nascent vesicles from the plasma membrane during endocytosis; however, it also participates in the formation of new vesicles from the Golgi network, vesicle trafficking, fusion processes and in the regulation of microtubule, and actin cytoskeleton dynamics. Over the last 8 years, more than 20 mutations in the dynamin-2 gene have been associated to two hereditary neuromuscular disorders: Charcot–Marie–Tooth neuropathy and centronuclear myopathy. Most of these mutations are grouped in the pleckstrin homology domain; however, there are no common mutations associated with both disorders, suggesting that they differently impact on dynamin-2 function in diverse tissues. In this review, we discuss the impact of these disease-related mutations on dynamin-2 function during vesicle trafficking and endocytotic processes.
Introduction
Dynamin was identified for the first time almost 25 years ago as a 100 kDa microtubule-associated protein that induced microtubule bundles and promoted microtubule sliding in vivo (1). As described by the same authors, the motor activity of dynamin required ATP and other co-purified polypeptides (1). A year later, the same group cloned and sequenced dynamin, and found that it contained a consensus GTP-binding site (2), and subsequently characterized its GTPase activity (3). At present, three dynamin isoforms encoded by three distinct genes (DNM1, DNM2, and DNM3) have been described in mammals (4). These exhibit approximately 80% homology in their sequences, yet they differ in their tissue expression pattern; dynamin-1 is mainly expressed in neuronal tissue, dynamin-2 is ubiquitously expressed, and dynamin-3 is expressed in brain, testis, and lungs (5). Of these three dynamin isoforms, only dynamin-2 appears to play a pleiotropic role during embryonic development (6). In fact, studies in knock-out animals show that deletion of dynamin-1 or -3 can be compensated by the other dynamin isoforms (7), while the deletion of dynamin-2 causes early embryonic lethality (8). Moreover, as discussed below, mutations in DMN2 result in severe hereditary neuropathies and myopathies in humans, strongly suggesting that dynamin-2 has more susceptible functions in the nervous and skeletal muscle tissues.
All dynamin isoforms exhibit at least four alternatively spliced variants, resulting in different dynamin proteins (5) that share a primary structure comprising: a large amino-terminal GTPase domain (G-domain) that binds and hydrolyzes GTP; a middle and a GTPase effector domains (GED) that form a “stalk” structurally essential region; a pleckstrin homology domain (PH) that binds inositol phospholipids and a carboxy-terminal proline and arginine rich-domain (PRD) that allows interaction with SH3-domain-containing-proteins (5) (Figure 1).
Figure 1
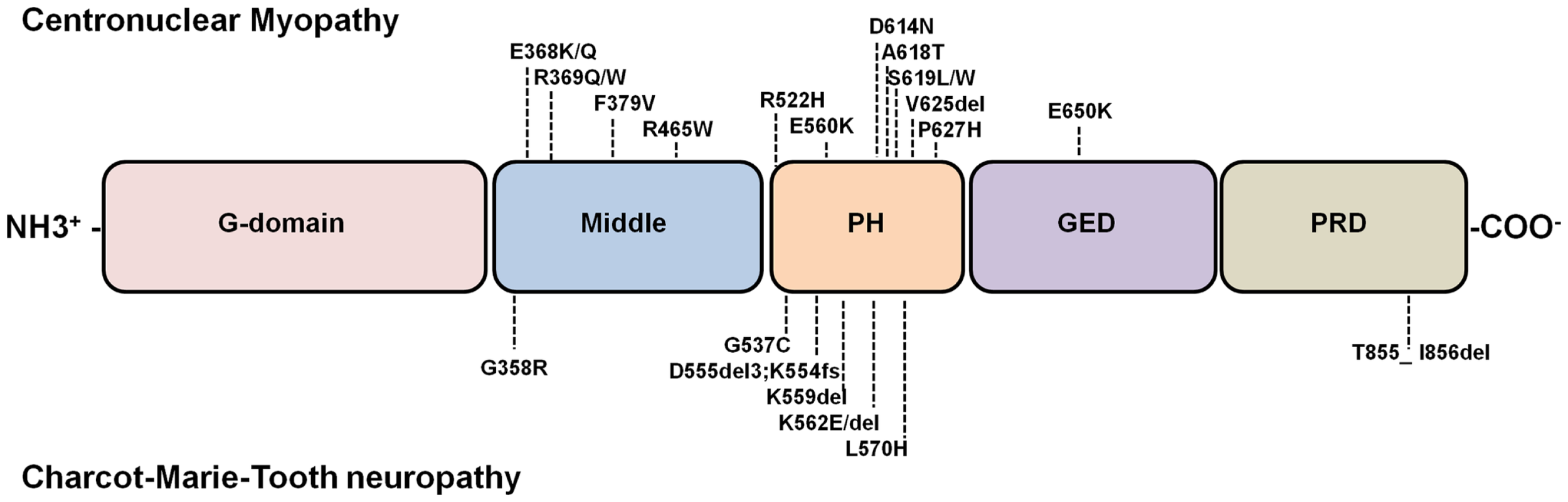
Diagram of dynamin structure and localization of dynamin-2 mutations linked to CNM and CMT. Dynamins are multimodular proteins comprising five highly conserved structural domains: a large N-terminal GTPase domain (G-domain), a middle domain, a PH domain that bind phosphoinositides, a GTPase effector domain (GED), and a C-terminal proline rich domain (PRD) that interacts with SH3-domain containing proteins. Most common disease-related dynamin-2 mutations are represented. Note that almost all dynamin-2 mutations identified in CNM and CMT patients are clustered into the PH domains; only one CNM-linked mutation has been found in GED, and one related to CMT has been identified in the PRD. The D555del3 mutation is one of the products of a 9-bp deletion in the exon 14 (1652_1659 + 1delATGAGGAGg) of the dynamin-2 gene (9). This gene deletion also results in a 65-kDa truncated protein (9). For that reason here is described as D555del3, K554fs. An updated database of DNM2 mutations is at the website www.umd.be/DNM2/ (10).
Dynamin function relies on its ability to form high order oligomers, and its self-assembly is necessary to promote its catalytic activity. Purified dynamin has been shown to spontaneously polymerize in the presence of negatively charged tubular templates such as lipid membranes (11), microtubules (3, 12), or actin bundles (13, 14) as well as after incubation in low ionic strength solutions (15). Over the last years several cryo-electron microscopy (16–18) and X-ray crystallographic studies (19, 20) of dynamin and its domains (21–24) have allowed a better understanding of the mechanisms mediating dynamin oligomerization. It appears that the stable dimers formed by the crossed interaction between the “stalk” regions of monomeric dynamins (20, 25) are the basic unit that allows dynamin polymerization (18), thus promoting the GTPase activation required for membrane remodeling and scission in different cellular processes.
In the present review, we discuss the different roles of dynamin during endocytosis, vesicle trafficking, and exocytosis, specially focusing in dynamin-2, and how disease-linked mutations in dynamin-2 gene alter such cellular processes.
Dynamin as a Key Component of Endocytosis and Vesicle Recycling
Dynamin is a GTPase that plays a crucial role in the recycling of secretory vesicle in neuroendocrine cells (26).
The first evidence suggesting a role for dynamin in endocytosis came from the mapping and characterization of the Drosophila shibire gene, which was identified to be the Drosophila homolog of mammal dynamin (27, 28). Drosophila bearing mutations in the shibire gene exhibited a rapid and reversible paralysis at temperatures exceeding 29°C (29). The first ultrastructural analyses of synaptic terminals of shibire mutants showed a decreased number of synaptic vesicles and accumulation of “collared pits” suggesting a blocked step in the endocytotic process (30, 31). These ultrastructural changes were also observed in garland cells, a type of cell considered to be very active in endocytosis, where horseradish peroxidase uptake activity was also reduced, thus confirming an alteration in endocytosis in the shibire mutants (32). The role of dynamin in endocytosis in mammalian cells was later demonstrated using dynamin mutants with reduced GTPase activity (33, 34). The use of the non-hydrolyzable GTP analog GTP-γ-S allowed the visualization of endocytotic pits with elongated necks, decorated by electron-dense rings positive for dynamin immunoreactivity, showing that dynamin oligomerizes around the neck of endocytotic pits (35). Moreover, the fact that the dynamin mutant K44A, defective in GTP binding and hydrolysis, specifically blocked the coated vesicle formation without affecting the coat assembly and invagination revealed that dynamin is required for the constriction and subsequent budding of the coated vesicles (36). However, later studies suggested that dynamin also plays a role during a pre-collar stage, when the clathrin lattice is growing (37, 38). In this stage, dynamin may function as a scaffolding molecule that interacts with SH3 domain-containing proteins that control coated pit assembly and maturation (37). The recruitment of dynamin at endocytosis sites (39) also depends on its interaction with SH3-domain-containing proteins and phosphoinositides present at the plasma membrane via its PRD (40) and PH domain, respectively (41).
Regarding the mechanism by which dynamin catalyzes membrane fission, it has been proposed that the assembly of dynamin into helical oligomers around the neck of clathrin-coated pit promotes the dimerization of G domains of adjacent helical rungs, leading to the hydrolysis of GTP (18). The GTP hydrolysis triggers a conformational change in the dynamin polymer, allowing the constriction of the dynamin ring (18, 42, 43). The ring constriction strength then drives the constriction of the membrane neck, increasing membrane curvature (44). Such change in membrane curvature raises the local elastic energy, reducing the energy barrier to fission, and subsequently triggering the spontaneous fission at the boundary between the dynamin ring and the bare membranes (44).
The ability of dynamin to catalyze membrane fission is not only required in CME, but is also needed in other types of endocytotic pathways that are independent of clathrin. For instance, dynamin is required for caveolae-mediated endocytosis (45), which is essential for the uptake of molecules such as the complement C5b-9 complex (46). Dynamin also participates in the internalization of the β-chain of the interleukin-2 receptor through a clathrin independent, but RhoA dependent process, which is inhibited by the overexpression of a dominant negative mutant of dynamin GTPase activity (47). The entry in host cells of many pathogens and toxins such as anthrax toxin (48), Ebola virus (49), HIV (50), or Hepatitis C virus (51) also require the participation of dynamin.
Finally, the role of dynamin in vesicle formation is not only restricted to the plasma membrane and, as discussed below; its function is also needed in intracellular compartments.
Role of Dynamin in Vesicle Trafficking and Golgi Function
Dynamin-mediated vesicle budding and membrane fission has also been reported in intracellular compartments such as endosomes (52, 53) and Golgi complex (54). Regarding dynamin-2 participation in vesicle trafficking from endosomes, this protein appears to play a role in two different steps: (1) the vesicle transport from late endosomes to the Golgi compartment (52) and (2) the recycling pathways from early endosomes (53). However, the establishment of dynamin-2 participation in the post-Golgi vesicle trafficking has been more controversial. Pioneer reports showed that the transport of vesicles from the Golgi to the cell surface or to lysosomes was independent of dynamin (36), and according to these results, no evidence of endogenous dynamin-2 localization in the Trans-Golgi network (TGN) nor of its participation in vesicle formation from this compartment were observed using different cell lines (55). Nevertheless, contemporary studies showed that ectopically expressed dynamin-2 localizes in the TGN in hepatocytes (54) and that the formation of clathrin-coated pits from Golgi membranes, in a cell-free assay, was inhibited in the presence of anti-dynamin antibodies, thus indicating the importance of dynamin at this level (54). Furthermore, canine kidney cells expressing a GTPase defective dynamin-2 mutant showed a restricted traffic of the protein p75 from the Golgi to the apical membrane (56). Also, the overexpression of a dominant negative mutant of dynamin-2 was shown to lead to the retention of proteins and accumulation of cisternae at the Golgi compartment, suggesting a role of dynamin-2 in keeping both the structure and function of the TGN (57).
In agreement with a role of dynamin-2 in Golgi vesicle formation, it was demonstrated that endogenous dynamin-2 localizes in the TGN in neuroendocrine mouse pituitary corticotrope cells (58), where it interacts with the βγ subunit of G-proteins via its PH domain. Interestingly, the overexpression of the purified PH domain induced the translocation of dynamin-2 from the Golgi complex to the plasma membrane, increasing receptor-mediated endocytosis but inhibiting basal and CRH-induced secretion of β-endorphins, suggesting a key role of dynamin-2 in the secretory pathway (58). Several studies have highlighted the importance of dynamin-2 to the proper traffic of nascent proteins from the TGN to the plasma membrane, a process that seems to be dependent on the actin cytoskeleton. In this regard, a subset of actin filaments anchored to the Golgi membrane via the small GTPase Arf-1 appears to form complexes with dynamin-2 and the actin-binding-protein cortactin, allowing the emergence and post-Golgi trafficking of secretory vesicles (59). Other proteins such as LimK1 and its substrate cofilin (60), syndapin, and the Wiskott–Aldrich-Syndrome-protein (WASP) (61) have been also involved in the regulation of the peri-Golgi actin cytoskeleton and in the dynamin-mediated transport of secretory vesicles from Golgi to the plasma membrane. Additionally, dynamin-2 function is necessary for the Golgi fragmentation and vesicle segregation induced by cholesterol in HeLa cells (62) and for the Golgi vesiculation induced by the c-SRC kinase activation (63) further supporting a pivotal role of dynamin in the Golgi dynamics along the secretory pathway.
Dynamin as a Facilitator of Membrane Fusion
Dynamins have been involved in different types of fusion processes. Among them regulated exocytosis in neuroendocrine cells (64–69), acrosomal reaction (70), cell-to-cell fusion (71, 72), and fusion of virus with host cells (73, 74).
The first evidences showing the involvement of dynamin in exocytosis came from experiments performed at the beginning of 2000, which suggested that dynamin was involved in kiss-and-run, a transient mode of exocytosis, in neuroendocrine cells (64–66). In this type of exocytosis, the vesicle partially releases its content, and then it is recovered intact (75, 76). It was then hypothesized that dynamin would allow the reclosure of the vesicle mouth, acting through a mechanism similar to that described for vesicle formation during endocytosis (77). However, the mechanism by which dynamin controls the quantal release of hormones still remains unclear. More intriguing is the mechanism by which dynamin facilitates the fusion process during exocytosis. For instance, dynamin-2 reportedly favors granule secretion in both natural killer (78) and insulin-secreting cells (79). More recently, it was reported that, in chromaffin cells, dynamin-1 speeds up the expansion of the fusion pore, an intermediate structure formed during exocytosis, in a GTPase activity-dependent fashion (68). Therefore, both dynamin-1 and -2 appear to be involved in fusion processes. A possible explanation for this function is that dynamin interacts with SNARE proteins or SNARE-interacting proteins. In this regard, dynamin-2 has been shown to associate to secretory granules in chromaffin cells (80) via its interaction with syntaxin (80) and synaptophysin (67). In mammalian sperm, dynamin-2 associates with the SNARE regulatory protein complexin I (81), where it favors membrane fusion events during acrosomal exocytosis (70). In yeast, the dynamin homolog Vps1p interacts with the t-SNARE Vamp3; the disruption of this association with an antibody against Vps1p inhibits the fusion reaction (82). More recently, Peters’ lab has demonstrated that Vps1 binds to the Qa SNARE Vamp3 and controls trans-SNARE formation, which is essential for membrane fusion in yeasts (83).
An alternative explanation is that dynamin controls fusion events via actin cytoskeleton dynamics. In this regard, cortical actin is dynamically rearranged during regulated exocytosis in neuroendocrine cells acting as a barrier as well as a carrier for the access of the secretory granules to the plasma membrane (84–86). Actin filaments also control the fusion pore expansion (87), as dynamin does (68). As we discuss below, given that dynamin regulates actin organization (13, 14, 88), it is plausible to assume that its actions on fusion processes relay on its ability to modulate actin dynamics. According to this idea, we have recently demonstrated that endogenous dynamin-2 directs a Ca2+-dependent polymerization of cortical actin in adrenal chromaffin cells. Interestingly, both cortical actin and dynamin-2 regulate the initial fusion pore expansion and quantal release of transmitters, suggesting a synergistic action during exocytosis (89). Corey Smith and Collaborators found that the fusion pore expansion in chromaffin cells is controlled by the association of dynamin-1 with syndapin (69), a protein that modulates actin polymerization through neural-Wiskott–Aldrich-syndrome-protein (N-WASP) (90).
The role of dynamin-2 in fusion processes has also been extended to the fusion of myoblasts to form multinucleated myotubes (71). Interestingly, the GTPase activity of dynamin-2 is required at a stage that follows hemifusion but precedes the expansion of the fusion pores (71). The underlying mechanism is still unclear, but it could explain the muscular dysfunction in centronuclear myopathies caused by dynamin-2 mutations. Dynamin-2 has also been involved in the cell-to-cell fusion triggered by HIV-1 virus infection (72) and in fusion between HIV-1 virus and endosomes (73, 74). The latter authors proposed that dynamin promotes the expansion of the fusion pore that connects the HIV-1 envelope and the endosomal membrane, but the possible mechanism remains to be clarified.
Taken together, these findings indicate a pleiotropic role of dynamin in membrane fusion. Although dynamin does not promote membrane fusion by itself, it seems to act after a hemifusion state, facilitating the expansion of the fusion pore (71). The underlying mechanism probably relies on dynamin ability to sense membrane curvature and remodel membranes (91).
Dynamin as a Regulator of Microtubule Instability
Although dynamin was first identified as a microtubules-associated protein (1) its specific role in microtubule integrity and dynamics is still unclear. Using papain-digestion experiments in vitro, Herskovits and Co-workers observed that the entire PRD constituted a microtubule-binding site (34). Later, it was demonstrated that only the PRD amino-terminal region was necessary for the association of dynamin to microtubules, whereas its C-terminus appeared to negatively regulate this interaction (92). Pioneering studies also described that dynamin polymerized around microtubules, interconnecting them and allowing bundle formation (1), and in turn, microtubules seemed to stimulate dynamin GTPase activity (12). Other findings showed that the middle domain of dynamin-2 binds to γ-tubulin (93) locating dynamin-2 to the centrosome, therefore suggesting its participation during centrosome cohesion (93). More recently, dynamin-2 was found to be enriched in microtubule bundles at the mitotic spindle of mitotic cells, playing a key role during the cell cycle progression (94). In 2009, Tanabe and Takei observed that depletion of dynamin-2 in COS-7 cells, led to an abnormal accumulation of stable microtubules, consequently inducing disturbance of the microtubules-dependent membrane trafficking. These authors did not find alterations in microtubule assembly in cells that expressed a specific interfering RNA against dynamin-2, but they did report increased stability of pre-existing microtubules, suggesting a role of dynamin-2 in microtubule dynamics instability (95). This dynamin role appears to be independent of its GTPase activity. On the other hand, a deletion of three amino acid residues located at the β3/β4 loops of the PH domain induced decoration of microtubules and accumulation of acetylated tubulin, indicating that this region is required for the correct bundling of microtubules (95). The authors also proposed that this action of dynamin-2 on microtubule instability is necessary for a proper formation of Golgi network and vesicle trafficking.
Dynamin as a Director of Actin Cytoskeleton Dynamics
Dynamin-2 is recruited to different actin-rich structures such as phagocytic cups (96), podosomes (97), Listeria actin (98), lamellipodia (99), and filopodia (88), supporting a connection between dynamin-2 function and actin cytoskeleton polymerization. Moreover, both actin and dynamin-2 have regulatory functions during T cells activation (100), phagocytosis (101), and clathrin-dependent endocytosis (102). In the latter, actin and dynamin-2 have been shown to exhibit a synergistic action, where one modulates the recruitment of the other, and both participate in membrane remodeling and scission (103, 104).
In all these processes, dynamin appears to promote actin assembly in a manner dependent on its GTPase activity (14). The mechanism is still unclear, yet it is probably a consequence of its association with several nucleation promoting factors (NPF) via a PRD-SH3 interaction; among them Abp1 (105), the N-WASP (106), and cortactin (13, 88), all of which have been described as dynamin-partners during actin polymerization.
In 2010, Gu and Co-workers reported a direct interaction between dynamin and short actin filaments (F-actin). The latter promoted dynamin oligomerization, and in turn dynamin induced F-actin elongation (14). These authors suggested a model wherein dynamin would favor actin polymerization by removing the “caping”-protein gelsolin of the “barbed” ends (14). More recently, Yamada et al. (88) demonstrated that dynamin induces actin bundle stabilization in a way dependent on its association with cortactin in growth cone filopodia, where they seem to form ring-structures along actin filaments that stabilize F-actin bundles (88). Since dynamin oligomerization enhances its catalytic activity (14), its association with cortactin during actin polymerization fits better with its role as a GTPase than that of a mere “uncapping” protein.
Impact of Disease-Related Dynamin-2 Mutations in Vesicle Trafficking and Endocytosis
Specific missense mutations and short deletions into the structural domains of dynamin-2 have been associated with two congenital autosomic neuromuscular disorders: Charcot–Marie–Tooth neuropathy (CMT) (9) and centronuclear myopathy (CNM) (107). While CMT is a demyelinating disease affecting peripheral nerves (108), CNM is characterized by a progressive weakness and atrophy of distal muscles, usually involving facial and extraocular musculature (109–111). Although most dynamin-2 mutations linked to CMT and CNM are located in the middle and PH domains, there are no common mutations to both disorders (112) and the molecular mechanisms that lead to the neuropathy or myopathy remain obscure. Figure 1 shows the localization of the CMT and CNM dynamin-2 mutations described until now, and Table 1 summarizes their impacts on different cellular processes.
Table 1
| Cellular process impaired | CNM mutations | CMT mutations |
|---|---|---|
| Intracellular calcium concentration in KI mice myofibers (113) | R465W | |
| Protein export from Golgi to plasma membrane in dynamin-2 (114) | E368K, R465W | D555del3, L570H |
| Clathrin-mediated endocytosis in: | ||
| COS-1 and/or COS-7 cells (95, 112, 115) | R465W, R522H, E560K, S619L/W, V625del, P627H | K558E, K562E |
| RT4 Schwann cells and NSC-34 motor neurons (116) | G358R, G537C, K562E, K559del, L570H | |
| Embryonic fibroblasts from homozygous KI mice (117) | R465W | |
| Clathrin-independent endocytosis (114) | E368K, R465W | D555del3, L570H |
| Raft-dependent endocytosis (114) | E368K, R465W | D555del3, L570H |
| Myelination in dorsal root ganglia (116) | G358R, G537C, K559del K562E, K562del, L570H | |
| Demyelination in dorsal root ganglia (116) | G358R, K562E | |
| Autophagy and autophagosome maturation (117) | R465W |
Cellular function alterations induced by CNM and CMT mutants.
Regarding how these disease-related mutations affect dynamin-2 oligomerization and catalytic properties, in vitro studies have revealed that the CNM-linked mutations E368K/Q, R369Q/W, and R465W located into the middle domain of dynamin-2 (107) as well as the mutations A618T, S619L/W, and V625del clustered into the PH domain (118) exhibit an increased stability of the oligomers and an enhanced GTPase activity (119, 120). However, how these and other dynamin-2 mutations impact on dynamin-dependent cellular processes is still under discussion.
Given that the best-known role of dynamin-2 is to catalyze membrane scission during endocytosis, most of the studies regarding the mechanism underlying CNM associated to mutations in dynamin-2 have been focused in this cellular process. However, the findings are contradictory. The overexpression of the CNM-linked mutants R465W, R522H, S619L, P627H V625del, and E650K impaired CME in COS-1 and COS-7 cells (112, 115). The fact that the CNM mutant D614N causes intracellular mislocalization of both dynamin-2 and clathrin (121) suggests that an impairment in CME could be a consequence of the anomalous distribution of dynamin-2 and other endocytotic proteins. In opposition to the aforementioned findings, fibroblasts from patients harboring the mutations R465W or S619L reportedly display normal CME (112). In a similar manner, embryonic fibroblasts from heterozygous knock-in (KI) mice carrying the mutation R465W exhibited no defects in CME (113). Unlike the heterozygous R465W KI mice, the homozygous animals displayed impaired receptor-mediated endocytosis, indicating that this mutation differentially impacts on CME according to the heterozygous or homozygous carrier state. In this regard, fibroblasts from patients harboring the homozygous mutation F379V, the first one linked to a lethal congenital syndrome, also exhibited dysfunctional endocytosis (6). In order to compare the effects of dynamin-2 mutations associated to CNM in a context that mimics the homozygous and heterozygous states, Schmid and Collaborators (114) established stable cell lines from dynamin-2 conditional null mouse fibroblasts and expressed two CNM-linked dynamin-2 mutants (E368K and R465W). Cells expressing comparable levels of wild-type dynamin-2 and the given mutant were selected to evaluate clathrin-dependent and independent endocytosis, and also vesicle trafficking. The authors showed that none of these mutations affected CME in the condition that mimics the heterozygous state; and they were able to fully rescue transferrin uptake in dynamin-2 KO cells. Nevertheless, all these mutants impaired clathrin-independent endocytosis of epidermal grow factor receptors and raft-dependent endocytosis of cholera toxin. Furthermore, also in the condition that mimics the heterozygous state, all the mutants disrupted trafficking of p75/neurotrophin receptor from Golgi to plasma membrane. It seems therefore plausible that the physiological dysfunctions in CNM patients harboring heterozygous mutations in dynamin-2 are not a consequence of defective CME, but other dynamin-2-dependent processes, such as clathrin-independent endocytosis or vesicle trafficking could be affected in these conditions.
Regarding the mechanism underlying CMT associated to dynamin-2 mutations, Sidiropoulos and Co-workers (116) suggested that an impairment of CME in Schwanm cells plays an important role in the pathogenesis of CMT neuropathy. They showed that the CMT mutants K562E, G358R, G537C, and L570H, but not the CNM mutants R465W and E560K, strongly reduced myelination in dorsal root ganglia explant cultures derived from heterozygous embryos carrying a dynamin-2 null allele, thus mimicking the heterozygous state. These CMT mutants also reduced transferrin internalization in Schwann cells and motor neurons. Conversely, none of the 11 CNM mutations evaluated in that work had any effect on transferrin internalization, neither in Schwann cells nor in motor neurons, suggesting that CMT and CNM mutations affect different cellular processes.
The aforementioned cellular impairments induced by CNM- and CMT-mutations, as well as others not previously mentioned, are summarized in the Table 1.
Concluding Remarks
The secretory pathway plays a pivotal role in mammalian cell function. Its disruption underlies many known diseases, such as those described herein. Dynamin-2 is critical in such mechanisms, as it regulates Golgi structure and function, cytoskeletal integrity, vesicle trafficking, and most interestingly, membrane fusion.
In the last 10 years, more than 20 disease-linked mutations have been reported in the dynamin-2 gene, affecting different dynamin-mediated cellular processes. Nevertheless, their impact on neuroendocrine tissues remains obscure. Also elusive are the mechanisms by which the said mutations affect actin cytoskeleton dynamics and exocytosis, processes which are dynamin-dependent and critical in the course of the secretory pathway. The latter opens an interesting research field that could prove useful to understand the mechanisms involved in the pathogenesis of dynamin-2 disease-related mutations.
Several diseases caused by genetic modifications remain as the final frontier in medicine, and although pivotal hallmarks such as the sequencing of the human genome have been attained, we still have much to learn about the cellular and molecular consequences of diverse gene mutations, specially, those that are common in many tissues. In this regard, dynamin-2 is a ubiquitous protein with diverse pleiotropic roles, whose mutations cause tissue-specific phenotypes. The existence of pertinent cell and animal models will be extremely useful for better understanding how dynamin-2 mutations impact on its properties and related functions, and consequently in the identification of potential therapeutic targets.
Statements
Acknowledgments
Fondecyt 1110552 (Chile); Proyecto Anillo ACT-1121 (CONICYT); Fondos ICM-ECONOMIA Project P09-022-F (Chile).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
Shpetner HS Vallee RB . Identification of dynamin, a novel mechanochemical enzyme that mediates interactions between microtubules. Cell (1989) 59:421–32.10.1016/0092-8674(89)90027-5
2
Obar RA Collins CA Hammarback JA Shpetner HS Vallee RB . Molecular cloning of the microtubule-associated mechanochemical enzyme dynamin reveals homology with a new family of GTP-binding proteins. Nature (1990) 347:256–61.10.1038/347256a0
3
Shpetner HS Vallee RB . Dynamin is a GTPase stimulated to high levels of activity by microtubules. Nature (1992) 355:733–5.10.1038/355733a0
4
Praefcke GJ McMahon HT . The dynamin superfamily: universal membrane tubulation and fission molecules?Nat Rev Mol Cell Biol (2004) 5:133–47.10.1038/nrm1313
5
Cao H Garcia F McNiven MA . Differential distribution of dynamin isoforms in mammalian cells. Mol Biol Cell (1998) 9:2595–609.10.1091/mbc.9.9.2595
6
Koutsopoulos OS Kretz C Weller CM Roux A Mojzisova H Böhm J et al Dynamin 2 homozygous mutation in humans with a lethal congenital syndrome. Eur J Hum Genet (2012) 21:637–42.10.1038/ejhg.2012.226
7
Raimondi A Ferguson SM Lou X Armbruster M Giovedi S Messa M et al Overlapping role of dynamin isoforms in synaptic vesicle endocytosis. Neuron (2011) 70:100–14.10.1016/j.neuron.2011.04.031
8
Ferguson SM Raimondi A Paradise S Mesaki K Ferguson A Destaing O et al Coordinated actions of actin and BAR proteins upstream of dynamin at endocytic clathrin-coated pits. Dev Cell (2009) 17:811–22.10.1016/j.devcel.2009.11.005.coordinated
9
Züchner S Noureddine M Kennerson M Verhoeven K Claeys K De Jonghe P et al Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet (2005) 37:289–94.10.1002/mrd.20660
10
Böhm J Biancalana V Dechene ET Bitoun M Pierson CR Schaefer E et al Mutation spectrum in the large GTPase dynamin 2, and genotype-phenotype correlation in autosomal dominant centronuclear myopathy. Hum Mutat (2012) 33:949–59.10.1002/humu.22067
11
Tuma PL Stachniak MC Collins CA . Activation of dynamin GTPase by acidic phospholipids and endogenous rat brain vesicles. J Biol Chem (1993) 268:17240–6.
12
Maeda K Nakata T Noda Y Sato-Yoshitake R Hirokawa N . Interaction of dynamin with microtubules: its structure and GTPase activity investigated by using highly purified dynamin. Mol Biol Cell (1992) 3:1181–94.10.1091/mbc.3.10.1181
13
Mooren OL Kotova TI Moore AJ Schafer DA . Dynamin2 GTPase and cortactin remodel actin filaments. J Biol Chem (2009) 284:23995–4005.10.1074/jbc.M109.024398
14
Gu C Yaddanapudi S Weins A Osborn T Reiser J Pollak M et al Direct dynamin-actin interactions regulate the actin cytoskeleton. EMBO J (2010) 29:3593–606.10.1038/emboj.2010.249
15
Hinshaw JE Schmid SL . Dynamin self-assembles into rings suggesting a mechanism for coated vesicle budding. Nature (1995) 374:190–2.10.1038/374190a0
16
Zhang P Hinshaw JE . Three-dimensional reconstruction of dynamin in the constricted state. Nat Cell Biol (2001) 3:922–6.10.1038/ncb1001-922
17
Danino D Moon KH Hinshaw JE . Rapid constriction of lipid bilayers by the mechanochemical enzyme dynamin. J Struct Biol (2004) 147:259–67.10.1016/j.jsb.2004.04.005
18
Chappie JS Mears JA Fang S Leonard M Schmid SL Milligan RA et al A pseudoatomic model of the dynamin polymer identifies a hydrolysis-dependent powerstroke. Cell (2011) 147:209–22.10.1016/j.cell.2011.09.003
19
Ford MG Jenni S Nunnari J . The crystal structure of dynamin. Nature (2011) 477:561–6.10.1038/nature10441
20
Faelber K Posor Y Gao S Held M Roske Y Schulze D et al Crystal structure of nucleotide-free dynamin. Nature (2011) 477:556–60.10.1038/nature10369
21
Timm D Salim K Gout I Guruprasad L Waterfield M Blundell T . Crystal structure of the pleckstrin homology domain from dynamin. Nat Struct Biol (1994) 1:782–8.10.1038/nsb1194-782
22
Niemann HH Knetsch ML Scherer A Manstein DJ Kull FJ . Crystal structure of a dynamin GTPase domain in both nucleotide-free and GDP-bound forms. EMBO J (2001) 20:5813–21.10.1093/emboj/20.21.5813
23
Reubold TF Eschenburg S Becker A Leonard M Schmid SL Vallee RB et al Crystal structure of the GTPase domain of rat dynamin 1. Proc Natl Acad Sci U S A (2005) 102:13093–8.10.1073/pnas.0506491102
24
Chappie JS Acharya S Liu Y Leonard M Pucadyil TJ Schmid SL . An intramolecular signaling element that modulates dynamin function in vitro and in vivo. Mol Biol Cell (2009) 20:3561–71.10.1091/mbc.E09
25
Chappie JS Acharya S Leonard M Schmid SL Dyda F . G domain dimerization controls dynamin’s assembly-stimulated GTPase activity. Nature (2010) 465:43540.10.1038/nature09032
26
Cárdenas AM Marengo FD . Rapid endocytosis and vesicle recycling in neuroendocrine cells. Cell Mol Neurobiol (2010) 30:1365–70.10.1007/s10571-010-9579-8
27
van der Bliek AM Meyerowitz EM . Dynamin-like protein encoded by the Drosophila shibire gene associated with vesicular traffic. Nature (1991) 351:411–4.10.1038/351411a0
28
Chen MS Obar RA Schroeder CC Austin TW Poodry CA Wadsworth SC et al Multiple forms of dynamin are encoded by shibire, a Drosophila gene involved in endocytosis. Nature (1991) 351:583–6.10.1038/351583a0
29
Grigliatti TA Hall L Rosenbluth R Suzuki DT . Temperature-sensitive mutations in Drosophila melanogaster. XIV. A selection of immobile adults. Mol Gen Genet (1973) 120:107–14.10.1007/BF00267238
30
Poodry CA Edgar L . Reversible alteration in the neuromuscular junctions of Drosophila melanogaster bearing a temperature-sensitive mutation, shibire. J Cell Biol (1979) 81:520–7.10.1083/jcb.81.3.520
31
Kosaka T Ikeda K . Possible temperature-dependent blockage of synaptic vesicle recycling induced by a single gene mutation in Drosophila. J Neurobiol (1983) 14:207–25.10.1002/neu.480140305
32
Kosaka T Ikeda K . Reversible blockage of membrane retrieval and endocytosis in the garland cell of the temperature-sensitive mutant of Drosophila melanogaster, shibirets1. J Cell Biol (1983) 97:499–507.10.1083/jcb.97.2.499
33
van der Bliek AM Redelmeier TE Damke H Tisdale EJ Meyerowitz EM Schmid SL . Mutations in human dynamin block an intermediate stage in coated vesicle formation. J Cell Biol (1993) 122:553–63.10.1083/jcb.122.3.553
34
Herskovits JS Burgess CC Obar RA Vallee RB . Effects of mutant rat dynamin on endocytosis. J Cell Biol (1993) 122:565–78.10.1083/jcb.122.3.565
35
Takei K McPherson PS Schmid SL De Camilli P . Tubular membrane invaginations coated by dynamin rings are induced by GTP-gamma S in nerve terminals. Nature (1995) 374:186–90.10.1038/374186a0
36
Damke H Baba T Warnock DE Schmid SL Jolla L . Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J Cell Biol (1994) 127:915–34.10.1083/jcb.127.4.915
37
Narayanan R Leonard M Song BD Schmid SL Ramaswami M . An internal GAP domain negatively regulates presynaptic dynamin in vivo: a two-step model for dynamin function. J Cell Biol (2005) 169:117–26.10.1083/jcb.200502042
38
Macia E Ehrlich M Massol R Boucrot E Brunner C Kirchhausen T . Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell (2006) 10:839–50.10.1016/j.devcel.2006.04.002
39
Merrifield CJ Perrais D Zenisek D . Coupling between clathrin-coated-pit invagination, cortactin recruitment, and membrane scission observed in live cells. Cell (2005) 121:593–606.10.1016/j.cell.2005.03.015
40
Shpetner HS Herskovits JS Vallee RB . A binding site for SH3 domains targets dynamin to coated pits. J Biol Chem (1996) 271:13–6.10.1074/jbc.271.1.13
41
Lee A Frank DW Marks MS Lemmon MA . Dominant-negative inhibition of receptor-mediated endocytosis by a dynamin-1 mutant with a defective pleckstrin homology domain. Curr Biol (1999) 9:261–4.10.1016/S0960-9822(99)80115-8
42
Roux A Uyhazi K Frost A De Camilli P . GTP-dependent twisting of dynamin implicates constriction and tension in membrane fission. Nature (2006) 441:528–31.10.1038/nature04718
43
Mears JA Ray P Hinshaw JE . A corkscrew model for dynamin constriction. Structure (2007) 15:1190–202.10.1016/j.str.2007.08.012
44
Morlot S Galli V Klein M Chiaruttini N Manzi J Humbert F et al Membrane shape at the edge of the dynamin helix sets location and duration of the fission reaction. Cell (2012) 151:619–29.10.1016/j.cell.2012.09.017
45
Henley JR Krueger EW Oswald BJ McNiven MA . Dynamin-mediated Internalization of caveolae. J Cell Biol (1998) 141:85–99.10.1083/jcb.141.1.85
46
Moskovich O Herzog LO Ehrlich M Fishelson Z . Caveolin-1 and dynamin-2 are essential for removal of the complement C5b-9 complex via endocytosis. J Biol Chem (2012) 287:19904–15.10.1074/jbc.M111.333039
47
Lamaze C Dujeancourt A Baba T Lo CG Benmerah A Dautry-Varsat A . Interleukin 2 receptors and detergent-resistant membrane domains define a clathrin-independent endocytic pathway. Mol Cell (2001) 7:661–71.10.1016/S1097-2765(01)00212-X
48
Abrami L Liu S Cosson P Leppla SH van der Goot FG . Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J Cell Biol (2003) 160:321–8.10.1083/jcb.200211018
49
Mulherkar N Raaben M de la Torre JC Whelan SP Chandran K . The Ebola virus glycoprotein mediates entry via a non-classical dynamin-dependent macropinocytic pathway. Virology (2011) 419:72–83.10.1016/j.virol.2011.08.009
50
Carter GC Bernstone L Baskaran D James W . HIV-1 infects macrophages by exploiting an endocytic route dependent on dynamin, Rac1 and Pak1. Virology (2011) 409:234–50.10.1128/JVI.01051-12
51
Abdulkarim AS Cao H Huang B McNiven MA . The large GTPase dynamin is required for hepatitis B virus protein secretion from hepatocytes. J Hepatol (2003) 38:76–83.10.1016/S0168-8278(02)00326-4
52
Nicoziani P Vilhardt F Llorente A Hilout L Courtoy PJ Sandvig K et al Role for dynamin in late endosome dynamics and trafficking of the cation-independent mannose 6-phosphate receptor. Mol Biol Cell (2000) 11:481–95.10.1091/mbc.11.2.481
53
Mesaki K Tanabe K Obayashi M Oe N Takei K . Fission of tubular endosomes triggers endosomal acidification and movement. PLoS One (2011) 6:e19764.10.1371/journal.pone.0019764
54
Jones SM Howell KE Henley JR Cao H McNiven MA . Role of dynamin in the formation of transport vesicles from the trans-Golgi network. Science (1998) 279:573–7.10.1126/science.279.5350.573
55
Kasai K Shin HW Shinotsuka C Murakami K Nakayama K . Dynamin II is involved in endocytosis but not in the formation of transport vesicles from the trans-Golgi network. J Biochem (1999) 125:780–9.10.1093/oxfordjournals.jbchem.a022349
56
Kreitzer G Marmorstein A Okamoto P Vallee R Rodriguez-Boulan E . Kinesin and dynamin are required for post-Golgi transport of a plasma-membrane protein. Nat Cell Biol (2000) 2:125–7.10.1038/35000081
57
Cao H Thompson HM Krueger EW McNiven MA . Disruption of Golgi structure and function in mammalian cells expressing a mutant dynamin. J Cell Sci (2000) 113:1993–2002.
58
Yang Z Li H Chai Z Fullerton MJ Cao Y Toh BH et al Dynamin II regulates hormone secretion in neuroendocrine cells. J Biol Chem (2001) 276:4251–60.10.1074/jbc.M006371200
59
Cao H Weller S Orth JD Chen J Huang B Chen JL et al Actin and Arf1-dependent recruitment of a cortactin-dynamin complex to the Golgi regulates post-Golgi transport. Nat Cell Biol (2005) 7:483–92.10.1038/ncb1246
60
Salvarezza SB Deborde S Schreiner R Campagne F Kessels MM Qualmann B et al LIM kinase 1 and cofilin regulate actin filament population required for dynamin-dependent apical carrier fission from the trans-Golgi network. Mol Biol Cell (2009) 20:438–51.10.1091/mbc.E08-08-0891
61
Kessels MM Dong J Leibig W Westermann P Qualmann B . Complexes of syndapin II with dynamin II promote vesicle formation at the trans-Golgi network. J Cell Sci (2006) 119:1504–16.10.1242/jcs.02877
62
Grimmer S Ying M Wälchli S van Deurs B Sandvig K . Golgi vesiculation induced by cholesterol occurs by a dynamin and cPLA2-dependent mechanism. Traffic (2005) 6:144–56.10.1111/j.1600-0854.2005.00258.x
63
Weller SG Capitani M Cao H Micaroni M Luini A Sallese M et al Src kinase regulates the integrity and function of the Golgi apparatus via activation of dynamin 2. Proc Natl Acad Sci U S A (2010) 107:5863–8.10.1073/pnas.0915123107
64
Artalejo CR Elhamdani A Palfrey HC . Sustained stimulation shifts the mechanism of endocytosis from dynamin-1-dependent rapid endocytosis to clathrin and dynamin-2-mediated slow endocytosis in chromaffin cells. Proc Natl Acad Sci U S A (2002) 99:6358–63.10.1073/pnas.082658499
65
Graham ME O’Callaghan DW McMahon HT Burgoyne RD . Dynamin-dependent and dynamin-independent processes contribute to the regulation of single vesicle release kinetics and quantal size. Proc Natl Acad Sci U S A (2002) 99:7124–9.10.1073/pnas.102645099
66
Holroyd P Lang T Wenzel D De Camilli P Jahn R . Imaging direct, dynamin-dependent recapture of fusing secretory granules on plasma membrane lawns from PC12 cells. Proc Natl Acad Sci U S A (2002) 99:16806–11.10.1073/pnas.222677399
67
González-Jamett AM Báez-Matus X Hevia MA Guerra MJ Olivares MJ Martínez AD et al The association of dynamin with synaptophysin regulates quantal size and duration of exocytotic events in chromaffin cells. J Neurosci (2010) 30:10683–91.10.1523/JNEUROSCI.5210-09.2010
68
Anantharam A Bittner MA Aikman RL Stuenkel EL Schmid SL Axelrod D et al A new role for the dynamin GTPase in the regulation of fusion pore expansion. Mol Biol Cell (2011) 22:1907–18.10.1091/mbc.E11-02-0101
69
Samasilp P Chan SA Smith C . Activity-dependent fusion pore expansion regulated by a calcineurin-dependent dynamin-syndapin pathway in mouse adrenal chromaffin cells. J Neurosci (2012) 32:10438–47.10.1523/JNEUROSCI.1299-12.2012
70
Reid AT Lord T Stanger SJ Roman SD McCluskey A Robinson PJ et al Dynamin regulates specific membrane fusion events necessary for acrosomal exocytosis in mouse spermatozoa. J Biol Chem (2012) 287:37659–72.10.1074/jbc.M112.392803
71
Leikina E Melikov K Sanyal S Verma SK Eun B Gebert C et al Extracellular annexins and dynamin are important for sequential steps in myoblast fusion. J Cell Biol (2013) 200:109–23.10.1083/jcb.201207012
72
Lai W Huang L Ho P Montefiori D Chen CH . The role of dynamin in HIV type 1 Env-mediated cell-cell fusion. AIDS Res Hum Retroviruses (2011) 27:1013–7.10.1089/AID.2010.0259
73
Miyauchi K Kim Y Latinovic O Morozov V Melikyan GB . HIV enters cell via endocytosis and dynamin-dependent fusion with endosomes. Cell (2010) 137:433–44.10.1016/j.cell.2009.02.046.HIV
74
de la Vega M Marin M Kondo N Miyauchi K Kim Y Epand RF et al Inhibition of HIV-1 endocytosis allows lipid mixing at the plasma membrane, but not complete fusion. Retrovirology (2011) 8:99.10.1186/1742-4690-8-99
75
Alés E Tabares L Poyato JM Valero V Lindau M Alvarez de Toledo G . High calcium concentrations shift the mode of exocytosis to the kiss-and-run mechanism. Nat Cell Biol (1999) 1:40–4.10.1038/9012
76
Perrais D Kleppe IC Taraska JW Almers W . Recapture after exocytosis causes differential retention of protein in granules of bovine chromaffin cells. J Physiol (2004) 560:413–28.10.1113/jphysiol.2004.064410
77
Tsuboi T McMahon HT Rutter GA . Mechanisms of dense core vesicle recapture following “kiss and run” (“cavicapture”) exocytosis in insulin-secreting cells. J Biol Chem (2004) 279:47115–24.10.1074/jbc.M408179200
78
Arneson LN Segovis CM Gomez TS Schoon RA Dick J Lou Z et al Dynamin 2 regulates granule exocytosis during NK cell-mediated cytotoxicity. J Immunol (2008) 181:6995–7001.
79
Min L Leung YM Tomas A Watson RT Gaisano HY Halban PA et al Dynamin is functionally coupled to insulin granule exocytosis. J Biol Chem (2007) 282:33530–6.10.1074/jbc.M703402200
80
Galas M Chasserot-golaz S Dirrig-grosch S Bader M . Presence of dynamin – syntaxin complexes associated with secretory granules in adrenal chromaffin cells. J Neurochem (2000) 75:1511–9.10.1046/j.1471-4159.2000.0751511.x
81
Zhao L Shi X Li L Miller DJ . Dynamin 2 associates with complexins and is found in the acrosomal region of mammalian sperm. Mol Reprod Dev (2007) 74:750–7.10.1002/mrd.20660
82
Peters C Baars TL Bühler S Mayer A . Mutual control of membrane fission and fusion proteins. Cell (2004) 119:667–78.10.1016/j.cell.2004.11.023
83
Alpadi K Kulkarni A Namjoshi S Srinivasan S Sippel KH Ayscough K et al Dynamin-SNARE interactions control trans-SNARE formation in intracellular membrane fusion. Nat Commun (2013) 4:1704.10.1038/ncomms2724
84
Gasman S Chasserot-Golaz S Malacombe M Way M Bader MF . Regulated exocytosis in neuroendocrine cells: a role for subplasmalemmal Cdc42/N-WASP-induced actin filaments. Mol Biol Cell (2004) 15:520–31.10.1091/mbc.E03-06-0402
85
Malacombe M Bader MF Gasman S . Exocytosis in neuroendocrine cells: new tasks for actin. Biochim Biophys Acta (2006) 1763:1175–83.10.1016/j.bbamcr.2006.09.004
86
Wollman R Meyer T . Coordinated oscillations in cortical actin and Ca2+ correlate with cycles of vesicle secretion. Nat Cell Biol (2012) 14:1261–9.10.1038/ncb2614
87
Berberian K Torres AJ Fang Q Kisler K Lindau M . F-actin and myosin II accelerate catecholamine release from chromaffin granules. J Neurosci (2009) 29:863–70.10.1523/JNEUROSCI.2818-08.2009
88
Yamada H Abe T Satoh A Okazaki N Tago S Kobayashi K et al Stabilization of actin bundles by a dynamin 1/cortactin ring complex is necessary for growth cone filopodia. J Neurosci (2013) 33:4514–26.10.1523/JNEUROSCI.2762-12.2013
89
González-Jamett AM Momboisse F Guerra MJ Ory S Báez-Matus X Barraza N et al Dynamin-2 regulates fusion pore expansion and quantal release through a mechanism that involves actin dynamics in neuroendocrine chromaffin cells. PLoS One (2013) 8:70638.10.1371/journal.pone.0070638
90
Dharmalingam E Haeckel A Pinyol R Schwintzer L Koch D Kessels MM et al F-BAR proteins of the syndapin family shape the plasma membrane and are crucial for neuromorphogenesis. J Neurosci (2009) 29:13315–27.10.1523/JNEUROSCI.3973-09.2009
91
Anantharam A Axelrod D Holz RW . Real-time imaging of plasma membrane deformations reveals pre-fusion membrane curvature changes and a role for dynamin in the regulation of fusion pore expansion. J Neurochem (2012) 122:661–71.10.1111/j.1471-4159.2012.07816.x
92
Hamao K Morita M Hosoya H . New function of the proline rich domain in dynamin-2 to negatively regulate its interaction with microtubules in mammalian cells. Exp Cell Res (2009) 315:1336–45.10.1016/j.yexcr.2009.01.025
93
Thompson HM Cao H Chen J Euteneuer U McNiven MA . Dynamin 2 binds gamma-tubulin and participates in centrosome cohesion. Nat Cell Biol (2004) 6:335–42.10.1038/ncb1112
94
Ishida N Nakamura Y Tanabe K Li S Takei K . Dynamin 2 associates with microtubules at mitosis and regulates cell cycle progression. Cell Struct Funct (2011) 36:145–54.10.1247/csf.10016
95
Tanabe K Takei K . Dynamic instability of microtubules requires dynamin 2 and is impaired in a Charcot-Marie-Tooth mutant. J Cell Biol (2009) 185:939–48.10.1083/jcb.200803153
96
Gold ES Underhill DM Morrissette NS Guo J McNiven MA Aderem A . Dynamin 2 is required for phagocytosis in macrophages. J Exp Med (1999) 190:1849–56.10.1084/jem.190.12.1849
97
Ochoa G Slepnev VI Neff L Ringstad N Takei K Daniell L et al A functional link between dynamin and the actin cytoskeleton at podosomes. J Cell Biol (2000) 150:377–89.10.1083/jcb.150.2.377
98
Lee E De Camilli P . Dynamin at actin tails. Proc Natl Acad Sci U S A (2002) 99:161–6.10.1073/pnas.012607799
99
Yamada H Abe T Li SA Masuoka Y Isoda M Watanabe M et al Dynasore, a dynamin inhibitor, suppresses lamellipodia formation and cancer cell invasion by destabilizing actin filaments. Biochem Biophys Res Commun (2009) 390:1142–8.10.1016/j.bbrc.2009.10.105
100
Gomez TS Hamann MJ McCarney S Savoy DN Lubking CM Heldebrant MP et al Dynamin 2 regulates T cell activation by controlling actin polymerization at the immunological synapse. Nat Immunol (2005) 6:261–70.10.1038/ni1168
101
Otsuka A Abe T Watanabe M Yagisawa H Takei K Yamada H . Dynamin 2 is required for actin assembly in phagocytosis in sertoli cells. Biochem Biophys Res Commun (2009) 378:478–82.10.1016/j.bbrc.2008.11.066
102
Merrifield CJ Feldman ME Wan L Almers W . Imaging actin and dynamin recruitment during invagination of single clathrin-coated pits. Nat Cell Biol (2002) 4:691–8.10.1038/ncb837
103
Itoh T Erdmann KS Roux A Habermann B Werner H De Camilli P . Dynamin and the actin cytoskeleton cooperatively regulate plasma membrane invagination by BAR and F-BAR proteins. Dev Cell (2005) 9:791–804.10.1016/j.devcel.2005.11.005
104
Taylor MJ Lampe M Merrifield CJ . A feedback loop between dynamin and actin recruitment during clathrin-mediated endocytosis. PLoS Biol (2012) 10:e1001302.10.1371/journal.pbio.1001302
105
Kessels MM Engqvist-Goldstein AE Drubin DG Qualmann B . Mammalian Abp1, a signal-responsive F-actin-binding protein, links the actin cytoskeleton to endocytosis via the GTPase dynamin. Sci Signal (2001) 153:351–66.10.1083/jcb.153.2.35
106
Shin N Lee S Ahn N Kim SA Ahn SG YongPark Z et al Sorting nexin 9 interacts with dynamin 1 and N-WASP and coordinates synaptic vesicle endocytosis. J Biol Chem (2007) 282:28939–50.10.1074/jbc.M700283200
107
Bitoun M Maugenre S Jeannet PY Lacène E Ferrer X Laforêt P et al Mutations in dynamin 2 cause dominant centronuclear myopathy. Nat Genet (2005) 37:1207–9.10.1038/ng1657
108
Tanabe K Takei K . Dynamin 2 in Charcot-Marie-Tooth disease. Acta Med Okayama (2012) 66:183–90.
109
Jeannet PY Bassez G Eymard B Laforêt P Urtizberea JA Rouche A et al Clinical and histologic findings in autosomal centronuclear myopathy. Neurology (2004) 62:1484–90.10.1212/01.WNL.0000124388.67003.56
110
Fisher J Towfighi J Darvish D Simmons Z . A case of hereditary inclusion body myopathy: 1 patient, 2 novel mutations. J Clin Neuromuscul Dis (2006) 7:179–84.10.1097/01.cnd.0000211406.94445.f0
111
Susman RD Quijano-Roy S Yang N Webster R Clarke NF Dowling J et al Expanding the clinical, pathological and MRI phenotype of DNM2-related centronuclear myopathy. Neuromuscul Disord (2010) 20:229–37.10.1016/j.nmd.2010.02.016
112
Koutsopoulos OS Koch C Tosch V Böhm J North KN Laporte J . Mild functional differences of dynamin 2 mutations associated to centronuclear myopathy and Charcot-Marie Tooth peripheral neuropathy. PLoS One (2011) 6:e27498.10.1371/journal.pone.0027498
113
Durieux AC Vignaud A Prudhon B Viou MT Beuvin M Vassilopoulos S et al A centronuclear myopathy-dynamin 2 mutation impairs skeletal muscle structure and function in mice. Hum Mol Genet (2010) 19:4820–36.10.1093/hmg/ddq413
114
Liu YW Lukiyanchuk V Schmid SL . Common membrane trafficking defects of disease-associated dynamin 2 mutations. Traffic (2011) 12:1620–33.10.1111/j.1600-0854.2011.01250.x
115
Bitoun M Durieux AC Prudhon B Bevilacqua JA Herledan A Sakanyan V et al Dynamin 2 mutations associated with human diseases impair clathrin-mediated receptor endocytosis. Hum Mutat (2009) 30:1419–27.10.1002/humu.21086
116
Sidiropoulos NM Miehe M Bock T Tinelli E Oertli CI . Dynamin 2 mutations in Charcot-Marie-Tooth clathrin-mediated endocytosis in myelination. Brain (2012) 135:1395–411.10.1093/brain/aws061
117
Durieux AC Vassilopoulos S Lainé J Fraysse B Briñas L Prudhon B et al A centronuclear myopathy – dynamin 2 mutation impairs autophagy in mice. Traffic (2012) 13:869–79.10.1111/j.1600-0854.2012.01348.x
118
Bitoun M Bevilacqua JA Prudhon B Maugenre S Taratuto AL Monges S et al Dynamin 2 mutations cause sporadic centronuclear myopathy with neonatal onset. Ann Neurol (2007) 62:666–70.10.1002/ana.21235
119
Wang L Barylko B Byers C Ross JA Jameson DM Albanesi JP . Dynamin 2 mutants linked to centronuclear myopathies form abnormally stable polymers. J Biol Chem (2010) 285:22753–7.10.1074/jbc.C110.130013
120
Kenniston JA Lemmon MA . Dynamin GTPase regulation is altered by PH domain mutations found in centronuclear myopathy patients. EMBO J (2010) 29:3054–67.10.1038/emboj.2010.187
121
Kierdaszuk B Berdynski M Karolczak J Redowicz MJ Zekanowski C Kaminska AM . A novel mutation in the DNM2 gene impairs dynamin 2 localization in skeletal muscle of a patient with late onset centronuclear myopathy. Neuromuscul Disord (2013) 23:219–28.10.1016/j.nmd.2012.12.007
Summary
Keywords
dynamin-2, endocytosis, exocytosis, actin, microtubules, mutations, Charcot–Marie–Tooth neuropathy, centronuclear myopathy
Citation
González-Jamett AM, Momboisse F, Haro-Acuña V, Bevilacqua JA, Caviedes P and Cárdenas AM (2013) Dynamin-2 Function and Dysfunction Along the Secretory Pathway. Front. Endocrinol. 4:126. doi: 10.3389/fendo.2013.00126
Received
29 May 2013
Accepted
31 August 2013
Published
18 September 2013
Volume
4 - 2013
Edited by
Stephane Gasman, Centre national de la Recherche Scientifique, France
Reviewed by
Jocelyn Laporte, Institut National de la Santé et de la Recherche Médicale, France; Fernando Diego Marengo, Universidad de Buenos Aires, Argentina
Copyright
© 2013 González-Jamett, Momboisse, Haro-Acuña, Bevilacqua, Caviedes and Cárdenas.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana María Cárdenas, Facultad de Ciencias, Centro Interdisciplinario de Neurociencia de Valparaíso, Universidad de Valparaíso, Gran Bretaña 1111, Playa Ancha 2360102, Valparaíso, Chile e-mail: ana.cardenas@uv.cl
This article was submitted to Neuroendocrine Science, a section of the journal Frontiers in Endocrinology.
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.