 Jia L. Zhuo
Jia L. Zhuo Fernanda M. Ferrao
Fernanda M. Ferrao Yun Zheng
Yun Zheng Xiao C. Li1
Xiao C. Li1
- 1Laboratory of Receptor and Signal Transduction, Department of Pharmacology and Toxicology, University of Mississippi Medical Center, Jackson, MS, USA
- 2Department of Medicine, Division of Nephrology, University of Mississippi Medical Center, Jackson, MS, USA
The renin-angiotensin system (RAS) is well-recognized as one of the oldest and most important regulators of arterial blood pressure, cardiovascular, and renal function. New frontiers have recently emerged in the RAS research well beyond its classic paradigm as a potent vasoconstrictor, an aldosterone release stimulator, or a sodium-retaining hormone. First, two new members of the RAS have been uncovered, which include the renin/(Pro)renin receptor (PRR) and angiotensin-converting enzyme 2 (ACE2). Recent studies suggest that prorenin may act on the PRR independent of the classical ACE/ANG II/AT1 receptor axis, whereas ACE2 may degrade ANG II to generate ANG (1–7), which activates the Mas receptor. Second, there is increasing evidence that ANG II may function as an intracellular peptide to activate intracellular and/or nuclear receptors. Third, currently there is a debate on the relative contribution of systemic versus intrarenal RAS to the physiological regulation of blood pressure and the development of hypertension. The objectives of this article are to review and discuss the new insights and perspectives derived from recent studies using novel transgenic mice that either overexpress or are deficient of one key enzyme, ANG peptide, or receptor of the RAS. This information may help us better understand how ANG II acts, both independently or through interactions with other members of the system, to regulate the kidney function and blood pressure in health and disease.
Introduction
Although Tigerstedt and Bergman discovered the rate-limiting enzyme renin about 115 years ago (1), the renin-angiotensin system (RAS) remains to be a remarkable subject for continuous research. Our current understanding of the RAS has greatly evolved from the classical renin/angiotensin-converting enzyme (ACE)/angiotensin II (ANG II)/AT1 receptor axis and its physiological roles in the regulation of cardiovascular and renal function, blood pressure, aldosterone biosynthesis and release, and body salt and fluid balance (2–14). However, new frontiers are continuously emerging from the RAS research in recent years, especially in uncovering new enzyme(s) and/or receptor(s) of the system, studying their novel roles, and elucidating their signaling transduction mechanisms. It is now recognized that the classical renin/ACE/ANG II/AT1 and AT2 axis is no longer the exclusive effector and signaling pathway for the system (15). Three new axes have been recently described to include the ACE2/ANG (1–7)/Mas receptor axis, the prorenin/PRR/MAP kinases ERK1/2 axis, and the ANG IV/AT4/IRAP (insulin-regulated aminopeptidase, IRAP) axis (Figure 1) (8, 12, 15–17). The notion that ANG II is the only active peptide of the RAS appears to be outdated, since ANG II can be hydrolyzed by various angiotensinases, ACE2, and neprilysin to generate ANG (1–7), ANG III, ANG IV, and ANG A (2, 16, 18). Prorenin and smaller ANG fragments, including ANG (1–7), ANG III, and ANG IV, can bind their respective receptors or act as an agonist for ANG II receptors to induce a physiological effect (2, 8, 17, 19–21). Indeed, in addition to AT1 and AT2 receptors that mediate the well-recognized effects of ANG II in the kidney and other tissues, new receptors for prorenin (PRR), ANG (1–7) (Mas receptor), and ANG IV (AT4 receptor) have been identified (21–23). Depending on the receptor activated, small ANG peptides may act as an agonist or an antagonist of ANG II. For example, appropriate concentrations of ANG (1–7), ANG III, and ANG IV may activate their respective Mas receptors (8, 9, 16), AT2 receptors (19, 24, 25), or AT4 receptors to oppose the known effects of ANG II (26, 27). Conversely, high concentrations of ANG (1–7), ANG III, and ANG IV may activate AT1 receptors to induce the well-recognized effects of ANG II (16, 20, 28–30). Furthermore, the renin/prorenin receptor, PRR, not only catalyzes prorenin to generate ANG II, but also induces intracellular responses in an ANG II-independent manner (13, 31, 32). Finally, the RAS is no longer considered to act only as an endocrine system, but also acts as a paracrine, autacrine, and intracrine system (33–37). It is likely that ANG II and its smaller ANG peptides may act as both endocrine, paracrine, and intracrine peptides by stimulating cell surface, cytoplasmic and nuclear receptors to exert biological, physiological, and nuclear effects.
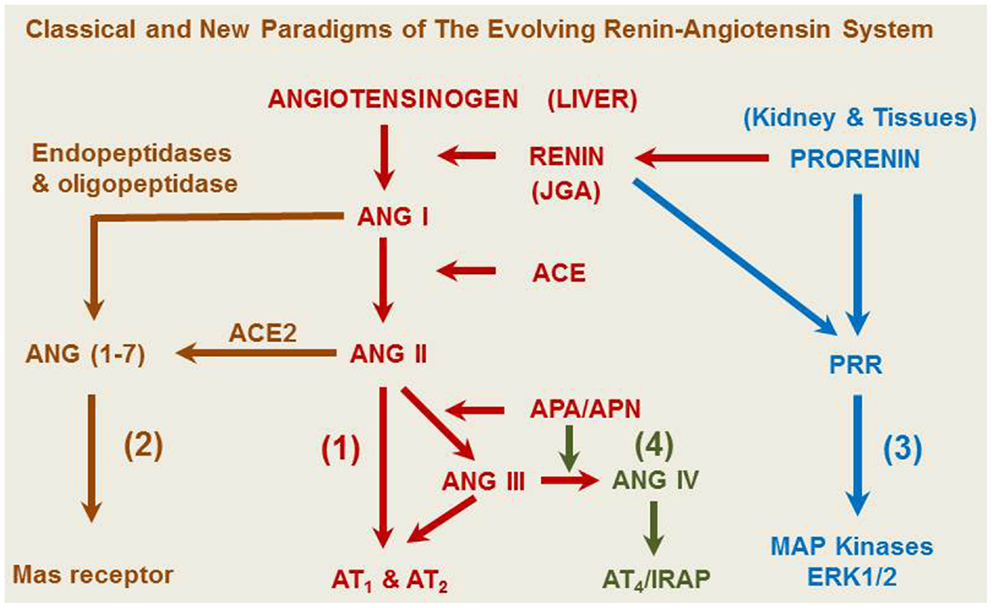
Figure 1. A representative overview of the evolving renin-angiotensin system. (1) The classical angiotensinogen/renin/ACE/ANG II/AT1 and AT2 receptor axis. (2) The prorenin/PRR/MAP kinases ERK 1/2 axis. (3) The ACE2/ANG (1–7)/Mas receptor axis. (4) The ANG IV/AT4/IRAP axis. ANG A, angiotensin A. ANG I, angiotensin I. ANG (1–7), angiotensin (1–7). ACE, angiotensin-converting enzyme. ACE2, angiotensin-converting enzyme 2. ANG II, angiotensin II. ANG III, angiotensin III. ANG IV, angiotensin (3–8). APA, aminopeptidase A; APN, aminopeptidase N; AT1, type 1 ANG II receptor; AT2, type 2 ANG II receptor; IRAP, insulin-regulated aminopeptidase or AT4 receptor; JGA, juxtaglomerular apparatus.
The major objective of this article is to review recent advances in biomedical research with a focus on the intrarenal RAS and its paracrine, autacrine, and intracrine roles. New insights, controversies, and perspectives will be discussed by reviewing recent in vitro and in vivo studies using innovative approaches or animal models including global and tissue-specific RAS transgenic animals. The review article will cover the classical ACE/ANG II/AT1 and AT2 receptor axis, the ACE2/ANG (1–7)/Mas receptor axis, the prorenin/PRR/MAP kinases ERK1/2 axis, and the ANG IV/AT4/IRAP axis. It is expected that this new information may further improve our understanding of physiological and pathophysiological roles of the RAS and help the development of new drugs or strategies to treat hypertension, diabetes, and cardiovascular and kidney diseases by targeting ANG II and other ANG peptides and/or their receptors.
Current Insights and Future Perspectives on the Roles of the Classical ACE/ANG II/AT1 and AT2 Receptor Axis in the Kidney
It is well established that the ACE/ANG II/AT1 and AT2 receptor axis may function as a circulating or endocrine and paracrine system to regulate cardiovascular, neural, adrenal, and renal function, contributing to normal blood pressure homeostasis and the development of hypertension. However, the specific role of and the extent to which the intrarenal ACE/ANG II/AT1 and AT2 receptor axis versus the systemic counterpart plays in normal blood pressure control and the development of hypertension remain an issue of continuous debate (10, 38–42). Now, there is a general consensus that all major components of the RAS necessary for generation of ANG II are expressed or present in the kidney (Figure 2) (2, 18, 43–45), and that the levels of ANG II in the kidney are much higher than in plasma (2, 44, 46–49). This is especially true that high ANG II levels have been demonstrated in interstitial and proximal tubular fluid of the kidney and intracellular endosomal compartment (46–48, 50–52).
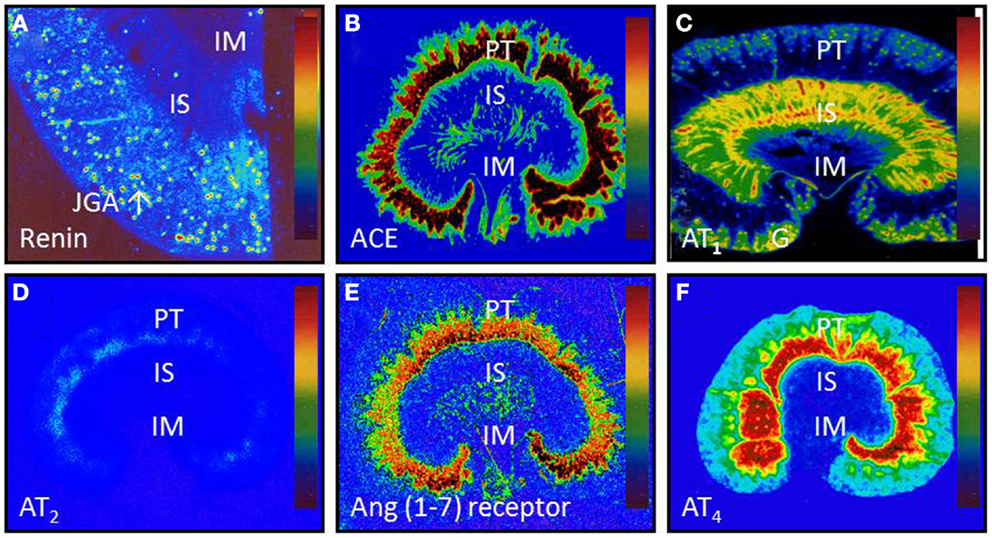
Figure 2. Intrarenal localization or expression of major components of the renin-angiotensin system. (A) Active renin binding in juxtaglomerular apparatus in the dog kidney using the radiolabeled renin inhibitor, 125I-H77. (B) ACE binding in the proximal tubule of the rat kidney using 125I-351A (C) AT1 receptor binding in the rat kidney in the presence of the AT2 receptor blocker PD123319. (D) AT2 receptor binding in the rat kidney in the presence of the AT1 receptor blocker losartan using 125I-[Sar1,Ile8]-Ang II. (E) Ang (1–7) receptor binding in the rat kidney using 125I-Ang (1–7) as the radioligand. And (F) Ang IV receptor binding in the rat kidney using 125I-Ang (3–8). The levels of binding are indicated by color calibration bars with red representing the highest, whereas blue showing the lowest levels of enzyme or receptor binding. G, glomerulus; IM, inner medulla; IS, inner stripe of the outer medulla; JGA, juxtaglomerular apparatus; P, proximal tubule. Reproduced from Li and Zhuo with permission (45).
The mechanisms underlying high levels of ANG II in the kidney are not well understood. In addition to the well-documented expression of all major components of the RAS in the kidney, two major mechanisms may play a critical role under physiological conditions and during the development of ANG II-dependent hypertension. The first is that AT1 receptors are abundantly expressed in the kidney, where AT1 (AT1a) receptor mediates the intracellular accumulation of ANG II especially in proximal tubules (48, 53–58). Classically, a receptor pharmacological dogma suggests that the purpose of G protein-coupled receptor (GPCR)-mediated internalization or endocytosis of an agonist or ligand is to desensitize the cellular responses to the agonist stimulation by moving the agonist/ligand into the cell for degradation in the lysosomal compartment (59–64). The receptor recycles back to the cell membrane to initiate a new round of biological response. However, we and others infused ANG II into rats and mice for 2 weeks, and found no desensitization of ANG II responses, because blood pressure continued to increase and hypertension persists as long as ANG II is infused (48, 53–58). Zhuo et al. reported that in ANG II-infused hypertensive rats, ANG II levels were about 10 times higher in renal cortical endosomes than in control rats via an AT1 receptor-mediated mechanism (48). Nishiyama et al. showed that renal interstitial fluid ANG II levels were substantially increased in ANG II-infused rats, an effect also mediated by AT1 receptors (65). In AT1a receptor-deficient mice (Agtr1a−/−), we further demonstrated that AT1 receptor-mediated increases in ANG II uptake in the kidney were largely abolished (57, 58). These studies suggest that AT1 (AT1a) receptor-mediated uptake of ANG II at least partly contributes to the demonstrated high levels of ANG II in the kidney.
The second classical dogma in the RAS field is that the expression and activity of the RAS is strictly regulated by a negative feedback mechanism by ANG II itself. An increase in the circulating and tissue ANG II is expected to suppress renin release from JGA cells and therefore the production of ANG II in the kidney. However, there is evidence that a positive feed-forward loop exists in the kidney during ANG II-dependent hypertension (43, 44, 66–69). Navar’s group has shown that prorenin and renin (68–70), angiotensinogen (43, 67), and ACE (66) are significantly augmented in response to long-term infusion of ANG II to induce hypertension in rats or mice. Renin and prorenin expression in the collecting ducts are also stimulated during ANG II infusion, likely contributing to increased urinary levels in ANG II-infused hypertensive rats (69–72). Taken together, these studies suggest that in ANG II-infused hypertensive animals, intrarenal ANG II production may be augmented due to increased expression of prorenin and renin, AGT, and ACE.
Currently, there is a great debate on whether AGT, ACE, and AT1 receptors in the kidney contribute to the normal blood pressure regulation and the development of hypertension (4, 10, 39–42, 73–77). The classical dogma is that the circulating RAS via the kidney derived renin, liver-derived AGT and vascular endothelial ACE, rather than the intrarenal RAS, plays an important role in the normal blood pressure control and the development of hypertension (78–82). To determine the roles of systemic/endothelial ACE versus tissue/kidney ACE in normal blood pressure and renal control, Bernstein’s group first used targeted homologous recombination to create mice, ACE 2/2, expressing a form of ACE that lacks the COOH-terminal half of ACE with normal or elevated circulating ACE without tissue-bound/kidney ACE (78). Homologous ACE 2/2 mice have significantly lower blood pressure, renal vascular thickening, urine concentrating defect, and significant increase in fractional proximal tubular reabsorption (78). These studies suggests that tissue-bound ACE, rather than circulating ACE, is important for maintaining normal blood pressure (78), and that ACE in the proximal tubule may not be necessary for maintaining normal proximal fluid reabsorption (80). The same group of investigators later generated the so-called ACE 3/3 mice, which is deficient of endothelial ACE in the lung, aorta, or any vascular structure (79). ACE activity in the kidney is about 14% that of wild-type mice, but hepatic ACE expression in ACE 3/3 mice is almost 90-fold that of wild-type. Interestingly, basal blood pressure, plasma ANG II levels, response to ACE inhibitors, and renal function of ACE 3/3 mice were similar to those of wild-type mice. The underlying conclusion of this study is that endothelial ACE is not required for maintaining normal blood pressure and renal function (79). Sen’s group also generated two different strains of mutant mice that express ACE either in vascular endothelial cells (Ts strain) or in renal proximal tubules (Gs strain) (81, 82). Both mutant mice show equivalent serum ACE and ANG II levels, normal kidney structure and fluid homeostasis. In contrast to Bernstein’s ACE3/3 mice (79), only those mutant mice that expressed ACE in vascular endothelial cells had normal blood pressure (81). Proximal fluid reabsorption was found to be normal in the chronic absence of proximal tubule ACE (82). Thus there is still a lack of consensus with respect to the precise roles of systemic/endothelial versus tissue/kidney ACE in normal blood pressure control.
Recently, Gonzalez-Villalobos et al. further determine the role of intrarenal ACE in the normal blood pressure regulation and the development of ANG II-induced hypertension (10, 75). First, Gonzalez-Villalobos et al. also used targeted homologous recombination to generate mice, ACE9/9, that express ACE only in the kidney tubules but not in other tissues (75), or mice with complete deficiency of the entire kidney ACE, ACE 10/10 (10). Similar to Sen’s Gs strain (82), ACE 9/9 mice had lower blood pressure, associated with reduced circulating ANG II, but maintained normal kidney ANG II levels. ACE 9/9 mice responded to chronic ANG I infusion to substantially increase blood pressure (75). In ACE 10/10 mice whose basal blood pressure was similar to wild-type mice, the blood pressure responses to 2-week of ANG II infusion were substantially attenuated in the kidney ACE-KO mice (10). The later study indicates that intrarenal ACE plays a key role in the development of ANG II-induced hypertension, whereas the absence of ACE in the kidney protects against hypertension (10).
However, a careful evaluation of these studies on different strains of ACE mutant mice evokes more questions than answers in the current debate on the relative roles of circulating and intrarenal ACE and therefore ANG II in the blood pressure regulation and the development of hypertension (39, 83). For example, mice with the lack of vascular endothelial ACE may be normotensive (79) or hypotensive (75, 81). Conversely, mice with the lack of kidney/proximal tubular ACE may be normotensive (10, 81). ACE/ANG II appear not to be necessary for maintaining normal proximal tubular fluid reabsorption in mice with overexpression or deficiency of ACE in the proximal tubule (79–82) or the entire kidney (10). Furthermore, circulating or kidney ANG II levels may be normal in these ACE transgenic mice despite of the lack of systemic/endothelial or kidney/proximal tubular ACE (10, 75, 79, 82). These contradictory biochemical, blood pressure, and proximal tubular transport phenotypes, as revealed in various mutant ACE-knockout mice, are difficult to reconcile with well-recognized roles of ACE in the formation of ANG II in the circulation and the kidney, in promoting sodium reabsorption in the proximal tubule and other tubular segments, and in maintaining normal blood pressure homeostasis. However, these diverse phenotypes may provide a new insight into an important role of AT1 (AT1a) receptor-mediated uptake of circulating ANG II by the kidney, especially in the proximal tubule, in maintaining normal levels of ANG II in the kidney of ACE9/9 and/or ACE10/10 mice (10, 75). As discussed previously, AT1 (AT1a) receptor-mediated uptake of circulating ANG II at least partly contributes to higher basal ANG II levels and increased ANG II levels in the kidney during ANG II-induced hypertension (48, 54, 57, 58, 84, 85). Another new insight derived from these mutant ACE mouse models is that blood pressure and proximal tubule phenotypes of these ACE-knockout mice are likely complicated by the fact that ACE is chiefly responsible for the metabolism of bradykinin, ANG (1–7), and many other vasoactive peptides such as substance P (8, 9, 18, 86). Knockout of systemic and/or kidney ACE would lead to marked decreases in circulating and intrarenal ANG II and generation of other vasodepressor substances in the circulation and kidney, which may alter blood pressure and renal responses to ANG II or other vasoactive substances under physiological as well as pathophysiological conditions.
Recent studies using mice with kidney or proximal tubule-specific knockout of AT1 receptors provide new insights and perspectives into the roles of the kidney or proximal tubular AT1a receptors in the normal blood pressure regulation and the development of hypertension (4, 38, 40–42, 77, 87). Coffman and Crowley’s group has been instrumental to use the kidney cross-transplantation approach between wild-type and global AT1a receptor-knockout mice (Agtr1a−/−) (4, 38, 87). These investigators transplanted the kidney of wild-type mice into Agtr1a−/− mice to generate systemic AT1a-KO mice, and conversely transplanted the kidney of Agtr1a−/− mice into wild-type mice to generate the kidney-specific AT1a-KO mice. Blood pressure and cardiac hypertrophic responses to ANG II infusion or high salt intake were compared in the systemic- and kidney-specific AT1a-KO mice (4, 38, 87). These elegant studies confirmed that the kidney AT1 receptors are absolutely required for the development of ANG II-dependent hypertension and cardiac hypertrophy, and systemic AT1 receptors is not sufficient for ANG II to induce hypertension or cardiac hypertrophy (38). Using the Cre/Lox strategy, Gurley et al. (40) and Li et al. (41) generated proximal tubule-specific AT1a-KO mice to determine the role of proximal tubule AT1a receptors in blood pressure regulation. Both studies demonstrated that deletion of AT1a receptor and its signaling in the proximal tubule alone is sufficient to significantly decrease basal blood pressure, despite intact systemic AT1a receptor expression and vascular responses (40, 41). Alternatively, we have recently produced adenoviral constructs encoding GFP-tagged AT1a receptor gene (AT1aR/GFP) (Figure 3), or an enhanced cyan fluorescent protein (ECFP)-tagged ANG II fusion protein, and a proximal tubule-specific sodium and glucose cotransporter 2 (sglt2) promoter (Figure 4) (42). We demonstrated that intrarenal transfer of AT1aR/GFP alone selectively in the proximal tubule was sufficient to increase systolic blood pressure by ∼12 mmHg 14 days after the gene transfer (42). Cotransfer of AT1aR/GFP with ECFP/ANG II increased blood pressure further to 18 mmHg. The increases in blood pressure were associated with twofold increases in phosphorylated MAP kinases ERK1/2, lysate and membrane NHE3 proteins in freshly isolated proximal tubules, and a decrease in 24 h urinary sodium excretion (42). Taken together, these elegant studies strongly suggest that the proximal tubule ACE/ANG II/AT1a receptor axis via promoting proximal tubular sodium and fluid reabsorption may contribute approximately 15 mmHg to basal blood pressure homeostasis in mice.
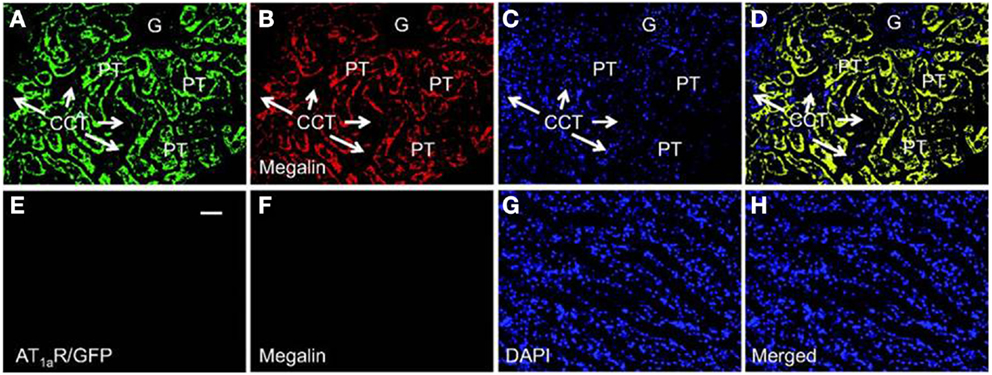
Figure 3. Proximal tubule-specific expression of AT1aR/GFP in a representative Agtr1a−/− mouse kidney 2 week after intrarenal adenoviral transfer. (A) AT1aR/GFP expression (green) in proximal tubules (PT). (B) Alexa Fluor 594-labeled megalin expression (red) in proximal tubules. (C) DAPI-stained nuclei (blue) in the same kidney section. (D) Merged image of (A–C), showing the colocalization of AT1aR/GFP and megalin expression (yellow) in proximal tubules. Only very low levels of AT1aR/GFP and megalin expression are visible in the glomerulus (G) and cortical collecting tubules (CCT). (E) AT1aR/GFP expression in the outer medulla. (F) Alexa Fluor 594-labeled megalin expression in the outer medulla. (G) DAPI-stained nuclei in the outer medulla. (H) Merged image of (E–G), showing the lack of AT1aR/GFP and megalin expression in the outer medulla. Magnification: ×40. Reproduced from Li and Zhuo with permission (42).
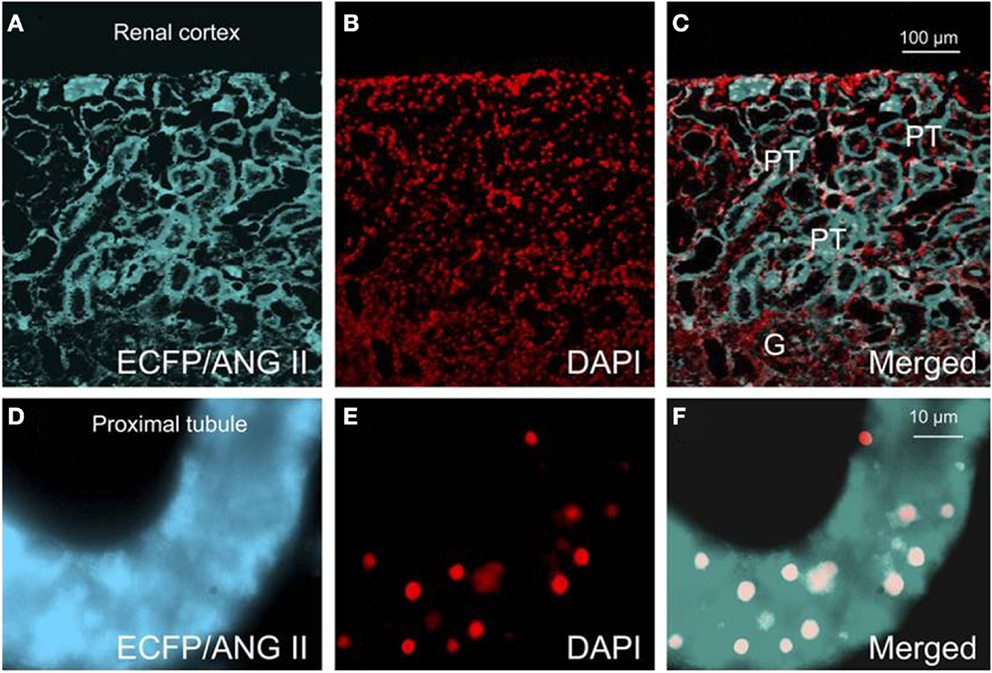
Figure 4. Effects of proximal tubule-specific, adenovirus-mediated transfer of ECFP/ANG II on ECFP/ANG II expression in the renal outer cortex and freshly isolated proximal tubule of mouse kidneys 2 wk after gene transfer. (A) ECFP expression (blue-green). (B) DAPI-stained nuclei (red). (C) Merged image of (A,B), respectively, in the outer renal cortex of a representative rat transferred with ECFP/ANG II selectively in proximal tubules. (D–F) Expression of ECFP/ANG II in a freshly isolated representative proximal convoluted tubule. Bars = 100 μm for the renal cortex and 10 μm for the isolated proximal tubule. G, glomerulus; PT, proximal tubule. Reproduced from Li et al. with permission (77).
Current Insights and Future Perspectives on the Roles of the ACE2/ANG (1–7)/Mas Receptor Axis in the Kidney
ANG (1–7) is the most extensively studied smaller ANG peptide in the RAS since 1970s (8, 9, 17, 18, 88). Early studies showed that structural deletion of either phenylalanine (position 8) or the dipeptide, Pro-Phe (positions 7 and 8) from ANG II completely removed the vasoconstrictor, central pressor, or thirst-stimulating actions of ANG II (89). The structural and activity studies suggested that ANG (1–7) may be an inactive component of the RAS. However, subsequent studies primarily from Ferrario’s group demonstrated that ANG (1–7) has significant vasodepressor and antihypertensive actions in hypertensive animals or humans, which may oppose the actions of ANG II either directly or indirectly by stimulation of prostaglandins and nitric oxide (8, 9, 17, 18, 88). The importance of this heptapeptide in cardiovascular, blood pressure, and renal control gains further recognition recently upon the molecular characterization of a GPCR using ANG (1–7) as a ligand, the Mas receptor (23). It is increasingly recognized that the new ACE2/ANG (1–7)/Mas receptor axis acts to counteract most of the known deleterious actions of the ACE/ANG II/AT1 receptor axis (8, 16, 17). However, recent studies on transgenic animals overexpressing ANG (1–7) have provided new insights and perspectives on whether ANG (1–7) plays beneficial cardiovascular, blood pressure, and renal hemodynamic effects (90–92).
The kidney is one of the key tissues in which ANG (1–7) is generated from the metabolism of ANG II by ACE2 with the proximal tubule exhibiting the most robust ACE2 activities (8, 49). ANG (1–7) can be easily detected in the proximal tubule and urine of rats, sheep, and humans, but it can be rapidly hydrolyzed to ANG (1–5) and ANG (1–4) by ACE and neprilysin (8, 49). Whether ANG (1–7) is primarily produced from the degradation of ANG II by ACE2 in the circulation and kidney remains an issue of continuous debate. An early study by Yamamoto et al. showed that infusion of ANG II in WKY or SHR rats was not accompanied by significantly increased plasma ANG (1–7) levels (93). Modrall et al. reported that in tissue ACE-knockout mice, intrarenal ANG I and ANG II levels were decreased by 70–80% compared with wild-type mice, but ANG (1–7) levels were surprisingly normal in the kidney (94). Thus a more balanced view may be that ANG (1–7) is derived from both the metabolism of ANG I via the endopeptidase-dependent pathway and the metabolism of ANG II by the ACE2-dependent pathway.
Both renal hemodynamic and tubular effects have been demonstrated although the signaling mechanisms involved are not fully understood (17). However, the current insight is that ANG (1–7) acts primarily to oppose the cardiovascular and renal effects of ANG II. For example, ANG II is known to increase blood pressure, induce renal vasoconstriction to decrease renal blood flow (RBF) and glomerular filtration rate (GFR), and induce antidiuresis and antinatriuresis (43, 95–98). By contrast, ANG (1–7) infusion generally opposes and attenuates these effects of ANG II (8, 16, 17, 36, 99). The diuretic/natriuretic effects of ANG (1–7) may be partly due to the renal vasodilatation as well as inhibition of sodium and water reabsorption along the nephron segments. Previous studies demonstrated that ANG (1–7) may be a potent inhibitor of Na+-K+-ATPase in the proximal tubule (16, 17). ANG (1–7) may inhibit Na+-K+-ATPase via AT2 receptor-mediated stimulation of the G(i/o) protein/cGMP/PKG signaling pathway (100, 101). Moreover, ANG (1–7) showed biphasic effects on the Na+/H+ exchanger activity in isolated proximal tubules mediated by the Mas receptor and changes in [Ca2+]i (30, 102). In rat inner medullary collecting ducts (IMCD), ANG (1–7) enhanced water transport via the vasopressin V2 receptor (103). However, some of renal effects induced by ANG (1–7) are very difficult to reconcile with the dogma on the potential roles of the ACE2/ANG (1–7)/Mas receptor axis to counteract with detrimental roles of the renin/ACE/ANG II/AT1 receptor axis. A careful review of the above-mentioned studies reveals that ANG (1–7) may also activate the well-recognized downstream ANG II/AT1 receptor signaling transduction to induce similar effects induced by ANG II.
New insights and perspectives into the physiological roles of ANG (1–7) acting via the Mas receptors in the cardiovascular, blood pressure, and renal regulation may be best inferred from transgenic animals with overexpression of ANG (1–7) (90, 91, 104) or ACE2 (105–107) to substantially increasing production of ANG (1–7) in the circulation or tissues or due to global or tissue-specific deletion of the Mas receptor. Santos’ group has generated transgenic rats that express an ANG (1–7)-producing fusion protein, TGR(A1–7)3292, in the testis (90). Expression of ANG (1–7) in the testis acts as an ANG (1–7) biological pump to increase the plasma ANG (1–7) concentration 2.5-fold. Surprisingly, overexpression of ANG (1–7) did not alter basal blood pressure levels in TGR(A1–7)3292 rats despite of significant increases in stroke volume and cardiac index and a decrease in total peripheral resistance (90, 104). While acute intravenous infusion of ANG (1–7) induces renal vasodilatation, diuresis, and natriuresis (17, 99), GFR and 24 h urinary sodium excretion in TGR(A1–7)3292 rats are similar to those in Sprague-Dawley rats, whereas 24 h urine excretion was decreased and osmolality increased, respectively (91). The results obtained from TGR(A1–7)3292 rats appear to be contradictory to the well-known vasodepressor, diuretic and natriuretic effects of ANG (1–7). In a different study, Rentzsch et al. generated transgenic rats on a SHRSP genetic background expressing the human ACE2 in vascular smooth muscle cells by the use of the SM22 promoter, SHRSP-ACE2 (105). SHRSP-ACE2 rats have significantly elevated circulating levels of ANG (1–7), which is associated with a 15 mmHg decrease in mean arterial blood pressure and significantly attenuated responses to ANG II (105). These data suggest that vascular ACE2 overexpression may be a novel therapeutic strategy in the treatment of hypertension. Liu et al. used the adenoviral gene delivery approach to overexpress ACE2 globally and found that blood pressure was not different between control and ACE2-overexpressing Wistar rats before and after streptozotocin treatment to induce diabetic nephropathy (106). Despite of these inconsistencies, global or tissue-specific overexpression of ACE2 has been reported to reduce blood pressure or hypertension-induced injury in SHR (108, 109), and protect from ischemia-induced cardiac injury (110), and attenuate diabetic nephropathy (106).
Although the GPCR Mas was reported to be the specific receptor for ANG (1–7) more than 10 years ago (23), there is surprisingly little progress that has been made in using these Mas receptor-deficient mice (Mas-KO) to determine the physiological roles of ANG (1–7) (111–114). Too often, the reported cardiovascular, blood pressure, and renal phenotypes are sometimes contradictory between studies. Botelho-Santos reported that mean arterial pressure in anesthetized Mas-KO mice (12–16 weeks old) was not different from that of WT mice, despite of significant decreases in stroke volume and cardiac index and marked increases in vascular resistance and a decrease in blood flow in the kidney (115). Walther et al. also confirmed that neither heart rate nor blood pressure was significantly different between Mas-KO mice and controls, although salt-induced increase in blood pressure was prevented in Mas-KO mice (116, 117). Subsequent studies from the same groups of investigators showed a significantly higher basal blood pressure in Mas-KO mice (112, 118). These differences may be explained by the difference in genetic backgrounds, in that the former Mas-KO mice were generated from mixed genetic background, 129 × C57BL/6, whereas the latter were generated from the FVB/N genetic background for seven generations (16, 119). Other studies supporting the counterregulatory roles of the ACE2/ANG (1–7)/Mas receptor axis against those of the ACE/ANG II/AT1 receptor axis in the kidney include the development of glomerular hyperfiltration and microalbuminuria in Mas-KO mice (120). However, Esteban et al. recently shown that ANG (1–7), via the Mas receptor, has proinflammatory properties at least as potent as those of ANG II and TNFα in the kidney (121). Clearly, controversies remain with respect to the specific roles of the Mas receptor in mediating the effects of ANG (1–7) in the kidney (122).
Current Insights and Future Perspectives on the Roles of the Prorenin/PRR/MAP Kinases ERK 1/2 Axis in the Kidney
A new frontier in the RAS research field emerges during recent years is the prorenin/PRR/MAP kinases ERK 1/2 axis. According to the classical dogma, prorenin is primarily synthesized in the juxtaglomerular (JGA) cells and is biologically inactive (123). Prorenin becomes active renin in JGA cells and is released in response to a decrease in blood pressure (hypotension), activation of renal sympathetic nerves, and sodium depletion. Renin released from JGA cells initiates the activation of the RAS by hydrolyzing circulating and tissue AGT to generate ANG I (123). This classical dogma may be subject to significant revisions as a result of recent progresses being made in the field.
There is strong evidence that prorenin may also be constitutively secreted from the kidney, and to a less extent from extrarenal tissues including eyes and adrenal glands (11–13, 22, 124–126). Whether prorenin is physiologically or pathophysiologically relevant remains an issue of intensive debate before and after Ngyuen et al. first cloned the prorenin/renin receptor (PRR) (22, 127). PRR has a single transmembrane domain and 350-amino acid (22, 127). It has specific binding site not only for the inactive precursor prorenin, but also for active renin, which is the key initiator of the ACE/ANG II/AT1 receptor axis. Thus it is difficult to determine whether it is prorenin or active renin that binds and activates PRR under physiological conditions and in cardiovascular, diabetic and renal diseases. However, it has been shown that prorenin has a “handle” region with higher affinity for PRR than renin, which binds to PRR to initiate the catalytic activity of prorenin, leading the activation of the prorenin/PRR/MAP kinases ERK1/2 axis (12, 22, 127). It has been further suggested that a decoy “handle” region peptide (HRP) may thus target this “handle” region by competitively inhibiting the binding of prorenin to the PRR, and produce pharmacological and therapeutical effects in treating cardiovascular, hypertensive, and diabetic diseases (31, 128, 129). Whether HRP may specifically block PRR to exert beneficial therapeutic effects remains highly controversial (13, 126, 130). Several studies have been unable to confirm the role(s) of prorenin and the effects of HRP in cultured cells and animals (131–133). Even if HRP is indeed effective in blocking prorenin and PRR interactions, its clinical relevance remains unknown due to its peptide properties. The renin-specific inhibitors have been developed to treat hypertension and cardiovascular and kidney diseases. Whether the renin inhibitors are therapeutically superior to classical ACE inhibitors or ARBs remains to be determined. If prorenin and PRR indeed play important physiological and pathophysiological roles in blood pressure regulation and pathologies of cardiovascular, renal, and diabetic diseases, the development of orally active PRR-specific inhibitors to block prorenin-induced activation of PRR will be highly necessary.
While prorenin and renin are present primarily in JGAs of the renal cortex under physiological conditions, PRR is reportedly expressed in glomerular mesangial cells and the subendothelium of renal arteries (22), and in the apical membrane of intercalated cells in collecting ducts (134). Activation of PRR by the rat recombinant prorenin has been shown to stimulate cyclooxygenase-2 (COX-2)-derived prostaglandins via MAP kinases 1/2 in rat renal inner medullary collecting duct cells (IMCD) (135). Furthermore, prorenin appears to activate the prorenin/PRR/MAP kinases ERK 1/2 axis to increase V-ATPase activity (vacuolar-type H+-ATPase) at nanomolar concentrations in intercalated cells, MDCK.C11 (136). PRR has been described as an accessory subunit for V-ATPase, and may function as a H+-ATPase subunit in distal nephron segments of the kidney (137). However, Oshima et al. reported that PRR may be necessary for the maintenance of normal podocyte structure and function (138).
Activation of PRR by prorenin may be implicated in the development and progression of renal diseases in animal models. Kaneshiro et al. generated transgenic rats with overexpression of human prorenin/renin, and showed that these rats slowly developed nephropathy via MAP Kinases ERK1/2 signaling through an ANG II-independent mechanism (139). Ichihara et al. showed that the prorenin/PRR/MAP kinases ERK1/2 axis plays a pivotal role in the development of diabetic nephropathy in ANG II AT1a receptor-deficient mice (129) and in diabetic rats (128). Furthermore, Prieto and Navar’ group has shown that prorenin and PRR expression are markedly increased in the collecting ducts of distal nephron in ANG II-induced and 2K1C renal hypertension, although the precise roles of prorenin and PRR as a byproduct or mediator of ANG II-dependent hypertension remain unknown (69, 72).
Overall, prorenin and PRR have been studied extensively during last several years and appear to play important roles under certain biological, physiological, and pathophysiological conditions or animal models (12, 140, 141). However, their specific roles in the physiological regulation of cardiovascular, blood pressure, and renal function and the development of cardiovascular, hypertensive, and renal diseases in humans remain to be confirmed (13, 126). Recently, Reudelhuber (13) and Campbell (126) have provided excellent critical reviews in these issues. One key issue is that mice is known to express abundant prorenin and PRR than rats and humans, but they do not develop hypertension or cardiovascular and renal diseases. Another issue is that it is difficult to prove the activation of PRR by prorenin independent from renin without genetic deletion of PRR in mice, which is lethal at present (142, 143). The third issue is that prorenin may be overexpressed in transgenic rats or mice with hundreds or even thousands of time higher than those in humans to manifest cardiovascular, blood pressure, and renal phenotypes, which is unlikely replicated in normal and diseased humans (125, 144, 145). Finally, some, if not all, prorenin-induced blood pressure and cardiovascular and renal responses remain to be ANG II/AT1 receptor-dependent (13, 32, 126).
Current Insights and Future Perspectives on the Roles of Intracrine or Intracellular ANG II in the Kidney
A new frontier in the RAS research field has recently gained increasing attention (33–37, 146). This is now recognized as an intracrine or intracellular RAS. Many tissues or cells may synthesize ANG II within the cells, wherein ANG II bind to intracellular and/or nuclear receptors, activate downstream signaling pathways, and induce cellular and/or nuclear responses independent of cell surface receptors (33, 147–150). Alternatively, we and others have shown that circulating, paracrine, and autacrine ANG II may enter cells via AT1 (AT1a) receptor-mediated uptake or internalization in the kidney, primarily in the proximal tubule (48, 52, 57, 58, 151, 152). There is substantial evidence that not all internalized ANG II are degraded in lysosomes as the classical receptor pharmacology dogma suggests, and ANG II may escape from degradation by lysosomes. For example, systemically infused 125I-labeled ANG I or 125I-ANG II have been identified and quantified in pig kidney cells (55, 56, 85) and rat kidney cells (153, 154). Imig et al. demonstrated ACE, ANG II and AT1a receptors in cortical endosomes of the rat kidney (52). In ANG II-infused rat kidney, we found that ANG II levels in the renal cortical light and heavy endosomes were up to 10-fold higher compared with control rats (48). Intracellular accumulation of ANG II in the proximal tubule of the kidney may be blocked by the AT1 receptor blockers candesartan (48), losartan or in AT1a-KO mice (57, 58). To further support the new intracellular ANG II paradigm, specific and functional AT1 (AT1a) and AT2 receptors have been demonstrated in rat renal cortical endosomes (48, 52), mouse kidney proximal tubule mitochondria (155), and rat or sheep renal cortical nuclei (156–159). Thus the localization of intracellular and/or nuclear ANG II and AT1/AT2 receptors provides evidence that ANG II may interact with AT1/AT2 receptors within the kidney cells to induce biological and physiological effects.
In the kidney, previous studies demonstrated that AT1a receptor-mediated endocytosis of ANG II is required for ANG II-stimulated proximal tubular sodium transport or uptake of 22Na+ (160–163). We also showed that AT1a receptor-mediated ANG II uptake was associated the inhibition of cAMP signaling (151), activation of NF-κB signaling (163), and increases in lysate and membrane phosphorylated NHE3 proteins in proximal tubule cells (164). However these studies by no means provide direct evidence to support the role of intracellular and/or nuclear ANG II in the regulation of renal function and blood pressure responses. Several recent proof-of-concept studies have provided some new insights and perspectives into the potential roles of intracellular ANG II in the kidney. First, we used the single cell microinjection approach as described by Haller et al. (149) to determine the role of intracellular ANG II and its receptors in mobilizing intracellular calcium responses in rabbit proximal tubule cells (150). While the cell surface AT1 receptors were blocked by losartan in the medium, ANG II was directly microinjected into single monolayer proximal tubule cells sub-cultured on glass coverslips with or without the AT1 receptor blocker losartan or the AT2 receptor blocker PD123319. Microinjection of ANG II evoked marked increases in intracellular calcium responses, which were largely blocked by concurrent microinjection of losartan, but not by PD123319, indicating an AT1 receptor-mediated response (150). In a subsequent proof-of-concept study, we isolated fresh nuclei from the renal cortex of the rat kidney and incubated the nuclei with ANG II in an in vitro transcriptional system to determine the transcriptional effects of ANG II (156). We demonstrated that ANG II directly stimulated nuclear AT1a receptors to increase in vitro transcription of mRNAs for TGF-β1, MCP-1, and NHE3, which are known to play important roles in cell proliferation and hypertrophy, tissue fibrosis, and sodium transport in the kidney. Again, these nuclear transcriptional responses to ANG II were blocked by losartan but not by PD123319, further underlying an important role of AT1 (AT1a) receptors in proximal tubule cells. In alternative proof-of-concept studies, Chappell’s group showed that ANG II and ANG (1–7) directly stimulated nuclear AT2 or ANG (1–7) receptors to increase NO production, and activated AT1 receptors to increase super oxide production in freshly isolated sheep kidney nuclei (157, 158, 165).
Although it has been hypothesized that intracellular ANG II may play a physiological role in the cardiovascular and renal systems and blood pressure regulation, there was no direct evidence supporting this role until recently. Cook’s group was instrumental in generating transgenic mice that globally express an ANG II fused downstream of ECFP in all tissues, and its expression was driven by the mouse metallothionein promoter (146). The fusion protein, ECFP/ANG II, lacks a secretory signal, so its expression is retained intracellularly. Although plasma ANG II was not altered in these transgenic mice, basal blood pressure was significantly increased by approximately 16 mmHg, and thrombotic microangiopathy or microthrombosis was developed within the glomerular capillaries and small vessels (146). This study demonstrated for the first time that overexpression of an intracellular ANG II fusion protein is sufficient to elevate basal blood pressure and induce renal pathology. To determine the role of intracellular ANG II in the regulation of proximal tubular reabsorption and blood pressure, we performed intrarenal transfer of the same ECFP/ANG II selectively in the proximal tubule of rats and mice (Figures 3 and 4) (42, 77, 166). We showed that proximal tubule-specific overexpression of intracellular ECFP/ANG II significantly increased blood pressure by approximately 15–20 mmHg in rats and C57BL/6J mice 7 days after the gene transfer, and the blood pressure responses were blocked by losartan treatment or in AT1a-KO mice (42, 166, 167). Furthermore, the hypertensive effects of proximal tubule-specific ECFP/ANG II expression were associated with decreases in 24 h urinary sodium excretion, increases in phosphorylated ERK1/2, lysate, and membrane NHE3 proteins in freshly isolated proximal tubules and decrease in fractional lithium excretion (42, 166, 167). These responses are consistently with the concept that intracellular ANG II may stimulate AT1 receptor to increase proximal tubular sodium and fluid reabsorption, which in turn contributes to the regulation of blood pressure.
Current Insights and Future Perspectives on the Roles of ANG III, ANG IV, or ANG A in the Kidney
Two other smaller ANG peptides, ANG III and ANG IV, have been reported to have significant effects on blood pressure and renal function (2, 18, 19, 24, 28, 168). ANG III, ANG (2–8), is derived from the metabolism of ANG II by aminopeptidase A. To date, there is no evidence for a specific ANG III receptor. In the kidney, ANG III normally binds to the AT1 receptor and AT2 receptors, and the reported natriuretic and antinatriuretic effects of ANG III may be dose-dependent on whether the AT1 or AT2 receptor is activated (2, 18, 28, 168). When centrally administrated, ANG III appears to enhance vasopressin release, thirst, and blood pressure (169). Most recently, Carey’s group has shown that intrarenal interstitial ANG III infusion induced natriuresis via the AT2 receptor/nitric oxide/cGMP-dependent mechanism (19, 24, 170).
In the kidney, ANG III can be further hydrolyzed by aminopeptidase N to generate ANG IV or ANG (3–8) (2, 18, 171, 172). The receptor for ANG IV, AT4, has been identified as an IRAP, associated with the M1 family of aminopeptidases and GLUT4 vesicles in insulin-responsive cells (21, 173). The AT4 receptor has been localized in different tissues in the brain, heart, blood vessels, and kidney (20, 26, 174, 175). It is worth mentioning that other peptides such as LVV-hemorphin 7 also bind the AT4 receptor (21, 175, 176), and ANG IV also stimulates the AT1 receptor (20, 177–179). ANG IV is implicated in the regulation of learning and memory in rodents and improves memory in animal models of amnesia, and has been suggested to be used to treat Alzheimer’s disease (21, 175, 176). Aminopeptidases A and N are particularly abundant in the kidney, especially in the glomeruli and proximal nephron segment (2, 18, 171, 172). We have previously shown that nanomolar concentrations of ANG IV may increase blood pressure and induce renal vasoconstriction via the AT1 receptor-activated signaling in anesthetized rats (20), but others showed increased renal cortical blood flow and decreases Na+ transport in isolated renal proximal tubules (26, 27). Furthermore, Ang IV infusion into the renal artery decreased RBF, without any change in blood pressure, suggesting an AT1-mediated constriction in renal vascular bed (180). Other Ang IV responses in different kidney cells appear to occur via AT1 receptor activation as well, such as Ca2+ mobilization in glomerular mesangial cells (20, 178), and in human proximal tubules cells (181). In wild-type and AT1a, AT1b, AT2 receptor and IRAP knockout mice, Ang IV was found to mediate blood pressure and renal vasoconstrictor effects via AT1a receptors (182, 183). Thus, the physiological roles of ANG IV/AT4 receptors in blood pressure and renal regulation remain uncertain, given that circulating and tissue ANG IV levels are unlikely to be higher than those of ANG II in health and disease and that ANG IV also binds and stimulates AT1 receptors.
Recently, an ANG peptide-derived fragment called ANG A (Ala-Arg-Val-Tyr-Ile-His-Pro-Phe) has been described in the plasma of healthy humans and with increased concentrations in end-stage renal failure patients (184–186). ANG A may be generated from ANG II by decarboxylation of Asp1 and have the same affinity for AT1 receptor as ANG II, and higher affinity for AT2 receptor (186, 187). In rats, ANG A and ANG II have similar hypertensive effects, but ANG A possesses a greater proliferative effect on vascular smooth muscle cells than ANG II (186, 187). In genetically modified mice and in normotensive and hypertensive rats, ANG A induces pressor and renal vasoconstrictor responses also in the AT1 receptor-dependent manner (186). The role(s) of ANG A and its receptor-mediated downstream signaling mechanisms remain incompletely understood. However, since the ANG II/AT1 receptor-dependent pathways are involved, the translational impact of the ANG A research may likely be limited because the available ARBs are expected to block the actions of ANG A in tissues.
Concluding Remarks
In summary, the RAS has evolved from a circulating and endocrine system to multiple endocrine, paracrine, and intracrine systems. At least four axes for the RAS have been identified in the kidney and other tissues (Figure 1) and their physiological and pathophysiological roles explored. These include the most-studied and recognized classical renin/ACE/ANG II/AT1 and AT2 receptor axis, and three new axes including the ACE2/ANG (1–7)/Mas receptor, the prorenin/PRR/MAP kinases ERK1/2, and the ANG IV/AT4/IRAP axis. Each of these axes has its own enzyme(s), substrate(s), agonist(s), or ligand(s), respective receptor and downstream signaling mechanisms. Thus the roles of the RAS have been extended far beyond the regulation of blood pressure, aldosterone synthesis, and body salt and fluid homeostasis by the AT1 and AT2 receptors. Indeed, novel actions have been described for each axis of the entire RAS, interactions of which undoubtedly contribute to the overall regulation of cardiovascular, neural, and renal function and blood pressure. It is now well understood that imbalance of actions induced by ANG II and its smaller metabolites, ANG (1–7), ANG III, and ANG IV in favoring increases in tissue ANG II formation and the activation of the ACE/ANG II/AT1 receptor axis may lead to the development of hypertension and ANG II-induced target organ injury and diseases. Conversely, genetic and pharmacological approaches to increase the production of ANG (1–7) via overexpression of ACE2 or ANG (1–7) fusion protein may partially oppose the well-recognized actions of ANG II through activation of the Mas receptor. However, despite of the great progress new challenges still remain in the RAS research field. For example, the challenges for studying the classical ACE/ANG II/AT1 receptor axis may include determining the roles of intracellular and nuclear ANG II and its receptors play in the nuclear and/or transcriptional responses to ANG II in various diseases, and developing novel molecular and pharmacological approaches or drugs to block the transcriptional actions of intracellular ANG II. Since ANG III, ANG IV, and ANG A may also function as potent agonists of the AT1 and/or AT2 receptor to alter blood pressure and renal function, their physiological and pathophysiological roles remain to be determined. Similarly, the challenges for studying the roles of the prorenin/PRR/MAP kinases ERK1/2 axis is how to better differentiate the ANG II/AT1 receptor-dependent and independent effects of prorenin/PRR activation, and whether blockade of prorenin activation provides additional and beneficial effects beyond renin and ACE inhibitors or AT1 receptor blockers. Finally, although the ACE/ANG (1–7)/Mas receptor axis may play a counterregulatory role to oppose the effects of the renin/ACE/ANG II/AT1 receptor axis, the development and clinical relevance of the orally active agonists or compounds that promote metabolism of ANG II to increase ANG (1–7) production or to activate the Mas receptor still await clinical trials.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors’ work was supported in part by National Institute of Diabetes, Digestive and Kidney Diseases grants (5RO1DK067299, 2R56DK067299, and 2RO1DK067299), American Society of Nephrology M. James Scherbenske grant, and Hearin Foundation Medical Research Scholar Award to Dr. Jia L. Zhuo. Ms. Fernanda Ferrao is an international exchange graduate student from the Federal University of Rio de Janeiro and supported by a fellowship from Brazilian National Research Council (CNPq), Brazil, whereas Dr. Yun Zheng was an international visiting scholar and supported by a fellowship from Chang Jiang University, China. Due to the focus of the article in intrarenal RAS, we are unable to include many other outstanding investigators’ work in this review.
References
1. Tigerstedt R, Bergman PG. Niere und Kreislauf. Scand Arch Physiol (1898) 8:223–71. doi: 10.1111/j.1748-1716.1898.tb00272.x
2. Carey RM, Siragy HM. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr Rev (2003) 24(3):261–71. doi:10.1210/er.2003-0001
3. Zhuo JL, Allen AM, Alcorn D, Aldred GP, MacGregor DP, Mendelsohn FAO. The distribution of angiotensin II receptors. In: Laragh JH, Brenner BM editors. Hypertension: Pathophysiology, Diagnosis, and Management. New York: Raven Press, Ltd (1995). p. 1739–62.
4. Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, et al. Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A (2006) 103(47):17985–90. doi:10.1073/pnas.0605545103
5. de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev (2000) 52(3):415–72.
6. De Mello WC, Danser AH. Angiotensin II and the heart: on the intracrine renin-angiotensin system. Hypertension (2000) 35(6):1183–8. doi:10.1161/01.HYP.35.6.1183
7. Dzau VJ. Theodore Cooper Lecture: tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension (2001) 37(4):1047–52. doi:10.1161/01.HYP.37.4.1047
8. Chappell MC. Emerging evidence for a functional angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS receptor axis: more than regulation of blood pressure? Hypertension (2007) 50(4):596–9. doi:10.1161/HYPERTENSIONAHA.106.076216
9. Ferrario CM. Angiotensin-converting enzyme 2 and angiotensin-(1-7): an evolving story in cardiovascular regulation. Hypertension (2006) 47(3):515–21. doi:10.1161/01.HYP.0000196268.08909.fb
10. Gonzalez-Villalobos RA, Janjoulia T, Fletcher NK, Giani JF, Nguyen MT, Riquier-Brison AD, et al. The absence of intrarenal ACE protects against hypertension. J Clin Invest (2013) 123(5):2011–23. doi:10.1172/JCI65460
11. Danser AH, Deinum J. Renin, proreinin and the putative (pro)renin receptor. Hypertension (2005) 46:1069–76. doi:10.1161/01.HYP.0000186329.92187.2e
12. Nguyen G. Renin, (pro)renin and receptor: an update. Clin Sci (Lond) (2011) 120(5):169–78. doi:10.1042/CS20100432
13. Reudelhuber TL. The interaction between prorenin, renin and the (pro)renin receptor: time to rethink the role in hypertension. Curr Opin Nephrol Hypertens (2012) 21(2):137–41. doi:10.1097/MNH.0b013e3283500927
14. Romero DG, Gomez-Sanchez EP, Gomez-Sanchez CE. Angiotensin II-regulated transcription regulatory genes in adrenal steroidogenesis. Physiol Genomics (2010) 42A(4):259–66. doi:10.1152/physiolgenomics.00098.2010
15. Rector FC Jr., Brunner FP, Seldin DW. Mechanism of glomerulotubular balance. I. Effect of aortic constriction and elevated ureteropelvic pressure on glomerular filtration rate, fractional reabsorption, transit time, and tubular size in the proximal tubule of the rat. J Clin Invest (1966) 45(4):590–602. doi:10.1172/JCI105373
16. Santos RA, Ferreira AJ, Verano-Braga T, Bader M. Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: new players of the renin-angiotensin system. J Endocrinol (2013) 216(2):R1–17. doi:10.1530/JOE-12-0341
17. Ferrario CM, Varagic J. The ANG-(1-7)/ACE2/mas axis in the regulation of nephron function. Am J Physiol Renal Physiol (2010) 298(6):F1297–305. doi:10.1152/ajprenal.00110.2010
18. Chappell MC. Nonclassical renin-angiotensin system and renal function. Compr Physiol (2012) 2(4):2733–52. doi:10.1002/cphy.c120002
19. Kemp BA, Bell JF, Rottkamp DM, Howell NL, Shao W, Navar LG, et al. Intrarenal angiotensin III is the predominant agonist for proximal tubule angiotensin type 2 receptors. Hypertension (2012) 60(2):387–95. doi:10.1161/HYPERTENSIONAHA.112.191403
20. Li XC, Campbell DJ, Ohishi M, Yuan S, Zhuo JL. AT1 receptor-activated signaling mediates angiotensin IV-induced renal cortical vasoconstriction in rats. Am J Physiol Renal Physiol (2006) 290(5):F1024–33. doi:10.1152/ajprenal.00221.2005
21. Albiston AL, McDowall SG, Matsacos D, Sim P, Clune E, Mustafa T, et al. Evidence that the angiotensin IV (AT(4)) receptor is the enzyme insulin- regulated aminopeptidase. J Biol Chem (2001) 276(52):48623–6. doi:10.1074/jbc.C100512200
22. Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T, Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest (2002) 109:1417–27. doi:10.1172/JCI0214276
23. Santos RA, Simoes E, Silva AC, Maric C, Silva DM, Machado RP, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci U S A (2003) 100(14):8258–63. doi:10.1073/pnas.1432869100
24. Padia SH, Kemp BA, Howell NL, Gildea JJ, Keller SR, Carey RM. Intrarenal angiotensin III infusion induces natriuresis and angiotensin type 2 receptor translocation in Wistar-Kyoto but not in spontaneously hypertensive rats. Hypertension (2009) 53(2):338–43. doi:10.1161/HYPERTENSIONAHA.108.124198
25. Carey RM. Update on the role of the AT2 receptor. Curr Opin Nephrol Hypertens (2005) 14(1):67–71. doi:10.1097/00041552-200501000-00011
26. Handa RK, Krebs LT, Harding JW, Handa SE. Angiotensin IV AT4-receptor system in the rat kidney. Am J Physiol (1998) 274(2 Pt 2):F290–9.
27. Coleman JK, Krebs LT, Hamilton TA, Ong B, Lawrence KA, Sardinia MF, et al. Autoradiographic identification of kidney angiotensin IV binding sites and angiotensin IV-induced renal cortical blood flow changes in rats. Peptides (1998) 19(2):269–77. doi:10.1016/S0196-9781(97)00291-X
28. Harris PJ, Zhuo JL, Skinner SL. Effects of angiotensins II and III on glomerulotubular balance in rats. Clin Exp Pharmacol Physiol (1987) 14(6):489–502. doi:10.1111/j.1440-1681.1987.tb01505.x
29. Khosla MC, Smeby RR, Bumpus FM. Structure-activity relationship in angiotensin analogs. In: Page IH, Bumpus FM editors. Handbook of Experimental Pharmacology XXXVII: Angiotensin. Berlin: Springer-Verlag (1974). p. 126–61.
30. Garcia NH, Garvin JL. Angiotensin 1-7 has a biphasic effect on fluid absorption in the proximal straight tubule. J Am Soc Nephrol (1994) 5(4):1133–8.
31. Nguyen G. The (pro)renin receptor: pathophysiological roles in cardiovascular and renal pathology. Curr Opin Nephrol Hypertens (2007) 16(2):129–33. doi:10.1097/MNH.0b013e328040bfab
32. Danser AH. (Pro)renin receptors: are they biologically relevant? Curr Opin Nephrol Hypertens (2009a) 18(1):74–8. doi:10.1097/MNH.0b013e3283196aaf
33. Carey RM. Functional intracellular renin-angiotensin systems: potential for pathophysiology of disease. Am J Physiol Regul Integr Comp Physiol (2012) 302(5):R479–81. doi:10.1152/ajpregu.00656.2011
34. Ellis B, Li XC, Miguel-Qin E, Gu V, Zhuo JL. Evidence for a functional intracellular angiotensin system in the proximal tubule of the kidney. Am J Physiol Regul Integr Comp Physiol (2012) 302(5):R494–509. doi:10.1152/ajpregu.00487.2011
35. Cook JL, Re RN. Lessons from in vitro studies and a related intracellular angiotensin II transgenic mouse model. Am J Physiol Regul Integr Comp Physiol (2012) 302(5):R482–93. doi:10.1152/ajpregu.00493.2011
36. Gwathmey TM, Alzayadneh EM, Pendergrass KD, Chappell MC. Novel roles of nuclear angiotensin receptors and signaling mechanisms. Am J Physiol Regul Integr Comp Physiol (2012) 302(5):R518–30. doi:10.1152/ajpregu.00525.2011
37. Kumar R, Yong QC, Thomas CM, Baker KM. Intracardiac intracellular angiotensin system in diabetes. Am J Physiol Regul Integr Comp Physiol (2012) 302(5):R510–7. doi:10.1152/ajpregu.00512.2011
38. Crowley SD, Gurley SB, Oliverio MI, Pazmino AK, Griffiths R, Flannery PJ, et al. Distinct roles for the kidney and systemic tissues in blood pressure regulation by the renin-angiotensin system. J Clin Invest (2005) 115(4):1092–9. doi:10.1172/JCI23378
39. Lu X, Roksnoer LC, Danser AH. The intrarenal renin-angiotensin system: does it exist? Implications from a recent study in renal angiotensin-converting enzyme knockout mice. Nephrol Dial Transplant (2013). doi:10.1093/ndt/gft333
40. Gurley SB, Riquier-Brison AD, Schnermann J, Sparks MA, Allen AM, Haase VH, et al. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab (2011) 13(4):469–75. doi:10.1016/j.cmet.2011.03.001
41. Li H, Weatherford ET, Davis DR, Keen HL, Grobe JL, Daugherty A, et al. Renal proximal tubule angiotensin AT1A receptors regulate blood pressure. Am J Physiol Regul Integr Comp Physiol (2011) 301(4):R1067–77. doi:10.1152/ajpregu.00124.2011
42. Li XC, Zhuo JL. Proximal tubule-dominant transfer of AT1a receptors induces blood pressure responses to intracellular angiotensin II in AT1a receptor-deficient mice. Am J Physiol Regul Integr Comp Physiol (2013) 304:R588–98. doi:10.1152/ajpregu.00338.2012
43. Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev (2007) 59(3):251–87. doi:10.1124/pr.59.3.3
44. Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intratubular renin-angiotensin system in hypertension. Hypertension (2011) 57(3):355–62. doi:10.1161/HYPERTENSIONAHA.110.163519
45. Zhuo JL, Li XC. New insights and perspectives on intrarenal renin-angiotensin system: focus on intracrine/intracellular angiotensin II. Peptides (2011) 32(7):1551–65. doi:10.1016/j.peptides.2011.05.012
46. Nishiyama A, Seth DM, Navar LG. Renal interstitial fluid concentrations of angiotensins I and II in anesthetized rats. Hypertension (2002) 39(1):129–34. doi:10.1161/hy0102.100536
47. Siragy HM, Howell NL, Ragsdale NV, Carey RM. Renal interstitial fluid angiotensin. Modulation by anesthesia, epinephrine, sodium depletion, and renin inhibition. Hypertension (1995) 25(5):1021–4. doi:10.1161/01.HYP.25.5.1021
48. Zhuo JL, Imig JD, Hammond TG, Orengo S, Benes E, Navar LG. Ang II accumulation in rat renal endosomes during Ang II-induced hypertension: role of AT1 receptor. Hypertension (2002) 39(1):116–21. doi:10.1161/hy0102.100780
49. Shaltout HA, Westwood BM, Averill DB, Ferrario CM, Figueroa JP, Diz DI, et al. Angiotensin metabolism in renal proximal tubules, urine, and serum of sheep: evidence for ACE2-dependent processing of angiotensin II. Am J Physiol Renal Physiol (2007) 292(1):F82–91. doi:10.1152/ajprenal.00139.2006
50. Braam B, Mitchell KD, Fox J, Navar LG. Proximal tubular secretion of angiotensin II in rats. Am J Physiol (1993) 264(5 Pt 2):F891–8.
51. Navar LG, Lewis L, Hymel A, Braam B, Mitchell KD. Tubular fluid concentrations and kidney contents of angiotensins I and II in anesthetized rats. J Am Soc Nephrol (1994) 5(4):1153–8.
52. Imig JD, Navar GL, Zou LX, O’Reilly KC, Allen PL, Kaysen JH, et al. Renal endosomes contain angiotensin peptides, converting enzyme, and AT1A receptors. Am J Physiol (1999) 277(2 Pt 2):F303–11.
53. Zou LX, Hymel A, Imig JD, Navar LG. Renal accumulation of circulating angiotensin II in angiotensin II-infused rats. Hypertension (1996) 27(3 Pt 2):658–62. doi:10.1161/01.HYP.27.3.658
54. Zou LX, Imig JD, Hymel A, Navar LG. Renal uptake of circulating angiotensin II in Val5-angiotensin II infused rats is mediated by AT1 receptor. Am J Hypertens (1998) 11(5):570–8. doi:10.1016/S0895-7061(97)00410-X
55. van Kats JP, Schalekamp MA, Verdouw PD, Duncker DJ, Danser AH. Intrarenal angiotensin II: interstitial and cellular levels and site of production. Kidney Int (2001) 60(6):2311–7. doi:10.1046/j.1523-1755.2001.00049.x
56. van Kats JP, van Meegen JR, Verdouw PD, Duncker DJ, Schalekamp MA, Danser AH. Subcellular localization of angiotensin II in kidney and adrenal. J Hypertens (2001) 19(3 Pt 2):583–9. doi:10.1097/00004872-200103001-00010
57. Li XC, Navar LG, Shao Y, Zhuo JL. Genetic deletion of AT1a receptors attenuates intracellular accumulation of angiotensin II in the kidney of AT1a receptor-deficient mice. Am J Physiol Renal Physiol (2007) 293:F586–93. doi:10.1152/ajprenal.00489.2006
58. Li XC, Zhuo JL. In vivo regulation of AT1a receptor-mediated intracellular uptake of [125I]-Val5-angiotensin II in the kidneys and adrenal glands of AT1a receptor-deficient mice. Am J Physiol Renal Physiol (2008a) 294:F293–302. doi:10.1152/ajprenal.00398.2007
59. Anborgh PH, Seachrist JL, Dale LB, Ferguson SS. Receptor/beta-arrestin complex formation and the differential trafficking and resensitization of beta2-adrenergic and angiotensin II type 1A receptors. Mol Endocrinol (2000) 14(12):2040–53. doi:10.1210/me.14.12.2040
60. Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev (2001) 53(1):1–24.
61. Hein L, Meinel L, Pratt RE, Dzau VJ, Kobilka BK. Intracellular trafficking of angiotensin II and its AT1 and AT2 receptors: evidence for selective sorting of receptor and ligand. Mol Endocrinol (1997) 11(9):1266–77. doi:10.1210/me.11.9.1266
62. Hunyady L, Catt KJ, Clark AJ, Gaborik Z. Mechanisms and functions of AT1 angiotensin receptor internalization. Regul Pept (2000) 91(1-3):29–44. doi:10.1016/S0167-0115(00)00137-3
63. Luttrell LM, Gesty-Palmer D. Beyond desensitization: physiological relevance of arrestin-dependent signaling. Pharmacol Rev (2010) 62(2):305–30. doi:10.1124/pr.109.002436
64. Thomas WG, Thekkumkara TJ, Baker KM. Molecular mechanisms of angiotensin II (AT1A) receptor endocytosis. Clin Exp Pharmacol Physiol Suppl (1996) 3:S74–80. doi:10.1111/j.1440-1681.1996.tb02817.x
65. Nishiyama A, Seth DM, Navar LG. Angiotensin II type 1 receptor-mediated augmentation of renal interstitial fluid angiotensin II in angiotensin II-induced hypertension. J Hypertens (2003) 21(10):1897–903. doi:10.1097/00004872-200310000-00017
66. Harrison-Bernard LM, Zhuo J, Kobori H, Ohishi M, Navar LG. Intrarenal AT1 receptor and ACE binding in ANG II-induced hypertensive rats. Am J Physiol Renal Physiol (2002) 282(1):F19–25.
67. Kobori H, Harrison-Bernard LM, Navar LG. Enhancement of angiotensinogen expression in angiotensin II-dependent hypertension. Hypertension (2001) 37(5):1329–35. doi:10.1161/01.HYP.37.5.1329
68. Prieto-Carrasquero MC, Kobori H, Ozawa Y, Gutierrez A, Seth D, Navar LG. AT1 receptor-mediated enhancement of collecting duct renin in angiotensin II-dependent hypertensive rats. Am J Physiol Renal Physiol (2005) 289(3):F632–7. doi:10.1152/ajprenal.00462.2004
69. Prieto MC, Williams DE, Liu L, Kavanagh KL, Mullins JJ, Mitchell KD. Enhancement of renin and prorenin receptor in collecting duct of Cyp1a1-Ren2 rats may contribute to development and progression of malignant hypertension. Am J Physiol Renal Physiol (2011) 300(2):F581–8. doi:10.1152/ajprenal.00433.2010
70. Liu L, Gonzalez AA, McCormack M, Seth DM, Kobori H, Navar LG, et al. Increased renin excretion associated with augmented urinary angiotensin (Ang) II levels in chronic angiotensin II-infused hypertensive rats. Am J Physiol Renal Physiol (2011) 301(6):F1195–201. doi:10.1152/ajprenal.00339.2011
71. Zhuo JL. Augmented intratubular renin and prorenin expression in the medullary collecting ducts of the kidney as a novel mechanism of angiotensin II-induced hypertension. Am J Physiol Renal Physiol (2011) 301(6):F1193–4. doi:10.1152/ajprenal.00555.2011
72. Prieto MC, Botros FT, Kavanagh K, Navar LG. Prorenin receptor in distal nephron segments of 2-kidney, 1-clip Goldblatt hypertensive rats. Ochsner J (2013) 13(1):26–32.
73. Matsusaka T, Niimura F, Shimizu A, Pastan I, Saito A, Kobori H, et al. Liver angiotensinogen is the primary source of renal angiotensin II. J Am Soc Nephrol (2012) 23(7):1181–9. doi:10.1681/ASN.2011121159
74. Navar LG, Satou R, Gonzalez-Villalobos RA. The increasing complexity of the intratubular Renin-Angiotensin system. J Am Soc Nephrol (2012) 23(7):1130–2. doi:10.1681/ASN.2012050493
75. Gonzalez-Villalobos RA, Billet S, Kim C, Satou R, Fuchs S, Bernstein KE, et al. Intrarenal angiotensin-converting enzyme induces hypertension in response to angiotensin I infusion. J Am Soc Nephrol (2011) 22(3):449–59. doi:10.1681/ASN.2010060624
76. Coffman TM. Under pressure: the search for the essential mechanisms of hypertension. Nat Med (2011) 17(11):1402–9. doi:10.1038/nm.2541
77. Li XC, Cook JL, Rubera I, Tauc M, Zhang F, Zhuo JL. Intrarenal transfer of an intracellular cyan fluorescent fusion of angiotensin II selectively in proximal tubules increases blood pressure in rats and mice. Am J Physiol Renal Physiol (2011) 300:F1076–88. doi:10.1152/ajprenal.00329.2010
78. Esther CR, Marino EM, Howard TE, Machaud A, Corvol P, Capecchi MR, et al. The critical role of tissue angiotensin-converting enzyme as revealed by gene targeting in mice. J Clin Invest (1997) 99:2375–85. doi:10.1172/JCI119419
79. Cole J, Quach DL, Sundaram K, Corvol P, Capecchi MR, Bernstein KE. Mice lacking endothelial angiotensin-converting enzyme have a normal blood pressure. Circ Res (2002) 90(1):87–92. doi:10.1161/hh0102.102360
80. Hashimoto S, Adams JW, Bernstein KE, Schnermann J. Micropuncture determination of nephron function in mice without tissue angiotensin-converting enzyme. Am J Physiol Renal Physiol (2005) 288(3):F445–52. doi:10.1152/ajprenal.00297.2004
81. Kessler SP, Hashimoto S, Senanayake PS, Gaughan C, Sen GC, Schnermann J. Nephron function in transgenic mice with selective vascular or tubular expression of Angiotensin-converting enzyme. J Am Soc Nephrol (2005) 16(12):3535–42. doi:10.1681/ASN.2005020151
82. Kessler SP, deS SP, Scheidemantel TS, Gomos JB, Rowe TM, Sen GC. Maintenance of normal blood pressure and renal functions are independent effects of angiotensin-converting enzyme. J Biol Chem (2003) 278(23):21105–12. doi:10.1074/jbc.M3023472000
83. Reudelhuber TL. Where hypertension happens. J Clin Invest (2013) 123(5):1934–6. doi:10.1172/JCI69296
84. Zou LX, Imig JD, von Thun AM, Hymel A, Ono H, Navar LG. Receptor-mediated intrarenal angiotensin II augmentation in angiotensin II-infused rats. Hypertension (1996) 28(4):669–77. doi:10.1161/01.HYP.28.4.669
85. van Kats JP, de Lannoy LM, Jan Danser AH, van Meegen JR, Verdouw PD, Schalekamp MA. Angiotensin II type 1 (AT1) receptor-mediated accumulation of angiotensin II in tissues and its intracellular half-life in vivo. Hypertension (1997) 30(1 Pt 1):42–9. doi:10.1161/01.HYP.30.1.42
86. Li P, Chappell MC, Ferrario CM, Brosnihan KB. Angiotensin-(1-7) augments bradykinin-induced vasodilation by competing with ACE and releasing nitric oxide. Hypertension (1997) 29(1 Pt 2):394–400. doi:10.1161/01.HYP.29.1.394
87. Crowley SD, Zhang J, Herrera M, Griffiths RC, Ruiz P, Coffman TM. The role of AT1 receptor-mediated salt retention in angiotensin II-dependent hypertension. Am J Physiol Renal Physiol (2011) 301(5):F1124–30. doi:10.1152/ajprenal.00305.2011
88. Ferrario CM, Brosnihan KB, Diz DI, Jaiswal N, Khosla MC, Milsted A, et al. Angiotensin-(1-7): a new hormone of the angiotensin system. Hypertension (1991) 18(5 Suppl):III126–33. doi:10.1161/01.HYP.18.5_Suppl.III126
89. Khosla MC, Smeby RR, Bumps FM. Structural-activity relationship in angiotensin II analogs. In: Page IH, Bumpus FM editors. Angiotensin. Handbook of Experimental Pharmacology XXXVII. Heidelberg: Springer-Verlag Berlin (1974). p. 126–61.
90. Botelho-Santos GA, Sampaio WO, Reudelhuber TL, Bader M, Campagnole-Santos MJ, Santos RA. Expression of an angiotensin-(1-7)-producing fusion protein in rats induced marked changes in regional vascular resistance. Am J Physiol Heart Circ Physiol (2007) 292(5):H2485–90. doi:10.1152/ajpheart.01245.2006
91. Ferreira AJ, Pinheiro SV, Castro CH, Silva GA, Silva AC, Almeida AP, et al. Renal function in transgenic rats expressing an angiotensin-(1-7)-producing fusion protein. Regul Pept (2006) 137(3):128–33. doi:10.1016/j.regpep.2006.06.005
92. Mercure C, Yogi A, Callera GE, Aranha AB, Bader M, Ferreira AJ, et al. Angiotensin(1-7) blunts hypertensive cardiac remodeling by a direct effect on the heart. Circ Res (2008) 103(11):1319–26. doi:10.1161/CIRCRESAHA.108.184911
93. Yamamoto K, Chappell MC, Brosnihan KB, Ferrario CM. In vivo metabolism of angiotensin I by neutral endopeptidase (EC 3.4.24.11) in spontaneously hypertensive rats. Hypertension (1992) 19(6 Pt 2):692–6. doi:10.1161/01.HYP.19.6.692
94. Modrall JG, Sadjadi J, Brosnihan KB, Gallagher PE, Yu CH, Kramer GL, et al. Depletion of tissue angiotensin-converting enzyme differentially influences the intrarenal and urinary expression of angiotensin peptides. Hypertension (2004) 43(4):849–53. doi:10.1161/01.HYP.0000121462.27393.f6
95. Harris PJ, Navar LG. Tubular transport responses to angiotensin II. Am J Physiol Renal Physiol (1985) 248:F621–30.
96. Navar LG, Inscho EW, Majid SA, Imig JD, Harrison-Bernard LM, Mitchell KD. Paracrine regulation of the renal microcirculation. Physiol Rev (1996) 76(2):425–536.
97. Hall JE. Regulation of glomerular filtration rate and sodium excretion by angiotensin II. Fed Proc (1986) 45(5):1431–7.
99. Ferrario CM, Chappell MC, Tallant EA, Brosnihan KB, Diz DI. Counterregulatory actions of angiotensin-(1-7). Hypertension (1997) 30(3 Pt 2):535–41. doi:10.1161/01.HYP.30.3.535
100. De Souza AM, Lopes AG, Pizzino CP, Fossari RN, Miguel NC, Cardozo FP, et al. Angiotensin II and angiotensin-(1-7) inhibit the inner cortex Na+-ATPase activity through AT2 receptor. Regul Pept (2004) 120(1-3):167–75. doi:10.1016/j.regpep.2004.03.005
101. Lara LS, Cavalcante F, Axelband F, De Souza AM, Lopes AG, Caruso-Neves C. Involvement of the Gi/o/cGMP/PKG pathway in the AT2-mediated inhibition of outer cortex proximal tubule Na+-ATPase by Ang-(1-7). Biochem J (2006) 395(1):183–90. doi:10.1042/BJ20051455
102. Castelo-Branco RC, Leite-Delova DC, de Mello-Aires M. Dose-dependent effects of angiotensin-(1-7) on the NHE3 exchanger and [Ca(2+)](i) in in vivo proximal tubules. Am J Physiol Renal Physiol (2013) 304(10):F1258–65. doi:10.1152/ajprenal.00401.2012
103. Magaldi AJ, Cesar KR, de AM, Simoes e Silva AC, Santos RA. Angiotensin-(1-7) stimulates water transport in rat inner medullary collecting duct: evidence for involvement of vasopressin V2 receptors. Pflugers Arch (2003) 447(2):223–30. doi:10.1007/s00424-003-1173-1
104. Santos RA, Ferreira AJ, Nadu AP, Braga AN, de Almeida AP, Campagnole-Santos MJ, et al. Expression of an angiotensin-(1-7)-producing fusion protein produces cardioprotective effects in rats. Physiol Genomics (2004) 17(3):292–9. doi:10.1152/physiolgenomics.00227.2003
105. Rentzsch B, Todiras M, Iliescu R, Popova E, Campos LA, Oliveira ML, et al. Transgenic angiotensin-converting enzyme 2 overexpression in vessels of SHRSP rats reduces blood pressure and improves endothelial function. Hypertension (2008) 52(5):967–73. doi:10.1161/HYPERTENSIONAHA.108.114322
106. Liu CX, Hu Q, Wang Y, Zhang W, Ma ZY, Feng JB, et al. Angiotensin-converting enzyme (ACE) 2 overexpression ameliorates glomerular injury in a rat model of diabetic nephropathy: a comparison with ACE inhibition. Mol Med (2011) 17(1-2):59–69. doi:10.2119/molmed.2010.00111
107. Nadarajah R, Milagres R, Dilauro M, Gutsol A, Xiao F, Zimpelmann J, et al. Podocyte-specific overexpression of human angiotensin-converting enzyme 2 attenuates diabetic nephropathy in mice. Kidney Int (2012) 82(3):292–303. doi:10.1038/ki.2012.83
108. Yamazato M, Yamazato Y, Sun C, Diez-Freire C, Raizada MK. Overexpression of angiotensin-converting enzyme 2 in the rostral ventrolateral medulla causes long-term decrease in blood pressure in the spontaneously hypertensive rats. Hypertension (2007) 49(4):926–31. doi:10.1161/01.HYP.0000259942.38108.20
109. Diez-Freire C, Vazquez J, Correa de Adjounian MF, Ferrari MF, Yuan L, Silver X, et al. ACE2 gene transfer attenuates hypertension-linked pathophysiological changes in the SHR. Physiol Genomics (2006) 27(1):12–9. doi:10.1152/physiolgenomics.00312.2005
110. Der SS, Grobe JL, Yuan L, Narielwala DR, Walter GA, Katovich MJ, et al. Cardiac overexpression of angiotensin converting enzyme 2 protects the heart from ischemia-induced pathophysiology. Hypertension (2008) 51(3):712–8. doi:10.1161/HYPERTENSIONAHA.107.100693
111. Qiao X, Li RS, Li H, Zhu GZ, Huang XG, Shao S, et al. Intermedin protects against renal ischemia-reperfusion injury by inhibition of oxidative stress. Am J Physiol Renal Physiol (2013) 304(1):F112–9. doi:10.1152/ajprenal.00054.2012
112. National Heart Lung and Blood Institute. The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Bethesda, MD: NIH Publication (2003).
113. Hummel SL, Seymour EM, Brook RD, Kolias TJ, Sheth SS, Rosenblum HR, et al. Low-sodium dietary approaches to stop hypertension diet reduces blood pressure, arterial stiffness, and oxidative stress in hypertensive heart failure with preserved ejection fraction. Hypertension (2012) 60(5):1200–6. doi:10.1161/HYPERTENSIONAHA.112.202705
114. Sacks FM, Svetkey LP, Vollmer WM, Appel LJ, Bray GA, Harsha D, et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N Engl J Med (2001) 344(1):3–10. doi:10.1056/NEJM200101043440101
115. Botelho-Santos GA, Bader M, Alenina N, Santos RA. Altered regional blood flow distribution in Mas-deficient mice. Ther Adv Cardiovasc Dis (2012) 6(5):201–11. doi:10.1177/1753944712461164
116. Walther T, Wessel N, Kang N, Sander A, Tschope C, Malberg H, et al. Altered heart rate and blood pressure variability in mice lacking the Mas protooncogene. Braz J Med Biol Res (2000) 33(1):1–9. doi:10.1590/S0100-879X2000000100001
117. Heringer-Walther S, Gembardt F, Perschel FH, Katz N, Schultheiss HP, Walther T. The genetic deletion of Mas abolishes salt induced hypertension in mice. Eur J Pharmacol (2012) 689(1-3):147–53. doi:10.1016/j.ejphar.2012.05.025
118. de Moura MM, dos Santos RA, Campagnole-Santos MJ, Todiras M, Bader M, Alenina N, et al. Altered cardiovascular reflexes responses in conscious Angiotensin-(1-7) receptor Mas-knockout mice. Peptides (2010) 31(10):1934–9. doi:10.1016/j.peptides.2010.06.030
119. Alenina N, Xu P, Rentzsch B, Patkin EL, Bader M. Genetically altered animal models for Mas and angiotensin-(1-7). Exp Physiol (2008) 93(5):528–37. doi:10.1113/expphysiol.2007.040345
120. Pinheiro SV, Ferreira AJ, Kitten GT, da Silveira KD, da Silva DA, Santos SH, et al. Genetic deletion of the angiotensin-(1-7) receptor Mas leads to glomerular hyperfiltration and microalbuminuria. Kidney Int (2009) 75(11):1184–93. doi:10.1038/ki.2009.61
121. Esteban V, Heringer-Walther S, Sterner-Kock A, de BR, van den Engel S, Wang Y, et al. Angiotensin-(1-7) and the G protein-coupled receptor MAS are key players in renal inflammation. PLoS One (2009) 4(4):e5406. doi:10.1371/journal.pone.0005406
122. Zimmerman D, Burns KD. Angiotensin-(1-7) in kidney disease: a review of the controversies. Clin Sci (Lond) (2012) 123(6):333–46. doi:10.1042/CS20120111
124. Danser AH, van den Dorpel MA, Deinum J, Derkx FH, Franken AA, Peperkamp E, et al. Renin, prorenin, and immunoreactive renin in vitreous fluid from eyes with and without diabetic retinopathy. J Clin Endocrinol Metab (1989) 68(1):160–7. doi:10.1210/jcem-68-1-160
125. Campbell DJ, Karam H, Menard J, Bruneval P, Mullins JJ. Prorenin contributes to angiotensin peptide formation in transgenic rats with rat prorenin expression targeted to the liver. Hypertension (2009) 54(6):1248–53. doi:10.1161/HYPERTENSIONAHA.109.138495
126. Campbell DJ. Critical review of prorenin and (pro)renin receptor research. Hypertension (2008) 51(5):1259–64. doi:10.1161/HYPERTENSIONAHA.108.110924
127. Nguyen G, Delarue F, Berrou J, Rondeau E, Sraer JD. Specific receptor binding of renin on human mesangial cells in culture increases plasminogen activator inhibitor-1 antigen. Kidney Int (1996) 50(6):1897–903. doi:10.1038/ki.1996.511
128. Ichihara A, Hayashi M, Kaneshiro Y, Suzuki F, Nakagawa T, Tada Y, et al. Inhibition of diabetic nephropathy by a decoy peptide corresponding to the “handle” region for nonproteolytic activation of prorenin. J Clin Invest (2004) 114(8):1128–35. doi:10.1172/JCI21398
129. Ichihara A, Suzuki F, Nakagawa T, Kaneshiro Y, Takemitsu T, Sakoda M, et al. Prorenin receptor blockade inhibits development of glomerulosclerosis in diabetic angiotensin II type 1a receptor-deficient mice. J Am Soc Nephrol (2006) 17(7):1950–61. doi:10.1681/ASN.2006010029
130. Luft FC. Renin and its putative receptor remain enigmas. J Am Soc Nephrol (2007) 18(7):1989–92. doi:10.1681/ASN.2007050558
131. Feldt S, Maschke U, Dechend R, Luft FC, Muller DN. The putative (pro)renin receptor blocker HRP fails to prevent (pro)renin signaling. J Am Soc Nephrol (2008) 19(4):743–8. doi:10.1681/ASN.2007091030
132. Feldt S, Batenburg WW, Mazak I, Maschke U, Wellner M, Kvakan H, et al. Prorenin and renin-induced extracellular signal-regulated kinase 1/2 activation in monocytes is not blocked by aliskiren or the handle-region peptide. Hypertension (2008) 51(3):682–8. doi:10.1161/HYPERTENSIONAHA.107.101444
133. Muller DN, Klanke B, Feldt S, Cordasic N, Hartner A, Schmieder RE, et al. (Pro)renin receptor peptide inhibitor “handle-region” peptide does not affect hypertensive nephrosclerosis in Goldblatt rats. Hypertension (2008) 51(3):676–81. doi:10.1161/HYPERTENSIONAHA.107.101493
134. Advani A, Kelly DJ, Cox AJ, White KE, Advani SL, Thai K, et al. The (Pro)renin receptor: site-specific and functional linkage to the vacuolar H+-ATPase in the kidney. Hypertension (2009) 54(2):261–9. doi:10.1161/HYPERTENSIONAHA.109.128645
135. Gonzalez AA, Luffman C, Bourgeois CR, Vio CP, Prieto MC. Angiotensin II-independent upregulation of cyclooxygenase-2 by activation of the (Pro)renin receptor in rat renal inner medullary cells. Hypertension (2013) 61(2):443–9. doi:10.1161/HYPERTENSIONAHA.112.196303
136. Lu X, Garrelds IM, Wagner CA, Danser AH, Meima ME. (Pro)renin receptor is required for prorenin-dependent and -independent regulation of vacuolar H+-ATPase activity in MDCK.C11 collecting duct cells. Am J Physiol Renal Physiol (2013) 305(3):F417–25. doi:10.1152/ajprenal.00037.2013
137. Kinouchi K, Ichihara A, Sano M, Sun-Wada GH, Wada Y, Kurauchi-Mito A, et al. The (pro)renin receptor/ATP6AP2 is essential for vacuolar H+-ATPase assembly in murine cardiomyocytes. Circ Res (2010) 107(1):30–4. doi:10.1161/CIRCRESAHA.110.224667
138. Oshima Y, Kinouchi K, Ichihara A, Sakoda M, Kurauchi-Mito A, Bokuda K, et al. Prorenin receptor is essential for normal podocyte structure and function. J Am Soc Nephrol (2011) 22(12):2203–12. doi:10.1681/ASN.2011020202
139. Kaneshiro Y, Ichihara A, Sakoda M, Takemitsu T, Nabi AH, Uddin MN, et al. Slowly progressive, angiotensin II-independent glomerulosclerosis in human (pro)renin receptor-transgenic rats. J Am Soc Nephrol (2007) 18(6):1789–95. doi:10.1681/ASN.2006091062
140. Nguyen G, Contrepas A. Physiology and pharmacology of the (pro)renin receptor. Curr Opin Pharmacol (2008) 8(2):127–32. doi:10.1016/j.coph.2007.12.009
141. Nguyen G. The (pro)renin receptor in health and disease. Ann Med (2010) 42(1):13–8. doi:10.3109/07853890903321567
142. Cruciat CM, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D, et al. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science (2010) 327(5964):459–63. doi:10.1126/science.1179802
143. Danser AH. (Pro)renin receptor and vacuolar H+-ATPase. Hypertension (2009b) 54(2):219–21. doi:10.1161/HYPERTENSIONAHA.109.135236
144. Mercure C, Prescott G, Lacombe MJ, Silversides DW, Reudelhuber TL. Chronic increases in circulating prorenin are not associated with renal or cardiac pathologies. Hypertension (2009) 53(6):1062–9. doi:10.1161/HYPERTENSIONAHA.108.115444
145. Peters B, Grisk O, Becher B, Wanka H, Kuttler B, Ludemann J, et al. Dose-dependent titration of prorenin and blood pressure in Cyp1a1ren-2 transgenic rats: absence of prorenin-induced glomerulosclerosis. J Hypertens (2008) 26(1):102–9. doi:10.1097/HJH.0b013e3282f0ab66
146. Redding KM, Chen BL, Singh A, Re RN, Navar LG, Seth DM, et al. Transgenic mice expressing an intracellular fluorescent fusion of angiotensin II demonstrate renal thrombotic microangiopathy and elevated blood pressure. Am J Physiol Heart Circ Physiol (2010) 298:H1807–18. doi:10.1152/ajpheart.00027.2010
147. Re RN. On the biological actions of intracellular angiotensin. Hypertension (2000) 35(6):1189–90. doi:10.1161/01.HYP.35.6.1189
148. Cook JL, Zhang Z, Re RN. In vitro evidence for an intracellular site of angiotensin action. Circ Res (2001) 89(12):1138–46. doi:10.1161/hh2401.101270
149. Haller H, Lindschau C, Erdmann B, Quass P, Luft FC. Effects of intracellular angiotensin II in vascular smooth muscle cells. Circ Res (1996) 79(4):765–72. doi:10.1161/01.RES.79.4.765
150. Zhuo JL, Li XC, Garvin JL, Navar LG, Carretero OA. Intracellular angiotensin II induces cytosolic Ca2+ mobilization by stimulating intracellular AT1 receptors in proximal tubule cells. Am J Physiol Renal Physiol (2006) 290:F1382–90. doi:10.1152/ajprenal.00269.2005
151. Li XC, Carretero OA, Navar LG, Zhuo JL. AT1 receptor-mediated accumulation of extracellular angiotensin II in proximal tubule cells: role of cytoskeleton microtubules and tyrosine phosphatases. Am J Physiol Renal Physiol (2006) 291:F375–83. doi:10.1152/ajprenal.00405.2005
152. Li XC, Hopfer U, Zhuo JL. AT1 receptor-mediated uptake of angiotensin II and NHE-3 expression in proximal tubule cells through the microtubule-dependent endocytic pathway. Am J Physiol Renal Physiol (2009) 297(5):F1342–52. doi:10.1152/ajprenal.90734.2008
153. Zhuo JL, Alcorn D, McCausland J, Casley D, Mendelsohn FA. In vivo occupancy of angiotensin II subtype 1 receptors in rat renal medullary interstitial cells. Hypertension (1994) 23(6 Pt 2):838–43. doi:10.1161/01.HYP.23.6.838
154. Zhuo JL, Alcorn D, Harris PJ, Mendelsohn FA. Localization and properties of angiotensin II receptors in rat kidney. Kidney Int Suppl (1993) 42:S40–6.
155. Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, et al. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci U S A (2011) 108(36):14849–54. doi:10.1073/pnas.1101507108
156. Li XC, Zhuo JL. Intracellular ANG II directly induces in vitro transcription of TGF-beta1, MCP-1, and NHE-3 mRNAs in isolated rat renal cortical nuclei via activation of nuclear AT1a receptors. Am J Physiol Cell Physiol (2008b) 294(4):C1034–45. doi:10.1152/ajpcell.00432.2007
157. Gwathmey T, Shaltout HA, Pendergrass KD, Pirro NT, Figueroa JP, Rose JC, et al. Nuclear angiotensin II – type 2 (AT2) receptors are functionally linked to nitric oxide production. Am J Physiol Renal Physiol (2009) 296:F1484–93. doi:10.1152/ajprenal.90766.2008
158. Gwathmey TM, Westwood BM, Pirro NT, Tang L, Rose JC, Diz DI, et al. Nuclear angiotensin-(1-7) receptor is functionally coupled to the formation of nitric oxide. Am J Physiol Renal Physiol (2010) 299(5):F983–90. doi:10.1152/ajprenal.00371.2010
159. Pendergrass KD, Averill DB, Ferrario CM, Diz DI, Chappell MC. Differential expression of nuclear AT1 receptors and angiotensin II within the kidney of the male congenic mRen2.Lewis rat. Am J Physiol Renal Physiol (2006) 290(6):F1497–506. doi:10.1152/ajprenal.00317.2005
160. Schelling JR, Linas SL. Angiotensin II-dependent proximal tubule sodium transport requires receptor-mediated endocytosis. Am J Physiol (1994) 266(3 Pt 1):C669–75.
161. Schelling JR, Hanson AS, Marzec R, Linas SL. Cytoskeleton-dependent endocytosis is required for apical type 1 angiotensin II receptor-mediated phospholipase C activation in cultured rat proximal tubule cells. J Clin Invest (1992) 90(6):2472–80. doi:10.1172/JCI116139
162. Thekkumkara TJ, Cookson R, Linas SL. Angiotensin (AT1A) receptor-mediated increases in transcellular sodium transport in proximal tubule cells. Am J Physiol (1998) 274:F897–905.
163. Zhuo JL, Carretero OA, Li XC. Effects of AT1 receptor-mediated endocytosis of extracellular Ang II on activation of nuclear factor-kappa B in proximal tubule cells. Ann N Y Acad Sci (2006) 1091:336–45. doi:10.1196/annals.1378.078
164. Li XC, Zhuo JL. Selective knockdown of AT1 receptors by RNA interference inhibits Val5-Ang II endocytosis and NHE-3 expression in immortalized rabbit proximal tubule cells. Am J Physiol Cell Physiol (2007) 293:C367–78. doi:10.1152/ajpcell.00463.2006
165. Gwathmey TM, Pendergrass KD, Reid SD, Rose JC, Diz DI, Chappell MC. Angiotensin-(1-7)-angiotensin-converting enzyme 2 attenuates reactive oxygen species formation to angiotensin II within the cell nucleus. Hypertension (2010) 55(1):166–71. doi:10.1161/HYPERTENSIONAHA.109.141622
166. Li XC, Hopfer U, Zhuo JL. Novel signaling mechanisms of intracellular angiotensin II-induced NHE3 expression and activation in mouse proximal tubule cells. Am J Physiol Renal Physiol (2012) 303(12):F1617–28. doi:10.1152/ajprenal.00219.2012
167. Harding JW, Wright JW, Swanson GN, Hanesworth JM, Krebs LT. AT4 receptors: specificity and distribution. Kidney Int (1994) 46(6):1510–2. doi:10.1038/ki.1994.432
168. Yugandhar VG, Clark MA. Angiotensin III: a physiological relevant peptide of the renin angiotensin system. Peptides (2013) 46:26–32. doi:10.1016/j.peptides.2013.04.014
169. Cesari M, Rossi GP, Pessina AC. Biological properties of the angiotensin peptides other than angiotensin II: implications for hypertension and cardiovascular diseases. J Hypertens (2002) 20(5):793–9. doi:10.1097/00004872-200205000-00002
170. Padia SH, Kemp BA, Howell NL, Siragy HM, Fournie-Zaluski MC, Roques BP, et al. Intrarenal aminopeptidase N inhibition augments natriuretic responses to angiotensin III in angiotensin type 1 receptor-blocked rats. Hypertension (2007) 49(3):625–30. doi:10.1161/01.HYP.0000254833.85106.4d
171. Ardaillou R, Chansel D. Synthesis and effects of active fragments of angiotensin II. Kidney Int (1997) 52(6):1458–68. doi:10.1038/ki.1997.476
172. Chansel D, Czekalski S, Vandermeersch S, Ruffet E, Fournie-Zaluski MC, Ardaillou R. Characterization of angiotensin IV-degrading enzymes and receptors on rat mesangial cells. Am J Physiol (1998) 275(4 Pt 2):F535–42.
173. Albiston AL, Yeatman HR, Pham V, Fuller SJ, Diwakarla S, Fernando RN, et al. Distinct distribution of GLUT4 and insulin regulated aminopeptidase in the mouse kidney. Regul Pept (2011) 166(1-3):83–9. doi:10.1016/j.regpep.2010.09.003
174. Wright JW, Harding JW. Important role for angiotensin III and IV in the brain renin-angiotensin system. Brain Res Brain Res Rev (1997) 25(1):96–124. doi:10.1016/S0165-0173(97)00019-2
175. Chai SY, Fernando R, Peck G, Ye SY, Mendelsohn FA, Jenkins TA, et al. The angiotensin IV/AT4 receptor. Cell Mol Life Sci (2004) 61(21):2728–37. doi:10.1007/s00018-004-4246-1
176. Lew RA, Mustafa T, Ye S, McDowall SG, Chai SY, Albiston AL. Angiotensin AT4 ligands are potent, competitive inhibitors of insulin regulated aminopeptidase (IRAP). J Neurochem (2003) 86(2):344–50. doi:10.1046/j.1471-4159.2003.01852.x
177. Gardiner SM, Kemp PA, March JE, Bennett T. Regional haemodynamic effects of angiotensin II (3-8) in conscious rats. Br J Pharmacol (1993) 110(1):159–62. doi:10.1111/j.1476-5381.1993.tb13786.x
178. Chansel D, Vandermeersch S, Oko A, Curat C, Ardaillou R. Effects of angiotensin IV and angiotensin-(1-7) on basal and angiotensin II-stimulated cytosolic Ca2+ in mesangial cells. Eur J Pharmacol (2001) 414(2-3):165–75. doi:10.1016/S0014-2999(01)00791-9
179. Loufrani L, Henrion D, Chansel D, Ardaillou R, Levy BI. Functional evidence for an angiotensin IV receptor in rat resistance arteries. J Pharmacol Exp Ther (1999) 291(2):583–8.
180. Fitzgerald SM, Evans RG, Bergstrom G, Anderson WP. Renal hemodynamic responses to intrarenal infusion of ligands for the putative angiotensin IV receptor in anesthetized rats. J Cardiovasc Pharmacol (1999) 34(2):206–11. doi:10.1097/00005344-199908000-00005
181. Handa RK. Characterization and signaling of the AT4 receptor in human proximal tubule epithelial (HK-2) cells. J Am Soc Nephrol (2001) 12(3):440–9.
182. Yang R, Walther T, Gembardt F, Smolders I, Vanderheyden P, Albiston AL, et al. Renal vasoconstrictor and pressor responses to angiotensin IV in mice are AT1a-receptor mediated. J Hypertens (2010) 28(3):487–94. doi:10.1097/HJH.0b013e3283343250
183. Yang R, Smolders I, De BD, Fouyn R, Halberg M, Demaegdt H, et al. Brain and peripheral angiotensin II type 1 receptors mediate renal vasoconstrictor and blood pressure responses to angiotensin IV in the rat. J Hypertens (2008) 26(5):998–1007. doi:10.1097/HJH.0b013e3282f5ed58
184. Coutinho DC, Foureaux G, Rodrigues KD, Salles RL, Moraes PL, Murca TM, et al. Cardiovascular effects of angiotensin A: a novel peptide of the renin-angiotensin system. J Renin Angiotensin Aldosterone Syst (2013). doi:10.1177/1470320312474856
185. Lautner RQ, Villela DC, Fraga-Silva RA, Silva N, Verano-Braga T, Costa-Fraga F, et al. Discovery and characterization of alamandine: a novel component of the renin-angiotensin system. Circ Res (2013) 112(8):1104–11. doi:10.1161/CIRCRESAHA.113.301077
186. Yang R, Smolders I, Vanderheyden P, Demaegdt H, Van EA, Vauquelin G, et al. Pressor and renal hemodynamic effects of the novel angiotensin A peptide are angiotensin II type 1A receptor dependent. Hypertension (2011) 57(5):956–64. doi:10.1161/HYPERTENSIONAHA.110.161836
Keywords: angiotensin 1-converting enzyme, ACE2, angiotensin II receptor, blood pressure, hypertension, kidney, proximal tubule, signal transduction
Citation: Zhuo JL, Ferrao FM, Zheng Y and Li XC (2013) New frontiers in the intrarenal renin-angiotensin system: a critical review of classical and new paradigms. Front. Endocrinol. 4:166. doi: 10.3389/fendo.2013.00166
Received: 30 August 2013; Paper pending published: 23 September 2013;
Accepted: 22 October 2013; Published online: 11 November 2013.
Edited by:
Walmor De Mello, University of Puerto Rico, USAReviewed by:
Norifumi Iijima, Yale University School of Medicine, USAMark Chappell, Wake Forest University School of Medicine, USA
Copyright: © 2013 Zhuo, Ferrao, Zheng and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jia L. Zhuo, Department of Pharmacology and Toxicology, University of Mississippi Medical Center, 2500 North State Street, Jackson, MS 39216-4505, USA e-mail:anpodW9AdW1jLmVkdQ==