 Mark C. Chappell
Mark C. Chappell Allyson C. Marshall
Allyson C. Marshall Ebaa M. Alzayadneh
Ebaa M. Alzayadneh Hossam A. Shaltout
Hossam A. Shaltout Debra I. Diz
Debra I. Diz- 1The Hypertension and Vascular Research Center, Wake Forest University School of Medicine, Winston-Salem, NC, USA
- 2Department of Obstetrics and Gynecology, Wake Forest University School of Medicine, Winston-Salem, NC, USA
- 3Department of Pharmacology and Toxicology, School of Pharmacy, Alexandria University, Alexandria, Egypt
The renin-angiotensin-system (RAS) constitutes an important hormonal system in the physiological regulation of blood pressure. Indeed, dysregulation of the RAS may lead to the development of cardiovascular pathologies including kidney injury. Moreover, the blockade of this system by the inhibition of angiotensin converting enzyme (ACE) or antagonism of the angiotensin type 1 receptor (AT1R) constitutes an effective therapeutic regimen. It is now apparent with the identification of multiple components of the RAS that the system is comprised of different angiotensin peptides with diverse biological actions mediated by distinct receptor subtypes. The classic RAS can be defined as the ACE-Ang II-AT1R axis that promotes vasoconstriction, sodium retention, and other mechanisms to maintain blood pressure, as well as increased oxidative stress, fibrosis, cellular growth, and inflammation in pathological conditions. In contrast, the non-classical RAS composed of the ACE2-Ang-(1–7)-Mas receptor axis generally opposes the actions of a stimulated Ang II-AT1R axis through an increase in nitric oxide and prostaglandins and mediates vasodilation, natriuresis, diuresis, and oxidative stress. Thus, a reduced tone of the Ang-(1–7) system may contribute to these pathologies as well. Moreover, the non-classical RAS components may contribute to the effects of therapeutic blockade of the classical system to reduce blood pressure and attenuate various indices of renal injury. The review considers recent studies on the ACE2-Ang-(1–7)-Mas receptor axis regarding the precursor for Ang-(1–7), the intracellular expression and sex differences of this system, as well as an emerging role of the Ang1-(1–7) pathway in fetal programing events and cardiovascular dysfunction.
Introduction
Over the past 20 years the concept of the renin-angiotensin-system (RAS) as a monolithic endocrine system reflected primarily by the interaction of the peptide Angiotensin II (Ang II) with the AT1-receptor subtype has undergone extensive revision. The emerging view of alternative pathways within the RAS that may functionally antagonize the Ang II-AT1-receptor axis may be traced back to both the characterization of the AT2 receptor subtype and the identification of the heptapeptide des-[Phe8]-Angiotensin II or Angiotensin-(1–7) [Ang-(1–7)] in the circulation and various tissues (1–4). Since that time, the elaboration of distinct biochemical components that comprise the “Ang-(1–7) axis” is now firmly established with the identification of a unique receptor for Ang-(1–7) – the G-protein coupled Mas receptor, selective antagonists and agonists for the receptor, and an angiotensin II converting enzyme (ACE2) that catalyzes the processing of Ang II to Ang-(1–7) (5–9). In addition to the identification of the components of the Ang-(1–7) system, there is the recognition of various signaling pathways including nitric oxide (NO), prostaglandins, and cellular phosphatases that are stimulated by the peptide (10, 11). Although the early studies of Ang-(1–7) primarily sought to establish a role for Ang-(1–7) in the regulation of blood pressure, particularly as endogenous levels of the peptide increase markedly following angiotensin converting enzyme (ACE) or AT1-receptor blockade; the pressure-independent actions of the Ang-(1–7) axis should be considered with perhaps equal importance (6, 12, 13). Indeed, the beneficial actions of Ang-(1–7) system encompass various pathologies from cancer and the anti-proliferative actions of the peptide to diabetes and the cellular effects on stem cells (8, 9, 14). In turn, deficiencies in Ang-(1–7) that contribute to autonomic dysfunction were apparent in hypertension (15) and aging (16); Ang-(1–7) deficiency in hypertension was restored by ACE inhibitor treatment (17) or chronic Ang-(1–7) replacement (18). The breadth of these effects is not surprising as the RAS is a tissue system whose protein and peptide components are expressed in essentially every organ and whose actions are implicated in numerous physiological events that influence renal, neuronal, cardiac, pancreatic, vascular, adrenal, pituitary, cognitive, aging, inflammatory, and reproductive functions (19). As the Ang II-AT1-receptor axis of the RAS is increasingly recognized as a key regulatory pathway in various tissues and cells, the counter-balancing Ang-(1–7) axis should be evident as well. In this review, we consider the current literature on the ACE2-Ang-(1–7)-Mas receptor axis regarding the sources for Ang-(1–7), the intracellular expression of this system, the emerging role of Ang1-(1–7) pathway in fetal programing events and cardiovascular dysfunction, and finally, the evidence for sex-dependent regulation and function of the Ang-(1–7) axis.
Sources of Angiotensin-(1–7)
Endopeptidases
Angiotensinogen, a glycosylated protein that is primarily synthesized and secreted by the liver as well as other tissues is the sole precursor for angiotensin peptides (20). The only known substrate for the aspartyl protease renin is angiotensinogen which releases the decapeptide Ang I from the amino-terminal portion of the protein (Figure 1). Ang I is then cleaved by ACE to form the bioactive peptide Ang II. Early studies revealed that endogenous levels of both Ang I and Ang-(1–7) were markedly increased following the administration of ACE inhibitors (21, 22). The augmented response in Ang-(1–7) suggested that the circulating peptide may contribute to the beneficial actions of the inhibition of ACE pathway in addition to that of reducing endogenous levels of Ang II. The increase in Ang-(1–7) in the presence of ACE blockade necessitates a processing pathway independent of the formation of Ang II. Several studies subsequently showed that the endopeptidase 3.4.24.11 (neprilysin) contributed to the circulating levels of Ang-(1–7) in animals chronically treated with various ACE inhibitors (23–26). Ang I infusion in normotensive WKY and hypertensive spontaneously hypertensive rat (SHR) treated with the ACE inhibitor lisinopril resulted in higher plasma levels of Ang-(1–7) and co-administration of the neprilysin inhibitor SCH39370 but not the prolyl oligopeptidase (POP) inhibitor z-prolyl prolinal abolished the circulating levels of the peptide (27). Moreover, acute infusion of a similar dose of Ang II did not increase circulating Ang-(1–7) in either control or lisinopril-treated WKY and SHR. The increase in Ang-(1–7) following ACE blockade reflects both a reduction in Ang-(1–7) metabolism and alternative processing of Ang I through tissue-specific endopeptidases (23). In this regard, Pereira et al. recently demonstrated that the endopeptidase EC3.4.24.15 (thimet oligopeptidase) may contribute to formation of Ang-(1–7) in the rat hippocampus (28). Interestingly, these investigators reported higher expression of this peptidase and the Mas receptor in a rat model of temporal lobe epilepsy suggesting a possible role of the Ang-(1–7)-Mas axis in this central pathology (28). Indeed, the study supports earlier reports of the direct processing of Ang I to Ang-(1–7) by thimet oligopeptidase in vascular smooth muscle cells and a rat hindlimb perfusion system (29, 30).
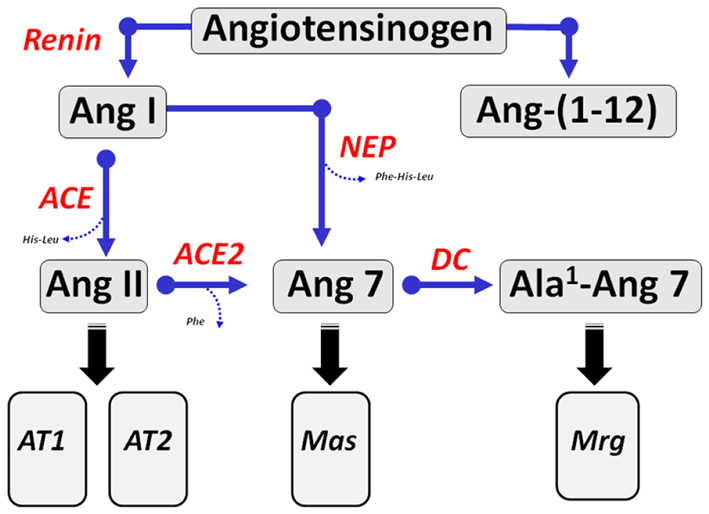
Figure 1. Enzymatic cascade of angiotensin peptide formation and metabolism. Renin cleaves the precursor protein angiotensinogen to angiotensin-(1–10) (Ang I) which is further processed to the biologically active peptides Ang-(1–8) (Ang II) by angiotensin converting enzyme (ACE) and Ang-(1–7) by endopeptidases such as neprilysin (NEP). Ang II undergoes further processing at the carboxyl terminus by the carboxypeptidase ACE2 to yield Ang-(1–7) (Ang 7). Ang-(1–7) undergoes decarboxylation (DC) of the aspartic acid residue to form Ala1-Ang-(1–7) (Ala1-Ang 7). The dodecapeptide Ang-(1–12) is derived from the hydrolysis of the Tyr12-Tyr13 bond of rat angiotensinogen by an unknown enzymatic pathway. Ang II recognizes both AT1 and AT2 receptors. Ang-(1–7) activates the Mas receptor and Ala1-Ang-(1–7) recognizes the Mas-D related receptor (Mrg).
ACE2
Apart from endopeptidases that process Ang I or Ang-(1–12) to Ang-(1–7), various mono-carboxypeptidases including prolyl carboxypeptidase (PCP), POP, and the ACE homolog ACE2 generate Ang-(1–7) directly from Ang II. It should be emphasized that PCP requires an acidic pH optimum for activity, but may contribute to lysosomal pathways for metabolism of internalized Ang II or to the processing of Ang II to Ang-(1–7) in urine (31). ACE2 continues to be of primary focus given its ability to effectively metabolize Ang II and generate Ang-(1–7) (32, 33). ACE2 exhibits an efficiency constant (Vmax/kM or kcat) for Ang II that is 10- to 100-fold higher than that of PC or POP. In this regard, the soluble form of ACE2 has been utilized as a therapeutic agent to reduce blood pressure and attenuate target organ damage in hypertensive and diabetic animal models (34–37). ACE2 mRNA expression was increased in the brain medulla following long-term AT1-receptor blockade (38). It is unclear whether the beneficial effects of ACE2 administration reflect the reduction in Ang II, the enhanced formation of Ang-(1–7) or the increased ratio of Ang-(1–7) to Ang II. Moreover, Turner and colleagues report that soluble ACE2 attenuated the integrin-dependent stimulation of focal adhesion kinase (FAK) and increased the expression of the Akt kinase suggesting the peptidase may have direct cellular effects apart from its peptidase activity (39).
In addition to the functional role of ACE2 that catalyzes the conversion of Ang II to Ang-(1–7), the peptidase may serve as a biomarker of renal and cardiac pathologies. Two studies in type I (streptozotocin-induced) and type II (db/db mice) diabetic models reported an early increase in the urinary excretion of ACE2 (40, 41). The enhanced excretion of ACE2 in db/db mice closely correlated to the increase in albuminuria or proteinuria. Moreover, chronic treatment with insulin-sensitizing agent rosiglitazone improved the metabolic balance in the db/db mice and reduced the excretion of both ACE2 and albumin (40). In contrast to the reduction in urinary levels of ACE2, the increased renal expression of ACE2 in the db/db mouse was not altered by rosiglitazone which may reflect an added therapeutic benefit to maintain the peptidase in the diabetic kidney (40). An important aspect of the two latter studies suggests that in the diabetic kidney, the development of tissue injury should not necessarily be interpreted as arising from a deficit in ACE2 expression. Indeed, the increase in tissue and urinary levels of ACE2 in pathological conditions may reflect a compensatory response to alter the balance of Ang II and Ang-(1–7) pathways within a particular tissue or cell type (7). In this regard, the deleterious effects of an ACE2 inhibitor or knockdown of the enzyme may be particularly evident under conditions of enhanced ACE2 expression. The circulating levels of ACE2, which are typically low to not detectable, are also increased in experimental conditions of diabetes. We show in a model of diabetic hypertension that circulating ACE2 activity increased over fivefold in female mRen2.Lewis rats (42). However, serum ACE activity also increased suggesting that the potential beneficial effects of higher ACE2 may be offset by ACE acting to increase Ang II and metabolize Ang-(1–7). Indeed, plasma levels of Ang-(1–7) were not changed in the diabetic mRen2.Lewis despite the marked increase in ACE2 activity. Moreover, circulating ACE activity was substantially higher than that of ACE2 when assessed under similar incubation and substrate conditions for each enzyme (42).
In the db/db mice, infusion of exogenous ACE2 that markedly increased serum levels of the enzyme did not alter urinary ACE2 suggesting that the enzyme is not readily filtered by the glomerulus (41). One mechanism for the increase in urinary excretion of ACE2 is the regulated shedding of the enzyme from the apical face of the proximal tubules (Figure 2). Studies by Lambert and colleagues originally reported that the disintegrin and metalloproteinase (ADAM17) secretase was responsible for the release of ACE2 (43). A subsequent report identified a specific sequence of the juxtamembrane stalk of ACE2 hydrolyzed that was by ADAM17 to release the peptidase from human pulmonary epithelial cells (44). In proximal epithelial cells of the db/db mouse kidney, there was extensive overlap of ACE2 and ADAM17 immunostaining (40). Moreover, rosiglitazone treatment attenuated ADAM17 expression which may contribute to the reduced shedding of ACE2 into the tubular fluid and subsequent excretion in the urine. In addition to the shedding of ACE2, ADAM17 may influence tissue damage by the release of the tethered inflammatory factors TNFα, EGF, and TGFα that subsequently activate their respective receptors in an autocrine or paracrine manner (45). If expression of ACE2 on the apical membrane of the tubules contributes to the regulation of the local concentrations of Ang II, an increase in ADAM17 may lead to inflammatory and fibrotic events through enhanced Ang II-AT1-receptor signaling, as well as increased cytokine and growth factor activation (Figure 2). Lazartigues and colleagues report that knockdown of ADAM17 in the brain of DOCA-salt mice reduced blood pressure, and increased the tissue expression of ACE2 (46). In this model of neurogenic hypertension, the benefit of ADAM17 knockdown may reflect a reduction of Ang II in brain; however, the direct effects on the release of EGF and other cytokines cannot be discounted. Indeed, the transactivation of the EGF receptor (EGFR) and signaling pathways is a key signaling event of the Ang II-AT1-receptor pathway (47). The increased shedding of ACE2 may also reduce levels of Ang-(1–7) and attenuate the inhibitory actions on the Ang II-AT1-receptor axis and other pro-inflammatory and pro-fibrotic pathways. Akhtar et al. recently reported that Ang-(1–7) attenuated EGFR activation in response to Ang II, as well as reduced the extent of renal injury in the diabetic SHR (48). Moreover, increasing evidence suggests that one of the primary pathways activated by Ang-(1–7) is the stimulation of various cellular phosphatases (PTP) including SHP-1 and DUSP-1 that may attenuate activated kinase-dependent pathways (49–53) (Figure 2).
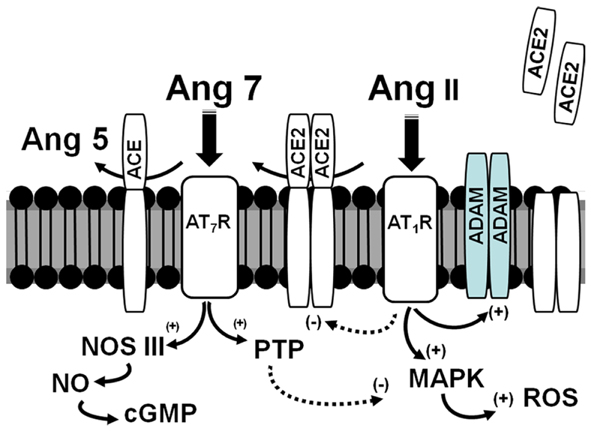
Figure 2. Scheme for the interaction of ACE2 and ADAMs on the apical surface of the proximal tubules in the diabetic kidney. Ang II binds to the AT1 receptor (AT1R) and stimulates MAP kinase (MAPK) pathways and production of reactive oxygen species (ROS). The Ang II-AT1-receptor axis may attenuate ACE2 expression but increase ADAM levels. ACE2 is anchored to the apical membrane and directly converts Ang II to Ang-(1–7) (Ang 7); ACE also anchored to the membrane metabolizes Ang-(1–7) to Ang-(1–5) (Ang 5). Ang-(1–7) recognizes the AT7/Mas receptor (AT7R) to antagonize the actions of the Ang II-AT1R by stimulation of protein phosphatases (PTP) and nitric oxide synthase (NOS) to form nitric oxide (NO) and cGMP. In pathological conditions, increased expression of ADAMs may hydrolyze ACE2 away from the apical surface to increase local concentrations of Ang II and reduce the levels of Ang-(1–7).
Angiotensin-(1–12)
Nagata and colleagues identified a novel endogenous angiotensin peptide termed Ang-(1–12) that contains the first 12 amino acids of the N-terminal sequence of rat angiotensinogen (Asp1-Arg2-Val3-Tyr4-Ile5-His6-Pro7-Phe8-His9-Leu10-Leu11-Tyr12) (54) (Figure 1). These investigators developed antibodies directed to the amino- and carboxyl-terminal sequences of Ang-(1–12) and demonstrated expression of Ang-(1–12) in essentially all tissues that contain Ang II with the highest levels in the intestine, brain, heart, plasma, and kidney of rat. Differential expression of Ang-(1–12) was evident in the heart and kidney of the SHR and the normotensive control Wistar Kyoto strain (WKY) (55). An antibody specific to the C-terminal sequence of rat Ang-(1–12) including Leu11-Tyr12 revealed selective staining in cardiac myocytes and proximal tubule cells of the kidney. The site of hydrolysis for formation of Ang-(1–12) from rat angiotensinogen occurs at residues Tyr12-Tyr13 which is distinct from the Leu10-Leu11 sequence cleaved by renin to form Ang I. Thus, the generation of Ang-(1–12) is likely through a non-renin dependent pathway and may be apparent in conditions of low or suppressed renin activity, particularly with the use of selective renin inhibitors. Similar to Ang I, Ang-(1–12) can be hydrolyzed at the Phe8-His9 bond by ACE or chymase to form Ang II (54, 56, 57). The conversion of Ang-(1–12) to Ang II by ACE in the circulation is consistent with the acute increase in blood pressure following an infusion of Ang-(1–12) in normotensive rats, as well as the blockade of the pressor response by either an ACE inhibitor or AT1-receptor antagonist. Arnold et al. also find that central Ang-(1–12) administration attenuated baroreflex sensitivity and the response was blocked by either an ACE inhibitor or AT1-receptor antagonist (57). Moreover, neutralization of Ang-(1–12) by intracerebroventricular (ICV) infusion of an affinity-purified antibody reduced blood pressure in the (mRen27)2 hypertensive rats consistent with the biochemical and immunocytochemical evidence for Ang-(1–12) in the rat brain (58). To our knowledge, the latter study by Isa and colleagues is the only report to date that demonstrates an endogenous role for Ang-(1–12).
As to the Ang-(1–7) axis, we recently demonstrated that Ang-(1–12) may be an alternative substrate for the generation Ang-(1–7) in the kidney (59). Isolated cortical membranes from the kidney of the hypertensive mRen2.Lewis rat processed Ang-(1–12) to Ang-(1–7) and Ang-(1–4). We observed a similar pattern of metabolism using the recombinant forms of mouse and human neprilysin. The selective neprilysin inhibitor SCH39370 abolished the formation of Ang-(1–7). We noted a peak corresponding to Ang I in the processing of Ang-(1–12) by the cortical membranes that was also abolished by the neprilysin inhibitor suggesting the peptide may be an intermediate in the processing of Ang-(1–12) to Ang-(1–7) (59). In these studies, we also show that circulating or renal renin did not metabolize Ang-(1–12) particularly in the presence of the ACE inhibitor lisinopril which implies that the peptide lacks the minimal sequence for recognition by renin (59). Bujak-Gizycka and colleagues demonstrated the generation of Ang-(1–12) in rat aorta homogenates by a serine peptidase using Ang-(1–14) as the substrate; however, the extent that this activity will process the angiotensinogen protein to Ang-(1–12) is not currently known (60). We did not detect the conversion of Ang-(1–12) to Ang-(1–7) in serum which would be consistent with the lack of soluble forms of neprilysin in the circulation, nor were there significant levels of Ang-(1–11) suggesting the absence of processing by ACE2 or other carboxypeptidases (59). It is feasible that Ang-(1–12) may be a potential substrate for Ang-(1–7) through the initial conversion to Ang-(1–11) by ACE2 and subsequent processing to Ang-(1–9) and Ang-(1–7) by ACE. However, ACE activity is far higher in the circulation than ACE2 and Ang-(1–7) formation from Ang-(1–12) or Ang I more likely reflects endopeptidase activity. Although further studies are required to discern the endogenous pathways for the formation and processing of Ang-(1–12), the peptide constitutes a potential substrate for the conversion to either the active products Ang II or Ang-(1–7).
Ala1-Angiotensin-(1–7) and Pro1-Glu2-Ang II
In addition to the precursors to Ang-(1–7), the peptide itself may serve as a precursor to other active forms. Santos and colleagues recently identified an endogenous analog of Ang-(1–7) in which the aspartic acid residue was decarboxylated to alanine (Ala) forming Ala1-Ang-(1–7) (Figure 1) (61). The Ala1-Ang-(1–7) analog (also termed almandine) may also potentially arise from the proteolytic processing of endogenous Ala1-Ang II (Ang A) by ACE2 (62). Similar to Ang-(1–7), Ala1-Ang-(1–7) induced the relaxation of isolated aortic vessels and chronic infusion of the analog lowered blood pressure. Interestingly, the vascular effects of Ala1-Ang-(1–7) were not blocked by the typical receptor antagonist D-Ala7-Ang-(1–7) (A779) against the Mas receptor, but were attenuated by D-Pro7-Ang-(1–7) and the AT2 receptor antagonist PD123319. This study further showed that Ala7-Ang-(1–7) stimulated the Mas-related receptor (MrgD) and did not interact with the Mas receptor. Identification of Ala1-Ang-(1–7) in the human circulation and in an isolated heart perfusion system was achieved by a HPLC-Mass spectrometry approach. It is worth noting that the available direct RIA or ELISA assays will not distinguish between the Asp1- and Ala1- forms of Ang-(1–7) since both isoforms share the identical C-terminal sequence that is typically recognized by the immunoreactive antibodies. Thus, an initial separation step such as HPLC combined with conventional immunoreactive assays will be required to routinely detect and quantify the different forms of Ang-(1–7) in the circulation and tissues. The potential importance of these findings may reflect the greater diversity of the Ang-(1–7) axis regarding the identification of both a novel ligand and receptor that contributes to vascular tone. Moreover, that the AT2 antagonist PD12319 antagonized the actions of Ala1-Ang-(1–7) at the MrgD receptor may explain the apparent interaction of Ang-(1–7) with the AT2 receptor noted in several studies (63–65).
Although distinct from either Ang-(1–7) or its Ala analog, Jankowski et al. identified another endogenous ligand to the AT7/Mas receptor in human serum termed angioprotectin (66). This peptide resembles the octapeptide Ang II but has substitutions of Pro and Glu at the first two N-terminal residues to form Pro1-Glu2-Ang II. Despite the fact that the angioprotectin contains both the Tyr4 and Phe8 residues considered to be essential to the actions of Ang II, the peptide lacked any vasoconstrictor activity in isolated aortic rings. However, the peptide induced a dose-dependent vasorelaxation of isolated vessels that was absent in vessels from the Mas-knockout mice, as well as acutely reduced blood pressure in the SHR. Moreover, Pro1-Glu2-Ang II stimulated NO formation in Mas-transfected CHO cells but not in the control cells. Finally, the study presented evidence for local formation of Pro1-Glu2-Ang II from Ang II in human endothelial cells that was enhanced by addition of exogenous proline and glutamic acid suggesting a post-transcriptional modification of Ang II. It is not known to what degree Pro1-Glu2-Ang II is processed by ACE2 or other carboxypeptidases to the Ang-(1–7) analog and whether Pro1-Glu2-Ang-(1–7) is functionally active at the either the Mas or MrgD receptors. It is also unclear the extent conventional immunoreactive assays for Ang II will detect endogenous Pro1-Glu2-Ang II in plasma or tissues given their identical C-terminal sequence. The circulating levels of Pro1-Glu2-Ang II were 15% of Ang II in humans, but the Ang II analog increased fivefold in patients with end-stage renal disease that may perhaps reflect a compensatory response in pathological conditions (66).
Ang-(1–7) Metabolism
The endogenous levels of Ang-(1–7) are influenced by access to processing enzymes such as the carboxypeptidase ACE2 or the endopeptidases neprilysin, thimet oligopeptidase, and prolyl endopeptidase (oligopeptidase). The levels of Ang-(1–7) are also dependent on peptidases that metabolize the peptide. Similar to bradykinin and substance P, ACE plays a significant role in the hydrolysis of Ang-(1–7) to the pentapeptide Ang-(1–5) in the circulation and the proximal tubules of the kidney cortex (Figure 2) (22, 67). ACE inhibition increased the half-life of Ang-(1–7) sixfold in the circulation and is necessary to demonstrate the accumulation of Ang-(1–7) from both Ang I- and Ang II-dependent pathways in the renal proximal tubules (67, 68). Thus, the mechanism for the increased levels of Ang-(1–7) following ACE inhibitor treatment reflects both protection of the peptide from ACE hydrolysis to Ang-(1–5) and shunting of Ang I to Ang-(1–7) through endopeptidase pathways such as neprilysin or thimet oligopeptidase (23). There is relatively little information on other peptidases that participate in the metabolism of Ang-(1–7) other than ACE. We recently detected an endopeptidase activity in the cerebrospinal fluid (CSF) of sheep that metabolized Ang-(1–7) at the Tyr4-Ile5 bond to yield Ang-(1–4) and constituted the majority of Ang-(1–7) degrading activity in CSF (69, 70). Although the identity of the peptidase is currently unknown, the activity was insensitive to inhibitors against neprilysin, thimet oligopeptidase, or neurolysin (EC3.4.24.26) (70). The Ang-(1–7) peptidase activity was abolished by the mercury-compounds p-chloromercuribenzoate (PCMB) and aminophenyl-mercuriacetate (APMA), as well as the chelating agents o-phenanthroline and EDTA, but not the cysteine epoxide inhibitor E-64 suggesting a metallopeptidase-like activity in CSF (70). The regulation of the CSF peptidase is described in the proceeding section on fetal programing.
Intracellular Ang-(1–7)-Mas Receptor System
The RAS was traditionally viewed as an endocrine system whereby circulating renin catalyzes an enzymatic cascade to form active peptide products; however, it is apparent that multiple tissues contain the necessary components for the local generation of angiotensin peptides (71, 72). These tissue systems may release the precursor angiotensinogen, the intermediate products Ang I and Ang-(1–12), or the active peptides Ang II and Ang-(1–7) to bind directly to cell surface receptors in an autocrine or paracrine manner. Robertson and Khairallah reported over 40 years ago the localization of Ang II binding sites on the chromatin fraction of vascular smooth muscle cells and cardiomyocytes suggesting an intracellular site of action for Ang II (73). Several laboratories subsequently identified Ang II receptors using classical receptor binding techniques on nuclei isolated from liver (74–76). Eggena and colleagues demonstrated that Ang II stimulated mRNA transcripts for angiotensinogen, renin, and PDGF from isolated liver nuclei suggesting that the nuclear binding sites were functional and capable of directly mediating gene expression (77, 78). Moreover, AT1 receptors were also evident on nuclei isolated from cortical and medullary areas of the rat kidney (79–81). Ang II-AT1-receptor stimulation on isolated renal nuclei increased mRNA expression of angiotensinogen, the sodium-hydrogen exchanger (NHE3) and the cytokine monocyte chemoattractant protein (MCP-1) (79). Ang II also elicited an immediate increase in calcium by isolated cortical nuclei or via microinjection of the peptide in intact epithelial cells (82). We find that Ang II directly stimulates reactive oxygen species (ROS) as demonstrated by the enhanced fluorescent signature of dichlorofluorescein (DCF); ROS formation was sensitive to the NAD(P)H oxidase inhibitor diphenyleneiodonium (DPI) and the AT1 antagonist losartan (83). Blockade of phosphoinositol 3-kinase (PI3K) and protein kinase C (PKC) abolished the Ang II-AT1-receptor-dependent stimulation of ROS in renal nuclei. In lieu of the nuclear localization of the NAD(P)H oxidase isoform NOX4, activation of AT1 receptors may acutely stimulate ROS by a PI3K-PKC pathway and subsequent phosphorylation of NOX4 (Figure 4) (83–87).
The studies demonstrating nuclear AT1 receptors within the kidney and other tissues clearly support an emerging view for the localization of various G-protein coupled receptors (GPRCs) to the nucleus (88–94). In regards to the Ang-(1–7)-Mas receptor system, O’Dowd and colleagues noted a canonical nuclear localization sequence on the Mas protein in their studies on AT1-receptor trafficking and localization in vascular smooth muscle cells (95). We undertook a series of studies to establish an intracellular role for Ang-(1–7) in the cortical tissue and proximal tubules isolated from the sheep kidney. Immunoblot analysis of nuclei isolated from sheep proximal tubules demonstrated a single immunoreactive band of 35 kDa utilizing an affinity-purified antibody against the human Mas protein (96). Receptor binding studies with the non-selective antagonist 125I-(Sarcosine1, Threonine8)-Ang II (Sarthran) revealed significant competition by the AT7/Mas receptor antagonist D-Ala7-Ang-(1–7) in nuclei isolated from the renal cortex. Functional assessment of the nuclear AT7 receptor was then assessed with the sensitive NO fluorophore DAF in the presence or absence of the NO synthase inhibitor L-NAME. Ang-(1–7) dose-dependently increased the fluorescent signature for NO which was abolished by prior treatment with L-NAME or the Ang-(1–7) antagonist, but not antagonists to the AT1 or AT2 receptors. Consistent with the stimulation of NO by Ang-(1–7), protein expression for endothelial nitric oxide synthase (eNOS) and soluble guanylate cyclase (sGC) was evident in the isolated nuclei of sheep proximal tubules (96). These data further support previous studies that localized eNOS and sGC to liver nuclei, as well as the stimulation of NO and cGMP by activation of the bradykinin B2 receptor (90, 97). The exact function of the Ang-(1–7) axis of the RAS within the nucleus is not known; however, we hypothesize this system may antagonize the intracellular actions of the Ang II-AT1-receptor pathway. To address this possibility, we assessed the influence of the selective ACE2 inhibitor MLN4760 and the Mas receptor antagonist on the activation of ROS by Ang II in renal cortical nuclei. The Ang II-AT1-receptor dependent increase in ROS was significantly augmented to a similar extent by treatment of nuclei with either the ACE2 inhibitor or the AT7 receptor antagonist (98). That both MLN4760 and D-Ala7-Ang-(1–7) increased the stimulation of ROS suggests that the conversion to Ang-(1–7) by ACE2 antagonizes the actions of the Ang II-AT1-receptor axis on the nucleus. It is possible that that simply blocking the degradation of Ang II with the ACE2 inhibitor may augment the actions of Ang II; however, the comparable effects of the AT7 receptor antagonist D-Ala7-Ang-(1–7) suggests a distinct role for Ang-(1–7). Since the Ang-(1–7) antagonist is a peptide and may potentially interact with ACE2, we further demonstrated that D-Ala7-Ang-(1–7) does not inhibit nuclear ACE2 activity as assessed by the HPLC-based conversion of Ang II to Ang-(1–7). Moreover, our studies suggest that the processing of Ang II to Ang-(1–7) by ACE2 on the nuclear membrane leads to the activation of signaling pathways distinct from that of Ang II (98). We do not know, however, whether the attenuation of ROS production by Ang-(1–7) involves the stimulation of NO or other signaling pathways. As previously discussed, Ang-(1–7) may attenuate the actions of Ang II and other growth hormones by the activation of intracellular phosphatases such as the dual specificity phosphatases MKP-1 and SHP-1 (50, 51). Several classes of phosphatases including MKP-1 traffic to the nucleus; however, it is unknown whether Ang-(1–7) can influence these enzymes to attenuate the actions of Ang II (99).
Clearly, one issue regarding the intracellular RAS and other peptidergic systems is the localization of the components within the cell. The nucleus is composed of two distinct bilayers termed the outer (OMN) and inner (INM) nuclear membranes. Nuclear pore proteins traverse both membrane domains and facilitate transport between the cytosol and the nuclear matrix which contains the chromatin-DNA complex. Portions of the ONM are continuous with the endoplasmic reticulum (ER) such that perinuclear space is shared with the ER. The nuclear envelope comprising both OMN and INM invaginates into the nuclear matrix creating a nuclear reticulum that is key in the regulated release of nuclear Ca2+ (100–102). Although various studies have localized GPRCs primarily to the nuclear envelope and matrix, it is currently unclear how the peptide ligands target the nuclear GPRCs, as well as the precise coupling of the receptors to their signaling pathways within the nucleus. Moreover, elucidation of the pathways that deliver peptide ligands to their respective intracellular receptors, as well as the intracellular regulation under normal and pathological conditions has not been established. As to the intracellular expression of angiotensins in the kidney, there is evidence for expression and uptake of angiotensinogen, as well as the uptake of Ang II and Ang-(1–7) by protein transporters such as megalin (71, 103–106). In addition, AT1-receptor mediated internalization of Ang II may contribute to the intracellular content of the peptide (72, 89). In this regard, intracellular peptidases such as ACE2 may potentially process the internalized Ang II to Ang-(1–7) as alternative pathway to attenuate AT1-receptor activity and stimulate the cellular actions of Ang-(1–7). Utilizing the renal epithelial NRK-52E cell line, we find evidence for the nuclear localization of angiotensinogen (Figure 3, left panels) consistent with earlier findings by Sherrod and colleagues regarding nuclear angiotensinogen in brain astrocytes and isolated nuclei of sheep proximal tubules (96, 107, 108). Interestingly, a second antibody directed to the Ang I sequence of angiotensinogen failed to detect the protein in the nucleus of the NRK-52E cells suggesting that enzymatic processing of the precursor may occur in this compartment (107). In support of an intracellular processing pathway, renin expression was also evident in the nucleus of the NRK cells (Figure 3, right panels). Isolated nuclei exhibited both renin and prorenin activity (following activation by trypsin) that was sensitive to the specific renin inhibitor aliskiren (Figure 3, bottom left panel), as well as immunoreactive levels of Ang II and Ang-(1–7) (107). In addition, peptide metabolism studies in isolated nuclei revealed the direct conversion of Ang I to Ang-(1–7) that was essentially abolished by a selective inhibitor (CPP) of the metalloendopeptidase thimet oligopeptidase (Figure 3, bottom right panel). Others have reported the nuclear expression of thimet oligopeptidase in brain, as well as the identification of a nuclear localization sequence for the human peptidase (109, 110). The NRK-52E cells may constitute a relevant cell model to establish the pathways that contribute to the intracellular generation and actions of Ang II and Ang-(1–7) within renal epithelial cells. As an alternative concept to intracellular formation, Ibarra and colleagues presented evidence for another model of nuclear signaling whereby the plasma membrane invaginates to the perinuclear area that facilitates presentation of intracellular signals (IGF receptor coupled to IP3 formation) discretely to the nucleus in cardiomyocytes (111). The apparent advantages of this system may reflect a more selective activation of the signaling cascade and independence from the intracellular generation of the peptide ligands (112). The latter study adds another potential mechanism to the complex pathways of the intracellular receptor system for angiotensins and other peptides, as well as emphasize the need for additional studies to elucidate their organization and function.
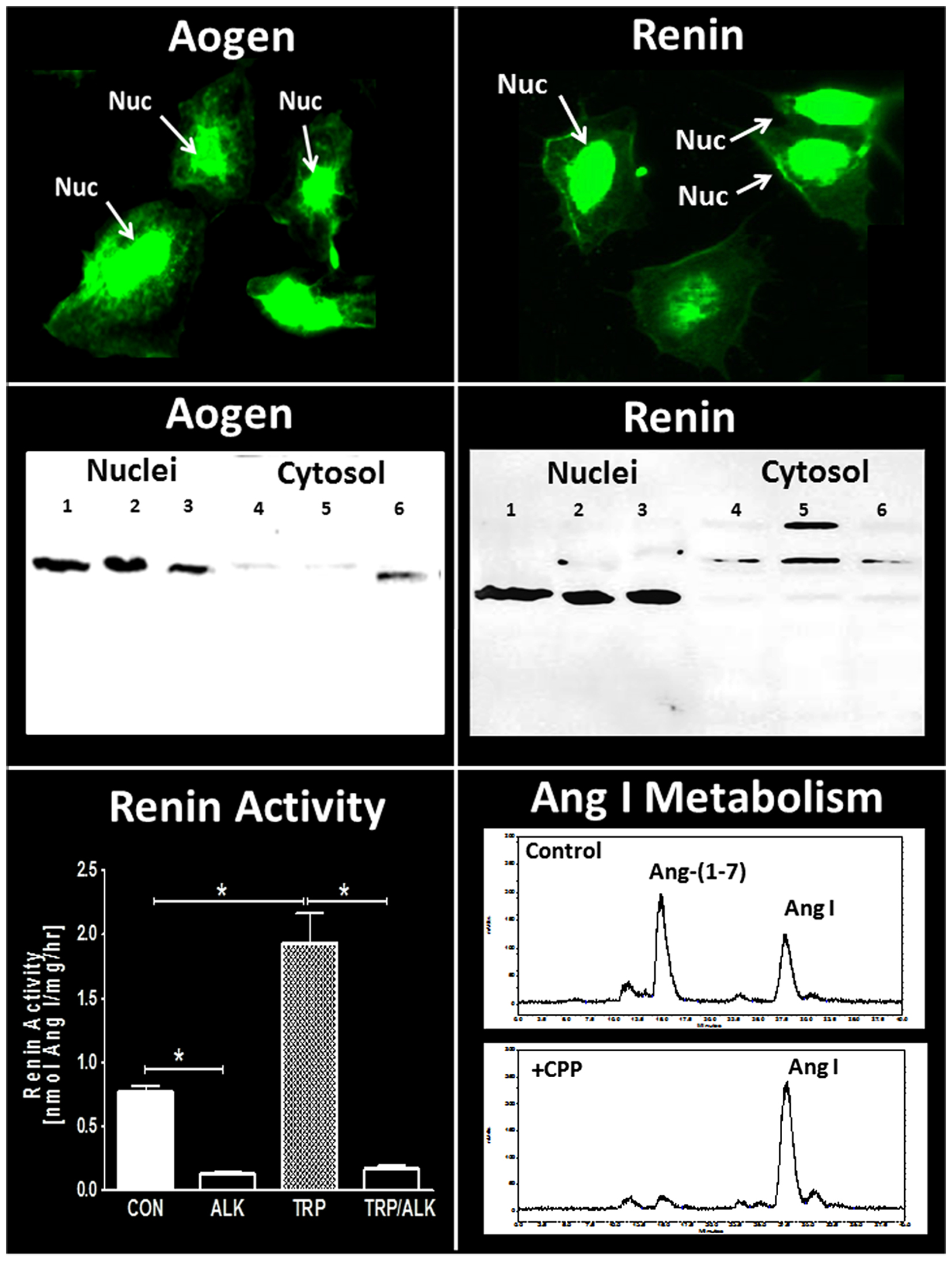
Figure 3. Expression of intracellular components of the renin-angiotensin system in NRK-52E renal epithelial cells. Immunofluorescent (IMF) staining and protein immunoblot for rat angiotensinogen (Aogen) and renin. Immunoblots of Aogen and renin in nuclei (lanes 1–3) and cytosol (lanes 4–6) were from three separate passages of NRK-52E cells. Major bands for Aogen and renin were identified at approximately 55 kDa. Renin activity (conversion of Aogen to Ang I) in isolated nuclear fractions was increased threefold following activation by trypsin (TRP) and was essentially abolished by the renin inhibitor aliskiren (ALK). Conversion of 125I-Ang I to 125I-Ang-(1–7) in the isolated nuclear fraction was predominantly blocked by the thimet oligopeptidase inhibitor CPP. Renin activity data are mean ± SEM; n = 4; *P < 0.05. Ang I metabolism representative of data from n = 4 separate cell passages. Adapted from Alzayadneh and Chappell (107).
In the endeavor to elucidate the intracellular pathways, the importance of robust biochemical and molecular techniques to characterize the RAS cannot be overly emphasized. Several reports have raised concerns regarding the specificity of commercial AT1 and AT2 antibodies widely utilized for western immunoblot and immunocytochemical distribution studies (113–115). Importantly, these studies find that receptor protein bands at the appropriate molecular weights were not abolished in AT1- or AT2-deleted cell and tissue samples. We have utilized antibodies to both AT1 and AT2 receptors to establish their molecular weight in the nuclear fraction as this pertains to the maturation or processing of the receptor protein. However, studies by our laboratory and others also incorporate peptide binding assays to quantitate receptor density and affinity, as well as various antagonists to identify the receptor subtype. The receptor binding assays also parallel the demonstration of functional signaling pathways (ROS, NO) on nuclei and the sensitivity to receptor antagonists. Reliance on the assessment of mRNA for the receptor may not equate to protein expression and certainly does not reveal the discrete intracellular distribution of the receptor. Antibodies to angiotensin receptors or other RAS components are useful and convenient tools to characterize this system; however, parallel approaches to establish the expression and regulation of the RAS particularly within the cell are clearly warranted.
Fetal Programing
Increasing evidence for the influence of early prenatal events in the fetus to induce a greater susceptibility to cardiovascular and metabolic pathologies is evident in both experimental models and in humans (116–119). Although the precise nature of fetal programing events is not known, alterations in the biochemical components and functional aspects of the RAS may constitute an important underlying mechanism (69, 120–128). Our recent studies utilize a sheep model of fetal programing in which pregnant ewes are administrated the glucocorticoid betamethasone at day 80 of gestation. This regimen parallels the dose and time that pregnant women are typically treated with glucocorticoids to enhance pulmonary function and reduce mortality of the fetus delivered preterm. Fetal exposure to glucocorticoids in sheep results in a significant reduction in the nephron number within the kidney, an increase in mean blood pressure, attenuation of the baroreflex response (BRS) in the control of heart rate and increased indices of metabolic dysfunction in adult animals (129) (Figure 4). In regards to the function of the RAS following glucocorticoid exposure, acute treatment with the AT1-receptor antagonist candesartan normalized blood pressure in the exposed sheep and improved the impaired BRS, but had no overall effect on pressure in the control or unexposed adult sheep (129). In contrast, administration of D-Ala7-Ang-(1–7) increased blood pressure and attenuated BRS in the control but not the betamethasone-exposed (BMS) sheep suggesting that the loss of Ang-(1–7) tone may be an additional consequence of fetal programing events (126). The protein expression of the AT7/Mas receptor was significantly lower in the brain medulla in both 6-month- and 1.8-year-old BMS sheep as compared to age-matched control sheep; however, the AT1-receptor protein expression was unchanged (122). We also find reduced CSF levels of Ang-(1–7) in the exposed sheep, as well as higher activities of the Ang-(1–7) peptidase (Figure 4) (69, 70). Indeed, the CSF content of Ang-(1–7) inversely correlated to Ang-(1–7) peptidase activity in the control and BMS sheep (Figure 4, lower right panel). Thus, the reduced expression of the AT7/Mas receptor and increased metabolism of Ang-(1–7) in brain may contribute to the loss of Ang-(1–7) tone in BMS sheep, as well as the enhanced responsiveness of the Ang II-AT1-receptor pathway in glucocorticoid-dependent programing without significant changes in the AT1-receptor levels.
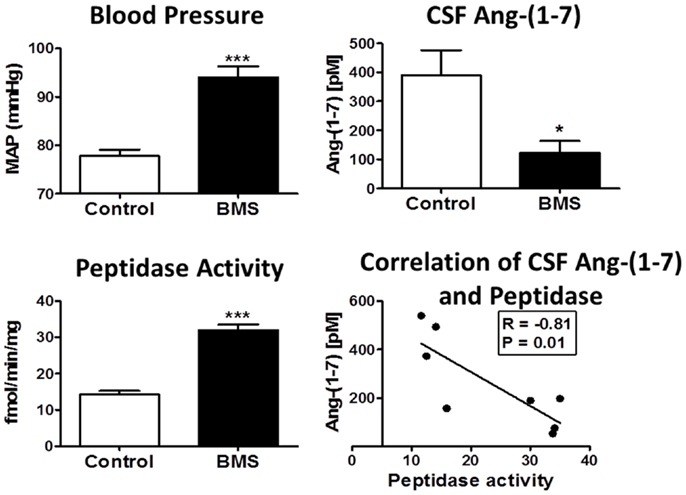
Figure 4. Betamethasone-exposed (BMS) offspring exhibit higher mean arterial pressure (MAP) and CSF endopeptidase activity than non-exposed sheep. Blood pressure (MAP) was higher in BMS animals at 6 months of age. CSF peptidase activity was twofold higher in BMS animals as compared to controls. CSF Ang-(1–7) peptide levels were lower in BMS animals. Ang-(1–7) peptide levels negatively correlate with peptidase activity in the CSF (r = −0.81, P = 0.01). Data are mean ± SEM; 4–5 per group; *P < 0.05 or ***P < 0.001 vs. controls. Adapted from Marshall et al. (70).
In regards to the renal RAS in fetal programing, Ang-(1–7)-dependent stimulation of sodium excretion was abrogated in the BMS sheep. Moreover, the anti-natriuretic response to Ang II was enhanced in the BMS sheep, as well as the reduction in renal plasma flow (130, 131). Consistent with the altered renal responses to Ang II and Ang-(1–7), expression of ACE2, the peptidase that contributes to the balance of Ang II to Ang-(1–7), was significantly reduced in the circulation, the proximal tubules and the urine of the BMS adult sheep (125). That both tubular and urinary forms of ACE2 were reduced in the BMS sheep suggests down regulation or reduced synthesis of the peptidase in the proximal tubules that may lead to the lower release or shedding of the enzyme from the apical membrane. Both ACE and neprilysin activities were readily detected in the proximal tubules and urine of adult sheep; however, their activities were not changed following betamethasone exposure. Moreover, circulating ACE activity increased while ACE2 activity decreased in the serum of BMS adult sheep (125). A kinetic analysis of ACE2 activity revealed a reduction in the maximal velocity (Vmax) of the enzyme rather than a change in substrate affinity (Km) suggesting reduced protein content in the circulation. These data further suggest that the soluble forms of the enzyme in serum exhibits similar kinetic characteristics as the native form, at least regarding the metabolism of Ang II to Ang-(1–7). The ratio of ACE to ACE2 also closely correlated with the mean blood pressure values in the control and BMS sheep (125).
In addition to the altered expression of ACE2, the balance of angiotensin receptor subtypes was changed as well (132). Moreover, the proportion of both AT2 and Mas receptor subtypes were lower in the renal cortex of the exposed group. However, the AT1 subtype was the predominant angiotensin receptor in the renal medulla and the receptor subtypes were unchanged between the control and exposed sheep. These data again emphasize the selective effects of fetal glucocorticoid exposure on expression of the RAS components within different tissue compartments. The intracellular studies on the Ang II and Ang-(1–7) signaling pathways further support the overall findings that the expression of angiotensin receptor subtypes were altered following glucocorticoid exposure. In isolated nuclei from the real cortex, the generation of NO by Ang-(1–7) was markedly reduced in the exposed sheep. In contrast, the Ang II-dependent stimulation of ROS via the AT1 receptor was augmented in renal nuclei from the BMS sheep. Furthermore, addition of the AT2 antagonist PD122319 exacerbated the production of ROS by Ang II, and this augmented response was particularly evident in the glucocorticoid-exposed group (132). It is not clear whether the AT2 receptor exhibits functional antagonism of the AT1 receptor to influence ROS formation or that the AT2 subtype sequesters Ang II from the AT1 receptor given the higher ratio of AT2 to AT1 sites in the sheep cortex.
Finally, fetal programing events may convey greater sensitivity to an additional stressor or insult, particularly as these animals age (119). Therefore, recent studies ascertained the renal responses to Ang-(1–7) in control and glucocorticoid-exposed adult sheep following removal of one kidney. In contrast to the intact, non-exposed sheep, Ang-(1–7) infusion reduced sodium excretion in uni-nephrectomized animals (133). The anti-natriuretic response to Ang-(1–7) was enhanced by the AT7 receptor antagonist D-Ala7-Ang-(1–7) and was subsequently blocked by the AT1 antagonist candesartan. Moreover, the exposed animals still exhibited an attenuated natriuretic response to the combination of Ang-(1–7) and candesartan in comparison to the unexposed control group. Similar hemodynamic responses were also observed for Ang-(1–7) in the uni-nephrectomized animals whereby the peptide alone reduced blood flow and the combination of Ang-(1–7)/candesartan increased flow; however, the overall vascular responses were similar between the control and exposed animals (133). Certainly, differences in species and sex, the primary or immortalized status of the cells, the dose of both peptide and antagonists, and the duration of treatment may influence the functional actions of Ang-(1–7) and the receptor(s) mediating these effects (134). In regards to the studies in the uni-nephrectomized sheep, the differential response in sodium handling to Ang-(1–7) likely reflects the compensatory mechanisms of the remaining kidney to accommodate the marked increase in cardiac output and fluid handling. The fascinating aspect of the renal studies is the plasticity in the Ang-(1–7) response that apparently encompasses an AT1 receptor interaction to reduce sodium excretion in the uni-nephrectomized animal, as well as the Mas receptor interaction to stimulate sodium excretion in the intact sheep. Moreover, the natriuretic response of Ang-(1–7) potentially mediated by the Mas receptor remains intact in the uni-nephrectomized sheep since it was unmasked by blockade with the AT1-receptor antagonist. However, the mechanism underlying the functional interaction of Ang-(1–7) with AT1 receptors in the single kidney is not known, as well as whether Ang-(1–7) stimulates signaling pathways identical to that of the Ang II-AT1-receptor axis to reduce sodium reabsorption and renal blood flow.
Sex Differences in the Ang-(1–7)-Mas Receptor Axis
Both experimental and clinical evidence suggest an important influence of sex on the development of cardiovascular disease that may reflect the regulation of the RAS by gonadal hormones including testosterone and estrogen. Women are generally thought to be protected from cardiovascular pathologies up to the time of menopause suggesting a beneficial effect of estradiol; however, several large clinical trials utilizing estrogen or combined estrogen/progesterone replacement in older women with underlying cardiovascular disease revealed adverse effects of either treatment. Experimental studies have largely focused on the role of estrogen to influence the ACE-Ang II-AT1-receptor axis of the RAS and generally reveal an inhibitory effect on the expression of ACE and the AT1 receptor (135–138). Estrogen depletion by ovariectomy in young mRen2.Lewis rats markedly exacerbated the hypertension and essentially abolished sex differences in blood pressure between the male and female congenics (139). In this model, estradiol replacement or treatment with the AT1-receptor antagonist olmesartan normalized blood pressure suggesting the loss of estrogen may lead to the dysregulation of the RAS. Indeed, circulating levels of Ang II and ACE activity were higher in the estrogen-depleted mRen2.Lewis while plasma levels of Ang-(1–7) were reduced, and the overall ratio of Ang II to Ang-(1–7) increased (139, 140). Brosnihan and colleagues originally proposed that the protective effects of estrogen may, in part, reflect a shift in the balance between circulating Ang II and Ang-(1–7) that may arise from the inhibitory effects of the steroid on ACE to promote Ang-(1–7) expression via increased synthesis and/or reduced metabolism of the peptide (141).
There are relatively few studies that have assessed tissue differences in Ang II and Ang-(1–7) in males and females. In the kidney of the mRen2.Lewis hypertensive rats, the tissue content of Ang II was twofold higher in the males (Figure 5). Conversely, renal levels of Ang-(1–7) were threefold lower in the males as compared to females. Interestingly, cortical ACE2 activity was 70% higher in the males perhaps suggesting a compensatory effect to buffer the higher Ang II content and blood pressure evident in the male mRen2.Lewis (Figure 5). In contrast, cortical neprilysin activity and protein expression were significantly higher in the female congenics as compared to males. The higher content of the endopeptidase may contribute to the differential expression of angiotensins in the female kidney to favor the enhanced conversion of Ang I to Ang-(1–7), as well as the metabolism of Ang II to Ang-(1–4). Cardiac ACE2 activity was also significantly higher in the male congenics; however, tissue levels of Ang II and Ang-(1–7) were not different between males and females. The renal content of Ang-(1–7) was also higher in female SHRs as compared to the males; however, tissue levels of Ang II were not different (142). Although this study did not determine peptidase expression in the SHR kidney as a potential mechanism for the higher Ang-(1–7) content, tissue levels of Ang-(1–7) were higher in females following Ang II treatment perhaps suggesting greater processing of Ang II to Ang-(1–7). Sandberg and colleagues assessed sex differences in ACE2 expression in the mouse kidney using the “four core” approach to distinguish the effect of sex chromosomes and ovarian steroids (143). Consistent with the results in the mRen2.Lewis rats, renal ACE2 activity and expression were higher in the male mice (143). Ovariectomy increased ACE2 expression in the female kidney, and estradiol replacement reduced the peptidase in both males and females; however, there was no influence of gonadal steroids on either cardiac or pulmonary ACE2 (143). These data suggest that at least under non-pathological conditions, estrogen exhibits an inhibitory influence on kidney ACE2 and raises the issue of whether this response contributes to the deleterious effects of estrogen replacement in older women. Cassis and colleagues reported the sex-dependent expression of circulating Ang-(1–7) in mice fed a high fat (HF) diet (144). Males exhibited a marked decline in plasma levels of Ang-(1–7) that was associated with higher circulating Ang II; however, plasma levels of Ang-(1–7) increased in the HF-fed females. Interestingly, ovariectomy of the HF-fed female mice reduced circulating Ang-(1–7) and ACE2 activity in adipose tissue but did not influence renal ACE2. The fall in circulating Ang-(1–7) was associated with a marked increase in nocturnal blood pressure. Moreover, administration of D-Ala7-Ang-(1–7) increased blood pressure in female mice maintained on the HF diet suggesting that the ACE2-Ang-(1–7)-Mas axis may buffer obesity-induced hypertension to a greater extent in females.
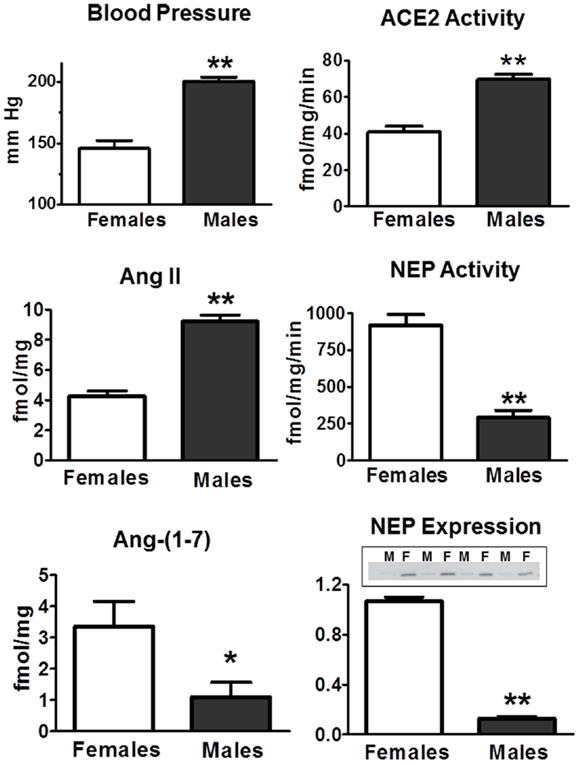
Figure 5. Sex differences in systolic blood pressure and RAS components in the renal cortex of 15-week-old hemizygous mRen2.Lewis congenic rats. Systolic blood pressure (mmHg) is higher in males. Intrarenal concentrations (femtomole peptide per milligram protein – fmol/mg) of Ang II are higher in males, but Ang-(1–7) content is lower. ACE2 activity (femtomole product per milligram protein per minute – fmol/mg/min) is higher in males, but neprilysin (NEP) activity is lower in males. NEP expression assessed by Western blot was lower in males (M) as compared to females (F). Data are mean ± SEM; n = 4–8 per group; *P < 0.05 or **P < 0.01. Adapted from Pendergrass et al. (81).
In regards to the Ang-(1–7) receptor, expression of the Mas receptor was increased in females but not in males following the infusion of Ang II which may explain the attenuated blood pressure to Ang II in the females (142). Pretreatment with the AT7 receptor antagonist D-Ala7-Ang-(1–7) enhanced the blood pressure response to Ang II in the female SHR. Similar findings were recently reported in the aldosterone salt-sensitive model where the mRNA levels for both ACE2 and the Mas receptor increased 1.5- and 5-fold, respectively, in the lamina terminalis (LT) of female rats (145). Chronic ICV treatment with the D-Ala7-Ang-(1–7) antagonist markedly augmented the blood pressure response in intact females treated with aldosterone and sodium chloride. Moreover, ovariectomy exacerbated the blood pressure response to aldosterone/salt; however, the mRNA expression of ACE2 or the Mas receptor in the LT area was not changed suggesting an inability to upregulate the central Ang-(1–7) axis contributes to the increase in blood pressure in this model (145). Denton and colleagues reported that D-Ala7-Ang-(1–7) alone decreased renal blood flow in female but not male normotensive Wistar rats, although the antagonist did not influence the renal response to acute Ang II infusion in either sex (64). In a separate study, the mRNA levels of the Mas protein were markedly higher in the kidney of adult female rats as compared to the males (146). Interestingly, Mas expression tended to decline in the male kidney while the mRNA levels of the receptor increased in females over the postnatal period (1–110 days). Our studies in the sheep model of fetal programing also provide evidence for sex differences in the responsiveness to Ang-(1–7). Although the exposure to betamethasone results in a similar increase in blood pressure and reduction in nephron number in the male and female sheep, the renal response to Ang-(1–7) or the antagonist differs with sex. An acute infusion of Ang-(1–7) results in a robust natriuretic response in control females but not the adult males (130). Moreover, betamethasone exposure was associated with reduced natriuresis in the males with or without Ang-(1–7) treatment, but significantly blunted the natriuretic actions of Ang-(1–7) in females. At this time, the mechanism underlying the sex-dependent effects of Ang-(1–7) on sodium excretion in control and betamethasone-exposed sheep are not known, but we speculate that it may involve the altered expression or signaling of the Mas receptor on the tubular elements of the kidney.
Summary
The current review has examined several aspects from the recent literature on the non-classical or alternative ACE2-Ang-(1–7)-Mas receptor axis of the RAS. The mounting biochemical and functional evidence clearly supports the tenet that this pathway may antagonize the ACE-Ang II-AT1-receptor arm of the RAS either directly through metabolism of Ang II to Ang-(1–7) by ACE2 or via distinct pathways that limit the activation of Ang II-AT1-receptor signaling. Indeed, the demonstration of an intracellular ACE2-Ang-(1–7)-Mas axis that attenuates the Ang II-dependent stimulation of ROS on renal nuclei is in keeping with the concept of a balanced RAS even within the cell and emphasizes the importance of targeting the intracellular system as a therapeutic approach to enhance the functional ratio of Ang-(1–7) to Ang II. The evidence that an altered Ang-(1–7) system within the brain and the kidney following antenatal glucocorticoid exposure implicates an interaction between the Ang II and Ang-(1–7) pathways that contribute or promote the cardiovascular dysfunction associated with fetal programing events. Finally, sex differences apparent in blood pressure regulation and cardiovascular pathologies may reflect alterations in the ACE2-Ang-(1–7)-Mas receptor axis of the RAS in addition to those effects typically associated with the ACE-Ang II-AT1-receptor pathway.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Chappell MC, Brosnihan KB, Diz DI, Ferrario CM. Identification of angiotensin-(1-7) in rat brain: evidence for differential processing of angiotensin peptides. J Biol Chem (1989) 264:16518–23.
2. de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger TH. The angiotensin II receptors. Pharmacol Rev (2000) 52:415–72.
3. Ichiki T, Inagami T. Expression, genomic organization, and transcription of the mouse angiotensin II type 2 receptor gene. Circ Res (1995) 76:693–700. doi: 10.1161/01.RES.76.5.693
4. Kambayashi Y, Bardhan S, Takahashi K, Tsuzuki S, Inui H, Hamakubo T, et al. Molecular cloning of a novel angiotensin II receptor isoform involved in phosphotyrosine phosphatase inhibition. J Biol Chem (1993) 268:24543–6.
5. Bader M, Ganten D. Update on tissue renin-angiotensin systems. J Mol Med (Berl) (2008) 86:615–21. doi:10.1007/s00109-008-0336-0
6. Chappell MC. Emerging evidence for a functional angiotensin-converting enzyme 2-angiotensin-(1-7) mas receptor axis; more than regulation of blood pressure? Hypertension (2007) 50:596–9. doi:10.1161/HYPERTENSIONAHA.106.076216
7. Chappell MC. Nonclassical renin-angiotensin system and renal function. Compr Physiol (2012) 2:2733–52. doi:10.1002/cphy.c120002
8. Ferrario CM, Ahmad S, Joyner J, Varagic J. Advances in the renin angiotensin system focus on angiotensin-converting enzyme 2 and angiotensin-(1-7). Adv Pharmacol (2010) 59:197–233. doi:10.1016/S1054-3589(10)59007-0
9. Santos RA, Ferreira AJ, Verano-Braga T, Bader M. Angiotensin-converting enzyme 2, angiotensin-(1-7) and Mas: new players of the renin-angiotensin system. J Endocrinol (2013) 216:R1–17. doi:10.1530/JOE-12-0341
10. Ferreira AJ, Santos RA, Raizada MK. Angiotensin-(1-7)/angiotensin-converting enzyme 2/mas receptor axis and related mechanisms. Int J Hypertens (2012) 2012:690785. doi:10.1155/2012/690785
11. Gomes ER, Santos RA, Guatimosim S. Angiotensin-(1-7)-mediated signaling in cardiomyocytes. Int J Hypertens (2012) 2012:493129. doi:10.1155/2012/493129
12. Ferreira AJ, Santos RA, Bradford CN, Mecca AP, Sumners C, Katovich MJ, et al. Therapeutic implications of the vasoprotective axis of the renin-angiotensin system in cardiovascular diseases. Hypertension (2010) 55:207–13. doi:10.1161/HYPERTENSIONAHA.109.140145
13. Ferreira AJ, Murca TM, Fraga-Silva RA, Castro CH, Raizada MK, Santos RA. New cardiovascular and pulmonary therapeutic strategies based on the Angiotensin-converting enzyme 2/angiotensin-(1-7)/mas receptor axis. Int J Hypertens (2012) 2012:147825. doi:10.1155/2012/147825
14. Santos RA, Ferreira AJ, Simoes e Silva AC. Recent advances in the angiotensin-converting enzyme 2-angiotensin(1-7)-Mas axis. Exp Physiol (2008) 93:519–27. doi:10.1113/expphysiol.2008.042002
15. Diz DI, Garcia-Espinosa MA, Gegick S, Ferrario CM, Tallant EA, Chappell MC, et al. ACE2 Inhibition in the solitary tract nucleus reduces baroreceptor reflex sensitivity for heart rate control. Exp Physiology (2008) 93:694–700. doi:10.1113/expphysiol.2007.040261
16. Sakima A, Averill DB, Gallagher PE, Kasper SO, Tommasi EN, Ferrario CM, et al. Impaired heart rate baroreflex in older rats: role of endogenous angiotensin-(1-7) at the nucleus tractus solitarii. Hypertension (2005) 46:333–40. doi:10.1161/01.HYP.0000178157.70142.33
17. Isa K, Arnold AC, Westwood BM, Chappell MC, Diz DI. Angiotensin-converting enzyme inhibition, but not AT(1) receptor blockade, in the solitary tract nucleus improves baroreflex sensitivity in anesthetized transgenic hypertensive (mRen2)27 rats. Hypertens Res (2011) 34:1257–62. doi:10.1038/hr.2011.110
18. Garcia-Espinosa T, Shaltout H, Gallagher PE, Chappell MC, Diz DI. In vivo expression of angiotensin-(1-7) lowers blood pressure and improves baroreflex function in transgenic (mRen2)27 rats. J Card Pharm (2012) 60:150–7. doi:10.1097/FJC.0b013e3182588b32
19. Paul M, Mehr AP, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev (2006) 86:747–803. doi:10.1152/physrev.00036.2005
20. Wu XC, Johns EJ. Nitric oxide modulation of neurally induced proximal tubular fluid reabsorption in the rat. Hypertension (2002) 39:790–3. doi:10.1161/hy0302.105681
21. Campbell DJ, Kladis A, Duncan AM. Effects of converting enzyme inhibitors on angiotensin and bradykinin peptides. Hypertension (1994) 23:439–49. doi:10.1161/01.HYP.23.4.439
22. Chappell MC, Pirro NT, Sykes A, Ferrario CM. Metabolism of angiotensin-(1-7) by angiotensin converting enzyme. Hypertension (1998) 31:362–7. doi:10.1161/01.HYP.31.1.362
23. Allred AJ, Diz DI, Ferrario CM, Chappell MC. Pathways for angiotensin-(1-7) metabolism in pulmonary and renal tissues. Am J Physiol (2000) 279:F841–50.
24. Anastasopoulos F, Leung R, Kladis A, James GM, Briscoe TA, Gorski TP, et al. Marked difference between angiotensin-converting enzyme and neutral endopeptidase inhibition in vivo by a dual inhibitor of both enzymes. J Pharmacol Exp Ther (1998) 284:799–805.
25. Campbell DJ, Anastasopoulos F, Duncan AM, James GM, Kladis A, Briscoe TA. Effects of neutral endopeptidase inhibition and combined angiotensin converting enzyme and neutral endopeptidase inhibition on angiotensin and bradykinin peptides in rats. J Pharmacol Exp Ther (1998) 287:567–77.
26. Iyer SN, Ferrario CM, Chappell MC. Angiotensin-(1-7) contributes to the antihypertensive effects of blockade of the renin-angiotensin system. Hypertension (1998) 31:356–61. doi:10.1161/01.HYP.31.1.356
27. Yamamoto K, Chappell MC, Brosnihan KB, Ferrario CM. In vivo metabolism of angiotensin I by neutral endopeptidase (EC 3.4.24.11) in spontaneously hypertensive rats. Hypertension (1992) 19:692–6. doi:10.1161/01.HYP.19.6.692
28. Pereira MG, Souza LL, Becari C, Duarte DA, Camacho FR, Oliveira JA, et al. Angiotensin II-independent angiotensin-(1-7) formation in rat hippocampus: involvement of thimet oligopeptidase. Hypertension (2013) 62:879–85. doi:10.1161/HYPERTENSIONAHA.113.01613
29. Chappell MC, Tallant EA, Brosnihan KB, Ferrario CM. Processing of angiotensin I to angiotensin-(1-7) by vascular smooth muscle cells. J Vasc Med Biol (1995) 5:129–37.
30. Chappell MC, Gomez MN, Pirro NT, Ferrario CM. Release of angiotensin-(1-7) from the rat hindlimb: influence of angiotensin-converting enzyme inhibition. Hypertension (2000) 35:348–52. doi:10.1161/01.HYP.35.1.348
31. Grobe N, Weir NM, Leiva O, Ong FS, Bernstein KE, Schmaier AH, et al. Identification of prolyl carboxypeptidase as an alternative enzyme for processing of renal angiotensin II using mass spectrometry. Am J Physiol Cell Physiol (2013) 304:C945–53. doi:10.1152/ajpcell.00346.2012
32. Rice GI, Thomas DA, Grant PJ, Turner AJ, Hooper NM. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J (2004) 383:45–51. doi:10.1042/BJ20040634
33. Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem (2002) 277:14838–43. doi:10.1074/jbc.M200581200
34. Oudit GY, Liu GC, Zhong J, Basu R, Chow FL, Zhou J, et al. Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes (2010) 59:529–38. doi:10.2337/db09-1218
35. Oudit GY, Penninger JM. Recombinant human angiotensin-converting enzyme 2 as a new renin-angiotensin system peptidase for heart failure therapy. Curr Heart Fail Rep (2011) 8:176–83. doi:10.1007/s11897-011-0063-7
36. Wysocki J, Ye M, Rodriguez E, Gonzalez-Pacheco FR, Barrios C, Evora K, et al. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension (2010) 55:90–8. doi:10.1161/HYPERTENSIONAHA.109.138420
37. Zhong J, Guo D, Chen CB, Wang W, Schuster M, Loibner H, et al. Prevention of angiotensin II-mediated renal oxidative stress, inflammation, and fibrosis by angiotensin-converting enzyme 2. Hypertension (2011) 57:314–21. doi:10.1161/HYPERTENSIONAHA.110.164244
38. Gallagher PE, Payne VS, Kasper SO, Tommasi EN, Westwood BM, Robbins ME, et al. Long-term systemic angiotensin II type 1 receptor blockade regulates mRNA expression of dorsomedial medulla renin-angiotensin system components. Physiol Genomics (2011) 43:829–35. doi:10.1152/physiolgenomics.00167.2010
39. Clarke NE, Fisher MJ, Porter KE, Lambert DW, Turner AJ. Angiotensin converting enzyme (ACE) and ACE2 bind integrins and ACE2 regulates integrin signalling. PLoS One (2012) 7:e34747. doi:10.1371/journal.pone.0034747
40. Chodavarapu H, Grobe N, Somineni HK, Salem ES, Madhu M, Elased KM. Rosiglitazone treatment of type 2 diabetic db/db mice attenuates urinary albumin and angiotensin converting enzyme 2 excretion. PLoS One (2013) 8:e62833. doi:10.1371/journal.pone.0062833
41. Wysocki J, Garcia-Halpin L, Ye M, Maier C, Sowers K, Burns KD, et al. Regulation of urinary ACE2 in diabetic mice. Am J Physiol Renal Physiol (2013) 305:F600–11. doi:10.1152/ajprenal.00600.2012
42. Yamaleyeva LM, Gilliam-Davis S, Almeida I, Brosnihan KB, Lindsey SH, Chappell MC. Differential regulation of circulating and renal ACE2 and ACE in hypertensive mRen2.Lewis rats with early-onset diabetes. Am J Physiol Renal Physiol (2012) 302:F1374–84. doi:10.1152/ajprenal.00656.2011
43. Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, et al. Tumor necrosis factor-a convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-Co-V) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem (2005) 280:30113–9. doi:10.1074/jbc.M505111200
44. Jia HP, Look DC, Tan P, Shi L, Hickey M, Gakhar L, et al. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am J Physiol Lung Cell Mol Physiol (2009) 297:L84–96. doi:10.1152/ajplung.00071.2009
45. Higashiyama S, Iwabuki H, Morimoto C, Hieda M, Inoue H, Matsushita N. Membrane-anchored growth factors, the epidermal growth factor family: beyond receptor ligands. Cancer Sci (2008) 99:214–20. doi:10.1111/j.1349-7006.2007.00676.x
46. Xia H, Sriramula S, Chahabra KH, Lazartigues E. Brain ACE2 shedding contributes to the development of neurogenic hypertension. Circ Res (2013) 113:1087–96. doi:10.1161/CIRCRESAHA.113.301811
47. Lautrette A, Li S, Alili R, Sunnarborg SW, Burtin M, Lee DC, et al. Angiotensin II and EGF receptor cross-talk in chronic kidney diseases: a new therapeutic approach. Nat Med (2005) 11:867–74. doi:10.1038/nm1275
48. Akhtar S, Yousif MH, Dhaunsi GS, Chandrasekhar B, Al-Farsi O, Benter IF. Angiotensin-(1-7) inhibits epidermal growth factor receptor transactivation via a Mas receptor-dependent pathway. Br J Pharmacol (2011) 165:1390–40. doi:10.1111/j.1476-5381.2011.01613.x
49. Cook KL, Metheny-Barlow LJ, Tallant EA, Gallagher PE. Angiotensin-(1-7) reduces fibrosis in orthotopic breast tumors. Cancer Res (2010) 70:8319–28. doi:10.1158/0008-5472.CAN-10-1136
50. Gallagher PE, Ferrario CM, Tallant EA. MAP kinase/phosphatase pathway mediates the regulation of ACE2 by angiotensin peptides. Am J Physiol Cell Physiol (2008) 295:C1169–74. doi:10.1152/ajpcell.00145.2008
51. Gava E, Samad-Zadeh A, Zimpelmann J, Bahramifarid N, Kitten GT, Santos RA, et al. Angiotensin-(1-7) activates a tyrosine phosphatase and inhibits glucose-induced signalling in proximal tubular cells. Nephrol Dial Transplant (2009) 24:1766–73. doi:10.1093/ndt/gfn736
52. McCollum LT, Gallagher PE, Ann TE. Angiotensin-(1-7) attenuates angiotensin II-induced cardiac remodeling associated with upregulation of dual-specificity phosphatase 1. Am J Physiol Heart Circ Physiol (2012) 302:H801–10. doi:10.1152/ajpheart.00908.2011
53. McCollum LT, Gallagher PE, Tallant EA. Angiotensin-(1-7) abrogates mitogen-stimulated proliferation of cardiac fibroblasts. Peptides (2012) 34:380–8. doi:10.1016/j.peptides.2012.01.020
54. Nagata S, Kato J, Sasaki K, Minamino N, Eto T, Kitamura K. Isolation and identification of proangiotensin-12, a possible component of the renin-angiotensin system. Biochem Biophys Res Commun (2006) 350:1026–31. doi:10.1016/j.bbrc.2006.09.146
55. Jessup JA, Trask AJ, Chappell MC, Nagata S, Kato J, Kitamura K, et al. Localization of the novel angiotensin peptide, angiotensin-12 [Ang-(1-12)], in heart and kidney of hypertensive and normotensive rats. Am J Physiol Heart Circ Physiol (2008) 294:H2242–7. doi:10.1152/ajpheart.91521.2007
56. Ahmad S, Simmons T, Varagic J, Moniwa N, Chappell MC, Ferrario CM. Chymase-dependent generation of angiotensin II from angiotensin-(1-12) in human atrial tissue. PLoS One (2011) 6:e28501. doi:10.1371/journal.pone.0028501
57. Arnold AC, Isa K, Shaltout HA, Nautiyal M, Ferrario CM, Chappell MC, et al. Angiotensin-(1-12) requires angiotensin converting enzyme and AT1 receptors for cardiovascular actions within the solitary tract nucleus. Am J Physiol Heart Circ Physiol (2010) 299:H763–71. doi:10.1152/ajpheart.00345.2010
58. Isa K, Garcia-Espinosa MA, Arnold AC, Pirro NT, Tommasi EN, Ganten D, et al. Chronic immunoneutralization of brain angiotensin-(1-12) lowers blood pressure in transgenic (mRen2)27 hypertensive rats. Am J Physiol Regul Integr Comp Physiol (2009). 297:R111–5. doi:10.1152/ajpregu.90588.2008
59. Westwood BM, Chappell MC. Divergent pathways for the angiotensin-(1-12) metabolism in the rat circulation and kidney. Peptides (2012) 35:190–5. doi:10.1016/j.peptides.2012.03.025
60. Bujak-Gizycka B, Olszanecki R, Suski M, Madek J, Stachowicz A, Korbut R. Angiotensinogen metabolism in rat aorta: robust formation of proangiotensin-12. J Physiol Pharmacol (2010) 61:679–82.
61. Lautner RQ, Villela DC, Fraga-Silva RA, Silva N, Verano-Braga T, Costa-Fraga F, et al. Discovery and characterization of almandine: a novel component of the renin-angiotensin system. Circ Res (2013) 112:1104–11. doi:10.1161/CIRCRESAHA.113.301077
62. Jankowski V, Vanholder R, van der Giet M, Tolle M, Karadogan S, Gobom J, et al. Mass-spectrometric identification of a novel angiotensin peptide in human plasma. Arterioscler Thromb Vasc Biol (2007) 27:297–302. doi:10.1161/01.ATV.0000253889.09765.5f
63. Bosnyak S, Widdop RE, Denton KM, Jones ES. Differential mechanisms of ang (1-7)-mediated vasodepressor effect in adult and aged candesartan-treated rats. Int J Hypertens (2012) 2012:192567. doi:10.1155/2012/192567
64. Safari T, Nematbakhsh M, Hilliard LM, Evans RG, Denton KM. Sex differences in the renal vascular response to angiotensin II involves the Mas receptor. Acta Physiol (Oxf) (2012) 206:150–6. doi:10.1111/j.1748-1716.2012.02468.x
65. Walters PE, Gaspari TA, Widdop RE. Angiotensin-(1-7) acts as a vasodepressor agent via angiotensin II type 2 receptors in conscious rats. Hypertension (2005) 45:960–6. doi:10.1161/01.HYP.0000160325.59323.b8
66. Jankowski V, Tolle M, Santos RA, Gunthner T, Krause E, Beyermann M, et al. Angioprotectin: an angiotensin II-like peptide causing vasodilatory effects. FASEB J (2011) 25:2987–95. doi:10.1096/fj.11-185470
67. Shaltout HA, Westwood B, Averill DB, Ferrario CM, Figueroa J, Diz DI, et al. Angiotensin metabolism in renal proximal tubules, urine and serum of sheep: evidence for ACE2-dependent processing of angiotensin II. Am J Physiol Renal Physiol (2006) 292:F82–91. doi:10.1152/ajprenal.00139.2006
68. Yamada K, Iyer SN, Chappell MC, Ganten D, Ferrario CM. Converting enzyme determines the plasma clearance of angiotensin-(1-7). Hypertension (1998) 98:496–502. doi:10.1161/01.HYP.32.3.496
69. Marshall AC, Shaltout HA, Pirro NT, Rose JC, Diz DI, Chappell MC. Antenatal betamethasone exposure is associated with lower Ang-(1-7) and increased ACE in the CSF of adult sheep. Am J Physiol Regul Integr Comp Physiol (2013) 305:R679–88. doi:10.1152/ajpregu.00321.2013
70. Marshall AC, Shaltout HA, Pirro NT, Rose JC, Diz DI, Chappell MC. Enhanced activity of an Angiotensin-(1-7) neuropeptidase in glucocorticoid-induced fetal programming. Peptides (Forthcoming 2014). doi:10.1016/j.peptides.2013.12.006
71. Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev (2007) 59:251–87. doi:10.1124/pr.59.3.3
72. Navar LG, Kobori H, Prieto MC, Gonzalez-Villalobos RA. Intrarenal renin-angiotensin system in hypertension. Hypertension (2011) 57:355–62. doi:10.1161/HYPERTENSIONAHA.110.163519
73. Robertson AL Jr., Khairallah PA. Angiotensin II: rapid localization in nuclei of smooth and cardiac muscle. Science (1971) 172:1138–9. doi:10.1126/science.172.3988.1138
74. Booz GW, Conrad KM, Hess AL, Singer HA, Baker KM. Angiotensin-II-binding sites on hepatocyte nuclei. Endocrinology (1992) 130:3641–9. doi:10.1210/en.130.6.3641
75. Re RN, Vizard DL, Brown J, LeGros L, Bryan SE. Angiotensin II receptors in chromatin. J Hypertens Suppl (1984) 2:S271–3.
76. Tang SS, Rogg H, Schumacher R, Dzau VJ. Characterization of nuclear angiotensin-II-binding sites in rat liver and comparison with plasma membrane receptors. Endocrinology (1992) 131:374–80. doi:10.1210/en.131.1.374
77. Eggena P, Zhu JH, Clegg K, Barrett JD. Nuclear angiotensin receptors induce transcription of renin and angiotensinogen mRNA. Hypertension (1993) 22:496–501. doi:10.1161/01.HYP.22.4.496
78. Eggena P, Zhu JH, Sereevinyayut S, Giordani M, Clegg K, Andersen PC, et al. Hepatic angiotensin II nuclear receptors and transcription of growth-related factors. J Hypertens (1996) 14:961–8.
79. Li XC, Zhuo JL. Intracellular ANG II directly induces in vitro transcription of TGF-beta1, MCP-1, and NHE-3 mRNAs in isolated rat renal cortical nuclei via activation of nuclear AT1a receptors. Am J Physiol Cell Physiol (2008) 294:C1034–45. doi:10.1152/ajpcell.00432.2007
80. Licea H, Walters MR, Navar G. Renal nuclear angiotensin II receptors in normal and hypertensive rats. Acta Physiol Hung (2002) 89:427–38. doi:10.1556/APhysiol.89.2002.4.3
81. Pendergrass KD, Averill DB, Ferrario CM, Diz DI, Chappell MC. Differential expression of nuclear AT1 receptors and angiotensin II within the kidney of the male congenic mRen2. Lewis rat. Am J Physiol Renal Physiol (2006) 290:F1497–506. doi:10.1152/ajprenal.00317.2005
82. Zhuo JL, Li XC, Garvin JL, Navar LG, Carretero OA. Intracellular ANG II induces cytosolic Ca2+ mobilization by stimulating intracellular AT1 receptors in proximal tubule cells. Am J Physiol Renal Physiol (2006) 290:F1382–90. doi:10.1152/ajprenal.00269.2005
83. Pendergrass KD, Gwathmey TM, Michalek RD, Grayson JM, Chappell MC. The angiotensin II-AT1 receptor stimulates reactive oxygen species within the cell nucleus. Biochem Biophys Res Commun (2009) 384:149–54. doi:10.1016/j.bbrc.2009.04.126
84. Ahmarani L, Avedanian L, Al-Khoury J, Perreault C, Jacques D, Bkaily G. Whole-cell and nuclear NADPH oxidases levels and distribution in human endocardial endothelial, vascular smooth muscle, and vascular endothelial cells. Can J Physiol Pharmacol (2013) 91:71–9. doi:10.1139/cjpp-2012-0265
85. Hilenski LL, Clempus RE, Quinn MT, Lambeth JD, Griendling KK. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol (2004) 24:677–83. doi:10.1161/01.ATV.0000112024.13727.2c
86. Kuroda J, Nakagawa K, Yamasaki T, Nakamura K, Takeya R, Kuribayashi F, et al. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells (2005) 10:1139–51. doi:10.1111/j.1365-2443.2005.00907.x
87. Spencer NY, Yan Z, Boudreau RL, Zhang Y, Luo M, Li Q, et al. Control of hepatic nuclear superoxide production by glucose 6-phosphate dehydrogenase and NADPH oxidase-4. J Biol Chem (2011) 286:8977–87. doi:10.1074/jbc.M110.193821
88. DeMello WC. Is an intracellular renin-angiotensin system involved in control of cell communication in heart? J Cardiovasc Pharmacol (1994) 23:640–6. doi:10.1097/00005344-199404000-00018
89. Ellis B, Li XC, Miguel-Qin E, Gu V, Zhuo JL. Evidence for a functional intracellular angiotensin system in the proximal tubule of the kidney. Am J Physiol Regul Integr Comp Physiol (2012) 302:R494–509. doi:10.1152/ajpregu.00487.2011
90. Gobeil F Jr., Zhu T, Brault S, Geha A, Vazquez-Tello A, Fortier A, et al. Nitric oxide signaling via nuclearized endothelial nitric-oxide synthase modulates expression of the immediate early genes iNOS and mPGES-1. J Biol Chem (2006) 281:16058–67. doi:10.1074/jbc.M602219200
91. Kumar R, Thomas CM, Yong QC, Chen W, Baker KM. The intracrine renin-angiotensin system. Clin Sci (Lond) (2012) 123:273–84. doi:10.1042/CS20120089
92. Re R. The nature of intracrine peptide hormone action. Hypertension (1999) 34:534–8. doi:10.1161/01.HYP.34.4.534
93. Gwathmey TM, Alzayadneh EB, Pendergrass KD, Chappell MC. Novel roles of nuclear angiotensin receptors and signaling mechanisms. Am J Physiol Regul Integr Comp Physiol (2012) 302(5):R518–30. doi:10.1152/ajpregu.00525.2011
94. Vaniotis G, Allen BG, Hebert TE. Nuclear GPCRs in cardiomyocytes: an insider’s view of beta-adrenergic receptor signaling. Am J Physiol Heart Circ Physiol (2011) 301:H1754–64. doi:10.1152/ajpheart.00657.2011
95. Lee DK, Lança AJ, Cheng R, Nguyen T, Ji XD, Gobeil F Jr., et al. Agonist-independent nuclear localization of the Apelin, angiotensin AT1, and bradykinin B2 receptors. J Biol Chem (2004) 279:7901–8. doi:10.1074/jbc.M306377200
96. Gwathmey TM, Westwood BM, Pirro NT, Tang L, Rose JC, Diz DI, et al. Nuclear angiotensin-(1-7) receptor is functionally coupled to the formation of nitric oxide. Am J Physiol Renal Physiol (2010) 299:F983–90. doi:10.1152/ajprenal.00371.2010
97. Savard M, Barbaz D, Belanger S, Muller-Esterl W, Bkaily G, Orleans-Juste P, et al. Expression of endogenous nuclear bradykinin B2 receptors mediating signaling in immediate early gene activation. J Cell Physiol (2008) 216:234–44. doi:10.1002/jcp.21398
98. Gwathmey-Williams T, Pendergrass KD, Rose JC, Diz DI, Chappell MC. Angiotensin-(1-7)-ACE2 attenuates reactive oxygen species formation to angiotensin II within the cell nucleus. Hypertension (2010) 55(1):166–71.
99. Planchon SM, Waite KA, Eng C. The nuclear affairs of PTEN. J Cell Sci (2008) 121:249–53. doi:10.1242/jcs.022459
100. Bootman MD, Fearnley C, Smyrinias I, MacDonald F, Roderick HL. An update on nuclear calcium signaling. J Cell Sci (2009) 122:2337–50. doi:10.1242/jcs.028100
101. Mauger JP. Role of the nuclear envelope in calcium signaling. Biol Cell (2012) 104:70–83. doi:10.1111/boc201100103
102. Malviya AN, Klein C. Mechanisms regulating nuclear calcium signaling. Can J Physiol Pharmacol (2006) 84:403–22. doi:10.1139/y05-130
103. Gonzalez-Villalobos R, Klassen RB, Allen PL, Navar LG, Hammond TG. Megalin binds and internalizes angiotensin II. Am J Physiol Renal Physiol (2005) 28:F420–7.
104. Gonzalez-Villalobos R, Klassen RB, Allen PL, Johanson K, Baker CB, Kobori H, et al. Megalin binds and internalizes angiotensin-(1-7). Am J Physiol Renal Physiol (2006) 290:F1270–5. doi:10.1152/ajprenal.00164.2005
105. Matsusaka T, Niimura F, Shimizu A, Pastan I, Saito A, Kobori H, et al. Liver angiotensinogen is the primary source of renal angiotensin II. J Am Soc Nephrol (2012) 23:1181–9. doi:10.1681/ASN.2011121159
106. Pohl M, Kaminski H, Castrop H, Bader M, Himmerkus N, Bleich M, et al. Intrarenal renin angiotensin system revisited: role of megalin-dependent endocytosis along the proximal nephron. J Biol Chem (2010) 285:41935–46. doi:10.1074/jbc.M110.150284
107. Alzayadneh EM, Chappell MC. Nuclear expression of renin-angiotensin system components in NRK-52E renal epithelial cells. J Renin Angiotensin Aldosterone Syst (Forthcoming 2014).
108. Sherrod M, Liu X, Zhang X, Sigmund CD. Nuclear localization of angiotensinogen in astrocytes. Am J Physiol Regul Integr Comp Physiol (2005) 288(2):R539–46. doi:10.1152/ajpregu.00594.2004
109. Massarelli EE, Casatti CA, Kato A, Camargo AC, Bauer JA, Glucksman MJ, et al. Differential subcellular distribution of neurolysin (EC 3.4.24.16) and thimet oligopeptidase (EC 3.4.24.15) in the rat brain. Brain Res (1999) 851:261–5. doi:10.1016/S0006-8993(99)02135-6
110. Thompson A, Huber G, Malherbe P. Cloning and functional expression of a metalloendopeptidase from human brain with the ability to cleave a beta-APP substrate peptide. Biochem Biophys Res Commun (1995) 213:66–73. doi:10.1006/bbrc.1995.2099
111. Ibarra C, Vicencio JM, Estrada M, Lin Y, Rocco P, Rebellato P, et al. Local control of nuclear calcium signaling in cardiac myocytes by perinuclear microdomains of sarcolemmal insulin-like growth factor 1 receptors. Circ Res (2013) 112:236–45. doi:10.1161/CIRCRESAHA.112.273839
112. Bers DM. Membrane receptor neighborhoods: snuggling up to the nucleus. Circ Res (2013) 112:224–6. doi:10.1161/CIRCRESAHA.112.300494
113. Benicky J, Hafko R, Sanchez-Lemus E, Aguilera G, Saavedra JM. Six commercially available angiotensin II AT1 receptor antibodies are non-specific. Cell Mol Neurobiol (2012) 32:1353–65. doi:10.1007/s10571-012-9862-y
114. Hafko R, Villapol S, Nostramo R, Symes A, Sabban EL, Inagami T, et al. Commercially available angiotensin II At(2) receptor antibodies are nonspecific. PLoS One (2013) 8:e69234. doi:10.1371/journal.pone.0069234
115. Herrera M, Sparks MA, Alfonso-Pecchio AR, Harrison-Bernard LM, Coffman TM. Lack of specificity of commercial antibodies leads to misidentification of angiotensin type-1 receptor protein. Hypertension (2013) 61(61):253–8. doi:10.1161/HYPERTENSIONAHA.112.203679
116. Dodic M, Abouantoun T, O’Connor A, Wintour EM, Moritz KM. Programming effects of short prenatal exposure to dexamethasone in sheep. Hypertension (2002) 40:729–34. doi:10.1161/01.HYP.0000036455.62159.7E
117. Doyle LW, Ford GW, Davis NM, Callanan C. Antenatal corticosteroid therapy and blood pressure at 14 years of age in preterm children. Clin Sci (Lond) (2000) 98:137–42. doi:10.1042/CS19990211
118. Seckl JR, Holmes MC. Mechanisms of disease: glucocorticoids, their placental metabolism and fetal ‘programming’ of adult pathophysiology. Nat Clin Pract Endocrinol Metab (2007) 3:479–88. doi:10.1038/ncpendmet0515
119. Zandi-Nejad K, Luyckx VA, Brenner BM. Adult hypertension and kidney disease: the role of fetal programming. Hypertension (2006) 47:502–8. doi:10.1161/01.HYP.0000198544.09909.1a
120. Alexander BT. Fetal programming of hypertension. Am J Physiol Regul Integr Comp Physiol (2006) 290:R1–10. doi:10.1152/ajpregu.00417.2005
121. Goyal R, Lister R, Leitzke A, Goyal D, Gheorghe CP, Longo LD. Antenatal maternal hypoxic stress: adaptations of the placental renin-angiotensin system in the mouse. Placenta (2011) 32:134–9. doi:10.1016/j.placenta.2010.11.004
122. Marshall AC, Shaltout HA, Nautiyal M, Rose JC, Chappell MC, Diz DI. Fetal betamethasone exposure attenuates angiotensin-(1-7)-Mas receptor expression in the dorsal medulla of adult sheep. Peptides (2013) 44:25–31. doi:10.1016/j.peptides.2013.03.018
123. Moritz KM, Johnson K, Douglas-Denton R, Wintour EM, Dodic M. Maternal glucocorticoid treatment programs alterations in the renin-angiotensin system of the ovine fetal kidney. Endocrinology (2002) 143:4455–63. doi:10.1210/en.2002-220534
124. Rasch R, Skriver E, Woods LL. The role of the RAS in programming of adult hypertension. Acta Physiol Scand (2004) 181:537–42. doi:10.1111/j.1365-201X.2004.01328.x
125. Shaltout HA, Figueroa JP, Rose JC, Diz DI, Chappell MC. Alterations in circulatory and renal angiotensin-converting enzyme and angiotensin-converting enzyme 2 in fetal programmed hypertension. Hypertension (2009) 53:404–8. doi:10.1161/HYPERTENSIONAHA.108.124339
126. Shaltout HA, Rose JC, Chappell MC, Diz DI. Angiotensin-(1-7) deficiency and baroreflex impairment precede the antenatal betamethasone exposure-induced elevation in blood pressure. Hypertension (2012) 59:453–8. doi:10.1161/HYPERTENSIONAHA.111.185876
127. Wintour EM, Moritz KM, Johnson K, Ricardo S, Samuel CS, Dodic M. Reduced nephron number in adult sheep, hypertensive as a result of prenatal glucocorticoid treatment. J Physiol (2003) 549:929–35. doi:10.1113/jphysiol.2003.042408
128. Wyrwoll CS, Mark PJ, Waddell BJ. Developmental programming of renal glucocorticoid sensitivity and the renin-angiotensin system. Hypertension (2007) 50:579–84. doi:10.1161/HYPERTENSIONAHA.107.091603
129. Shaltout HA, Rose JC, Figueroa JP, Chappell MC, Diz DI, Averill DB. Acute AT(1)-receptor blockade reverses the hemodynamic and baroreflex impairment in adult sheep exposed to antenatal betamethasone. Am J Physiol Heart Circ Physiol (2010) 299:H541–7. doi:10.1152/ajpheart.00100.2010
130. Tang L, Carey LC, Bi J, Valego N, Sun X, Deibel P, et al. Gender differences in the effects of antenatal betamethasone exposure on renal function in adult sheep. Am J Physiol Regul Integr Comp Physiol (2009) 296:R309–17. doi:10.1152/ajpregu.90645.2008
131. Tang L, Bi J, Valego N, Carey L, Figueroa J, Chappell M, et al. Prenatal betamethasone exposure alters renal function in immature sheep: sex differences in effects. Am J Physiol Regul Integr Comp Physiol (2010) 299:R793–803. doi:10.1152/ajpregu.00590.2009
132. Gwathmey TM, Shaltout HA, Rose JC, Diz DI, Chappell MC. Glucocorticoid-induced fetal programming alters the functional complement of angiotensin receptor subtypes within the kidney. Hypertension (2011) 57:620–6. doi:10.1161/HYPERTENSIONAHA.110.164970
133. Bi J, Contag SA, Carey LC, Tang L, Valego NK, Chappell MC, et al. Antenatal betamethasone exposure alters renal responses to angiotensin-(1-7) in uninephrectomized adult male sheep. J Renin Angiotensin Aldosterone Syst (2013) 14:290–8. doi:10.1177/1470320312465217
134. Zimmerman D, Burns KD. Angiotensin-(1-7) in kidney disease: a review of the controversies. Clin Sci (Lond) (2012) 123:333–46. doi:10.1042/CS20120111
135. Bachmann J, Feldmer M, Ganten U, Stock G, Ganten D. Sexual dimorphism of blood pressure: possible role of the renin-angiotensin system. J Steroid Biochem (1991) 40:511–5. doi:10.1016/0960-0760(91)90270-F
136. Brosnihan KB, Senanayake PS, Li P, Ferrario CM. Bi-directional actions of estrogen on the renin-angiotensin system. Braz J Med Biol Res (1999) 32:373–81.
137. Reckelhoff JF, Zhang H, Srivastava K. Gender differences in development of hypertension in spontaneously hypertensive rats: role of the renin-angiotensin system. Hypertension (2000) 35:480–3. doi:10.1161/01.HYP.35.1.480
138. Sullivan JC. Sex and the renin-angiotensin system: inequality between the sexes in response to RAS stimulation and inhibition. Am J Physiol Regul Integr Comp Physiol (2008) 294:R1220–6. doi:10.1152/ajpregu.00864.2007
139. Chappell MC, Gallagher PE, Averill DB, Ferrario CM, Brosnihan KB. Estrogen or the AT1 antagonist olmesartan reverses the development of profound hypertension in the congenic mRen2. Lewis rat. Hypertension (2003) 42:781–6. doi:10.1161/01.HYP.0000085210.66399.A3
140. Chappell MC, Westwood BM, Yamaleyeva LM. Differential effects of sex steroids in young and aged female mRen2.Lewis rats: a model of estrogen and salt-sensitive hypertension. Gend Med (2008) 5(Suppl A):S65–75. doi:10.1016/j.genm.2008.03.007
141. Brosnihan KB, Li P, Ganten D, Ferrario CM. Estrogen protects transgenic hypertensive rats by shifting the vasoconstrictor-vasodilator balance of RAS. Am J Physiol (1997) 273:R1908–15.
142. Sullivan JC, Bhatia K, Yamamoto T, Elmarakby AA. Angiotensin (1-7) receptor antagonism equalizes angiotensin II-induced hypertension in male and female spontaneously hypertensive rats. Hypertension (2010) 56:658–66. doi:10.1161/HYPERTENSIONAHA.110.153668
143. Liu J, Ji H, Zheng W, Wu X, Zhu JJ, Arnold AP, et al. Sex differences in renal angiotensin converting enzyme 2 (ACE2) activity are 17beta-oestradiol-dependent and sex chromosome-independent. Biol Sex Differ (2010) 1:6. doi:10.1186/2042-6410-1-6
144. Gupte M, Thatcher SE, Boustany-Kari CM, Shoemaker R, Yiannikouris F, Zhang X, et al. Angiotensin converting enzyme 2 contributes to sex differences in the development of obesity hypertension in C57BL/6 mice. Arterioscler Thromb Vasc Biol (2012) 32:1392–9. doi:10.1161/ATVBAHA.112.248559
145. Xue B, Zhang Z, Johnson RF, Guo F, Hay M, Johnson AK. Central endogenous angiotensin-(1-7) protects against aldosterone/NaCl-induced hypertension in female rats. Am J Physiol Heart Circ Physiol (2013) 305:H699–705. doi:10.1152/ajpheart.00193.2013
Keywords: Ang-(1–7), Ala1-Ang-(1–7), ACE2, ACE, Mas receptor, Mas-related receptor D, fetal programing
Citation: Chappell MC, Marshall AC, Alzayadneh EM, Shaltout HA and Diz DI (2014) Update on the angiotensin converting enzyme 2-angiotensin (1–7)-Mas receptor axis: fetal programing, sex differences, and intracellular pathways. Front. Endocrinol. 4:201. doi: 10.3389/fendo.2013.00201
Received: 09 September 2013; Paper pending published: 30 September 2013;
Accepted: 18 December 2013; Published online: 09 January 2014.
Edited by:
Walmor De Mello, University of Puerto Rico, USAReviewed by:
Nicola J. Smith, Victor Chang Cardiac Research Institute, AustraliaMichael Bader, Max Delbrück Center, Germany
Copyright: © 2014 Chappell, Marshall, Alzayadneh, Shaltout and Diz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Debra I. Diz, The Hypertension and Vascular Research Center, Wake Forest University School of Medicine, Medical Center Blvd, Winston-Salem, NC 27157-1032, USA e-mail:ZGRpekB3YWtlaGVhbHRoLmVkdQ==