 Beatriz Morte
Beatriz Morte Juan Bernal
Juan Bernal- Instituto de Investigaciones Biomédicas “Alberto Sols”, Consejo Superior de Investigaciones Científicas, Center for Biomedical Research on Rare Diseases (CIBERER), Universidad Autónoma de Madrid, Madrid, Spain
Thyroid hormone (TH) action is exerted mainly through regulation of gene expression by binding of T3 to the nuclear receptors. T4 plays an important role as a source of intracellular T3 in the central nervous system via the action of the type 2 deiodinase (D2), expressed in the astrocytes. A model of T3 availability to neural cells has been proposed and validated. The model contemplates that brain T3 has a double origin: a fraction is available directly from the circulation, and another is produced locally from T4 in the astrocytes by D2. The fetal brain depends almost entirely on the T3 generated locally. The contribution of systemic T3 increases subsequently during development to account for approximately 50% of total brain T3 in the late postnatal and adult stages. In this article, we review the experimental data in support of this model, and how the factors affecting T3 availability in the brain, such as deiodinases and transporters, play a decisive role in modulating local TH action during development.
The Model
Thyroid hormone (TH) action is exerted mainly through regulation of gene expression by binding of T3 to the nuclear receptors (1, 2). T4 plays an important role as a source of intracellular T3 in the central nervous system via the action of the type 2 deiodinase (D2). This process is regulated physiologically. D2 activity in brain increases during development in correlation with the more T3 sensitive developmental period. D2 activity and T3 concentrations are synchronized in a spatial and temporal fashion to determine critical brain processes such as myelination, neuronal migration, glial differentiation, and neurogenesis. On the other hand, D2 activity is inversely regulated by T4, so that brain T3 levels fluctuates less than circulating TH levels, due to changes in D2 activity in response to changes in T4 availability at least at postnatal and adult stages (3).
Although neurons are the primary target of T3 actions, Guadaño-Ferraz et al. (4) demonstrated that D2 expression takes place predominantly, if not exclusively, in glial cells: the tanycytes (5) lining part of the third ventricle surface and in the astrocytes throughout the brain. The Dio2 mRNA was not restricted to the cell body but was also present along the cellular processes. This observation indicated an important role for glial cells in TH homeostasis in the brain and a close coupling between glial cells and neurons in TH metabolism. According to these observations, the authors suggested a model of T3 availability to neural cells. On the one hand, circulating T4 and T3 would enter the brain through the blood–brain barrier (BBB). T4, upon entering the brain would reach the astrocytes through their end-feet in contact with the capillaries, and produce additional T3 by D2-mediated deiodination.
Evidence from Transcriptomic Data
Support for the glial specificity of Dio2 expression came from transcriptomic studies by Cahoy et al. (6). These authors performed transcriptomic studies in primary neural cells isolated from the mouse brain, without further manipulations to establish the patterns of gene expression representative from the different cell types in vivo. Dio2 was enriched up to 50 times in the astrocytes, and therefore may be considered as a highly specific astrocyte gene. Despite this cellular specificity, there is evidence that Dio2 is also expressed in cells other than the astrocytes in some situations. For example, in profound hypothyroidism in the rat, Dio2 expression was also observed in a fraction of cerebral cortex interneurons (7). It was more recently demonstrated that astrocyte-specific inactivation of Dio2 (GFAP–Cre–D2KO mice) reduced D2 activity in brain to less than 10% of the control (8).
Evidence from Paracrine Interactions In vitro
Freitas et al. (9) studied in vitro the evidence for a paracrine interaction between astrocytes and neurons. The goal was to check whether the T3 generated in glial cells was able to activate neuronal gene expression. This experimental system was based on an in vitro co-culture system of H4 human glioma cells expressing D2 and neuroblastoma cells. The two cell types were located in two adjacent compartments bathed with the same culture media. The authors demonstrated that upon incubation with T4, D2 activity in glial cells resulted in increased T3 production that reached neurons and promoted TH-responsive gene expression.
Evidence from Transporter Pathophysiology: Direct Functional Demonstration of the Paracrine Model In vivo
A large body of evidence has accumulated in recent years demonstrating the crucial importance of transporters in mediating the cellular uptake of THs through the cell membranes (10). The most specific and physiologically relevant transporters for THs identified so far are the monocarboxylate transporter 8 (MCT8, SLC16A2), and the organic anion transporting polypeptide 1C1 (OATP1C1, SLCO1C1). MCT8 mutations cause an X-linked syndrome with severe psychomotor retardation and elevated serum T3 levels, indicating the importance of this transporter in TH availability to the brain (11, 12). Other transporters may also contribute to this process although their specific roles are less clear.
MCT8 transports T4 and T3 (13) and OATP1C1 exhibits a remarkable affinity and specificity toward T4 and rT3 (14, 15). Roberts et al. (16) reported the presence of Oatp1c1 protein in the abluminal side of endothelial capillary cells forming the BBB. The Oatp1c1 signal overlapped partially with aquaporin 4, a marker of astrocytes’ end-feet, which are in contact with brain micro capillaries. Mct8/MCT8 is also expressed in the micro capillaries in rodent and human, but not in the astrocytes’ end-feet. In addition, Mct8 is also expressed in neurons and choroid plexus (17).
Based on the preferences for TH transport and on the expression patterns of these two transporters, delivery of circulating T4 and T3 to the brain may be formulated in the following way: circulating T3 and T4, crossing the BBB through Mct8 would be delivered to the extracellular fluid, reaching directly the neural cells in the proximity of the micro capillaries. On the other hand, T4, but not T3, would be delivered directly to the astrocytes after Oatp1c1-mediated transport through the BBB. It is important to notice that OATP1C1 is poorly expressed in the BBB of human fetus (16) and adult primates (18) and therefore in the human brain, T4 transport is dependent on MCT8.
We provided experimental evidence in support for this model in rodents by measuring the relative effects of T4 and T3 on brain gene expression in Mct8 knockout mice (Mct8KO) (19). T3 or T4 were administered to wild-type (WT) and to Mct8KO mice previously made hypothyroid, and expression of two neuronal target genes was measured in the cerebellum and striatum. Whereas T4 and T3 were similarly active in WT mice, the Mct8KO mice only responded to T4. The data suggested that the critical restriction to T3 transport in the absence of Mct8 is located at the BBB rather than at the plasma membrane of individual neurons, where other transporters can substitute for Mct8. The similarity in the effects of T4 in the Mct8-deficient and in the WT mice, suggested that T4 can reach the astrocytes in the Mct8-deficient mice through a different transporter, most probably Oatp1c1, and produce enough T3 to regulate neuronal gene expression through a paracrine interaction. Actually, the increased D2 activity present in the Mct8KO mice (20, 21), would facilitate this pathway in face of lower circulating T4, and restricted brain uptake of T3. The consequence is that the Mct8KO mice maintain brain gene expression similar to WT mice, with a few exceptions. Direct proof that D2 activity was indeed responsible for normal expression of most brain TH-regulated genes in the absence of Mct8 was also provided (22). In the Mct8KO mice, inactivation of D2 led to patterns of brain gene expression that were similar to that of severely hypothyroid mice, highlighting the importance of D2.
Therefore, the studies on the Mct8-deficient mice evidenced that the factors affecting T3 availability in the brain, namely deiodinases and transporters, play a decisive role in modulating local TH action. These studies also gave a functional direct demonstration of the critical role of astrocytes in regulating the amount of T3 available for neuronal uptake in vivo. The model also explains why the double inactivation of Mct8 and Oatp1c1 transporters (23) leads to a situation in the brain similar to hypothyroidism, as in the double Mct8 and Dio2 KO.
The Relative Contributions of Direct T3 Uptake from the Circulation and the Local T3 Production in the Astrocytes
Postnatal and Adult Rodents
In the D2-deficient mice, brain T3 concentrations at postnatal day 15 are about half of normal, suggesting that at least from this postnatal age onward, 50% of the total brain T3 derives from direct T3 uptake from the circulation and another 50% derives from local T3 production in the astrocytes (24) (Figure 1). In support of this, Mct8 inactivation also leads to roughly 50% reduction of brain T3 (21).
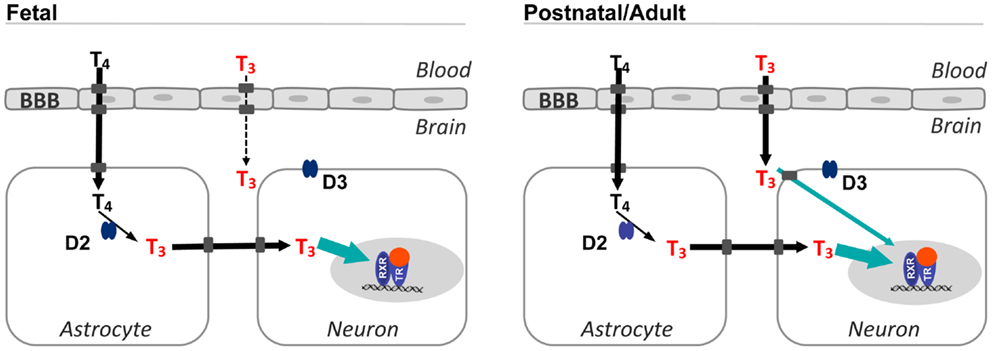
Figure 1. Sources of T3 in the fetal and postnatal brain. T4 from the circulation crosses the BBB and reaches the astrocytes and is converted to T3 by type 2 deiodinase. This is the main source of T3 in the fetal brain. In postnatal and adult animals, T3 from the circulation can also access the brain. The proportion of T3 from the circulation increases up to 50% in the late postnatal stages. In situ T3 production accounts for a high occupancy of thyroid hormone receptors (green arrow), and is also important for expression of genes regulated negatively by thyroid hormone. Transporters in the cell membranes are represented by gray squares, without naming any specific transporter. Although in the rodent brain, the BBB expresses Mct8 and Oatp1c1, the latter is not present in the primate BBB. T4 enters the astrocytes most likely through Oatp1c1. Passage of T3 from astrocytes to neurons is facilitated by Mct8 and also other transporters, since in Mct8 KO mice there is no apparent restriction for the passage of astrocytic T3 to neurons. Type 3 deiodinase is localized in the plasma membrane of neurons. BBB, blood–brain barrier; D2, type 2 deiodinase; D3, type 3 deiodinase; TR, thyroid hormone receptor; RXR, 9-cis retinoic acid receptor.
Local production of T3 accounts for a much higher occupancy of brain nuclear receptors compared to the liver. Crantz et al. (25) estimated that local conversion of T4 to T3 accounted for 77% of the nuclear T3 receptor occupancy in the cerebral cortex and up to 37% in the cerebellum. Circulating T3 was responsible for about 20–25% of nuclear receptor occupancy. These authors concluded that the nuclear receptors were almost saturated by T3 in the cortex, and at 60% of total receptor capacity in the cerebellum. Full receptor saturation under euthyroid conditions may explain why TH-responsive genes in the cortex of euthyroid animals do not respond or do so modestly above the normal expression level after administration of excess T3. In studies of T3 administration to Dio3 KO mice, we found that the response to high doses of T3 was higher in D3-deficient mice than in the WT euthyroid mice (26). Therefore, nuclear occupancy was not a limiting factor in gene responses to T3, and the reason why the euthyroid brain does not respond genomically to excess T3 is due to the protective action of D3.
Despite the importance of D2 for the local generation of T3, D2-deficient mice showed normal expression of several genes positively regulated by T3 (24). We confirmed these observations for additional genes, and showed that in the absence of D2, the expression levels of positively regulated genes are maintained by T3 from the circulation (22). However, expression of the negatively regulated genes is more frequently affected by D2 inactivation, suggesting that they are sensitive to the source of T3. The mechanism underlying these observations is unknown, and somehow the source of T3 may influence directly or indirectly the cellular T3 content.
Thyroid Hormone Transport in the Fetal Brain
The fetal brain depends almost entirely from T4 monodeiodination, so that most brain T3 is produced locally from T4 (Figure 1). In the human fetal brain, despite the presence of T3 receptors and T4 in tissues by the second trimester, and even before, only the brain showed a significant T3 concentration in the nucleus as compared with other tissues (27). This observation indicates that the main source of T3 in the fetal brain is local T4 deiodination. In support of this idea, D2 activity rises in the human cerebral cortex during the second trimester in parallel with T3 concentrations (28).
In the rat (29, 30), physiological doses of T4 administered to hypothyroid pregnant rats could normalize T3 concentrations in the brain and increase neuronal gene expression. In contrast, administration of even large doses of T3 to the mother failed to increase T3 concentration in the fetal brain despite reaching other tissues, and was not able to normalize fetal gene expression. The rat fetal brain contains significant D2 activity (31) making it plausible that most if not all brain T3 is produced locally.
Why Does Circulating T3 Cannot Enter the Fetal Brain?
The reason why the fetal brain is apparently impermeable to circulating T3 is unknown. A possible explanation could be a lack of expression of the Mct8 transporter. To address this issue, the expression of Mct8 and of the specific T4 transporter Oatp1c1 were analyzed by confocal microscopy in the prenatal rat cortex. Both proteins were present in the brain capillaries and in the epithelial cells of the choroid plexus (30).
If T3 cannot enter the brain despite expression of Mct8, what is the role of this transporter in the fetal brain? It is possible that Mct8 at early stages of development has a more prominent role in TH efflux from the brain (32, 33) as proposed in the liver for Mct8 and Mct10 (34). Another possibility is that circulating T3 crossing the BBB through Mct8-mediated transport is deposited directly in the extracellular fluid where it could be rapidly degraded by neuronal D3 activity (35). D3 is abundantly expressed in fetal tissues including the brain (36, 37), where it is expressed in neurons (38, 39) and serves as a fine regulator of tissue TH concentrations. For example, in the human fetal brain, D3 activity in the cerebellum prevents the accumulation of T3 during mid-gestation (28), at the same time that D2 activity is responsible for T3 accumulation in the cortex. Hernandez et al. (40) demonstrated that D3 plays a critical role in maintaining low levels of TH during fetal and early neonatal life in mice. During postnatal development, disruption of the Dio3 gene increases basal expression of the T3-target gene Hr (41). The catalytic site of D3 was earlier proposed to face the extracellular fluid (42, 43). If this was true, T3 in the extracellular fluid of the fetal brain would be easily accessible to D3-mediated degradation. However, more recent evidence derived from Dio3 transfection experiments indicates that D3 substrates need to be internalized for degradation (44). Even so, it may be speculated that the topography of D3 localization in the plasma membrane could allow easier and faster degradation of substrates crossing the neuronal membrane from the extracellular fluid. Within this context, one attractive, but still speculative hypothesis is that the increased proportion of T3 entering the brain from the circulation, taking place from fetal to postnatal stages, is due a decrease of D3 activity. The contribution of systemic T3 would then increase in parallel to account for approximately 50% of the total brain T3.
Local Generation of T3 from T4 in the Context of Astroglial Maturation
The contention that in the fetal brain most T3 derives from T4 has to be examined in the context of D2 activity and maturation of D2-expressing astrocytes during development. The main surge of D2 activity in the rat brain is postnatal, with a peak around postnatal day 15, a time in which the highest T3 concentration in the brain is reached. In the fetal brain, D2 activity is low, and shows a discrete peak just before birth, at prenatal days 18–21 (31, 45). Consequently, brain T3 concentrations during the perinatal period are low, about half to one-third of adult rats.
Some aspects of the model are not well-understood, and should be refined to take into account several factors. One is astrocyte development, which is mainly postnatal (46), increasing in number during postnatal stages, and following a pattern similar to D2 activity. Although the role of astrocytes during the fetal and early postnatal periods has been questioned (47), native astrocytes isolated from the P1 mouse brain already contain a high concentration of Dio2 mRNA (6). It may be that the small population of astrocytes present in the last few days of fetal stages has an important role in the local formation of T3 in the brain. Another difficulty is to extrapolate the model to the human situation. The human brain expresses little OATP1C1 in the BBB, and T4 and T3 transport apparently rely exclusively on MCT8. Therefore, entry of T4 to the astrocytes has to take place through a different transporter. Specifically addressing the patterns of thyroid hormone transporter, and deiodinases in the fetal and postnatal human brain should shed light on these issues.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Bernal J, Guadano-Ferraz A, Morte B. Perspectives in the study of thyroid hormone action on brain development and function. Thyroid (2003) 13:1005–12. doi: 10.1089/105072503770867174
2. Bernal J. Thyroid hormone receptors in brain development and function. Nat Clin Pract Endocrinol Metab (2007) 3:249–59. doi:10.1038/ncpendmet0424
3. Croteau W, Davey JC, Galton VA, St Germain DL. Cloning of the mammalian type II iodothyronine deiodinase. A selenoprotein differentially expressed and regulated in human and rat brain and other tissues. J Clin Invest (1996) 98:405–17. doi:10.1172/JCI118806
4. Guadaño-Ferraz A, Obregon MJ, St Germain DL, Bernal J. The type 2 iodothyronine deiodinase is expressed primarily in glial cells in the neonatal rat brain. Proc Natl Acad Sci U S A (1997) 94:10391–6. doi:10.1073/pnas.94.19.10391
5. Tu HM, Kim SW, Salvatore D, Bartha T, Legradi G, Larsen PR, et al. Regional distribution of type 2 thyroxine deiodinase messenger ribonucleic acid in rat hypothalamus and pituitary and its regulation by thyroid hormone. Endocrinology (1997) 138:3359–68. doi:10.1210/endo.138.8.5318
6. Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci (2008) 28:264–78. doi:10.1523/JNEUROSCI.4178-07.2008
7. Guadaño-Ferraz A, Escamez MJ, Rausell E, Bernal J. Expression of type 2 iodothyronine deiodinase in hypothyroid rat brain indicates an important role of thyroid hormone in the development of specific primary sensory systems. J Neurosci (1999) 19:3430–9.
8. Fonseca TL, Werneck-De-Castro JP, Castillo M, Bocco BM, Fernandes GW, Mcaninch EA, et al. Tissue-specific inactivation of type 2 deiodinase reveals multilevel control of fatty acid oxidation by thyroid hormone in the mouse. Diabetes (2014) 63:1594–604. doi:10.2337/db13-1768
9. Freitas BC, Gereben B, Castillo M, Kallo I, Zeold A, Egri P, et al. Paracrine signaling by glial cell-derived triiodothyronine activates neuronal gene expression in the rodent brain and human cells. J Clin Invest (2010) 120:2206–17. doi:10.1172/JCI41977
10. Friesema EC, Jansen J, Milici C, Visser TJ. Thyroid hormone transporters. Vitam Horm (2005) 70:137–67. doi:10.1016/S0083-6729(05)70005-4
11. Dumitrescu AM, Liao XH, Best TB, Brockmann K, Refetoff S. A novel syndrome combining thyroid and neurological abnormalities is associated with mutations in a monocarboxylate transporter gene. Am J Hum Genet (2004) 74:168–75. doi:10.1086/380999
12. Friesema EC, Grueters A, Biebermann H, Krude H, von Moers A, Reeser M, et al. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet (2004) 364:1435–7. doi:10.1016/S0140-6736(04)17226-7
13. Friesema EC, Ganguly S, Abdalla A, Manning Fox JE, Halestrap AP, Visser TJ. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem (2003) 278:40128–35. doi:10.1074/jbc.M300909200
14. Pizzagalli F, Hagenbuch B, Stieger B, Klenk U, Folkers G, Meier PJ. Identification of a novel human organic anion transporting polypeptide as a high affinity thyroxine transporter. Mol Endocrinol (2002) 16:2283–96. doi:10.1210/me.2001-0309
15. Sugiyama D, Kusuhara H, Taniguchi H, Ishikawa S, Nozaki Y, Aburatani H, et al. Functional characterization of rat brain-specific organic anion transporter (Oatp14) at the blood-brain barrier: high affinity transporter for thyroxine. J Biol Chem (2003) 278:43489–95. doi:10.1074/jbc.M306933200
16. Roberts LM, Woodford K, Zhou M, Black DS, Haggerty JE, Tate EH, et al. Expression of the thyroid hormone transporters monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood-brain barrier. Endocrinology (2008) 149:6251–61. doi:10.1210/en.2008-0378
17. Heuer H, Maier MK, Iden S, Mittag J, Friesema EC, Visser TJ, et al. The monocarboxylate transporter 8 linked to human psychomotor retardation is highly expressed in thyroid hormone-sensitive neuron populations. Endocrinology (2005) 146:1701–6. doi:10.1210/en.2004-1179
18. Ito K, Uchida Y, Ohtsuki S, Aizawa S, Kawakami H, Katsukura Y, et al. Quantitative membrane protein expression at the blood-brain barrier of adult and younger cynomolgus monkeys. J Pharm Sci (2011) 100:3939–50. doi:10.1002/jps.22487
19. Ceballos A, Belinchon MM, Sanchez-Mendoza E, Grijota-Martinez C, Dumitrescu AM, Refetoff S, et al. Importance of monocarboxylate transporter 8 for the blood-brain barrier-dependent availability of 3,5,3’-triiodo-l-thyronine. Endocrinology (2009) 150:2491–6. doi:10.1210/en.2008-1616
20. Dumitrescu AM, Liao XH, Weiss RE, Millen K, Refetoff S. Tissue-specific thyroid hormone deprivation and excess in monocarboxylate transporter (mct) 8-deficient mice. Endocrinology (2006) 147:4036–43. doi:10.1210/en.2006-0390
21. Trajkovic M, Visser TJ, Mittag J, Horn S, Lukas J, Darras VM, et al. Abnormal thyroid hormone metabolism in mice lacking the monocarboxylate transporter 8. J Clin Invest (2007) 117:627–35. doi:10.1172/JCI28253
22. Morte B, Ceballos A, Diez D, Grijota-Martinez C, Dumitrescu AM, Di Cosmo C, et al. Thyroid hormone-regulated mouse cerebral cortex genes are differentially dependent on the source of the hormone: a study in monocarboxylate transporter-8- and deiodinase-2-deficient mice. Endocrinology (2010) 151:2381–7. doi:10.1210/en.2009-0944
23. Mayerl S, Muller J, Bauer R, Richert S, Kassmann CM, Darras VM, et al. Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J Clin Invest (2014) 124:1987–99. doi:10.1172/JCI70324
24. Galton VA, Wood ET, St Germain EA, Withrow CA, Aldrich G, St Germain GM, et al. Thyroid hormone homeostasis and action in the type 2 deiodinase-deficient rodent brain during development. Endocrinology (2007) 148:3080–8. doi:10.1210/en.2006-1727
25. Crantz FR, Silva JE, Larsen PR. An analysis of the sources and quantity of 3,5,3’-triiodothyronine specifically bound to nuclear receptors in rat cerebral cortex and cerebellum. Endocrinology (1982) 110:367–75. doi:10.1210/endo-110-2-367
26. Hernandez A, Morte B, Belinchon MM, Ceballos A, Bernal J. Critical role of types 2 and 3 deiodinases in the negative regulation of gene expression by T(3)in the mouse cerebral cortex. Endocrinology (2012) 153:2919–28. doi:10.1210/en.2011-1905
27. Bernal J, Pekonen F. Ontogenesis of the nuclear 3,5,3’-triiodothyronine receptor in the human fetal brain. Endocrinology (1984) 114:677–9. doi:10.1210/endo-114-2-677
28. Kester MH, Martinez de Mena R, Obregon MJ, Marinkovic D, Howatson A, Visser TJ, et al. Iodothyronine levels in the human developing brain: major regulatory roles of iodothyronine deiodinases in different areas. J Clin Endocrinol Metab (2004) 89:3117–28. doi:10.1210/jc.2003-031832
29. Calvo R, Obregon MJ, Ruiz de Ona C, Escobar del Rey F, Morreale de Escobar G. Congenital hypothyroidism, as studied in rats. Crucial role of maternal thyroxine but not of 3,5,3’-triiodothyronine in the protection of the fetal brain. J Clin Invest (1990) 86:889–99. doi:10.1172/JCI114790
30. Grijota-Martinez C, Diez D, Morreale de Escobar G, Bernal J, Morte B. Lack of action of exogenously administered T3 on the fetal rat brain despite expression of the monocarboxylate transporter 8. Endocrinology (2011) 152(4):1713–21. doi:10.1210/en.2010-1014
31. Ruiz de Ona C, Obregon MJ, Escobar del Rey F, Morreale de Escobar G. Developmental changes in rat brain 5’-deiodinase and thyroid hormones during the fetal period: the effects of fetal hypothyroidism and maternal thyroid hormones. Pediatr Res (1988) 24:588–94. doi:10.1203/00006450-198811000-00010
32. Ferrara AM, Liao XH, Gil-Ibanez P, Marcinkowski T, Bernal J, Weiss RE, et al. Changes in thyroid status during perinatal development of MCT8-deficient male mice. Endocrinology (2013) 154:2533–41. doi:10.1210/en.2012-2031
33. Núñez B, Martínez de Mena R, Obregon MJ, Font-Llitjós M, Nunes V, Palacín M, et al. Cerebral cortex hyperthyroidism of newborn Mct8-deficient mice transiently suppressed by Lat2 inactivation. Plos One (2014) 9(5):e96915. doi:10.1371/journal.pone.0096915
34. Muller J, Mayerl S, Visser TJ, Darras VM, Boelen A, Frappart L, et al. Tissue-specific alterations in thyroid hormone homeostasis in combined Mct10 and Mct8 deficiency. Endocrinology (2014) 155:315–25. doi:10.1210/en.2013-1800
35. Hernandez A. Structure and function of the type 3 deiodinase gene. Thyroid (2005) 15:865–74. doi:10.1089/thy.2005.15.865
36. Kaplan MM, Yaskoski KA. Maturational patterns of iodothyronine phenolic and tyrosyl ring deiodinase activities in rat cerebrum, cerebellum, and hypothalamus. J Clin Invest (1981) 67:1208–14. doi:10.1172/JCI110136
37. Galton VA, Martinez E, Hernandez A, St Germain EA, Bates JM, St Germain DL. Pregnant rat uterus expresses high levels of the type 3 iodothyronine deiodinase. J Clin Invest (1999) 103:979–87. doi:10.1172/JCI6073
38. Tu HM, Legradi G, Bartha T, Salvatore D, Lechan RM, Larsen PR. Regional expression of the type 3 iodothyronine deiodinase messenger ribonucleic acid in the rat central nervous system and its regulation by thyroid hormone. Endocrinology (1999) 140:784–90. doi:10.1210/endo.140.2.6486
39. Escamez MJ, Guadano-Ferraz A, Cuadrado A, Bernal J. Type 3 iodothyronine deiodinase is selectively expressed in areas related to sexual differentiation in the newborn rat brain. Endocrinology (1999) 140:5443–6. doi:10.1210/endo.140.11.7244
40. Hernandez A, Martinez ME, Fiering S, Galton VA, St Germain D. Type 3 deiodinase is critical for the maturation and function of the thyroid axis. J Clin Invest (2006) 116:476–84. doi:10.1172/JCI26240
41. Hernandez A, Quignodon L, Martinez ME, Flamant F, St Germain DL. Type 3 deiodinase deficiency causes spatial and temporal alterations in brain T3 signaling that are dissociated from serum thyroid hormone levels. Endocrinology (2010) 151:5550–8. doi:10.1210/en.2010-0450
42. Gereben B, Zavacki AM, Ribich S, Kim BW, Huang SA, Simonides WS, et al. Cellular and molecular basis of deiodinase-regulated thyroid hormone signaling. Endocr Rev (2008) 29:898–938. doi:10.1210/er.2008-0019
43. Baqui M, Botero D, Gereben B, Curcio C, Harney JW, Salvatore D, et al. Human type 3 iodothyronine selenodeiodinase is located in the plasma membrane and undergoes rapid internalization to endosomes. J Biol Chem (2003) 278:1206–11. doi:10.1074/jbc.M210266200
44. Jansen J, Friesema EC, Kester MH, Schwartz CE, Visser TJ. Genotype-phenotype relationship in patients with mutations in thyroid hormone transporter MCT8. Endocrinology (2008) 149:2184–90. doi:10.1210/en.2007-1475
45. Obregon MJ, Ruiz de Ona C, Calvo R, Escobar del Rey F, Morreale de Escobar G. Outer ring iodothyronine deiodinases and thyroid hormone economy: responses to iodine deficiency in the rat fetus and neonate. Endocrinology (1991) 129:2663–73. doi:10.1210/endo-129-5-2663
46. Bushong EA, Martone ME, Ellisman MH. Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. Int J Dev Neurosci (2004) 22:73–86. doi:10.1016/j.ijdevneu.2003.12.008
Keywords: thyroid hormone, type 2 deiodinase, astrocytes, fetal and postnatal brain, thyroid hormone transporters, T3 availability
Citation: Morte B and Bernal J (2014) Thyroid hormone action: astrocyte–neuron communication. Front. Endocrinol. 5:82. doi: 10.3389/fendo.2014.00082
Received: 01 April 2014; Paper pending published: 22 April 2014;
Accepted: 16 May 2014; Published online: 30 May 2014.
Edited by:
Noriyuki Koibuchi, Gunma University Graduate School of Medicine, JapanReviewed by:
Veerle M. Darras, Katholieke Universiteit Leuven, BelgiumHeike Heuer, Leibniz Institute for Age Research-Fritz Lipmann Institute, Germany
Copyright: © 2014 Morte and Bernal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beatriz Morte and Juan Bernal, Instituto de Investigaciones Biomédicas “Alberto Sols”, Consejo Superior de Investigaciones Científicas, Center for Biomedical Research on Rare Diseases (CIBERER), Universidad Autónoma de Madrid, Arturo Duperier 4, Madrid 28029, Spain e-mail:Ym1vcnRlQGlpYi51YW0uZXM=;amJlcm5hbEBpaWIudWFtLmVz