 Beina Teng
Beina Teng Michelle Duong
Michelle Duong Irini Tossidou
Irini Tossidou Xuejiao Yu
Xuejiao Yu Mario Schiffer
Mario Schiffer- Department of Medicine/Nephrology, Hannover Medical School, Hannover, Germany
The early glomerular changes in diabetes include a podocyte phenotype with loss of slit diaphragm proteins, changes in the actin cytoskeleton and foot process architecture. This review focuses on the role of the protein kinase C (PKC) family in podocytes and points out the differential roles of classical, novel, and atypical PKCs in podocytes. Some PKC isoforms are indispensable for proper glomerular development and slit diaphragm maintenance, whereas others might be harmful when activated in the diabetic milieu. Therefore, some might be interesting treatment targets in the early phase of diabetes.
Introduction
Diabetic nephropathy (DN) is the most common cause of the end-stage renal disease (ESRD) (1). The glomerular changes in DN are characterized by excessive accumulation of extracellular matrix (ECM) with thickening of glomerular and tubular basement membranes and mesangial expansion, which ultimately progress primarily to glomerulosclerosis and secondarily to tubulointerstitial fibrosis. The various cell types involved include glomerular endothelial cells, mesangial cells, podocytes, and tubular epithelia, which are all targets of hyperglycemic injury. However, accumulating evidence suggests that the extent of injury and loss of podocytes is a major prognostic determinant in both, type I and type II DN (2–6). As the terminally differentiated podocytes are believed to play a critical role in maintaining the integrity of the glomerular filtration barrier, effaced podocytes may contribute to the development of albuminuria, a hallmark of DN. Although primarily structure proteins were thought to be the key elements that compose the slit diaphragm initially, it has become clear that the slit diaphragm protein complex is a highly dynamic functional protein complex and is able to initiate cascades of signaling pathways that affect podocyte function (7). More recent data indicate that podocytes express receptors for many circulating hormones and growth factors, which also suggest that a more complex cross-talk between the kidney and other organs affected by diabetes may occur in health and disease (8).
Among various signaling kinases, the protein kinase C (PKC) family seems to play a critical role in the pathogenesis of DN (9). The activation of PKCs in the kidney is a well-known pathway of the diabetic milieu. The PKC family is involved in a variety of signal transduction pathways, cell proliferation, differentiation, cell cycle, and apoptosis. However, the role of PKCs on podocytes in DN has not yet been fully defined. This present review will give an overview of the general role of PKCs and summarize the recent research into the regulatory role of PKCs in podocytes under diabetic conditions.
PKC Subfamilies and Isoforms
All PKC isoforms contain a highly conserved catalytic domain and a regulatory domain. The catalytic domain consists of several motifs and is essential for the ATP/substrate-binding and catalysis, whereas the N-terminal regulatory domain contains an auto inhibitory pseudo-substrate domain and two discrete membrane targeting modules, C1 and C2. The sequence of pseudo-substrate contains an alanine in place of the serine/threonine phospho-acceptor site, but otherwise resembles a PKC substrate (10).
Protein kinase C isoforms are subdivided into three subfamilies based on differences of structure in their N-terminal regulatory domain. The isoforms α, βI, βII, and γ belong to conventional PKCs (cPKCs). The regulatory domains of cPKCs contain a C1 domain that functions as diacylglycerol (DAG)/phorbol 12-myristat 13-acetat (PMA) binding motif and a C2 domain that binds anionic phospholipids in a calcium-dependent manner (10). The novel PKCs (nPKCsδ, ε, η, and θ) also have two C1 domains and a C2 domain. The nPKC C2 domains lack the critical calcium-coordinating acidic residues, which is the distinct difference between cPKC and nPKC. The nPKCs can be maximally activated by agonists that promote DAG accumulation or by PMA, but they are insensitive to calcium. Atypical PKCs (aPKCs, ζ, and λ/ι) lack a calcium-sensitive C2 domain; however, they contain an atypical C1 domain, which binds PIP3 or ceramide, but not DAG or PMA. The activity of aPKCs is primarily regulated by protein–protein interaction and phosphorylation catalyzed by phosphoinositide-dependent kinase-1 (PDK-1). Although a few PKC isoforms are expressed in a tissue-specific manner, most are ubiquitously expressed.
Regulation of PKCs
Unless it is post-translationally or co-translationally phosphorylated, PKCs are incapable of being activated by DAG or other cofactors (11, 12). PKC is translated as a catalytically inactive protein, is converted into an active enzyme by an initial phosphate addition and then into a mature form by further phosphorylation (11, 13, 14). In addition to phosphorylation on serine and threonine residues, PKCs also undergo phosphorylation by tyrosine kinases. PKCs are regulated by two sequential and equally critical mechanisms: phosphorylation triggered by PDK-1 and binding to DAG and/or other cofactors such as phosphatidylserine (PS) or phorbol ester (PE). Each mechanism regulates the structure, subcellular localization, and function of PKCs (15).
As a consequence of increased glycolytic flux, chronic hyperglycemia elevates the de novo synthesis of DAG and thus leads to an increased activation of DAG dependent classic and novel PKC isoforms in cultured bovine aortic endothelial cells and smooth muscle cells (16). Furthermore, high-glucose induced cellular levels lead to an increased generation of advanced glycation end products (AGEs), which initiate several signaling events by activating PKC, MAP kinase, and transcription factors such as nuclear factor-κB (NF-κB). This would increase the activity of various growth factors, such as TGF-β, and thereby alter expression of ECM proteins (17). In addition, under high-glucose conditions PKC is activated by higher concentrations of reactive oxygen species (ROS) generated following AGE:RAGE (AGE receptor) interactions (15, 18). In turn, the ROS are generated via NADPH-oxidase activated by PKC, ROS/PKC therefore can act in a cyclical manner to activate one another (19).
Conventional PKCs in Diabetic Nephropathy
Among all the PKC isoforms, the role of PKCα in the pathogenesis of DN has been investigated intensively, and several studies have demonstrated that PKCα-deficient mice show a better outcome after streptozotocin (STZ) induced diabetes with less proteinuria and preserved nephrin expression (20, 21). Studies from our group underline the involvement of PKCα in proteinuria development in DN. The expression of PKCα in podocytes of patients with DN was increased. Mice were treated after STZ-induced diabetes with the synthetic PKCα inhibitor (GÖ6976), which prevented proteinuria development and led to preserved nephrin expression. Furthermore, we could show a central role for PKCα in endocytosis of the slit diaphragm component nephrin (22, 23). Quack et al. further concluded that proteinuria of diabetic mice is as a result of increased endocytosis of nephrin, which is mediated by a complex consisting of PKCα, protein interacting with c kinase-1 (PICK1), and beta-arrestin2. They found rising glucose levels go along with increased binding of beta-arrestin to nephrin in vitro as well as in in vivo, but only with preceding PKCα phosphorylating activity on nephrin. This fact and the indispensability of PICK1 suggest PKCα and PICK1 as possible drug targets in early stages of DN (23). These studies show that expression of PKCα is regulated by glucose concentration in the external milieu of the podocyte and that PKCα is directly involved in the maintenance of the slit diaphragm. High levels of PKCα in podocytes led to enhanced endocytosis of nephrin and instability of the slit diaphragm.
In addition, Menne and colleagues demonstrated that the high-glucose induced downregulation of nephrin is probably caused by the PKCα mediated reduction of the transcriptional factor Wilms tumor 1 (WT-1), which has previously been described as a directly interacting binding partner of the nephrin promoter (24, 25). These findings are consistent with earlier studies, suggesting one of the PKC isoforms might be pivotal for the regulation of nephrin transcription and expression in podocytes (26), but not for CD2AP and Podocin as they remain at levels similar to those in non-diabetic kidneys (27).
Langham et al. showed that in diabetic patients treated with perindopril, an ACE inhibitor, nephrin levels preserved resembling those of non-diabetic subjects (28) and subsequently could be one factor contributing to the anti-proteinuric effects of ACE inhibitors. Another study suggests that the AGEs, which can be inhibited by aminoguanidine, are also implicated in the downregulation of nephrin in diabetes (29). Interestingly, several groups could demonstrate the reduction of PKC activity under the treatment with ACE inhibitors and aminoguanidine in diabetic subjects that could explain the nephrin protective effects (30, 31).
Most previous studies with specific PKCβ inhibitor ruboxistaurin (LX333531) in vivo and in vitro indicated that PKCβ isoform is primarily responsible for the high-glucose-induced renal effects in diabetes (32–36). Meier et al. tested this hypothesis by inducing DN in PKCβ deficient mice and did not find a significant preventive effect of PKCβ deficiency on albuminuria. In contrast to non-albuminuric diabetic PKCα−/− mice, the loss of the basement membrane proteoglycan perlecan and the podocyte slit diaphragm protein nephrin were not prevented in the PKCβ−/− mice under diabetic conditions (20, 37). However, the hyperglycemia-induced renal and glomerular hypertrophy as well as increased expression of ECM proteins was reduced in PKCβ deficiency diabetic mice.
In summary, the two important physiological features of DN, renal hypertrophy and albuminuria, are regulated through different PKC isoforms; PKCα is involved in the development of albuminuria and maintenance the glomerular filtration barrier structure, whereas the PKCβ-isoform contributes to hyperglycemia-induced renal fibrosis.
Another study by Menne et al. combined the findings about PKCα and PKCβ and demonstrated that a dual inhibition of both isoforms has a synergistic effect and is capable of preventing the development of experimental DN in streptozocin-induced diabetic mice (38). Blocking both isoforms has a beneficial effect on the development of renal hypertrophy and albuminuria in mice after 8 weeks of diabetes. A pharmacological approach with CGP41252, an inhibitor of PKCα and PKCβ showed that the occurrence of albuminuria could be avoided and preexisting albuminuria could be diminished in both type I and type II diabetic mice (38). But the treatment had little impact on the development of renal hypertrophy. Higher doses treatment also increased mortality.
Essential Role of Novel and Atypical PKCs
Only little is known about PKCε in renal function, especially about its role in podocytes, although several previous studies showed increased expression and activation of PKCε isoform in experimental DN (39, 40). Meier et al. investigated the functional role of PKCε in renal physiology using PKCε-knockout mice and found a renal phenotype with an elevated occurrence of tubulointerstitial fibrosis and glomerulosclerosis, whereas a systemic profibrotic phenotype was not observed (41). Moreover, they demonstrated an increased level of albuminuria in knockout mice whereas the kidney/body ratio remained normal in comparison to non-diabetic wild type mice, indicating that PKCε is probably less implicated in development of renal hypertrophy. Experiments on diabetic mice further showed that knockout of PKCε can exacerbate the renal phenotype with a significantly increased urinary albumin/creatinine ratio and expression of ECM proteins. These data suggest that a rising level of PKCε has a protective function in kidney injury, rather than inducing profibrotic changes.
The atypical isoforms PKCλ and PKCι are both highly expressed in podocytes. Although it is not clear whether both isotypes perform distinct roles in the cell, aPKCs are well-known to be important in the establishment of cell polarity (42, 43). They form an evolutionarily conserved complex consisting of aPKC and PAR3 and PAR6, two PDZ domain containing scaffold proteins (44). Studies showed that the Par3–Par6–aPKC complex interacts with nephrin–podocin through the direct connection of Par3 to nephrin, an essential structural component for the maintenance of integrity of the glomerular filter as well as for the signal transduction (45). Interestingly, our own studies of the glomerular development in regard to the Par polarity complex and slit diaphragm molecules show that the PAR3–PAR6–aPKCλ complex translocates together with other tight junction proteins like ZO-1 from the apical to the basolateral side of the cell preceding the targeting of slit diaphragm components such as nephrin and podocin to the basal membrane, development of foot processes, and the construction of slit diaphragms (46).
The very recent study of Satoh and colleagues shows the necessity of aPKC in the exocytosis of newly produced nephrin and its localization on the surface of podocytes (47). This finding could also partially explain the diminished nephrin expression in proteinuria patients with diabetes (48). Under high-glucose conditions, activation of aPKC presents a protective effect on the nephrin expression (48).
The activation of aPKC is required for the insulin-induced glucose transport and thus defective aPKC activation in muscle and adipocytes has been shown previously in type II diabetic rats, monkeys, and human beings, which leads to a disturbed glucose uptake into muscle and the whole body glucose transfer (49). The knockout of the other isoform PKCζ has no specific renal phenotype but seems to be able compensate for partial functions of aPKCλ loss as the double-knockout leads to glomerular developmental defects with no secondary foot processes (50). This is most likely because of the regulatory cytoskeletal functions of aPKCs with direct influence on small GTPase activation of aPKCs in podocytes (51). Nevertheless, the role of aPKC in DN still remains unclear.
PKC Up-Regulated Growth Factor Expression in Diabetic Nephropathy
Podocytes are the major site of vascular endothelial growth factor (VEGF) production in the human kidney (52), and the expression of VEGF is increased in podocytes in diabetic rats and human beings (53, 54). Thus, the up-regulation of VEGF plays a critical role in the progression of DN. Hoshi et al. have shown that under high-glucose conditions, VEGF expression was up-regulated mediated through activation of PKC and extracellular signal-regulated kinase (ERK) in podocytes (55). PKC and ERK are known to regulate activator protein-1 (AP-1) activation (56–58), which promotes the binding of AP-1 to the promoter region of the VEGF gene (59, 60). In addition, AGEs upregulate VEGF mRNA levels through transcription factors such as NF-κB and AP-1. AGEs are also able to activate PKCs, which further increase the expression of VEGF. Although VEGF is required for normal glomerulogenesis and essential for maintenance of glomerular filtration barrier, podocyte-specific overexpression of VEGF164 or VEGF165 isoform in animals leads to structural and functional renal changes similar to those abnormalities seen in DN, including proteinuria, glomerular hypertrophy, glomerular basement membrane thickening, mesangial expansion, loss of slit diaphragms, and podocyte effacement (61, 62). In DN, the activation of TGF-β1 has been demonstrated to promote podocyte apoptosis and the development of glomerulosclerosis. A reduced expression level of the profibrotic cytokine TGF-β1 was detected in diabetic PKCβ−/− mice, while the alteration was not observed in diabetic PKCα−/− mice (20). This observation suggested that PKCβ isoform is more important in the up-regulation of TGF-β and for the development of glomerular fibrosis under diabetic conditions, whereas PKCα shows its critical role in the integrity of glomerular filtration barrier (37). However, PKCα plays a key role in the signaling response after stimulation with TGF-β1. An enhanced and prolonged activation of PI3K/AKT and ERK1/2 as well as a reduced proapoptotic signaling via p38MAPK in PKCα-knockout podocytes compared to wild type podocytes was detected after TGF-β1 treatment, which indicated the involvement of PKCα in the TGF-β mediated apoptosis (63).
Of note, deletion of PKCε signaling not only leads to increased expression of TGF-β1 but also induces activation of the TGF-β1 signaling pathway in glomeruli (41). PKCι might also mediate a high-glucose-induced increase in TGF-β receptor II (TGF-βRII) promoter activity, which leads to the up-regulation of TGF-βRII and fibronectin (64).
PKC Regulated Structure Proteins in Podocytes
P-cadherin as member of the classical cadherins, a superfamily of glycoproteins, is known to be a basic scaffold for the glomerular slit diaphragm (65). Xu et al. have first demonstrated decreased expression of P-cadherin mRNA and protein in experimental diabetic glomeruli and in high-glucose stimulated podocytes, which suggests that a potential role for P-cadherin loss in the development of proteinuria in early DN (66). They also found that PKC inhibitors could ameliorate the decrement of P-cadherin in podocytes under high-glucose conditions. Thus, activation of PKC regulated pathways seems to be involved in the regulation of P-cadherin expression and contributes to the disruption of podocyte integrity (66).
Nevertheless, it seems likely that the molecular changes of the slit diaphragm complex, but not one single slit diaphragm-associated molecule contribute to the pathogenesis of glomerular filtration barrier in DN. P-cadherin, α-, β-, and γ-catenin, and ZO-1 are described to compose adherens junctions at the slit diaphragm and establish the link with the actin cytoskeleton (65, 67). However, the role and regulation of β-catenin with P-cadherin and actin cytoskeletal proteins have not been thoroughly explored. In the situation of the podocyte injury induced by high glucose, β-catenin will be released from the destruction complex because of the activation of the Wnt-pathway. Afterwards β-catenin translocates into the nucleus to activate the downstream genes via the aggregation of a transcriptional complex with TCF/LEF, which leads to proteinuria and glomerulosclerosis (68, 69). Several groups demonstrated that PKC activation phosphorylated N-terminal serine residues of β-catenin, which promoted β-catenin degradation (70, 71). In contrast, our ongoing work was focused on several PKC isoforms, whose activation is able to dephosphorylate β-catenin to prevent β-catenin from degradation. It is still unknown, whether this process is protective for the high-glucose-induced cell adherens junction reduction. Nevertheless, PKC activation may be a novel mechanism in regulating β-catenin in glomerular injury, which will be further investigated by our laboratory.
Conclusion
Podocyte injury or podocyte loss is a hallmark for the pathogenesis of DN. Based on the above described findings, PKC activation seems to be the most critical pathway involved in the progression of glomerular injury. Obviously, there is a fine balance between activation and inactivation of the different PKC isoforms and their cross-talk involving foot process cytoskeletal architecture, cellular junction formation, and orchestration of turnover and surface expression of slit diaphragm components (Figure 1). Therefore, PKCs and their involved pathways are potential therapeutic targets in podocytes to prevent the progression of diabetic glomerulopathy.
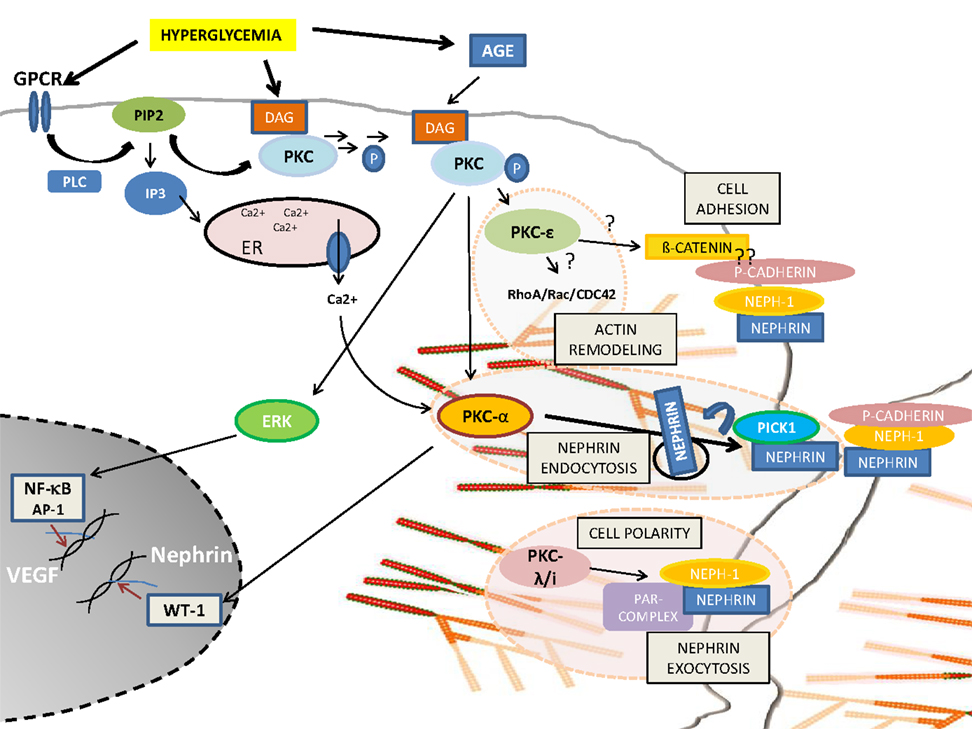
Figure 1. Schematic overview of PKC-isoform functions in podocytes. DAG or calcium activated PKCα induces nephrin endocytosis via the adaptor molecule Pick1. PKCλ/ι is required for foot process formation, cell polarity, and nephrin exocytosis. PKCε might be involved in actin remodeling via small GTPases and might orchestrate cell adhesion and cell–cell contact formation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Fioretto P, Caramori ML, Mauer M. The kidney in diabetes: dynamic pathways of injury and repair. The Camillo Golgi Lecture 2007. Diabetologia (2008) 51:1347–55. doi: 10.1007/s00125-008-1051-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Meyer TW, Bennett PH, Nelson RG. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with type II diabetes and microalbuminuria. Diabetologia (1999) 42:1341–4. doi:10.1007/s001250051447
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes (2006) 55:225–33. doi:10.2337/diabetes.55.01.06.db05-0894
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Toyoda M, Najafian B, Kim Y, Caramori ML, Mauer M. Podocyte detachment and reduced glomerular capillary endothelial fenestration in human type 1 diabetic nephropathy. Diabetes (2007) 56:2155–60. doi:10.2337/db07-0019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Verzola D, Gandolfo MT, Ferrario F, Rastaldi MP, Villaggio B, Gianiorio F, et al. Apoptosis in the kidneys of patients with type II diabetic nephropathy. Kidney Int (2007) 72:1262–72. doi:10.1038/sj.ki.5002531
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. White KE, Bilous RW, Marshall SM, El NM, Remuzzi G, Piras G, et al. Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes (2002) 51:3083–9. doi:10.2337/diabetes.51.10.3083
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Benzing T. Signaling at the slit diaphragm. J Am Soc Nephrol (2004) 15:1382–91. doi:10.1097/01.ASN.0000130167.30769.55
8. Diez-Sampedro A, Lenz O, Fornoni A. Podocytopathy in diabetes: a metabolic and endocrine disorder. Am J Kidney Dis (2011) 58:637–46. doi:10.1053/j.ajkd.2011.03.035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Li J, Gobe G. Protein kinase C activation and its role in kidney disease. Nephrology (Carlton) (2006) 11:428–34. doi:10.1111/j.1440-1797.2006.00673.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev (2008) 88:1341–78. doi:10.1152/physrev.00034.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Pears C, Stabel S, Cazaubon S, Parker PJ. Studies on the phosphorylation of protein kinase C-alpha. Biochem J (1992) 283(Pt 2):515–8.
12. Dietrich A, Rose-John S, Marks F. Expression of the kinase domain of mouse protein kinase C in E. coli. Biochem Int (1989) 19:163–72.
13. Cazaubon SM, Parker PJ. Identification of the phosphorylated region responsible for the permissive activation of protein kinase C. J Biol Chem (1993) 268:17559–63.
14. Borner C, Filipuzzi I, Wartmann M, Eppenberger U, Fabbro D. Biosynthesis and posttranslational modifications of protein kinase C in human breast cancer cells. J Biol Chem (1989) 264:13902–9.
15. Liu WS, Heckman CA. The sevenfold way of PKC regulation. Cell Signal (1998) 10:529–42. doi:10.1016/S0898-6568(98)00012-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Inoguchi T, Battan R, Handler E, Sportsman JR, Heath W, King GL. Preferential elevation of protein kinase C isoform beta II and diacylglycerol levels in the aorta and heart of diabetic rats: differential reversibility to glycemic control by islet cell transplantation. Proc Natl Acad Sci U S A (1992) 89:11059–63. doi:10.1073/pnas.89.22.11059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Jakus V, Rietbrock N. Advanced glycation end-products and the progress of diabetic vascular complications. Physiol Res (2004) 53:131–42.
18. Meinhardt G, Roth J, Totok G. Protein kinase C activation modulates pro- and anti-apoptotic signaling pathways. Eur J Cell Biol (2000) 79:824–33. doi:10.1078/0171-9335-00100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Lee HB, Yu MR, Yang Y, Jiang Z, Ha H. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J Am Soc Nephrol (2003) 14:S241–5. doi:10.1097/01.ASN.0000077410.66390.0F
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Menne J, Park JK, Boehne M, Elger M, Lindschau C, Kirsch T, et al. Diminished loss of proteoglycans and lack of albuminuria in protein kinase C-alpha-deficient diabetic mice. Diabetes (2004) 53:2101–9. doi:10.2337/diabetes.53.8.2101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Menne J, Meier M, Park JK, Boehne M, Kirsch T, Lindschau C, et al. Nephrin loss in experimental diabetic nephropathy is prevented by deletion of protein kinase C alpha signaling in-vivo. Kidney Int (2006) 70:1456–62. doi:10.1038/sj.ki.5001830
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Tossidou I, Teng B, Menne J, Shushakova N, Park JK, Becker JU, et al. Podocytic PKC-alpha is regulated in murine and human diabetes and mediates nephrin endocytosis. PLoS One (2010) 5:e10185. doi:10.1371/journal.pone.0010185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Quack I, Woznowski M, Potthoff SA, Palmer R, Konigshausen E, Sivritas S, et al. PKC alpha mediates beta-arrestin2-dependent nephrin endocytosis in hyperglycemia. J Biol Chem (2011) 286:12959–70. doi:10.1074/jbc.M110.204024
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Guo G, Morrison DJ, Licht JD, Quaggin SE. WT1 activates a glomerular-specific enhancer identified from the human nephrin gene. J Am Soc Nephrol (2004) 15:2851–6. doi:10.1097/01.ASN.0000143474.91362.C4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Wagner N, Wagner KD, Xing Y, Scholz H, Schedl A. The major podocyte protein nephrin is transcriptionally activated by the Wilms’ tumor suppressor WT1. J Am Soc Nephrol (2004) 15:3044–51. doi:10.1097/01.ASN.0000146687.99058.25
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Wang SX, Mene P, Holthofer H. Nephrin mRNA regulation by protein kinase C. J Nephrol (2001) 14:98–103.
27. Benigni A, Gagliardini E, Tomasoni S, Abbate M, Ruggenenti P, Kalluri R, et al. Selective impairment of gene expression and assembly of nephrin in human diabetic nephropathy. Kidney Int (2004) 65:2193–200. doi:10.1111/j.1523-1755.2004.00636.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Langham RG, Kelly DJ, Cox AJ, Thomson NM, Holthofer H, Zaoui P, et al. Proteinuria and the expression of the podocyte slit diaphragm protein, nephrin, in diabetic nephropathy: effects of angiotensin converting enzyme inhibition. Diabetologia (2002) 45:1572–6. doi:10.1007/s00125-002-0946-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Dempsey EC, Newton AC, Mochly-Rosen D, Fields AP, Reyland ME, Insel PA, et al. Protein kinase C isozymes and the regulation of diverse cell responses. Am J Physiol Lung Cell Mol Physiol (2000) 279:L429–38.
30. Osicka TM, Yu Y, Panagiotopoulos S, Clavant SP, Kiriazis Z, Pike RN, et al. Prevention of albuminuria by aminoguanidine or ramipril in streptozotocin-induced diabetic rats is associated with the normalization of glomerular protein kinase C. Diabetes (2000) 49:87–93. doi:10.2337/diabetes.49.1.87
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Thallas-Bonke V, Lindschau C, Rizkalla B, Bach LA, Boner G, Meier M, et al. Attenuation of extracellular matrix accumulation in diabetic nephropathy by the advanced glycation end product cross-link breaker ALT-711 via a protein kinase C-alpha-dependent pathway. Diabetes (2004) 53:2921–30. doi:10.2337/diabetes.53.11.2921
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Meier M, King GL. Protein kinase C activation and its pharmacological inhibition in vascular disease. Vasc Med (2000) 5:173–85. doi:10.1177/1358836X0000500307
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Ishii H, Jirousek MR, Koya D, Takagi C, Xia P, Clermont A, et al. Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science (1996) 272:728–31. doi:10.1126/science.272.5262.728
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Koya D, Jirousek MR, Lin YW, Ishii H, Kuboki K, King GL. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J Clin Invest (1997) 100:115–26. doi:10.1172/JCI119503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Koya D, Haneda M, Nakagawa H, Isshiki K, Sato H, Maeda S, et al. Amelioration of accelerated diabetic mesangial expansion by treatment with a PKC beta inhibitor in diabetic db/db mice, a rodent model for type 2 diabetes. FASEB J (2000) 14:439–47.
36. Kelly DJ, Zhang Y, Hepper C, Gow RM, Jaworski K, Kemp BE, et al. Protein kinase C beta inhibition attenuates the progression of experimental diabetic nephropathy in the presence of continued hypertension. Diabetes (2003) 52:512–8. doi:10.2337/diabetes.52.2.512
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Meier M, Park JK, Overheu D, Kirsch T, Lindschau C, Gueler F, et al. Deletion of protein kinase C-beta isoform in vivo reduces renal hypertrophy but not albuminuria in the streptozotocin-induced diabetic mouse model. Diabetes (2007) 56:346–54. doi:10.2337/db06-0891
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Menne J, Shushakova N, Bartels J, Kiyan Y, Laudeley R, Haller H, et al. Dual inhibition of classical protein kinase C-alpha and protein kinase C-beta isoforms protects against experimental murine diabetic nephropathy. Diabetes (2013) 62:1167–74. doi:10.2337/db12-0534
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Kang N, Alexander G, Park JK, Maasch C, Buchwalow I, Luft FC, et al. Differential expression of protein kinase C isoforms in streptozotocin-induced diabetic rats. Kidney Int (1999) 56:1737–50. doi:10.1046/j.1523-1755.1999.00725.x
40. Whiteside CI, Dlugosz JA. Mesangial cell protein kinase C isozyme activation in the diabetic milieu. Am J Physiol Renal Physiol (2002) 282:F975–80. doi:10.1152/ajprenal.00014.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Meier M, Menne J, Park JK, Holtz M, Gueler F, Kirsch T, et al. Deletion of protein kinase C-epsilon signaling pathway induces glomerulosclerosis and tubulointerstitial fibrosis in vivo. J Am Soc Nephrol (2007) 18:1190–8. doi:10.1681/ASN.2005070694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Henrique D, Schweisguth F. Cell polarity: the ups and downs of the Par6/aPKC complex. Curr Opin Genet Dev (2003) 13:341–50. doi:10.1016/S0959-437X(03)00077-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Macara IG. Par proteins: partners in polarization. Curr Biol (2004) 14:R160–2. doi:10.1016/j.cub.2004.01.048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Suzuki A, Yamanaka T, Hirose T, Manabe N, Mizuno K, Shimizu M, et al. Atypical protein kinase C is involved in the evolutionarily conserved par protein complex and plays a critical role in establishing epithelia-specific junctional structures. J Cell Biol (2001) 152:1183–96. doi:10.1083/jcb.152.6.1183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Hartleben B, Schweizer H, Lubben P, Bartram MP, Moller CC, Herr R, et al. Neph-Nephrin proteins bind the Par3-Par6-atypical protein kinase C (aPKC) complex to regulate podocyte cell polarity. J Biol Chem (2008) 283:23033–8. doi:10.1074/jbc.M803143200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Huber TB, Hartleben B, Winkelmann K, Schneider L, Becker JU, Leitges M, et al. Loss of podocyte aPKClambda/iota causes polarity defects and nephrotic syndrome. J Am Soc Nephrol (2009) 20:798–806. doi:10.1681/ASN.2008080871
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Satoh D, Hirose T, Harita Y, Daimon C, Harada T, Kurihara H, et al. aPKClambda maintains the integrity of the glomerular slit diaphragm through trafficking of nephrin to the cell surface. J Biochem (2014) 156:115–28. doi:10.1093/jb/mvu022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Doublier S, Salvidio G, Lupia E, Ruotsalainen V, Verzola D, Deferrari G, et al. Nephrin expression is reduced in human diabetic nephropathy: evidence for a distinct role for glycated albumin and angiotensin II. Diabetes (2003) 52:1023–30. doi:10.2337/diabetes.52.4.1023
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Sajan MP, Farese RV. Insulin signalling in hepatocytes of humans with type 2 diabetes: excessive production and activity of protein kinase C-iota (PKC-iota) and dependent processes and reversal by PKC-iota inhibitors. Diabetologia (2012) 55:1446–57. doi:10.1007/s00125-012-2477-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Hartleben B, Widmeier E, Suhm M, Worthmann K, Schell C, Helmstadter M, et al. aPKClambda/iota and aPKCzeta contribute to podocyte differentiation and glomerular maturation. J Am Soc Nephrol (2013) 24:253–67. doi:10.1681/ASN.2012060582
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Worthmann K, Leitges M, Teng B, Sestu M, Tossidou I, Samson T, et al. Def-6, a novel regulator of small GTPases in podocytes, acts downstream of atypical protein kinase C (aPKC) lambda/iota. Am J Pathol (2013) 183:1945–59. doi:10.1016/j.ajpath.2013.08.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Brown LF, Berse B, Tognazzi K, Manseau EJ, Van de Water L, Senger DR, et al. Vascular permeability factor mRNA and protein expression in human kidney. Kidney Int (1992) 42:1457–61. doi:10.1038/ki.1992.441
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Cha DR, Kim NH, Yoon JW, Jo SK, Cho WY, Kim HK, et al. Role of vascular endothelial growth factor in diabetic nephropathy. Kidney Int Suppl (2000) 77:S104–12. doi:10.1046/j.1523-1755.2000.07717.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Lee EY, Shim MS, Kim MJ, Hong SY, Shin YG, Chung CH. Angiotensin II receptor blocker attenuates overexpression of vascular endothelial growth factor in diabetic podocytes. Exp Mol Med (2004) 36:65–70. doi:10.1038/emm.2004.9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Hoshi S, Nomoto K, Kuromitsu J, Tomari S, Nagata M. High glucose induced VEGF expression via PKC and ERK in glomerular podocytes. Biochem Biophys Res Commun (2002) 290:177–84. doi:10.1006/bbrc.2001.6138
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Janknecht R, Ernst WH, Houthaeve T, Nordheim A. C-terminal phosphorylation of the serum-response factor. Eur J Biochem (1993) 216:469–75. doi:10.1111/j.1432-1033.1993.tb18165.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem (1995) 270:16483–6. doi:10.1074/jbc.270.28.16483
58. Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol (1997) 9:240–6. doi:10.1016/S0955-0674(97)80068-3
59. Shima DT, Kuroki M, Deutsch U, Ng YS, Adamis AP, D’Amore PA. The mouse gene for vascular endothelial growth factor. Genomic structure, definition of the transcriptional unit, and characterization of transcriptional and post-transcriptional regulatory sequences. J Biol Chem (1996) 271:3877–83.
60. Tischer E, Mitchell R, Hartman T, Silva M, Gospodarowicz D, Fiddes JC, et al. The human gene for vascular endothelial growth factor. Multiple protein forms are encoded through alternative exon splicing. J Biol Chem (1991) 266:11947–54.
61. Veron D, Reidy KJ, Bertuccio C, Teichman J, Villegas G, Jimenez J, et al. Overexpression of VEGF-A in podocytes of adult mice causes glomerular disease. Kidney Int (2010) 77:989–99. doi:10.1038/ki.2010.64
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Liu E, Morimoto M, Kitajima S, Koike T, Yu Y, Shiiki H, et al. Increased expression of vascular endothelial growth factor in kidney leads to progressive impairment of glomerular functions. J Am Soc Nephrol (2007) 18:2094–104. doi:10.1681/ASN.2006010075
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Tossidou I, Starker G, Kruger J, Meier M, Leitges M, Haller H, et al. PKC-alpha modulates TGF-beta signaling and impairs podocyte survival. Cell Physiol Biochem (2009) 24:627–34. doi:10.1159/000257518
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Chuang LY, Guh JY, Liu SF, Hung MY, Liao TN, Chiang TA, et al. Regulation of type II transforming-growth-factor-beta receptors by protein kinase C iota. Biochem J (2003) 375:385–93. doi:10.1042/BJ20030522
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol (2000) 11:1–8.
66. Xu ZG, Ryu DR, Yoo TH, Jung DS, Kim JJ, Kim HJ, et al. P-Cadherin is decreased in diabetic glomeruli and in glucose-stimulated podocytes in vivo and in vitro studies. Nephrol Dial Transplant (2005) 20:524–31. doi:10.1093/ndt/gfh642
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Hendriksen J, Jansen M, Brown CM, van der Velde H, van HM, Galjart N, et al. Plasma membrane recruitment of dephosphorylated beta-catenin upon activation of the Wnt pathway. J Cell Sci (2008) 121:1793–802. doi:10.1242/jcs.025536
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Kato H, Gruenwald A, Suh JH, Miner JH, Barisoni-Thomas L, Taketo MM, et al. Wnt/beta-catenin pathway in podocytes integrates cell adhesion, differentiation, and survival. J Biol Chem (2011) 286:26003–15. doi:10.1074/jbc.M111.223164
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Dai C, Stolz DB, Kiss LP, Monga SP, Holzman LB, Liu Y. Wnt/beta-catenin signaling promotes podocyte dysfunction and albuminuria. J Am Soc Nephrol (2009) 20:1997–2008. doi:10.1681/ASN.2009010019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Gwak J, Cho M, Gong SJ, Won J, Kim DE, Kim EY, et al. Protein-kinase-C-mediated beta-catenin phosphorylation negatively regulates the Wnt/beta-catenin pathway. J Cell Sci (2006) 119:4702–9. doi:10.1242/jcs.03256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Raab MS, Breitkreutz I, Tonon G, Zhang J, Hayden PJ, Nguyen T, et al. Targeting PKC: a novel role for beta-catenin in ER stress and apoptotic signaling. Blood (2009) 113:1513–21. doi:10.1182/blood-2008-05-157040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: proteinuria, diabetic podocytopathy, glomerulosclerosis, nephrin endocytosis, effacement
Citation: Teng B, Duong M, Tossidou I, Yu X and Schiffer M (2014) Role of protein kinase C in podocytes and development of glomerular damage in diabetic nephropathy. Front. Endocrinol. 5:179. doi: 10.3389/fendo.2014.00179
Received: 16 July 2014; Paper pending published: 19 September 2014;
Accepted: 06 October 2014; Published online: 05 November 2014.
Edited by:
Barbara Lewko, Medical University of Gdansk, PolandReviewed by:
Undurti Narasimha Das, UND Life Sciences, USAChris R. J. Kennedy, Ottawa Hospital Research Institute, Canada
Copyright: © 2014 Teng, Duong, Tossidou, Yu and Schiffer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mario Schiffer, Department of Medicine/Nephrology, Hannover Medical School, Carl Neuberg Street 1-OE6840, Hannover 30625, Germany e-mail:c2NoaWZmZXIubWFyaW9AbWgtaGFubm92ZXIuZGU=