 Kazunari Nohara
Kazunari Nohara Seung-Hee Yoo
Seung-Hee Yoo Zheng (Jake) Chen
Zheng (Jake) Chen- Department of Biochemistry and Molecular Biology, The University of Texas Health Science Center at Houston, Houston, TX, USA
Modernization of human society parallels an epidemic of metabolic disorders including obesity. Apart from excess caloric intake, a 24/7 lifestyle poses another important challenge to our metabolic health. Recent research under both laboratory and epidemiological settings has indicated that abnormal temporal organization of sleep and wakeful activities including food intake is a significant risk factor for metabolic disease. The circadian clock system is our intrinsic biological timer that regulates internal rhythms such as the sleep/wake cycle and also responses to external stimuli including light and food. Initially thought to be mainly involved in the timing of sleep, the clock, and/or clock genes may also play a role in sleep architecture and homeostasis. Importantly, an extensive body of evidence has firmly established a master regulatory role of the clock in energy balance. Together, a close relationship between well-timed circadian/sleep cycles and metabolic health is emerging. Exploiting this functional connection, an important holistic strategy toward curbing the epidemic of metabolic disorders (e.g., obesity) involves corrective measures on the circadian clock and sleep. In addition to behavioral and environmental interventions including meal timing and light control, pharmacological agents targeting sleep and circadian clocks promise convenient and effective applications. Recent studies, for example, have reported small molecules targeting specific clock components and displaying robust beneficial effects on sleep and metabolism. Furthermore, a group of clock-amplitude-enhancing small molecules (CEMs) identified via high-throughput chemical screens are of particular interest for future in vivo studies of their metabolic and sleep efficacies. Elucidating the functional relationship between clock, sleep, and metabolism will also have far-reaching implications for various chronic human diseases and aging.
Introduction
Our bodily functions are temporally coordinated to achieve optimal fitness by an intrinsic biological timer called the circadian clock (1). The clock has evolved in response to the daily rotation of Earth, functioning to drive cycles of metabolism, physiology, and behavior. Among the most fundamental biological cycles is the sleep/wake cycle, in part characterized by profound metabolic changes and alternating energy flux between sleep/fasting and wakefulness/feeding (2, 3). Epidemiological studies of shift workers have provided initial evidence for a greater risk of metabolic disorders correlated with circadian/sleep disruption (4, 5). Furthermore, during natural aging, reduced energy metabolism is accompanied by sleep fragmentation and dampened circadian rhythms in hormone secretion, body temperature, and circadian gene expression (6–8). Such associative evidence strongly suggests a close, reciprocal relationship among the circadian clock, sleep, and metabolism.
Outstanding questions remain concerning the integration of the clock, sleep, and metabolic health. Whereas increasing evidence has demonstrated a complex interplay of molecular mechanisms that the circadian clock employs to regulate metabolic processes (9, 10), the role of sleep in circadian metabolic regulation is less clear (11, 12). Specifically, how sleep disturbances influence energy balance is not well-understood at the molecular level. From the translational perspective, much work is required to fully exploit clock and sleep based interventions against metabolic disorders. In the current review, we will describe temporal control of energy metabolism under both physiological and pathological settings and discuss behavioral, environmental, and pharmacological strategies of targeting circadian/sleep cycles as preventive or therapeutic measures against metabolic disease (Figure 1). We primarily limit our discussion to mammalian species unless otherwise noted.
Figure 1. Biological rhythms-based intervention for metabolic benefits. Energy metabolism is regulated by both sleep and circadian cycles. Phasic misalignment (e.g., shift work) and/or dampened amplitude of daily behavior (e.g., sleep, food intake) can profoundly impair metabolic homeostasis. Therefore, as opposed to direct manipulation of metabolic regulators, interventional strategies can aim to restore normal phase and amplitude of these biological cycles in order to improve metabolic health. Such interventions may involve behavioral (e.g., meal timing) and environmental (e.g., lighting) means as well as pharmacological agents capable of correcting cycle phase and/or amplitude (e.g., clock-enhancing small molecules, or CEMs). Such rhythm-based interventions may in turn lead to metabolic improvement including favorable energy balance, reduced body weight, and enhanced insulin sensitivity.
Sleep, Circadian Clock, and Metabolism
Sleep and Circadian Cycles
Sleep is essential for health and survival (13). Sleep deprivation, depending on duration and severity, can lead to acute impairment of cognitive and physiological functions, increased risks of chronic diseases such as cardiovascular and metabolic diseases, and ultimately fatality (14–17). Despite potentially life-threatening consequences for animals in their natural habitats, sleep clearly serves a vital, evolutionarily conserved function. Mammalian sleep architecture has been characterized in detail by electroencephalograms (EEGs), consisting of rapid eye movement (REM) and non-REM sleep (18). The latter is further divided into stages 1–3, with the deepest slow-wave sleep occurring during stage 3. Sleep is traditionally considered to be regulated by two overlapping processes, the sleep drive (homeostasis, process S) and the circadian timing of sleep (rhythmic sleep propensity, process C) (2, 19). Paradoxically, very little is known about the molecular function of sleep and genetic regulation of sleep homeostasis, which is typically measured by the delta power of slow-wave sleep. Sleep has been proposed to play a role in memory consolidation and metabolic homeostasis involving removal of deleterious byproducts and/or restoration of essential metabolites (3, 18, 20). Interestingly, a recent study demonstrated β-amyloid (Aβ), a neurotoxic peptide accumulated in Alzheimer’s disease, was actively cleared from sleeping brain, highlighting a critical function of the sleep/wake cycle for metabolic detoxification (21).
Timing of the sleep/wake cycle, as well as numerous other physiological processes, is known to be regulated by the circadian clock, our intrinsic biological timer (22–24). The clock is self-sustained, but subjected to resetting by external cues including light and food (25). In mammals, light is transmitted through the retinohypothalamic tract (RHT) to the suprachiasmatic nuclei (SCN), the master pacemaker of the clock system. Through neuronal and hormonal signals such as TGF-α and prokinectin-2, the SCN synchronizes and orchestrates peripheral clocks in individual tissues throughout the body (26). The basic clock unit is the molecular oscillator, ubiquitously present in almost every cell in our body (1). The oscillator is composed of interlocked feedback loops. In the core loop, positive factors (CLOCK/NPAS2, BMAL1) drive the transcription of genes encoding the negative components CRYs and PERs (CRYPTOCHROME1/2, PERIOD1/2), which in turn heterodimerize to inhibit their own transcription. Bmal1 transcription is further regulated by competing nuclear hormone receptors including retinoid-related orphan receptors (RORs) as positive regulators and reverse-ErbA (REV-ERBs) as negative regulators in the secondary stabilization loop, ultimately generating 24-h molecular oscillation. The molecular oscillator drives expression of the so-called clock-controlled genes (CCGs) in a tissue-dependent manner, which subsequently controls metabolic, physiological, and behavioral outputs (27). Genetic studies in recent years have also provided evidence for important roles of clock genes in sleep homeostasis (22) (see below), indicating a possible interdependence of processes S and C via clock genes.
Melatonin is a pineal gland-derived hormone playing an important role at the interface of the sleep/wake cycle and the circadian clock (28). Melatonin levels display a clear circadian pattern, peaking at night to promote sleep and reaching the trough in the morning and remaining low during the day (29). In accordance, the melatonin biosynthesis pathway, including the key enzyme aralkylamine N-acetyltransferase (AANAT), has been shown to be subjected to clock control (30). Furthermore, light exposure also suppresses melatonin synthesis and secretion, mediated by the superior cervical ganglion through the SCN (31). In blind subjects devoid of light perception and exhibiting longer free-running rhythms with an average of 24.5 h, rhythmic melatonin treatment was able to entrain them to approximately 24.0-h periods and concomitantly improve their sleep amount and quality (32). These studies strongly suggest the importance of melatonin for both circadian periodicity and sleep timing, amount, and quality.
Temporal Control of Energy Metabolism
The necessity to temporally coordinate metabolic events is most apparent in photosynthetic organisms such as cyanobacteria and plants where oxygen-generating photosynthesis must be segregated from oxygen-sensitive nitrogen fixation (33). In fact, redox cycles have recently been shown to operate in diverse phylogenetic branches, presumably evolved following the great oxygenation event (GOE) approximately 2.5 billion years ago (34–36). Particularly, much evidence has recently emerged concerning temporal control of energy metabolism in mammals (37). Epidemiological studies have shown increased risk of weight gain and insulin resistance in shift workers (4, 5, 38). In accordance, human subjects placed under controlled quasi-circadian schedules (28-h days; sleep:wake = 1:2) in a laboratory setting also suffered metabolic deficits characteristic of a prediabetic state within only 10 days of exposure (39), providing a dramatic example of acute influence of misaligned circadian cycles on metabolic well-being. In mice, disruption of circadian cycles, via either genetic or environmental means, has been shown to elicit metabolic disorders (9). For example, ClockΔ19/Δ19 mutant mice were hyperglycemic and prone to body weight gain either under high-fat diet challenge or later in life (40). Importantly, these mice also displayed disrupted circadian rhythms in eating and activity, concordant with the compromising effects of the mutation on the circadian oscillator (41). Furthermore, Bmal1-null mice were also found to be obesity prone when fed with high-fat diet at a young age before confounding phenotypes emerged (42). Consistent with these genetic mutation phenotypes, mice that were artificially subjected to lighting conditions designed to cause misalignment and/or amplitude dampening exhibited body weight gain and other metabolic consequences (43). Together, these studies strongly indicate a close correlation between circadian cycle disruption and metabolic disorders including obesity.
Consistent with the above loss-of-function evidence indicating a temporal regulation of metabolism, profiling studies have supplied high-resolution views of a prevalent circadian oscillation of mRNA, protein, and metabolites in various tissues (27, 44–47). One important finding is that the clock controls metabolic pathways by selectively targeting key steps and components (48). Numerous mechanistic studies since have provided molecular evidence for this mode of “rate-limiting step” regulation. For example, levels of NAD+, an important metabolite for redox balance and a key cofactor for metabolic regulators including SIRT1 and Poly(ADP-ribose) polymerase 1, oscillate in a circadian manner (49). Molecular analysis subsequently showed that the promoter of the gene encoding nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme catalyzing the formation of nicotinamide mononucleotide from nicotinamide and 5′-phosphoribosyl-pyrophosphate during NAD+ biosynthesis, contains a canonical E-box recognized by CLOCK/BMAL1 and thus tightly controlled by the circadian clock (50–52). Interestingly, NAD+ rhythms have been shown to control mitochondrial oxidative metabolism such as fatty acid oxidation, in part through deacetylation of oxidative enzymes by SIRT3 (52).
The circadian metabolic regulation also involves systemic control by the central nervous system including the hypothalamus and the brainstem (25, 53). In the hypothalamus, multiple cell bodies have been shown to play an important role in endocrine regulation and energy homeostasis, including arcuate nucleus (ARC), paraventricular nucleus (PVN), lateral hypothalamic area (LHA), and dorsomedial hypothalamus (DMH) (54, 55). Importantly, the SCN master pacemaker interacts with these extra-SCN energy centers to exert central control over circadian metabolism. For example, SCN neurons form reciprocal connections with ARC neurons expressing orexigenic neuropeptide Y/Agouti-related protein (NPY/AgRP) and anorexignenic pro-opiomelanocortin/cocaine and amphetamine-regulated transcript (POMC/CART) peptides (56, 57). Furthermore, SCN neurons and nutrient-sensing neurons in the ARC and ventromedial hypothalamus (VMH) project to DMH either directly or indirectly, and employ the melanocortin system to regulate thermogenesis, sleep, corticosteroid secretion, wakefulness, and feeding (53, 55). In addition, SCN neurons also innervate the LHA where orexin (ORX, also known as hypocretin) neurons are located (53). ORX is a neuropeptide hormone which stimulates arousal and energy expenditure (37, 58). In particular, the ORX signaling pathway serves a pivotal function in the balancing action of the ventrolateral preoptic nucleus (VLPO)/extended VLPO (eVLPO) of the hypothalamus and the tuberomammillary nucleus (TMN)/raphe/locus coeruleus (LC) in the hypothalamus and the brainstem (55). ORX-deficient mice suffered from narcolepsy, and interestingly also became obese due to reduced energy expenditure despite a concomitant impairment in food intake (59).
Sleep Restriction as a Risk Factor for Obesity and Energy Imbalance
A growing body of evidence supports a functional correlation between sleep restriction (shorter duration and/or poor quality) and metabolic disorders, particularly obesity and insulin resistance (4, 38, 60, 61). As mentioned above, epidemiological studies have unveiled an increased prevalence of metabolic syndrome in night/shift workers who stay up at night and sleep during the day (4). For example, among female nurses studied, rotating shift-work duration positively correlated with a trend of increase in body mass index (BMI) (62) and development of type 2 diabetes (63). Under laboratory conditions, partial or total sleep deprivation over a short time period was found to increase energy expenditure (64, 65); however, insufficient sleep provoked hyperphagia to override the enhanced energy need and thus increase the risk of exaggerated body weight gain and obesity (65).
An important physiological substrate mediating sleep regulation of energy balance is the neuroendocrine system (55). For example, during slow-wave sleep, brain glucose metabolism is diminished, which tightly correlates with a reduction in the whole body glucose metabolism (66). This in turn corresponds to attenuated sympathetic nervous activity and reciprocally augmented secretion of growth hormones. Conversely, deprivation of nocturnal sleep in normal humans led to lower post-sleep/-bed time increase of insulin secretion rate (ISR) and also reduction in growth hormone secretion relative to subjects with normal sleep (67). A similar decrease of ISR was also observed during daytime recovery sleep compared with nocturnal sleep (67), suggesting poor sleep quality associated with abnormal timing (diurnal sleep) can negatively impact hormonal regulation of energy homeostasis (68). Furthermore, sleep time restriction has also been shown to have deleterious effects on glucose homeostasis (64, 67). For example, sleep limitation at 4-h time-in-bed (TIB) for six nights was found to compromise glucose tolerance (12, 61). Interestingly, even two nights of 4-h TIB was sufficient to cause significantly elevated ghrelin:leptin ratio (by approximately 70%), indicating a strong propensity for greater food intake (69). Despite certain experimental incongruence (64), a general role of sleep in neuroendocrine regulation of energy metabolism is well accepted.
Many important questions remain to be further investigated. For example, we still lack mechanistic understanding at the molecular or cellular level. In particular, little is known with regard to the relationship between sleep restriction and circadian disruption in the context of metabolic disease. Specific manipulation in the circadian clockwork can lead to defined circadian phenotypes, and consequently a clear causal relationship between circadian cycle and sleep (see below). However, due to limited molecular understanding of the sleep circuit, sleep restriction by behavioral and environmental means is invariably accompanied with altered circadian rhythms, precluding an unequivocal delineation of functional causality. Furthermore, the functional consequence specific for sleep shortening or impairment is not always clear, especially in epidemiological studies (12).
Core Clock Genes in the Regulation of Sleep and Metabolism
Molecular, genetic, and physiological characterization of core clock genes have revealed profound influence of circadian timing on both sleep and metabolism (22, 23, 53). An elegant example of circadian gene function in sleep disorder was illustrated by studies of familial advanced sleep phase disorder (FASPD, previously called FASP syndrome) (23). Affected individuals display Mendelian circadian rhythm and sleep phenotypes characterized by excessive phase advances in sleep/wake cycles (e.g., going to bed at 7:30 p.m.). Two mutations have been identified as genetic basis for FASPD, including a serine-to-glycine mutation in human PER2 (70, 71) and a threonine to alanine mutation in casein kinase Iδ (CKIδ), a kinase known to phosphorylate PER2 prior to ubiquitin-mediated PER2 degradation (72–74). Interestingly, the Per2 mutation site serine, S662, was the first serine in a five-serine phospho-cluster, and appeared not to be a substrate site for CKI kinases. More recent work on Drosophila PER protein, to which PER2 is most homologous, identified NEMO as the priming kinase for dPER, functioning to promote subsequent phosphorylation events by the Drosophila casein kinase double-time (DBT) and ultimately proteasomal degradation (75). Together, these genetic and molecular studies highlight a key role of Per2 in the regulation of sleep phase. Interestingly, the mammalian Per3 gene, homologous to Per2 (76), has also been shown to play a role in sleep phase and homeostasis control in mouse knockout and human polymorphism studies (77, 78).
More recently, familial natural short sleepers (FNSS) were found to harbor a mutation in the gene encoding the circadian transcriptional repressor DEC2 (79). DEC2 and its homolog DEC1 were initially found to regulate Per1 gene transcription (80), and mouse studies have provided evidence for their role in circadian phase, resetting in response to light pulses (80–82). The P385R DEC2 mutation identified in FNSS was shown to diminish its transcriptional repression in reporter assays. Importantly, whereas FASPD patients do not exhibit deficits in sleep homeostasis (83), FNSS suffers clear sleep deprivation (79), suggesting a role of circadian genes in sleep homeostasis. This study thus adds to a growing body of genetic evidence implicating various clock genes in the regulation of the homeostatic process of sleep (22, 84, 85).
Pioneering studies showed that SCN lesion led to sleep fragmentation, consistent with a role of the clock in sleep timing and architecture (86). However, the homeostatic recovery subsequent to sleep deprivation appeared not to be affected, suggesting that clock gene expression in the SCN may not affect sleep homeostasis. Considering the close relationship of both sleep and peripheral (non-SCN) clocks with metabolic well-being, it has been postulated that metabolism may form the physiological basis for peripheral clock regulation of sleep homeostasis (22, 53). In other words, whereas the circadian clock can directly govern sleep timing (Process C), the homeostatic control (Process S) may be regulated by the clock indirectly via circadian metabolic pathways (10). Alternatively, it is also possible that different clock genes may in fact serve distinct non-clock functions to regulate sleep. This notion is supported by divergent phenotypes in circadian mouse models that are not easily compatible in the strict context of the clock. One notable case is the severe phenotype observed in Bmal1 knockout mice (87). In addition to a loss of circadian rhythmicity, the mice displayed profound developmental, metabolic, and pre-mature aging phenotypes not characteristic of other arrhythmic mouse models (76, 88–90).
Genetic background and the nature of the genetic mutation should also be taken into account when considering the role of clock genes in sleep. CLOCK and NPAS2 are paralogous basic helix–loop–helix PAS domain-containing transcription factors (88, 91). The classical ClockΔ19/Δ19 mutant mice, expressing a dominant negative version of CLOCK deficient in transactivation yet still able to interact with its heterodimeric partner BMAL1, displayed severely compromised circadian rhythms and suffered from hyperphagia and defective glucose and lipid homeostasis (40). Interestingly, these mice also exhibited altered sleep homeostasis under both baseline conditions and sleep recovery following deprivation; in particular, REM sleep rebound over 24 h following sleep deprivation was reduced by half in ClockΔ19/Δ19 mice (92). When compared with wild-type (WT) mice, ClockΔ19/Δ19 mice showed normal or even accelerated food anticipatory activities (FAA) in response to restricted feeding (93) (Figure 2A), and time-course analysis of core clock gene expression under the restricted feeding condition revealed concordantly accelerated phase shift (Figure 2B). In comparison, Npas2-deficient mice were largely normal in circadian behavioral rhythmicity (90). However, these mice showed a unique phenotype wherein the siesta sleep in the second half of the active period was largely absent as indicated by actogram recording (90). Detailed analysis further revealed a role of Npas2 in maintaining normal NREM sleep time (94). In accordance with an important metabolic sensing function of NPAS2 (10), Npas2-deficient mice were ill-adaptive to restricted feeding, as indicated by a delayed FAA response (90). It remains unclear whether CLOCK possesses a metabolic sensing function similar to that of NPAS2 or whether the distinct FAA responses in ClockΔ19/Δ19 and Npas2-deficient mice were due to differences in circadian rhythmicity or other gene-specific effects such as target gene expression (95). Overall, future research is needed to delineate the roles of specific clock genes in sleep regulation, particularly in the context of metabolic function of the clock.
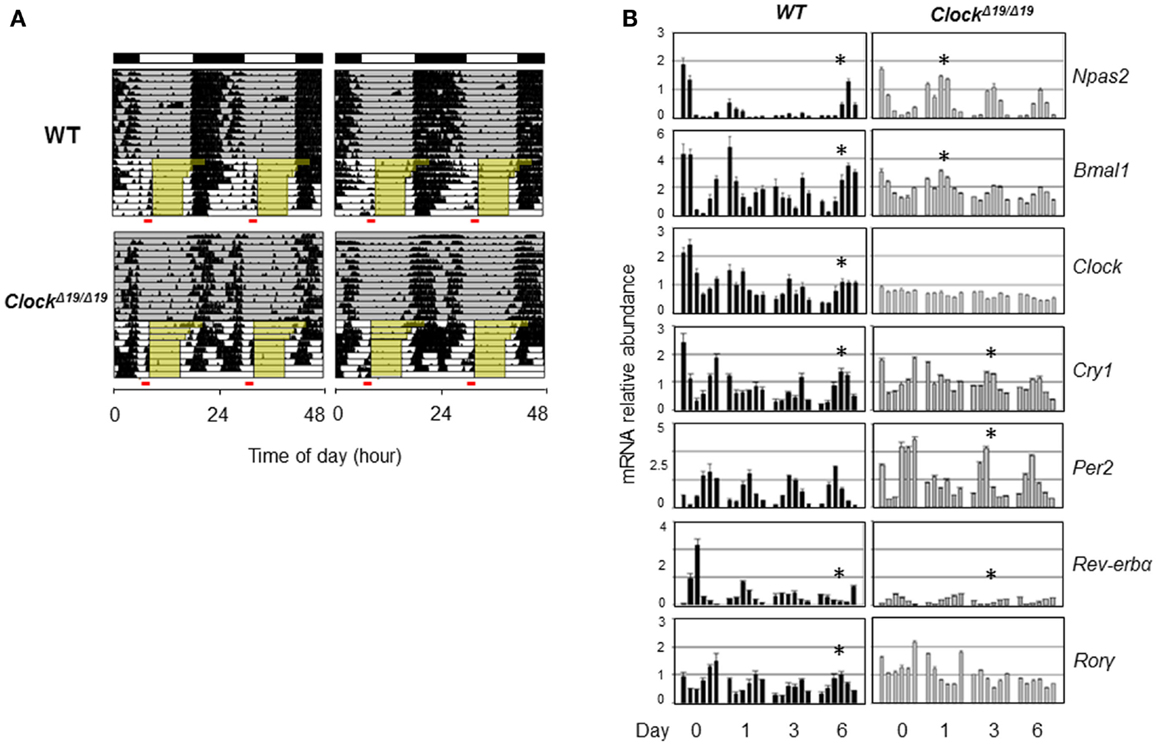
Figure 2. Behavioral and molecular responses of ClockΔ19/Δ19 to restricted feeding. (A) Double-plotted actograms of C57B/6 wild-type (WT) and ClockΔ19/Δ19 mice under 12L:12D conditions as denoted in the bars at the top. Ad lib food availability is indicated by the gray shading. Step-wise food restriction was carried out from 12 to 8 h (indicated by yellow shading), and the food anticipatory activities (FAA) were detected just prior to food availability (marked by red bars). Two representative actograms are shown for each genotype. (B) Mice were sacrificed on the indicated days of restricted feeding as in (A). Liver samples were collected from three mice per genotype on each of the indicated six time points, every 4 h across the circadian cycle starting from ZT0 (light on). Total RNAs were extracted from these samples and expression of circadian core clock genes are quantified by real-time qPCR analysis. Asterisks indicate the time when circadian phase was reversed by restricted feeding.
Modulating Sleep and Circadian Cycles for Metabolic Benefits
Behavioral and Environmental Interventions
Dieting and physical exercise are common interventions to restore energy balance favoring expenditure over intake, thereby guarding against obesity and other metabolic disorders (96). Importantly, meal timing is increasingly appreciated as an important temporal parameter for hormonal balance and energy expenditure. In shift workers, sustained disruption of the intrinsic/environmental synchrony leads to abnormally timed meals. Although the total daily caloric intake remained constant, glucose and lipid homeostasis was disrupted, correlated with increased risks of obesity in shift workers (97). Furthermore, in night-eating syndrome (NES), late night eating, and consequently significant nighttime calorie intake leads to disruption of the sleep/wake cycle; importantly, patients are also prone to developing obesity (98). Laboratory rodent studies have also provided experimental evidence in support of a key role of meal timing and metabolic health. For example, daytime-limited feeding in nocturnal mice resulted in greater body weight gain compared with normal nighttime feeding (99).
More recent mouse studies further underscore an interventional role of meal timing. In one study, time-restricted feeding (TRF) of high-fat diet for 8 h daily during the dark phase was compared with ad lib conditions (100). Although the calorie intake was comparable, the TRF regimen conferred a striking protective function against metabolic disorders including obesity and hyperinsulinemia. Importantly, TRF was found to enhance nutrient mobilization and energy expenditure, consistent with the notion that the obesity risk associated with abnormal meal timing may result from the inability for efficient and timely energy expenditure. The authors further explored the metabolic efficacy of TRF using mice fed with different obesogenic diets, including high-fat, high-fructose, and high-fat/high-sucrose diets (101). Remarkably, in both preventive and therapeutic experimental designs, TRF demonstrated robust efficacy in reducing body weight gain and generally protecting mice from potential or existing metabolic disorders. Several important questions remain to be answered, including the underlying mechanisms likely involving diet-specific circadian pathways, the efficacy in humans and importantly the effects on sleep and other behavioral rhythms. Regardless, these studies highlight a simple behavioral intervention potentially applicable to a diverse array of metabolic challenges.
For millennia, human daily life was dictated by the “natural” light and dark cycles. However, technological and social modernization over the past century drastically altered our lifestyle, and one of the most profound consequences is constant exposure to artificial bright light. On the one hand, bright light therapy is widely used to guard against mood disorders (102). On the other, light can entrain not only the central circadian pacemaker but also peripheral clocks via the sympathetic nervous system (103). Therefore, light exposure in the evening can result in misalignment between the sleep/wake cycle and the intrinsic circadian rhythms in night/shift workers (104), which may in turn increase the incidence rate of the metabolic syndrome. Interestingly, even dim light exposure at night for nocturnal rodents was found to carry significant metabolic consequences (105). Specifically, male mice exposed to dim light at night (LAN) displayed altered meal timing over the circadian cycle, consuming significantly elevated calories during the daytime. These mice consequently showed greater body weight gain, an effect reversible by restricted feeding during the active phase under the LAN conditions (43, 106). In addition to circadian timing, light wavelength is also important; for example, the circadian rhythm of melatonin is more sensitive to photic resetting at a short wavelength of 460 nm compared with 555 nm (107). These studies together suggest that an optimal lighting condition (source and timing) may play an important role in behavioral circadian rhythms and concomitantly prevent metabolic disease.
Metabolic Effects of Common Hypnotics
Although behavioral interventions are usually relatively inexpensive and easy to implement, the outcome requires high patient compliance. Specifically, for metabolic disease, lifestyle changes focusing on improving dietary quality, physical activity, and sleep habits can serve as a first-line treatment but are often unsuccessful clinically due to poor compliance (108). Therefore, it is important to develop pharmacological agents, which normally do not require drastic and prolonged lifestyle modification and thus promise greater patient motivation (109).
Table 1 lists several major classes of sleep medicine and their reported metabolic effects. Besides playing an important role for circadian/sleep cycles, melatonin is also a key metabolic regulator, previously shown to regulate pancreatic insulin secretion and glucose transport (110). In accordance, human studies have provided association evidence supporting a direct relationship between melatonin levels and the risk of developing type 2 diabetes (111), suggesting a causal relationship between reduced melatonin levels, either by sleep debt or shift-work, and metabolic disease. As a result, recent studies using animal models and humans have begun to explore the therapeutic efficacy of melatonin for the metabolic syndrome (112, 113). A number of small-molecule agonists for melatonin receptors have also been identified (114, 115). Interestingly, consistent with the reported suppressive effects of melatonin on body weight gain, newly identified melatonin agonists were also found to improve energy metabolism and insulin sensitivity (114–116). For example, in obese rats fed with high-fat/high-sucrose diets for 5 months, treatment of NEU-P11 via intra-peritoneal injection was able to blunt weight gain, reduce abdominal fat, and improve glucose homeostasis (114, 115) (Table 1). This metabolic efficacy is concordant with a genome-wide association study (GWAS) of European subjects where a single nucleotide polymorphism (SNP) rs1387153, localized near the melatonin receptor 2 (MTNR1B) locus, was identified as a key modulator of fasting blood glucose (117), together suggesting an important role of melatonin signaling in mediating the metabolic regulation by the sleep/circadian clock. In comparison, olanzapine, a promiscuous ligand for both gamma-aminobutyric acid receptor type A (GABAA) and histamine H1 receptors, was found to adversely affect energy metabolism, including body weight gain and altered plasma glucose and lipid levels (118) (Table 1). Therefore, metabolic efficacy, beneficial, or adverse, of sleep medicines should be characterized for individual drugs.

Table 1. Metabolic effects of commonly used hypnotic drugs.
A number of endogenous metabolites, including adenosine, have been shown to induce sleep in rodents (18, 20, 134). Moreover, high-throughput screening using behavioral assays has also led to identification of novel small-molecule modulators of sleep. In a fly based sleep screen of 1280 bioactive compounds (135), reserpine, an inhibitor of the vesicular monoamine transporter (VMAT) functioning in the formation of monoamine-containing presynaptic vesicles, displayed a sleep-inducing activity based on locomoter behavior assays. In accordance, VMAT mutant flies showed enhanced sleep and arousal threshold levels. Likewise, a similar rest/wake locomotor behavioral assay in larval zebrafish was adopted to screen 5648 structurally divergent compounds (136). Strikingly, approximately 10% of the compounds were found to impact certain sleep parameters to varying degrees. In addition to conserved sleep-regulatory targets such as adrenergic receptors, this study also identified novel molecular targets including Ether-a-go-go Related Gene (ERG) potassium channels. Future studies will be needed to address metabolic functions and adverse effects of the newly identified sleep modulating compounds. Given the divergence of sleep between mammalian and non-mammalian species, it remains to be seen whether the identified novel targets and/or small-molecule modulators can be extrapolated to mammals. Ultimately, a direct mammalian sleep screen, albeit challenging, will likely yield unique insight.
Circadian Clock-Modulating Small Molecules
With greater appreciation of a broad and fundamental role of the circadian clock in various pathophysiologies, several recent studies have reported identification of clock-modulating small molecules (1, 137–139). Compared with the traditional chronotherapy where dosing of drugs is purposely aligned to a certain circadian time window to maximize the therapeutic index, such small molecules allow direct molecular manipulation of the circadian clock for beneficial effects in physiology and behavior.
Clock-modulating small molecules can be developed either by targeting a defined clock component of interest or via circadian phenotypic screening (138, 139). Several circadian components have been exploited for specific ligand development, most notably CKI and the antagonizing nuclear hormone receptors REV-ERBs and RORs. A number of CKI inhibitors have previously been characterized, displaying cross-inhibition of multiple CKI isoforms (e.g., CKIδ and CKIε) (140, 141). Surprisingly, an isoform-specific inhibitor of CKIε, PF-4800567, was found to exert minimal effects on circadian periodicity (142, 143), in support of a major role of CKIδ in clock regulation. These studies serve as proof of principle to further exploit the CKIε-specific ligand in clock-related physiology, particularly in light of a demonstrated CKIε function in sleep (144).
A number of ligands for REV-ERBs and RORs have been reported in recent years (139, 145). A series of studies focused on SR9011, a REV-ERB agonist developed through targeted chemical modification (146). SR9011 was found to alter clock gene expression in metabolically active tissues and transiently repress circadian wheel-running behavior. SR9011 treatment in diet-induced obese mice led to improved metabolic parameters including body weight and serum lipid and glucose levels (146). More recent work further demonstrated that SR9011 regulates sleep, increasing wakefulness in treated mice and reciprocally suppressing REM and slow-wave sleep (147). The latter study also reported an anxiolytic effect of SR9011 in WT but not Rev-erbβ knockout mice. These findings together highlight a promising potential of REV-ERB agonists in modulating circadian/sleep-related metabolism and behavior. Small molecules targeting RORs and other clock components such as the melanopsin receptor have also been described (139, 148).
Small-molecule identification via phenotypic screens entails high-throughput based circadian reporter assays, most commonly driven by Per2 and Bmal1 promoters (138, 141, 149). Given the robust precision in circadian periodicity measurement, a great majority of clock-modulating small molecules were identified based on their effects on circadian period length (138). Highlighting a predominant regulatory role of CKI in circadian periodicity, the most pronounced period-lengthening effects are primarily associated with CKI inhibitory compounds (140, 141, 150). These studies complement the molecular genetic finding that PER protein half-life is a key determinant of the period length (74). Likewise, CRY proteins, dimerizing with PERs in the negative arm of the core loop, are also subjected to elaborate protein stability control, and mutations in two paralogous E3 ligases for CRYs have been discovered in mouse forward genetic screens to cause period changes (151). KL001, a period-lengthening compound identified by unbiased phenotypic screen, was recently shown to directly interact with CRY proteins and consequently interfere with their degradation (152).
Recent studies have also revealed a group of clock-amplitude-enhancing small molecules (CEMs) dubbed (138, 150). For example, four CEMs were previously found to enhance cellular and tissue reporter rhythms in both WT and ClockΔ19/+ heterozygous mutant background (150). These molecules were not able to reinvigorate fully, or at least severely, disrupted rhythms in ClockΔ19/Δ19 homozygous or Bmal1-null cells (138). For practical purposes, however, this is not a major limitation as null clock phenotypes are rare in human populations. Interestingly, one of the CEMs exhibits a unique quality of enhancing SCN rhythms (150), known to be highly recalcitrant to genetic and environmental perturbations due to robust neuronal coupling (153, 154). While acute jet-lag can be best remedied when the clock is disrupted or with low amplitude (155), for chronic diseases and aging known to display dampened circadian rhythms (7, 8, 156–158), it is tantalizing to speculate that CEMs can help retard or even reverse physiological decline and improve metabolic and physiological well-being in animals and humans. For example, sleep fragmentation is a hallmark of aging and also frequently observed in age-related diseases including Alzheimer’s disease (159). It is characterized by multiple short periods of sleep during normal sleep timing and sleep during usual active phase, thus exemplifying dampened amplitude of the sleep/wake cycle. It is of great interest to investigate a putative role of CEMs in sleep and also metabolism. At the mechanistic level, the molecular basis for clock amplitude, or robustness, is not as well-understood compared with circadian period and phase regulation (160). Besides the well-characterized nuclear hormone receptors in the stabilization loop of the core oscillator (REV-ERBs and RORs) (25), genomic siRNA screen studies have revealed hundreds of genes that, when knocked down, enhanced or repressed the oscillatory amplitude of reporter rhythms, indicating a broad systemic control of clock amplitude (161).
Concluding Remarks
Exciting advances in the past decade or so have revealed much molecular insight into the circadian regulatory mechanism of energy metabolism (10, 37). Existing evidence also supports a causal role of sleep disruption or restriction in the development of the metabolic syndrome including obesity (12), although the mechanistic basis is not well-understood. As the research community continues to decipher genetic and molecular function and mechanism of sleep, behavioral, environmental, and pharmacological intervention strategies are being actively pursued to manipulate circadian and sleep rhythms to optimize cellular energetics (Figure 1). This novel approach exploits the translational potential of biological rhythms to combat the continuing rise of the global metabolic disease epidemic in our modern society, complementing the more “direct” strategy aiming to correct particular metabolic regulators or pathways.
The metabolic regulation by biological rhythms is important beyond the realm of the metabolic disease. Other pathologies are also characterized by strong temporal components and intimately associated with dysregulated metabolism (1). For example, the morning surge of blood pressure is a well-documented culprit for cardiovascular disease related sudden death (162, 163). The circadian/sleep cycles also play an important role in cardiometabolic function, as illustrated by increased hypertensive risks in circadian clock mutant mice, forced desynchrony in laboratory studies, as well as shift workers (163–165). No disease has a greater penetrance than aging – we all age. In addition to the aforementioned sleep and circadian deficits, age-associated decline also encompasses deterioration of energy homeostasis, physical performance and cognitive function, which may eventually manifest in chronic and degenerative diseases (6–8, 156, 166). Application of biological rhythm-based intervention strategies to these disease targets is of great potential and will likely lead to exciting advances in the near future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was in part supported by the Robert A. Welch Foundation (AU-1731), American Heart Association (11SDG7600045), and NIH/NIA (R01 AG045828) to ZC, and Texas Medical Center Digestive Diseases Center (TMC DDC) P/F Awards to SH-Y and ZC (NIDDK Center Grant P30-DK056338).
References
1. Takahashi JS, Hong HK, Ko CH, McDearmon EL. The genetics of mammalian circadian order and disorder: implications for physiology and disease. Nat Rev Genet (2008) 9:764–75. doi: 10.1038/nrg2430
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Borbely AA, Achermann P. Sleep homeostasis and models of sleep regulation. J Biol Rhythms (1999) 14:557–68.
3. Andretic R, Franken P, Tafti M. Genetics of sleep. Annu Rev Genet (2008) 42:361–88. doi:10.1146/annurev.genet.42.110807.091541
4. Karlsson B, Knutsson A, Lindahl B. Is there an association between shift work and having a metabolic syndrome? Results from a population based study of 27,485 people. Occup Environ Med (2001) 58:747–52. doi:10.1136/oem.58.11.747
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Morikawa Y, Nakagawa H, Miura K, Soyama Y, Ishizaki M, Kido T, et al. Effect of shift work on body mass index and metabolic parameters. Scand J Work Environ Health (2007) 33:45–50. doi:10.5271/sjweh.1063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Kondratova AA, Kondratov RV. The circadian clock and pathology of the ageing brain. Nat Rev Neurosci (2012) 13:325–35. doi:10.1038/nrn3208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Gibson EM, Williams WP III, Kriegsfeld LJ. Aging in the circadian system: considerations for health, disease prevention and longevity. Exp Gerontol (2009) 44:51–6. doi:10.1016/j.exger.2008.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Brown SA, Pagani L, Cajochen C, Eckert A. Systemic and cellular reflections on ageing and the circadian oscillator: a mini-review. Gerontology (2011) 57:427–34. doi:10.1159/000320673
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Bass J, Takahashi JS. Circadian integration of metabolism and energetics. Science (2010) 330:1349–54. doi:10.1126/science.1195027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Rutter J, Reick M, McKnight SL. Metabolism and the control of circadian rhythms. Annu Rev Biochem (2002) 71:307–31. doi:10.1146/annurev.biochem.71.090501.142857
11. Laposky AD, Bass J, Kohsaka A, Turek FW. Sleep and circadian rhythms: key components in the regulation of energy metabolism. FEBS Lett (2008) 582:142–51. doi:10.1016/j.febslet.2007.06.079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Spiegel K, Tasali E, Leproult R, Van Cauter E. Effects of poor and short sleep on glucose metabolism and obesity risk. Nat Rev Endocrinol (2009) 5:253–61. doi:10.1038/nrendo.2009.23
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Mignot E. Why we sleep: the temporal organization of recovery. PLoS Biol (2008) 6:e106. doi:10.1371/journal.pbio.0060106
14. Rechtschaffen A, Bergmann BM, Everson CA, Kushida CA, Gilliland MA. Sleep deprivation in the rat: X. Integration and discussion of the findings. Sleep (1989) 12:68–87.
15. Shaw PJ, Tononi G, Greenspan RJ, Robinson DF. Stress response genes protect against lethal effects of sleep deprivation in Drosophila. Nature (2002) 417:287–91. doi:10.1038/417287a
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Liu Y, Wheaton AG, Chapman DP, Croft JB. Sleep duration and chronic diseases among U.S. adults age 45 years and older: evidence from the 2010 behavioral risk factor surveillance system. Sleep (2013) 36:1421–7. doi:10.5665/sleep.3028
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. von Ruesten A, Weikert C, Fietze I, Boeing H. Association of sleep duration with chronic diseases in the European prospective investigation into cancer and nutrition (EPIC)-potsdam study. PLoS One (2012) 7:e30972. doi:10.1371/journal.pone.0030972
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Sehgal A, Mignot E. Genetics of sleep and sleep disorders. Cell (2011) 146:194–207. doi:10.1016/j.cell.2011.07.004
19. Daan S, Beersma DG, Borbely AA. Timing of human sleep: recovery process gated by a circadian pacemaker. Am J Physiol (1984) 246:R161–83.
20. Siegel JM. Clues to the functions of mammalian sleep. Nature (2005) 437:1264–71. doi:10.1038/nature04285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science (2013) 342:373–7. doi:10.1126/science.1241224
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Franken P, Dijk DJ. Circadian clock genes and sleep homeostasis. Eur J Neurosci (2009) 29:1820–9. doi:10.1111/j.1460-9568.2009.06723.x
23. Jones CR, Huang AL, Ptacek LJ, Fu YH. Genetic basis of human circadian rhythm disorders. Exp Neurol (2013) 243:28–33. doi:10.1016/j.expneurol.2012.07.012
24. Schwartz WJ. Circadian rhythms: a tale of two nuclei. Curr Biol (2009) 19:R460–2. doi:10.1016/j.cub.2009.04.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Liu AC, Lewis WG, Kay SA. Mammalian circadian signaling networks and therapeutic targets. Nat Chem Biol (2007) 3:630–9. doi:10.1038/nchembio.2007.37
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Welsh DK, Takahashi JS, Kay SA. Suprachiasmatic nucleus: cell autonomy and network properties. Annu Rev Physiol (2010) 72:551–77. doi:10.1146/annurev-physiol-021909-135919
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Green CB, Takahashi JS, Bass J. The meter of metabolism. Cell (2008) 134:728–42. doi:10.1016/j.cell.2008.08.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Morris CJ, Aeschbach D, Scheer FA. Circadian system, sleep and endocrinology. Mol Cell Endocrinol (2012) 349:91–104. doi:10.1016/j.mce.2011.09.003
29. Reiter RJ. The pineal gland and melatonin in relation to aging: a summary of the theories and of the data. Exp Gerontol (1995) 30:199–212. doi:10.1016/0531-5565(94)00045-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Iuvone PM, Tosini G, Pozdeyev N, Haque R, Klein DC, Chaurasia SS. Circadian clocks, clock networks, arylalkylamine N-acetyltransferase, and melatonin in the retina. Prog Retin Eye Res (2005) 24:433–56. doi:10.1016/j.preteyeres.2005.01.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Lewy AJ, Wehr TA, Goodwin FK, Newsome DA, Markey SP. Light suppresses melatonin secretion in humans. Science (1980) 210:1267–9. doi:10.1126/science.7434030
32. Sack RL, Brandes RW, Kendall AR, Lewy AJ. Entrainment of free-running circadian rhythms by melatonin in blind people. N Engl J Med (2000) 343:1070–7. doi:10.1056/NEJM200010123431503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Chen Z, McKnight SL. A conserved DNA damage response pathway responsible for coupling the cell division cycle to the circadian and metabolic cycles. Cell Cycle (2007) 6:2906–12. doi:10.4161/cc.6.23.5041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Edgar RS, Green EW, Zhao Y, van Ooijen G, Olmedo M, Qin X, et al. Peroxiredoxins are conserved markers of circadian rhythms. Nature (2012) 485:459–64. doi:10.1038/nature11088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Chen Z, Odstrcil EA, Tu BP, McKnight SL. Restriction of DNA replication to the reductive phase of the metabolic cycle protects genome integrity. Science (2007) 316:1916–9. doi:10.1126/science.1140958
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. O’Neill JS, Reddy AB. Circadian clocks in human red blood cells. Nature (2011) 469:498–503. doi:10.1038/nature09702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Bass J. Circadian topology of metabolism. Nature (2012) 491:348–56. doi:10.1038/nature11704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Knutson KL, Spiegel K, Penev P, Van Cauter E. The metabolic consequences of sleep deprivation. Sleep Med Rev (2007) 11:163–78. doi:10.1016/j.smrv.2007.01.002
39. Scheer FA, Hilton MF, Mantzoros CS, Shea SA. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc Natl Acad Sci U S A (2009) 106:4453–8. doi:10.1073/pnas.0808180106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, et al. Obesity and metabolic syndrome in circadian clock mutant mice. Science (2005) 308:1043–5. doi:10.1126/science.1108750
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Antoch MP, Song EJ, Chang AM, Vitaterna MH, Zhao Y, Wilsbacher LD, et al. Functional identification of the mouse circadian clock gene by transgenic BAC rescue. Cell (1997) 89:655–67. doi:10.1016/S0092-8674(00)80246-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Shi SQ, Ansari TS, McGuinness OP, Wasserman DH, Johnson CH. Circadian disruption leads to insulin resistance and obesity. Curr Biol (2013) 23:372–81. doi:10.1016/j.cub.2013.01.048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Fonken LK, Workman JL, Walton JC, Weil ZM, Morris JS, Haim A, et al. Light at night increases body mass by shifting the time of food intake. Proc Natl Acad Sci U S A (2010) 107:18664–9. doi:10.1073/pnas.1008734107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Hogenesch JB, Ueda HR. Understanding systems-level properties: timely stories from the study of clocks. Nat Rev Genet (2011) 12:407–16. doi:10.1038/nrg2972
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, et al. Harmonics of circadian gene transcription in mammals. PLoS Genet (2009) 5:e1000442. doi:10.1371/journal.pgen.1000442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Reddy AB, Karp NA, Maywood ES, Sage EA, Deery M, O’Neill JS, et al. Circadian orchestration of the hepatic proteome. Curr Biol (2006) 16:1107–15. doi:10.1016/j.cub.2006.04.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Kasukawa T, Sugimoto M, Hida A, Minami Y, Mori M, Honma S, et al. Human blood metabolite timetable indicates internal body time. Proc Natl Acad Sci U S A (2012) 109:15036–41. doi:10.1073/pnas.1207768109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell (2002) 109:307–20. doi:10.1016/S0092-8674(02)00722-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Asher G, Reinke H, Altmeyer M, Gutierrez-Arcelus M, Hottiger MO, Schibler U. Poly(ADP-ribose) polymerase 1 participates in the phase entrainment of circadian clocks to feeding. Cell (2010) 142:943–53. doi:10.1016/j.cell.2010.08.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science (2009) 324:654–7. doi:10.1126/science.1170803
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science (2009) 324:651–4. doi:10.1126/science.1171641
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science (2013) 342:1243417. doi:10.1126/science.1243417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Huang W, Ramsey KM, Marcheva B, Bass J. Circadian rhythms, sleep, and metabolism. J Clin Invest (2011) 121:2133–41. doi:10.1172/JCI46043
54. Sanchez-Lasheras C, Konner AC, Bruning JC. Integrative neurobiology of energy homeostasis-neurocircuits, signals and mediators. Front Neuroendocrinol (2010) 31:4–15. doi:10.1016/j.yfrne.2009.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature (2005) 437:1257–63. doi:10.1038/nature04284
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Saeb-Parsy K, Lombardelli S, Khan FZ, McDowall K, Au-Yong IT, Dyball RE. Neural connections of hypothalamic neuroendocrine nuclei in the rat. J Neuroendocrinol (2000) 12:635–48. doi:10.1046/j.1365-2826.2000.00503.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Yi CX, van der Vliet J, Dai J, Yin G, Ru L, Buijs RM. Ventromedial arcuate nucleus communicates peripheral metabolic information to the suprachiasmatic nucleus. Endocrinology (2006) 147:283–94. doi:10.1210/en.2005-1051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Mieda M, Yanagisawa M. Sleep, feeding, and neuropeptides: roles of orexins and orexin receptors. Curr Opin Neurobiol (2002) 12:339–45. doi:10.1016/S0959-4388(02)00331-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, et al. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron (2001) 30:345–54. doi:10.1016/S0896-6273(01)00293-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Taheri S, Lin L, Austin D, Young T, Mignot E. Short sleep duration is associated with reduced leptin, elevated ghrelin, and increased body mass index. PLoS Med (2004) 1:e62. doi:10.1371/journal.pmed.0010062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Spiegel K, Leproult R, Van Cauter E. Impact of sleep debt on metabolic and endocrine function. Lancet (1999) 354:1435–9. doi:10.1016/S0140-6736(99)01376-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Tada Y, Kawano Y, Maeda I, Yoshizaki T, Sunami A, Yokoyama Y, et al. Association of body mass index with lifestyle and rotating shift work in Japanese female nurses. Obesity (2014) 22:2489–93. doi:10.1002/oby.20908
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Pan A, Schernhammer ES, Sun Q, Hu FB. Rotating night shift work and risk of type 2 diabetes: two prospective cohort studies in women. PLoS Med (2011) 8:e1001141. doi:10.1371/journal.pmed.1001141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Nedeltcheva AV, Scheer FA. Metabolic effects of sleep disruption, links to obesity and diabetes. Curr Opin Endocrinol Diabetes Obes (2014) 21:293–8. doi:10.1097/MED.0000000000000082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Markwald RR, Melanson EL, Smith MR, Higgins J, Perreault L, Eckel RH, et al. Impact of insufficient sleep on total daily energy expenditure, food intake, and weight gain. Proc Natl Acad Sci U S A (2013) 110:5695–700. doi:10.1073/pnas.1216951110
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Boyle PJ, Scott JC, Krentz AJ, Nagy RJ, Comstock E, Hoffman C. Diminished brain glucose metabolism is a significant determinant for falling rates of systemic glucose utilization during sleep in normal humans. J Clin Invest (1994) 93:529–35. doi:10.1172/JCI117003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Scheen AJ, Byrne MM, Plat L, Leproult R, Van Cauter E. Relationships between sleep quality and glucose regulation in normal humans. Am J Physiol (1996) 271:E261–70.
68. Van Cauter E. Sleep disturbances and insulin resistance. Diabet Med (2011) 28:1455–62. doi:10.1111/j.1464-5491.2011.03459.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Spiegel K, Tasali E, Penev P, Van Cauter E. Brief communication: sleep curtailment in healthy young men is associated with decreased leptin levels, elevated ghrelin levels, and increased hunger and appetite. Ann Intern Med (2004) 141:846–50. doi:10.7326/0003-4819-141-11-200412070-00008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Xu Y, Toh KL, Jones CR, Shin JY, Fu YH, Ptacek LJ. Modeling of a human circadian mutation yields insights into clock regulation by PER2. Cell (2007) 128:59–70. doi:10.1016/j.cell.2006.11.043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, et al. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science (2001) 291:1040–3. doi:10.1126/science.1057499
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Xu Y, Padiath QS, Shapiro RE, Jones CR, Wu SC, Saigoh N, et al. Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature (2005) 434:640–4. doi:10.1038/nature03453
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Vanselow K, Vanselow JT, Westermark PO, Reischl S, Maier B, Korte T, et al. Differential effects of PER2 phosphorylation: molecular basis for the human familial advanced sleep phase syndrome (FASPS). Genes Dev (2006) 20:2660–72. doi:10.1101/gad.397006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Gallego M, Virshup DM. Post-translational modifications regulate the ticking of the circadian clock. Nat Rev Mol Cell Biol (2007) 8:139–48. doi:10.1038/nrm2106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Chiu JC, Ko HW, Edery I. NEMO/NLK phosphorylates PERIOD to initiate a time-delay phosphorylation circuit that sets circadian clock speed. Cell (2011) 145:357–70. doi:10.1016/j.cell.2011.04.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Bae K, Jin X, Maywood ES, Hastings MH, Reppert SM, Weaver DR. Differential functions of mPer1, mPer2, and mPer3 in the SCN circadian clock. Neuron (2001) 30:525–36. doi:10.1016/S0896-6273(01)00302-6
77. Hasan S, van der Veen DR, Winsky-Sommerer R, Dijk DJ, Archer SN. Altered sleep and behavioral activity phenotypes in PER3-deficient mice. Am J Physiol Regul Integr Comp Physiol (2011) 301:R1821–30. doi:10.1152/ajpregu.00260.2011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Hida A, Kitamura S, Katayose Y, Kato M, Ono H, Kadotani H, et al. Screening of clock gene polymorphisms demonstrates association of a PER3 polymorphism with morningness-eveningness preference and circadian rhythm sleep disorder. Sci Rep (2014) 4:6309. doi:10.1038/srep06309
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. He Y, Jones CR, Fujiki N, Xu Y, Guo B, Holder JL Jr, et al. The transcriptional repressor DEC2 regulates sleep length in mammals. Science (2009) 325:866–70. doi:10.1126/science.1174443
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Honma S, Kawamoto T, Takagi Y, Fujimoto K, Sato F, Noshiro M, et al. Dec1 and Dec2 are regulators of the mammalian molecular clock. Nature (2002) 419:841–4. doi:10.1038/nature01123
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Kon N, Hirota T, Kawamoto T, Kato Y, Tsubota T, Fukada Y. Activation of TGF-beta/activin signalling resets the circadian clock through rapid induction of Dec1 transcripts. Nat Cell Biol (2008) 10:1463–9. doi:10.1038/ncb1806
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Rossner MJ, Oster H, Wichert SP, Reinecke L, Wehr MC, Reinecke J, et al. Disturbed clockwork resetting in Sharp-1 and Sharp-2 single and double mutant mice. PLoS One (2008) 3:e2762. doi:10.1371/journal.pone.0002762
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Chong SY, Ptacek LJ, Fu YH. Genetic insights on sleep schedules: this time, it’s PERsonal. Trends Genet (2012) 28:598–605. doi:10.1016/j.tig.2012.08.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. Rosenwasser AM. Circadian clock genes: non-circadian roles in sleep, addiction, and psychiatric disorders? Neurosci Biobehav Rev (2010) 34:1249–55. doi:10.1016/j.neubiorev.2010.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Striz M, O’Hara BF. Clock genes and sleep homeostasis: a fundamental link within the two-process model? Sleep (2013) 36:301–2. doi:10.5665/sleep.2430
86. Trachsel L, Edgar DM, Seidel WF, Heller HC, Dement WC. Sleep homeostasis in suprachiasmatic nuclei-lesioned rats: effects of sleep deprivation and triazolam administration. Brain Res (1992) 589:253–61. doi:10.1016/0006-8993(92)91284-L
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core componentof the circadian clock. Genes Dev (2006) 20:1868–73. doi:10.1101/gad.1432206
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. DeBruyne JP, Weaver DR, Reppert SM. CLOCK and NPAS2 have overlapping roles in the suprachiasmatic circadian clock. Nat Neurosci (2007) 10:543–5. doi:10.1038/nn1884
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell (2002) 111:41–50. doi:10.1016/S0092-8674(02)01223-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Dudley CA, Erbel-Sieler C, Estill SJ, Reick M, Franken P, Pitts S, et al. Altered patterns of sleep and behavioral adaptability in NPAS2-deficient mice. Science (2003) 301:379–83. doi:10.1126/science.1082795
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Reick M, Garcia JA, Dudley C, McKnight SL. NPAS2: an analog of clock operative in the mammalian forebrain. Science (2001) 293:506–9. doi:10.1126/science.1060699
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Naylor E, Bergmann BM, Krauski K, Zee PC, Takahashi JS, Vitaterna MH, et al. The circadian clock mutation alters sleep homeostasis in the mouse. J Neurosci (2000) 20:8138–43.
93. Pitts S, Perone E, Silver R. Food-entrained circadian rhythms are sustained in arrhythmic Clk/Clk mutant mice. Am J Physiol Regul Integr Comp Physiol (2003) 285:R57–67. doi:10.1152/ajpregu.00023.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Franken P, Dudley CA, Estill SJ, Barakat M, Thomason R, O’Hara BF, et al. NPAS2 as a transcriptional regulator of non-rapid eye movement sleep: genotype and sex interactions. Proc Natl Acad Sci U S A (2006) 103:7118–23. doi:10.1073/pnas.0602006103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, et al. Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science (2012) 338:349–54. doi:10.1126/science.1226339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Partonen T. Obesity = physical activity + dietary intake + sleep stages + light exposure. Ann Med (2014) 46:245–6. doi:10.3109/07853890.2014.932921
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Antunes LC, Levandovski R, Dantas G, Caumo W, Hidalgo MP. Obesity and shift work: chronobiological aspects. Nutr Res Rev (2010) 23:155–68. doi:10.1017/S0954422410000016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Howell MJ, Schenck CH, Crow SJ. A review of nighttime eating disorders. Sleep Med Rev (2009) 13:23–34. doi:10.1016/j.smrv.2008.07.005
99. Arble DM, Vitaterna MH, Turek FW. Rhythmic leptin is required for weight gain from circadian desynchronized feeding in the mouse. PLoS One (2011) 6:e25079. doi:10.1371/journal.pone.0025079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab (2012) 15:848–60. doi:10.1016/j.cmet.2012.04.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Chaix A, Zarrinpar A, Miu P, Panda S. Time-restricted feeding is a preventative and therapeutic intervention against diverse nutritional challenges. Cell Metab (2014) 20:991–1005. doi:10.1016/j.cmet.2014.11.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Golden RN, Gaynes BN, Ekstrom RD, Hamer RM, Jacobsen FM, Suppes T, et al. The efficacy of light therapy in the treatment of mood disorders: a review and meta-analysis of the evidence. Am J Psychiatry (2005) 162:656–62. doi:10.1176/appi.ajp.162.4.656
103. Ishida A, Mutoh T, Ueyama T, Bando H, Masubuchi S, Nakahara D, et al. Light activates the adrenal gland: timing of gene expression and glucocorticoid release. Cell Metab (2005) 2:297–307. doi:10.1016/j.cmet.2005.09.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Czeisler CA, Allan JS, Strogatz SH, Ronda JM, Sanchez R, Rios CD, et al. Bright light resets the human circadian pacemaker independent of the timing of the sleep-wake cycle. Science (1986) 233:667–71. doi:10.1126/science.3726555
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Fonken LK, Nelson RJ. The effects of light at night on circadian clocks and metabolism. Endocr Rev (2014) 35:648–70. doi:10.1210/er.2013-1051
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Fonken LK, Aubrecht TG, Melendez-Fernandez OH, Weil ZM, Nelson RJ. Dim light at night disrupts molecular circadian rhythms and increases body weight. J Biol Rhythms (2013) 28:262–71. doi:10.1177/0748730413493862
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Lockley SW, Brainard GC, Czeisler CA. High sensitivity of the human circadian melatonin rhythm to resetting by short wavelength light. J Clin Endocrinol Metab (2003) 88:4502–5. doi:10.1210/jc.2003-030570
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Portero McLellan KC, Wyne K, Villagomez ET, Hsueh WA. Therapeutic interventions to reduce the risk of progression from prediabetes to type 2 diabetes mellitus. Ther Clin Risk Manag (2014) 10:173–88. doi:10.2147/TCRM.S39564
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Tran K, Levin RM, Mousa SA. Behavioral intervention versus pharmacotherapy or their combinations in the management of overactive bladder dysfunction. Adv Urol (2009) 2009:345324. doi:10.1155/2009/345324
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Peschke E, Muhlbauer E. New evidence for a role of melatonin in glucose regulation. Best Pract Res Clin Endocrinol Metab (2010) 24:829–41. doi:10.1016/j.beem.2010.09.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. McMullan CJ, Schernhammer ES, Rimm EB, Hu FB, Forman JP. Melatonin secretion and the incidence of type 2 diabetes. JAMA (2013) 309:1388–96. doi:10.1001/jama.2013.2710
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Pai SA, Majumdar AS. Protective effects of melatonin against metabolic and reproductive disturbances in polycystic ovary syndrome in rats. J Pharm Pharmacol (2014) 66:1710–21. doi:10.1111/jphp.12297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Rubio-Sastre P, Scheer FA, Gomez-Abellan P, Madrid JA, Garaulet M. Acute melatonin administration in humans impairs glucose tolerance in both the morning and evening. Sleep (2014) 37:1715–9. doi:10.5665/sleep.4088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. She M, Deng X, Guo Z, Laudon M, Hu Z, Liao D, et al. NEU-P11, a novel melatonin agonist, inhibits weight gain and improves insulin sensitivity in high-fat/high-sucrose-fed rats. Pharmacol Res (2009) 59:248–53. doi:10.1016/j.phrs.2009.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. She M, Hu X, Su Z, Zhang C, Yang S, Ding L, et al. Piromelatine, a novel melatonin receptor agonist, stabilizes metabolic profiles and ameliorates insulin resistance in chronic sleep restricted rats. Eur J Pharmacol (2014) 727:60–5. doi:10.1016/j.ejphar.2014.01.037
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Prunet-Marcassus B, Desbazeille M, Bros A, Louche K, Delagrange P, Renard P, et al. Melatonin reduces body weight gain in Sprague Dawley rats with diet-induced obesity. Endocrinology (2003) 144:5347–52. doi:10.1210/en.2003-0693
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Bouatia-Naji N, Bonnefond A, Cavalcanti-Proenca C, Sparso T, Holmkvist J, Marchand M, et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet (2009) 41:89–94. doi:10.1038/ng.277
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Almeras N, Despres JP, Villeneuve J, Demers MF, Roy MA, Cadrin C, et al. Development of an atherogenic metabolic risk factor profile associated with the use of atypical antipsychotics. J Clin Psychiatry (2004) 65:557–64. doi:10.4088/JCP.v65n0417
119. Coccurello R, Caprioli A, Conti R, Ghirardi O, Borsini F, Carminati P, et al. Olanzapine (LY170053, 2-methyl-4-(4-methyl-1-piperazinyl)-10H-thieno[2,3-b][1,5] benzodiazepine), but not the novel atypical antipsychotic ST2472 (9-piperazin-1-ylpyrrolo[2,1-b][1,3]benzothiazepine), chronic administration induces weight gain, hyperphagia, and metabolic dysregulation in mice. J Pharmacol Exp Ther (2008) 326:905–11. doi:10.1124/jpet.108.137240
120. Zai CC, Tiwari AK, Chowdhury NI, Brandl EJ, Shaikh SA, Freeman N, et al. Association study of GABAA alpha2 receptor subunit gene variants in antipsychotic-associated weight gain. J Clin Psychopharmacol (2015) 35:7–12. doi:10.1097/JCP.0000000000000261
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Murphy HM, Ihekoronze C, Wideman CH. Zolpidem-induced changes in activity, metabolism, and anxiety in rats. Pharmacol Biochem Behav (2011) 98:81–6. doi:10.1016/j.pbb.2010.12.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Ratliff JC, Barber JA, Palmese LB, Reutenauer EL, Tek C. Association of prescription H1 antihistamine use with obesity: results from the national health and nutrition examination survey. Obesity (2010) 18:2398–400. doi:10.1038/oby.2010.176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Raveendran VV, Kassel KM, Smith DD, Luyendyk JP, Williams KJ, Cherian R, et al. H1-antihistamines exacerbate high-fat diet-induced hepatic steatosis in wild-type but not in apolipoprotein E knockout mice. Am J Physiol Gastrointest Liver Physiol (2014) 307:G219–28. doi:10.1152/ajpgi.00027.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Lian J, Huang XF, Pai N, Deng C. Preventing olanzapine-induced weight gain using betahistine: a study in a rat model with chronic olanzapine treatment. PLoS One (2014) 9:e104160. doi:10.1371/journal.pone.0104160
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Wolden-Hanson T, Mitton DR, McCants RL, Yellon SM, Wilkinson CW, Matsumoto AM, et al. Daily melatonin administration to middle-aged male rats suppresses body weight, intraabdominal adiposity, and plasma leptin and insulin independent of food intake and total body fat. Endocrinology (2000) 141:487–97. doi:10.1210/endo.141.2.7311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Kitagawa A, Ohta Y, Ohashi K. Melatonin improves metabolic syndrome induced by high fructose intake in rats. J Pineal Res (2012) 52:403–13. doi:10.1111/j.1600-079X.2011.00955.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Kozirog M, Poliwczak AR, Duchnowicz P, Koter-Michalak M, Sikora J, Broncel M. Melatonin treatment improves blood pressure, lipid profile, and parameters of oxidative stress in patients with metabolic syndrome. J Pineal Res (2011) 50:261–6. doi:10.1111/j.1600-079X.2010.00835.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Oxenkrug GF, Summergrad P. Ramelteon attenuates age-associated hypertension and weight gain in spontaneously hypertensive rats. Ann N Y Acad Sci (2010) 1199:114–20. doi:10.1111/j.1749-6632.2009.05355.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Zhou L, Sutton GM, Rochford JJ, Semple RK, Lam DD, Oksanen LJ, et al. Serotonin 2C receptor agonists improve type 2 diabetes via melanocortin-4 receptor signaling pathways. Cell Metab (2007) 6:398–405. doi:10.1016/j.cmet.2007.10.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Fidler MC, Sanchez M, Raether B, Weissman NJ, Smith SR, Shanahan WR, et al. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab (2011) 96:3067–77. doi:10.1210/jc.2011-1256
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Haynes AC, Chapman H, Taylor C, Moore GB, Cawthorne MA, Tadayyon M, et al. Anorectic, thermogenic and anti-obesity activity of a selective orexin-1 receptor antagonist in ob/ob mice. Regul Pept (2002) 104:153–9. doi:10.1016/S0167-0115(01)00358-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Steiner MA, Sciarretta C, Pasquali A, Jenck F. The selective orexin receptor 1 antagonist ACT-335827 in a rat model of diet-induced obesity associated with metabolic syndrome. Front Pharmacol (2013) 4:165. doi:10.3389/fphar.2013.00165
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Girault EM, Foppen E, Ackermans MT, Fliers E, Kalsbeek A. Central administration of an orexin receptor 1 antagonist prevents the stimulatory effect of olanzapine on endogenous glucose production. Brain Res (2013) 1527:238–45. doi:10.1016/j.brainres.2013.06.034
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Saper CB, Romanovsky AA, Scammell TE. Neural circuitry engaged by prostaglandins during the sickness syndrome. Nat Neurosci (2012) 15:1088–95. doi:10.1038/nn.3159
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Nall AH, Sehgal A. Small-molecule screen in adult Drosophila identifies VMAT as a regulator of sleep. J Neurosci (2013) 33:8534–40. doi:10.1523/JNEUROSCI.0253-13.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Rihel J, Prober DA, Arvanites A, Lam K, Zimmerman S, Jang S, et al. Zebrafish behavioral profiling links drugs to biological targets and rest/wake regulation. Science (2010) 327:348–51. doi:10.1126/science.1183090
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Hirota T, Kay SA. High-throughput screening and chemical biology: new approaches for understanding circadian clock mechanisms. Chem Biol (2009) 16:921–7. doi:10.1016/j.chembiol.2009.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Chen Z, Yoo SH, Takahashi JS. Small molecule modifiers of circadian clocks. Cell Mol Life Sci (2013) 70:2985–98. doi:10.1007/s00018-012-1207-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Kojetin DJ, Burris TP. REV-ERB and ROR nuclear receptors as drug targets. Nat Rev Drug Discov (2014) 13:197–216. doi:10.1038/nrd4100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Isojima Y, Nakajima M, Ukai H, Fujishima H, Yamada RG, Masumoto KH, et al. CKIepsilon/delta-dependent phosphorylation is a temperature-insensitive, period-determining process in the mammalian circadian clock. Proc Natl Acad Sci U S A (2009) 106:15744–9. doi:10.1073/pnas.0908733106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Hirota T, Lee JW, Lewis WG, Zhang EE, Breton G, Liu X, et al. High-throughput chemical screen identifies a novel potent modulator of cellular circadian rhythms and reveals CKIalpha as a clock regulatory kinase. PLoS Biol (2010) 8:e1000559. doi:10.1371/journal.pbio.1000559
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Walton KM, Fisher K, Rubitski D, Marconi M, Meng QJ, Sladek M, et al. Selective inhibition of casein kinase 1 epsilon minimally alters circadian clock period. J Pharmacol Exp Ther (2009) 330:430–9. doi:10.1124/jpet.109.151415
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Meng QJ, Maywood ES, Bechtold DA, Lu WQ, Li J, Gibbs JE, et al. Entrainment of disrupted circadian behavior through inhibition of casein kinase 1 (CK1) enzymes. Proc Natl Acad Sci U S A (2010) 107:15240–5. doi:10.1073/pnas.1005101107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Zhou L, Bryant CD, Loudon A, Palmer AA, Vitaterna MH, Turek FW. The circadian clock gene Csnk1e regulates rapid eye movement sleep amount, and nonrapid eye movement sleep architecture in mice. Sleep (2014) 37:793A–793A. doi:10.5665/sleep.3590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Trump RP, Bresciani S, Cooper AW, Tellam JP, Wojno J, Blaikley J, et al. Optimized chemical probes for REV-ERBalpha. J Med Chem (2013) 56:4729–37. doi:10.1021/jm400458q
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Solt LA, Wang Y, Banerjee S, Hughes T, Kojetin DJ, Lundasen T, et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature (2012) 485:62–8. doi:10.1038/nature11030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Banerjee S, Wang Y, Solt LA, Griffett K, Kazantzis M, Amador A, et al. Pharmacological targeting of the mammalian clock regulates sleep architecture and emotional behaviour. Nat Commun (2014) 5:5759. doi:10.1038/ncomms6759
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Jones KA, Hatori M, Mure LS, Bramley JR, Artymyshyn R, Hong SP, et al. Small-molecule antagonists of melanopsin-mediated phototransduction. Nat Chem Biol (2013) 9:630–5. doi:10.1038/nchembio.1333
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, et al. PERIOD2:LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci U S A (2004) 101:5339–46. doi:10.1073/pnas.0308709101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Chen Z, Yoo SH, Park YS, Kim KH, Wei S, Buhr E, et al. Identification of diverse modulators of central and peripheral circadian clocks by high-throughput chemical screening. Proc Natl Acad Sci U S A (2012) 109:101–6. doi:10.1073/pnas.1118034108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Yoo SH, Mohawk JA, Siepka SM, Shan Y, Huh SK, Hong HK, et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell (2013) 152:1091–105. doi:10.1016/j.cell.2013.01.055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Hirota T, Lee JW, St John PC, Sawa M, Iwaisako K, Noguchi T, et al. Identification of small molecule activators of cryptochrome. Science (2012) 337:1094–7. doi:10.1126/science.1223710
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Liu AC, Welsh DK, Ko CH, Tran HG, Zhang EE, Priest AA, et al. Intercellular coupling confers robustness against mutations in the SCN circadian clock network. Cell (2007) 129:605–16. doi:10.1016/j.cell.2007.02.047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Buhr ED, Yoo SH, Takahashi JS. Temperature as a universal resetting cue for mammalian circadian oscillators. Science (2010) 330:379–85. doi:10.1126/science.1195262
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Yamaguchi Y, Suzuki T, Mizoro Y, Kori H, Okada K, Chen Y, et al. Mice genetically deficient in vasopressin V1a and V1b receptors are resistant to jet lag. Science (2013) 342:85–90. doi:10.1126/science.1238599
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Schroeder AM, Colwell CS. How to fix a broken clock. Trends Pharmacol Sci (2013) 34:605–19. doi:10.1016/j.tips.2013.09.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Lefta M, Wolff G, Esser KA. Circadian rhythms, the molecular clock, and skeletal muscle. Curr Top Dev Biol (2011) 96:231–71. doi:10.1016/B978-0-12-385940-2.00009-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Vitaterna MH, Ko CH, Chang AM, Buhr ED, Fruechte EM, Schook A, et al. The mouse clock mutation reduces circadian pacemaker amplitude and enhances efficacy of resetting stimuli and phase-response curve amplitude. Proc Natl Acad Sci U S A (2006) 103:9327–32. doi:10.1073/pnas.0603601103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Ju YE, Lucey BP, Holtzman DM. Sleep and Alzheimer disease pathology – a bidirectional relationship. Nat Rev Neurol (2014) 10:115–9. doi:10.1038/nrneurol.2013.269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Hogenesch JB, Herzog ED. Intracellular and intercellular processes determine robustness of the circadian clock. FEBS Lett (2011) 585:1427–34. doi:10.1016/j.febslet.2011.04.048
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Zhang EE, Liu AC, Hirota T, Miraglia LJ, Welch G, Pongsawakul PY, et al. A genome-wide RNAi screen for modifiers of the circadian clock in human cells. Cell (2009) 139:199–210. doi:10.1016/j.cell.2009.08.031
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Muller JE, Tofler GH, Stone PH. Circadian variation and triggers of onset of acute cardiovascular disease. Circulation (1989) 79:733–43. doi:10.1161/01.CIR.79.4.733
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Zuber AM, Centeno G, Pradervand S, Nikolaeva S, Maquelin L, Cardinaux L, et al. Molecular clock is involved in predictive circadian adjustment of renal function. Proc Natl Acad Sci U S A (2009) 106:16523–8. doi:10.1073/pnas.0904890106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Curtis AM, Fitzgerald GA. Central and peripheral clocks in cardiovascular and metabolic function. Ann Med (2006) 38:552–9. doi:10.1080/07853890600995010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Scheer FA, Hu K, Evoniuk H, Kelly EE, Malhotra A, Hilton MF, et al. Impact of the human circadian system, exercise, and their interaction on cardiovascular function. Proc Natl Acad Sci U S A (2010) 107:20541–6. doi:10.1073/pnas.1006749107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
166. St-Onge MP, O’Keeffe M, Roberts AL, RoyChoudhury A, Laferrere B. Short sleep duration, glucose dysregulation and hormonal regulation of appetite in men and women. Sleep (2012) 35:1503–10. doi:10.5665/sleep.2198
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: sleep, circadian clock, metabolic disease, obesity, intervention, small molecules
Citation: Nohara K, Yoo S-H and Chen Z (2015) Manipulating the circadian and sleep cycles to protect against metabolic disease. Front. Endocrinol. 6:35. doi: 10.3389/fendo.2015.00035
Received: 20 January 2015; Accepted: 03 March 2015;
Published online: 23 March 2015.
Edited by:
Sridhar R. Gumpeny, Endocrine and Diabetes Centre, IndiaReviewed by:
Timo Dirk Mueller, University of Cincinnati Metabolic Diseases Institute, USATamara Castañeda, German Diabetes Center, Germany
Michael T. Sellix, University of Rochester School of Medicine and Dentistry, USA
Copyright: © 2015 Nohara, Yoo and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zheng (Jake) Chen, Department of Biochemistry and Molecular Biology, The University of Texas Health Science Center at Houston, 6431 Fannin Street, MSB 6.200, Houston, TX 77030, USA e-mail:emhlbmcuY2hlbi4xQHV0aC50bWMuZWR1