 Susan M. Farabaugh
Susan M. Farabaugh David N. Boone
David N. Boone Adrian V. Lee
Adrian V. Lee- 1Department of Pharmacology and Chemical Biology, University of Pittsburgh, Pittsburgh, PA, USA
- 2Women’s Cancer Research Center, Magee-Womens Research Institute, University of Pittsburgh Cancer Institute, University of Pittsburgh, Pittsburgh, PA, USA
- 3Department of Human Genetics, University of Pittsburgh, Pittsburgh, PA, USA
Insulin-like growth factor (IGF) signaling is fundamental for growth and survival. A large body of evidence (laboratory, epidemiological, and clinical) implicates the exploitation of this pathway in cancer. Up to 50% of breast tumors express the activated form of the type 1 insulin-like growth factor receptor (IGF1R). Breast cancers are categorized into subtypes based upon hormone and ERRB2 receptor expression and/or gene expression profiling. Even though IGF1R influences tumorigenic phenotypes and drug resistance across all breast cancer subtypes, it has specific expression and function in each. In some subtypes, IGF1R levels correlate with a favorable prognosis, while in others it is associated with recurrence and poor prognosis, suggesting different actions based upon cellular and molecular contexts. In this review, we examine IGF1R expression and function as it relates to breast cancer subtype and therapy-acquired resistance. Additionally, we discuss the role of IGF1R in stem cell maintenance and lineage differentiation and how these cell fate influences may alter the differentiation potential and cellular composition of breast tumors.
IGF Signaling
Insulin-like growth factor 1 and 2 (IGF1 and IGF2) are peptides that act as circulating endocrine hormones critical for normal body growth. Pituitary-derived growth hormone (GH) stimulates the liver to express IGF1, which is secreted and affects growth of multiple cell types (1, 2). Other tissues also produce IGF ligands that act in a paracrine or autocrine manner (3). IGF ligands are bound by insulin-like growth factor binding proteins (IGFBP1-6) (4). While IGFBP binding increases ligand stability, IGFBP binding also decreases ligand bioavailability and competes with ligand-receptor binding.
Insulin-like growth factor 1 or IGF2 stimulates downstream signaling events primarily by binding and activating the type 1 insulin-like growth factor receptor (IGF1R). Insulin-like growth factor type 2 receptor (IGF2R), which is homologous to the mannose-6-phosphate receptor, does not seem to have a signaling function and may act as a sink to modulate IGF2 ligand bioavailability. IGF1R has high similarity to the insulin receptor (InsR) (5, 6). Indeed, although the affinities are much lower, IGF1 can bind and activate the InsR while, in turn, insulin can bind and activate IGF1R (7). IGF2 binds both receptors with similar affinities. In addition, a fetal form of InsR that has alternate splicing, termed as insulin receptor isoform A (InsR-A), has a high affinity for IGF2. IGF1R and InsR exist primarily as heterotetramers. However, hybrid IGF1R/InsR receptors, consisting of subunits from both receptors, can form and bind all three ligands (8–13). Therefore, there is much crosstalk and overlap in receptor signaling downstream of IGFs and insulin.
Binding of IGF1 or IGF2 to IGF1R results in auto-phosphorylation of the IGF1R kinase domain and activation of intracellular signaling cascades. Insulin receptor substrates 1 and 2 (IRS1 and IRS2), the main signaling adaptors for both IGF1R and InsR, are recruited to the receptor and act as docking sites (14). IRS1 and IRS2 link the activated receptors to numerous intracellular adaptor proteins and downstream signaling cascades such as PI3K/AKT and RAS/MAPK/ERK1 (15).
Type 1 insulin-like growth factor receptor signaling cascades regulate cell growth, survival, and motility. The IGF1R pathway mediates strong anti-apoptotic signals through three known pathways. The PI3K/AKT and RAS/MAPK pathways both facilitate IGF1R-induced resistance to apoptosis (16). Additionally, IGF1R interacts with 14.3.3 proteins to induce the mitochondrial translocation of Raf (17). All three of these pathways converge to phosphorylate BAD and block apoptosis. Additionally, IGF1R mediates the cell cycle through MAPK/ERK activation. ERK induces proliferation through phosphorylation of transcription factors such as c-Fos and Ets-like transcription factor 1 (Elk-1) (18). IGF1R signaling also activates c-Myc, JNK, and c-Jun (19). Cell cycle progression is promoted by IGF1R-mediated increases in ribosome activity (20) and expression of cyclins A, B, and D1 (21, 22). These robust anti-apoptotic properties of IGF1R and cell cycle regulation both play critical roles in promoting IGF1R-mediated tumorigenesis.
IGF1 Signaling and Mammary Gland Function
Insulin-like growth factor 1 is critical for mammary development. In the mammary gland, GH stimulates IGF1 production from the stroma (23), and this is enhanced by estradiol (24, 25). IGF-I acts in a paracrine manner to stimulate terminal end bud (TEB) growth and form the ductal structures that extend through the mammary fat pad. Mammary gland development is decreased in IGF1 (−/−) null females (26). Interestingly, IGF1 produced within the mammary gland is more potent at stimulating mammary growth than circulating IGF1 derived from the liver (27). The IGF1 deficiency in IGF1 (−/−) null mice can be rescued by continuous 5-day injections of the des-IGF1 variant. Longer treatments with IGF1 plus estradiol enhance TEB formation and ductal morphogenesis. Interestingly, treatment with GH and estradiol does not restore development in IGF1-null mice, indicating the necessity of IGF1 for the full function and stimulation by these hormones (26).
Similar to the critical role for IGF1 in mammary gland development, IGF1R is also required for mammary gland morphogenesis. Reconstitution assays using IGF1R-deficient embryonic mammary buds demonstrate decreased growth potential and cell proliferation during morphogenesis (28). The reduction in mammary gland morphogenesis observed upon loss of IGF1/IGF1R signaling is a result of decreased cell cycle progression and increased apoptosis (29–31).
Although critical for mammary gland development, IGF1 needs to function in cooperation with growth factors such as epidermal growth factor (EGF) and transforming growth factor beta (TGF-B) as well as the IGF1 receptors and binding proteins for efficient signaling and full mammary function (32).
The IGF Pathway in Cancer
As IGF1 plays such a critical role in cell growth, survival, and migration, it is not surprising that alterations in the IGF1 signaling pathway are linked to the development and progression of multiple cancers including breast, lung, osteosarcoma, gynecological, prostate, and gastrointestinal cancers. Many different alterations in the IGF system promote tumorigenesis: increased IGF1 and IGF2 expression (33–36), decreased levels of circulating IGFBPs (which increase ligand bioavailability) (37–39), and changes in receptor expression (40, 41).
Recent large genomic analyses now allow a comprehensive examination of genomic and molecular changes in the IGF pathway in cancer. Analysis of data from The Cancer Genome Atlas (TCGA) using the cBIO portal across most cancer types shows genomic alterations in IGF ligands (IGF1, 2), receptors (IGF1R, IGF2R), binding proteins (IGFBP1–6), and IRSs (IRS1, 2, 4); this representing 18 genes (Figure 1A). The greatest change is in stomach cancer where 43% of tumors show a molecular alteration (amplification, deletion, or base pair mutation) in one or more of the 18 genes of the IGF family. Interestingly, different tumor types show distinct molecular changes. For example, sarcomas exhibit copy number changes such as DNA amplification and deletion, whereas pancreas, melanoma, and lung cancers are dominated by base pair mutations. Some tumor types display a very low to absent mutation rate such as thyroid, acute myelogenous leukemia, and glioblastoma.
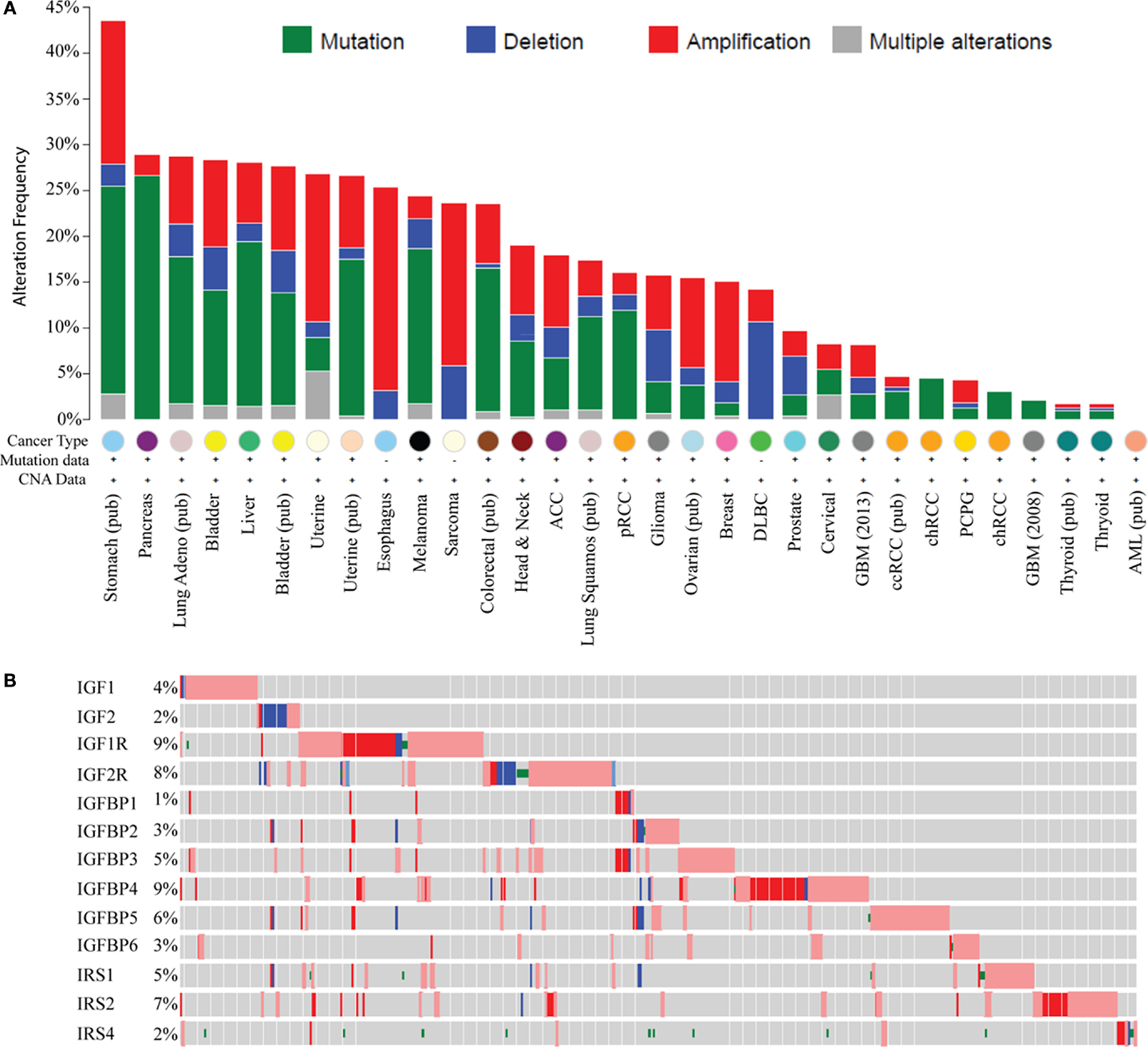
Figure 1. Genomic and transcriptomic alterations in the IGF pathway in cancer. Data are from The Cancer Genome Atlas at cBio (http://www.cbioportal.org/) in February 2015. For several cancers, the data are published (pub) while for others it is provisional and in progress. (A) Histogram showing genomic changes (somatic base pair mutation and copy number alteration) in 13 members of the IGF pathway [notated on the y-axis in (B)]. Each bar represents the percentage of tumors, which show an alteration in these genes and each cancer type is indicated on the X-axis. (B) Genomic and transcriptomic changes (mRNA levels) specifically in breast cancer for 13 genes indicated on the y-axis. Each row represents a different gene. Each gray box represents an individual tumor. The percentage of tumors showing an alteration for each gene is shown on the Y -axis next to the gene. A red box indicates the gene is amplified and blue is deleted. A box with a red outline represents RNA overexpression compared to normal breast and blue is underexpression. A green dot represents a somatic base pair mutation. Tumors without an alteration in any of the IGF family members have been removed for visualization.
In breast cancer, 15% of TCGA-documented breast cancers (42) contain genomic alterations in the IGF pathway. These alterations consist mainly of amplification and are generally rare with only IGF1R and IRS2 showing amplification in >5% of cases. When considering mRNA levels (using the TCGA provisional data on 962 breast cancers which includes RNA-seq), 45.3% of breast cancers (n = 436/962) show a molecular alteration in at least one IGF family member. IGFIR is amplified, overexpressed, or somatically mutated in 9% of tumors. Other notable changes are amplification or overexpression of IGFBP4 (9%), IGFBP5 (6%), and IRS2 (7%) (Figure 1B).
While somatic mutation of IGF family members has only recently been comprehensively described, a large literature has reported on germline polymorphisms. In particular, germline polymorphisms in IGF1, IGF1R, and IGFBP3 are associated with increased risk of breast (43–45), prostate (46–48), lung (49), and pancreatic (50) cancers.
While many studies have examined alterations in IGF pathway components, few measured pathway activity. For example, tumor tissue microarrays show that 87% of primary breast tumors express IGF1R (51); however, the active phosphorylated form of IGF1R/InsR, as measured by immunohistochemistry (IHC), is only present in roughly 50% of breast cancers where it correlates with poor survival (52). We examined IGF pathway activity by combining IGF-regulated mRNA levels into an “IGF gene signature.” Breast tumors expressing the IGF1 gene signature significantly correlate with numerous poor prognostic factors and expression of this signature is one of the strongest indicators of poor disease outcome (53).
IGF1R Across Breast Cancer Subtypes
Breast tumors display tremendous heterogeneity among different patients due at least in part to varying molecular alterations and divergent cells of origin. In recent years, gene expression profiling has helped to define breast cancer subtypes. Molecular profiling divides breast tumors into six major subtypes, which are related to the known major drivers (and targetable biomarkers): estrogen receptor α (ERα), progesterone receptor (PR), and erbb2 receptor tyrosine kinase 2 (ERBB2/HER2). The current molecular subtypes include luminal A, luminal B, ERBB2-like, triple negative/basal-like, claudin-low, and normal-like (54, 55). Each subtype can also be further classified into more defined subgroups.
The expression and role of IGF1R in various breast cancer subtypes, and in particular its role in causing resistance to targeted therapies, has been extensively studied. By identifying in which tumor types the IGF1R pathway actively drives tumor initiation and progression, we can better define the subtype(s) that may benefit from anti-IGF1R therapies. For an overview of IGF pathway expression across the breast cancer molecular subtypes, we analyzed TCGA data (42) (Figure 2). Most IGF pathway members, including IGF1R itself, tend to be more highly expressed in luminal A and luminal B tumors and comparatively underexpressed in basal and ERBB2+ tumor types (Figure 2). This expression only denotes mRNA levels and does not demonstrate pathway activation.
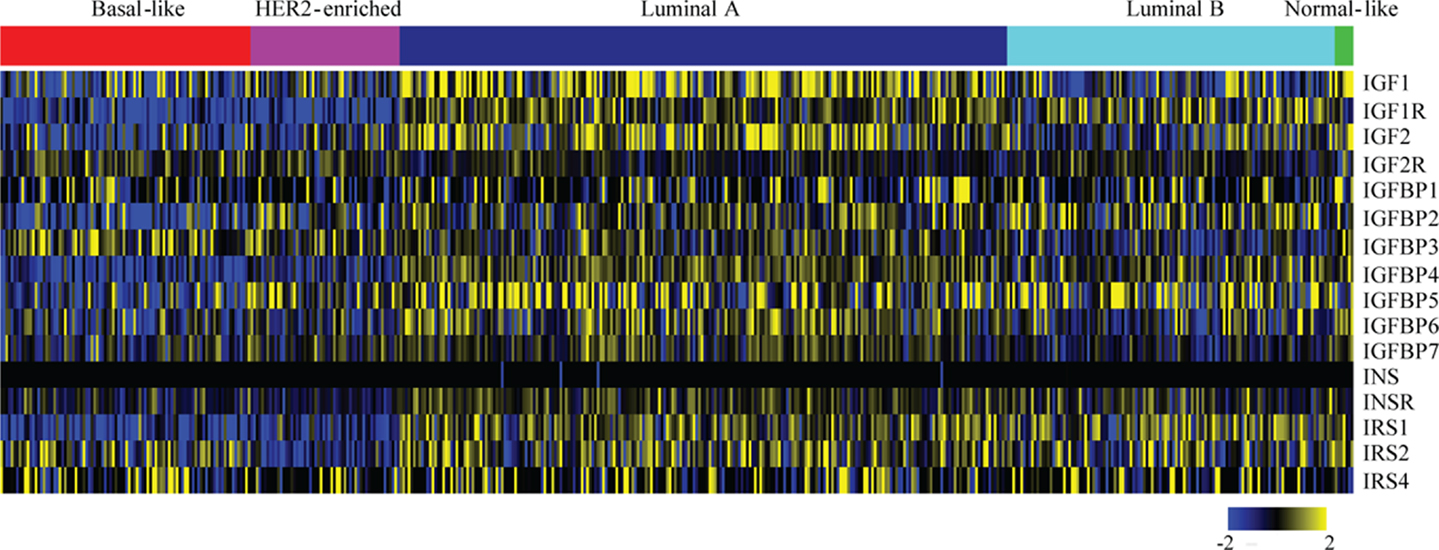
Figure 2. Expression levels of IGF pathway components across breast cancer subtypes. Level 3, IlluminaHiSeq_RNASeqV2 normalized gene expression values for all breast cancer tumors were downloaded from The Cancer Genome Atlas. The expressions of the indicated genes from tumors (columns) with calculated PAM50 scores (42) were extracted, log2 transformed, median centered for each gene (rows), and a heatmap was generated using MeV. The PAM50 subtype clusters are shown above with the indicated colors.
Below, we describe the main breast cancer molecular subtypes and the role IGF1R is believed to play in each.
Luminal
The majority of breast cancers fall into the luminal classification. Luminal subtypes tend to be hormone receptor positive (ER+, PR+) and comprise ~50% (luminal A) and ~10–20% (luminal B) of all breast cancer cases (56). Luminal A tumors have the best overall prognosis. Luminal B tumors are similar to luminal A tumors, but are more aggressive (higher grade) and are typically diagnosed at a younger age with higher reoccurrence rates. Hormone therapies (e.g., tamoxifen and aromatase inhibitors) have greatly increased the overall prognosis of the luminal subtype.
Estrogen receptor α is a major regulator of IGF signaling, due in part to transcriptional activation of IGF1R and many other IGF signaling components such as IRS1 (57–59). Consistent with this, the hormonally driven luminal subtypes tend to have higher levels of IGF1R and IRS expression as opposed to tumors that are less hormonally driven (triple negative and ERBB2+) (60, 61). IGF1R is expressed in 52 and 84% of Luminal A and 57.5 and 76% of Luminal B tumors, respectively (62, 63). IGF1R expression does not affect breast cancer specific survival in luminal A tumors. Interestingly, luminal B tumors with higher total IGF1R levels have significantly better prognosis than those with low levels of IGF1R (62). Law et al. demonstrated that roughly 50% of all luminal tumors show phosphorylated, and presumably active, IGF1R (52). As IGF1R is upregulated by ERα, the better prognosis of IGF1R-expressing luminal tumors may be associated with the use of ER-targeted therapies.
Estrogen receptor α and the IGF pathway show dynamic and intricate crosstalk, resulting in bidirectional regulation of expression and activity (64). ERα transcriptionally upregulates IGF1R expression. IGF1R transcriptionally upregulates ERα in an mTOR/S6K1-dependent manner and increases ERα phosphorylation to stimulate transcriptional activity (65, 66). Importantly, during endocrine resistance, converging growth factor signaling on the PI3K/AKT and MAPK/ERK pathways bypass the need for ERα activity (67–69). Surprisingly, however, use of anti-IGF1R therapy in the setting of endocrine resistance does not improve prognosis (70).
Consistent with its ERα-dependent regulation, IGF1R levels are reduced in many tamoxifen- and aromatase inhibitor-resistant cell and mouse tumor models (71–73) as well as patient tumors (74). However, the remaining IGF1R is strongly phosphorylated with hyperactive IGF1R/InsR/PI3K/AKT/mTOR signaling beyond pre-resistance levels, suggesting that the cells/tumors acquire resistance through an IGF1R-directed mechanism even in cases of reduced IGF1R levels (69, 73). Furthermore, the Yee lab has shown that while IGF1R is reduced in tamoxifen resistant breast cancer cells, InsR is still expressed and able to signal via insulin to promote growth (75).
Recently, the G protein estrogen receptor 1 (CPER/GPR30) has been identified as a potential mediator of rapid estrogens response. Increased GPER expression is associated with increased risk of metastasis and poor survival (76). Both IGF1 and insulin upregulate GPER expression through the c-Fos/AP1 pathway. IGF1 and insulin transactivate GPER to promote migration and proliferation (77, 78). ERα is required for IGF1-induced transactivation of GPER (77). Interestingly, GPER expression increases in tumors treated with tamoxifen (79) and correlates with a poorer prognosis specifically in tamoxifen treated patients (79–82). These results suggest that GPER may be a potential pathway for IGF1- and insulin-induced tamoxifen resistance.
Given basic signaling mechanisms linking the IGF and ERα pathways, combined targeted therapy has been suggested as a potential unique therapeutic strategy in breast cancer. For this reason, and following on from several preclinical studies, several trials targeting IGF1R in luminal breast cancer were performed. Unfortunately, anti-IGF1R therapy provided little benefit in this setting. It should be noted, however, that therapy was given to all patients independent of whether the IGF pathway was present or active (e.g., in the absence of biomarkers). Several in vitro studies suggest that biomarker selection is critical for identifying the tumors that will respond to IGF1R inhibitors (70, 83).
A major hurdle to anti-IGF1R therapy is the intricate network of feedback that occurs in this and related pathways. For example, the PI3K/Akt/mTOR/S6K pathway exerts a major negative feedback upon IGF1R/IRSs and when any part of this pathway is inhibited there is a concomitant increase in IGF1R activity. This was noted in Phase I trials of an mTOR inhibitor and validated in many preclinical studies (84). For example, inhibition of AKT in long-term estrogen deprived cell lines results in positive feedback that upregulates several upstream growth factor proteins through FoxO and ERα-regulated transcription, including IGF1R and IGF ligands (85). Combined inhibition of IGF1R/IR along with AKT inhibition and ER deprivation enhances the anti-tumor effect in vivo (85). The ability of the pathway to autoregulate and compensate for ER downregulation appears to be the cause for endocrine therapeutic resistance. The only way to impede resistance may be through inhibition of the overarching converging system, targeting multiple intercrossing growth signaling pathways to limit compensation on as many levels as possible.
ERBB2-Like
ERBB2 (HER2)-like tumors comprise ~15% of breast cancers and are typically hormone receptor negative with a 40% probability of p53 mutation (56). The ERBB2-like subtype shows poorer prognosis than luminal tumors, with early age of onset, higher tumor grade, and lymph node positivity. Patients with ERBB2-like tumors tend to have early recurrence and a poorer prognosis.
Accumulating evidence indicates crosstalk between ERBB2 and IGF signaling in breast tumorigenesis. About 10–20% of ERBB2+ tumors express IGF1R protein (62, 63). Active phosphorylated IGF1R/IR is found in 49% (86) and 64% (52) of ERBB2+ tumors. Brown et al. found phosphorylated IGF1R/IR does not correlate with prognosis in trastuzumab-treated ERBB2+ tumors (86). However, Yerushalmi et al. observed that ERBB2+ tumors expressing higher total IGF1R protein levels have decreased breast cancer specific survival compared to the lower IGF1R-expressing ERBB2+ counterparts (62). In this study, these ERBB2-enriched tumors are the only subtype presenting a low patient prognosis in correlation with IGF1R expression (62).
The risk of recurrence is higher for ERBB2 positive breast cancers than for ERBB2 negative breast cancers. This anti-ERBB2 therapy resistance is often due to activation of alternative growth factor receptor pathways. ERBB2-postive tumors expressing strong IGF1R membrane staining are less likely to respond to trastuzumab and vinorelbine than those with negative or low IGF1R protein expression (87). Other studies do not indicate a correlation between IGF1R protein expression and trastuzumab response (86, 88, 89) unless IGF1R expression is combined expression of downstream IGF1R signaling effectors, such as PI3K or mTOR (88).
Unlike clinical data, in vitro breast cancer cell line data suggest a strong correlation between increased IGF1R activity and trastuzumab-resistance (90–93). Interestingly, miRNAs, which typically inhibit IGF1R, show decreased expression upon trastuzumab-resistance (93, 94) and providing one possible mechanism of IGF1R upregulation. In trastuzumab-resistant cell line models, IGF1R forms a complex with ERBB2, and even a triplex with ERBB3 (95–97). This heterodimer/trimer promotes crosstalk between the growth receptor pathways. For example, IGF1-induced IGF1R phosphorylation leads to ligand-independent phosphorylation of ERBB2, which circumvents trastuzumab antibody inhibition and leads to an ERBB2-based mechanism of resistance (95, 98). In trastuzumab-resistant cells, IGF1R-promoted ERBB2 phosphorylation and IGF1R-induced invasion are mediated by Src and FoxM1 (98). Co-targeting ERBB2 and IGF1R reduces Erk/AKT activation, cell proliferation, in vitro invasion, and xenograft tumor growth to a greater extent than targeting either receptor individually (98, 99). Interestingly, treating trastuzumab-resistant cells with metformin re-sensitizes cells by disrupting the ERBB/IGF1R complexes (97), again strongly suggesting that a combined therapy would hold promise for patients with ERBB2+ breast tumors.
Triple Negative Breast Cancer
The triple negative breast cancer (TNBC) subtype accounts for up to 10–20% of breast cancers. Approximately 75% of TNBCs have a basal-like phenotype (56). Most BRCA-mutant tumors fall into this subtype (100). TNBCs are defined by the absence of ER, PR, and ERBB2. Driver mutations and subsequent targeted therapies are currently unknown. These tumors tend to be high grade and poorly differentiated with high rates of recurrence and poor prognosis. Intriguingly, TNBCs respond well to neoadjuvant chemotherapy with high rates of pathological complete response as compared to response in other subtypes. However, TNBCs still trend toward a poorer prognosis with increased rates of recurrence. The disproportion between response and outcome suggests neoadjuvant therapies are not capable of abolishing the driving tumorigenic cell types, underlining the need for identifiable and targetable driver mutations in TNBC.
Women of African descent are three times more likely to have TNBC: 30% of breast cancers diagnosed in African-American women are TN as compared to 11–13% of non-African-American women (101–103). Interestingly, African-American women have higher IGF1R expression in normal breast tissue while Caucasian-Americans have higher levels of IGF2R (104). This differential IGF1R/IGF2R expression may explain the increased occurrence of the more aggressive TNBC subtype in African-American women. Although IGF1R levels are similar between normal and malignant African-American breast tissues, phosphorylation of IGF1R and its downstream effectors are significantly higher in the malignant samples (104). Consequently, IGF1 signaling and proliferation (detected by gene expression profiling) are higher in TNBCs from African-American women compared to European-American (51). These studies underscore the significance of IGF1R in TNBC.
About 22–46% of TNBCs express IGF1R protein (52, 62, 63) and this expression correlates with shorter survival (105). The IGF1 gene signature correlates with expression signatures of TNBC tumors and cell lines (106) where both sample types are responsive to IGF1 signaling, promoting proliferation, and cell survival (107).
Laboratory studies analyzing anti-IGF1R therapeutic response typically demonstrate a favorable response to TNBC therapies. We demonstrated that TNBC cell lines and a primary tumor xenograft are sensitive to the anti-IGF-IR/InsR tyrosine kinase inhibitor BMS-754807 (106). Surprisingly, expression of a dominant-negative IGF1R during MMTV-Wnt1-mediated tumorigenesis accelerates mammary tumor formation and promotes aggressiveness (108). Interestingly, these tumors possess IGF2 signaling as well as a suggested role for InsR signaling. Additional studies demonstrate that IGF1R inhibition does not abrogate IGF-induced phenotypes in the presence of increased IGF2/IGF2R signaling (109). In TCGA patient data, IGF2R expression is significantly higher in basal-like tumors as compared to luminal tumors (Figure 2) (p value <0.001, t-test). Taken together, these studies suggest that IGF1R inhibition may be beneficial in some triple negative breast cancers but that the benefit will be very context-dependent.
Recently, the G protein estrogen receptor 1 (GPER/GPR30) has been identified as a potential growth regulator of TNBCs (110, 111). GPER is believed to mediate rapid estrogen response independently of ER; and thus, can drive estrogen-responsive growth even in ER-negative cells. As mentioned above, IGF1 signaling induces GPER expression and GPER promotes IGF1-induced migration and proliferation (77, 78). More work need to be completed in this area to determine if GPER could be a potential biomarker for anti-IGF1R-responsive TNBCs.
Most BRCA1 tumors phenocopy TNBC (100). In line with BRCA1-mediated repression of the IGF1R promoter (112, 113), BRCA1-mutant tumors show elevated IGFIR and IGF1 levels, leading to reduced apoptosis, and enhanced survival (113–115). Importantly, inhibition of the IGF1R/PI3K/AKT pathway decreases proliferation in BRCA1-deficient cells (116). These studies suggest IGF1R signaling significantly contributes to tumor cell proliferation and survival in BRCA1-deficient breast cancers.
The Influence of IGF1R on Cell Potential and Cell Fate
IGF1R Signaling and Stemness
The IGF system regulates stem cell maintenance in normal tissue processes. In human embryonic stem cells, the stem cell niche produces IGF2, which is required for survival and expansion (117). In neural stem cells, IGF2 is believed to bind and act through the InsR-A rather than IGF1R (118). Conversely, the human embryonic niche relies on the IGF2/IGF1R axis for self renewal and stem cell expansion (117), suggesting the necessity of IGF1R-promoted signaling in maintaining the stem cell population. In the hematopoietic and muscular system, expression of a skeleton muscle-localized IGF1 transgene enhances skeletal muscle regeneration in irradiated mice in part by recruiting proliferating bone marrow-derived cells, increasing stem cell marker populations, and accelerating myogenic differentiation (119, 120). Additionally in the hematopoietic system, IGF1R levels in newborn umbilical cord blood correlates positively to the total number of hematopoietic stem and progenitor cells (121). These studies demonstrate a role for IGF1R in regulation of stem and progenitor cell populations.
A recent review published by Roberta Malaguarnera and Antonio Belfiore summarized in great detail the known pathways and links between the IGF1R pathway and the epithelial-to-mesenchymal transition (EMT) and stem cell-related processes across several tissue types, both normal and cancerous (122). EMT is a naturally occurring process for remodeling of tissues and wound healing where polarized epithelial cells lose adherence and gain mesenchymal characteristics, including enhanced mobility and matrix invasion. Tumors often undergo EMT in an out-of-context manner. Interestingly, cells undergoing EMT also acquire stem cell-associated characteristics such as the capacity for self-renewal, gain of specific gene expression changes and cell surface markers, and ability to initiate tumorigenesis (123). The gain of these stem cell-associated properties suggests an overlap between EMT and stem cell mechanisms. In cancerous tissues, these overlapping mechanisms, now activated out-of-context, may play key roles in metastatic spread and resistance to cancer treatments.
Insulin-like growth factor 1 signaling is an integral part of both the EMT and stem cell-related processes in normal and cancerous tissues. Numerous in vitro studies demonstrate IGF1R as a driver of self renewal, stem cell surface markers, migration, and invasion in both normal and cancerous tissues and tumor initiation in hepatic, lung, prostate, and breast cancers (122). Each of these studies has begun to reveal the mechanisms of IGF1R-regulated EMT and stem phenotypes. Stem-promoting signaling pathways such as Wnt/B-catenin (124–127), Notch (128–130), and Shh (131, 132) act upstream to increase IGF1R expression with cross talk and regulation at the IGF1R promoter level by Sp1 and HMGA1 (133–136). In addition to upstream regulation by these master stem cell master controllers, IGF1R promotes positive downstream feedback through regulation and interaction with the well known EMT and stemness-linked transcription factors Zeb1 (137), NFκB (138), Snail (138), Twist (139), and p53, Sox2, Oct4, Nanog (140–142). Additionally, the tumor suppressor p53, known for inhibition of many stem cell regulators, inhibits IGF1R (143), which in turn act to downregulate p53. Interaction with these numerous stem-related pathways and factors strongly supports the central role of IGF1R in both the induction and the maintenance of stemness and EMT.
This role for IGF1R in promotion of stem-like characteristics has been specifically demonstrated in primary breast cancer. In human primary breast cancer xenografts, total and phosphorylated IGF1R expression is significantly higher in the CD44+CD24− sorted breast cancer stem cell population compared to the non-CD44+CD24− population (144). IGF1R expression and upregulated AKT activity are required for maintenance of this population. IGF1R inhibition reduces the aldehyde dehydrogenase+ stem-like population and suppresses mammosphere-forming capacity. Notably, silencing of IGF1R reduces tumor initiating ability of the xenografts. (144). Together this data demonstrate an active role for IGF1R in driving mammary stem-like phenotypes in vitro and in vivo.
IGF1R and Lineage Fate
In addition to stem cell maintenance, two new studies suggest a link between IGF1R and cell lineage fate. In thyroid tissues, cell lineage differentiation is associated with IGF pathway activation (145). A similar trend is observed in neural cells. The lineage restricted neural progenitors primarily express IGF1R while the neural stems cells appear to rely more heavily on IGF2 and IR-A signaling (118). These studies suggest that IGF1R may be responsible for promoting cell fate, or at the very least, be restricted to maintaining differentiated cells of a specific lineage.
Morphogenesis and homeostasis of the mammary gland relies on stem cell function for the production and maintenance of the myoepithelial and luminal lineages. The TEBs contain the mammary gland stem cell niche and is where lineage differentiation and ductal morphogenesis occurs. IGF1 signaling promotes the development of the TEB. TEB formation and ductal outgrowth are grossly impaired in IGF1−/− mice (26), signifying that IGF1 signaling is significant for development and/or maintenance of the mammary stem cell niche. While IGF1R-null mice die postnatally before mammary gland development, mammary gland transplantation of embryonic IGF1R-null mammary buds shows reduced ductal growth similar to IGF1-null mice (28). The IGF pathway is also important in pregnancy and lactation where luminal differentiation is vital. During early pregnancy, alveolar differentiation is reduced in heterozygous IGF1 mice (146). This same lack of alveolar budding and decreased alveolar density is observed in transgenic mice containing a pregnancy-induced kinase-dead IGF1R (147). Thus, the IGF pathway seems to have a role not only in mammary gland stem cell maintenance but also in lineage specification throughout the many stages of mammary gland development.
IGF1R and Breast Cancer Lineages
Breast tumors show both intra- and inter-tumor heterogeneity, suggesting distinct tumor initiation and progression pathways for each tumor type. Breast tumor heterogeneity likely results from a combination of both clonal outgrowth and aberrant differentiation of progeny. Defining the breast cancer cell of origin is a topic of major interest. Based on the cell of origin hypothesis, the heterogeneity of the tumor is restricted by a strictly linear differentiating breast cell hierarchy. Basically, the characteristics and differentiation potential of the tumor cells are restricted to the characteristics and differentiation potential of the cell of origin, which, in this case, is typically considered as a stem or progenitor cell. A variation on this cell of origin concept is that the tumor phenotype and potential is at least partially, if not primarily, determined by the genetic alterations acquired and not solely on the mammary lineage of origin. This variation suggests that the gained alterations activate or deactivate pathways out of context to alter a cell’s capabilities for differentiation and cell fate. Whether the heterogeneity is determined by the cell of origin or the genetic alterations or a mixture of explanations, the underlying importance is in understanding the potential and fate of the individual tumor cells.
Type 1 insulin-like growth factor receptor’s role in mammary stem cell maintenance and necessity for lineage differentiation suggests that aberrantly expressed IGF1R may be capable of enhancing cell potential and altering cell fate in a tumor, perhaps even in tumors composed of fully differentiated cells. As discussed above, IGF1R expression is essential for driving luminal alveolar differentiation, linking IGF1R to the luminal lineage. In breast cancer, IGF1R expression correlates most strongly with luminal breast cancers (60, 61). This expression may be a result of ER-driven growth through IGF1R rather than a causative link between IGF1R and the luminal lineage; nevertheless, the presence of IGF1R may still affect cell signaling and perhaps cell lineage. In addition to luminal tumors, IGF1R actively promotes tumor growth and survival in p53 and BRCA1-mutant tumors, which usually emulate the basal-like subtype (113–115). Although basal-like breast cancers are defined by basal and myoepithelial marker expression (148), they present with a luminal progenitor gene signature (148–152). In support of this luminal link, recent studies suggest BRCA1-associated basal-like tumors derive from a luminal progenitor cell of origin rather than a basally positioned cell (150, 153). Elevated IGF1R expression and signaling in these basal-like tumors appear to have active roles in tumor promotion. Thus, as IGF1R is associated with cell lineage fate, IGF1R signaling may be influencing the gain and loss of lineage markers and phenotypes in these tumors. Taken all together, these data suggest that IGF1R may be connected with driving lineage fate, particularly luminal-associated fate.
Therapeutic Targeting of IGF1R as an Anti-Cancer Therapy
Based upon extensive basic, preclinical, and clinical evidence, a range of anti-IGF1R therapeutic strategies have been developed, including humanized monoclonal antibodies, which prevent ligand binding and small-molecule inhibitors that inhibit the tyrosine kinase domain. Early clinical data from anti-IGF1R trials were very promising. In a Phase I trial of the monoclonal antibody, AMG 479, a patient with chemo-refractory Ewing sarcoma had complete remission (154). In a Phase II trial, 14% of 125 patients with recurrent or refractory sarcoma responded to the monoclonal IGF1R antibody, R1507 (155, 156). In both of these trials, therapy was well tolerated. In contrast, two large NSCLC Phase III trials of figitumumab in combination with either carboplatin and paclitaxel or Tarceva, respectively, were terminated due to lack of response and severe toxicities (157, 158).
The lack of response in anti-IGF1R trials has been a large concern. The failure may be a consequence of trial design and not the efficacy of the IGF1R inhibitors themselves (159). In the Phase III trials, patients were not screened for IGF1R expression, limiting the percent of patients that even had a chance to respond to the therapy. Additionally, monoclonal antibody inhibition is specific to IGF1R and does not inhibit the InsR, which can also stimulate tumorigenesis. InsR signaling increases upon IGF1R inhibition, suggesting pathway compensation (160–163). As touched on throughout the above review, there is also substantial crosstalk between many of the growth factor signaling pathways, including IGFR, EGFR, ErbB2, and ERα. A combinatory therapy approach may be needed for efficient suppression without compensation or resistance.
Before continuing anti-IGF1R therapies, it is necessary to define additional IGF1R-based biomarkers to more accurately predict anti-tumor response and identify responsive tumors. A better understanding of the important components of the IGF1R signaling pathway and the instances where crosstalk and compensation can occur is required. Only then can we pinpoint the cancer subtypes that will benefit from IGF1R therapeutics, alone or in combination with other inhibitors.
Overview
Type 1 insulin-like growth factor receptor plays a key role in cancer promotion, resistance, and recurrence across breast cancer subtypes. Understanding the role of IGF1R in individual subtypes is critical to better targeting. The current research highlights IGF1R as necessary for stem cell maintenance and expansion. In turn, additional studies demonstrate the need and expression of IGF1R in more differentiated populations. Taken together, these studies suggest that IGF1R may not only be driving stem cell characteristics, leading to increased therapy resistance, but also that altered IGF1R signaling in tumor cells may influence expression of lineage-linked traits and direct lineage potential and fate, contributing to the heterogeneity of tumors.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the University of Pittsburgh. This work was supported in part by the National Cancer Institute of the National Institutes of Health under award number R01CA94118 (AL), a postdoctoral fellowship from the Department of Defense W81XWH-14-1-0063 (SF), and a postdoctoral fellowship from Susan G. Komen for the Cure (DB). AL is a recipient of a Scientific Advisory Council award from Susan G. Komen for the Cure, and a Hillman Foundation Fellow. The authors acknowledge support from the University of Pittsburgh Cancer Institute and UPMC.
References
1. Thissen JP, Ketelslegers JM, Underwood LE. Nutritional regulation of the insulin-like growth factors. Endocr Rev (1994) 15:80–101. doi: 10.1210/edrv-15-1-80
2. Tatar M, Bartke A, Antebi A. The endocrine regulation of aging by insulin-like signals. Science (2003) 299:1346–51. doi:10.1126/science.1081447
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Ellis MJ, Jenkins S, Hanfelt J, Redington ME, Taylor M, Leek R, et al. Insulin-like growth factors in human breast cancer. Breast Cancer Res Treat (1998) 52:175–84. doi:10.1023/A:1006127621512
4. Furstenberger G, Senn HJ. Insulin-like growth factors and cancer. Lancet Oncol (2002) 3:298–302. doi:10.1016/S1470-2045(02)00731-3
5. Munshi S, Hall DL, Kornienko M, Darke PL, Kuo LC. Structure of apo, unactivated insulin-like growth factor-1 receptor kinase at 1.5 A resolution. Acta Crystallogr D Biol Crystallogr (2003) 59:1725–30. doi:10.1107/S0907444903015415
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
6. Chitnis MM, Yuen JS, Protheroe AS, Pollak M, Macaulay VM. The type 1 insulin-like growth factor receptor pathway. Clin Cancer Res (2008) 14:6364–70. doi:10.1158/1078-0432.CCR-07-4879
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Danielsen A, Larsen E, Gammeltoft S. Chromaffin cells express two types of insulin-like growth factor receptors. Brain Res (1990) 518:95–100. doi:10.1016/0006-8993(90)90958-E
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Moxham CP, Duronio V, Jacobs S. Insulin-like growth factor I receptor beta-subunit heterogeneity. Evidence for hybrid tetramers composed of insulin-like growth factor I and insulin receptor heterodimers. J Biol Chem (1989) 264:13238–44.
9. Soos MA, Siddle K. Immunological relationships between receptors for insulin and insulin-like growth factor I. Evidence for structural heterogeneity of insulin-like growth factor I receptors involving hybrids with insulin receptors. Biochem J (1989) 263:553–63.
10. Treadway JL, Morrison BD, Goldfine ID, Pessin JE. Assembly of insulin/insulin-like growth factor-1 hybrid receptors in vitro. J Biol Chem (1989) 264:21450–3.
11. Pandini G, Vigneri R, Costantino A, Frasca F, Ippolito A, Fujita-Yamaguchi Y, et al. Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: evidence for a second mechanism of IGF-I signaling. Clin Cancer Res (1999) 5:1935–44.
12. Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, Belfiore A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem (2002) 277:39684–95. doi:10.1074/jbc.M202766200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Boone DN, Lee AV. Targeting the insulin-like growth factor receptor: developing biomarkers from gene expression profiling. Crit Rev Oncog (2012) 17:161–73. doi:10.1615/CritRevOncog.v17.i2.30
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Myers MG Jr, Sun XJ, Cheatham B, Jachna BR, Glasheen EM, Backer JM, et al. IRS-1 is a common element in insulin and insulin-like growth factor-I signaling to the phosphatidylinositol 3’-kinase. Endocrinology (1993) 132:1421–30. doi:10.1210/endo.132.4.8384986
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Dearth RK, Cui X, Kim HJ, Kuiatse I, Lawrence NA, Zhang X, et al. Mammary tumorigenesis and metastasis caused by overexpression of insulin receptor substrate 1 (IRS-1) or IRS-2. Mol Cell Biol (2006) 26:9302–14. doi:10.1128/MCB.00260-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Shelton JG, Steelman LS, White ER, McCubrey JA. Synergy between PI3K/Akt and Raf/MEK/ERK pathways in IGF-1R mediated cell cycle progression and prevention of apoptosis in hematopoietic cells. Cell Cycle (2004) 3:372–9. doi:10.4161/cc.3.2.624
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Peruzzi F, Prisco M, Morrione A, Valentinis B, Baserga R. Anti-apoptotic signaling of the insulin-like growth factor-I receptor through mitochondrial translocation of c-Raf and Nedd4. J Biol Chem (2001) 276:25990–6. doi:10.1074/jbc.M103188200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Buchwalter G, Gross C, Wasylyk B. Ets ternary complex transcription factors. Gene (2004) 324:1–14. doi:10.1016/j.gene.2003.09.028
19. Mittelstadt PR, Salvador JM, Fornace AJ Jr, Ashwell JD. Activating p38 MAPK: new tricks for an old kinase. Cell Cycle (2005) 4:1189–92. doi:10.4161/cc.4.9.2043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Sunami T, Byrne N, Diehl RE, Funabashi K, Hall DL, Ikuta M, et al. Structural basis of human p70 ribosomal S6 kinase-1 regulation by activation loop phosphorylation. J Biol Chem (2010) 285:4587–94. doi:10.1074/jbc.M109.040667
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Mahmoudabady M, Mathieu M, Touihri K, Hadad I, Da Costa AM, Naeije R, et al. Cardiac insulin-like growth factor-1 and cyclins gene expression in canine models of ischemic or overpacing cardiomyopathy. BMC Cardiovasc Disord (2009) 9:49. doi:10.1186/1471-2261-9-49
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Tognon CE, Sorensen PH. Targeting the insulin-like growth factor 1 receptor (IGF1R) signaling pathway for cancer therapy. Expert Opin Ther Targets (2012) 16:33–48. doi:10.1517/14728222.2011.638626
23. Kleinberg DL, Ruan W, Catanese V, Newman CB, Feldman M. Non-lactogenic effects of growth hormone on growth and insulin-like growth factor-I messenger ribonucleic acid of rat mammary gland. Endocrinology (1990) 126:3274–6. doi:10.1210/endo-126-6-3274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Ruan W, Catanese V, Wieczorek R, Feldman M, Kleinberg DL. Estradiol enhances the stimulatory effect of insulin-like growth factor-I (IGF-I) on mammary development and growth hormone-induced IGF-I messenger ribonucleic acid. Endocrinology (1995) 136:1296–302. doi:10.1210/endo.136.3.7867584
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Walden PD, Ruan W, Feldman M, Kleinberg DL. Evidence that the mammary fat pad mediates the action of growth hormone in mammary gland development. Endocrinology (1998) 139:659–62. doi:10.1210/endo.139.2.5718
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Ruan W, Kleinberg DL. Insulin-like growth factor I is essential for terminal end bud formation and ductal morphogenesis during mammary development. Endocrinology (1999) 140:5075–81. doi:10.1210/endo.140.11.7095
27. Richards RG, Klotz DM, Walker MP, Diaugustine RP. Mammary gland branching morphogenesis is diminished in mice with a deficiency of insulin-like growth factor-I (IGF-I), but not in mice with a liver-specific deletion of IGF-I. Endocrinology (2004) 145:3106–10. doi:10.1210/en.2003-1112
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Bonnette SG, Hadsell DL. Targeted disruption of the IGF-I receptor gene decreases cellular proliferation in mammary terminal end buds. Endocrinology (2001) 142:4937–45. doi:10.1210/endo.142.11.8500
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Hadsell DL, Bonnette SG, Lee AV. Genetic manipulation of the IGF-I axis to regulate mammary gland development and function. J Dairy Sci (2002) 85:365–77. doi:10.3168/jds.S0022-0302(02)74083-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Lee AV, Zhang P, Ivanova M, Bonnette S, Oesterreich S, Rosen JM, et al. Developmental and hormonal signals dramatically alter the localization and abundance of insulin receptor substrate proteins in the mammary gland. Endocrinology (2003) 144:2683–94. doi:10.1210/en.2002-221103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Stull MA, Rowzee AM, Loladze AV, Wood TL. Growth factor regulation of cell cycle progression in mammary epithelial cells. J Mammary Gland Biol Neoplasia (2004) 9:15–26. doi:10.1023/B:JOMG.0000023585.95430.f4
32. Kleinberg DL, Ruan W. IGF-I, GH, and sex steroid effects in normal mammary gland development. J Mammary Gland Biol Neoplasia (2008) 13:353–60. doi:10.1007/s10911-008-9103-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Scott J, Cowell J, Robertson ME, Priestley LM, Wadey R, Hopkins B, et al. Insulin-like growth factor-II gene expression in Wilms’ tumour and embryonic tissues. Nature (1985) 317:260–2. doi:10.1038/317260a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Wu HK, Squire JA, Catzavelos CG, Weksberg R. Relaxation of imprinting of human insulin-like growth factor II gene, IGF2, in sporadic breast carcinomas. Biochem Biophys Res Commun (1997) 235:123–9. doi:10.1006/bbrc.1997.6744
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Chan JM, Stampfer MJ, Giovannucci E, Gann PH, Ma J, Wilkinson P, et al. Plasma insulin-like growth factor-I and prostate cancer risk: a prospective study. Science (1998) 279:563–6. doi:10.1126/science.279.5350.563
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B, et al. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet (1998) 351:1393–6. doi:10.1016/S0140-6736(97)10384-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Renehan AG, Zwahlen M, Minder C, O’Dwyer ST, Shalet SM, Egger M. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: systematic review and meta-regression analysis. Lancet (2004) 363:1346–53. doi:10.1016/S0140-6736(04)16044-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Shi R, Yu H, McLarty J, Glass J. IGF-I and breast cancer: a meta-analysis. Int J Cancer (2004) 111:418–23. doi:10.1002/ijc.20233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Sugumar A, Liu YC, Xia Q, Koh YS, Matsuo K. Insulin-like growth factor (IGF)-I and IGF-binding protein 3 and the risk of premenopausal breast cancer: a meta-analysis of literature. Int J Cancer (2004) 111:293–7. doi:10.1002/ijc.20253
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Yu H, Rohan T. Role of the insulin-like growth factor family in cancer development and progression. J Natl Cancer Inst (2000) 92:1472–89. doi:10.1093/jnci/92.18.1472
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Gallagher EJ, LeRoith D. Minireview: IGF, insulin, and cancer. Endocrinology (2011) 152:2546–51. doi:10.1210/en.2011-0231
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. TCGA. Comprehensive molecular portraits of human breast tumors. Nature (2012) 490:61–70. doi:10.1038/nature11412
43. Yu H, Li BD, Smith M, Shi R, Berkel HJ, Kato I. Polymorphic CA repeats in the IGF-I gene and breast cancer. Breast Cancer Res Treat (2001) 70:117–22. doi:10.1023/A:1012947027213
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Wagner K, Hemminki K, Israelsson E, Grzybowska E, Soderberg M, Pamula J, et al. Polymorphisms in the IGF-1 and IGFBP 3 promoter and the risk of breast cancer. Breast Cancer Res Treat (2005) 92:133–40. doi:10.1007/s10549-005-2417-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Deming SL, Ren Z, Wen W, Shu XO, Cai Q, Gao YT, et al. Genetic variation in IGF1, IGF-1R, IGFALS, and IGFBP3 in breast cancer survival among Chinese women: a report from the Shanghai Breast Cancer Study. Breast Cancer Res Treat (2007) 104:309–19. doi:10.1007/s10549-006-9420-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Tsuchiya N, Wang L, Horikawa Y, Inoue T, Kakinuma H, Matsuura S, et al. CA repeat polymorphism in the insulin-like growth factor-I gene is associated with increased risk of prostate cancer and benign prostatic hyperplasia. Int J Oncol (2005) 26:225–31. doi:10.3892/ijo.26.1.225
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Cheng I, Stram DO, Penney KL, Pike M, Le Marchand L, Kolonel LN, et al. Common genetic variation in IGF1 and prostate cancer risk in the Multiethnic Cohort. J Natl Cancer Inst (2006) 98:123–34. doi:10.1093/jnci/djj013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Habuchi T. Common genetic polymorphisms and prognosis of sporadic cancers: prostate cancer as a model. Future Oncol (2006) 2:233–45. doi:10.2217/14796694.2.2.233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Moon JW, Chang YS, Ahn CW, Yoo KN, Shin JH, Kong JH, et al. Promoter -202 A/C polymorphism of insulin-like growth factor binding protein-3 gene and non-small cell lung cancer risk. Int J Cancer (2006) 118:353–6. doi:10.1002/ijc.21339
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Dong X, Javle M, Hess KR, Shroff R, Abbruzzese JL, Li D. Insulin-like growth factor axis gene polymorphisms and clinical outcomes in pancreatic cancer. Gastroenterology (2010) 139:464–73.e1–3. doi:10.1053/j.gastro.2010.04.042
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Nielsen TO, Andrews HN, Cheang M, Kucab JE, Hsu FD, Ragaz J, et al. Expression of the insulin-like growth factor I receptor and urokinase plasminogen activator in breast cancer is associated with poor survival: potential for intervention with 17-allylamino geldanamycin. Cancer Res (2004) 64:286–91. doi:10.1158/0008-5472.CAN-03-1242
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Law JH, Habibi G, Hu K, Masoudi H, Wang MY, Stratford AL, et al. Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res (2008) 68:10238–46. doi:10.1158/0008-5472.CAN-08-2755
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Creighton CJ, Casa A, Lazard Z, Huang S, Tsimelzon A, Hilsenbeck SG, et al. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol (2008) 26:4078–85. doi:10.1200/JCO.2007.13.4429
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A (2001) 98:10869–74. doi:10.1073/pnas.191367098
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Kittaneh M, Montero AJ, Gluck S. Molecular profiling for breast cancer: a comprehensive review. Biomark Cancer (2013) 5:61–70. doi:10.4137/BIC.S9455
56. Eroles P, Bosch A, Perez-Fidalgo JA, Lluch A. Molecular biology in breast cancer: intrinsic subtypes and signaling pathways. Cancer Treat Rev (2012) 38:698–707. doi:10.1016/j.ctrv.2011.11.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Lee AV, Jackson JG, Gooch JL, Hilsenbeck SG, Coronado-Heinsohn E, Osborne CK, et al. Enhancement of insulin-like growth factor signaling in human breast cancer: estrogen regulation of insulin receptor substrate-1 expression in vitro and in vivo. Mol Endocrinol (1999) 13:787–96. doi:10.1210/mend.13.5.0274
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Salerno M, Sisci D, Mauro L, Guvakova MA, Ando S, Surmacz E. Insulin receptor substrate 1 is a target for the pure antiestrogen ICI 182,780 in breast cancer cells. Int J Cancer (1999) 81:299–304. doi:10.1002/(SICI)1097-0215(19990412)81:2<299::AID-IJC21>3.3.CO;2-#
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Maor S, Mayer D, Yarden RI, Lee AV, Sarfstein R, Werner H, et al. Estrogen receptor regulates insulin-like growth factor-I receptor gene expression in breast tumor cells: involvement of transcription factor Sp1. J Endocrinol (2006) 191:605–12. doi:10.1677/joe.1.07016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Bhargava R, Beriwal S, McManus K, Dabbs DJ. Insulin-like growth factor receptor-1 (IGF-1R) expression in normal breast, proliferative breast lesions, and breast carcinoma. Appl Immunohistochem Mol Morphol (2011) 19:218–25. doi:10.1097/PAI.0b013e3181ffc58c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Kolacinska A, Chalubinska J, Zawlik I, Szymanska B, Borowska-Garganisz E, Nowik M, et al. Apoptosis-, proliferation, immune function-, and drug resistance-related genes in ER positive, HER2 positive and triple negative breast cancer. Neoplasma (2012) 59:424–32. doi:10.4149/neo_2012_055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Yerushalmi R, Gelmon KA, Leung S, Gao D, Cheang M, Pollak M, et al. Insulin-like growth factor receptor (IGF-1R) in breast cancer subtypes. Breast Cancer Res Treat (2012) 132:131–42. doi:10.1007/s10549-011-1529-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Shin SJ, Gong G, Lee HJ, Kang J, Bae YK, Lee A, et al. Positive expression of insulin-like growth factor-1 receptor is associated with a positive hormone receptor status and a favorable prognosis in breast cancer. J Breast Cancer (2014) 17:113–20. doi:10.4048/jbc.2014.17.2.113
64. Hawsawi Y, El-Gendy R, Twelves C, Speirs V, Beattie J. Insulin-like growth factor – oestradiol crosstalk and mammary gland tumourigenesis. Biochim Biophys Acta (2013) 1836:345–53. doi:10.1016/j.bbcan.2013.10.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
65. Becker MA, Ibrahim YH, Cui X, Lee AV, Yee D. The IGF pathway regulates ERalpha through a S6K1-dependent mechanism in breast cancer cells. Mol Endocrinol (2011) 25:516–28. doi:10.1210/me.2010-0373
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Aaltonen KE, Rosendahl AH, Olsson H, Malmstrom P, Hartman L, Ferno M. Association between insulin-like growth factor-1 receptor (IGF1R) negativity and poor prognosis in a cohort of women with primary breast cancer. BMC Cancer (2014) 14:794. doi:10.1186/1471-2407-14-794
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Zhang Y, Moerkens M, Ramaiahgari S, De Bont H, Price L, Meerman J, et al. Elevated insulin-like growth factor 1 receptor signaling induces antiestrogen resistance through the MAPK/ERK and PI3K/Akt signaling routes. Breast Cancer Res (2011) 13:R52. doi:10.1186/bcr2883
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Baxi SM, Tan W, Murphy ST, Smeal T, Yin MJ. Targeting 3-phosphoinoside-dependent kinase-1 to inhibit insulin-like growth factor-I induced AKT and p70 S6 kinase activation in breast cancer cells. PLoS One (2012) 7:e48402. doi:10.1371/journal.pone.0048402
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Miller TW. Endocrine resistance: what do we know? Am Soc Clin Oncol Educ Book (2013). doi:10.1200/EdBook_AM.2013.33.e37
70. Robertson JF, Ferrero JM, Bourgeois H, Kennecke H, De Boer RH, Jacot W, et al. Ganitumab with either exemestane or fulvestrant for postmenopausal women with advanced, hormone-receptor-positive breast cancer: a randomised, controlled, double-blind, Phase 2 trial. Lancet Oncol (2013) 14:228–35. doi:10.1016/S1470-2045(13)70026-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Brockdorff BL, Heiberg I, Lykkesfeldt AE. Resistance to different antiestrogens is caused by different multi-factorial changes and is associated with reduced expression of IGF receptor Ialpha. Endocr Relat Cancer (2003) 10:579–90. doi:10.1677/erc.0.0100579
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Knowlden JM, Hutcheson IR, Barrow D, Gee JM, Nicholson RI. Insulin-like growth factor-I receptor signaling in tamoxifen-resistant breast cancer: a supporting role to the epidermal growth factor receptor. Endocrinology (2005) 146:4609–18. doi:10.1210/en.2005-0247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res (2008) 68:826–33. doi:10.1158/0008-5472.CAN-07-2707
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. Arnedos M, Drury S, Afentakis M, A’Hern R, Hills M, Salter J, et al. Biomarker changes associated with the development of resistance to aromatase inhibitors (AIs) in estrogen receptor-positive breast cancer. Ann Oncol (2014) 25:605–10. doi:10.1093/annonc/mdt575
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
75. Fagan DH, Uselman RR, Sachdev D, Yee D. Acquired resistance to tamoxifen is associated with loss of the type I insulin-like growth factor receptor: implications for breast cancer treatment. Cancer Res (2012) 72:3372–80. doi:10.1158/0008-5472.CAN-12-0684
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Arias-Pulido H, Royce M, Gong Y, Joste N, Lomo L, Lee SJ, et al. GPR30 and estrogen receptor expression: new insights into hormone dependence of inflammatory breast cancer. Breast Cancer Res Treat (2010) 123:51–8. doi:10.1007/s10549-009-0631-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. De Marco P, Bartella V, Vivacqua A, Lappano R, Santolla MF, Morcavallo A, et al. Insulin-like growth factor-I regulates GPER expression and function in cancer cells. Oncogene (2013) 32:678–88. doi:10.1038/onc.2012.97
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. De Marco P, Romeo E, Vivacqua A, Malaguarnera R, Abonante S, Romeo F, et al. GPER1 is regulated by insulin in cancer cells and cancer-associated fibroblasts. Endocr Relat Cancer (2014) 21:739–53. doi:10.1530/ERC-14-0245
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Ignatov A, Ignatov T, Weissenborn C, Eggemann H, Bischoff J, Semczuk A, et al. G-protein-coupled estrogen receptor GPR30 and tamoxifen resistance in breast cancer. Breast Cancer Res Treat (2011) 128:457–66. doi:10.1007/s10549-011-1584-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Ignatov A, Ignatov T, Roessner A, Costa SD, Kalinski T. Role of GPR30 in the mechanisms of tamoxifen resistance in breast cancer MCF-7 cells. Breast Cancer Res Treat (2010) 123:87–96. doi:10.1007/s10549-009-0624-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Mo Z, Liu M, Yang F, Luo H, Li Z, Tu G, et al. GPR30 as an initiator of tamoxifen resistance in hormone-dependent breast cancer. Breast Cancer Res (2013) 15:R114. doi:10.1186/bcr3581
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Sjostrom M, Hartman L, Grabau D, Fornander T, Malmstrom P, Nordenskjold B, et al. Lack of G protein-coupled estrogen receptor (GPER) in the plasma membrane is associated with excellent long-term prognosis in breast cancer. Breast Cancer Res Treat (2014) 145:61–71. doi:10.1007/s10549-014-2936-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Shang Y, Mao Y, Batson J, Scales SJ, Phillips G, Lackner MR, et al. Antixenograft tumor activity of a humanized anti-insulin-like growth factor-I receptor monoclonal antibody is associated with decreased AKT activation and glucose uptake. Mol Cancer Ther (2008) 7:2599–608. doi:10.1158/1535-7163.MCT-07-2401
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
84. O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res (2006) 66:1500–8. doi:10.1158/0008-5472.CAN-05-2925
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
85. Fox EM, Kuba MG, Miller TW, Davies BR, Arteaga CL. Autocrine IGF-I/insulin receptor axis compensates for inhibition of AKT in ER-positive breast cancer cells with resistance to estrogen deprivation. Breast Cancer Res (2013) 15:R55. doi:10.1186/bcr3449
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Browne BC, Eustace AJ, Kennedy S, O’Brien NA, Pedersen K, McDermott MS, et al. Evaluation of IGF1R and phosphorylated IGF1R as targets in HER2-positive breast cancer cell lines and tumours. Breast Cancer Res Treat (2012) 136:717–27. doi:10.1007/s10549-012-2260-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
87. Harris LN, You F, Schnitt SJ, Witkiewicz A, Lu X, Sgroi D, et al. Predictors of resistance to preoperative trastuzumab and vinorelbine for HER2-positive early breast cancer. Clin Cancer Res (2007) 13:1198–207. doi:10.1158/1078-0432.CCR-06-1304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
88. Smith BL, Chin D, Maltzman W, Crosby K, Hortobagyi GN, Bacus SS. The efficacy of herceptin therapies is influenced by the expression of other erbB receptors, their ligands and the activation of downstream signalling proteins. Br J Cancer (2004) 91:1190–4. doi:10.1038/sj.bjc.6602090
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
89. Kostler WJ, Hudelist G, Rabitsch W, Czerwenka K, Muller R, Singer CF, et al. Insulin-like growth factor-1 receptor (IGF-1R) expression does not predict for resistance to trastuzumab-based treatment in patients with Her-2/neu overexpressing metastatic breast cancer. J Cancer Res Clin Oncol (2006) 132:9–18. doi:10.1007/s00432-005-0038-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Jerome L, Alami N, Belanger S, Page V, Yu Q, Paterson J, et al. Recombinant human insulin-like growth factor binding protein 3 inhibits growth of human epidermal growth factor receptor-2-overexpressing breast tumors and potentiates herceptin activity in vivo. Cancer Res (2006) 66:7245–52. doi:10.1158/0008-5472.CAN-05-3555
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Oliveras-Ferraros C, Vazquez-Martin A, Martin-Castillo B, Perez-Martinez MC, Cufi S, Del Barco S, et al. Pathway-focused proteomic signatures in HER2-overexpressing breast cancer with a basal-like phenotype: new insights into de novo resistance to trastuzumab (herceptin). Int J Oncol (2010) 37:669–78. doi:10.3892/ijo_00000716
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Nahta R. Deciphering the role of insulin-like growth factor-I receptor in trastuzumab resistance. Chemother Res Pract (2012) 2012:648965. doi:10.1155/2012/648965
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Corcoran C, Rani S, Breslin S, Gogarty M, Ghobrial IM, Crown J, et al. miR-630 targets IGF1R to regulate response to HER-targeting drugs and overall cancer cell progression in HER2 over-expressing breast cancer. Mol Cancer (2014) 13:71. doi:10.1186/1476-4598-13-71
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Ye XM, Zhu HY, Bai WD, Wang T, Wang L, Chen Y, et al. Epigenetic silencing of miR-375 induces trastuzumab resistance in HER2-positive breast cancer by targeting IGF1R. BMC Cancer (2014) 14:134. doi:10.1186/1471-2407-14-134
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res (2005) 65:11118–28. doi:10.1158/0008-5472.CAN-04-3841
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Huang X, Gao L, Wang S, McManaman JL, Thor AD, Yang X, et al. Heterotrimerization of the growth factor receptors erbB2, erbB3, and insulin-like growth factor-I receptor in breast cancer cells resistant to herceptin. Cancer Res (2010) 70:1204–14. doi:10.1158/0008-5472.CAN-09-3321
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Liu B, Fan Z, Edgerton SM, Yang X, Lind SE, Thor AD. Potent anti-proliferative effects of metformin on trastuzumab-resistant breast cancer cells via inhibition of erbB2/IGF-1 receptor interactions. Cell Cycle (2011) 10:2959–66. doi:10.4161/cc.10.17.16359
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Sanabria-Figueroa E, Donnelly S, Foy K, Buss M, Castellino RC, Paplomata E, et al. Insulin-like growth factor-1 receptor signaling increases the invasive potential of HER2-overexpressing breast cancer cells via Src-FAK and FoxM1. Mol Pharmacol (2015) 87(2):150–61. doi:10.1124/mol.114.095380
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Chen C, Zhang Y, Li J, Tsao SW, Zhang MY. Superior antitumor activity of a novel bispecific antibody cotargeting human epidermal growth factor receptor 2 and type I insulin-like growth factor receptor. Mol Cancer Ther (2014) 13:90–100. doi:10.1158/1535-7163.MCT-13-0558
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Turner NC, Reis-Filho JS, Russell AM, Springall RJ, Ryder K, Steele D, et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene (2007) 26:2126–32. doi:10.1038/sj.onc.1210014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
101. Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA (2006) 295:2492–502. doi:10.1001/jama.295.21.2492
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
102. Stead LA, Lash TL, Sobieraj JE, Chi DD, Westrup JL, Charlot M, et al. Triple-negative breast cancers are increased in black women regardless of age or body mass index. Breast Cancer Res (2009) 11:R18. doi:10.1186/bcr2242
103. Boyle P. Triple-negative breast cancer: epidemiological considerations and recommendations. Ann Oncol (2012) 23(Suppl 6):vi7–12. doi:10.1093/annonc/mds187
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Kalla Singh S, Tan QW, Brito C, De Leon M, De Leon D. Insulin-like growth factors I and II receptors in the breast cancer survival disparity among African-American women. Growth Horm IGF Res (2010) 20:245–54. doi:10.1016/j.ghir.2010.03.001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Hartog H, Horlings HM, Van Der Vegt B, Kreike B, Ajouaou A, Van De Vijver MJ, et al. Divergent effects of insulin-like growth factor-1 receptor expression on prognosis of estrogen receptor positive versus triple negative invasive ductal breast carcinoma. Breast Cancer Res Treat (2011) 129:725–36. doi:10.1007/s10549-010-1256-6
106. Litzenburger BC, Creighton CJ, Tsimelzon A, Chan BT, Hilsenbeck SG, Wang T, et al. High IGF-IR activity in triple-negative breast cancer cell lines and tumorgrafts correlates with sensitivity to anti-IGF-IR therapy. Clin Cancer Res (2011) 17:2314–27. doi:10.1158/1078-0432.CCR-10-1903
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Davison Z, De Blacquiere GE, Westley BR, May FE. Insulin-like growth factor-dependent proliferation and survival of triple-negative breast cancer cells: implications for therapy. Neoplasia (2011) 13:504–15. doi:10.1593/neo.101590
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Rota LM, Albanito L, Shin ME, Goyeneche CL, Shushanov S, Gallagher EJ, et al. IGF1R inhibition in mammary epithelia promotes canonical Wnt signaling and Wnt1-driven tumors. Cancer Res (2014) 74:5668–79. doi:10.1158/0008-5472.CAN-14-0970
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Mancini M, Gariboldi MB, Taiana E, Bonzi MC, Craparotta I, Pagin M, et al. Co-targeting the IGF system and HIF-1 inhibits migration and invasion by (triple-negative) breast cancer cells. Br J Cancer (2014) 110:2865–73. doi:10.1038/bjc.2014.269
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
110. Girgert R, Emons G, Grundker C. Inactivation of GPR30 reduces growth of triple-negative breast cancer cells: possible application in targeted therapy. Breast Cancer Res Treat (2012) 134:199–205. doi:10.1007/s10549-012-1968-x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
111. Steiman J, Peralta EA, Louis S, Kamel O. Biology of the estrogen receptor, GPR30, in triple negative breast cancer. Am J Surg (2013) 206:698–703. doi:10.1016/j.amjsurg.2013.07.014
112. Abramovitch S, Glaser T, Ouchi T, Werner H. BRCA1-Sp1 interactions in transcriptional regulation of the IGF-IR gene. FEBS Lett (2003) 541:149–54. doi:10.1016/S0014-5793(03)00315-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Maor S, Yosepovich A, Papa MZ, Yarden RI, Mayer D, Friedman E, et al. Elevated insulin-like growth factor-I receptor (IGF-IR) levels in primary breast tumors associated with BRCA1 mutations. Cancer Lett (2007) 257:236–43. doi:10.1016/j.canlet.2007.07.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Abramovitch S, Werner H. Functional and physical interactions between BRCA1 and p53 in transcriptional regulation of the IGF-IR gene. Horm Metab Res (2003) 35:758–62. doi:10.1055/s-2004-814154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Hudelist G, Wagner T, Rosner M, Fink-Retter A, Gschwantler-Kaulich D, Czerwenka K, et al. Intratumoral IGF-I protein expression is selectively upregulated in breast cancer patients with BRCA1/2 mutations. Endocr Relat Cancer (2007) 14:1053–62. doi:10.1677/ERC-06-0075
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Kang HJ, Yi YW, Kim HJ, Hong YB, Seong YS, Bae I. BRCA1 negatively regulates IGF-1 expression through an estrogen-responsive element-like site. Cell Death Dis (2012) 3:e336. doi:10.1038/cddis.2012.78
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Bendall SC, Stewart MH, Menendez P, George D, Vijayaragavan K, Werbowetski-Ogilvie T, et al. IGF and FGF cooperatively establish the regulatory stem cell niche of pluripotent human cells in vitro. Nature (2007) 448:1015–21. doi:10.1038/nature06027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Ziegler AN, Chidambaram S, Forbes BE, Wood TL, Levison SW. Insulin-like growth factor-II (IGF-II) and IGF-II analogs with enhanced insulin receptor-a binding affinity promote neural stem cell expansion. J Biol Chem (2014) 289:4626–33. doi:10.1074/jbc.M113.537597
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
119. Musaro A, Giacinti C, Borsellino G, Dobrowolny G, Pelosi L, Cairns L, et al. Stem cell-mediated muscle regeneration is enhanced by local isoform of insulin-like growth factor 1. Proc Natl Acad Sci U S A (2004) 101:1206–10. doi:10.1073/pnas.0303792101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Mourkioti F, Rosenthal N. IGF-1, inflammation and stem cells: interactions during muscle regeneration. Trends Immunol (2005) 26:535–42. doi:10.1016/j.it.2005.08.002
121. Savarese TM, Strohsnitter WC, Low HP, Liu Q, Baik I, Okulicz W, et al. Correlation of umbilical cord blood hormones and growth factors with stem cell potential: implications for the prenatal origin of breast cancer hypothesis. Breast Cancer Res (2007) 9:R29. doi:10.1186/bcr1674
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Malaguarnera R, Belfiore A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front Endocrinol (Lausanne) (2014) 5:10. doi:10.3389/fendo.2014.00010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell (2008) 133:704–15. doi:10.1016/j.cell.2008.03.027
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Playford MP, Bicknell D, Bodmer WF, Macaulay VM. Insulin-like growth factor 1 regulates the location, stability, and transcriptional activity of beta-catenin. Proc Natl Acad Sci U S A (2000) 97:12103–8. doi:10.1073/pnas.210394297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Desbois-Mouthon C, Cadoret A, Blivet-Van Eggelpoel MJ, Bertrand F, Cherqui G, Perret C, et al. Insulin and IGF-1 stimulate the beta-catenin pathway through two signalling cascades involving GSK-3beta inhibition and Ras activation. Oncogene (2001) 20:252–9. doi:10.1038/sj.onc.1204064
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Vanamala J, Reddivari L, Radhakrishnan S, Tarver C. Resveratrol suppresses IGF-1 induced human colon cancer cell proliferation and elevates apoptosis via suppression of IGF-1R/Wnt and activation of p53 signaling pathways. BMC Cancer (2010) 10:238. doi:10.1186/1471-2407-10-238
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Ye P, Hu Q, Liu H, Yan Y, D’Ercole AJ. Beta-catenin mediates insulin-like growth factor-I actions to promote cyclin D1 mRNA expression, cell proliferation and survival in oligodendroglial cultures. Glia (2010) 58:1031–41. doi:10.1002/glia.20984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Eliasz S, Liang S, Chen Y, De Marco MA, Machek O, Skucha S, et al. Notch-1 stimulates survival of lung adenocarcinoma cells during hypoxia by activating the IGF-1R pathway. Oncogene (2010) 29:2488–98. doi:10.1038/onc.2010.7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Hsu HJ, Drummond-Barbosa D. Insulin signals control the competence of the Drosophila female germline stem cell niche to respond to Notch ligands. Dev Biol (2011) 350:290–300. doi:10.1016/j.ydbio.2010.11.032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Medyouf H, Gusscott S, Wang H, Tseng JC, Wai C, Nemirovsky O, et al. High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. J Exp Med (2011) 208:1809–22. doi:10.1084/jem.20110121
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Rao G, Pedone CA, Del Valle L, Reiss K, Holland EC, Fults DW. Sonic hedgehog and insulin-like growth factor signaling synergize to induce medulloblastoma formation from nestin-expressing neural progenitors in mice. Oncogene (2004) 23:6156–62. doi:10.1038/sj.onc.1207818
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
132. Fernandez C, Tatard VM, Bertrand N, Dahmane N. Differential modulation of Sonic-hedgehog-induced cerebellar granule cell precursor proliferation by the IGF signaling network. Dev Neurosci (2010) 32:59–70. doi:10.1159/000274458
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
133. Foti D, Iuliano R, Chiefari E, Brunetti A. A nucleoprotein complex containing Sp1, C/EBP beta, and HMGI-Y controls human insulin receptor gene transcription. Mol Cell Biol (2003) 23:2720–32. doi:10.1128/MCB.23.8.2720-2732.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
134. Werner H, Maor S. The insulin-like growth factor-I receptor gene: a downstream target for oncogene and tumor suppressor action. Trends Endocrinol Metab (2006) 17:236–42. doi:10.1016/j.tem.2006.06.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Aiello A, Pandini G, Sarfstein R, Werner H, Manfioletti G, Vigneri R, et al. HMGA1 protein is a positive regulator of the insulin-like growth factor-I receptor gene. Eur J Cancer (2010) 46:1919–26. doi:10.1016/j.ejca.2010.02.050
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Sarfstein R, Belfiore A, Werner H. Identification of insulin-like growth factor-I receptor (IGF-IR) gene promoter-binding proteins in estrogen receptor (ER)-positive and ER-depleted breast cancer cells. Cancers (Basel) (2010) 2:233–61. doi:10.3390/cancers2020233
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Graham TR, Zhau HE, Odero-Marah VA, Osunkoya AO, Kimbro KS, Tighiouart M, et al. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res (2008) 68:2479–88. doi:10.1158/0008-5472.CAN-07-2559
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Kim HJ, Litzenburger BC, Cui X, Delgado DA, Grabiner BC, Lin X, et al. Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-kappaB and snail. Mol Cell Biol (2007) 27:3165–75. doi:10.1128/MCB.01315-06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Dupont J, Fernandez AM, Glackin CA, Helman L, Leroith D. Insulin-like growth factor 1 (IGF-1)-induced twist expression is involved in the anti-apoptotic effects of the IGF-1 receptor. J Biol Chem (2001) 276:26699–707. doi:10.1074/jbc.M102664200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Takahashi K, Suzuki K. Association of insulin-like growth-factor-I-induced DNA synthesis with phosphorylation and nuclear exclusion of p53 in human breast cancer MCF-7 cells. Int J Cancer (1993) 55:453–8. doi:10.1002/ijc.2910550322
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Shan J, Shen J, Liu L, Xia F, Xu C, Duan G, et al. Nanog regulates self-renewal of cancer stem cells through the insulin-like growth factor pathway in human hepatocellular carcinoma. Hepatology (2012) 56:1004–14. doi:10.1002/hep.25745
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Xu C, Xie D, Yu SC, Yang XJ, He LR, Yang J, et al. Beta-catenin/POU5F1/SOX2 transcription factor complex mediates IGF-I receptor signaling and predicts poor prognosis in lung adenocarcinoma. Cancer Res (2013) 73:3181–9. doi:10.1158/0008-5472.CAN-12-4403
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Girnita L, Girnita A, Larsson O. Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor 1 receptor. Proc Natl Acad Sci U S A (2003) 100:8247–52. doi:10.1073/pnas.1431613100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Chang WW, Lin RJ, Yu J, Chang WY, Fu CH, Lai A, et al. The expression and significance of insulin-like growth factor-1 receptor and its pathway on breast cancer stem/progenitors. Breast Cancer Res (2013) 15:R39. doi:10.1186/bcr3423
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Malaguarnera R, Frasca F, Garozzo A, Giani F, Pandini G, Vella V, et al. Insulin receptor isoforms and insulin-like growth factor receptor in human follicular cell precursors from papillary thyroid cancer and normal thyroid. J Clin Endocrinol Metab (2011) 96:766–74. doi:10.1210/jc.2010-1255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Loladze AV, Stull MA, Rowzee AM, Demarco J, Lantry JH III, Rosen CJ, et al. Epithelial-specific and stage-specific functions of insulin-like growth factor-I during postnatal mammary development. Endocrinology (2006) 147:5412–23. doi:10.1210/en.2006-0427
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Sun Z, Shushanov S, Leroith D, Wood TL. Decreased IGF type 1 receptor signaling in mammary epithelium during pregnancy leads to reduced proliferation, alveolar differentiation, and expression of insulin receptor substrate (IRS)-1 and IRS-2. Endocrinology (2011) 152:3233–45. doi:10.1210/en.2010-1296
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Cheang MC, Voduc D, Bajdik C, Leung S, Mckinney S, Chia SK, et al. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res (2008) 14:1368–76. doi:10.1158/1078-0432.CCR-07-1658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Cheang MC, Van De Rijn M, Nielsen TO. Gene expression profiling of breast cancer. Annu Rev Pathol (2008) 3:67–97. doi:10.1146/annurev.pathmechdis.3.121806.151505
150. Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med (2009) 15:907–13. doi:10.1038/nm.2000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res (2010) 12:R68. doi:10.1186/bcr2635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Perou CM, Borresen-Dale AL. Systems biology and genomics of breast cancer. Cold Spring Harb Perspect Biol (2011) 3:a003293. doi:10.1101/cshperspect.a003293
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Molyneux G, Geyer FC, Magnay FA, Mccarthy A, Kendrick H, Natrajan R, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell (2010) 7:403–17. doi:10.1016/j.stem.2010.07.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Tolcher AW, Sarantopoulos J, Patnaik A, Papadopoulos K, Lin CC, Rodon J, et al. Phase I, pharmacokinetic, and pharmacodynamic study of AMG 479, a fully human monoclonal antibody to insulin-like growth factor receptor 1. J Clin Oncol (2009) 27:5800–7. doi:10.1200/JCO.2009.23.6745
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Pappo AS, Patel SR, Crowley J, Reinke DK, Kuenkele KP, Chawla SP, et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: results of a Phase II Sarcoma Alliance for Research through Collaboration study. J Clin Oncol (2011) 29:4541–7. doi:10.1200/JCO.2010.34.0000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Pappo AS, Vassal G, Crowley JJ, Bolejack V, Hogendoorn PC, Chugh R, et al. A Phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: results of a Sarcoma Alliance for Research Through Collaboration study. Cancer (2014) 120:2448–56. doi:10.1002/cncr.28728
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Yang Y, Yee D. Targeting insulin and insulin-like growth factor signaling in breast cancer. J Mammary Gland Biol Neoplasia (2012) 17:251–61. doi:10.1007/s10911-012-9268-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Langer CJ, Novello S, Park K, Krzakowski M, Karp DD, Mok T, et al. Randomized, Phase III trial of first-line figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in patients with advanced non-small-cell lung cancer. J Clin Oncol (2014) 32:2059–66. doi:10.1200/JCO.2013.54.4932
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Zhang H, Pelzer AM, Kiang DT, Yee D. Down-regulation of type I insulin-like growth factor receptor increases sensitivity of breast cancer cells to insulin. Cancer Res (2007) 67:391–7. doi:10.1158/0008-5472.CAN-06-1712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
161. Buck E, Gokhale PC, Koujak S, Brown E, Eyzaguirre A, Tao N, et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol Cancer Ther (2010) 9:2652–64. doi:10.1158/1535-7163.MCT-10-0318
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Dinchuk JE, Cao C, Huang F, Reeves KA, Wang J, Myers F, et al. Insulin receptor (IR) pathway hyperactivity in IGF-IR null cells and suppression of downstream growth signaling using the dual IGF-IR/IR inhibitor, BMS-754807. Endocrinology (2010) 151:4123–32. doi:10.1210/en.2010-0032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Ulanet DB, Ludwig DL, Kahn CR, Hanahan D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proc Natl Acad Sci U S A (2010) 107:10791–8. doi:10.1073/pnas.0914076107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: IGF1R, breast cancer subtypes, lineages, luminal, ERBB2+, ER+, triple negative
Citation: Farabaugh SM, Boone DN and Lee AV (2015) Role of IGF1R in breast cancer subtypes, stemness, and lineage differentiation. Front. Endocrinol. 6:59. doi: 10.3389/fendo.2015.00059
Received: 15 February 2015; Accepted: 07 April 2015;
Published: 24 April 2015
Edited by:
Antonino Belfiore, University Magna Graecia of Catanzaro, ItalyReviewed by:
Marcello Maggiolini, University of Calabria, ItalyFrancesca Peruzzi, LSU Health Sciences Center, USA
Douglas Yee, University of Minnesota, USA
Copyright: © 2015 Farabaugh, Boone and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adrian V. Lee, Magee-Womens Research Institute, University of Pittsburgh Cancer Institute, 204 Craft Avenue, Room A412, Pittsburgh, PA 15213, USAbGVlYXZAdXBtYy5lZHU=