 Fumihiko Hakuno1,2
Fumihiko Hakuno1,2 Toshiaki Fukushima3,4Yosuke Yoneyama1,2
Toshiaki Fukushima3,4Yosuke Yoneyama1,2 Hiroyasu Kamei1,2Atsufumi Ozoe1,2Hidehito Yoshihara5Daisuke Yamanaka1,2
Hiroyasu Kamei1,2Atsufumi Ozoe1,2Hidehito Yoshihara5Daisuke Yamanaka1,2 Takashi Shibano1,2Meri Sone-Yonezawa1,2
Takashi Shibano1,2Meri Sone-Yonezawa1,2 Bu-Chin Yu1,2
Bu-Chin Yu1,2 Kazuhiro Chida1,2
Kazuhiro Chida1,2 Shin-Ichiro Takahashi1,2*
Shin-Ichiro Takahashi1,2*- 1Department of Animal Sciences, Graduate School of Agriculture and Life Sciences, The University of Tokyo, Tokyo, Japan
- 2Department of Applied Biological Chemistry, Graduate School of Agriculture and Life Sciences, The University of Tokyo, Tokyo, Japan
- 3Laboratory of Biomedical Chemistry, Basic Life Sciences, Institute of Biomedical and Health Sciences, Hiroshima University, Hiroshima, Japan
- 4Department of Biological Sciences, Faculty of Bioscience and Biotechnology, Tokyo Institute of Technology, Kanagawa, Japan
- 5Laboratory of Protein Metabolism, Tokyo Metropolitan Institute of Medical Science, Tokyo, Japan
Insulin-like peptides, such as insulin-like growth factors (IGFs) and insulin, induce a variety of bioactivities, such as growth, differentiation, survival, increased anabolism, and decreased catabolism in many cell types and in vivo. In general, IGFs or insulin bind to IGF-I receptor (IGF-IR) or insulin receptor (IR), activating the receptor tyrosine kinase. Insulin receptor substrates (IRSs) are known to be major substrates of receptor kinases, mediating IGF/insulin signals to direct bioactivities. Recently, we discovered that IRSs form high-molecular-mass complexes (referred to here as IRSomes) even without IGF/insulin stimulation. These complexes contain proteins (referred to here as IRSAPs; IRS-associated proteins), which modulate tyrosine phosphorylation of IRSs by receptor kinases, control IRS stability, and determine intracellular localization of IRSs. In addition, in these complexes, we found not only proteins that are involved in RNA metabolism but also RNAs themselves. Thus, IRSAPs possibly contribute to modulation of IGF/insulin bioactivities. Since it is established that disorder of modulation of insulin-like activities causes various age-related diseases including cancer, we could propose that the IRSome is an important target for treatment of these diseases.
Introduction
Bioactivities of Insulin-Like Peptides
Insulin-like peptides, such as insulin-like growth factors (IGF-I and IGF-II) and insulin, are a family of peptide hormones that are highly homologous in structure and function. Both IGFs and insulin exert a variety of bioactivities that mainly increase anabolism and decrease catabolism.
Insulin-like growth factor-I is produced in a wide range of tissues including liver, a major IGF-producing tissue. Throughout life, IGF-I levels are positively regulated by growth hormone and insulin and negatively controlled by glucocorticoid and malnutrition (1). Meanwhile, IGF-II exhibits its highest levels in the fetal stage declining after birth. Both IGFs are constitutively secreted and thus do not show apparent diurnal variation. IGFs exert biological effects on most cell types [except for liver and fat tissues, in which IGF-I receptor (IGF-IR) declines after birth]. Therefore, IGFs can act by autocrine and paracrine mechanisms as well as by endocrine mechanisms. Many studies have analyzed bioactivities of IGFs using a variety of cell types including primary cultured cells and established cell lines, and have revealed that IGFs exert relatively chronic effects including induction of cell proliferation, differentiation, survival and migration, and maintenance of cell functions (2). Together with other reports (3), IGFs are believed to be indispensable for growth and development.
One of the characteristics of insulin is that this hormone plays a major role in metabolic actions (4–6). In response to elevated blood glucose levels, insulin is transiently secreted from pancreatic β cells, and it promotes glucose uptake and glycogen synthesis and inhibits glycogenolysis and gluconeogenesis in the target tissues such as muscle, adipose, and liver. In addition to these actions involved in glucose homeostasis, insulin also promotes uptake of amino acids, protein synthesis, and lipid metabolism.
These insulin-like activities are well regulated by a variety of hormones, growth factors, and cytokines under various physiological conditions (7–10). The fine regulation makes normal follicle development, implantation, development, growth, maturation, metabolic control, and aging possible (3, 11). Whenever dysregulation of insulin-like activities is induced, growth disorders and many types of age-related diseases including diabetes, cancer, neurodegenerative disease, arteriosclerosis, and osteoporosis develop (12–14). Especially, in the past several decades, evidence has accumulated that deregulation of insulin-like activities is involved in diseases including cancer. Using many types of cancer cells and genetically engineered mice, it has been demonstrated that IGFs and insulin promote cancer cell proliferation, motility, invasion, and survival, and augmentation of these activities is proposed as a mechanism for cancer development, growth, and metastasis (15–17).
Signal Transduction of Insulin-Like Peptides
Insulin-like peptides transduce signals by binding to IGF-IR and insulin receptors (IR) on the plasma membrane of target cells. IGF-IR and IR are similar to each other and belong to the receptor tyrosine kinase family. These receptors form disulfide-linked hetero-tetramers composed of two α-subunits and two β-subunits, which possess extracellular domains interacting with hormones and intracellular domains with tyrosine kinase activity. IGFs bind to IGF-IR with high affinity, but to IR with low affinity. In the same manner, insulin binds to IR with high affinity, whereas to IGF-IR with low affinity. Moreover, IGF-IR forms hybrid receptors with IR in cells that equally express both IGF-IR and IR. These hybrid receptors are shown to possess high affinity for IGF-I (18). IR exists as two isoforms, IR-A and IR-B, which result from alternative splicing of IR pre-mRNA. IR-B mRNA includes exon 11, whereas IR-A mRNA lacks exon 11 that encodes the 12 amino-acid region of the C-terminus of the α-subunit. IR-B binds to insulin, whereas IR-A binds to not only insulin but also IGF-II. Because IR-A binds to IGF-II and is highly expressed in several cancer cells and tissues, it is likely to be involved in oncogenic transformation (19).
Binding of ligands to their receptors (IGF-IR and IR) on the plasma membrane activates their receptor tyrosine kinase activities. The activated receptors tyrosine-phosphorylate themselves (auto-phosphorylation) and their specific substrates, some of which are insulin receptor substrate (IRS) proteins (Figure 1A). Tyrosine-phosphorylated IRSs are recognized by Src-homology (SH) 2-containing proteins (Figure 1B), one of which is phosphoinositide 3-kinase (PI3K). Phosphorylation of phosphoinositide in membranes by PI3K results in activation of serine/threonine kinase Akt. The activated Akt phosphorylates many substrates to promote various bioactivities. The other signaling molecule recognizing tyrosine-phosphorylated IRSs is Grb2. Grb2 is reported to form a complex with SOS to activate small GTPase Ras. The activated Ras induces the MAPK/ERK pathway leading to mitogenic activities (20). IRSs are associated with other proteins even without their tyrosine phosphorylation (Figure 1C), as described in detail below.
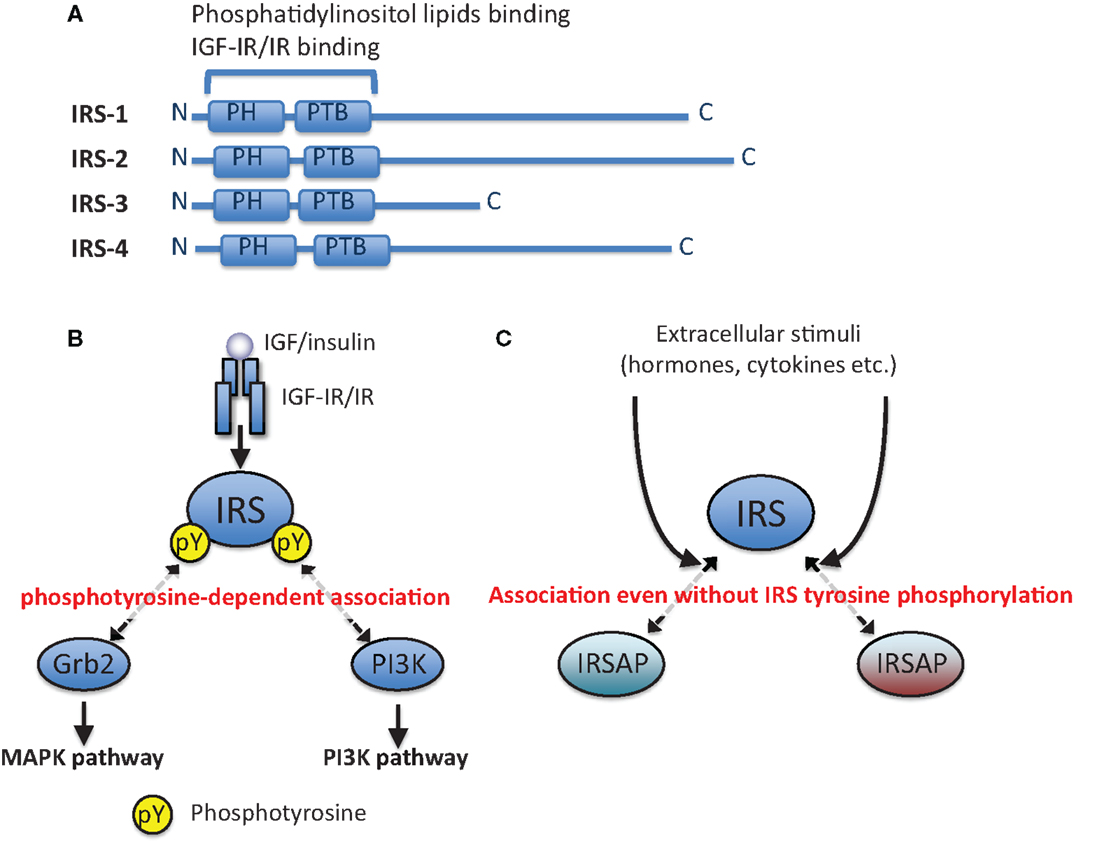
Figure 1. Phosphotyrosine-dependent and -independent association of IRS with other proteins. (A) Schematic illustration of IRS-1, IRS-2, IRS-3, and IRS-4. (B) Canonical IGF/insulin signaling pathways. IRSs are tyrosine-phosphorylated by IGF-IR/IR tyrosine kinase, and then they are associated with downstream signaling molecules in a phosphotyrosine (pY)-dependent manner. (C) IRSs are associated with other proteins (IRSAPs) even without their tyrosine phosphorylation, and this interaction can be changed by extracellular stimuli in addition to IGF/insulin stimulation.
Insulin-Like Peptides and Cancer
Accumulated evidence suggests that excess IGF/insulin activity contributes to cancer development (Figure 2). IGFs are often produced in cancer cells and/or the surrounding stromal cells. IGF-IR (21, 22) and IR (especially IR-A) (22–25) are also overexpressed in many cancer cells. In cell cultures, IGF/insulin promotes proliferation, survival, and migration of various cancer cells. IGF is also known to mediate the transforming action of many oncogenes (26). Recently, IGF/insulin is proposed to play roles in the maintenance of cancer cell stemness (27) as well as cancer metabolism (28).
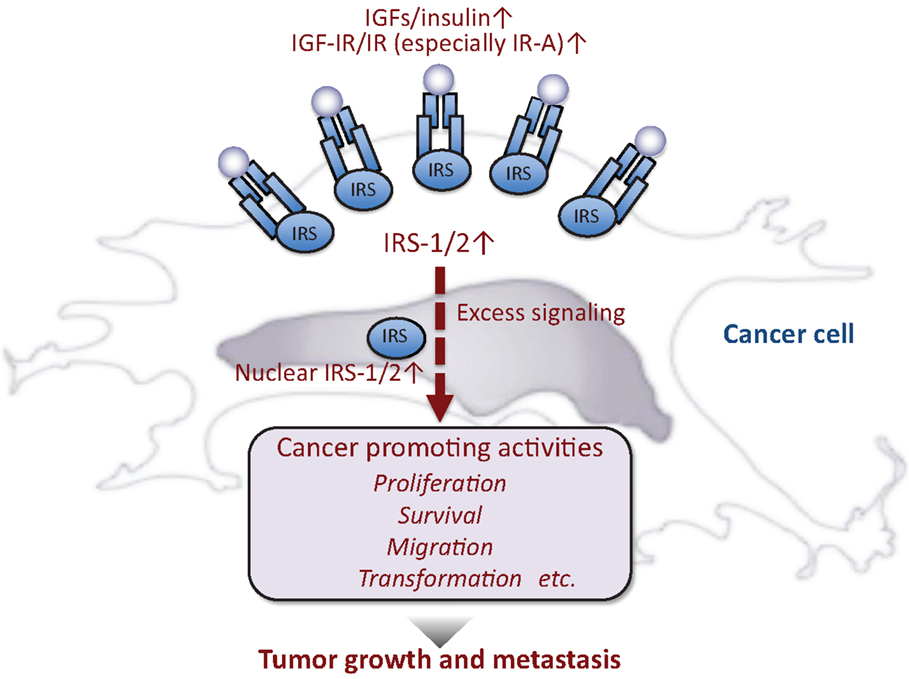
Figure 2. Insulin-like activities and cancer. In general, cancer cells produce or are exposed to high levels of insulin-like peptides and/or IGF/insulin signals are enhanced in these cells, resulting in maintenance of cancer phenotypes.
Genetic and pharmacological manipulation clearly shows important roles of IGF/insulin and their receptors not only in cultured cancer cells but also in mouse tumor models. For example, injection of neutralizing antibody against IGF-I and IGF-II into mice suppressed the development of bone tumors (29, 30) and liver metastasis of colorectal cancers (31). Liver-specific IGF-I knockout mice showed delayed-onset of chemically and genetically induced mammary tumors (32), and neutralizing antibody against IGF-IR and IGF-IR kinase inhibitor showed anti-tumor effects in mouse xenograft models (33–35). These studies demonstrate roles of IGFs and IGF-IR in tumorigenesis and following studies show roles of insulin and IR. IR knockdown inhibited breast cancer cell proliferation, angiogenesis, and metastasis in nude mice (36), and pancreatic β cell-specific IR knockout impaired tumorigenesis in genetic mouse models with pancreatic neuroendocrine tumor (37). Moreover, combined action of IGF-IR/IR was also investigated. Pharmacological inhibition of both IGF-IR/IR kinases inhibited the growth of tumors transplanted from constitutively active IGF-IR-transgenic mice (38). The dual inhibitor also reversed tumor-promoting effects of type-2 diabetes in breast cancer mouse models (39). An antibody with neutralizing activity against both IGF-IR and IGF-I/insulin-hybrid receptors showed more potent anti-tumor effects than antibodies targeting only the IGF-IR or hybrid receptors in xenograft models (40). The combination of IR gene deficiency and IGF-IR-neutralizing antibody treatment suppresses tumor growth in a transgenic mouse model of pancreatic neuroendocrine carcinogenesis. However, the IGF-IR antibody does not significantly impair tumor growth in the model with the intact IR gene (37).
In humans, population-based studies show that high circulating IGF-I levels are associated with the risk of cancer development (41), and hyperinsulinemia is related to poor cancer prognosis (42, 43). Recently, a potential clinical benefit of IGF-IR-neutralizing antibodies was reported, but it was limited to a small subset of cancers [Ewing’s sarcoma (44), non-small cell lung carcinoma (45), and some other chemotherapy-resistant solid tumors (46)]. The failure of IGF-IR-targeted therapy in many other studies points out the putative importance of combinatory target therapy toward IGF-IR and IR (47).
Insulin Receptor Substrates
Characteristics and Physiological Significance of IRSs
The genes encoding IRS proteins are evolutionarily conserved through the animal kingdom, from worm to human, and a single or multiple IRS paralogous genes are found within a species. In vertebrates, three to four types of genes encoding structurally related IRS proteins (IRS-1, -2, -3, and -4) have been found in many species (48, 49). From previous reports and current genome database analyses, the human genome harbors IRS-1, -2, and -4 genes, but it apparently lost the IRS-3 gene during evolution though the mouse genome contains all four IRS genes. It is noteworthy that the most analyzable vertebrate genomes from fish to primates contain both IRS-1 and -2 genes (http://www.ensembl.org/index.html), implying roles for these genes in the regulation of fundamental physiological events conserved throughout chordate evolution. In invertebrate model species, the chico in fly and the ist-1 in worm are known as functional IRS orthologous genes (50, 51).
Common structural features of IRS proteins are the pleckstrin homology (PH) and phosphotyrosine binding (PTB) domains in the amino-terminal region (Figure 1A). Since IRSs lack any enzymatic activities in their primary sequences, they are regarded as adaptor or docking proteins that mediate signaling. As we explained above, the IRSs interact with tyrosine-phosphorylated IGF-IR/IR through the PTB domains, and the tyrosine residues in IRSs are subsequently phosphorylated via the receptor tyrosine kinases. IRS-2, which has a unique structural feature, also interacts with the tyrosine kinase domain of IRs through its specific region called as KRLB region (52, 53). The tyrosine-phosphorylated IRS recruits major IGF/insulin signaling molecules to the membrane through its binding to membrane with its PH domain, which leads to the activation of PI3K–Akt and/or Ras–MAPK signaling cascades. Consequently, IRS serves a major branching node of IGF/insulin signaling (Figure 1B).
For IGF/insulin-dependent PI3K–Akt activation, several specific tyrosine residues in the binding motif of the SH2 domains of class I PI3K p85 regulatory subunit (YxxM; where X is any amino acid) are typically important. The phosphotyrosine residues at 608, 628, 658, and 939 in IRS-1 (54–56) and at 649, 671, 734, and 814 in IRS-2 were demonstrated to be involved in PI3K p85 binding (57). The tyrosine 891 in IRS-1 is known to be the Grb2-binding site phosphorylated by IGF/insulin-stimuli, which is involved in Ras–MAPK cascade activation (58). In addition, the serine/threonine phosphorylation and the post-translational modifications of lysine residues in IRS also play major roles in fine tuning the IGF/insulin signaling. S6K phosphorylates serine/threonine residues of IRS, which in turn leads to the degradation of IRS protein and attenuation of IGF/insulin signaling (20, 59). It has been reported that S6K directly phosphorylates IRS-1 on multiple serine residues including IRS-1 serine 307 to inhibit insulin signaling, and these serine phosphorylations were increased in adipose tissues in obese mice (60). By contrast, a recent study shows that IRS-1 serine 307 phosphorylation promotes insulin sensitivity (61). The serine/threonine phosphorylation of IRSs could be complex but an important clue for deciphering the pathology of IGF/insulin signaling. The ubiquitination on lysine residues in IRS causes degradation of the IRSs (59, 62, 63). Also, the IRS-2 acetylation on its lysine residue decreases IGF-induced MAPK signaling in neuron and protects neurons from oxidative damage induced cell death (64).
The physiological significance of IRS proteins has been studied by using gene knockout mice (65–80). The study using the Irs-1 gene null mice or using the cells from the knockout animal have established the notion that Irs-1 plays crucial roles in growth and development while Irs-2 is indispensable for maintaining systemic glucose metabolism rather than mediating signals for body growth (65, 75). The physiological roles of IRSs are not limited to glucose metabolism and growth (50, 69, 72, 73, 78, 81–87). IRS-1 maintains vascular health, and IRS-1 and IRS-2 governed bone turnover and adipocyte differentiation (72, 78). The massive increase in hepatic IRS-2 under protein malnutrition condition suggests that IRS-2 coordinates liver function with changing amino-acid nutrition in vivo (83). It is also noteworthy that loss of the Irs gene in both rodent and fly models resulted in extended lifespan (50, 85–87). Intriguingly, it has been recently reported that IRS-1 interacts with RNA molecules (88, 89). These facts underscore the multifunctional characteristics of IRS proteins in various pathophysiological contexts.
IRSs and Cancer
IRSs contribute to in cancer development (Figure 2). High expression levels of IRS-1/2 are reported in various types of cancer cells (90). In addition, intense tyrosine phosphorylation of IRS-1 is found in a variety of solid tumors (91). Exceptionally, downregulation of IRS-1 is found in advanced breast cancer (92) and non-small cell lung cancer (93). From the clinical point of view, it should be noted that IRSs are often increased by the treatment of cancer cells with anti-cancer drugs targeting signaling pathways downstream of IRSs (94, 95). The possible underlying mechanism is that the drug-induced inhibition of the downstream signaling suppresses the negative feedback regulation that reduces IRS levels, thereby increasing IRS levels (96). This phenomenon may decrease the anti-cancer activity of the drugs. Interestingly, treatment of mice with novel compounds that promote IRS degradation significantly inhibited the growth of melanoma, ovarian, and prostate cancers (95).
Many studies have shown roles for IRS-1/2 in cancer development. In various cancer cells, overexpression/knockdown experiments indicate that IRS-1/2 promotes cell proliferation, survival, migration, and/or transformation (90, 94, 97–103). Transgenic mice with tissue-specific IRS-1/2 overexpression showed increased tumorigenesis and metastasis (104, 105). Knockout mice studies also reveal that IRS-1 deficiency decreases incidence and growth of several tumors (106, 107). IRS-2 deficiency suppresses metastasis (108, 109). Taken together, many studies indicate that IRS-1 and IRS-2 play redundant roles in cancer development, but some reports clearly show their distinct roles: IRS-1 tends to promote cancer proliferation whereas IRS-2 preferentially promotes cell migration (104). Moreover, in some cases, IRS-1 unexpectedly functions as a negative factor in cancer development (110–113). Roles of IRS-1/2 in cancer development may be dependent on tissues, oncogenic stimulations, and/or environmental factors.
Mechanistically, it is widely accepted that IRSs can regulate proliferation, survival, and migration of cancer cells through the activation of the downstream PI3K pathway and MAPK pathway. One of the still unsolved issues is how IRS-1 and IRS-2 play distinct roles in some models. Another issue is the molecular mechanism underlying IRS-promoted cell transformation. In addition, recent studies highlight nuclear IRSs observed in cancer, and their interaction with several nuclear proteins (114). In order to reveal mechanisms of IRS-driven cancer development, it is becoming more important to study proteins that interact with IRSs in isoform-specific and context-dependent manners.
High-Molecular-Mass Complexes Containing IRSs
As described above, tyrosine phosphorylation of IRSs are important for transmission of IGF/insulin signals (Figure 1B). However, recently, we have reported that IRSs interact with various proteins even without IRS tyrosine phosphorylation (Figure 1C). Such IRS-associated proteins were referred to here as IRSAPs and their function of IRSAPs were reviewed below.
Roles of Proteins Co-Immunoprecipitated with IRSs in Modulation of Tyrosine Phosphorylation-Dependent Signals of Insulin-Like Peptides
We had previously reported that pretreatment of FRTL-5 thyrocytes with thyrotropin (TSH) or other cAMP-generating agents potentiated IGF-dependent tyrosine phosphorylation of IRS-2, leading to enhancement of cell proliferation in response to IGF-I (115). To investigate the mechanisms in the potentiation of tyrosine phosphorylation of IRS-2, we performed in vitro tyrosine phosphorylation assays. IRS-2 was prepared by immunoprecipitation using anti-IRS-2 antibody from cells treated with or without dibutyryl cAMP, and subjected to in vitro tyrosine phosphorylation assay using IGF-IR as the tyrosine kinase. Tyrosine phosphorylation by the IGF-IR of IRS-2 prepared from FRTL-5 cells treated with dibutyryl cAMP was higher than IRS-2 prepared from control cells (116), suggesting that long-term cAMP stimulus increased the availability of IRS-2 as a substrate to be phosphorylated more intensely by IGF-IR kinase. In addition, when the immunoprecipitated IRS-2 was washed with a high-salt buffer solution to dissociate protein components prior to performing the phosphorylation reaction, the cAMP-induced potentiation of tyrosine phosphorylation of IRS-2 was abolished (116). These results indicated that protein components co-immunoprecipitated with IRS-2 are necessary for the potentiation of tyrosine phosphorylation of IRS-2 by IGF-IR kinase in FRTL-5 cells. We also observed that the IRS-1 immunoprecipitated from tumor necrosis factor (TNF)-α-treated cells was little phosphorylated by IR tyrosine kinase in vitro and protein components co-immunoprecipitated with IRS-1 are necessary for TNF-α-induced decreases in the availability of IRS-1 to receptor tyrosine kinase (116). Based on these results, we concluded that proteins associated with IRSs play important roles in the modulation of IRS tyrosine phosphorylation by IGF-IR or IR.
Forming High-Molecular-Mass Complex Containing IRSs
We then analyzed the molecular mass of complexes containing IRSs in several types of cells without IGF-I/insulin stimulation. Blue native-polyacrylamide gel electrophoresis (BN-PAGE) in which protein complexes are separated under native conditions was used, followed by immunoblotting with anti-IRS-1 antibody. In 3T3-L1 adipocytes, IRS-1 was detected at approximately 400–1,300 kDa (mainly 400 kDa) (116). In MCF-7 breast cancer cells, IRS-1 was detected at 400 and 1,300 kDa (88). We also analyzed the molecular mass of complexes containing IRS-2. In FRTL-5 thyroid cells, IRS-2 was detected at 400 kDa, and in MCF-7 cells IRS-2 was detected at 500, 800, and 1,300 kDa (88, 116). Given that IRS-1 and IRS-2 are usually detected at 150–180 kDa in SDS-PAGE, these results suggest that IRSs form high-molecular-mass complexes even without IGF/insulin stimulation. The molecular mass of the complexes differs with cell types and IRS isoforms.
Interestingly, the molecular mass changes not only with IGF-I/insulin stimulation but also with stimulation by other cytokines/hormones that modulate IGF-I/insulin signaling (Figure 1C). For example, stimulation of FRTL-5 cells with TSH or other cAMP-generating agents potentiates IGF-I-dependent IRS-2 tyrosine phosphorylation and cell proliferation (115), and cAMP stimulation increases the molecular mass of complexes containing IRS-2 from 400 kDa up to 800 kDa (116). It is possible that some molecules that modulate IRS-mediated signaling associate/dissociate with the complexes in response to stimulation by various cytokines/hormones, as described below. These complexes were referred to here as IRSome.
Functions of Proteins in the IRSome
There are a few papers reporting that IRSs are associated with various proteins. We have identified the proteins in IRSomes by yeast two-hybrid screening using IRSs as baits and LC-MS/MS analysis of proteins co-immunoprecipitated with IRSs. We succeeded in identifying at least 50 proteins that associate with IRSs in a tyrosine phosphorylation-independent manner. These complexes contain proteins modulating tyrosine phosphorylation of IRSs by receptor kinase, proteins controlling stability of IRSs, proteins determining intracellular localization of IRSs, and proteins mediating insulin-like activities. In addition, we found not only proteins that are involved in RNA metabolism but also RNAs themselves in these complexes (Figure 3).
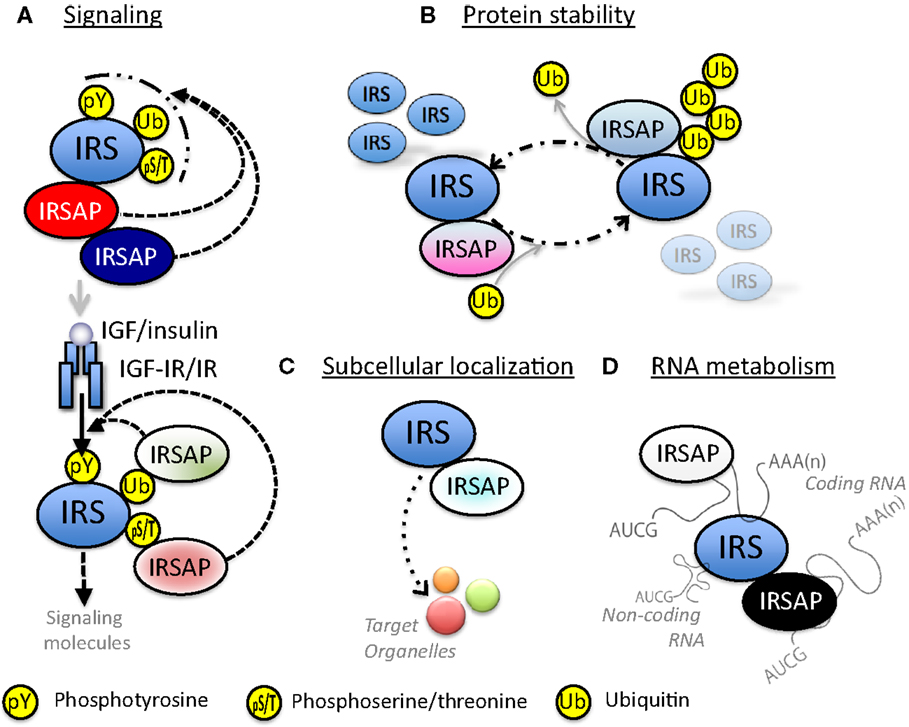
Figure 3. Possible functions of IRS-associated proteins (IRSAPs). (A) IRS-associated proteins modulate IGF/insulin signal intensity through various mechanisms. Some IRSAPs regulate post-translational modification of IRSs. Other IRSAPs interact with IRSs through recognition of post-translationally modified IRSs. These modification and/or protein-interaction regulate the availability to IGF-IR or IR. (B) IRS-associated proteins control stability of IRSs proteins. (C) IRS-associated proteins transport IRSs to specific organelles. (D) IRSs interact with RNA directly or through RNA-binding protein. This complex might control RNA metabolism.
Control of IRS Tyrosine Phosphorylation and Binding to PI3K
IRS-associated proteins can modulate IGF/insulin signal intensity through various mechanisms (Table 1; Figure 3A). One important role of IRS-associated proteins is to regulate the intensity of IGF/insulin signaling. IRSs are tyrosine-phosphorylated by the IGF-IR, IR, and other tyrosine kinases (117–123), resulting in activation of downstream signaling. Several tyrosine phosphatases are shown to dephosphorylate IRSs (124–128). The IRS dephosphorylation reaction can be modulated by other IRS-associated proteins. The mediator of ErbB2-driven cell motility 1 (MEMO1) binds to IRS-1 and competes with SH-PTP2 tyrosine phosphatase for IRS-1 interaction, thereby preventing IRS-1 dephosphorylation (129). SH2B1, a SH2 domain-containing adaptor protein, binds to IRS-1/2 and enhances insulin signaling by inhibiting tyrosine dephosphorylation of IRS-1/2 (130).
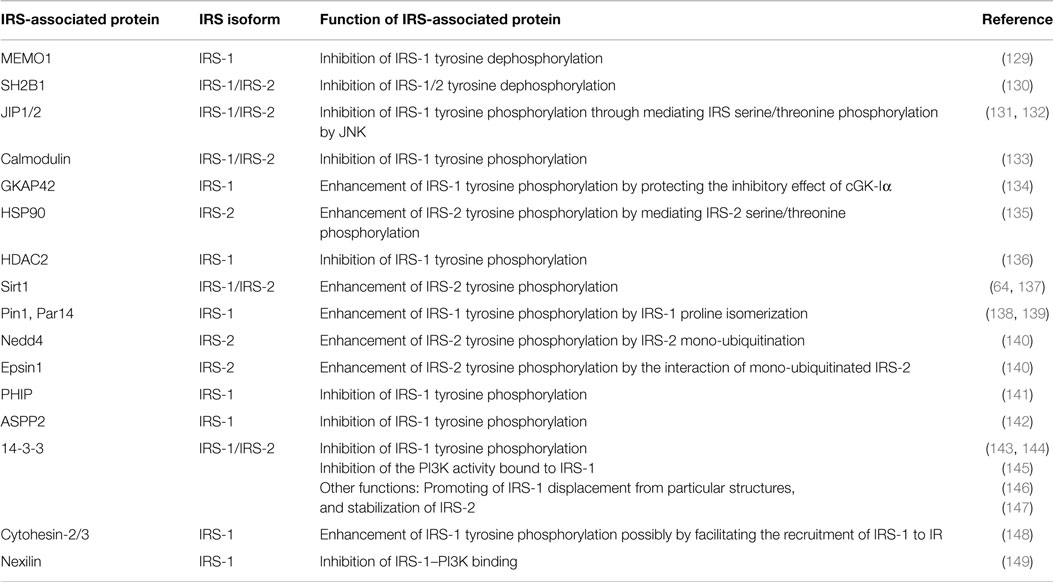
Table 1. IRS-associated proteins that control IRS tyrosine phosphorylation and binding to PI3K.
Many serine/threonine kinases are reported to phosphorylate IRSs at serine/threonine residues, which suppress IRS tyrosine phosphorylation (14). Some IRS-associated proteins modulate IRS tyrosine phosphorylation through the control of IRS serine/threonine phosphorylation. It is well known that JNK can induce IRS-1/2 serine phosphorylation, leading to the suppression of IRS tyrosine phosphorylation and insulin resistance (150). JIP1/2, JNK-interacting proteins, interact with IRS-1/2, and JIP deficiency causes increased IRS-1 tyrosine phosphorylation and abnormal glucose homeostasis in mice (131, 132), indicating that JIPs function as a scaffold linking IRSs and JNK. Calmodulin-dependent kinases (CAMKs) are known to induce IRS-1/2 serine phosphorylation (150), and the binding protein calmodulin interacts with IRS-1/2 (133). Calmodulin overexpression decreased insulin-induced IRS-1 tyrosine phosphorylation (133), suggesting that calmodulin may function as a scaffold linking IRSs and CAMKs. Recently, we identified GKAP42, a cGMP-dependent protein kinase (cGK) anchoring protein, as an IRS-1-associated protein (134). GKAP42 knockdown suppressed insulin-induced IRS-1 tyrosine phosphorylation, which was completely restored by the additional knockdown of cGK-Iα (134), suggesting that cGK-Iα inhibits IRS-1 tyrosine phosphorylation and GKAP42 protects the inhibitory effect of cGK-Iα. In some cases, IRS serine/threonine phosphorylation enhances IRS tyrosine phosphorylation. We reported that HSP90 interacts with IRS-2, and increases IRS-2 tyrosine phosphorylation through mediating IRS-2 serine/threonine phosphorylation by unknown kinase(s) in response to the activation of the cAMP pathway (135).
IRS-associated proteins can also affect IRS post-translational modifications other than serine/threonine phosphorylation. Histone deacetylase 2 (HDAC2) interacts with IRS-1, and overexpression of this protein suppresses insulin-induced IRS-1 tyrosine phosphorylation (136). Another deacetylase Sirt1 interacts with IRS-1 and IRS-2 (137), and inhibition of Sirt1 activity prevented deacetylation of IRS-2 and suppressed IRS-2 tyrosine phosphorylation (64, 137). Pin1 and Par14, peptidyl-prolyl cis/trans isomerases, interact with IRS-1 (138, 139). Pin1 enhances insulin-induced IRS-1 tyrosine phosphorylation through its isomerase activity, and Pin1 knockout mice exhibit impaired insulin signaling with glucose intolerance (138, 139). Similarly, Par14 enhances insulin-induced IRS-1 tyrosine phosphorylation, and Par14 knockdown exacerbated the glucose intolerance in Pin1 knockout mice (138, 139). Prolyl cis/trans isomerization in IRS-1 may be critical for efficient IRS-1 tyrosine phosphorylation. Recently, we found that ubiquitin ligase Nedd4 interacts with IRS-2 and induces IRS-2 ubiquitination (140). As described below, many ubiquitin ligases ubiquitinate IRS-1/2 and induce their proteasomal degradation, resulting in decreased IGF/insulin signaling. In contrast, Nedd4 increases IGF-I-dependent IRS-2 tyrosine phosphorylation and cell proliferation. Our subsequent study revealed that Nedd4 monoubiquitinates IRS-2, which promotes its association with Epsin1, a ubiquitin-binding protein localized at the plasma membrane. Nedd4 recruits IRS-2 to the plasma membrane, likely through promoting Epsin1 binding, and enhanced IRS-2 tyrosine phosphorylation by IGF-IR kinase (Figure 4). Nedd4 overexpression in zebrafish embryo accelerates the body growth and IRS-2 knockdown suppresses the effects of Nedd4 overexpression, indicating that Nedd4–IRS-2 complexes play important roles in body growth.
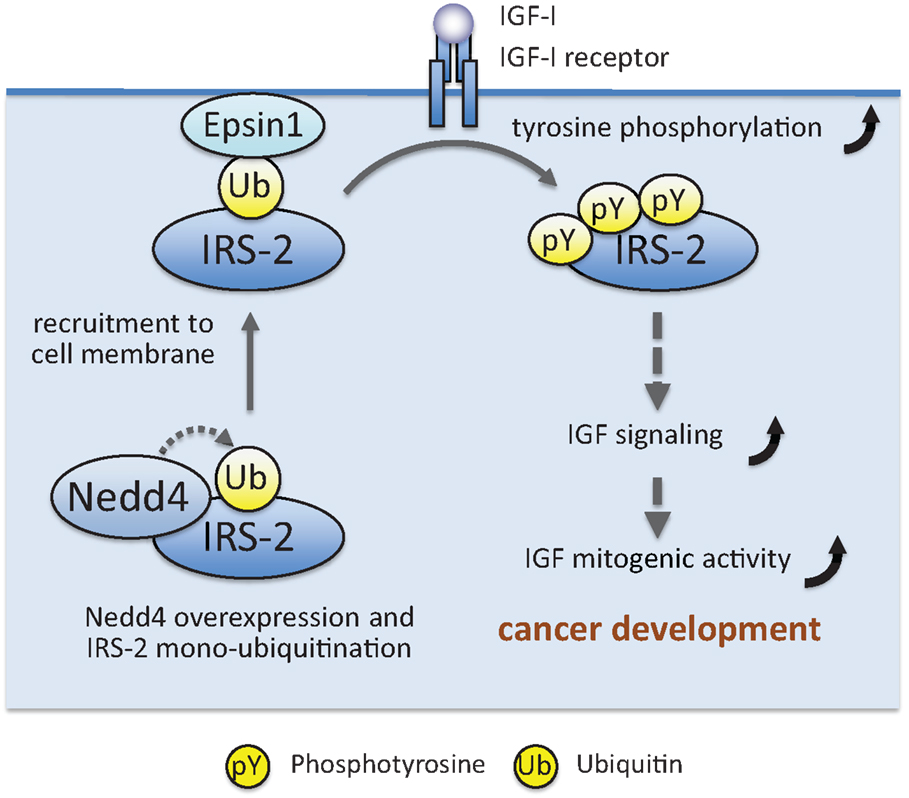
Figure 4. Working model showing how Nedd4 enhances IGF signaling and mitogenic activity, leading to maintenance of cancer phenotype. A detailed explanation is given in the text.
Some IRS-associated proteins can regulate the interaction between IGF-IR/IR and IRS, resulting in the modulation of IRS tyrosine phosphorylation. PHIP, a protein with largely unknown function, interacts with IRS-1 PH domain (141). ASPP2, a p53-binding protein, also interacts with the IRS-1 PTB domain (142). Overexpression of these proteins (or truncated proteins containing IRS binding sites) suppresses insulin-induced IRS tyrosine phosphorylation (141, 142). These observations support the hypothesis that these proteins impair the availability of IRSs to IR by binding to the PH/PTB domain. 14-3-3s interact with IRS-1/2 (143, 145, 151, 152) and suppress insulin-induced IRS-1 tyrosine phosphorylation possibly by inhibiting the interaction of the IRS-1 PTB domain with IR (143). Because 14-3-3–IRS association is dependent on PI3K pathway activation (145, 152), 14-3-3s may participate in the negative feedback regulation of IRS tyrosine phosphorylation. Other reports also show that 14-3-3 binding can modulate IRS-1 tyrosine phosphorylation through inhibiting the activity of PKCα (144), and can modulate the PI3K activity bound to IRS-1 (145), IRS-1 localization (146), as well as IRS-2 stability (147). Cytohesin-2/3, multi-domain proteins with guanine nucleotide exchange factor activity for ARF family GTPases, also interacts with IRS-1 and is proposed to facilitate the recruitment of IRS-1 to IR, thereby enhancing IRS-1 tyrosine phosphorylation (148).
An IRS-associated protein can modulate insulin signal intensity, not by changing IRS tyrosine phosphorylation but by affecting the interaction of IRS with the downstream effector. Nexilin, an actin binding protein, associates with IRS-1 under basal conditions, and this complex dissociates in response to insulin stimulation (149). Nexilin knockdown does not affect IRS-1 tyrosine phosphorylation but enhances IRS-1–PI3K interaction and Akt phosphorylation, whereas Nexilin overexpression inhibits Akt phosphorylation (149), suggesting that Nexilin suppresses IRS-1–PI3K interaction and the downstream insulin signaling.
Control of Stability of IRSs
Recent studies have shown that the stability of IRSs is controlled by IRS-associated E3 ligases and deubiquitinating enzymes (DUBs) via the ubiquitin-proteasome pathway (Table 2; Figure 3B). These E3 ligases and DUBs associate with IRSs under various conditions, resulting in dramatically changed IRS protein levels and induction of insulin-like activities according to surrounding environments.
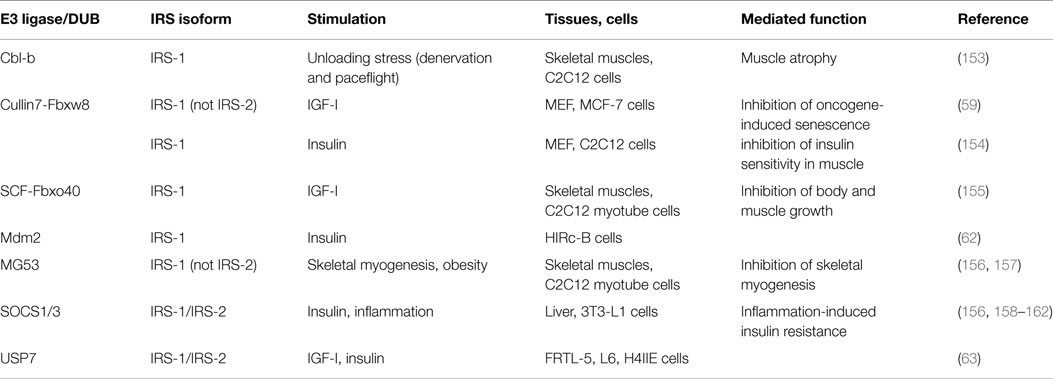
Table 2. IRS-associated proteins that control IRS protein stability.
It is well known that ubiquitination and degradation of IRSs are induced by prolonged IGF/insulin stimulation, which is one of the important mechanisms for the desensitization of IGF/insulin signaling (163–166). Several E3 ligases have been reported to induce the degradation such as cullin7 (Cul7)–Fbxw8 E3 ligase complex, SCF (Skp1, cullin1 and Rbx1)–Fbxo40 E3 ligase complex, SOCS1 and 3 complexes, and Mdm2 (59, 62, 155, 158). The degradation mechanisms of these E3 ligases were analyzed by using downstream pathway inhibitors such as PI3K inhibitor and ERK inhibitor; however, these results were often different. Some suggested that Cul7–Fbxw8 ubiquitinated and degraded IRS-1, but not IRS-2, via phosphorylation of IRS-1 by mTOR/S6K in MCF-7 cells (59), while ubiquitination by SCF–Fbxo40 required only direct signaling from IGF-IR in skeletal myotubes (155). And other reports suggested that activation of PI3K but not mTOR is required for IRS-1 degradation (167). In addition, it was reported that expression of Fbxo40 was muscle specific and was upregulated during differentiation, and the E3 ligases other than SCF–Fbxo40 did not regulate the IGF-I mediated degradation of IRS-1 in skeletal myotubes (155). These E3 ligases were also related to different bioactivities. Whereas embryonic fibroblasts of Cul7−/− mice showed increased IRS-1 protein levels and activation of downstream Akt and MEK/ERK pathways, the null cells showed oncogene-induced senescence (59). And heterozygosity of either Cul7 or Fbxw8 increased PI3K/Akt activation in skeletal muscle tissue upon insulin stimulation, and these mice displayed enhanced insulin sensitivity and plasma glucose clearance (154). Knockdown of Fbxo40 also increased IRS-1 protein levels and activation of downstream pathways; however, knockdown of Fbxo40 induced hypertrophy of myofibers and Fbxo40−/− mice showed enhanced body and muscle size during the growth phase unlike Cul7 (155). These observations suggested that each E3 ligase ubiquitinated IRSs via different signals and had different functions in specific tissues.
Other than IGF/insulin stimulation, various stimulation factors and physiological conditions affect IRS protein levels. So far, several E3 ligases had been identified as responsible factors for the degradation of IRS-1/2 under some conditions. Obesity and inflammatory cytokines secreted from adipose tissues induce IRS-1/2 degradation in insulin target tissues, which is thought to be one of the mechanisms for the onset of insulin resistance and type 2 diabetes. SOCS1/3 and E3 ligase MG53 expression, also known as TRIM72, were elevated by inflammation and obesity, which may induce insulin resistance in adipose tissue, liver, and skeletal muscle (156, 159, 160). Unloading stress such as denervation and spaceflight induces IRS-1 degradation and activates the FOXO3-dependent induction of atrogin-1/MAFbx in muscle, which causes unloading-mediated muscle atrophy. It was reported that unloading stress activated the E3 ligase Casitas B cell lymphoma-b (Cbl-b), and Cbl-b ubiquitinated and degraded IRS-1. And a pentapeptide mimetic of tyrosine 608-phosphorylated IRS-1 prevented the association with Cbl-b and IRS-1 and denervation-induced muscle atrophy in mice (153). Proper IRS-1 expression is important for IGF-induced myogenesis (168). MG53 is increased during myogenesis, and induces IRS-1 ubiquitination and degradation, thereby negatively regulating muscle differentiation (157).
Recently, we identified the deubiquitinating enzyme, ubiquitin-specific protease 7 (USP7) as an IRS-1/2 associated protein and showed that the deubiquitinase activity of USP7 plays important roles in IRS-1/2 stabilization through preventing their proteasomal degradation. In addition, IGF-I/insulin treatment dissociated USP7 from IRS-1/2 via the PI3K pathway activation (63). These findings suggested that the dissociation of USP7 from IRS-1/2 upon IGF/insulin stimulation may be involved in the feedback inhibition mechanism of IGF/insulin signaling by allowing the ubiquitination of IRS-1/2. Whereas protein degradation via the ubiquitin-proteasome pathway is regulated by the balance between ubiquitination and deubiquitination of target proteins, the understanding of DUBs remains less clear compared to E3 ligases. To elucidate the stability mechanism of IRSs, it is necessary to reveal not only how E3 ligases ubiquitinate IRSs but also how DUB(s) deubiquitinate IRSs. Taken together, these proteins controlling stability of IRS may present a novel approach for the treatment of diseases that are associated with insulin-like activities.
Control of Intracellular Localization of IRSs
IRS-associated proteins can control IRS intracellular localization (Table 3; Figure 3C). It has been suggested that tyrosine phosphorylation of IRSs occurs at the cell surface because IGF-IR/IR is activated at the cell surface. Despite this claim, early investigations using subcellular fractionation revealed that phosphorylated IRS and its associated PI3K activity are abundant in the intracellular membrane compartments but less so in the plasma membrane (169–171). Another observation was that other growth factors such as platelet-derived growth factor (PDGF) that equally activate PI3K do not significantly induce glucose uptake. Since PDGF stimulates PI3K mainly in the plasma membrane (172), the IRS localization to intracellular membrane compartments is thought to be an important component of IGF/insulin-specific actions. However, IRSs are cytosolic proteins without post-translational modification that target it to the membrane structures. Thus, they rely instead on interaction with membrane-associated factors (Table 3; Figure 3C). Two clathrin adaptor complexes, AP-1 and AP-3, have been reported to regulate the subcellular localization of IRS-1 (173, 174). It is well established that clathrin mediates the intracellular trafficking of cargos between different membrane compartments through the formation of the coated-vesicles (175). AP complexes are heterotetrameric and function in cargo selectivity and the initiation of clathrin vesicle formation (176). In particular, AP-1 sorts cargo proteins between endosomes and the trans-Golgi network, whereas AP-3 is involved in the transport to lysosomes or related organelles. AP-1 interacts with IRS-1 through its μ1A subunit, which recognizes the YxxΦ-type motifs in the cargo proteins. While IRS-1 is predominantly localized to vesicular structures, an IRS-1 mutant lacking the binding sites to μ1A is mislocalized to the late endosomes that are positive for cation-independent mannose-6-phosphate receptor, suggesting that IRS-1 is transported from late endosomes to peripheral vesicles through the interaction with the AP-1 complex. Furthermore, depletion of AP-1 binding sites in IRS-1 impaired IGF-I-induced cell proliferation, accompanied by reduced IRS-1–PI3K signaling. Interestingly, AP-1 binding sites in IRS-1 overlap PI3K binding sites when phosphorylated by IGF-IR/IR (55, 177). By contrast, it has been structurally demonstrated that AP-1 cannot bind to the phosphorylated YxxΦ motif (178). These observations offer a model in which IRS-1 is sorted by AP-1 and subsequently dissociates from AP-1, and then is targeted for IGF/insulin-mediated tyrosine phosphorylation and downstream signaling activation in a spatially compartmentalized manner. IRS-1 also interacts with σ3 subunit of AP-3 (174). σ3 has been shown to possess an activity to translocate IRS-1 from the cytosol to membrane fractions in vitro. From the investigations of other AP complexes, AP-1 and AP-2, σ3 together with the δ subunit is expected to recognize the di-leucine motif (179), implying that the interaction mode with AP-2 is different from that with AP-1.
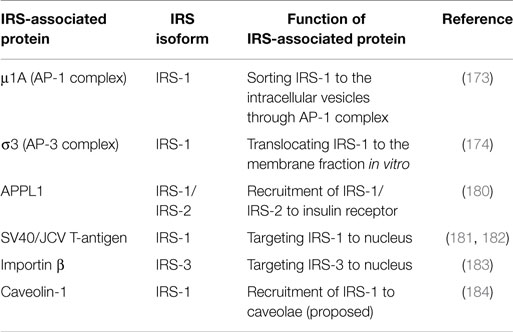
Table 3. Proteins involved in the subcellular localization of IRS.
Components other than AP-1 and AP-3 complexes have been reported to regulate the targeting of IRS-1 to the intracellular membrane compartments (Table 3). Early investigation of the IRS-1-enriched membrane fraction using electron microscopy implied that IRS-1 is localized to the intracellular membrane compartments through the cytoskeletal proteins such as actin (169, 185). Recently, IRS-1 and IRS-2 have been reported to interact with APPL1, which regulates the endocytosis from plasma membrane to early endosomes (180). APPL1 also interacts with IR and deletion of APPL1 reduced the IR-IRS signaling, suggesting that APPL1 serves as the platform for the interaction between IRS and the receptor. Given that IRS localized in endosomes is phosphorylated, IGF-IR/IR should be internalized after ligand stimulation. In fact, several reports suggest that internalization of IGF-IR/IR is required for proper signaling (186–189). How the trafficking of IGF-IR/IR and IRS localization are coordinated to activate the downstream signaling remains an open question that needs to be addressed in further studies.
In addition to the targeting to the intracellular membrane compartment, IRSs have been reported to localize to other sites in the cells. For example, IRS-1 has been found in nucleus (181, 182, 190). Nuclear targeting of IRS-1 is mediated by the interaction with viral T-antigens such as SV40 and its human equivalent (181, 182). Furthermore, nuclear IRS-1 is suggested to function in ribosomal RNA (rRNA) biosynthesis and the transcription of cell cycle genes such as c-myc and cyclin D1 (191, 192). Nuclear IRS-1 is also reported to interact with Yes-associated protein (YAP1), an oncogenic transcription factor negatively regulated by the Hippo pathway (193). IRS-3, another IRS family protein, is also localized to the nucleus (194). IRS-3 interacts with importin β, which together with importin α mediates the protein import through the nuclear pore complex (183). In vitro binding analyses revealed that the PTB domain of IRS-3 interacts with importin β, contains the sequence required for its nuclear targeting (183, 194), and is not conserved in those of IRS-1 and IRS-2, both of which are predominantly localized to the cytosolic portion. In addition, it has been shown using Gal4 chimeric transcription assay that among IRS proteins, only IRS-3 possesses the transcriptional activity (194). This activity partially accounted for the evidence that IRS-3 interacts with Bcl-3, which forms a complex with p50 NF-κB, and enhances NF-κB-dependent anti-apoptotic gene expression (84).
As mentioned above, there is the possibility that the tyrosine phosphorylation of IRSs in the plasma membrane does not contribute to the downstream signaling. However, several reports suggested that IRS localizing at specific regions of the plasma membrane called caveolae functions in insulin signaling. Caveolae are the invaginated microdomains of the plasma membrane enriched in cholesterol and sphingolipids. Both IR and IRS-1 were found in caveolae in 3T3-L1 adipocytes (16, 195, 196). Among the various components of caveolae, IRS-1 has been shown to interact with caveolin-1 (184). Investigations using electron microscopy revealed that IR is concentrated in the neck of caveolae in response to insulin (195, 197), suggesting that the caveolae function as microdomains for insulin signal transduction. The primary cilium has been recently shown as another microdomain in the plasma membrane where a portion of IRS-1 is targeted (198). Phosphorylation of a portion of IRS-1 and Akt was found in the cilia, indicating that the IRS-mediated signaling is activated at the cilia. Interestingly, HSP90, which is known to interact with IRS-2 (135), was identified as a chaperone that forms the neck of the cilia, implying that HSP90 represents IGF/insulin signaling platform in the cilia.
Control of RNA Metabolism and Translation
In seeking to identify IRS-interacting proteins, we found that proteins related to RNA translation, such as poly(A) binding protein (PABP), and eukaryotic initiation factors (eIF) 4E and 4G, associate with IRS-1 in MCF-7 human breast cancer cells (88) (Table 4; Figure 3D). Moreover, the formation of the high-molecular-mass complex containing IRS-1 (IRS-1 complex) is abolished by treatment with RNase but not the IRS-2 complex, indicating that only the IRS-1 complex contains RNAs. Indeed, we found that poly(A)+ RNAs are contained in the IRS-1 complex, and the interactions between IRS-1 and PABP/eIF4E/4G are mediated by RNA (88). Although IGF and insulin have been shown to activate protein synthesis through the IRS-1–mTORC1 pathway (199), it has not yet been reported that the messenger ribonucleoprotein (mRNP) complex included IRS-1. Notably, in the absence of growth factors, the IRS-1 complex seems to associate with monosomes, but in the highly proliferating cells that are stimulated with serum, IRS-1 complexes increase their molecular mass associating with polysomes (88). Together, these findings suggest that RNA-containing IRS-1 complexes have some function in the cells, which raises a new possibility that IRS-1 plays an unrevealed role as a platform for the translation of mRNA species.
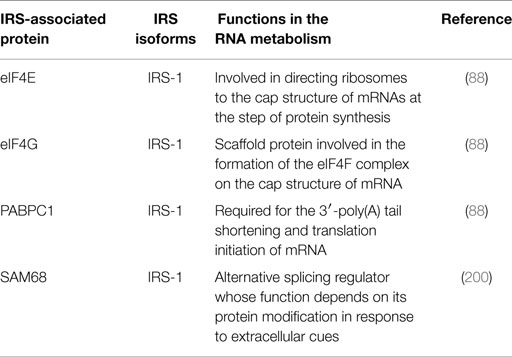
Table 4. IRS-associated proteins that are involved in RNA metabolism.
In addition, IRS-1 could regulate the translation of mRNAs surrounding itself; however, the mechanism of the activation requires much additional investigation. Further, the specific mRNA regulated by this complex needs to be established for full appreciation of this new model of IRS-1 action. It will be also important to reveal how mRNAs are recruited to the IRS-1 complex. Since IRSs do not have a canonical RNA-binding domain, it is possible that some undefined RNA-binding proteins are contained in IRS-1 complex and these proteins tether the selected mRNAs to IRS-1.
In relation to the novel possible function of IRS-1 in mRNA translation, as estimated by the interaction of IRS-1 with mRNP components like PABP, eIF4E, and eIF4G (88), we found that IRS-1 also forms a complex with U96A small nucleolar RNA (snoRNA) (89), which functions in the post-transcriptional rRNA modification in the nucleus (201, 202). Cross-linking immunoprecipitation (CLIP), which is a method to analyze protein–RNA interaction, revealed that IRS-1 somehow interacts with U96A snoRNA in close proximity (89). Moreover, the amount of U96A snoRNA is lower in the IRS-1-deficient cells compared to wild-type cells, which suggests that IRS-1 plays a role in the snoRNA biogenesis. Since modification of rRNA has been shown to be required for a specific class of mRNA translation, such as internal ribosome entry site (IRES) containing mRNA (203, 204) in addition to the translation of the general mRNA translation (205), it is possible that IRS-1 takes part in upregulating a specific type of snoRNA abundance in cells, which results in the sequential rRNA maturation and ribosome function in the selected mRNA translation as well. Previous studies have been shown that IRSs are localized in the nucleus under various conditions (181, 190, 206, 207), and function in the expression of genes encoding the factors that drive cell proliferation including rRNAs (191, 208). Thus, IRS-1 seems to function in both transcription and modification of rRNA, thereby regulating cell proliferation in a multilayer fashion. Determining precisely how IRS-1 localizes in the nucleus and binds to snoRNA is an essential step to further the understanding of these complex interactions.
As described in the sections above, the proteins that interact with IRSs determine their phosphorylation status, stability, and intracellular localizations. It is not clear, however, if all their functions are induced simultaneously in the same IRS–RNA complex or if individual complexes specifically modulate the IRS-mediated RNA translation. Further analysis is needed to resolve these issues; however, the ability of IRSs to control RNA metabolism and translation as well as the signaling outcomes via differences in the association with their partners is a fascinating new model of IGF/insulin-mediated protein synthesis.
Possible Functions of IRSome in Physiology and Diseases Including Cancer
A lot of IRS-associated proteins are related to the physiological regulation of insulin-like activity. As described above, IGF bioactivity is often potentiated by other hormones, by which appropriate IGF activities will be expressed in vivo (209). In thyroid epithelial cells, TSH stimulation and the following activation of cAMP pathway enhance IGF-I-induced IRS-2 tyrosine phosphorylation and cell proliferation (209), which is thought to be important for thyroid morphogenesis and thyroid hormone homeostasis (210). We reported that HSP90 and Nedd4 mediate cAMP-dependent enhancement of IRS-2 tyrosine phosphorylation and cell proliferation (135, 140). Notably, Nedd4–IRS-2 interaction is one of the important tuning points in this signal crosstalk, because Nedd4–IRS-2 interaction is upregulated in response to cAMP and it triggers the mechanisms by which cAMP signaling enhances IGF signaling (140). Conversely, IGF/insulin signaling can modulate other signaling pathways through IRS-associated proteins. IRS-1 interacts with the inhibitory G protein Gi in response to insulin in platelets, which may enhance cAMP signaling and lead to the attenuation of platelet activation (211). IRS-1/2 interacts with and stabilizes Disheveled, thereby promoting Wnt/beta-catenin signaling (212). IRS-1/2 interacts with sarco-endoplasmic reticulum calcium ATPase (SERCA), and may regulate Ca2+ signaling (213, 214). IRS-1/2 can also interact with Bcl-2, and may modulate apoptosis signaling (215).
IRS-associated proteins are relevant to insulin resistance and abnormal glucose metabolism. Obesity increases inflammatory cytokines including TNF-α. TNF-α decreases GKAP42, an IRS-associated protein that enhances IRS-1-mediated signaling (134), and also induces the association of IRS-1 with calmodulin that suppresses IRS-1-mediated signaling (133, 216). Cytokine-induced JNK activation causes IRS-1 serine phosphorylation and impairs insulin signaling, which is mediated by IRS-associated protein JIP (131, 132). Obesity and inflammatory cytokines increase ubiquitin ligase SOCS1/3 and MG53, thereby decreasing IRS-1/2 levels (156, 159, 160). Obesity seems to impair APPL1 function as a linker of IRSs to IR (180). These events could contribute to the induction of insulin resistance and diabetes. On the other hand, food intake increases Pin1, an IRS-associated protein that enhances IRS-1-mediated signaling, thereby leading to increased insulin sensitivity and adiposity (138). Several Sirt1 activators have beneficial effects on glucose homeostasis and insulin sensitivity in obese mice (217), which is partly explained by the function of Sirt1 as an IRS-associated protein that enhances IRS-2-mediated signaling (64, 137).
IRS-associated proteins may also contribute to the maintenance of the cancer phenotype. Nedd4 and Epsin1, which coordinately enhance IGF-I-induced IRS-2 tyrosine phosphorylation, are often overexpressed in cancer cells (218, 219). Nedd4 is known to function as an oncogenic protein (218), and we showed that Nedd4 is required for IGF-I-induced proliferation of prostate cancer cells (140) (Figure 4). Another IRS-associated protein MEMO1 is often increased in aggressive cancers, and is reported to induce epithelial–mesenchymal transition in breast cancer cells and enhance their metastasis. The underlying mechanism partly involves preventing IRS-1 dephosphorylation and increasing IGF signaling (129). Protein levels of IRSs are often upregulated in various cancer cells. IRS-associated ubiquitin ligases and DUBs may contribute to this upregulation. Nuclear IRSs are also often observed in cancer, and many IRS-associated proteins are suggested to mediate cancer-related nuclear IRS functions (114). T-antigens, v-src, and estrogen receptors can interact with IRSs and mediate IRS nuclear translocation. In nuclei, Rad51 recombinase interacts with IRSs, and the interaction is proposed to impair DNA repair. UBF-1, a key regulator of RNA polymerase-I, also interacts with nuclear IRSs, and the interaction is proposed to enhance rRNA synthesis, thereby promoting cell growth. A transcriptional factor β-catenin also interacts with nuclear IRSs, which enhance the expression of the β-catenin target genes that promote cell proliferation. Through these interactions, nuclear IRSs may participate in cancer development (114). In addition, as described above, IRSs possess cell transformation promoting ability. Although the molecular mechanism how IRSs promote cell transformation remains largely unclear, the observation that IRS can interact with oncoproteins including SV40 T-antigen, JCV T-antigen, Ret, NPM–ALK, and ETV6–NTRK3 (122, 182, 220–223) provide clues to the mechanism.
Conclusion
We and others discovered that IRSs are associated with a variety of proteins. IRSs form high-molecular-mass complexes even without their tyrosine phosphorylation (Figure 1C). These complexes contain proteins modulating insulin-like signals of IRSs triggered by receptor tyrosine kinase, proteins controlling stability of IRSs, proteins determining intracellular localization of IRSs, proteins involved in RNA metabolism, and RNAs themselves (Figure 3). Based on these results, we propose that IRSs function not only as signaling mediators through tyrosine phosphorylation but also as scaffold proteins independent of tyrosine phosphorylation in mediation of novel biological activities or modulation of insulin-like activities (Figures 1 and 3).
However, these proteins seem not to function independently. In other words, they are likely to interlock. For example, after IRSs are synthesized, IRSs are recognized by sorting proteins and recruited to specific intracellular compartments. IRSs are associated with the specific proteins there. Another example is intramolecular modification of IRSs. IRSs are post-translationally modified by IRS-associated proteins, such as serine/threonine kinases or phosphatases, E3 ubiquitin ligases or DUBs, acetylases or deacetylases, which are activated by various extracellular factors, such as hormones and cytokines and various physiological states. And then, differentially modified IRSs are recognized by different IRSAPs, which mediate novel biological activities or modulate insulin-like activities. For these reasons, the phenotypes of mice or cells in which IRSs were knocked out or knocked down should be reconsidered from the point of view of IRS-associated proteins.
As mentioned in the Introduction, when dysregulation of insulin-like activities is induced, many types of age-related diseases including diabetes, cancer, neurodegenerative disease, arteriosclerosis, and osteoporosis will develop (12–14). Based on our and others’ recent data about IRSAPs, IRSAPs would be expected to contribute to the modulation of insulin-like activities. Thus, the disorder of modulation of insulin-like activities that underlies several age-related diseases including cancer is possibly explained by IRSome function. The IRSome is an important new target for the treatment of these diseases by controlling insulin-like activities.
Author Contributions
Wrote, read, and approved the final manuscript: FH, TF, YY, HK, AO, HY, DY, TS, MS-Y, BY, KC, S-IT.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Leonard Girnita (Karolinska Hospital, Stockholm, Sweden), Dr. Ignacio Torres-Aleman (Cajal Institute, Madrid, Spain), Dr. Yusuke Higashi (The University of Missouri University, Columbia, MO, USA), Dr. Cunming Duan (The University of Michigan, Ann Abor, MI, USA), and Dr. Asako Takenaka (Meiji University, Kawasaki, Japan) for helpful discussions. We appreciate Dr. Akihiro Ito, Dr. Minoru Yoshida (Chemical Genetics Laboratory, RIKEN Advanced Science Institute, Saitama), Dr. Yasushi Yasushi Saeki, Dr. Keiji Tanaka (Laboratory of Protein Metabolism, Tokyo Metropolitan Institute of Medical Science, Setagaya-ku, Tokyo), Dr. Shun-Ichiro Iemura, Dr. Tohru Natsume (Molecular Profiling Research Center for Drug Discovery, National Institute of Advanced Industrial Science and Technology), Dr. Naoyuki Kataoka (Laboratory for Malignancy Control Research, Medical Innovation Center, Graduate School of Medicine, Kyoto University, Kyoto), and Dr. Tomoichiro Asano (Department of Medical Science, Graduate School of Medicine, Hiroshima University, Hiroshima) to support identification and function analysis of IRS-associated proteins. We also exceptionally acknowledge the help of Dr. Susan Hall (The University of North Carolina at Chapel Hill, NC, USA). Funding: this work was partially supported by Grant-in-Aid [(A)(2)#16208028, (A) #22248030, (A) #25252047, (S)#25221204], Challenging Exploratory Research (#20658065, #25660210, #15K14840), core-to-core program from (the Japan Society for the Promotion of Science (JSPS) and Program for Promotion of Basic and Applied Researches for Innovations in Bio-oriented Industry (#25006A) to S-IT and by Grant-in-Aid [(B)#243801521] to FH. This work was also partially supported by Grant-in-Aid for Young Scientists [(B)#24790928] and Grant-in Aid [(C)#26460369] to TF and Grant-in-Aid for Young Scientists [(B)#25850220] to HK from JSPS. AO, YY, HY, and BCY are recipients of a JSPS Research Fellowship for Young Scientists and for foreign Scientists. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
BN-PAGE, blue native-polyacrylamide gel electrophoresis; CLIP, UV cross-linking and immunoprecipitation; IGF-IR, IGF-I receptor; IGFs, insulin-like growth factors; IR, insulin receptor; IRES, internal ribosome entry site; IRSAPs, IRS-associated proteins; IRSome, high-molecular-mass complexes containing IRSs; IRSs, insulin receptor substrates; mRNP, messenger ribonucleoprotein; PH, pleckstrin homology; PI3K, phosphoinositide 3-kinase; PTB, phosphotyrosine binding; rRNA, ribosomal RNA; SH, Src-homology; snoRNA, small nucleolar RNA; TNF, tumor necrosis factor; TSH, thyrotropin.
References
1. Thissen JP, Ketelslegers JM, Underwood LE. Nutritional regulation of the insulin-like growth factors. Endocr Rev (1994) 15(1):80–101. doi:10.1210/edrv-15-1-80
2. Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev (1995) 16(1):3–34. doi:10.1210/er.16.1.3
3. Nakae J, Kido Y, Accili D. Distinct and overlapping functions of insulin and IGF-I receptors. Endocr Rev (2001) 22(6):818–35. doi:10.1210/edrv.22.6.0452
4. Rui L. Energy metabolism in the liver. Compr Physiol (2014) 4(1):177–97. doi:10.1002/cphy.c130024
5. Newsholme P, Cruzat V, Arfuso F, Keane K. Nutrient regulation of insulin secretion and action. J Endocrinol (2014) 221(3):R105–20. doi:10.1530/JOE-13-0616
6. Rhodes CJ, White MF. Molecular insights into insulin action and secretion. Eur J Clin Invest (2002) 32(Suppl 3):3–13. doi:10.1046/j.1365-2362.32.s3.2.x
7. Qureshi FG, Tchorzewski MT, Duncan MD, Harmon JW. EGF and IGF-I synergistically stimulate proliferation of human esophageal epithelial cells. J Surg Res (1997) 69(2):354–8. doi:10.1006/jsre.1997.5080
8. Stewart AJ, Johnson MD, May FE, Westley BR. Role of insulin-like growth factors and the type I insulin-like growth factor receptor in the estrogen-stimulated proliferation of human breast cancer cells. J Biol Chem (1990) 265(34):21172–8.
9. Takahashi S, Conti M, Van Wyk JJ. Thyrotropin potentiation of insulin-like growth factor-I dependent deoxyribonucleic acid synthesis in FRTL-5 cells: mediation by an autocrine amplification factor(s). Endocrinology (1990) 126(2):736–45. doi:10.1210/endo-126-2-736
10. Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci U S A (1994) 91(11):4854–8. doi:10.1073/pnas.91.11.4854
11. Dupont J, Holzenberger M. Biology of insulin-like growth factors in development. Birth Defects Res C Embryo Today (2003) 69(4):257–71. doi:10.1002/bdrc.10022
12. Piriz J, Muller A, Trejo JL, Torres-Aleman I. IGF-I and the aging mammalian brain. Exp Gerontol (2011) 46(2–3):96–9. doi:10.1016/j.exger.2010.08.022
13. Higashi Y, Sukhanov S, Anwar A, Shai SY, Delafontaine P. Aging, atherosclerosis, and IGF-1. J Gerontol A Biol Sci Med Sci (2012) 67(6):626–39. doi:10.1093/gerona/gls102
14. Laviola L, Natalicchio A, Perrini S, Giorgino F. Abnormalities of IGF-I signaling in the pathogenesis of diseases of the bone, brain, and fetoplacental unit in humans. Am J Physiol Endocrinol Metab (2008) 295(5):E991–9. doi:10.1152/ajpendo.90452.2008
15. Belardi V, Gallagher EJ, Novosyadlyy R, LeRoith D. Insulin and IGFs in obesity-related breast cancer. J Mammary Gland Biol Neoplasia (2013) 18(3–4):277–89. doi:10.1007/s10911-013-9303-7
16. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer (2008) 8(12):915–28. doi:10.1038/nrc2536
17. Samani AA, Yakar S, LeRoith D, Brodt P. The role of the IGF system in cancer growth and metastasis: overview and recent insights. Endocr Rev (2007) 28(1):20–47. doi:10.1210/er.2006-0001
18. Slaaby R, Schaffer L, Lautrup-Larsen I, Andersen AS, Shaw AC, Mathiasen IS, et al. Hybrid receptors formed by insulin receptor (IR) and insulin-like growth factor I receptor (IGF-IR) have low insulin and high IGF-1 affinity irrespective of the IR splice variant. J Biol Chem (2006) 281(36):25869–74. doi:10.1074/jbc.M605189200
19. Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev (2009) 30(6):586–623. doi:10.1210/er.2008-0047
20. Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia (2012) 55(10):2565–82. doi:10.1007/s00125-012-2644-8
21. Pollak MN, Perdue JF, Margolese RG, Baer K, Richard M. Presence of somatomedin receptors on primary human breast and colon carcinomas. Cancer Lett (1987) 38(1–2):223–30. doi:10.1016/0304-3835(87)90218-7
22. Pandini G, Vigneri R, Costantino A, Frasca F, Ippolito A, Fujita-Yamaguchi Y, et al. Insulin and insulin-like growth factor-I (IGF-I) receptor overexpression in breast cancers leads to insulin/IGF-I hybrid receptor overexpression: evidence for a second mechanism of IGF-I signaling. Clin Cancer Res (1999) 5(7):1935–44.
23. Cox ME, Gleave ME, Zakikhani M, Bell RH, Piura E, Vickers E, et al. Insulin receptor expression by human prostate cancers. Prostate (2009) 69(1):33–40. doi:10.1002/pros.20852
24. Law JH, Habibi G, Hu K, Masoudi H, Wang MY, Stratford AL, et al. Phosphorylated insulin-like growth factor-I/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res (2008) 68(24):10238–46. doi:10.1158/0008-5472.CAN-08-2755
25. Vella V, Sciacca L, Pandini G, Mineo R, Squatrito S, Vigneri R, et al. The IGF system in thyroid cancer: new concepts. Mol Pathol (2001) 54(3):121–4. doi:10.1136/mp.54.3.121
26. Sell C, Rubini M, Rubin R, Liu JP, Efstratiadis A, Baserga R. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc Natl Acad Sci U S A (1993) 90(23):11217–21. doi:10.1073/pnas.90.23.11217
27. Malaguarnera R, Belfiore A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front Endocrinol (Lausanne) (2014) 5:10. doi:10.3389/fendo.2014.00010
28. Shaw LM. The insulin receptor substrate (IRS) proteins: at the intersection of metabolism and cancer. Cell Cycle (2011) 10(11):1750–6. doi:10.4161/cc.10.11.15824
29. Goya M, Miyamoto S, Nagai K, Ohki Y, Nakamura K, Shitara K, et al. Growth inhibition of human prostate cancer cells in human adult bone implanted into nonobese diabetic/severe combined immunodeficient mice by a ligand-specific antibody to human insulin-like growth factors. Cancer Res (2004) 64(17):6252–8. doi:10.1158/0008-5472.CAN-04-0919
30. Araki K, Sangai T, Miyamoto S, Maeda H, Zhang SC, Nakamura M, et al. Inhibition of bone-derived insulin-like growth factors by a ligand-specific antibody suppresses the growth of human multiple myeloma in the human adult bone explanted in NOD/SCID mouse. Int J Cancer (2006) 118(10):2602–8. doi:10.1002/ijc.21653
31. Miyamoto S, Nakamura M, Shitara K, Nakamura K, Ohki Y, Ishii G, et al. Blockade of paracrine supply of insulin-like growth factors using neutralizing antibodies suppresses the liver metastasis of human colorectal cancers. Clin Cancer Res (2005) 11(9):3494–502. doi:10.1158/1078-0432.CCR-04-1701
32. Wu Y, Cui K, Miyoshi K, Hennighausen L, Green JE, Setser J, et al. Reduced circulating insulin-like growth factor I levels delay the onset of chemically and genetically induced mammary tumors. Cancer Res (2003) 63(15):4384–8.
33. Arteaga CL, Kitten LJ, Coronado EB, Jacobs S, Kull FC Jr, Allred DC, et al. Blockade of the type I somatomedin receptor inhibits growth of human breast cancer cells in athymic mice. J Clin Invest (1989) 84(5):1418–23. doi:10.1172/JCI114315
34. Gansler T, Furlanetto R, Gramling TS, Robinson KA, Blocker N, Buse MG, et al. Antibody to type I insulin-like growth factor receptor inhibits growth of Wilms’ tumor in culture and in athymic mice. Am J Pathol (1989) 135(6):961–6.
35. Ji QS, Mulvihill MJ, Rosenfeld-Franklin M, Cooke A, Feng L, Mak G, et al. A novel, potent, and selective insulin-like growth factor-I receptor kinase inhibitor blocks insulin-like growth factor-I receptor signaling in vitro and inhibits insulin-like growth factor-I receptor dependent tumor growth in vivo. Mol Cancer Ther (2007) 6(8):2158–67. doi:10.1158/1535-7163.MCT-07-0070
36. Zhang H, Fagan DH, Zeng X, Freeman KT, Sachdev D, Yee D. Inhibition of cancer cell proliferation and metastasis by insulin receptor downregulation. Oncogene (2010) 29(17):2517–27. doi:10.1038/onc.2010.17
37. Ulanet DB, Ludwig DL, Kahn CR, Hanahan D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to IGF-1R targeted therapy. Proc Natl Acad Sci U S A (2010) 107(24):10791–8. doi:10.1073/pnas.0914076107
38. Haluska P, Carboni JM, Loegering DA, Lee FY, Wittman M, Saulnier MG, et al. In vitro and in vivo antitumor effects of the dual insulin-like growth factor-I/insulin receptor inhibitor, BMS-554417. Cancer Res (2006) 66(1):362–71. doi:10.1158/0008-5472.CAN-05-1107
39. Novosyadlyy R, Lann DE, Vijayakumar A, Rowzee A, Lazzarino DA, Fierz Y, et al. Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Res (2010) 70(2):741–51. doi:10.1158/0008-5472.CAN-09-2141
40. Pandini G, Wurch T, Akla B, Corvaia N, Belfiore A, Goetsch L. Functional responses and in vivo anti-tumour activity of h7C10: a humanised monoclonal antibody with neutralising activity against the insulin-like growth factor-1 (IGF-1) receptor and insulin/IGF-1 hybrid receptors. Eur J Cancer (2007) 43(8):1318–27. doi:10.1016/j.ejca.2007.03.009
41. Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer (2004) 4(7):505–18. doi:10.1038/nrc1387
42. Ma J, Li H, Giovannucci E, Mucci L, Qiu W, Nguyen PL, et al. Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: a long-term survival analysis. Lancet Oncol (2008) 9(11):1039–47. doi:10.1016/S1470-2045(08)70235-3
43. Goodwin PJ, Ennis M, Pritchard KI, Trudeau ME, Koo J, Madarnas Y, et al. Fasting insulin and outcome in ear ly-stage breast cancer: results of a prospective cohort study. J Clin Oncol (2002) 20(1):42–51. doi:10.1200/JCO.20.1.42
44. Olmos D, Postel-Vinay S, Molife LR, Okuno SH, Schuetze SM, Paccagnella ML, et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohort study. Lancet Oncol (2010) 11(2):129–35. doi:10.1016/S1470-2045(09)70354-7
45. Gualberto A, Dolled-Filhart M, Gustavson M, Christiansen J, Wang YF, Hixon ML, et al. Molecular analysis of non-small cell lung cancer identifies subsets with different sensitivity to insulin-like growth factor I receptor inhibition. Clin Cancer Res (2010) 16(18):4654–65. doi:10.1158/1078-0432.CCR-10-0089
46. Atzori F, Tabernero J, Cervantes A, Prudkin L, Andreu J, Rodriguez-Braun E, et al. A phase I pharmacokinetic and pharmacodynamic study of dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res (2011) 17(19):6304–12. doi:10.1158/1078-0432.CCR-10-3336
47. Janssen JA, Varewijck AJ. IGF-IR targeted therapy: past, present and future. Front Endocrinol (Lausanne) (2014) 5:224. doi:10.3389/fendo.2014.00224
48. Bugner V, Aurhammer T, Kuhl M. Xenopus laevis insulin receptor substrate IRS-1 is important for eye development. Dev Dyn (2011) 240(7):1705–15. doi:10.1002/dvdy.22659
49. White MF. The IRS-signalling system: a network of docking proteins that mediate insulin action. Mol Cell Biochem (1998) 182(1–2):3–11. doi:10.1023/A:1006806722619
50. Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, et al. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science (2001) 292(5514):104–6. doi:10.1126/science.1057991
51. Wolkow CA, Munoz MJ, Riddle DL, Ruvkun G. Insulin receptor substrate and p55 orthologous adaptor proteins function in the Caenorhabditis elegans daf-2/insulin-like signaling pathway. J Biol Chem (2002) 277(51):49591–7. doi:10.1074/jbc.M207866200
52. Sawka-Verhelle D, Baron V, Mothe I, Filloux C, White MF, Van Obberghen E. Tyr624 and Tyr628 in insulin receptor substrate-2 mediate its association with the insulin receptor. J Biol Chem (1997) 272(26):16414–20. doi:10.1074/jbc.272.26.16414
53. Wu J, Tseng YD, Xu CF, Neubert TA, White MF, Hubbard SR. Structural and biochemical characterization of the KRLB region in insulin receptor substrate-2. Nat Struct Mol Biol (2008) 15(3):251–8. doi:10.1038/nsmb.1388
54. Rordorf-Nikolic T, Van Horn DJ, Chen D, White MF, Backer JM. Regulation of phosphatidylinositol 3’-kinase by tyrosyl phosphoproteins. Full activation requires occupancy of both SH2 domains in the 85-kDa regulatory subunit. J Biol Chem (1995) 270(8):3662–6.
55. Sun XJ, Crimmins DL, Myers MG Jr, Miralpeix M, White MF. Pleiotropic insulin signals are engaged by multisite phosphorylation of IRS-1. Mol Cell Biol (1993) 13(12):7418–28.
56. Valverde AM, Arribas M, Mur C, Navarro P, Pons S, Cassard-Doulcier AM, et al. Insulin-induced up-regulated uncoupling protein-1 expression is mediated by insulin receptor substrate 1 through the phosphatidylinositol 3-kinase/Akt signaling pathway in fetal brown adipocytes. J Biol Chem (2003) 278(12):10221–31. doi:10.1074/jbc.M209363200
57. Landis J, Shaw LM. Insulin receptor substrate 2-mediated phosphatidylinositol 3-kinase signaling selectively inhibits glycogen synthase kinase 3beta to regulate aerobic glycolysis. J Biol Chem (2014) 289(26):18603–13. doi:10.1074/jbc.M114.564070
58. Myers MG Jr, Wang LM, Sun XJ, Zhang Y, Yenush L, Schlessinger J, et al. Role of IRS-1-GRB-2 complexes in insulin signaling. Mol Cell Biol (1994) 14(6):3577–87.
59. Xu X, Sarikas A, Dias-Santagata DC, Dolios G, Lafontant PJ, Tsai SC, et al. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Mol Cell (2008) 30(4):403–14. doi:10.1016/j.molcel.2008.03.009
60. Zhang J, Gao Z, Yin J, Quon MJ, Ye J. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2. J Biol Chem (2008) 283(51):35375–82. doi:10.1074/jbc.M806480200
61. Copps KD, Hancer NJ, Opare-Ado L, Qiu W, Walsh C, White MF. Irs1 serine 307 promotes insulin sensitivity in mice. Cell Metab (2010) 11(1):84–92. doi:10.1016/j.cmet.2009.11.003
62. Usui I, Imamura T, Huang J, Satoh H, Shenoy SK, Lefkowitz RJ, et al. beta-arrestin-1 competitively inhibits insulin-induced ubiquitination and degradation of insulin receptor substrate 1. Mol Cell Biol (2004) 24(20):8929–37. doi:10.1128/MCB.24.20.8929-8937.2004
63. Yoshihara H, Fukushima T, Hakuno F, Saeki Y, Tanaka K, Ito A, et al. Insulin/insulin-like growth factor (IGF) stimulation abrogates an association between a deubiquitinating enzyme USP7 and insulin receptor substrates (IRSs) followed by proteasomal degradation of IRSs. Biochem Biophys Res Commun (2012) 423(1):122–7. doi:10.1016/j.bbrc.2012.05.093
64. Li Y, Xu W, McBurney MW, Longo VD. SirT1 inhibition reduces IGF-I/IRS-2/Ras/ERK1/2 signaling and protects neurons. Cell Metab (2008) 8(1):38–48. doi:10.1016/j.cmet.2008.05.004
65. Araki E, Lipes MA, Patti ME, Bruning JC, Haag B III, Johnson RS, et al. Alternative pathway of insulin signalling in mice with targeted disruption of the IRS-1 gene. Nature (1994) 372(6502):186–90. doi:10.1038/372186a0
66. Burks DJ, Font de Mora J, Schubert M, Withers DJ, Myers MG, Towery HH, et al. IRS-2 pathways integrate female reproduction and energy homeostasis. Nature (2000) 407(6802):377–82. doi:10.1038/35030105
67. Fantin VR, Wang Q, Lienhard GE, Keller SR. Mice lacking insulin receptor substrate 4 exhibit mild defects in growth, reproduction, and glucose homeostasis. Am J Physiol Endocrinol Metab (2000) 278(1):E127–33.
68. Hoshi K, Ogata N, Shimoaka T, Terauchi Y, Kadowaki T, Kenmotsu S, et al. Deficiency of insulin receptor substrate-1 impairs skeletal growth through early closure of epiphyseal cartilage. J Bone Miner Res (2004) 19(2):214–23. doi:10.1359/JBMR.0301221
69. Kulkarni RN, Winnay JN, Daniels M, Bruning JC, Flier SN, Hanahan D, et al. Altered function of insulin receptor substrate-1-deficient mouse islets and cultured beta-cell lines. J Clin Invest (1999) 104(12):R69–75. doi:10.1172/JCI8339
70. Laustsen PG, Michael MD, Crute BE, Cohen SE, Ueki K, Kulkarni RN, et al. Lipoatrophic diabetes in Irs1(-/-)/Irs3(-/-) double knockout mice. Genes Dev (2002) 16(24):3213–22. doi:10.1101/gad.1034802
71. Liu SC, Wang Q, Lienhard GE, Keller SR. Insulin receptor substrate 3 is not essential for growth or glucose homeostasis. J Biol Chem (1999) 274(25):18093–9. doi:10.1074/jbc.274.25.18093
72. Miki H, Yamauchi T, Suzuki R, Komeda K, Tsuchida A, Kubota N, et al. Essential role of insulin receptor substrate 1 (IRS-1) and IRS-2 in adipocyte differentiation. Mol Cell Biol (2001) 21(7):2521–32. doi:10.1128/MCB.21.7.2521-2532.2001
73. Ogata N, Chikazu D, Kubota N, Terauchi Y, Tobe K, Azuma Y, et al. Insulin receptor substrate-1 in osteoblast is indispensable for maintaining bone turnover. J Clin Invest (2000) 105(7):935–43. doi:10.1172/JCI9017
74. Previs SF, Withers DJ, Ren JM, White MF, Shulman GI. Contrasting effects of IRS-1 versus IRS-2 gene disruption on carbohydrate and lipid metabolism in vivo. J Biol Chem (2000) 275(50):38990–4. doi:10.1074/jbc.M006490200
75. Tamemoto H, Kadowaki T, Tobe K, Yagi T, Sakura H, Hayakawa T, et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature (1994) 372(6502):182–6. doi:10.1038/372182a0
76. Withers DJ, Burks DJ, Towery HH, Altamuro SL, Flint CL, White MF. Irs-2 coordinates Igf-1 receptor-mediated beta-cell development and peripheral insulin signalling. Nat Genet (1999) 23(1):32–40. doi:10.1038/12631
77. Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature (1998) 391(6670):900–4. doi:10.1038/36116
78. Yamauchi T, Tobe K, Tamemoto H, Ueki K, Kaburagi Y, Yamamoto-Honda R, et al. Insulin signalling and insulin actions in the muscles and livers of insulin-resistant, insulin receptor substrate 1-deficient mice. Mol Cell Biol (1996) 16(6):3074–84.
79. Sadagurski M, Dong XC, Myers MG Jr, White MF. Irs2 and Irs4 synergize in non-LepRb neurons to control energy balance and glucose homeostasis. Mol Metab (2014) 3(1):55–63. doi:10.1016/j.molmet.2013.10.004
80. Sadagurski M, Leshan RL, Patterson C, Rozzo A, Kuznetsova A, Skorupski J, et al. IRS2 signaling in LepR-b neurons suppresses FoxO1 to control energy balance independently of leptin action. Cell Metab (2012) 15(5):703–12. doi:10.1016/j.cmet.2012.04.011
81. Akune T, Ogata N, Hoshi K, Kubota N, Terauchi Y, Tobe K, et al. Insulin receptor substrate-2 maintains predominance of anabolic function over catabolic function of osteoblasts. J Cell Biol (2002) 159(1):147–56. doi:10.1083/jcb.200204046
82. Abe H, Yamada N, Kamata K, Kuwaki T, Shimada M, Osuga J, et al. Hypertension, hypertriglyceridemia, and impaired endothelium-dependent vascular relaxation in mice lacking insulin receptor substrate-1. J Clin Invest (1998) 101(8):1784–8. doi:10.1172/JCI1594
83. Toyoshima Y, Tokita R, Taguchi Y, Akiyama-Akanishi N, Takenaka A, Kato H, et al. Tissue-specific effects of protein malnutrition on insulin signaling pathway and lipid accumulation in growing rats. Endocr J (2014) 61(5):499–512. doi:10.1507/endocrj.EJ13-0514
84. Kabuta T, Hakuno F, Cho Y, Yamanaka D, Chida K, Asano T, et al. Insulin receptor substrate-3, interacting with Bcl-3, enhances p50 NF-kappaB activity. Biochem Biophys Res Commun (2010) 394(3):697–702. doi:10.1016/j.bbrc.2010.03.054
85. Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, Gurnani P, et al. Suppression of aging in mice by the hormone klotho. Science (2005) 309(5742):1829–33. doi:10.1126/science.1112766
86. Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science (2001) 292(5514):107–10. doi:10.1126/science.1057987
87. Tu MP, Epstein D, Tatar M. The demography of slow aging in male and female Drosophila mutant for the insulin-receptor substrate homologue chico. Aging Cell (2002) 1(1):75–80. doi:10.1046/j.1474-9728.2002.00010.x
88. Ozoe A, Sone M, Fukushima T, Kataoka N, Arai T, Chida K, et al. Insulin receptor substrate-1 (IRS-1) forms a ribonucleoprotein complex associated with polysomes. FEBS Lett (2013) 587(15):2319–24. doi:10.1016/j.febslet.2013.05.066
89. Ozoe A, Sone M, Fukushima T, Kataoka N, Chida K, Asano T, et al. Insulin receptor substrate-1 associates with small nucleolar RNA which contributes to ribosome biogenesis. Front Endocrinol (Lausanne) (2014) 5:24. doi:10.3389/fendo.2014.00024
90. Mardilovich K, Pankratz SL, Shaw LM. Expression and function of the insulin receptor substrate proteins in cancer. Cell Commun Signal (2009) 7:14. doi:10.1186/1478-811X-7-14
91. Chang Q, Li Y, White MF, Fletcher JA, Xiao S. Constitutive activation of insulin receptor substrate 1 is a frequent event in human tumors: therapeutic implications. Cancer Res (2002) 62(21):6035–8.
92. Schnarr B, Strunz K, Ohsam J, Benner A, Wacker J, Mayer D. Down-regulation of insulin-like growth factor-I receptor and insulin receptor substrate-1 expression in advanced human breast cancer. Int J Cancer (2000) 89(6):506–13. doi:10.1002/1097-0215(20001120)89:6<506::AID-IJC7>3.0.CO;2-F
93. Han CH, Cho JY, Moon JT, Kim HJ, Kim SK, Shin DH, et al. Clinical significance of insulin receptor substrate-I down-regulation in non-small cell lung cancer. Oncol Rep (2006) 16(6):1205–10. doi:10.3892/or.16.6.1205
94. Oliveira JC, Souza KK, Dias MM, Faria MC, Ropelle ER, Flores MB, et al. Antineoplastic effect of rapamycin is potentiated by inhibition of IRS-1 signaling in prostate cancer cells xenografts. J Cancer Res Clin Oncol (2008) 134(8):833–9. doi:10.1007/s00432-008-0359-5
95. Reuveni H, Flashner-Abramson E, Steiner L, Makedonski K, Song R, Shir A, et al. Therapeutic destruction of insulin receptor substrates for cancer treatment. Cancer Res (2013) 73(14):4383–94. doi:10.1158/0008-5472.CAN-12-3385
96. O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res (2006) 66(3):1500–8. doi:10.1158/0008-5472.CAN-05-2925
97. Dearth RK, Cui X, Kim HJ, Hadsell DL, Lee AV. Oncogenic transformation by the signaling adaptor proteins insulin receptor substrate (IRS)-1 and IRS-2. Cell Cycle (2007) 6(6):705–13. doi:10.4161/cc.6.6.4035
98. Sugita H, Kaneki M, Furuhashi S, Hirota M, Takamori H, Baba H. Nitric oxide inhibits the proliferation and invasion of pancreatic cancer cells through degradation of insulin receptor substrate-1 protein. Mol Cancer Res (2010) 8(8):1152–63. doi:10.1158/1541-7786.MCR-09-0472
99. Chang KW, Chu TH, Gong NR, Chiang WF, Yang CC, Liu CJ, et al. miR-370 modulates insulin receptor substrate-1 expression and inhibits the tumor phenotypes of oral carcinoma. Oral Dis (2013) 19(6):611–9. doi:10.1111/odi.12046
100. de Blaquiere GE, May FE, Westley BR. Increased expression of both insulin receptor substrates 1 and 2 confers increased sensitivity to IGF-1 stimulated cell migration. Endocr Relat Cancer (2009) 16(2):635–47. doi:10.1677/ERC-08-0216
101. Ma Y, Kong Q, Hua H, Luo T, Jiang Y. Aflatoxin B1 up-regulates insulin receptor substrate 2 and stimulates hepatoma cell migration. PLoS One (2012) 7(10):e47961. doi:10.1371/journal.pone.0047961
102. Giles KM, Brown RA, Epis MR, Kalinowski FC, Leedman PJ. miRNA-7-5p inhibits melanoma cell migration and invasion. Biochem Biophys Res Commun (2013) 430(2):706–10. doi:10.1016/j.bbrc.2012.11.086
103. Kim B, Feldman EL. Insulin receptor substrate (IRS)-2, not IRS-1, protects human neuroblastoma cells against apoptosis. Apoptosis (2009) 14(5):665–73. doi:10.1007/s10495-009-0331-0
104. Dearth RK, Cui X, Kim HJ, Kuiatse I, Lawrence NA, Zhang X, et al. Mammary tumorigenesis and metastasis caused by overexpression of insulin receptor substrate 1 (IRS-1) or IRS-2. Mol Cell Biol (2006) 26(24):9302–14. doi:10.1128/MCB.00260-06
105. Longato L, de la Monte S, Kuzushita N, Horimoto M, Rogers AB, Slagle BL, et al. Overexpression of insulin receptor substrate-1 and hepatitis Bx genes causes premalignant alterations in the liver. Hepatology (2009) 49(6):1935–43. doi:10.1002/hep.22856
106. Ramocki NM, Wilkins HR, Magness ST, Simmons JG, Scull BP, Lee GH, et al. Insulin receptor substrate-1 deficiency promotes apoptosis in the putative intestinal crypt stem cell region, limits Apcmin/+ tumors, and regulates Sox9. Endocrinology (2008) 149(1):261–7. doi:10.1210/en.2007-0869
107. Nakamura A, Tajima K, Zolzaya K, Sato K, Inoue R, Yoneda M, et al. Protection from non-alcoholic steatohepatitis and liver tumorigenesis in high fat-fed insulin receptor substrate-1-knockout mice despite insulin resistance. Diabetologia (2012) 55(12):3382–91. doi:10.1007/s00125-012-2703-1
108. Nagle JA, Ma Z, Byrne MA, White MF, Shaw LM. Involvement of insulin receptor substrate 2 in mammary tumor metastasis. Mol Cell Biol (2004) 24(22):9726–35. doi:10.1128/MCB.24.22.9726-9735.2004
109. Szabolcs M, Keniry M, Simpson L, Reid LJ, Koujak S, Schiff SC, et al. Irs2 inactivation suppresses tumor progression in Pten± mice. Am J Pathol (2009) 174(1):276–86. doi:10.2353/ajpath.2009.080086
110. Ma Z, Gibson SL, Byrne MA, Zhang J, White MF, Shaw LM. Suppression of insulin receptor substrate 1 (IRS-1) promotes mammary tumor metastasis. Mol Cell Biol (2006) 26(24):9338–51. doi:10.1128/MCB.01032-06
111. Luo X, Fan S, Huang W, Zhai S, Ma Z, Li P, et al. Downregulation of IRS-1 promotes metastasis of head and neck squamous cell carcinoma. Oncol Rep (2012) 28(2):659–67. doi:10.3892/or.2012.1846
112. Cao M, Li Y, Lu H, Meng Q, Wang L, Cai L, et al. MiR-23a-mediated migration/invasion is rescued by its target, IRS-1, in non-small cell lung cancer cells. J Cancer Res Clin Oncol (2014) 140(10):1661–70. doi:10.1007/s00432-014-1725-0
113. Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med (2010) 16(2):219–23. doi:10.1038/nm.2084
114. Reiss K, Del Valle L, Lassak A, Trojanek J. Nuclear IRS-1 and cancer. J Cell Physiol (2012) 227(8):2992–3000. doi:10.1002/jcp.24019
115. Ariga M, Nedachi T, Akahori M, Sakamoto H, Ito Y, Hakuno F, et al. Signalling pathways of insulin-like growth factor-I that are augmented by cAMP in FRTL-5 cells. Biochem J (2000) 348(2):409–16. doi:10.1042/0264-6021:3480409
116. Fukushima T, Arai T, Ariga-Nedachi M, Okajima H, Ooi Y, Iijima Y, et al. Insulin receptor substrates form high-molecular-mass complexes that modulate their availability to insulin/insulin-like growth factor-I receptor tyrosine kinases. Biochem Biophys Res Commun (2011) 404(3):767–73. doi:10.1016/j.bbrc.2010.12.045
117. Sun XJ, Pons S, Asano T, Myers MJ, Glasheen E, White MF. The Fyn tyrosine kinase binds Irs-1 and forms a distinct signaling complex during insulin stimulation. J Biol Chem (1996) 271(18):10583–7. doi:10.1074/jbc.271.18.10583
118. Miranda C, Greco A, Miele C, Pierotti MA, Van Obberghen E. IRS-1 and IRS-2 are recruited by TrkA receptor and oncogenic TRK-T1. J Cell Physiol (2001) 186(1):35–46. doi:10.1002/1097-4652(200101)186:1<35::AID-JCP1003>3.3.CO;2-O
119. Lebrun P, Mothe-Satney I, Delahaye L, Van Obberghen E, Baron V. Insulin receptor substrate-1 as a signaling molecule for focal adhesion kinase pp125(FAK) and pp60(src). J Biol Chem (1998) 273(48):32244–53. doi:10.1074/jbc.273.48.32244
120. Jiang H, Harris MB, Rothman P. IL-4/IL-13 signaling beyond JAK/STAT. J Allergy Clin Immunol (2000) 105(6):1063–70. doi:10.1067/mai.2000.107604
121. Iwanishi M, Czech MP, Cherniack AD. The protein-tyrosine kinase fer associates with signaling complexes containing insulin receptor substrate-1 and phosphatidylinositol 3-kinase. J Biol Chem (2000) 275(50):38995–9000. doi:10.1074/jbc.M006665200
122. Bonvini P, Gastaldi T, Falini B, Rosolen A. Nucleophosmin-anaplastic lymphoma kinase (NPM-ALK), a novel Hsp90-client tyrosine kinase: down-regulation of NPM-ALK expression and tyrosine phosphorylation in ALK(+) CD30(+) lymphoma cells by the Hsp90 antagonist 17-allylamino,17-demethoxygeldanamycin. Cancer Res (2002) 62(5):1559–66.
123. Knowlden JM, Jones HE, Barrow D, Gee JM, Nicholson RI, Hutcheson IR. Insulin receptor substrate-1 involvement in epidermal growth factor receptor and insulin-like growth factor receptor signalling: implication for Gefitinib (‘Iressa’) response and resistance. Breast Cancer Res Treat (2008) 111(1):79–91. doi:10.1007/s10549-007-9763-9
124. Noguchi T, Matozaki T, Horita K, Fujioka Y, Kasuga M. Role of SH-PTP2, a protein-tyrosine phosphatase with Src homology 2 domains, in insulin-stimulated Ras activation. Mol Cell Biol (1994) 14(10):6674–82.
125. Ali MS, Schieffer B, Delafontaine P, Bernstein KE, Ling BN, Marrero MB. Angiotensin II stimulates tyrosine phosphorylation and activation of insulin receptor substrate 1 and protein-tyrosine phosphatase 1D in vascular smooth muscle cells. J Biol Chem (1997) 272(19):12373–9. doi:10.1074/jbc.272.19.12373
126. Calera MR, Vallega G, Pilch PF. Dynamics of protein-tyrosine phosphatases in rat adipocytes. J Biol Chem (2000) 275(9):6308–12. doi:10.1074/jbc.275.9.6308
127. Dromard M, Bompard G, Glondu-Lassis M, Puech C, Chalbos D, Freiss G. The putative tumor suppressor gene PTPN13/PTPL1 induces apoptosis through insulin receptor substrate-1 dephosphorylation. Cancer Res (2007) 67(14):6806–13. doi:10.1158/0008-5472.CAN-07-0513
128. Koh A, Lee MN, Yang YR, Jeong H, Ghim J, Noh J, et al. C1-ten is a protein tyrosine phosphatase of IRS-1, regulating IRS-1 stability and muscle atrophy. Mol Cell Biol (2013) 33(8):1608–20. doi:10.1128/MCB.01447-12
129. Sorokin AV, Chen J. MEMO1, a new IRS1-interacting protein, induces epithelial-mesenchymal transition in mammary epithelial cells. Oncogene (2013) 32(26):3130–8. doi:10.1038/onc.2012.327
130. Morris DL, Cho KW, Zhou Y, Rui L. SH2B1 enhances insulin sensitivity by both stimulating the insulin receptor and inhibiting tyrosine dephosphorylation of IRS proteins. Diabetes (2009) 58(9):2039–47. doi:10.2337/db08-1388
131. Jaeschke A, Czech MP, Davis RJ. An essential role of the JIP1 scaffold protein for JNK activation in adipose tissue. Genes Dev (2004) 18(16):1976–80. doi:10.1101/gad.1216504
132. Standen CL, Kennedy NJ, Flavell RA, Davis RJ. Signal transduction cross-talk mediated by JIP and IRS scaffold protein complexes. Mol Cell Biol (2009) 29(17):4831–40. doi:10.1128/MCB.00155-09
133. Li Z, Joyal JL, Sacks DB. Binding of IRS proteins to calmodulin is enhanced in insulin resistance. Biochemistry (2000) 39(17):5089–96. doi:10.1021/bi992623z
134. Ando Y, Shinozawa Y, Iijima Y, Yu BC, Sone M, Ooi Y, et al. TNF-alpha-induced repression of GKAP42 protein levels through cGK-Ialpha causes insulin resistance in 3T3-L1 adipocytes. J Biol Chem (2015) 290(9):5881–92. doi:10.1074/jbc.M114.624759
135. Fukushima T, Okajima H, Yamanaka D, Ariga M, Nagata S, Ito A, et al. HSP90 interacting with IRS-2 is involved in cAMP-dependent potentiation of IGF-I signals in FRTL-5 cells. Mol Cell Endocrinol (2011) 344(1–2):81–9. doi:10.1016/j.mce.2011.06.029
136. Kaiser C, James SR. Acetylation of insulin receptor substrate-1 is permissive for tyrosine phosphorylation. BMC Biol (2004) 2:23. doi:10.1186/1741-7007-2-23
137. Zhang J. The direct involvement of SirT1 in insulin-induced IRS-2 tyrosine phosphorylation. J Biol Chem (2007) 282(47):34356–64. doi:10.1074/jbc.M706644200
138. Nakatsu Y, Sakoda H, Kushiyama A, Zhang J, Ono H, Fujishiro M, et al. Peptidyl-prolyl cis/trans isomerase NIMA-interacting 1 associates with insulin receptor substrate-1 and enhances insulin actions and adipogenesis. J Biol Chem (2011) 286(23):20812–22. doi:10.1074/jbc.M110.206904
139. Zhang J, Nakatsu Y, Shinjo T, Guo Y, Sakoda H, Yamamotoya T, et al. Par14 protein associates with insulin receptor substrate 1 (IRS-1), thereby enhancing insulin-induced IRS-1 phosphorylation and metabolic actions. J Biol Chem (2013) 288(28):20692–701. doi:10.1074/jbc.M113.485730
140. Fukushima T, Yoshihara H, Furuta H, Kamei H, Hakuno F, Luan J, et al. Nedd4-induced mono-ubiquitination of IRS-2 enhances IGF signaling and mitogenic activity. Nat Commun (2015) 6:6780. doi:10.1038/ncomms7780
141. Farhang-Fallah J, Yin X, Trentin G, Cheng AM, Rozakis-Adcock M. Cloning and characterization of PHIP, a novel insulin receptor substrate-1 pleckstrin homology domain interacting protein. J Biol Chem (2000) 275(51):40492–7. doi:10.1074/jbc.C000611200
142. Hakuno F, Kurihara S, Watson RT, Pessin JE, Takahashi S. 53BP2S, interacting with insulin receptor substrates, modulates insulin signaling. J Biol Chem (2007) 282(52):37747–58. doi:10.1074/jbc.M702472200
143. Ogihara T, Isobe T, Ichimura T, Taoka M, Funaki M, Sakoda H, et al. 14-3-3 protein binds to insulin receptor substrate-1, one of the binding sites of which is in the phosphotyrosine binding domain. J Biol Chem (1997) 272(40):25267–74. doi:10.1074/jbc.272.40.25267
144. Oriente F, Andreozzi F, Romano C, Perruolo G, Perfetti A, Fiory F, et al. Protein kinase C-alpha regulates insulin action and degradation by interacting with insulin receptor substrate-1 and 14-3-3epsilon. J Biol Chem (2005) 280(49):40642–9. doi:10.1074/jbc.M508570200
145. Kosaki A, Yamada K, Suga J, Otaka A, Kuzuya H. 14-3-3beta protein associates with insulin receptor substrate 1 and decreases insulin-stimulated phosphatidylinositol 3’-kinase activity in 3T3L1 adipocytes. J Biol Chem (1998) 273(2):940–4. doi:10.1074/jbc.273.2.940
146. Xiang X, Yuan M, Song Y, Ruderman N, Wen R, Luo Z. 14-3-3 facilitates insulin-stimulated intracellular trafficking of insulin receptor substrate 1. Mol Endocrinol (2002) 16(3):552–62. doi:10.1210/mend.16.3.0790
147. Neukamm SS, Ott J, Dammeier S, Lehmann R, Haring HU, Schleicher E, et al. Phosphorylation of serine 1137/1138 of mouse IRS2 regulates cAMP-dependent binding to 14-3-3 proteins and IRS2 protein degradation. J Biol Chem (2013) 288(23):16403–15. doi:10.1074/jbc.M113.474593
148. Hafner M, Schmitz A, Grune I, Srivatsan SG, Paul B, Kolanus W, et al. Inhibition of cytohesins by SecinH3 leads to hepatic insulin resistance. Nature (2006) 444(7121):941–4. doi:10.1038/nature05415
149. Lee A, Hakuno F, Northcott P, Pessin JE, Rozakis Adcock M. Nexilin, a cardiomyopathy-associated F-actin binding protein, binds and regulates IRS1 signaling in skeletal muscle cells. PLoS One (2013) 8(1):e55634. doi:10.1371/journal.pone.0055634
150. Boura-Halfon S, Zick Y. Serine kinases of insulin receptor substrate proteins. Vitam Horm (2009) 80:313–49. doi:10.1016/S0083-6729(08)00612-2
151. Craparo A, Freund R, Gustafson TA. 14-3-3 (epsilon) interacts with the insulin-like growth factor I receptor and insulin receptor substrate I in a phosphoserine-dependent manner. J Biol Chem (1997) 272(17):11663–9. doi:10.1074/jbc.272.17.11663
152. Neukamm SS, Toth R, Morrice N, Campbell DG, Mackintosh C, Lehmann R, et al. Identification of the amino acids 300-600 of IRS-2 as 14-3-3 binding region with the importance of IGF-1/insulin-regulated phosphorylation of Ser-573. PLoS One (2012) 7(8):e43296. doi:10.1371/journal.pone.0043296
153. Nakao R, Hirasaka K, Goto J, Ishidoh K, Yamada C, Ohno A, et al. Ubiquitin ligase Cbl-b is a negative regulator for insulin-like growth factor 1 signaling during muscle atrophy caused by unloading. Mol Cell Biol (2009) 29(17):4798–811. doi:10.1128/MCB.01347-08
154. Scheufele F, Wolf B, Kruse M, Hartmann T, Lempart J, Muehlich S, et al. Evidence for a regulatory role of Cullin-RING E3 ubiquitin ligase 7 in insulin signaling. Cell Signal (2014) 26(2):233–9. doi:10.1016/j.cellsig.2013.11.005
155. Shi J, Luo L, Eash J, Ibebunjo C, Glass DJ. The SCF-Fbxo40 complex induces IRS1 ubiquitination in skeletal muscle, limiting IGF1 signaling. Dev Cell (2011) 21(5):835–47. doi:10.1016/j.devcel.2011.09.011
156. Song R, Peng W, Zhang Y, Lv F, Wu HK, Guo J, et al. Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature (2013) 494(7437):375–9. doi:10.1038/nature11834
157. Yi JS, Park JS, Ham YM, Nguyen N, Lee NR, Hong J, et al. MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat Commun (2013) 4:2354. doi:10.1038/ncomms3354
158. Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem (2002) 277(44):42394–8. doi:10.1074/jbc.C200444200
159. Emanuelli B, Peraldi P, Filloux C, Chavey C, Freidinger K, Hilton DJ, et al. SOCS-3 inhibits insulin signaling and is up-regulated in response to tumor necrosis factor-alpha in the adipose tissue of obese mice. J Biol Chem (2001) 276(51):47944–9. doi:10.1074/jbc.M104602200
160. Ueki K, Kadowaki T, Kahn CR. Role of suppressors of cytokine signaling SOCS-1 and SOCS-3 in hepatic steatosis and the metabolic syndrome. Hepatol Res (2005) 33(2):185–92. doi:10.1016/j.hepres.2005.09.032
161. He F, Stephens JM. Induction of SOCS-3 is insufficient to confer IRS-1 protein degradation in 3T3-L1 adipocytes. Biochem Biophys Res Commun (2006) 344(1):95–8. doi:10.1016/j.bbrc.2006.03.142
162. Jiang Y, Zhang Q, Soderland C, Steinle JJ. TNFalpha and SOCS3 regulate IRS-1 to increase retinal endothelial cell apoptosis. Cell Signal (2012) 24(5):1086–92. doi:10.1016/j.cellsig.2012.01.003
163. Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, et al. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol Endocrinol (2000) 14(6):783–94. doi:10.1210/mend.14.6.0446
164. Tzatsos A, Kandror KV. Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol (2006) 26(1):63–76. doi:10.1128/MCB.26.1.63-76.2006
165. Lee AV, Gooch JL, Oesterreich S, Guler RL, Yee D. Insulin-like growth factor I-induced degradation of insulin receptor substrate 1 is mediated by the 26S proteasome and blocked by phosphatidylinositol 3’-kinase inhibition. Mol Cell Biol (2000) 20(5):1489–96. doi:10.1128/MCB.20.5.1489-1496.2000
166. Rui L, Fisher TL, Thomas J, White MF. Regulation of insulin/insulin-like growth factor-1 signaling by proteasome-mediated degradation of insulin receptor substrate-2. J Biol Chem (2001) 276(43):40362–7. doi:10.1074/jbc.M105332200
167. Zhande R, Mitchell JJ, Wu J, Sun XJ. Molecular mechanism of insulin-induced degradation of insulin receptor substrate 1. Mol Cell Biol (2002) 22(4):1016–26. doi:10.1128/MCB.22.4.1016-1026.2002
168. Hakuno F, Yamauchi Y, Kaneko G, Yoneyama Y, Nakae J, Chida K, et al. Constitutive expression of insulin receptor substrate (IRS)-1 inhibits myogenic differentiation through nuclear exclusion of Foxo1 in L6 myoblasts. PLoS One (2011) 6(10):e25655. doi:10.1371/journal.pone.0025655
169. Clark SF, Martin S, Carozzi AJ, Hill MM, James DE. Intracellular localization of phosphatidylinositide 3-kinase and insulin receptor substrate-1 in adipocytes: potential involvement of a membrane skeleton. J Cell Biol (1998) 140(5):1211–25. doi:10.1083/jcb.140.5.1211
170. Heller-Harrison RA, Morin M, Czech MP. Insulin regulation of membrane-associated insulin receptor substrate 1. J Biol Chem (1995) 270(41):24442–50. doi:10.1074/jbc.270.41.24442
171. Kelly KL, Ruderman NB. Insulin-stimulated phosphatidylinositol 3-kinase. Association with a 185-kDa tyrosine-phosphorylated protein (IRS-1) and localization in a low density membrane vesicle. J Biol Chem (1993) 268(6):4391–8.
172. Ricort JM, Tanti JF, Van Obberghen E, Le Marchand-Brustel Y. Different effects of insulin and platelet-derived growth factor on phosphatidylinositol 3-kinase at the subcellular level in 3T3-L1 adipocytes. A possible explanation for their specific effects on glucose transport. Eur J Biochem (1996) 239(1):17–22. doi:10.1111/j.1432-1033.1996.0017u.x
173. Yoneyama Y, Matsuo M, Take K, Kabuta T, Chida K, Hakuno F, et al. The AP-1 complex regulates intracellular localization of insulin receptor substrate 1, which is required for insulin-like growth factor I-dependent cell proliferation. Mol Cell Biol (2013) 33(10):1991–2003. doi:10.1128/MCB.01394-12
174. VanRenterghem B, Morin M, Czech MP, Heller-Harrison RA. Interaction of insulin receptor substrate-1 with the sigma3A subunit of the adaptor protein complex-3 in cultured adipocytes. J Biol Chem (1998) 273(45):29942–9. doi:10.1074/jbc.273.45.29942
175. Traub LM, Bonifacino JS. Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harb Perspect Biol (2013) 5(11):a016790. doi:10.1101/cshperspect.a016790
176. Nakatsu F, Ohno H. Adaptor protein complexes as the key regulators of protein sorting in the post-Golgi network. Cell Struct Funct (2003) 28(5):419–29. doi:10.1247/csf.28.419
177. Myers MG Jr, Zhang Y, Aldaz GA, Grammer T, Glasheen EM, Yenush L, et al. YMXM motifs and signaling by an insulin receptor substrate 1 molecule without tyrosine phosphorylation sites. Mol Cell Biol (1996) 16(8):4147–55.
178. Owen DJ, Evans PR. A structural explanation for the recognition of tyrosine-based endocytotic signals. Science (1998) 282(5392):1327–32. doi:10.1126/science.282.5392.1327
179. Doray B, Lee I, Knisely J, Bu G, Kornfeld S. The gamma/sigma1 and alpha/sigma2 hemicomplexes of clathrin adaptors AP-1 and AP-2 harbor the dileucine recognition site. Mol Biol Cell (2007) 18(5):1887–96. doi:10.1091/mbc.E07-01-0012
180. Ryu J, Galan AK, Xin X, Dong F, Abdul-Ghani MA, Zhou L, et al. APPL1 potentiates insulin sensitivity by facilitating the binding of IRS1/2 to the insulin receptor. Cell Rep (2014) 7(4):1227–38. doi:10.1016/j.celrep.2014.04.006
181. Lassak A, Del Valle L, Peruzzi F, Wang JY, Enam S, Croul S, et al. Insulin receptor substrate 1 translocation to the nucleus by the human JC virus T-antigen. J Biol Chem (2002) 277(19):17231–8. doi:10.1074/jbc.M110885200
182. Fei ZL, D’Ambrosio C, Li S, Surmacz E, Baserga R. Association of insulin receptor substrate 1 with simian virus 40 large T antigen. Mol Cell Biol (1995) 15(8):4232–9.
183. Kabuta T, Take K, Kabuta C, Hakuno F, Takahashi S. Differential subcellular localization of insulin receptor substrates depends on C-terminal regions and importin beta. Biochem Biophys Res Commun (2008) 377(3):741–6. doi:10.1016/j.bbrc.2008.09.106
184. Panetta D, Biedi C, Repetto S, Cordera R, Maggi D. IGF-I regulates caveolin 1 and IRS1 interaction in caveolae. Biochem Biophys Res Commun (2004) 316(1):240–3. doi:10.1016/j.bbrc.2004.02.037
185. Thomas EC, Zhe Y, Molero JC, Schmitz-Peiffer C, Ramm G, James DE, et al. The subcellular fractionation properties and function of insulin receptor substrate-1 (IRS-1) are independent of cytoskeletal integrity. Int J Biochem Cell Biol (2006) 38(10):1686–99. doi:10.1016/j.biocel.2006.03.009
186. Burgess JW, Wada I, Ling N, Khan MN, Bergeron JJ, Posner BI. Decrease in beta-subunit phosphotyrosine correlates with internalization and activation of the endosomal insulin receptor kinase. J Biol Chem (1992) 267(14):10077–86.
187. Hsu D, Knudson PE, Zapf A, Rolband GC, Olefsky JM. NPXY motif in the insulin-like growth factor-I receptor is required for efficient ligand-mediated receptor internalization and biological signaling. Endocrinology (1994) 134(2):744–50. doi:10.1210/endo.134.2.8299569
188. Romanelli RJ, LeBeau AP, Fulmer CG, Lazzarino DA, Hochberg A, Wood TL. Insulin-like growth factor type-I receptor internalization and recycling mediate the sustained phosphorylation of Akt. J Biol Chem (2007) 282(31):22513–24. doi:10.1074/jbc.M704309200
189. Kublaoui B, Lee J, Pilch PF. Dynamics of signaling during insulin-stimulated endocytosis of its receptor in adipocytes. J Biol Chem (1995) 270(1):59–65. doi:10.1074/jbc.270.1.59
190. Prisco M, Santini F, Baffa R, Liu M, Drakas R, Wu A, et al. Nuclear translocation of insulin receptor substrate-1 by the simian virus 40 T antigen and the activated type 1 insulin-like growth factor receptor. J Biol Chem (2002) 277(35):32078–85. doi:10.1074/jbc.M204658200
191. Tu X, Batta P, Innocent N, Prisco M, Casaburi I, Belletti B, et al. Nuclear translocation of insulin receptor substrate-1 by oncogenes and Igf-I. Effect on ribosomal RNA synthesis. J Biol Chem (2002) 277(46):44357–65. doi:10.1074/jbc.M208001200
192. Wu A, Chen J, Baserga R. Nuclear insulin receptor substrate-1 activates promoters of cell cycle progression genes. Oncogene (2008) 27(3):397–403. doi:10.1038/sj.onc.1210636
193. Fernandez A, Northcott PA, Dalton J, Fraga C, Ellison D, Angers S, et al. YAP1 is amplified and up-regulated in hedgehog-associated medulloblastomas and mediates Sonic hedgehog-driven neural precursor proliferation. Genes Dev (2009) 23(23):2729–41. doi:10.1101/Gad.1824509
194. Kabuta T, Hakuno F, Asano T, Takahashi S. Insulin receptor substrate-3 functions as transcriptional activator in the nucleus. J Biol Chem (2002) 277(9):6846–51. doi:10.1074/jbc.M107058200
195. Karlsson M, Thorn H, Danielsson A, Stenkula KG, Ost A, Gustavsson J, et al. Colocalization of insulin receptor and insulin receptor substrate-1 to caveolae in primary human adipocytes. Cholesterol depletion blocks insulin signalling for metabolic and mitogenic control. Eur J Biochem (2004) 271(12):2471–9. doi:10.1111/j.1432-1033.2004.04177.x
196. Stenkula KG, Thorn H, Franck N, Hallin E, Sauma L, Nystrom FH, et al. Human, but not rat, IRS1 targets to the plasma membrane in both human and rat adipocytes. Biochem Biophys Res Commun (2007) 363(3):840–5. doi:10.1016/j.bbrc.2007.09.065
197. Foti M, Porcheron G, Fournier M, Maeder C, Carpentier JL. The neck of caveolae is a distinct plasma membrane subdomain that concentrates insulin receptors in 3T3-L1 adipocytes. Proc Natl Acad Sci U S A (2007) 104(4):1242–7. doi:10.1073/pnas.0610523104
198. Wang H, Zou X, Wei Z, Wu Y, Li R, Zeng R, et al. Hsp90alpha forms a stable complex at the cilium neck for the interaction of signalling molecules in IGF-1 receptor signalling. J Cell Sci (2015) 128(1):100–8. doi:10.1242/jcs.155101
199. Holz MK, Ballif BA, Gygi SP, Blenis J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell (2005) 123(4):569–80. doi:10.1016/j.cell.2005.10.024
200. Quintana-Portillo R, Canfran-Duque A, Issad T, Sanchez-Margalet V, Gonzalez-Yanes C. Sam68 interacts with IRS1. Biochem Pharmacol (2012) 83(1):78–87. doi:10.1016/j.bcp.2011.09.030
201. Vitali P, Royo H, Seitz H, Bachellerie JP, Huttenhofer A, Cavaille J. Identification of 13 novel human modification guide RNAs. Nucleic Acids Res (2003) 31(22):6543–51. doi:10.1093/nar/gkg849
202. Kiss T. Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions. Cell (2002) 109(2):145–8. doi:10.1016/S0092-8674(02)00718-3
203. Yoon A, Peng G, Brandenburger Y, Zollo O, Xu W, Rego E, et al. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science (2006) 312(5775):902–6. doi:10.1126/science.1123835
204. Jack K, Bellodi C, Landry DM, Niederer RO, Meskauskas A, Musalgaonkar S, et al. rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells. Mol Cell (2011) 44(4):660–6. doi:10.1016/j.molcel.2011.09.017
205. Liang XH, Liu Q, Fournier MJ. rRNA modifications in an intersubunit bridge of the ribosome strongly affect both ribosome biogenesis and activity. Mol Cell (2007) 28(6):965–77. doi:10.1016/j.molcel.2007.10.012
206. Chen J, Wu A, Sun H, Drakas R, Garofalo C, Cascio S, et al. Functional significance of type 1 insulin-like growth factor-mediated nuclear translocation of the insulin receptor substrate-1 and beta-catenin. J Biol Chem (2005) 280(33):29912–20. doi:10.1074/jbc.M504516200
207. Morelli C, Garofalo C, Sisci D, del Rincon S, Cascio S, Tu X, et al. Nuclear insulin receptor substrate 1 interacts with estrogen receptor alpha at ERE promoters. Oncogene (2004) 23(45):7517–26. doi:10.1038/sj.onc.1208014
208. Sun H, Tu X, Prisco M, Wu A, Casiburi I, Baserga R. Insulin-like growth factor I receptor signaling and nuclear translocation of insulin receptor substrates 1 and 2. Mol Endocrinol (2003) 17(3):472–86. doi:10.1210/me.2002-0276
209. Van Wyk JJ, Conti M, Del Monte P, Takahashi SI. The role of somatomedins in the growth and function of endocrine tissues. In: Sizonenko PC, Aubert ML, editors. Dev Endocrinol. New York, NY: Raven Press (1990). p. 127–40.
210. Muller K, Fuhrer D, Mittag J, Kloting N, Bluher M, Weiss RE, et al. TSH compensates thyroid-specific IGF-I receptor knockout and causes papillary thyroid hyperplasia. Mol Endocrinol (2011) 25(11):1867–79. doi:10.1210/me.2011-0065
211. Ferreira IA, Eybrechts KL, Mocking AI, Kroner C, Akkerman JW. IRS-1 mediates inhibition of Ca2+ mobilization by insulin via the inhibitory G-protein Gi. J Biol Chem (2004) 279(5):3254–64. doi:10.1074/jbc.M305474200
212. Geng Y, Ju Y, Ren F, Qiu Y, Tomita Y, Tomoeda M, et al. Insulin receptor substrate 1/2 (IRS1/2) regulates Wnt/beta-catenin signaling through blocking autophagic degradation of dishevelled2. J Biol Chem (2014) 289(16):11230–41. doi:10.1074/jbc.M113.544999
213. Algenstaedt P, Antonetti DA, Yaffe MB, Kahn CR. Insulin receptor substrate proteins create a link between the tyrosine phosphorylation cascade and the Ca2+-ATPases in muscle and heart. J Biol Chem (1997) 272(38):23696–702. doi:10.1074/jbc.272.38.23696
214. Borge PD, Wolf BA. Insulin receptor substrate 1 regulation of sarco-endoplasmic reticulum calcium ATPase 3 in insulin-secreting beta -cells. J Biol Chem (2003) 278(13):11359–68. doi:10.1074/jbc.M209521200
215. Ueno H, Kondo E, Yamamoto HR, Tobe K, Nakamoto T, Sasaki K, et al. Association of insulin receptor substrate proteins with Bcl-2 and their effects on its phosphorylation and antiapoptotic function. Mol Biol Cell (2000) 11(2):735–46. doi:10.1091/mbc.11.2.735
216. Worrall DS, Olefsky JM. The effects of intracellular calcium depletion on insulin signaling in 3T3-L1 adipocytes. Mol Endocrinol (2002) 16(2):378–89. doi:10.1210/mend.16.2.0776
217. Liang F, Kume S, Koya D. SIRT1 and insulin resistance. Nat Rev Endocrinol (2009) 5(7):367–73. doi:10.1038/nrendo.2009.101
218. Ye X, Wang L, Shang B, Wang Z, Wei W. NEDD4: a promising target for cancer therapy. Curr Cancer Drug Targets (2014) 14(6):549–56. doi:10.2174/1568009614666140725092430
219. Tessneer KL, Cai X, Pasula S, Dong Y, Liu X, Chang B, et al. Epsin family of endocytic adaptor proteins as oncogenic regulators of cancer progression. J Can Res Updates (2013) 2(3):144–50. doi:10.6000/1929-2279.2013.02.03.2
220. Khalili K, Del Valle L, Wang JY, Darbinian N, Lassak A, Safak M, et al. T-antigen of human polyomavirus JC cooperates with IGF-IR signaling system in cerebellar tumors of the childhood-medulloblastomas. Anticancer Res (2003) 23(3A):2035–41.
221. Melillo RM, Carlomagno F, De Vita G, Formisano P, Vecchio G, Fusco A, et al. The insulin receptor substrate (IRS)-1 recruits phosphatidylinositol 3-kinase to Ret: evidence for a competition between Shc and IRS-1 for the binding to Ret. Oncogene (2001) 20(2):209–18. doi:10.1038/sj.onc.1204049
222. Hennige AM, Lammers R, Arlt D, Hoppner W, Strack V, Niederfellner G, et al. Ret oncogene signal transduction via a IRS-2/PI 3-kinase/PKB and a SHC/Grb-2 dependent pathway: possible implication for transforming activity in NIH3T3 cells. Mol Cell Endocrinol (2000) 167(1–2):69–76. doi:10.1016/S0303-7207(00)00283-5
223. Lannon CL, Martin MJ, Tognon CE, Jin W, Kim SJ, Sorensen PH. A highly conserved NTRK3 C-terminal sequence in the ETV6-NTRK3 oncoprotein binds the phosphotyrosine binding domain of insulin receptor substrate-1: an essential interaction for transformation. J Biol Chem (2004) 279(8):6225–34. doi:10.1074/jbc.M307388200
Keywords: insulin-like growth factor, insulin, insulin receptor substrate, IRS-associated protein, IRSome, cancer
Citation: Hakuno F, Fukushima T, Yoneyama Y, Kamei H, Ozoe A, Yoshihara H, Yamanaka D, Shibano T, Sone-Yonezawa M, Yu B-C, Chida K and Takahashi S-I (2015) The novel functions of high-molecular-mass complexes containing insulin receptor substrates in mediation and modulation of insulin-like activities: emerging concept of diverse functions by IRS-associated proteins. Front. Endocrinol. 6:73. doi: 10.3389/fendo.2015.00073
Received: 04 March 2015; Accepted: 25 April 2015;
Published: 26 May 2015
Edited by:
Antonino Belfiore, University Magna Graecia of Catanzaro, ItalyReviewed by:
Marco Falasca, Curtin University, AustraliaTeresa Wood, Rutgers Biomedical and Health Sciences, USA
Iulian Oprea, Karolinska Institute, Sweden
Copyright: © 2015 Hakuno, Fukushima, Yoneyama, Kamei, Ozoe, Yoshihara, Yamanaka, Shibano, Sone-Yonezawa, Yu, Chida and Takahashi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shin-Ichiro Takahashi, Laboratory of Cell Regulation, Departments of Animal Sciences and Applied Biological Chemistry, Graduate School of Agriculture and Life Science, The University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan,YXRrc2hpbkBtYWlsLmVjYy51LXRva3lvLmFjLmpw