 Xiaoyong Yang1,2,3*
Xiaoyong Yang1,2,3* Hai-Bin Ruan1,2
Hai-Bin Ruan1,2
- 1Program in Integrative Cell Signaling and Neurobiology of Metabolism, Yale University School of Medicine, New Haven, CT, USA
- 2Section of Comparative Medicine, Yale University School of Medicine, New Haven, CT, USA
- 3Department of Cellular and Molecular Physiology, Yale University School of Medicine, New Haven, CT, USA
The obesity epidemic continues rising as a global health challenge, despite the increasing public awareness and the use of lifestyle and medical interventions. The biomedical community is urged to develop new treatments to obesity. Excess energy is stored as fat in white adipose tissue (WAT), dysfunction of which lies at the core of obesity and associated metabolic disorders. By contrast, brown adipose tissue (BAT) burns fat and dissipates chemical energy as heat. The development and activation of “brown-like” adipocytes, also known as beige cells, result in WAT browning and thermogenesis. The recent discovery of brown and beige adipocytes in adult humans has sparked the exploration of the development, regulation, and function of these thermogenic adipocytes. The central nervous system drives the sympathetic nerve activity in BAT and WAT to control heat production and energy homeostasis. This review provides an overview of the integration of thermal, hormonal, and nutritional information on hypothalamic circuits in thermoregulation.
A Trio of Fat
Obesity is a rising global epidemic. Excess adiposity is a major risk factor for type 2 diabetes, cardiovascular disease, and hypertension. Overweight and obesity arise when energy intake exceeds energy expenditure and consequently excessive calories are stored in the adipose tissue. The adipose organ comprises white adipose tissue (WAT) and brown adipose tissue (BAT). WAT primarily stores energy as triglycerides, whereas BAT dissipates chemical energy as heat, a process mediated by uncoupling protein 1 (UCP1) (1–3). WAT excess and dysfunction lie at the core of obesity and associated metabolic disorders. By contrast, BAT-mediated adaptive thermogenesis serves to maintain body temperature during cold, contributes to fever during infection, as well as counteracts obesity and related metabolic dysfunction (4). Here, adaptive thermogenesis is defined as non-shivering heat production in response to changes in environmental and physiological settings, such as cold, diet, fever, and stress. Adaptive thermogenesis in BAT is believed to be solely dependent on UCP1 (1–6). However, UCP1-independent thermogenesis in other tissues such as WAT has also been suggested (7, 8).
In 2009, metabolically active BAT was “re-discovered” in adult humans (9–13). The activity of BAT in humans responds to cold challenge and is inversely associated with body mass index (BMI) and age (14). Yet in its infancy, increasing the mass and activity of BAT is considered as a therapeutic option for human obesity.
A “brown-like” type of adipocytes, called beige cells, has been recently discovered in specific WAT depots. Although different from classic brown adipocytes in their origin and molecular identity, beige adipocytes express Ucp1, contribute to thermoregulation, and prevent metabolic dysfunction in mice (15, 16). The process of recruiting and activating beige adipocytes is referred to as “browning.” Although classical brown adipocytes are present in adult humans (9, 17, 18), growing evidence suggests that adult human BAT is mainly composed of beige adipocytes (19–21). Considering the fact that interscapular BAT in human infants consists of bona fide brown adipocytes and that BAT depots in newborns and adults are located at different sites (22, 23), we speculate that classic brown adipocytes degenerate whereas beige adipocytes gradually prevail with age in humans. Understanding the molecular mechanisms under this transition is important for future therapeutics designed to boost BAT function. It is also intriguing and important to determine the existence and identity of beige adipocytes in other WAT depots, such as subcutaneous fat in humans.
The potential of brown and beige adipocytes as anti-obesity targets attracts extensive interest. The last decade has seen an explosion in our understanding of the development, regulation, and pathophysiology of these distinct adipocytes. Chronic cold exposure, by stimulating the sympathetic nervous system (SNS), is a major and potent activator of BAT thermogenesis and WAT browning. Agonists to β3-adrenergic receptors (AR) that are selectively expressed in brown and beige adipocytes stimulate thermogenesis in both rodents and humans (24). In addition, numerous intrinsic proteins and secreted factors have been shown to affect the development and function of brown and/or beige adipocytes (15, 16, 25). The role of the central nervous system (CNS) in controlling adipose tissue thermogenesis has been an area of intense investigation (26–31). In this review, we summarize the recent progress in our understanding of the central regulation of thermogenesis in brown and beige cells.
Hypothalamus Orchestrates Metabolism
The hypothalamus acts as to orchestrate homeostatic functions such as food intake, energy expenditure, glucose metabolism, and circadian rhythm. Homeostasis is achieved through the complex crosstalk between the hypothalamus and peripheral tissues in response to environmental cues. The arcuate nucleus (ARC) of the hypothalamus is considered to be a primary integrator of peripheral signals, including hormones and nutrients. Two extensively studied populations of neurons in the ARC are orexigenic neurons expressing agouti-related protein (AgRP)/neuropeptide Y (NPY) and anorexigenic neurons expressing proopiomelanocortin (POMC). These neurons are sensitive, mostly in opposite ways, to hormones such as leptin, insulin, and ghrelin, as well as nutrients such as glucose, amino acids, and fatty acids (32–34). Melanocortin peptides produced by POMC neurons are agonists whereas AgRP is an antagonist of melanocortin-3 and -4 receptors (MC3R and MC4R) that are expressed on the second-order neurons. Located largely in the lateral hypothalamus (LH) and the paraventricular nucleus (PVN) of the hypothalamus, these downstream neurons receive projections from the ARC as well as direct inputs from peripheral signals. Together with the ARC, the LH and PVN function as a metabolic integrator and regulator by projecting to high-order neurons in the CNS and secreting various neuropeptides, for example orexin, melanin concentrating hormone (MCH), cocaine- and amphetamine-regulated transcript (CART), and corticotropin-releasing hormone (CRH).
In addition to their neuroendocrine role, hypothalamic neurons also project to the SNS to control peripheral metabolism (Figure 1). Both BAT and WAT are extensively innervated by the sympathetic fibers that can be tracked back to the hypothalamus (35). By using the neurotropic pseudorabies virus (PRV), a number of studies have described the neuroanatomy of the sympathetic control of adipose tissues in rodents. Although BAT and WAT are anatomically and functionally distinct, common brain areas with efferent projections to both adipose tissues have been identified, including the ARC, LH, and PVN of the hypothalamus and other neuronal sites discussed below (35). Release of catecholamine, particularly norepinephrine (NE), from the sympathetic fibers and subsequent activation of β-AR signaling in adipocytes are necessary for the initiation of lipolysis and the activation of thermogenesis (36). It is thus reasonable to speculate that a “command” neural network dictates these two processes in BAT and WAT (35). It should be noted that only a small portion (about 5–15%) of individual neurons in these common brain regions projects both to BAT and WAT and distinct sympathetic circuits project to different WAT depots (35, 36). It is conceivable that the anatomic architecture of neuronal projections to adipose tissues is evolved to allow differential sympathetic drive across fat depots in response to different lipolytic/thermogenic stimuli (37). Generally speaking, sympathetic drive to BAT is more intense than that to WAT depots, demonstrated by more sympathetic nerve endings on adipocytes, higher NE levels and NE turnover rates, and increased expression levels of tyrosine hydroxylase (37–39). Moreover, there are differential sympathetic activities between various WAT depots at the basal, cold-induced, and fasting-induced conditions (37, 39). It will be interesting to determine what factors control such differential effects of the SNS on adipose tissues.
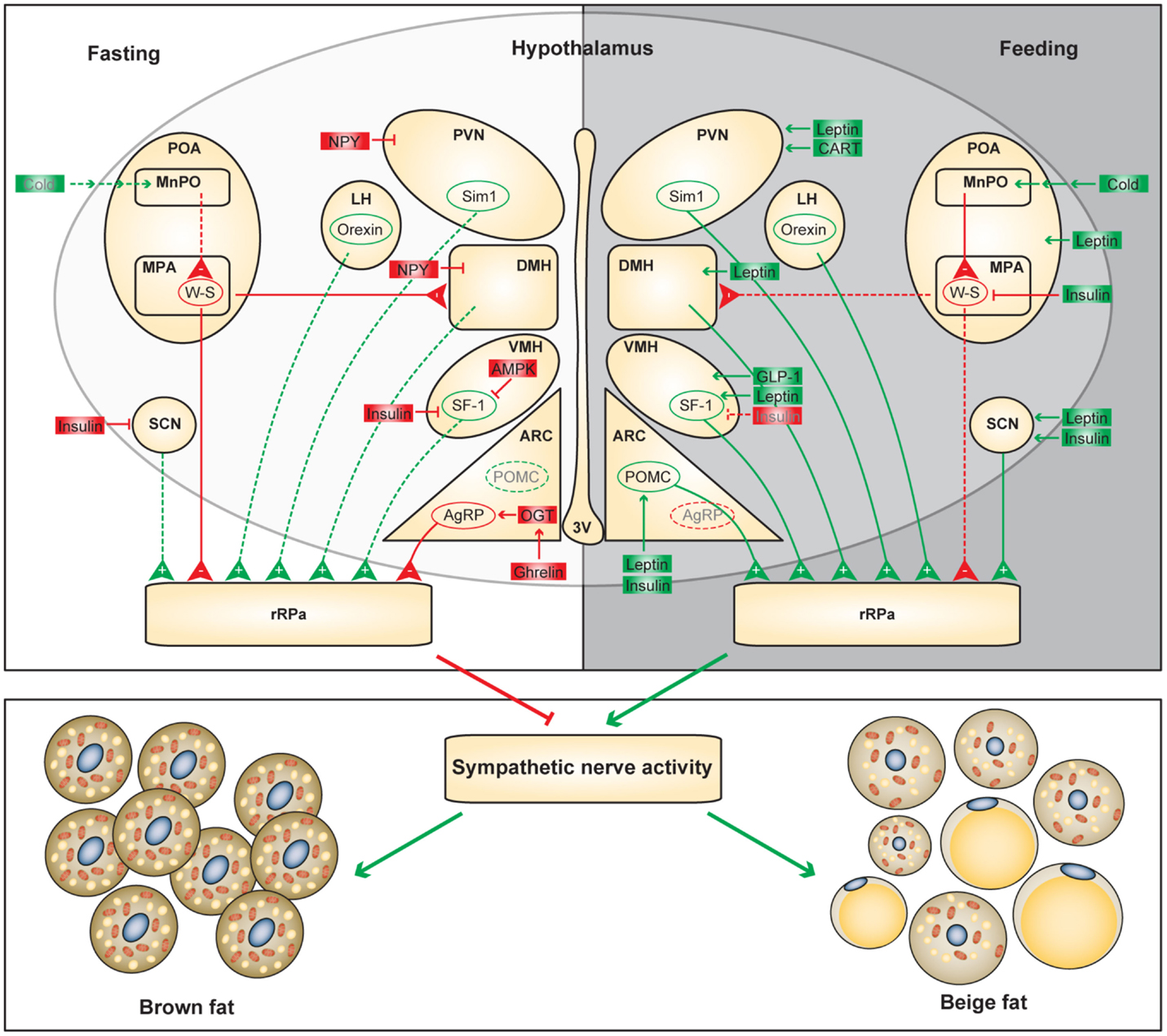
Figure 1. Functional neuroanatomical model of the hypothalamic control of adaptive thermogenesis. Differential regulation of the hypothalamic circuits during fasting and feeding is shown. Positive and negative regulators of the adaptive thermogenesis are shown in green and red, respectively. Dashed lines and gray letters indicate inactive or diminished signals.
Along with well-characterized leptin resistance and insulin resistance, catecholamine resistance develops in obesity, which is characterized by impaired catecholamine synthesis and/or sensitivity (40–42). Catecholamine-induced lipolysis and thermogenesis are compromised in WAT of obese animals and patients through several proposed mechanisms, including leptin resistance, α2-AR expression, and inflammation (43, 44). Yet the contribution of catecholamine resistance to the defects in thermoregulation awaits future investigation.
The Preoptic Area Integrates Thermal Information
Cold exposure and subsequent SNS activation have a profound effect on brown and beige adipocytes. Upon cold exposure, cutaneous transient receptor potential (TRP) cation channels, e.g. TRPM8, sense skin temperature and transmit signals to primary sensory neurons in the dorsal root ganglia (28). This thermal information is delivered to third-order sensory neurons in the lateral parabrachial nucleus (LPB) and then finally to the median preoptic (MnPO) subnucleus of the hypothalamic preoptic area (POA). The medial preoptic area (MPA) of the POA contains warm-sensitive neurons, which receive inhibitory projections from the MnPO (26, 30). Cooling of both distal skin and local hypothalamus reduces the discharge of these warm-sensitive neurons, suggesting that the POA is a central temperature sensor.
Neurons in the POA control SNS activity and thermogenesis by projecting to the dorsomedial hypothalamic area (DMH) and rostral raphe pallidus (rRPa) in the rostral ventromedial medulla, and finally to the sympathetic preganglionic neurons in the intermediolateral nucleus (IML). The neuroanatomical blueprint and neurotransmitters involved in POA regulation of thermogenesis have been reviewed in detail elsewhere (26, 28, 30). Direct cooling of the POA elicits BAT activation (45). Glutamatergic activation of the MnPO and lesions in the inhibitory MPA region both evoke thermogenesis in BAT (46, 47). Conversely, inhibition of MnPO blocks BAT thermogenesis induced by skin cooling (47).
These data demonstrate that the POA integrates thermal information to regulate cold-induced thermogenesis. Febrile responses are also mediated by the POA, which will not be discussed in this review (26, 30). The POA provides efferent signals to WAT depots as well (35); however, roles of the POA in the regulation of WAT browning have not been characterized.
Hypothalamic Hormone Sensing in Thermoregulation
In the late 1970s, seminal experiments performed by Rothwell and Stock demonstrated that cafeteria diet increased the activity of SNS and BAT (48). Diet-induced thermogenesis has since been considered as an important compensatory mechanism that offsets energy surplus. A series of diets such as cafeteria diet and high-fat diet (HFD) induce metabolic inefficiency and BAT recruitment, thus being called as “recruiting diets” (1). Several factors have been proposed to be important for the thermogenic effect of recruiting diets, including the protein-dilution effect, adipocyte-derived leptin, and hypothalamic neurons (1). Parasagittal hypothalamic knife-cuts and medial hypothalamic lesions in rats impaired cafeteria diet-induced thermogenesis in BAT (49, 50). The PVN was later demonstrated as one of the important hypothalamic nuclei mediating diet-induced thermogenesis (51).
In addition, caloric restriction and daily feeding–fasting cycles also control body temperature and thermogenesis in BAT and beige fat (52, 53, 39). Importantly, cold-induced thermogenesis induces and requires food consumption (39, 54). These data suggest that feeding or sufficient nutrient supply provides permissive signals to adaptive thermogenesis. Leptin secreted by adipocytes in proportion to the fat mass and insulin secreted by pancreatic β cells in response to blood glucose have been proposed as these permissive signals.
Preoptic Area
The POA, as a temperature central sensor, is also under the hormonal regulation. Leptin-responsive neurons are abundant in the POA (55). Cold induces the activity of the leptin receptor (LepR)-expressing neurons in the POA, which project to rRPa to regulate sympathetic BAT inputs (56). The insulin receptor is also detectable in the POA. Insulin injection into the POA decreases the firing rate of warm-sensitive neurons, which results in BAT thermogenesis and elevated core body temperature (57). To understand the molecular mechanisms underlying the integration of various thermogenic cues in the POA will be a promising avenue for future research.
Arcuate Nucleus
The ARC is considered as the primary mediator of leptin signaling in the regulation of energy balance and glucose metabolism (33). Injection of leptin into the ARC increases sympathetic drive to BAT (58), and GABAergic RIP-Cre neurons in the ARC may mediate the ability of leptin to stimulate thermogenesis (59). Orexigenic neuropeptides AgRP and NPY inhibit BAT function, while anorexigenic α-MSH increases SNS activity and BAT function (60, 61).
However, it was not known whether neuronal circuits in the ARC also control WAT browning until recently. Acute activation of hunger-promoting AgRP neurons in the hypothalamus suppresses the browning process. O-linked β-N-acetylglucosamine (O-GlcNAc) modification of cytoplasmic and nuclear proteins is a nutrient-sensitive pathway (62, 63). The levels of O-GlcNAc modification and O-GlcNAc transferase (OGT) are enriched in AgRP neurons and are elevated by fasting and the hunger hormone Ghrelin (39). Genetic ablation of OGT in AgRP neurons inhibits neuronal excitability, promotes WAT browning, and protects mice against diet-induced obesity and insulin resistance (39). On the other hand, insulin and leptin act synergistically on POMC neurons to promote WAT browning and prevent diet-induced obesity (64). Phosphatases PTP1B and TCPTP attenuate leptin and insulin signaling in POMC neurons, and double knockout of PTP1B and TCPTP in POMC neurons promotes WAT browning (64). These two complimentary stories demonstrate that the hunger and satiety neurons in the ARC control browning of fat depending on the body’s energy state. Activation of POMC neurons during caloric intake protects against diet-induced obesity whereas activation of AgRP neurons informs the body to store energy during fasting.
Dorsomedial Hypothalamic Area
Neurons in the DMH receive GABAergic inputs from warm-sensitive neurons in the MPA (28). Derepression of DMH neurons by infusing an antagonist to the GABAA receptor rapidly and profoundly increases BAT and core body temperature (65, 66), which is dependent on the activation of downstream rRPa neurons (67, 68).
A population of neurons in the DMH expresses the LepR. The intra-DMH injection of leptin increases BAT temperature, in a β3-AR-dependent manner (56, 69). Moreover, blockage of leptin signaling in the DMH blunts the increase in BAT temperature elicited by intraperitoneal injection of leptin (69). Recently, Rezai-Zadeh and colleagues demonstrated that selective activation of DMH neurons by DREADD technique promotes BAT thermogenesis and decreases body weight (70). Conversely, the deficiency of the LepR in the DMH reduces thermogenesis and promotes weight gain (70).
Neuropeptide Y is also expressed by neurons within the DMH, besides the ARC of the hypothalamus. Npy expression in the DMH is induced in the conditions where animals demand more energy, for example, chronic food restriction and exercise (71, 72). Similar to that in the ARC, NPY in the DMH promotes food intake and body weight (73). Using viral-mediated knockdown approach in rat, the same group recently showed that DMH knockdown of Npy increases BAT activation and the browning of WAT through the SNS (74). Collectively, these data suggest that the DMH is another hypothalamic locus where orexigenic and anorexigenic signals converge to regulate thermogenesis in fat tissue.
Ventromedial Hypothalamic Area
The ventromedial hypothalamic area (VMH) is one of the first hypothalamic sites that have been identified to regulate thermogenesis. Although the anatomical linkage between the VMH and adipose tissue is controversial (35), electrical and glutamate stimulation of the VMH has been extensively shown to activate BAT thermogenesis via the SNS (30, 75, 76). Moreover, genetic manipulation of steroidogenic factor-1 (SF-1) neurons, the major population in the VMH, has also demonstrated the importance of the VMH in thermoregulation (77, 78).
Leptin microinjection into the VMH increases glucose uptake in BAT, although thermogenic effect was not tested in the study (79). Selective deletion of the LepR in SF-1 neurons reduces Ucp1 expression in BAT, suppresses thermogenesis, and produces obesity (80, 81). It has been suggested that the action of leptin on BAT thermogenesis is preferentially mediated by the PI3K/AKT/FOXOI pathway (82, 83). By contrast, insulin suppresses the firing frequency of SF-1 neurons in the VMH (84). Microinjection of insulin to the VMH suppresses BAT thermogenesis in response to cold and glutamate stimulation (85, 86). More interestingly, the magnitude of the suppression shows diurnal rhythm, which is greater at noon than at night in rats (87).
Fatty acid metabolism is essential for neuronal function (88). AMPK-regulated lipogenesis in the VMH is an important regulator of BAT thermogenesis and controller of body weight (89). Recently, Lopez and colleagues further showed that various hormones, including thyroid hormones, estradiol, and glucagon-like peptide-1 (GLP-1), activate BAT thermogenesis through relieving the suppression on lipogenic genes by AMPK in the VMH (89–91). These studies strongly argue that the VMH senses hormonal cues to regulate BAT function; however, the involvement of the VMH in the regulation of WAT browning has not been explored.
Paraventricular Nucleus
The PVN is strongly susceptible to trans-synaptic infection by RPV from BAT (35); however, its effect on thermogenesis has been controversial. Electrical stimulation of the PVN does not affect BAT function (92). Microinjection of N-methyl-d-aspartate (NMDA) or bicuculline to activate neurons in the PVN blocks sympathetic drive to BAT induced by cold, indicating that the PVN negatively regulates thermogenesis (93). Paradoxically, stimulation of the PVN with glutamate is shown to activate BAT thermogenesis (94).
There is a large population of MC4R-expressing neurons in the PVN, and the MC4R agonist melanotan II (MTII) induces BAT thermogenesis (95). Single-minded 1 (Sim1) is necessary for development of the PVN, and ablation of Sim1 neurons reduces Ucp1 expression, BAT temperature, and energy expenditure (96). Restoration of MC4R in the Sim1 neurons fails to rescue reduced energy expenditure in Mc4r knockout mice, suggesting that MC4R elsewhere controls energy expenditure (97). However, a recent study of Mc4r deletion in Sim1 neurons showed that Sim1 neurons are important in the regulation of energy expenditure (98).
Leptin regulates the synaptic activity of neurons in the PVN that project to BAT (99). Viral administration of leptin in the PVN stimulates the expression of Ucp1 in BAT (100). The orexigenic peptide NPY reduces energy expenditure; however, its site of action was not identified until recently (101). Shi et al. demonstrated that ARC-derived NPY decreases tyrosine hydroxinase (TH) expression in the PVN to down-regulate BAT Ucp1 expression and energy expenditure (61). On the other hand, the anorexigenic peptide CART, when injected in the PVN, induces the Ucp1 expression in both BAT and WAT (102).
Suprachiasmatic Nucleus
Core body temperature, like all vital aspects of physiology and metabolism, shows circadian rhythm (103). Thermogenic plasticity in BAT is also rhythmic and under the control of cell-autonomous clock machinery (104–107). Directly entrained by light, the suprachiasmatic nucleus (SCN) in the hypothalamus has been established as a circadian pacemaker that synchronizes peripheral clocks across the body to adjust behavior and physiology in accordance with the day/night cycle (108, 109). Both BAT and WAT receive sympathetic flow that can be tracked back to the SCN (35, 110). Glutamate injection to activate the neurons in the SCN stimulates BAT thermogenesis, and this effect is greater during the dark phase (111). Nevertheless, it is still unknown whether the SCN controls the circadian rhythm of thermogenesis and if WAT browning is also under circadian regulation.
LepR-expressing neurons exist in the SCN, and leptin phase-advances the circadian rhythm of SCN on brain slices (112). In vivo analysis revealed that leptin modulates clock gene expression in the SCN and the sleep/wake cycle (113). It will be of interest to determine whether leptin in the SCN regulates the circadian rhythm of thermogenesis. In addition, intra-SCN injection of insulin to rats decreases the sympathetic activity on BAT at noon (fasted state) and increases the activity at night (fed state), indicating a circadian control of BAT function by insulin action on the SCN (87). These data suggest that the SCN may receive peripheral satiety cues to modulate the circadian rhythm of energy expenditure to maintain homeostasis.
Psychological Regulation of Thermogenesis
Psychological fever, one of the most common psychological diseases, is characterized by acute or persistent increase in body temperature when patients are psychologically stressed (101). It was suggested that SNS-activated BAT thermogenesis is responsible for stress-induced hyperthermia (114, 115). Orexin neurons in the hypothalamus control multiple physiological processes, including arousal, wakefulness, and appetite. The orexin neurons are considered important for inflammatory fever and the defense against cold (116). It has also been shown that the orexin neurons are indispensable for the stress-induced Ucp1 expression and thermogenesis in BAT and resultant hyperthermia (117). Recently, a functional neuroanatomical study using an optogenetic approach has identified a DMH-medullary raphe circuit that drives psychological stress-activated BAT thermogenesis and hyperthermia (118).
Environmental enrichment is the stimulation of the brain by physical and social surroundings. This regimen supports neurogenesis and could aid the treatment of neurodegenerative diseases (119). Intriguingly, environmental enrichment in rodents induces WAT browning and decreases adiposity (120). Hypothalamic brain-derived neurotrophic factor (BDNF) has been shown to increase thermogenesis and energy expenditure by acting on neurons in the PVN and VMH (121, 122). Environmental enrichment up-regulates hypothalamic BDNF, and the inhibition of BDNF blocks the environmental enrichment-induced WAT browning (120).
Summary and Perspective
Yet to be fully defined, the developmental and functional identities of brown and beige adipocytes are distinct (16). Separate populations of neurons in common areas of the hypothalamus projecting to BAT and different WAT depots may contribute to the functional and regulatory differences between fat depots (35). The blueprint of the neuroanatomical regulation in BAT thermogenesis is becoming apparent; however, we just begin to explore the central regulation of the browning process in WAT (39, 64, 123).
It is not surprising that the hypothalamic areas that control food intake also modulate adaptive thermogenesis. In the hunger state, orexigenic neurons attenuate SNS-mediated heat production in brown and beige fat to preserve energy; conversely, in the satiety state, anorexigenic neurons promote energy expenditure to maintain homeostasis (Figure 1). However, adaptive thermogenesis is not efficacious to offset long-term energy imbalance, which partially explains the susceptibility to diet-induced obesity and the difficulty of weight loss by dietary interventions in humans. It is important to unravel the biological logic of distinct hypothalamic efferent outputs. In addition to mapping the neuronal circuits controlling thermogenesis in BAT and WAT, there is an urgent need to identify intracellular mechanisms by which neuronal activities are dictated by thermogenic cues. Understanding the molecular and cellular basis of neuronal regulation of adaptive thermogenesis will lay a foundation for future therapeutics against obesity. For example, GLP-1 receptor agonists, which have been widely used to treat diabetes, can be administrated centrally to stimulate brown adipose thermogenesis (90). ARC-specific administration of OGT inhibitors and PTP1B inhibitors suppress AgRP neurons and stimulate POMC neurons, respectively, to promote energy expenditure in brown and beige adipocytes (39, 64).
Hypothalamic circuits respond to both thermal information and nutritional cues to modulate cold- and diet-induced thermogenesis. It has been demonstrated that feeding provides permissive signals for the activation of brown and beige adipocytes upon cold challenge (39, 54). It is conceivable that yet-to-be-found neuronal circuits exist in the hypothalamus serve as an integrator of various thermogenic stimuli. Characterization of such integration sites will help us understand how mammals, including human beings, tradeoff between life-history variables such as hunger and cold during the evolution.
Adaptive thermogenesis is thought to evolve slowly (in days) to adjust to changes in temperature and food availability/composition; however, the activation or silencing of CNS and SNS respond immediately (in minutes) to stimuli, followed by delayed (in hours) changes in Ucp1 expression. Appropriate tools to manipulate neurons should be utilized to better characterize neuronal networks in thermoregulation. Studies using the chemical/electrical stimulation and anatomical lesions are difficult to control efficacy and often lead to off-target effects. Genetically modified animal models can continue to be used to reveal neuron population-specific function. However, the caution should be taken because genetically modified animals may exhibit developmental defects due to non-specific Cre expression and observed phenotypes are often the results of the long-term and/or secondary effects. Recent advances in optogenetics and chemogenetics produce powerful tools to acutely and precisely control neuronal activity (124). These tools in combination with classical transgenic approaches will accelerate the progress in defining the functional neuronal architecture in thermoregulation.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by National Institutes of Health R01DK089098, National Institutes of Health P01 DK057751, State of Connecticut Department of Public Health 2014-0139, and Ellison Medical Foundation to XY, and American Heart Association 14SDG20120052 to H-BR.
References
1. Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev (2004) 84:277–359. doi:10.1152/physrev.00015.2003
3. Tchkonia T, Thomou T, Zhu Y, Karagiannides I, Pothoulakis C, Jensen MD, et al. Mechanisms and metabolic implications of regional differences among fat depots. Cell Metab (2013) 17:644–56. doi:10.1016/j.cmet.2013.03.008
4. Schulz TJ, Tseng YH. Brown adipose tissue: development, metabolism and beyond. Biochem J (2013) 453:167–78. doi:10.1042/BJ20130457
5. Nedergaard J, Golozoubova V, Matthias A, Asadi A, Jacobsson A, Cannon B. UCP1: the only protein able to mediate adaptive non-shivering thermogenesis and metabolic inefficiency. Biochim Biophys Acta (2001) 1504:82–106. doi:10.1016/S0005-2728(00)00247-4
6. Feldmann HM, Golozoubova V, Cannon B, Nedergaard J. UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab (2009) 9:203–9. doi:10.1016/j.cmet.2008.12.014
7. Granneman JG, Burnazi M, Zhu Z, Schwamb LA. White adipose tissue contributes to UCP1-independent thermogenesis. Am J Physiol Endocrinol Metab (2003) 285:E1230–6. doi:10.1152/ajpendo.00197.2003
8. Ukropec J, Anunciado RP, Ravussin Y, Hulver MW, Kozak LP. UCP1-independent thermogenesis in white adipose tissue of cold-acclimated Ucp1-/- mice. J Biol Chem (2006) 281:31894–908. doi:10.1074/jbc.M606114200
9. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med (2009) 360:1509–17. doi:10.1056/NEJMoa0810780
10. Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: effects of cold exposure and adiposity. Diabetes (2009) 58:1526–31. doi:10.2337/db09-0530
11. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med (2009) 360:1500–8. doi:10.1056/NEJMoa0808718
12. Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, et al. Functional brown adipose tissue in healthy adults. N Engl J Med (2009) 360:1518–25. doi:10.1056/NEJMoa0808949
13. Zingaretti MC, Crosta F, Vitali A, Guerrieri M, Frontini A, Cannon B, et al. The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB J (2009) 23:3113–20. doi:10.1096/fj.09-133546
14. Tam CS, Lecoultre V, Ravussin E. Brown adipose tissue: mechanisms and potential therapeutic targets. Circulation (2012) 125:2782–91. doi:10.1161/CIRCULATIONAHA.111.042929
15. Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat Med (2013) 19:1252–63. doi:10.1038/nm.3361
16. Wu J, Cohen P, Spiegelman BM. Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev (2013) 27:234–50. doi:10.1101/gad.211649.112
17. Cypess AM, White AP, Vernochet C, Schulz TJ, Xue R, Sass CA, et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat Med (2013) 19:635–9. doi:10.1038/nm.3112
18. Jespersen NZ, Larsen TJ, Peijs L, Daugaard S, Homoe P, Loft A, et al. A classical brown adipose tissue mRNA signature partly overlaps with brite in the supraclavicular region of adult humans. Cell Metab (2013) 17:798–805. doi:10.1016/j.cmet.2013.04.011
19. Sharp LZ, Shinoda K, Ohno H, Scheel DW, Tomoda E, Ruiz L, et al. Human BAT possesses molecular signatures that resemble beige/brite cells. PLoS One (2012) 7:e49452. doi:10.1371/journal.pone.0049452
20. Wu J, Bostrom P, Sparks LM, Ye L, Choi JH, Giang AH, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell (2012) 150:366–76. doi:10.1016/j.cell.2012.05.016
21. Shinoda K, Luijten IH, Hasegawa Y, Hong H, Sonne SB, Kim M, et al. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nat Med (2015) 21:389–94. doi:10.1038/nm.3819
22. van Marken Lichtenbelt W. Human brown fat and obesity: methodological aspects. Front Endocrinol (2011) 2:52. doi:10.3389/fendo.2011.00052
23. Lidell ME, Betz MJ, Dahlqvist Leinhard O, Heglind M, Elander L, Slawik M, et al. Evidence for two types of brown adipose tissue in humans. Nat Med (2013) 19:631–4. doi:10.1038/nm.3017
24. Cypess AM, Weiner LS, Roberts-Toler C, Franquet Elia E, Kessler SH, Kahn PA, et al. Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab (2015) 21:33–8. doi:10.1016/j.cmet.2014.12.009
25. Sidossis L, Kajimura S. Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis. J Clin Invest (2015) 125:478–86. doi:10.1172/JCI78362
26. Morrison SF, Nakamura K. Central neural pathways for thermoregulation. Front Biosci (Landmark Ed) (2011) 16:74–104. doi:10.2741/3677
27. Clapham JC. Central control of thermogenesis. Neuropharmacology (2012) 63:111–23. doi:10.1016/j.neuropharm.2011.10.014
28. Morrison SF, Madden CJ, Tupone D. Central neural regulation of brown adipose tissue thermogenesis and energy expenditure. Cell Metab (2014) 19:741–56. doi:10.1016/j.cmet.2014.02.007
29. Celi FS, Le TN, Ni B. Physiology and relevance of human adaptive thermogenesis response. Trends Endocrinol Metab (2015) 26:238–47. doi:10.1016/j.tem.2015.03.003
30. Contreras C, Gonzalez F, Ferno J, Dieguez C, Rahmouni K, Nogueiras R, et al. The brain and brown fat. Ann Med (2015) 47:150–68. doi:10.3109/07853890.2014.919727
31. Richard D. Cognitive and autonomic determinants of energy homeostasis in obesity. Nat Rev Endocrinol (2015) 11:489–501. doi:10.1038/nrendo.2015.103
32. Lenard NR, Berthoud HR. Central and peripheral regulation of food intake and physical activity: pathways and genes. Obesity (Silver Spring) (2008) 16(Suppl 3):S11–22. doi:10.1038/oby.2008.511
33. Belgardt BF, Okamura T, Bruning JC. Hormone and glucose signalling in POMC and AgRP neurons. J Physiol (2009) 587:5305–14. doi:10.1113/jphysiol.2009.179192
34. Dietrich MO, Horvath TL. Hypothalamic control of energy balance: insights into the role of synaptic plasticity. Trends Neurosci (2013) 36:65–73. doi:10.1016/j.tins.2012.12.005
35. Stefanidis A, Wiedmann NM, Adler ES, Oldfield BJ. Hypothalamic control of adipose tissue. Best Pract Res Clin Endocrinol Metab (2014) 28:685–701. doi:10.1016/j.beem.2014.08.001
36. Bartness TJ, Liu Y, Shrestha YB, Ryu V. Neural innervation of white adipose tissue and the control of lipolysis. Front Neuroendocrinol (2014) 35:473–93. doi:10.1016/j.yfrne.2014.04.001
37. Brito NA, Brito MN, Bartness TJ. Differential sympathetic drive to adipose tissues after food deprivation, cold exposure or glucoprivation. Am J Physiol Regul Integr Comp Physiol (2008) 294:R1445–52. doi:10.1152/ajpregu.00068.2008
38. Trayhurn P, Ashwell M. Control of white and brown adipose tissues by the autonomic nervous system. Proc Nutr Soc (1987) 46:135–42. doi:10.1079/PNS19870017
39. Ruan HB, Dietrich MO, Liu ZW, Zimmer MR, Li MD, Singh JP, et al. O-GlcNAc transferase enables AgRP neurons to suppress browning of white fat. Cell (2014) 159:306–17. doi:10.1016/j.cell.2014.09.010
40. Connacher AA, Bennet WM, Jung RT, Bier DM, Smith CC, Scrimgeour CM, et al. Effect of adrenaline infusion on fatty acid and glucose turnover in lean and obese human subjects in the post-absorptive and fed states. Clin Sci (Lond) (1991) 81:635–44. doi:10.1042/cs0810635
41. Horowitz JF, Klein S. Whole body and abdominal lipolytic sensitivity to epinephrine is suppressed in upper body obese women. Am J Physiol Endocrinol Metab (2000) 278:E1144–52.
42. Jocken JW, Blaak EE. Catecholamine-induced lipolysis in adipose tissue and skeletal muscle in obesity. Physiol Behav (2008) 94:219–30. doi:10.1016/j.physbeh.2008.01.002
43. Yehuda-Shnaidman E, Buehrer B, Pi J, Kumar N, Collins S. Acute stimulation of white adipocyte respiration by PKA-induced lipolysis. Diabetes (2010) 59:2474–83. doi:10.2337/db10-0245
44. Mowers J, Uhm M, Reilly SM, Simon J, Leto D, Chiang SH, et al. Inflammation produces catecholamine resistance in obesity via activation of PDE3B by the protein kinases IKKepsilon and TBK1. Elife (2013) 2:e01119. doi:10.7554/eLife.01119
45. Imai-Matsumura K, Matsumura K, Nakayama T. Involvement of ventromedial hypothalamus in brown adipose tissue thermogenesis induced by preoptic cooling in rats. Jpn J Physiol (1984) 34:939–43. doi:10.2170/jjphysiol.34.939
46. Szymusiak R, Satinoff E. Acute thermoregulatory effects of unilateral electrolytic lesions of the medial and lateral preoptic area in rats. Physiol Behav (1982) 28:161–70. doi:10.1016/0031-9384(82)90118-4
47. Nakamura K, Morrison SF. Preoptic mechanism for cold-defensive responses to skin cooling. J Physiol (2008) 586:2611–20. doi:10.1113/jphysiol.2008.152686
48. Rothwell NJ, Stock MJ. A role for brown adipose tissue in diet-induced thermogenesis. Nature (1979) 281:31–5. doi:10.1038/281031a0
49. Coscina DV, Chambers JW, Park I, Hogan S, Himms-Hagen J. Impaired diet-induced thermogenesis in brown adipose tissue from rats made obese with parasagittal hypothalamic knife-cuts. Brain Res Bull (1985) 14:585–93. doi:10.1016/0361-9230(85)90108-X
50. Hogan S, Himms-Hagen J, Coscina DV. Lack of diet-induced thermogenesis in brown adipose tissue of obese medial hypothalamic-lesioned rats. Physiol Behav (1985) 35:287–94. doi:10.1016/0031-9384(85)90351-8
51. De Luca B, Monda M, Amaro S, Pellicano MP, Cioffi LA. Lack of diet-induced thermogenesis following lesions of paraventricular nucleus in rats. Physiol Behav (1989) 46:685–91. doi:10.1016/0031-9384(89)90352-1
52. Apfelbaum M, Bostsarron J, Lacatis D. Effect of caloric restriction and excessive caloric intake on energy expenditure. Am J Clin Nutr (1971) 24:1405–9.
53. Shibata H, Bukowiecki LJ. Regulatory alterations of daily energy expenditure induced by fasting or overfeeding in unrestrained rats. J Appl Physiol (1985) (1987) 63:465–70.
54. Cannon B, Nedergaard J. Nonshivering thermogenesis and its adequate measurement in metabolic studies. J Exp Biol (2011) 214:242–53. doi:10.1242/jeb.050989
55. Frontini A, Giordano A. Leptin-sensitive neurons in mouse preoptic area express alpha 1A- and alpha 2A-adrenergic receptor isoforms. Neurosci Lett (2010) 471:83–8. doi:10.1016/j.neulet.2010.01.016
56. Zhang Y, Kerman IA, Laque A, Nguyen P, Faouzi M, Louis GW, et al. Leptin-receptor-expressing neurons in the dorsomedial hypothalamus and median preoptic area regulate sympathetic brown adipose tissue circuits. J Neurosci (2011) 31:1873–84. doi:10.1523/JNEUROSCI.3223-10.2011
57. Sanchez-Alavez M, Tabarean IV, Osborn O, Mitsukawa K, Schaefer J, Dubins J, et al. Insulin causes hyperthermia by direct inhibition of warm-sensitive neurons. Diabetes (2010) 59:43–50. doi:10.2337/db09-1128
58. Harlan SM, Morgan DA, Agassandian K, Guo DF, Cassell MD, Sigmund CD, et al. Ablation of the leptin receptor in the hypothalamic arcuate nucleus abrogates leptin-induced sympathetic activation. Circ Res (2011) 108:808–12. doi:10.1161/CIRCRESAHA.111.240226
59. Kong D, Tong Q, Ye C, Koda S, Fuller PM, Krashes MJ, et al. GABAergic RIP-Cre neurons in the arcuate nucleus selectively regulate energy expenditure. Cell (2012) 151:645–57. doi:10.1016/j.cell.2012.09.020
60. Yasuda T, Masaki T, Kakuma T, Yoshimatsu H. Hypothalamic melanocortin system regulates sympathetic nerve activity in brown adipose tissue. Exp Biol Med (Maywood) (2004) 229:235–9.
61. Shi YC, Lau J, Lin Z, Zhang H, Zhai L, Sperk G, et al. Arcuate NPY controls sympathetic output and BAT function via a relay of tyrosine hydroxylase neurons in the PVN. Cell Metab (2013) 17:236–48. doi:10.1016/j.cmet.2013.01.006
62. Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature (2007) 446:1017–22. doi:10.1038/nature05815
63. Ruan HB, Singh JP, Li MD, Wu J, Yang X. Cracking the O-GlcNAc code in metabolism. Trends Endocrinol Metab (2013) 24:301–9. doi:10.1016/j.tem.2013.02.002
64. Dodd GT, Decherf S, Loh K, Simonds SE, Wiede F, Balland E, et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell (2015) 160:88–104. doi:10.1016/j.cell.2014.12.022
65. Zaretskaia MV, Zaretsky DV, Shekhar A, Dimicco JA. Chemical stimulation of the dorsomedial hypothalamus evokes non-shivering thermogenesis in anesthetized rats. Brain Res (2002) 928:113–25. doi:10.1016/S0006-8993(01)03369-8
66. Dimicco JA, Zaretsky DV. The dorsomedial hypothalamus: a new player in thermoregulation. Am J Physiol Regul Integr Comp Physiol (2007) 292:R47–63. doi:10.1152/ajpregu.00498.2006
67. Cao WH, Fan W, Morrison SF. Medullary pathways mediating specific sympathetic responses to activation of dorsomedial hypothalamus. Neuroscience (2004) 126:229–40. doi:10.1016/j.neuroscience.2004.03.013
68. Cao WH, Morrison SF. Glutamate receptors in the raphe pallidus mediate brown adipose tissue thermogenesis evoked by activation of dorsomedial hypothalamic neurons. Neuropharmacology (2006) 51:426–37. doi:10.1016/j.neuropharm.2006.03.031
69. Enriori PJ, Sinnayah P, Simonds SE, Garcia Rudaz C, Cowley MA. Leptin action in the dorsomedial hypothalamus increases sympathetic tone to brown adipose tissue in spite of systemic leptin resistance. J Neurosci (2011) 31:12189–97. doi:10.1523/JNEUROSCI.2336-11.2011
70. Rezai-Zadeh K, Yu S, Jiang Y, Laque A, Schwartzenburg C, Morrison CD, et al. Leptin receptor neurons in the dorsomedial hypothalamus are key regulators of energy expenditure and body weight, but not food intake. Mol Metab (2014) 3:681–93. doi:10.1016/j.molmet.2014.07.008
71. Bi S, Robinson BM, Moran TH. Acute food deprivation and chronic food restriction differentially affect hypothalamic NPY mRNA expression. Am J Physiol Regul Integr Comp Physiol (2003) 285:R1030–6. doi:10.1152/ajpregu.00734.2002
72. Kawaguchi M, Scott KA, Moran TH, Bi S. Dorsomedial hypothalamic corticotropin-releasing factor mediation of exercise-induced anorexia. Am J Physiol Regul Integr Comp Physiol (2005) 288:R1800–5. doi:10.1152/ajpregu.00805.2004
73. Yang L, Scott KA, Hyun J, Tamashiro KL, Tray N, Moran TH, et al. Role of dorsomedial hypothalamic neuropeptide Y in modulating food intake and energy balance. J Neurosci (2009) 29:179–90. doi:10.1523/JNEUROSCI.4379-08.2009
74. Chao PT, Yang L, Aja S, Moran TH, Bi S. Knockdown of NPY expression in the dorsomedial hypothalamus promotes development of brown adipocytes and prevents diet-induced obesity. Cell Metab (2011) 13:573–83. doi:10.1016/j.cmet.2011.02.019
75. Perkins MN, Rothwell NJ, Stock MJ, Stone TW. Activation of brown adipose tissue thermogenesis by the ventromedial hypothalamus. Nature (1981) 289:401–2. doi:10.1038/289401a0
76. Minokoshi Y, Saito M, Shimazu T. Sympathetic denervation impairs responses of brown adipose tissue to VMH stimulation. Am J Physiol (1986) 251:R1005–8.
77. Musatov S, Chen W, Pfaff DW, Mobbs CV, Yang XJ, Clegg DJ, et al. Silencing of estrogen receptor alpha in the ventromedial nucleus of hypothalamus leads to metabolic syndrome. Proc Natl Acad Sci U S A (2007) 104:2501–6. doi:10.1073/pnas.0610787104
78. Kim KW, Zhao L, Donato J Jr, Kohno D, Xu Y, Elias CF, et al. Steroidogenic factor 1 directs programs regulating diet-induced thermogenesis and leptin action in the ventral medial hypothalamic nucleus. Proc Natl Acad Sci U S A (2011) 108:10673–8. doi:10.1073/pnas.1102364108
79. Minokoshi Y, Haque MS, Shimazu T. Microinjection of leptin into the ventromedial hypothalamus increases glucose uptake in peripheral tissues in rats. Diabetes (1999) 48:287–91. doi:10.2337/diabetes.48.2.287
80. Dhillon H, Zigman JM, Ye C, Lee CE, Mcgovern RA, Tang V, et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron (2006) 49:191–203. doi:10.1016/j.neuron.2005.12.021
81. Bingham NC, Anderson KK, Reuter AL, Stallings NR, Parker KL. Selective loss of leptin receptors in the ventromedial hypothalamic nucleus results in increased adiposity and a metabolic syndrome. Endocrinology (2008) 149:2138–48. doi:10.1210/en.2007-1200
82. Xu Y, Hill JW, Fukuda M, Gautron L, Sohn JW, Kim KW, et al. PI3K signaling in the ventromedial hypothalamic nucleus is required for normal energy homeostasis. Cell Metab (2010) 12:88–95. doi:10.1016/j.cmet.2010.05.002
83. Kim KW, Donato J Jr, Berglund ED, Choi YH, Kohno D, Elias CF, et al. FOXO1 in the ventromedial hypothalamus regulates energy balance. J Clin Invest (2012) 122:2578–89. doi:10.1172/JCI62848
84. Klockener T, Hess S, Belgardt BF, Paeger L, Verhagen LA, Husch A, et al. High-fat feeding promotes obesity via insulin receptor/PI3K-dependent inhibition of SF-1 VMH neurons. Nat Neurosci (2011) 14:911–8. doi:10.1038/nn.2847
85. Amir S, Lagiorgia M, Pollock R. Intra-ventromedial hypothalamic injection of insulin suppresses brown fat thermogenesis in the anaesthetized rat. Brain Res (1989) 480:340–3. doi:10.1016/0006-8993(89)90204-7
86. Amir S, Schiavetto A, Pollock R. Insulin co-injection suppresses the thermogenic response to glutamate microinjection into the VMH in rats. Brain Res (1990) 527:326–9. doi:10.1016/0006-8993(90)91153-8
87. Sakaguchi T, Takahashi M, Bray GA. Diurnal changes in sympathetic activity. Relation to food intake and to insulin injected into the ventromedial or suprachiasmatic nucleus. J Clin Invest (1988) 82:282–6. doi:10.1172/JCI113584
88. Panov A, Orynbayeva Z, Vavilin V, Lyakhovich V. Fatty acids in energy metabolism of the central nervous system. Biomed Res Int (2014) 2014:472459. doi:10.1155/2014/472459
89. Lopez M, Varela L, Vazquez MJ, Rodriguez-Cuenca S, Gonzalez CR, Velagapudi VR, et al. Hypothalamic AMPK and fatty acid metabolism mediate thyroid regulation of energy balance. Nat Med (2010) 16:1001–8. doi:10.1038/nm.2207
90. Beiroa D, Imbernon M, Gallego R, Senra A, Herranz D, Villarroya F, et al. GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes (2014) 63:3346–58. doi:10.2337/db14-0302
91. Martinez de Morentin PB, Gonzalez-Garcia I, Martins L, Lage R, Fernandez-Mallo D, Martinez-Sanchez N, et al. Estradiol regulates brown adipose tissue thermogenesis via hypothalamic AMPK. Cell Metab (2014) 20:41–53. doi:10.1016/j.cmet.2014.03.031
92. Holt SJ, Wheal HV, York DA. Response of brown adipose tissue to electrical stimulation of hypothalamic centres in intact and adrenalectomized Zucker rats. Neurosci Lett (1988) 84:63–7. doi:10.1016/0304-3940(88)90338-2
93. Madden CJ, Morrison SF. Neurons in the paraventricular nucleus of the hypothalamus inhibit sympathetic outflow to brown adipose tissue. Am J Physiol Regul Integr Comp Physiol (2009) 296:R831–43. doi:10.1152/ajpregu.91007.2008
94. Amir S. Stimulation of the paraventricular nucleus with glutamate activates interscapular brown adipose tissue thermogenesis in rats. Brain Res (1990) 508:152–5. doi:10.1016/0006-8993(90)91129-5
95. Song CK, Vaughan CH, Keen-Rhinehart E, Harris RB, Richard D, Bartness TJ. Melanocortin-4 receptor mRNA expressed in sympathetic outflow neurons to brown adipose tissue: neuroanatomical and functional evidence. Am J Physiol Regul Integr Comp Physiol (2008) 295:R417–28. doi:10.1152/ajpregu.00174.2008
96. Xi D, Gandhi N, Lai M, Kublaoui BM. Ablation of Sim1 neurons causes obesity through hyperphagia and reduced energy expenditure. PLoS One (2012) 7:e36453. doi:10.1371/journal.pone.0036453
97. Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell (2005) 123:493–505. doi:10.1016/j.cell.2005.08.035
98. Xu Y, Wu Z, Sun H, Zhu Y, Kim ER, Lowell BB, et al. Glutamate mediates the function of melanocortin receptor 4 on Sim1 neurons in body weight regulation. Cell Metab (2013) 18:860–70. doi:10.1016/j.cmet.2013.11.003
99. Jiang Y, Munzberg H, Derbenev A, Zsombok A. Leptin regulates synaptic activity of brown adipose tissue-related pre-sympathetic neurons in the paraventricular nucleus of the mice. FASEB J (2015) 29:1057.1.
100. Bagnasco M, Dube MG, Katz A, Kalra PS, Kalra SP. Leptin expression in hypothalamic PVN reverses dietary obesity and hyperinsulinemia but stimulates ghrelin. Obes Res (2003) 11:1463–70. doi:10.1038/oby.2003.196
101. Oka T, Oka K, Hori T. Mechanisms and mediators of psychological stress-induced rise in core temperature. Psychosom Med (2001) 63:476–86. doi:10.1097/00006842-200105000-00018
102. Wang C, Billington CJ, Levine AS, Kotz CM. Effect of CART in the hypothalamic paraventricular nucleus on feeding and uncoupling protein gene expression. Neuroreport (2000) 11:3251–5. doi:10.1097/00001756-200009280-00040
103. Refinetti R, Menaker M. The circadian rhythm of body temperature. Physiol Behav (1992) 51:613–37. doi:10.1016/0031-9384(92)90188-8
104. Redlin U, Nuesslein B, Schmidt I. Circadian changes of brown adipose tissue thermogenesis in juvenile rats. Am J Physiol (1992) 262:R504–8.
105. Chappuis S, Ripperger JA, Schnell A, Rando G, Jud C, Wahli W, et al. Role of the circadian clock gene per2 in adaptation to cold temperature. Mol Metab (2013) 2:184–93. doi:10.1016/j.molmet.2013.05.002
106. Gerhart-Hines Z, Feng D, Emmett MJ, Everett LJ, Loro E, Briggs ER, et al. The nuclear receptor Rev-erbalpha controls circadian thermogenic plasticity. Nature (2013) 503:410–3. doi:10.1038/nature12642
107. Nam D, Guo B, Chatterjee S, Chen MH, Nelson D, Yechoor VK, et al. The adipocyte clock controls brown adipogenesis through the TGF-beta and BMP signaling pathways. J Cell Sci (2015) 128:1835–47. doi:10.1242/jcs.167643
108. Welsh DK, Takahashi JS, Kay SA. Suprachiasmatic nucleus: cell autonomy and network properties. Annu Rev Physiol (2010) 72:551–77. doi:10.1146/annurev-physiol-021909-135919
109. Partch CL, Green CB, Takahashi JS. Molecular architecture of the mammalian circadian clock. Trends Cell Biol (2014) 24:90–9. doi:10.1016/j.tcb.2013.07.002
110. Bartness TJ, Song CK, Demas GE. SCN efferents to peripheral tissues: implications for biological rhythms. J Biol Rhythms (2001) 16:196–204. doi:10.1177/074873040101600302
111. Amir S, Shizgal P, Rompre PP. Glutamate injection into the suprachiasmatic nucleus stimulates brown fat thermogenesis in the rat. Brain Res (1989) 498:140–4. doi:10.1016/0006-8993(89)90409-5
112. Prosser RA, Bergeron HE. Leptin phase-advances the rat suprachiasmatic circadian clock in vitro. Neurosci Lett (2003) 336:139–42. doi:10.1016/S0304-3940(02)01234-X
113. Mendoza J, Lopez-Lopez C, Revel FG, Jeanneau K, Delerue F, Prinssen E, et al. Dimorphic effects of leptin on the circadian and hypocretinergic systems of mice. J Neuroendocrinol (2011) 23:28–38. doi:10.1111/j.1365-2826.2010.02072.x
114. Hayashida S, Oka T, Mera T, Tsuji S. Repeated social defeat stress induces chronic hyperthermia in rats. Physiol Behav (2010) 101:124–31. doi:10.1016/j.physbeh.2010.04.027
115. Lkhagvasuren B, Nakamura Y, Oka T, Sudo N, Nakamura K. Social defeat stress induces hyperthermia through activation of thermoregulatory sympathetic premotor neurons in the medullary raphe region. Eur J Neurosci (2011) 34:1442–52. doi:10.1111/j.1460-9568.2011.07863.x
116. Takahashi Y, Zhang W, Sameshima K, Kuroki C, Matsumoto A, Sunanaga J, et al. Orexin neurons are indispensable for prostaglandin E2-induced fever and defence against environmental cooling in mice. J Physiol (2013) 591:5623–43. doi:10.1113/jphysiol.2013.261271
117. Zhang W, Sunanaga J, Takahashi Y, Mori T, Sakurai T, Kanmura Y, et al. Orexin neurons are indispensable for stress-induced thermogenesis in mice. J Physiol (2010) 588:4117–29. doi:10.1113/jphysiol.2010.195099
118. Kataoka N, Hioki H, Kaneko T, Nakamura K. Psychological stress activates a dorsomedial hypothalamus-medullary raphe circuit driving brown adipose tissue thermogenesis and hyperthermia. Cell Metab (2014) 20:346–58. doi:10.1016/j.cmet.2014.05.018
119. Baroncelli L, Braschi C, Spolidoro M, Begenisic T, Sale A, Maffei L. Nurturing brain plasticity: impact of environmental enrichment. Cell Death Differ (2010) 17:1092–103. doi:10.1038/cdd.2009.193
120. Cao L, Choi EY, Liu X, Martin A, Wang C, Xu X, et al. White to brown fat phenotypic switch induced by genetic and environmental activation of a hypothalamic-adipocyte axis. Cell Metab (2011) 14:324–38. doi:10.1016/j.cmet.2011.06.020
121. Wang C, Bomberg E, Billington C, Levine A, Kotz CM. Brain-derived neurotrophic factor in the hypothalamic paraventricular nucleus increases energy expenditure by elevating metabolic rate. Am J Physiol Regul Integr Comp Physiol (2007) 293:R992–1002. doi:10.1152/ajpregu.00516.2006
122. Wang C, Bomberg E, Billington CJ, Levine AS, Kotz CM. Brain-derived neurotrophic factor (BDNF) in the hypothalamic ventromedial nucleus increases energy expenditure. Brain Res (2010) 1336:66–77. doi:10.1016/j.brainres.2010.04.013
123. McGlashon JM, Gorecki MC, Kozlowski AE, Thirnbeck CK, Markan KR, Leslie KL, et al. Central serotonergic neurons activate and recruit thermogenic brown and beige fat and regulate glucose and lipid homeostasis. Cell Metab (2015) 21:692–705. doi:10.1016/j.cmet.2015.04.008
Keywords: brown adipose tissue, white adipose tissue, beige fat, thermogenesis, hypothalamus, sympathetic nervous system, Ucp1, obesity
Citation: Yang X and Ruan H-B (2015) Neuronal control of adaptive thermogenesis. Front. Endocrinol. 6:149. doi: 10.3389/fendo.2015.00149
Received: 28 July 2015; Accepted: 07 September 2015;
Published: 25 September 2015
Edited by:
Jun Wu, University of Michigan, USAReviewed by:
Lei Sun, Duke-NUS Graduate Medical School, SingaporeYun-Hee Lee, Yonsei University, Korea
Copyright: © 2015 Yang and Ruan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyong Yang, Section of Comparative Medicine, Yale University School of Medicine, P.O. Box 208016, New Haven, CT 06520-8016, USA,eGlhb3lvbmcueWFuZ0B5YWxlLmVkdQ==