 Nada El Ghorayeb
Nada El Ghorayeb Isabelle Bourdeau
Isabelle Bourdeau André Lacroix
André Lacroix- Department of Medicine, Division of Endocrinology, Centre de Recherche du Centre hospitalier de l’Université de Montréal (CRCHUM), Université de Montréal, Montréal, QC, Canada
The major physiological regulators of aldosterone production from the adrenal zona glomerulosa are potassium and angiotensin II; other acute regulators include adrenocorticotropic hormone (ACTH) and serotonin. Their interactions with G-protein coupled hormone receptors activate cAMP/PKA pathway thereby regulating intracellular calcium flux and CYP11B2 transcription, which is the specific steroidogenic enzyme of aldosterone synthesis. In primary aldosteronism (PA), the increased production of aldosterone and resultant relative hypervolemia inhibits the renin and angiotensin system; aldosterone secretion is mostly independent from the suppressed renin–angiotensin system, but is not autonomous, as it is regulated by a diversity of other ligands of various eutopic or ectopic receptors, in addition to activation of calcium flux resulting from mutations of various ion channels. Among the abnormalities in various hormone receptors, an overexpression of the melanocortin type 2 receptor (MC2R) could be responsible for aldosterone hypersecretion in aldosteronomas. An exaggerated increase in plasma aldosterone concentration (PAC) is found in patients with PA secondary either to unilateral aldosteronomas or bilateral adrenal hyperplasia (BAH) following acute ACTH administration compared to normal individuals. A diurnal increase in PAC in early morning and its suppression by dexamethasone confirms the increased role of endogenous ACTH as an important aldosterone secretagogue in PA. Screening using a combination of dexamethasone and fludrocortisone test reveals a higher prevalence of PA in hypertensive populations compared to the aldosterone to renin ratio. The variable level of MC2R overexpression in each aldosteronomas or in the adjacent zona glomerulosa hyperplasia may explain the inconsistent results of adrenal vein sampling between basal levels and post ACTH administration in the determination of source of aldosterone excess. In the rare cases of glucocorticoid remediable aldosteronism, a chimeric CYP11B2 becomes regulated by ACTH activating its chimeric CYP11B1 promoter of aldosterone synthase in bilateral adrenal fasciculate-like hyperplasia. This review will focus on the role of ACTH on excess aldosterone secretion in PA with particular focus on the aberrant expression of MC2R in comparison with other aberrant ligands and their GPCRs in this frequent pathology.
Introduction
Primary aldosteronism (PA) was first described in patients with unilateral aldosterone-producing adenomas (1). It is characterized by increased aldosterone secretion causing salt retention, increased urinary potassium excretion, relative hypervolemia, suppressed plasma renin activity (PRA), and hypertension. PA is the most common curable form of secondary hypertension as it affects 4.3% of the general hypertensive population, 9.5% of patients referred to hypertension clinics (2), and up to 20% of those with resistant hypertension (3). PA is most often secondary to bilateral adrenal hyperplasia (BAH; 50–70% of cases) or to an aldosterone-producing adenoma (APA; 30–50% of cases) (4). The classical concept that a unique unilateral aldosteronoma is the causative lesion responsible for a high proportion of this surgically curable form of PA was recently challenged by the identification of zona glomerulosa (ZG) hyperplasia and nodulation adjacent to aldosteronomas when resected adrenals are examined carefully (5–7).
In order to prevent cardiovascular, metabolic and renal morbidities, early diagnosis and management of PA are mandatory (8–10). Unilateral adrenalectomy (UA) provides superior benefit compared to medical therapy in lateralized PA in terms of cardiovascular outcomes (11, 12), quality of life (12), and all-cause mortality (13); however, in BAH, pharmacological blockade of aldosterone excess using mineralocorticoid receptor antagonists, such as spironolactone or eplerenone, is the recommended treatment (4). Therefore, subtyping of PA is required to direct patients to surgical vs. medical therapy (4). To date, adrenal vein sampling (AVS) is the gold standard to differentiate lateralized from bilateral sources of PA (4) because adrenal imaging provides poor specificity in detecting lateralized cases (14) except in patients <35 years old (15).
In PA excess, plasma aldosterone concentration (PAC), despite suppressed renin activity, is not really autonomous, as frequently stated. It could be autonomous if it was solely or constitutively regulated by somatic and germline mutations of various ion channels genes regulating intracellular ionic homeostasis and cell membrane potential as reviewed elsewhere (16). In fact, several autocrine/paracrine hormones and regulatory mechanisms (17) activate variable levels of aberrant eutopic or ectopic receptors (18), which regulate aldosterone secretion either in unilateral adenomas or in BAH. In this review, we will focus on the role of one of these hormones, the adrenocorticotropic hormone (ACTH), in stimulating aldosterone secretion in normal and pathologic conditions and briefly mention others which play similar roles in PA.
Normal Physiology of the Renin–Angiotensin System
Renin is an enzyme produced primarily by the juxtaglomerular apparatus of the kidney and its release is the rate-limiting step in the regulation of the RAS (19, 20); it is controlled by four factors: (1) the macula densa comprises chemoreceptors for monitoring the sodium and chloride loads present in the distal tubule, (2) juxtaglomerular cells acting as pressure transducers that sense stretch of the afferent arteriolar wall and thus renal perfusion pressure, (3) the sympathetic nervous system (SNS), which increases the release of renin, particularly in response to upright posture, in addition to (4) inhibiting factors, including K+, Ca++, angiotensin II, and atrial natriuretic peptides (19).
The action of renin on angiotensinogen, synthesized in the liver, generates angiotensin I (19). Angiotensin-converting enzyme (ACE), localized in cell membranes particularly of the lung, cleaves angiotensin I into angiotensin II, which is the main biologically active angiotensin (19). Angiotensin II functions through the AT-1 receptor (AT1R) to maintain normal extracellular volume and blood pressure by increasing aldosterone secretion from the ZG via increased transcription of CYP11B2 (aldosterone synthase) (Figure 1) as well as constricting vascular smooth muscle, releasing norepinephrine, and epinephrine from the adrenal medulla, enhancing the activity of the SNS and finally promoting the release of vasopressin (19).
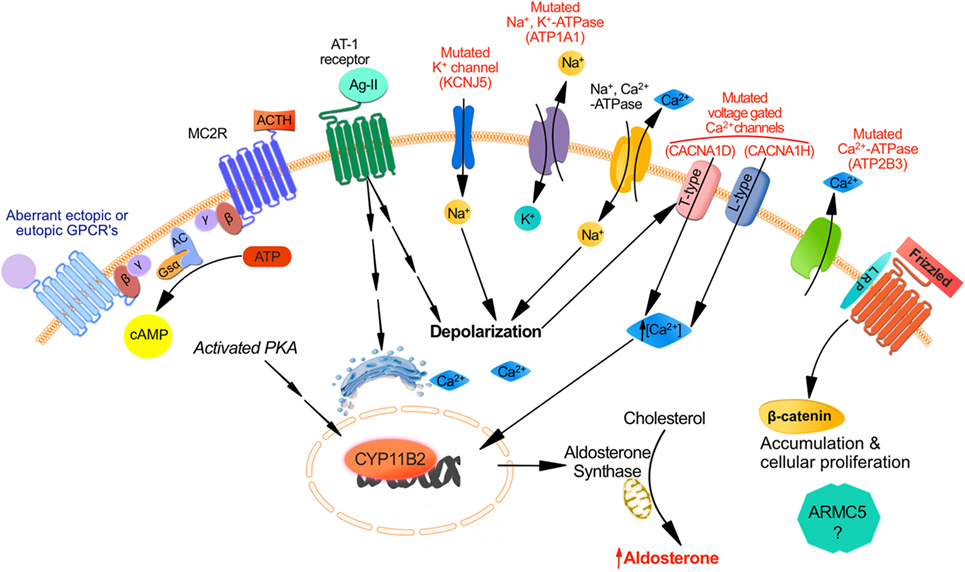
Figure 1. Mechanisms responsible for aldosterone synthesis in zona glomerulosa cells under normal physiological conditions and excess production in primary aldosteronism. The strongly negative resting membrane potential of zona glomerulosa (ZG) cells under resting physiological conditions is maintained by the concentration gradient of K+ between the intracellular and extracellular space, which is generated by the activity of the Na+, K+-ATPase. Angiotensin II and increased K+ lead to cell membrane depolarization, which opens voltage-dependent Ca2+ channels. Furthermore, Angiostensin II acts through the Angiotensin II type 1 receptor (AT1R) inducing Ca2+ release from the endoplasmic reticulum. Consequently, the increase in intracellular Ca2+ concentration activates the calcium signaling pathway, which triggers activation of CYP11B2 transcription. The role for ACTH in the regulation of aldosterone secretion whether in normal physiology or in PA is in part determined by the level of expression of ACTH receptors (MC2R) in ZG cells. MC2R which is a GPCR coupled to the stimulatory Gsα subunit may induce an increase of intracellular cAMP concentration which activates protein kinase A thereby increasing CREB phosphorylation and CYP11B2 transcription. Aberrant expression of other GPCR may also be responsible for aldosterone excess despite a suppressed renin angiotensin system: eutopic GPCR include those for serotonin (5-HT4R); ectopic GPCR include those for glucose-dependent insulinotropic peptide (GIPR), luteinizing hormone/human chorionic gonadotropin (LH–hCG R), β-adrenergic receptors (β-AR), vasopressin (V1-AVPR) glucagon (glucagon receptor), TRH (TRH R), and Endothelin-1 ETA and ETB receptors. Other mechanisms implicated in PA involve somatic and germline mutations in ion channels genes regulating intracellular ionic homeostasis and cell membrane potentials: increase intracellular Na+ concentrations and cell membrane depolarization result from KCNJ5 gain-of-function mutations affecting GIRK4 and ATP1A1 mutations of the Na+, K+-ATPase. Direct increase of intracellular Ca2+ concentrations could also result from mutations in ATP2B3 encoding for the plasma membrane Ca2+-ATPase, mutations in CACNA1D affecting the Cav1.3 subunit of the L-type voltage-gated calcium channel or CACNA1H affecting the Cav3.2 subunit of the voltage-gated calcium channel. Finally dysregulation in cellular proliferation/apoptosis accelerating adenoma formation could be due either to activation of the Wnt/β-catenin pathway or to gene mutations such as ARMC5 although the mechanism of the latter mutation is not fully elucidated.
Zona glomerulosa cells are organized in rosette structures that spontaneously generate periodic depolarizing changes in membrane potential that are modulated in frequency by angiotensin II and extracellular K+ (21, 22); Angiotensin II induces cell membrane depolarization most probably due to a Gi-mediated shift in the voltage dependence of channel activation toward more negative potentials thereby increasing intracellular Ca2+ signal, which stimulates hormone-sensitive lipase and steroidogenic acute regulatory protein (StAR). Another mechanism by which angiotensin II binding to AT1R stimulates aldosterone secretion implicates activating the phospholipase C/inositol 1,4,5-trisphosphate pathway, releasing Ca2+ stores from the endoplasmic reticulum, and activation of T-type voltage-gated Ca2+ channels (23) (Figure 1).
Dopamine, atrial natriuretic peptide, and heparin inhibit aldosterone secretion. The secretion of aldosterone is restricted to the ZG because of zone-specific expression of aldosterone synthase (CYP11B2), which is regulated by the activation of calcium signaling (24).
Pathophysiology of Primary Aldosteronism
The binding of free aldosterone to the mineralocorticoid receptor in the cytosol of epithelial cells (24), principally in the kidney, controls potassium homeostasis and maintains normal intravascular volume by increasing intestinal and renal Na+ and Cl− absorption and reabsorption, respectively. Increased production of aldosterone in PA results in sodium retention, hypertension, and can also result in hypokalemia (20). In addition to the two most common subtypes of PA (BAH in 50–70% of the cases and APA in 30–50%), less frequent causes include primary (unilateral) adrenal hyperplasia (5%), aldosterone-producing adrenocortical carcinoma (<1%), familial hyperaldosteronism (1%), and ectopic aldosterone-producing adenoma or carcinoma (<0.1%). The mechanisms implicated in the pathophysiology of PA are not fully elucidated. Somatic and germline mutations in ion channels genes regulating intracellular ionic homeostasis and cell membrane potential were described in sporadic APA and type-III familial PA (25–28) (Figure 1). Somatic mutations in the potassium channel gene KCNJ5 are found in almost 30–40% of aldosteronomas and alter channel selectivity allowing enhanced Na+ conductance. Na+ influx results in cell depolarization, the activation of voltage-gated Ca2+ channels, aldosterone production, and cell proliferation (25, 29). Somatic and germline mutations in CACNA1D gene encoding a voltage-gated calcium channel result in channel activation and less depolarized potentials causing increased Ca2+ influx, aldosterone production and cell proliferation in affected ZG cells (27, 30). Mutations in ATP1A1 gene (encoding the Na+/K+ ATPase α subunit) and ATP2B3 gene (encoding the plasma membrane Ca2+ ATPase) were identified in 5.2 and 1.6%, respectively of patients in a series of APA (26). Mutations in CACNA1H gene, which encodes a voltage-gated calcium channel (Cav3.2) were discovered in children with PA; they result in impaired channel inactivation and activation at more hyperpolarized potentials, producing increased intracellular Ca2+ and aldosterone excess (31). Different mutations in the genes described above are found in different aldosterone-producing nodules from the same adrenal, suggesting that somatic mutations are independent events (32, 33).
No mutations of any of the above ion channel genes were found in BAH or in ZG hyperplasia adjacent to the dominant aldosteronomas (26, 29, 32, 33); these findings suggest that nodule formation and excess aldosterone production are two dissociated events, implying a two-hit hypothesis for APA formation (16, 34). The first hit causing a unilateral aldosteronoma or a dominant nodule adjacent to ZG hyperplasia may result from a somatic mutation in one of the genes described above, at least in approximately 60% of cases. Possible causes of the second hit that results in dysregulation in cellular proliferation/apoptosis accelerating adenoma formation could be due either to activation of the Wnt/β-catenin pathway (35, 36), PKA pathway, or to gene mutations such as ARMC5 (37) (Figure 1). However, the pathophysiology of progression from normal adrenal to APA and the causes of diffuse bilateral hyperplasias, either as BAH or in mild form adjacent to the dominant aldosteronoma, are still unknown. Aldosterone-producing cell clusters (APCCs), which have increased expression of CYP11B2, are nests of cells below the adrenal capsule. They protrude into cortisol-producing cells that are usually negative for CYP11B2 expression. Nishimoto et al. found that APCCs are common in normal adrenals, and they harbor a different mutational spectrum compared to APA suggesting that APCCs could be a precursor for APA (38). In addition, several hormones activating variable levels of eutopic or ectopic aberrant receptors (18) (Figure 1) as well as autocrine and paracrine regulatory mechanisms (17) can increase aldosterone secretion in PA (either APA or BAH) independently from the suppressed RAS (see later section).
Role of ACTH in Aldosterone Production in Normal Physiology
Adrenocorticotropic hormone can stimulate aldosterone secretion acutely and transiently under normal conditions, but to a lesser extent than angiotensin II and potassium. ACTH is a 39-amino-acid peptide, which results from the cleavage of its proopiomelanocortin (POMC) precursor by prohormone convertases PC1/3 and may be further cleaved by PC2 to generate α-melanocyte-stimulating hormone (α-MSH) (39, 40). It is mainly produced in the anterior pituitary corticotropes, but is also produced in brain, adrenal medulla, skin, and placenta (41–43). ACTH can induce aldosterone production at lower doses than the ones needed for cortisol and DHEA production (44). Furthermore, ACTH stimulates aldosterone production acutely and sometimes chronically.
Acute Effects of ACTH
The initial binding of ACTH to its specific melanocortin type 2 receptor (MC2R) stimulates both cortisol and aldosterone secretion. MC2R (45) is a seven transmembrane domain receptor that belongs to the family of melanocortin receptors (MCRs) (45). Five MCRs constitute a distinct family of G-protein coupled hormone receptors (GPCR); MC2R is the smallest MCR and GPCR (45, 46). MC2R is expressed in zona fasciculata (ZF) and ZG cells (47). The binding of ACTH to its MC2R induces the dissociation of Gs-α subunit and activation of adenylate cyclase (AC) that generates cAMP from ATP (48) (Figure 1). cAMP molecules bind to specific domains of the regulatory subunits of protein kinase A (PKA) thereby dissociating the tetramer and releasing the catalytic subunit (PRKACA) from its inactivating regulatory subunits. Activated PRKACA phosphorylates and activates steroidogenic acute regulatory protein (StAR) as well as cAMP response element binding protein (CREB), thereby increasing StAR expression. On the other hand, activation of the PKA pathway induces a slow but sustained calcium influx through the L-type calcium channels. The subsequent increase in intracellular calcium activates calcium/calmodulin-dependent protein kinase and steroidogenesis (49, 50).
Chronic Effects of ACTH
In contrast to in vivo studies that suggest that ACTH is a short-term stimulator of aldosterone production, in vitro studies showed that ACTH can act as a major stimulus of aldosterone secretion. Continuous intravenous infusion of ACTH leads to a transient stimulation of aldosterone secretion, whereas its pulsatile administration leads to a sustained stimulation of aldosterone up to 72 h (51). Moreover, chronic exposure to ACTH (2 days or more) leads to transformation of ZG cells into ZF-like cells with elongated mitochondria with lamellar and tubular cristae becoming round with ovoid cristae; at the functional level, the synthesis of angiotensin II receptors, steroidogenic enzymes, and their products is altered (52–56).
Role of ACTH in Excess Aldosterone Secretion in PA
Diurnal Rhythmicity of Aldosterone
In recumbent normal subjects on a regular diet, the circadian rhythm of PAC is regulated by the activity of plasma renin independently of ACTH (57). In contrast, patients with PA have a circadian rhythm of PAC mediated by changes in ACTH rather than by the suppressed plasma renin–angiotensin II levels (58). Several groups described that PAC falls following overnight sleep when ACTH levels are low despite upright posture or angiotensin II infusion. Similarly, they noted a marked increase in PAC shortly after ACTH administration (59–63), which was higher compared to normal controls or patients with essential hypertension (64, 65). Furthermore, abolition of diurnal rhythm by dexamethasone in PA demonstrates the impact of ACTH on adrenal steroidogenesis (66). Administration of dexamethasone 0.75–2.0 mg per day for 2 days decreased aldosterone levels by a mean of 49% in a group of 15 patients with aldosteronomas; in 33%, the suppression was greater than 80% (67).
ACTH Role in Familial Hyperaldosteronism
Familial hyperaldosteronism (FH) type-1, previously known as glucocorticoid-remediable aldosteronism (GRA), was first described as a form of hyperaldosteronism relieved by dexamethasone (68). It is suspected in young PA patients whose relatives suffer from cerebrovascular accidents. It is an autosomal dominant disease whereby the promoter of the chimeric 11β-hydroxylase/aldosterone synthase gene belongs to the 5′ end of CYP11 B1 (11β hydroxylase) and drives the expression of the 3′ end of CYP11 B2 (aldosterone synthase) ectopically in ZF cells under the main regulation by ACTH (69); in these patients, dexamethasone usually decreases aldosterone secretion by more than 80% or to <4 ng/dL (67), but the diagnosis is now performed using genetic analysis. In contrast to FH type-1, FH type-2 is defined as PA in a patient with a first-degree relative (parent/sibling/offspring) with established PA but without FH type-1 gene rearrangement. Linkage analysis has mapped FH type-2 to chromosome 7p22 but no responsible gene has been identified yet (70). The prevalence of FH type-2 in PA is higher (1.2–6%) than FH type-1 (≤1%). The FH type-3 and -4 are not regulated by ACTH stimulation, but they are caused by germline mutations in KCNJ5 (71) and CACNA1D/CACNA1H (30, 31) genes, respectively.
ACTH Suppression or Stimulation Tests Can Reveal the Presence of PA
Based on the rationale that ACTH plays a more important role in PA than in normal subjects or those with essential hypertension, investigators in Athens compared the classical saline infusion test (SIT) to postdexamethasone SIT in 151 patients with single adrenal adenomas and detected almost double rate of aldosterone hypersecretion following dexamethasone administration (24 vs. 12%) (72). Similarly, they used a combined fludrocortisone–dexamethasone suppression test (FDST), which is a modification of the classic confirmatory fludrocortisone suppression test (FST) for the diagnosis of PA; it involves the administration of dexamethasone to hypertensives patients at midnight of the last day of the FST in order to eliminate the stimulatory effect of ACTH on aldosterone secretion. They demonstrated that the prevalence of PA rises from 5 to 13% with the usual diagnostic tests to 28.7–31% when using FDST; mineralocorticoid receptor blockade resulted in significant improvement in blood pressure in these patients (73–75). The same group administered an ultralow-dose (0.03 μg) ACTH to 113 hypertensives without PA: the 30 patients (27%) who exhibited an aldosterone hyperresponse had significantly higher PAC, ARR, and PAC/ACTH ratio in the treadmill test; normalization of blood pressure by mineralocorticoid antagonists in these patients was also evident compared to the group of hypertensive not sensitive to ACTH/stress (76). Therefore, the benefit of mineralocorticoid blockade could extend even to hypertensive patients without confirmed PA who present an aldosterone hyperreseponse to ACTH/stress, this category of hypertenives harboring a mild form of BAH. In contrast, another group examining the diagnostic accuracy of ACTH test in 158 hypertensive patients found that it was not very effective in differentiating between APA patients and non-PA patients (77).
Use of ACTH to Identify the Source of Aldosterone Excess
Many efforts were conducted to find an easier and cheaper test than adrenal vein sampling (AVS), which is available only in tertiary care center with experienced angioradiologists to distinguish between lateralized and bilateral sources of aldosterone. Differential increase in PAC during upright posture was suggested to be a valuable tool to distinguish APA from BAH, but further studies showed that several APA and BAH had similar rise in PAC to upright posture (78). APA whether responsive or not to angiotensin II was found to be more sensitive to ACTH stimulation resulting in larger increase of PAC than in patients with BAH or essential hypertension (79–81). BAH patients also displayed increased response of PAC to ACTH administration than normal subjects or patients with essential hypertension (82). PAC increased more after ACTH bolus in the APA group compared with BAH group, which had an intermediate increase compared to normal controls (18, 66). A study in which patients received dexamethasone (1 mg) the evening before receiving i.v. injection of 50 IU of ACTH showed that the exaggerated PAC response was higher after 120 min in patients with APA than in BAH (83). It was suggested that this could be used for identifying the etiology subtype; however, significant overlap was present between APA, unilateral hyperplasia, and BAH cases and using an optimal cutoff value of the aldosterone >78 ng/dL for APA, provided a sensitivity of 76.8% and a specificity of 87.2% (83).
Kline et al. studied 65 patients with confirmed PA who were divided by histology into confirmed lateralized and non-lateralized; PAC in inferior vena cava (IVC) sampled during AVS before and after cosyntropin infusion was analyzed. Baseline and peak IVC aldosterone was higher in lateralized patients (APA) but incremental aldosterone rise was much greater in subjects with bilateral hyperplasia (84). This shows that ACTH can regulate APA as well as BAH, but that the effects are more pronounced in APA.
Role of ACTH Stimulation during Adrenal Venous Sampling
The usefulness of ACTH stimulation in the conduct of AVS procedure is controversial and remains a matter of debate because of conflicting results. Some centers use cosyntropin infusion or bolus in order to minimize stress-induced or spontaneous fluctuations in aldosterone secretion when performing sequential non-simultaneous AVS, to maximize the gradient of cortisol from the adrenal vein to the inferior vena cava, and to maximize aldosterone secretion from an APA (85). In contrast, other groups found that ACTH-stimulation of aldosterone production from the contralateral gland or adjacent hyperplasia may reduce the gradient of aldosterone production resulting in incorrect lateralization (86, 87). The effect of both continuous ACTH infusion and bolus on the performance and interpretation of AVS in confirmed PA patients was investigated (88). Both methods lead to a significant increase in selectivity index for the right adrenal vein and ACTH bolus for the left adrenal vein. Lateralization index was not significantly affected after continuous ACTH infusion and i.v. bolus. In 88 and 78% of the patients, the diagnosis obtained was the same before and after ACTH infusion and i.v. bolus, respectively (88). Recently, our group demonstrated that ACTH increased selectivity on both sides from 66.7% in basal samples to 91.8% poststimulation. A discordance of lateralization between basal and post-ACTH values was observed in 28% of cases, mostly lateralized cases basally that became bilateral post ACTH (87). The variation in the response to ACTH stimulation could be due to the variable expression of MC2R in APA (see later) (18). Careful examination of the levels of aldosterone in the adrenal vein contralateral to the dominant or lateralized APA and pathology confirmed the frequent presence of bilateral background hyperplasia and this could predict less favorable post-operative outcome with residual hypertension (35, 87).
Increased but Variable Expression of MC2R in PA
The explanation for the increased role of ACTH in the regulation of aldosterone in PA may be secondary to the overexpression and function of MC2R in this condition. The expression of MC2R mRNA was shown to be upregulated in human adrenocortical neoplasms specifically in functional adenomas in contrast to non-functioning adenomas and carcinomas (89). More specifically, a few studies have demonstrated increased eutopic expression of MC2R assessed by RT-PCR or transcriptome studies in resected aldosteronomas as compared to cortisol-secreting adenomas, non-functional adenomas, and adrenocortical carcinomas (90–92). A particularly pertinent informative study included 15 adrenal tumors (14 APA and 1 BAH); MC2R mRNA levels were increased by a mean of 3.9-fold in those tissues compared to normal adrenal (18). However great variability existed in the level of expression in each tumor as 4 had lower levels than normal (0.3-fold to 0.7-fold), while those with increased expression varied between 1.4- and 20.6-fold compared to normal. The data are limited to mRNA expression without available measurements at the protein levels (no good specific MC2R antibody), but correlated well with the in vivo increased response to ACTH administration. There is almost no data on MC2R expression in BAH as those patients are usually not operated, but in the only case with BAH studied by this group MC2R was 20-fold increased. These data appear to be compatible with the findings that the majority of patients stimulated with ACTH during AVS will have concordant results before and after ACTH as the majority overexpress MC2R; however, the 28% of discordant results we found (87) may be explained by cases where MC2R are relatively decreased in the dominant adenoma but is present in adjacent hyperplasia. This hypothesis remains to be validated in prospective studies.
Other Hormones and Aberrant Receptors Regulating Aldosterone Secretion in Primary Aldosteronism
Adrenocorticotropin hormone is not the exclusive trigger of aldosterone secretion since several other hormones have a role in the pathophysiology of PA in addition to the ion channels mutations. Serotonin plays a significant role in aldosterone synthesis in normal physiological and in PA. The administration of serotonin 5-HT4 agonists such as metoclopramide, cisapride, and tegaserod resulted in higher stimulation of aldosterone in PA as compared to the physiological moderate increase in normal individuals (18, 93, 94). Whereas non-specific inhibitors of 5-HT such as cyproheptadine and ketanserin produced only minor and transient inhibition of aldosterone secretion in aldosteronomas (95, 96), specific 5-HT4R antagonists such as GR113808 were potent inhibitors of basal- and cisapride-induced aldosterone secretion (93). Chromaffin cells, endothelial cells, nerve terminals, and cells of the immune system are localized in the immediate vicinity of ZG cells and can secrete various factors to control aldosterone secretion (97). Local release of 5-HT by perivascular mast cells (MC) can activate 5-HT4R expressed in ZG cells and consequently stimulate aldosterone production (98). A role of MC in tumorigenesis was proposed (99, 100). The density of MC was found to be increased in APA tissues compared with normal adrenals (101). As the 5-HT4R have been found to be overexpressed in the majority of APA (but variable as MC2R) (17, 18, 102) and the ligand may be locally overexpressed also, a paracrine loop of regulation of aldosterone production appears to be present.
The compelling evidence supporting that various aberrant GPCR are frequently expressed in bilateral macronodular adrenal hyperplasia and Cushing’s syndrome (103) led many researchers to investigate the presence of aberrant GPCR in PA. Adrenal production of aldosterone in APA and BAH was found to be under the influence of aberrant GPCR and their ligands, as demonstrated by in vivo and in vitro studies (104, 105). The expression of ectopic receptors, which are usually not expressed at significant levels in normal ZG cells include those for glucose-dependent insulinotropic peptide (GIPR) (106), luteinizing hormone/human chorionic gonadotropin (LH–hCG R) (18, 106–112), β-adrenergic receptors (β-AR) (113), vasopressin (V1-AVPR) (18, 106, 114, 115), glucagon (glucagon receptor), TRH (TRH R) (18, 112, 116) and Endothelin-1 ETA and ETB receptors (117). Using a microarray approach in 10 aldosteronomas compared with five normal adrenals and 13 cortisol-secreting adenomas, the six GCPRs with highest increase in expression included LHCGR, 5-HT4R, GnRHR, glutamate receptor metabotropic 3, endothelin receptor ETB receptors, and MC2R (92). Table 1 summarizes the different types of aberrant eutopic or ectopic GPCR involved in aldosterone excess in PA. Co-expression of multiple aberrant GPCR was also reported; renin-independent stimulation of aldosterone secretion was observed in vivo following mixed meal, oral glucose, or administration of GIP, vasopressin, and tegaserod in a patient with unilateral source of PA (106). On the other hand, co-secretion of aldosterone and cortisol due to aberrant expression of GPCR was noted; in a patient with BMAH and β-AR-aberrant expression, isoproterenol stimulated both cortisol and aldosterone production (113).
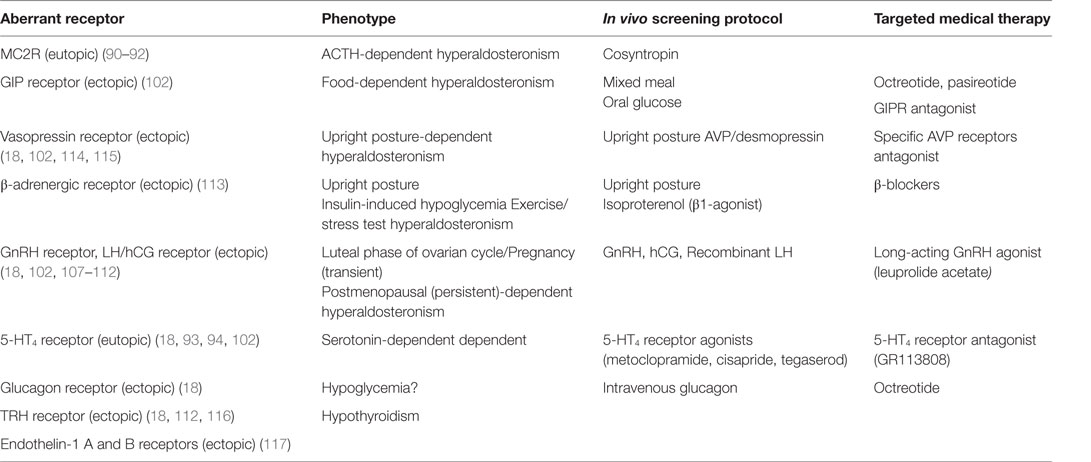
Table 1. Types of GPCR involved in aldosterone hypersecretion in patients with PA.
Activating somatic CTNNB1 (β-catenin) mutations have now been identified in tumors of three women with APAs, two who presented during pregnancy and one after menopause (118). All three had heterozygous activating mutations of CTNNB1 and expressed aberrant LHCG and GNRH receptors at levels 100-fold higher than in other APAs. It was shown that the CTNNB1 mutation led to activation of the WNT pathway; it was suggested that could be the cause of dedifferentiation of gonadal progenitor cells present in the adrenal tissues with increased expression of gonadal receptors. It is thought that the high levels of endogenous human chorionic gonadotropin (hCG) during pregnancy and of gonadotropin-releasing hormone (GnRH) and luteinizing hormone (LH) after menopause led to the identification of APAs in these patients (118).
It is currently unclear whether these aberrant regulatory secretory mechanisms by ACTH and other hormones and the overexpression of their GPCR in PA are secondary to unknown proliferative mechanisms or are primary and at least partially responsible for the abnormal proliferation, the initiation of diffuse BAH. However, they clearly play a role in aldosterone secretion which is not autonomous.
Conclusion
Our understanding of the increased occurrence and complexity of molecular etiology and unique signature in each case of PA has progressed greatly in recent years. The increased role of ACTH, of the variable expression of MC2R, and of other aberrant GPCR in PA should receive further attention in the future. The development of effective antagonists to MC2R and other aberrant GPCR could eventually offer interesting alternatives in patients with bilateral sources of excess aldosterone in combination with better antagonists of the mineralocorticoid receptor.
Author Contributions
NG, IB, and AL contributed to the conception and design of the manuscript as well as to drafting the review article and they all provided final approval of the version to be published.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work is supported by grant 201209NMD from the Canadian Institutes of Health Research to AL and IB. NG fellowship support was also provided in part by Novartis Canada. IB receives a salary Grant from FRQS (Fonds de la recherche du Québec-Santé).
Abbreviations
β-AR, β-adrenergic receptors; 5-HT4R, serotonin receptor; AC, adenylate cyclase; ACTH, adrenocorticotropin hormone; APA, aldosterone-producing adenoma; APA, aldosterone-producing adenoma; APCCs, aldosterone-producing cell clusters; AT-1 receptor, angiotensin II type 1 receptor; ATP, adenosine triphosphate; AVS, adrenal venous sampling; BAH, bilateral adrenal hyperplasia; cAMP, cyclic adenosine monophosphate; CREB, cAMP response element binding protein; CYP11B1, cytochrome P450 family 11 subfamily B member 1 encodes 11-beta hydroxylase; CYP11B2, cytochrome P450 family 11 subfamily B member 2 encodes aldosterone synthetase; ET, endothelin-1; FH, familial hyperaldosteronism; FST, fludrocortisone suppression test; GIP, glucose-dependent insulinotropic peptide; GPCR, G-protein-coupled receptor; GRA, glucocorticoid-remediable aldosteronism; Gs-α, G stimulatory α subunit; LH–hCG, luteinizing hormone/human chorionic gonadotropin; MC2R, melanocortin type 2 receptor; mRNA, messenger ribonucleic acid; PA, primary aldosteronsim; PAC, plasma aldosterone; PKA, protein kinase; RAS, rennin–angiotensin system; RT-PCR, reverese transcriptase-polymerase chain reaction; StAR, steroidogenic acute regulatory protein; TRH, thyrotropin releasing hormone; UA, unilateral adrenalectomy; V1-AVPR, vasopressin receptor type 1; ZF, zona fasciculata; ZG, zona glomerulosa.
References
1. Conn JW. Presidential address. I. Painting background. II. Primary aldosteronism, a new clinical syndrome. J Lab Clin Med (1955) 45(1):3–17.
2. Hannemann A, Wallaschofski H. Prevalence of primary aldosteronism in patient’s cohorts and in population-based studies – a review of the current literature. Horm Metab Res (2012) 44(3):157–62. doi:10.1055/s-0031-1295438
3. Calhoun DA. Is there an unrecognized epidemic of primary aldosteronism? Pro. Hypertension (2007) 50(3):447–453; discussion 447–453. doi:10.1161/HYPERTENSIONAHA.106.086116
4. Funder JW, Carey RM, Fardella C, Gomez-Sanchez CE, Mantero F, Stowasser M, et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. J Clin Endocrinol Metab (2008) 93(9):3266–81. doi:10.1210/jcem.93.9.9998
5. Boulkroun S, Samson-Couterie B, Dzib JF, Lefebvre H, Louiset E, Amar L, et al. Adrenal cortex remodeling and functional zona glomerulosa hyperplasia in primary aldosteronism. Hypertension (2010) 56(5):885–92. doi:10.1161/HYPERTENSIONAHA.110.158543
6. Nishimoto K, Nakagawa K, Li D, Kosaka T, Oya M, Mikami S, et al. Adrenocortical zonation in humans under normal and pathological conditions. J Clin Endocrinol Metab (2010) 95(5):2296–305. doi:10.1210/jc.2009-2010
7. Duan K, Mete O. Clinicopathologic correlates of primary aldosteronism. Arch Pathol Lab Med (2015) 139(7):948–54. doi:10.5858/arpa.2014-0156-RS
8. Catena C, Colussi G, Nadalini E, Chiuch A, Baroselli S, Lapenna R, et al. Cardiovascular outcomes in patients with primary aldosteronism after treatment. Arch Intern Med (2008) 168(1):80–5. doi:10.1001/archinternmed.2007.33
9. Mulatero P, Monticone S, Bertello C, Viola A, Tizzani D, Iannaccone A, et al. Long-term cardio- and cerebrovascular events in patients with primary aldosteronism. J Clin Endocrinol Metab (2013) 98(12):4826–33. doi:10.1210/jc.2013-2805
10. Savard S, Amar L, Plouin PF, Steichen O. Cardiovascular complications associated with primary aldosteronism: a controlled cross-sectional study. Hypertension (2013) 62(2):331–6. doi:10.1161/HYPERTENSIONAHA.113.01060
11. Catena C, Colussi G, Lapenna R, Nadalini E, Chiuch A, Gianfagna P, et al. Long-term cardiac effects of adrenalectomy or mineralocorticoid antagonists in patients with primary aldosteronism. Hypertension (2007) 50(5):911–8. doi:10.1161/HYPERTENSIONAHA.107.095448
12. Ahmed AH, Gordon RD, Sukor N, Pimenta E, Stowasser M. Quality of life in patients with bilateral primary aldosteronism before and during treatment with spironolactone and/or amiloride, including a comparison with our previously published results in those with unilateral disease treated surgically. J Clin Endocrinol Metab (2011) 96(9):2904–11. doi:10.1210/jc.2011-0138
13. Reincke M, Fischer E, Gerum S, Merkle K, Schulz S, Pallauf A, et al. Observational study mortality in treated primary aldosteronism: the German Conn’s registry. Hypertension (2012) 60(3):618–24. doi:10.1161/HYPERTENSIONAHA.112.197111
14. Magill SB, Raff H, Shaker JL, Brickner RC, Knechtges TE, Kehoe ME, et al. Comparison of adrenal vein sampling and computed tomography in the differentiation of primary aldosteronism. J Clin Endocrinol Metab (2001) 86(3):1066–71. doi:10.1210/jc.86.3.1066
15. Lim V, Guo Q, Grant CS, Thompson GB, Richards ML, Farley DR, et al. Accuracy of adrenal imaging and adrenal venous sampling in predicting surgical cure of primary aldosteronism. J Clin Endocrinol Metab (2014) 99(8):2712–9. doi:10.1210/jc.2013-4146
16. Lalli E, Barhanin J, Zennaro MC, Warth R. Local control of aldosterone production and primary aldosteronism. Trends Endocrinol Metab (2016). doi:10.1016/j.tem.2016.01.003
17. Lefebvre H, Prévost G, Louiset E. Autocrine/paracrine regulatory mechanisms in adrenocortical neoplasms responsible for primary adrenal hypercorticism. Eur J Endocrinol (2013) 169(5):R115–38. doi:10.1530/EJE-13-0308
18. Zwermann O, Suttmann Y, Bidlingmaier M, Beuschlein F, Reincke M. Screening for membrane hormone receptor expression in primary aldosteronism. Eur J Endocrinol (2009) 160(3):443–51. doi:10.1530/EJE-08-0711
19. Hall JE. Historical perspective of the renin-angiotensin system. Mol Biotechnol (2003) 24(1):27–39. doi:10.1385/MB:24:1:27
20. Spat A, Hunyady L. Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev (2004) 84(2):489–539. doi:10.1152/physrev.00030.2003
21. Hu C, Rusin CG, Tan Z, Guagliardo NA, Barrett PQ. Zona glomerulosa cells of the mouse adrenal cortex are intrinsic electrical oscillators. J Clin Invest (2012) 122(6):2046–53. doi:10.1172/JCI61996
22. Barrett PQ, Guagliardo NA, Klein PM, Hu C, Breault DT, Beenhakker MP. Role of voltage-gated calcium channels in the regulation of aldosterone production from zona glomerulosa cells of the adrenal cortex. J Physiol (2016). doi:10.1113/JP271896
23. Spät A, Hunyady L, Szanda G. Signaling interactions in the adrenal cortex. Front Endocrinol (2016) 7:17. doi:10.3389/fendo.2016.00017
24. Arlt W, Stewart PM. Adrenal corticosteroid biosynthesis, metabolism, and action. Endocrinol Metab Clin North Am (2005) 34(2):293–313, viii. doi:10.1016/j.ecl.2005.01.002
25. Choi M, Scholl UI, Yue P, Björklund P, Zhao B, Nelson-Williams C, et al. K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science (2011) 331(6018):768–72. doi:10.1126/science.1198785
26. Beuschlein F, Boulkroun S, Osswald A, Wieland T, Nielsen HN, Lichtenauer UD, et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat Genet (2013) 45(4):.e441–2. doi:10.1038/ng.2550
27. Scholl UI, Goh G, Stölting G, de Oliveira RC, Choi M, Overton JD, et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet (2013) 45(9):1050–4. doi:10.1038/ng.2695
28. Fernandes-Rosa FL, Williams TA, Riester A, Steichen O, Beuschlein F, Boulkroun S, et al. Genetic spectrum and clinical correlates of somatic mutations in aldosterone-producing adenoma. Hypertension (2014) 64(2):354–61. doi:10.1161/HYPERTENSIONAHA.114.03419
29. Boulkroun S, Beuschlein F, Rossi GP, Golib-Dzib JF, Fischer E, Amar L, et al. Prevalence, clinical, and molecular correlates of KCNJ5 mutations in primary aldosteronism. Hypertension (2012) 59(3):592–8. doi:10.1161/HYPERTENSIONAHA.111.186478
30. Scholl UI, Nelson-Williams C, Yue P, Grekin R, Wyatt RJ, Dillon MJ, et al. Hypertension with or without adrenal hyperplasia due to different inherited mutations in the potassium channel KCNJ5. Proc Natl Acad Sci U S A (2012) 109(7):2533–8. doi:10.1073/pnas.1121407109
31. Scholl UI, Stölting G, Nelson-Williams C, Vichot AA, Choi M, Loring E, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife (2015) 4:e06315. doi:10.7554/eLife.06315
32. Fernandes-Rosa F, Giscos-Douriez I, Amar L, Gomez-Sanchez CE, Meatchi T, Boulkroun S, et al. PP.09.27: genetic abnormalities in lateralized multinodular primary aldosteronism. J Hypertens (2015) 33:e215–6. doi:10.1097/01.hjh.0000468004.04564.fb
33. Fernandes-Rosa FL, Giscos-Douriez I, Amar L, Gomez-Sanchez CE, Meatchi T, Boulkroun S, et al. Different somatic mutations in multinodular adrenals with aldosterone-producing adenoma. Hypertension (2015) 66(5):1014–22. doi:10.1161/HYPERTENSIONAHA.115.05993
34. Gomez-Sanchez CE, Gomez-Sanchez EP. Mutations of the potassium channel KCNJ5 causing aldosterone-producing adenomas: one or two hits? Hypertension (2012) 59(2):196–7. doi:10.1161/HYPERTENSIONAHA.111.186205
35. Boulkroun S, Samson-Couterie B, Golib-Dzib JF, Amar L, Plouin PF, Sibony M, et al. Aldosterone-producing adenoma formation in the adrenal cortex involves expression of stem/progenitor cell markers. Endocrinology (2011) 152(12):4753–63. doi:10.1210/en.2011-1205
36. El Wakil A, Lalli E. The Wnt/beta-catenin pathway in adrenocortical development and cancer. Mol Cell Endocrinol (2011) 332(1–2):32–7. doi:10.1016/j.mce.2010.11.014
37. Zilbermint M, Xekouki P, Faucz FR, Berthon A, Gkourogianni A, Schernthaner-Reiter MH, et al. Primary aldosteronism and ARMC5 variants. J Clin Endocrinol Metab (2015) 100(6):E900–9. doi:10.1210/jc.2014-4167
38. Nishimoto K, Tomlins SA, Kuick R, Cani AK, Giordano TJ, Hovelson DH, et al. Aldosterone-stimulating somatic gene mutations are common in normal adrenal glands. Proc Natl Acad Sci U S A (2015) 112(33):E4591–9. doi:10.1073/pnas.1505529112
39. Raffin-Sanson ML, de Keyzer Y, Bertagna X. Proopiomelanocortin, a polypeptide precursor with multiple functions: from physiology to pathological conditions. Eur J Endocrinol (2003) 149(2):79–90. doi:10.1530/eje.0.1490079
40. Dores RM, Londraville RL, Prokop J, Davis P, Dewey N, Lesinski N. Molecular evolution of GPCRs: melanocortin/melanocortin receptors. J Mol Endocrinol (2014) 52(3):T29–42. doi:10.1530/JME-14-0050
41. Vrezas I, Willenberg HS, Mansmann G, Hiroi N, Fritzen R, Bornstein SR. Ectopic adrenocorticotropin (ACTH) and corticotropin-releasing hormone (CRH) production in the adrenal gland: basic and clinical aspects. Microsc Res Tech (2003) 61(3):308–14. doi:10.1002/jemt.10340
42. Bicknell AB. The tissue-specific processing of pro-opiomelanocortin. J Neuroendocrinol (2008) 20(6):692–9. doi:10.1111/j.1365-2826.2008.01709.x
43. Evans JF, Fernando A, Ragolia L. Functional melanocortin-2 receptors are expressed by mouse aorta-derived mesenchymal progenitor cells. Mol Cell Endocrinol (2012) 355(1):60–70. doi:10.1016/j.mce.2012.01.019
44. Daidoh H, Morita H, Mune T, Murayama M, Hanafusa J, Ni H, et al. Responses of plasma adrenocortical steroids to low dose ACTH in normal subjects. Clin Endocrinol (Oxf) (1995) 43(3):311–5. doi:10.1111/j.1365-2265.1995.tb02037.x
45. Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science (1992) 257(5074):1248–51. doi:10.1126/science.1325670
46. Cone RD, Mountjoy KG, Robbins LS, Nadeau JH, Johnson KR, Roselli-Rehfuss L, et al. Cloning and functional characterization of a family of receptors for the melanotropic peptides. Ann N Y Acad Sci (1993) 680:342–63. doi:10.1111/j.1749-6632.1993.tb19694.x
47. Chan LF, Metherell LA, Clark AJ. Effects of melanocortins on adrenal gland physiology. Eur J Pharmacol (2011) 660(1):171–80. doi:10.1016/j.ejphar.2010.11.041
48. de Joussineau C, Sahut-Barnola I, Levy I, Saloustros E, Val P, Stratakis CA, et al. The cAMP pathway and the control of adrenocortical development and growth. Mol Cell Endocrinol (2012) 351(1):28–36. doi:10.1016/j.mce.2011.10.006
49. Hattangady NG, Olala LO, Bollag WB, Rainey WE. Acute and chronic regulation of aldosterone production. Mol Cell Endocrinol (2012) 350(2):151–62. doi:10.1016/j.mce.2011.07.034
50. Gallo-Payet N. Adrenal and extra-adrenal functions of ACTH. J Mol Endocrinol (2016) 56(4):135–56. doi:10.1530/JME-15-0257
51. Seely EW, Conlin PR, Brent GA, Dluhy RG. Adrenocorticotropin stimulation of aldosterone: prolonged continuous versus pulsatile infusion. J Clin Endocrinol Metab (1989) 69(5):1028–32. doi:10.1210/jcem-69-5-1028
52. Aguilera G, Fujita K, Catt KJ. Mechanisms of inhibition of aldosterone secretion by adrenocorticotropin. Endocrinology (1981) 108(2):522–8. doi:10.1210/endo-108-2-522
53. Crivello JF, Gill GN. Induction of cultured bovine adrenocortical zona glomerulosa cell 17-hydroxylase activity by ACTH. Mol Cell Endocrinol (1983) 30(1):97–107. doi:10.1016/0303-7207(83)90204-6
54. Bird IM, Pasquarette MM, Rainey WE, Mason JI. Differential control of 17 alpha-hydroxylase and 3 beta-hydroxysteroid dehydrogenaseg expression in human adrenocortical H295R cells. J Clin Endocrinol Metab (1996) 81(6):2171–8. doi:10.1210/jc.81.6.2171
55. Corander MP, Coll AP. Melanocortins and body weight regulation: glucocorticoids, Agouti-related protein and beyond. Eur J Pharmacol (2011) 660(1):111–8. doi:10.1016/j.ejphar.2010.10.103
56. Gallo-Payet N, Battista MC. Steroidogenesis-adrenal cell signal transduction. Compr Physiol (2014) 4(3):889–964. doi:10.1002/cphy.c130050
57. Williams GH, Tuck ML, Rose LI, Dluhy RG, Underwood RH. Studies of the control of plasma aldosterone concentration in normal man. 3. Response to sodium chloride infusion. J Clin Invest (1972) 51(10):2645–52. doi:10.1172/JCI106974
58. Kem DC, Gomez-Sanchez C, Kramer NJ, Holland OB, Higgins JR. Plasma aldosterone and renin activity response to ACTH infusion in dexamethasone-suppressed normal and sodium-depleted man. J Clin Endocrinol Metab (1975) 40(1):116–24. doi:10.1210/jcem-40-1-116
59. Wolfe LK, Gordon RD, Island DP, Liddle GW. An analysis of factors determining the circadian pattern of aldosterone excretion. J Clin Endocrinol Metab (1966) 26(11):1261–6. doi:10.1210/jcem-26-11-1261
60. Newton MA, Laragh JH. Effects of glucocorticoid administration on aldosterone excretion and plasma renin in normal subjects, in essential hypertension and in primary aldosteronism. J Clin Endocrinol Metab (1968) 28(7):1014–22. doi:10.1210/jcem-28-7-1006
61. Schambelan M, Brust NL, Chang BC, Slater KL, Biglieri EG. Circadian rhythm and effect of posture on plasma aldosterone concentration in primary aldosteronism. J Clin Endocrinol Metab (1976) 43(1):115–31. doi:10.1210/jcem-43-1-115
62. Guthrie GP Jr. Multiple plasma steroid responses to graded ACTH infusions in patients with primary aldosteronism. J Lab Clin Med (1981) 98(3):364–73.
63. Stowasser M, Gordon RD, Klemm SA, Tunny TJ. Renin-aldosterone response to dexamethasone in glucocorticoid-suppressible hyperaldosteronism is altered by coexistent renal artery stenosis. J Clin Endocrinol Metab (1993) 77(3):800–4. doi:10.1210/jcem.77.3.8396580
64. Kem DC, Weinberger MH, Higgins JR, Kramer NJ, Gomez-Sanchez C, Holland OB. Plasma aldosterone response to ACTH in primary aldosteronism and in patients with low renin hypertension. J Clin Endocrinol Metab (1978) 46(4):552–60. doi:10.1210/jcem-46-4-552
65. Saruta T, Okuno T, Eguchi T, Nakamura R, Saito I, Kondo K, et al. Responses of aldosterone-producing adenomas to ACTH and angiotensins. Acta Endocrinol (Copenh) (1979) 92(4):702–9.
66. Sonoyama T, Sone M, Tamura N, Honda K, Taura D, Kojima K, et al. Role of endogenous ACTH on circadian aldosterone rhythm in patients with primary aldosteronism. Endocr Connect (2014) 3(4):173–9. doi:10.1530/EC-14-0086
67. Litchfield WR, New MI, Coolidge C, Lifton RP, Dluhy RG. Evaluation of the dexamethasone suppression test for the diagnosis of glucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab (1997) 82(11):3570–3. doi:10.1210/jcem.82.11.4381
68. Sutherland DJ, Ruse JL, Laidlaw JC. Hypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasone. Can Med Assoc J (1966) 95(22):1109–19.
69. Lifton RP, Dluhy RG, Powers M, Rich GM, Gutkin M, Fallo F, et al. Hereditary hypertension caused by chimaeric gene duplications and ectopic expression of aldosterone synthase. Nat Genet (1992) 2(1):66–74. doi:10.1038/ng0992-66
70. Sukor N, Mulatero P, Gordon RD, So A, Duffy D, Bertello C, et al. Further evidence for linkage of familial hyperaldosteronism type II at chromosome 7p22 in Italian as well as Australian and South American families. J Hypertens (2008) 26(8):1577–82. doi:10.1097/HJH.0b013e3283028352
71. Geller DS, Zhang J, Wisgerhof MV, Shackleton C, Kashgarian M, Lifton RP. A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab (2008) 93(8):3117–23. doi:10.1210/jc.2008-0594
72. Piaditis GP, Kaltsas GA, Androulakis II, Gouli A, Makras P, Papadogias D, et al. High prevalence of autonomous cortisol and aldosterone secretion from adrenal adenomas. Clin Endocrinol (Oxf) (2009) 71(6):772–8. doi:10.1111/j.1365-2265.2009.03551.x
73. Gouli A, Kaltsas G, Tzonou A, Markou A, Androulakis II, Ragkou D, et al. High prevalence of autonomous aldosterone secretion among patients with essential hypertension. Eur J Clin Invest (2011) 41(11):1227–36. doi:10.1111/j.1365-2362.2011.02531.x
74. Markou A, Pappa T, Kaltsas G, Gouli A, Mitsakis K, Tsounas P, et al. Evidence of primary aldosteronism in a predominantly female cohort of normotensive individuals: a very high odds ratio for progression into arterial hypertension. J Clin Endocrinol Metab (2013) 98(4):1409–16. doi:10.1210/jc.2012-3353
75. Papanastasiou L, Markou A, Pappa T, Gouli A, Tsounas P, Fountoulakis S, et al. Primary aldosteronism in hypertensive patients: clinical implications and target therapy. Eur J Clin Invest (2014) 44(8):697–706. doi:10.1111/eci.12286
76. Markou A, Sertedaki A, Kaltsas G, Androulakis II, Marakaki C, Pappa T, et al. Stress-induced aldosterone hyper-secretion in a substantial subset of patients with essential hypertension. J Clin Endocrinol Metab (2015) 100(8):2857–64. doi:10.1210/jc.2015-1268
77. Terui K, Kageyama K, Nigawara T, Moriyama T, Sakihara S, Takayasu S, et al. Evaluation of the (1-24) adrenocorticotropin stimulation test for the diagnosis of primary aldosteronism. J Renin Angiotensin Aldosterone Syst (2016) 17(1). doi:10.1177/1470320315625703
78. Tunny TJ, Klemm SA, Stowasser M, Gordon RD. Angiotensin-responsive aldosterone-producing adenomas: postoperative disappearance of aldosterone response to angiotensin. Clin Exp Pharmacol Physiol (1993) 20(5):306–9. doi:10.1111/j.1440-1681.1993.tb01690.x
79. Mantero F, Gion M, Armanini D, Opocher G. Aldosterone regulation in primary aldosteronism: differences between adenoma and bilateral hyperplasia. Clin Sci Mol Med Suppl (1976) 3:329s–32s.
80. Stowasser M, Klemm SA, Tunny TJ, Gordon RD. Plasma aldosterone response to ACTH in subtypes of primary aldosteronism. Clin Exp Pharmacol Physiol (1995) 22(6–7):460–2. doi:10.1111/j.1440-1681.1995.tb02044.x
81. Sonoyama T, Sone M, Miyashita K, Tamura N, Yamahara K, Park K, et al. Significance of adrenocorticotropin stimulation test in the diagnosis of an aldosterone-producing adenoma. J Clin Endocrinol Metab (2011) 96(9):2771–8. doi:10.1210/jc.2011-0573
82. Biglieri EG, Irony I, Kater CE. Identification and implications of new types of mineralocorticoid hypertension. J Steroid Biochem (1989) 32(1B):199–204. doi:10.1016/0022-4731(89)90164-7
83. Jiang Y, Zhang C, Wang W, Su T, Zhou W, Jiang L, et al. Diagnostic value of ACTH stimulation test in determining the subtypes of primary aldosteronism. J Clin Endocrinol Metab (2015) 100(5):1837–44. doi:10.1210/jc.2014-3551
84. Kline GA, Pasieka JL, Harvey A, So B, Dias VC. A marked proportional rise in IVC aldosterone following cosyntropin administration during AVS is a signal to the presence of adrenal hyperplasia in primary aldosteronism. J Hum Hypertens (2014) 28(5):298–302. doi:10.1038/jhh.2013.116
85. Young WF, Stanson AW. What are the keys to successful adrenal venous sampling (AVS) in patients with primary aldosteronism? Clin Endocrinol (Oxf) (2009) 70(1):14–7. doi:10.1111/j.1365-2265.2008.03450.x
86. Seccia TM, Miotto D, De Toni R, Pitter G, Mantero F, Pessina AC, et al. Adrenocorticotropic hormone stimulation during adrenal vein sampling for identifying surgically curable subtypes of primary aldosteronism: comparison of 3 different protocols. Hypertension (2009) 53(5):761–6. doi:10.1161/HYPERTENSIONAHA.108.128553
87. El Ghorayeb N, Mazzuco TL, Bourdeau I, Mailhot JP, Shi Zhu P, Therasse E, et al. Basal and post ACTH aldosterone and its ratios are useful during adrenal vein sampling in primary aldosteronism. J Clin Endocrinol Metab (2016) 101(4):1826–35. doi:10.1210/jc.2015-3915
88. Monticone S, Satoh F, Giacchetti G, Viola A, Morimoto R, Kudo M, et al. Effect of adrenocorticotropic hormone stimulation during adrenal vein sampling in primary aldosteronism. Hypertension (2012) 59(4):840–6. doi:10.1161/HYPERTENSIONAHA.111.189548
89. Reincke M, Beuschlein F, Latronico AC, Arlt W, Chrousos GP, Allolio B. Expression of adrenocorticotrophic hormone receptor mRNA in human adrenocortical neoplasms: correlation with P450scc expression. Clin Endocrinol (Oxf) (1997) 46(5):619–26. doi:10.1046/j.1365-2265.1997.1991009.x
90. Arnaldi G, Mancini V, Costantini C, Giovagnetti M, Petrelli M, Masini A, et al. ACTH receptor mRNA in human adrenocortical tumors: overexpression in aldosteronomas. Endocr Res (1998) 24(3–4):845–9. doi:10.3109/07435809809032695
91. Schubert B, Fassnacht M, Beuschlein F, Zenkert S, Allolio B, Reincke M. Angiotensin II type 1 receptor and ACTH receptor expression in human adrenocortical neoplasms. Clin Endocrinol (Oxf) (2001) 54(5):627–32. doi:10.1046/j.1365-2265.2001.01253.x
92. Ye P, Mariniello B, Mantero F, Shibata H, Rainey WE. G-protein-coupled receptors in aldosterone-producing adenomas: a potential cause of hyperaldosteronism. J Endocrinol (2007) 195(1):39–48. doi:10.1677/JOE-07-0037
93. Lefebvre H, Cartier D, Duparc C, Lihrmann I, Contesse V, Delarue C, et al. Characterization of serotonin(4) receptors in adrenocortical aldosterone-producing adenomas: in vivo and in vitro studies. J Clin Endocrinol Metab (2002) 87(3):1211–6. doi:10.1210/jcem.87.3.8327
94. Cartier D, Jégou S, Parmentier F, Lihrmann I, Louiset E, Kuhn JM, et al. Expression profile of serotonin4 (5-HT4) receptors in adrenocortical aldosterone-producing adenomas. Eur J Endocrinol (2005) 153(6):939–47. doi:10.1530/eje.1.02051
95. Gross MD, Grekin RJ, Gniadek TC, Villareal JZ. Suppression of aldosterone by cyproheptadine in idiopathic aldosteronism. N Engl J Med (1981) 305(4):181–5. doi:10.1056/NEJM198107233050401
96. Mantero F, Rocco S, Opocher G, Armanini D, Boscaro M, D’Agostino D. Effect of ketanserin in primary aldosteronism. J Cardiovasc Pharmacol (1985) 7(Suppl 7):S172–5. doi:10.1097/00005344-198500077-00048
97. Ehrhart-Bornstein M, Hinson JP, Bornstein SR, Scherbaum WA, Vinson GP. Intraadrenal interactions in the regulation of adrenocortical steroidogenesis. Endocr Rev (1998) 19(2):101–43. doi:10.1210/edrv.19.2.0326
98. Lefebvre H, Compagnon P, Contesse V, Delarue C, Thuillez C, Vaudry H, et al. Production and metabolism of serotonin (5-HT) by the human adrenal cortex: paracrine stimulation of aldosterone secretion by 5-HT. J Clin Endocrinol Metab (2001) 86(10):5001–7. doi:10.1210/jcem.86.10.7917
99. Soucek L, Lawlor ER, Soto D, Shchors K, Swigart LB, Evan GI. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat Med (2007) 13(10):1211–8. doi:10.1038/nm1649
100. Maltby S, Khazaie K, McNagny KM. Mast cells in tumor growth: angiogenesis, tissue remodelling and immune-modulation. Biochim Biophys Acta (2009) 1796(1):19–26. doi:10.1016/j.bbcan.2009.02.001
101. Duparc C, Moreau L, Dzib JF, Boyer HG, Tetsi Nomigni M, Boutelet I, et al. Mast cell hyperplasia is associated with aldosterone hypersecretion in a subset of aldosterone-producing adenomas. J Clin Endocrinol Metab (2015) 100(4):jc20143660. doi:10.1210/jc.2014-3660
102. Lampron A, Bourdeau I, Oble S, Godbout A, Schürch W, Arjane P, et al. Regulation of aldosterone secretion by several aberrant receptors including for glucose-dependent insulinotropic peptide in a patient with an aldosteronoma. J Clin Endocrinol Metab (2009) 94(3):750–6. doi:10.1210/jc.2008-1340
103. El Ghorayeb N, Bourdeau I, Lacroix A. Multiple aberrant hormone receptors in Cushing’s syndrome. Eur J Endocrinol (2015) 173(4):M45–60. doi:10.1530/EJE-15-0200
104. Lacroix A, Bourdeau I, Lampron A, Mazzuco TL, Tremblay J, Hamet P. Aberrant G-protein coupled receptor expression in relation to adrenocortical overfunction. Clin Endocrinol (Oxf) (2010) 73(1):1–15. doi:10.1111/j.1365-2265.2009.03689.x
105. Mazzuco TL, Grunenwald S, Lampron A, Bourdeau I, Lacroix A. Aberrant hormone receptors in primary aldosteronism. Horm Metab Res (2010) 42(6):416–23. doi:10.1055/s-0029-1243602
106. Lampronti I, Bianchi N, Zuccato C, Dall’acqua F, Vedaldi D, Viola G, et al. Increase in gamma-globin mRNA content in human erythroid cells treated with angelicin analogs. Int J Hematol (2009) 90(3):318–27. doi:10.1007/s12185-009-0422-2
107. Sealey JE, Itskovitz-Eldor J, Rubattu S, James GD, August P, Thaler I, et al. Estradiol- and progesterone-related increases in the renin-aldosterone system: studies during ovarian stimulation and early pregnancy. J Clin Endocrinol Metab (1994) 79(1):258–64. doi:10.1210/jcem.79.1.8027239
108. Chidambaram M, Duncan JA, Lai VS, Cattran DC, Floras JS, Scholey JW, et al. Variation in the renin angiotensin system throughout the normal menstrual cycle. J Am Soc Nephrol (2002) 13(2):446–52.
109. Saner-Amigh K, Mayhew BA, Mantero F, Schiavi F, White PC, Rao CV, et al. Elevated expression of luteinizing hormone receptor in aldosterone-producing adenomas. J Clin Endocrinol Metab (2006) 91(3):1136–42. doi:10.1210/jc.2005-1298
110. Szmuilowicz ED, Adler GK, Williams JS, Green DE, Yao TM, Hopkins PN, et al. Relationship between aldosterone and progesterone in the human menstrual cycle. J Clin Endocrinol Metab (2006) 91(10):3981–7. doi:10.1210/jc.2006-1154
111. Albiger NM, Sartorato P, Mariniello B, Iacobone M, Finco I, Fassina A, et al. A case of primary aldosteronism in pregnancy: do LH and GNRH receptors have a potential role in regulating aldosterone secretion? Eur J Endocrinol (2011) 164(3):405–12. doi:10.1530/EJE-10-0879
112. Nicolini G, Balzan S, Morelli L, Iacconi P, Sabatino L, Ripoli A, et al. LH, progesterone, and TSH can stimulate aldosterone in vitro: a study on normal adrenal cortex and aldosterone producing adenoma. Horm Metab Res (2014) 46(5):318–21. doi:10.1055/s-0033-1358733
113. Lacroix A, Tremblay J, Rousseau G, Bouvier M, Hamet P. Propranolol therapy for ectopic beta-adrenergic receptors in adrenal Cushing’s syndrome. N Engl J Med (1997) 337(20):1429–34. doi:10.1056/NEJM199711133372004
114. Mune T, Murase H, Yamakita N, Fukuda T, Murayama M, Miura A, et al. Eutopic overexpression of vasopressin v1a receptor in adrenocorticotropin-independent macronodular adrenal hyperplasia. J Clin Endocrinol Metab (2002) 87(12):5706–13. doi:10.1210/jc.2002-020067
115. Perraudin V, Delarue C, Lefebvre H, Do Rego JL, Vaudry H, Kuhn JM. Evidence for a role of vasopressin in the control of aldosterone secretion in primary aldosteronism: in vitro and in vivo studies. J Clin Endocrinol Metab (2006) 91(4):1566–72. doi:10.1210/jc.2005-1453
116. Fommei E, Iervasi G. The role of thyroid hormone in blood pressure homeostasis: evidence from short-term hypothyroidism in humans. J Clin Endocrinol Metab (2002) 87(5):1996–2000. doi:10.1210/jcem.87.5.8464
117. Rossi GP, Ganzaroli C, Cesari M, Maresca A, Plebani M, Nussdorfer GG, et al. Endothelin receptor blockade lowers plasma aldosterone levels via different mechanisms in primary aldosteronism and high-to-normal renin hypertension. Cardiovasc Res (2003) 57(1):277–83. doi:10.1016/S0008-6363(02)00658-2
Keywords: ACTH, aldosterone regulation, melanocortin type 2 receptor, aberrant G-protein coupled receptors, primary aldosteronism
Citation: El Ghorayeb N, Bourdeau I and Lacroix A (2016) Role of ACTH and Other Hormones in the Regulation of Aldosterone Production in Primary Aldosteronism. Front. Endocrinol. 7:72. doi: 10.3389/fendo.2016.00072
Received: 08 April 2016; Accepted: 09 June 2016;
Published: 27 June 2016
Edited by:
James A. Carr, Texas Tech University, USAReviewed by:
Chris J. Van Koppen, ElexoPharm GmbH, GermanyAlessandro Cannavo, Temple University, USA
Copyright: © 2016 El Ghorayeb, Bourdeau and Lacroix. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: André Lacroix, YW5kcmUubGFjcm9peEB1bW9udHJlYWwuY2E=