 Terry W. Moody1*
Terry W. Moody1* Nicole Tashakkori1
Nicole Tashakkori1 Samuel A. Mantey2Paola Moreno2Irene Ramos-Alvarez2
Samuel A. Mantey2Paola Moreno2Irene Ramos-Alvarez2 Marcello Leopoldo3
Marcello Leopoldo3 Robert T. Jensen2
Robert T. Jensen2
- 1Department of Health and Human Services, National Cancer Institute, Center for Cancer Research, Bethesda, MD, United States
- 2National Institute of Diabetes, Digestive and Kidney Disease, Digestive Diseases Branch, Bethesda, MD, United States
- 3Dipartimento di Farmacia, Scienze del Farmaco, Università degli Studi di Bari Aldo Moro, Bari, Italy
While peptide antagonists for the gastrin-releasing peptide receptor (BB2R), neuromedin B receptor (BB1R), and bombesin (BB) receptor subtype-3 (BRS-3) exist, there is a need to develop non-peptide small molecule inhibitors for all three BBR. The BB agonist (BA)1 binds with high affinity to the BB1R, BB2R, and BRS-3. In this communication, small molecule BBR antagonists were evaluated using human lung cancer cells. AM-37 and ST-36 inhibited binding to human BB1R, BB2R, and BRS-3 with similar affinity (Ki = 1.4–10.8 µM). AM-13 and AM-14 were approximately an order of magnitude less potent than AM-37 and ST-36. The ability of BA1 to elevate cytosolic Ca2+ in human lung cancer cells transfected with BB1R, BB2R, and BRS-3 was antagonized by AM-37 and ST-36. BA1 increased tyrosine phosphorylation of the EGFR and ERK in lung cancer cells, which was blocked by AM-37 and ST-36. AM-37 and ST-36 reduced the growth of lung cancer cells that have BBR. The results indicate that AM-37 and ST-36 function as small molecule BB receptor antagonists.
Introduction
The bombesin (BB) family of peptides is biologically active in the central nervous system (CNS) and periphery. BB, a 14 amino acid peptide isolated from frog skin, has 9 of the 10 same C-terminal amino acids as does human gastrin-releasing peptide (GRP), a 27 amino acid peptide (1). GRP binds with high affinity to the BB2R, which regulates pruritus, lung development, and gastrin secretion. Neuromedin B (NMB) is a 10 amino acid peptide with 70% sequence homology to the C-terminal of BB. NMB binds with high affinity to the BB1R and causes satiety, hypothermia, and thyrotropin (TSH) secretion from the pituitary (2). BB receptor subtype-3 (BRS-3) is an orphan receptor with homology to the BB1R and BB2R, and binds the universal agonist, BB agonist (BA)1, with high affinity as does the BB1R and BB2R (3). Because BRS-3 knockout mice have impaired energy balance, glucose homeostasis, and increased body weight, BRS-3 agonists may function as satiety agents (4). In the CNS, GRP and NMB may act in a paracrine manner being released from brain neurons in the hypothalamus and dentate gyrus, respectively, activating BB2R and BB1R in adjacent cells (5).
In numerous cancers, including lung cancer, GRP and NMB function in an autocrine manner to stimulate cellular proliferation. Small cell lung cancer (SCLC), a neuroendocrine tumor, has high levels of GRP (6, 7). GRP is secreted from SCLC and binds to cell surface BB2R resulting in increased cellular proliferation (8). NMB is present in both SCLC and non-small cell lung cancer (NSCLC) cells, and after secretion it binds to cell surface BB1R stimulating proliferation (9). Because many lung cancer cells have BB1R, BB2R, and/or BRS-3 there is a need to develop antagonists that block all three receptors of the BB family.
The human BB1R, BB2R, and BRS-3 contain 390, 384, and 399 amino acids and have approximately 50% sequence homology. The BB1R, BB2R, and BRS-3 are members of the rhodopsin β group G protein-coupled receptors (GPCR) family, and they interact with Gq causing phosphatidylinositol (PI) turnover (10). PI-4,5-bisphosphate (PIP2) is metabolized to diacylglycerol, which activates protein kinase C and inositol-trisphosphate (IP3) which causes elevated cytosolic Ca2+. Neuropeptide receptors regulate the transactivation of the epidermal growth factor (EGF) receptor leading to NSCLC proliferation (11). The proliferation of NSCLC cells caused by BA1 can be inhibited by the tyrosine kinase inhibitor (TKI) gefitinib or BBR antagonists. The actions of BA1 on BB1R, BB2R, and BRS-3 are antagonized selectively by PD168368, PD176252, and Bantag-1, respectively (12).
In the present study, small molecules were synthesized and their ability to antagonize BB1R, BB2R, and BRS-3 in lung cancer cells evaluated. The results indicate that AM-37 and ST-36 are useful agents to inhibit the growth of NSCLC cells which have BB1R, BB2R, or BRS-3.
Materials and Methods
Cell Culture
Non-small cell lung cancer cell line NCI-H1299 (ATCC, Manassas, VA, USA) was stably transfected with BB1R, BB2R, and BRS-3. The transfected cells were grown in RPMI-1640 containing 10% fetal bovine serum (FBS) with 0.3 mg/ml geneticin (Invitrogen, Grand Island, NY, USA). The transfected cells, which contained approximately 100,000 receptors/cell, were weekly split using trypsin/EDTA (13). In addition, lung cancer cell lines NCI-H727, H1299, and H1975 were purchased from ATCC and cultured in RPMI-1640, which contained 10% FBS. The cell types were derived from different human biopsy specimens. These studies were approved by the NIDDK biospecimens and biosafety committees.
Ligand Synthesis
The small molecules were synthesized as described previously (14). Figure 1D shows the structural formula of AM-37, (R)-3-(1H-indol-3-yl)-2-[3-(4-methoxyphenyl)ureido]-N-[[1-(3-pyridinyl)cyclohexyl]methyl]propanamide, and of its S-enantiomer ST-36. Figure 1E shows the structural formula of AM-13, (R)-N-[[1-(4-fluorophenyl)cyclohexyl]methyl]-3-(1H-indol-3-yl)-2-[3-(4-methoxyphenyl)ureido]propanamide, and its S-enantiomer AM-14. The molecular weight of AM-37 and ST-36 is 525.6 Da, whereas the molecular weight of AM-13 and AM-14 is 542.2 Da.
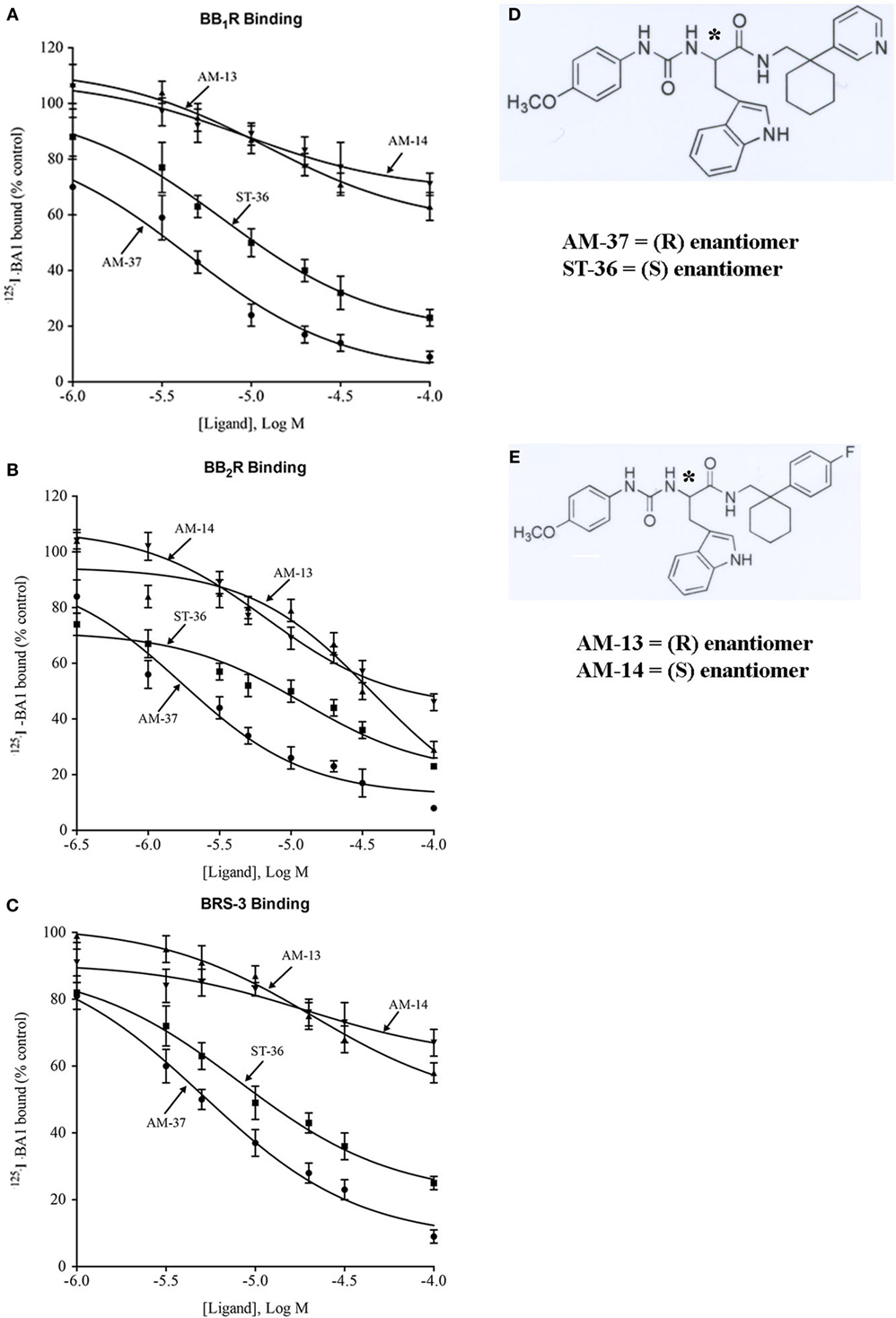
Figure 1. Binding. The ability of varying concentrations of AM-37 (●), ST-36 (■), AM-13 (▲), and AM-14 (▼) to inhibit specific 125I-BA1 binding was investigated using (A) BB1R-, (B) BB2R-, and (C) BRS-3-transfected NCI-H1299 cells. The mean value ± SD of three determinations each repeated in duplicate is shown. (D) The structure of AM-37 and ST-36 is shown. (E) The structure of AM-13 and AM-14 is shown; *indicates the optically active site.
Receptor Binding
The ability of AM-37, ST-36, AM-13, and AM-14 to inhibit specific 125I-BA1 binding to NSCLC cells transfected stably with BB1R, BB2R, and BRS-3 was investigated. NSCLC cells were placed in 24 well plates. When confluent, the cells were washed three times with PBS. The cells were incubated with binding buffer (PBS containing 0.25% bovine serum albumin and 0.025% bacitracin, Sigma-Aldrich, St. Louis, MO, USA). Various concentrations of AM-37, ST-36, AM-13, or AM-14 were added to the cells for 10 min, followed by 100,000 cpm of 125I-BA1 (0.16 nM) and incubated at 37°C for 30 min when equilibrium of binding was reached. The cells were rinsed three times with binding buffer for 2 min at 4°C. The cells that contained bound peptide dissolved in 0.2 N NaOH and counted in a Wallac 1470 γ-counter. The Ki was calculated as described (15).
Cytosolic Ca2+
The ability of AM-37, ST-36, AM-13, and AM-14 to function as BBR antagonists was investigated. NSCLC cells transfected with BB1R, BB2R, and BRS-3 were harvested and loaded with Fura-2AM (Calbiochem, La Jolla, CA, USA) as described previously (16). The excitation ratio was determined at 340 and 380 nm with an emission wavelength of 510 nm. The lung cancer cellular calcium response was determined after the addition of AM-37, ST-36, AM-13, or AM-14 followed by 10 nM BA1.
Tyrosine Phosphorylation
The tyrosine phosphorylation of the EGFR and ERK was investigated by western blot. NSCLC cells transfected with BB1R, BB2R, and BRS-3 were placed in 10 cm dishes. When the cells were confluent, they were placed in SIT medium (RPMI-1640 containing 3 × 10−8 M sodium selenite, 5 µg/ml bovine insulin, and 10 µg/ml apo-transferrin; Sigma-Aldrich, St. Louis, MO, USA) for 3 h. AM-37, ST-36, AM-13, or AM-14 were added for 30 min followed by 100 nM BA1 for 2 min. Cell extracts were made as described previously (16), and 600 µg of protein extract was immunoprecipitated with 4 µg anti-phosphotyrosine antibody (Becton Dickenson, USA). The immunoprecipitates were fractionated using a 4–20% polyacrylamide gel (Novex, San Diego, CA, USA). Proteins were transferred to a nitrocellulose membrane and incubated with 2 µg anti-EGFR or anti-ERK antibody (Cell Signaling Technologies, Danvers, MA, USA). After washing the blot, it was incubated with enhanced chemiluminescence detection reagent (Thermo Scientific) for 5 min and exposed to Biomax XAR film (Carestream, Rochester, NY, USA). The band intensity was determined using a Kodak image station 440 densitometer. Alternatively, 20 µg of protein extract was loaded onto polyacrylamide gels and after transfer to nitrocellulose, the blot was probed with anti-PY1,068-EGFR, anti-EGFR, anti-PY204ERK, or anti-ERK (Cell Signaling Technologies, Danvers, MA, USA).
Proliferation
The proliferation of NSCLC cells was investigated using the 3-(4,5-demethylthiazol-2-yl)-2,3-diphenyl-2H-tetrazolium bromide (MTT) assay as described previously (16). NCI-H727, H1299, and H1975 cells were placed in SIT medium and varying concentration of AM-37, ST-36, AM-13, or AM-14 added. After 2 days, 0.1% MTT solution (15 µl) was added. After 4 h, DMSO (150 µl) was added and the absorbance at 570 nm was determined.
Statistical Analysis
The results are expressed as the mean ± SD. Statistical significance of differences was performed by a one-way or two-way repeated measures of variance. The binding curves were drawn using PRISM.
Results
Receptor Binding
The ability of the small molecules to bind to BB1R, BB2R, and BRS-3 was investigated. AM-37 (R-enantiomer) inhibited specific 125I-BA1 binding to BB1R, BB2R, and BRS-3 in a dose-dependent manner with Ki values 3.6, 1.4, and 5.5 µM, respectively (Figure 1). ST-36 (S-enantiomer) inhibited specific 125I-BA1 binding to BB1R, BB2R, and BRS-3 with Ki values of 7.9, 6.9, and 10.8 µM, respectively (Figure 1). In contrast, AM-13 (R-enantiomer) and AM-14 (S-enantiomer) inhibited specific 125I-BA1 binding to BB1R, BB2R, and BRS-3 with Ki > 20 µM. The results indicate that AM-37and ST-36 bind to BB1R, BB2R, and BRS-3 with greater affinity than does AM-13 and AM-14.
The specificity of binding was investigated. Table 1 shows that BA1 bound with high affinity (Ki = 0.002, 0.0005, and 0.004 µM) to BB1R, BB2R, and BRS-3. AM-37, ST-36, AM-13, and AM-14 inhibited specific 125I-BA1 binding (Ki = 1.4, 6.9, 27, and 45 µM) to BB2R. ST-36 inhibited specific 125I-BA1 binding (Ki = 7.9 and 10.8 µM) to BB1R and BRS-3, respectively. AM-13 and AM-14 bind with low affinity to BB1R and BRS-3 (Ki > 100 µM and >100 μM, respectively).
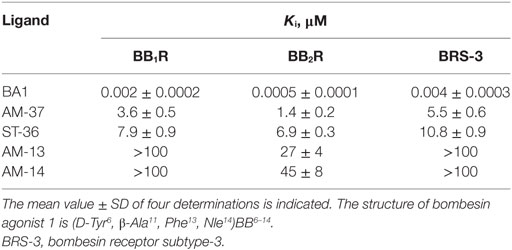
Table 1. Binding to lung cancer cells transfected with human bombesin receptors.
Cytosolic Ca2+
The ability of the small molecules to function as BB1R, BB2R, and BRS-3 antagonists was investigated. Addition of 10 nM BA1 to NCI-H1299 cells transfected with BB1R increased the cytosolic Ca2+ from 160 to 178 nM within seconds (Figure 2A). The response was transient and returned to baseline after 1 min. Addition of 30 µM AM-37 to NCI-H1299 cells transfected with BB1R had no effect on the basal cytosolic Ca2+ but blocked the increase in cytosolic Ca2+ caused by BA1 (Figure 2B). Addition of 30 µM AM-14 had no effect of basal cytosolic Ca2+ but partially blocked the increase caused by 10 nM BA1 (Figure 2C). Table 2 shows that AM-37 and AM-14 significantly inhibited the ability of BA1 to increase cytosolic Ca2+ after addition to NCI-H1299 cells transfected with BB1R. Addition of 10 nM BA1 to NCI-H1299 cells transfected with BB2R increased the cytosolic Ca2+ from 160 to 186 nM (Figure 2D). Addition of 30 µM ST-36 to NCI-H1299 cells transfected with BB2R had no effect on the basal cytosolic Ca2+ but blocked the increase in cytosolic Ca2+ caused by BA1 (Figure 2E). Addition of 30 µM AM-14 had no effect of basal cytosolic Ca2+ but partially blocked the increase caused by 10 nM BA1 (Figure 2F). Table 2 shows that ST-36 and AM-14 significantly decreased the ability of 10 nM BA1 to elevate cytosolic Ca2+ in NCI-H1299 cells transfected with BB2R. Addition of 10 nM BA1 to NCI-H1299 cells transfected with BRS-3 increased the cytosolic Ca2+ from 170 to 194 nM (Figure 2G). Addition of 30 µM ST-36 to NCI-H1299 cells transfected with BRS-3 had no effect on the basal cytosolic Ca2+ but blocked the increase in cytosolic Ca2+ caused by BA1 (Figure 2H). Addition of 30 µM AM-13 had no effect of basal cytosolic Ca2+ but partially blocked the increase caused by 10 nM BA1 (Figure 2I). Table 2 shows that ST-36 and AM-13 significantly decreased the ability of 10 nM BA1 to elevate cytosolic Ca2+ in NCI-H1299 cells transfected with BRS-3. The results indicate that AM-37 and ST-36 are antagonists for BB1R, BB2R, and BRS-3. In contrast, AM-13 and AM-14 are weak antagonists for the BBR family.
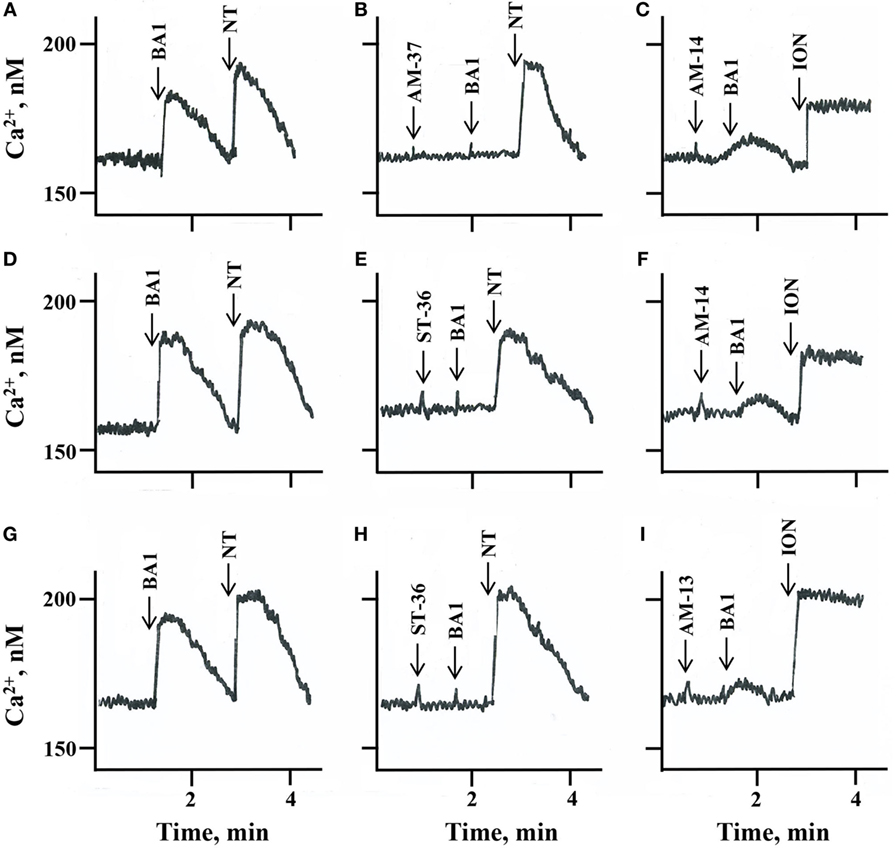
Figure 2. Cytosolic Ca2+. The ability of (A) 10 nM bombesin agonist 1 (BA)1 and 10 nM neurotensin (NT), (B) 30 µM AM-37 followed by 10 nM BA1 and 10 nM NT, and (C) 30 µM AM-14 followed by 10 nM BA1 and 5 µg/ml ionomycin (ION) to increase cytosolic Ca2+ was determined as a function of time after the addition to NCI-H1299 cells transfected with BB1R. The ability of (D) 10 nM BA1 and 10 nM NT, (E) 30 µM ST-36 followed by 10 nM BA1 and 10 nM NT, and (F) 30 µM AM-14 followed by 10 nM BA1 and 5 µg/ml ION to increase cytosolic Ca2+ was determined as a function of time after the addition to NCI-H1299 cells transfected with BB2R. The ability of (G) 10 nM BA1 and 10 nM NT, (H) 30 µM ST-36 followed by 10 nM BA1 and 10 nM NT, and (I) 30 µM AM-13 followed by 10 nM BA1 and 5 µg/ml ION to increase cytosolic Ca2+ was determined as a function of time after the addition to NCI-H1299 cells transfected with bombesin receptor subtype-3. This experiment is representative of three others.
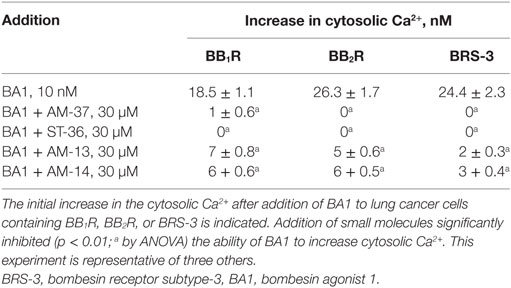
Table 2. Increases in cytosolic Ca2+ using human lung cancer cells transfected with bombesin receptors.
The specificity of AM-37, ST-36, AM-13, and AM-14 was investigated. 10 nM neurotensin (NT) or 5 µg/ml ionomycin (ION) strongly increased the cytosolic Ca2+ in NSCLC cells. AM-37 or ST-36 had no effect on the ability of NT to increase cytosolic Ca2+ in NSCLC cells. AM-13 or AM-14 had no effect on the ability of ION to increase Ca2+ in NSCLC cells. Therefore, AM-36 and ST-37 are antagonists for the BBR but not the NTR.
Tyrosine Phosphorylation
The ability of the small molecules to impair EGFR transactivation was investigated. Previously, we found that the BB1R and BRS-3 regulate EGFR tyrosine phosphorylation (13, 16). Figure 3 shows that addition of 100 nM BA1 to NCI-H1299 cells transfected with BB2R increased significantly the EGFR tyrosine phosphorylation to 326%. If the cells were pretreated with 10 µM AM-37 or ST-36, addition of BA1 had little effect. In contrast, if the cells were treated with 10 µM AM-13, BA1 increased strongly EGFR tyrosine phosphorylation. Similarly, BA1 addition to NCI-H1299 cells transfected with BB2R increased ERK tyrosine phosphorylation to 277%. This increase in ERK tyrosine phosphorylation was decreased significantly in the cells pretreated with AM-37 or ST-36 but not AM-13. Similarly, AM-14 had little effect on EGFR or ERK tyrosine phosphorylation (data not shown). The results indicate that AM-37 and ST-36 antagonize the ability of the BB2R to regulate tyrosine phosphorylation of the EGFR and ERK. Similar transactivation results were obtained for NSCLC cells transfected with BB1R or BRS-3 (data not shown).
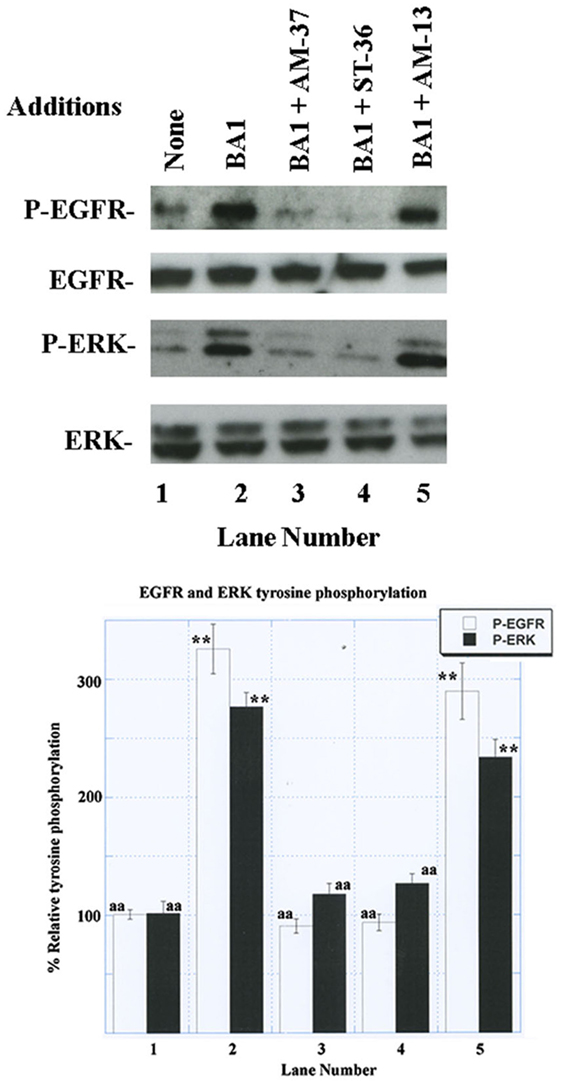
Figure 3. Western blot. (Top) The ability of 100 nM bombesin agonist (BA)1 to increase EGFR and ERK tyrosine phosphorylation was investigated using NCI-H1299 cells transfected with BB2R in the presence of 10 µM AM-37, 10 µM ST-36, and 10 µM AM-13. (Bottom) BA1 or BA1 plus AM-13 increased significantly EGFR and ERK tyrosine phosphorylation relative to the control, whereas total ERK and EGFR were unaltered; p < 0.01; ** by ANOVA. The control, BA1 + AM-37, and BA1 + ST-36 were significantly reduced relative to BA1; p < 0.01, aa by ANOVA. The experiment is representative of three others.
Proliferation
The ability of the small molecules to inhibit lung cancer proliferation was investigated. AM-37 inhibited NCI-H1299 proliferation in a dose-dependent manner. Figure 4 shows that AM-37 had little effect at 3 µM but strongly inhibited proliferation at 30 µM. The IC50 for AM-37 was 16 µM. Similarly, ST-36 had an IC50 of 22 µM, whereas AM-14 was less potent (IC50 > 50 µM).
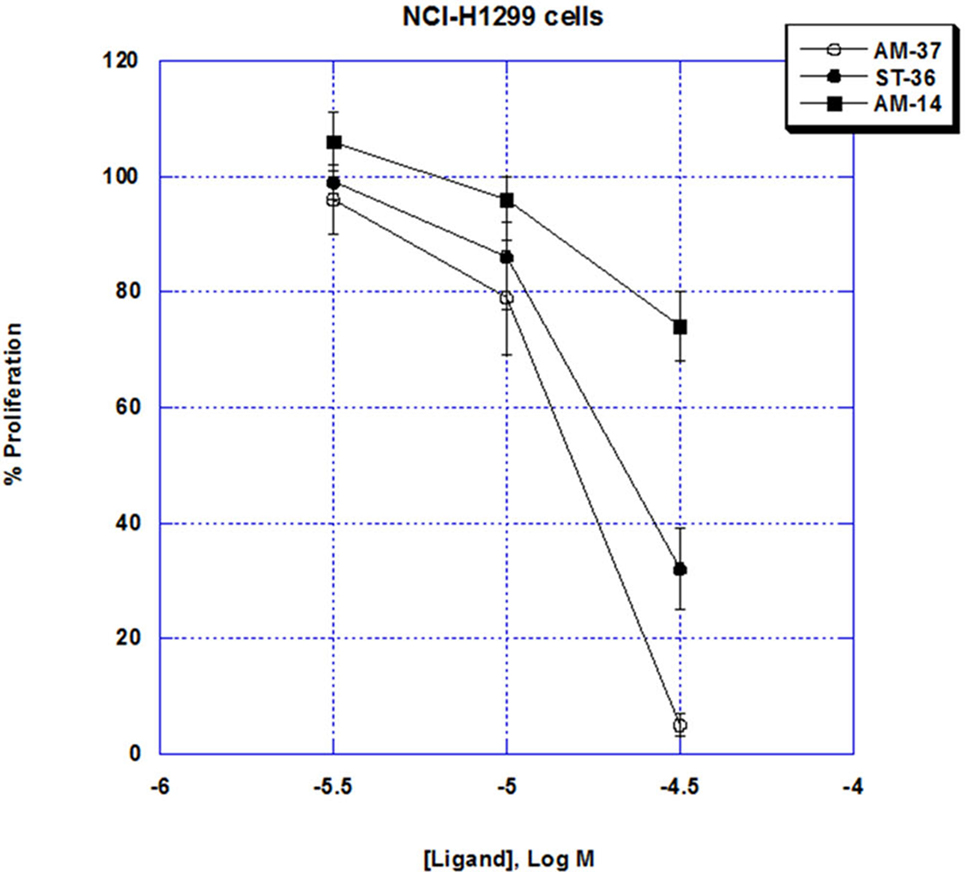
Figure 4. MTT assay. The ability of varying doses of AM-37 (○), ST-36 (●), and AM-14 (■), to inhibit the proliferation of NCI-H1299 cells is shown. The mean value ± SD of eight determinations is indicated. This experiment is representative of two others.
The specificity of the small molecules was investigated. Table 3 shows that AM-37, ST-36, AM-13, and AM-14 (50 µM) inhibited significantly the proliferation of NCI-H727 cells, which have mRNA for BB1R, BB2R, and BRS-3. In contrast, AM-37, ST-36, AM-13, and AM-14 had little effect on NCI-H1975 cells, which lack BB1R, BB2R, and BRS-3. These results indicate that the BBR is essential for AM-37, ST-36, AM-13, or AM-14 to inhibit cancer cellular proliferation.

Table 3. MTT proliferation assay using human lung cancer cell lines.
Discussion
While NSCLC patients are traditionally treated with combination chemotherapy, the 5-year survival rate is only 16% (17). Some NSCLC patients (13%) have L858R EGFR mutations, and these patients respond to TKI such as gefitinib or erlotinib; however, secondary EGFR mutations can occur such as T790M resulting in TKI resistance (18). Numerous GPCR are expressed in lung cancer cell lines and biopsy specimens. BB2R mRNA is expressed in 46–67% of the lung cancer cell lines examined (19). BB1R mRNA is present in 81% of the NSCLC cell lines examined (20). Using autoradiographic techniques, BRS-3 binding sites were detected in 40% of the lung cancer biopsy specimens examined (21). The EGFR is abundant on NSCLC (approximately 100,000 EGFR/cell), whereas BBR are present on most native NSCLC cells (approximately 2,000 BBR/cell) (22).
Addition of GRP to NSCLC cells causes transactivation of the EGFR (23). The effects of GRP on NSCLC tyrosine phosphorylation of the EGFR are impaired by gefitinib, a TKI, and PD176252, a peptoid BB2R antagonist. Because the ERK and EGFR tyrosine phosphorylation caused by GRP was impaired by marimastat, GM6001 and antibodies to TGFα, matrix metalloproteases may regulate the cellular shedding of TGFα from NSCLC cells. The TGFα may then bind to the EGFR causing its tyrosine phosphorylation. The results indicate that the BB2R regulates EGFR transactivation in NSCLC cells.
The BB1R regulates EGFR transactivation (16). The increase in EGFR and ERK tyrosine phosphorylation caused by NMB addition to NSCLC cells was impaired by PD168368, a BB1R peptoid antagonist, as well as gefitinib. The increase in EGFR tyrosine phosphorylation caused by NMB was impaired by N-acetyl cysteine (NAC), an antioxidant, or tiron, a superoxide scavenger. NMB increased reactive oxygen species (ROS) in NSCLC cells, and the increase was inhibited by Tiron. It remains to be determined if the ROS impair protein tyrosine phosphatases in NSCLC cells, which remove phosphate from the P-EGFR. Activation of BRS-3 with BA1 increased EGFR and ERK tyrosine phosphorylation (13). The increase in EGFR tyrosine phosphorylation caused by BA1 is impaired by NAC, tiron, and diphenyleneiodonium, an inhibitor of NADPH oxidase enzymes.
ML-18 is a small molecule that prefers BRS-3 relative to BB1R or BB2R (24). ML-18, an S-enantiomer, inhibits 125I-BA1 binding to BRS-3, BB2R, and BB1R with IC50 values of 4.8, 16, and >100 μM, respectively, whereas the R-enantiomer EMY-98 is inactive. ML-18 is a BRS-3 antagonist, which inhibits the ability of BA1 to increase cytosolic Ca2+, increase ERK and EGFR tyrosine phosphorylation (24). Also, ML-18 inhibited NSCLC growth and increased the cytotoxicity of gefitinib. His107 is important for BRS-3 to bind antagonists with high affinity (25). Tyr101 of the BB2R is important for binding of non-peptide antagonists (26). Similarly, this Tyr is conserved in the BB1R and BRS-3. It remains to be determined if this Tyr is essential for binding of AM-37 to the BB1R, BB2R, or BRS-3. ST-36, which is an S-enantiomer, inhibited specific 125I-BA1 binding to BB1R, BB2R, and BRS-3 with IC50 values of 7.9, 6.9, and 10.8 µM, respectively. It is surprising that AM-37, which is the R-enantiomer, binds with slightly higher affinity to BBR than does ST-36. Previously, the BB1R was found to prefer PD168,368, which is an S-isomer, relative to the R-isomer (27).
(D-Arg1, D-Trp5,7,9, Leu11)substance P (SP) is an inhibitor of signal transduction and growth of SCLC cells (28). (D-Arg1, D-Trp5,7,9, Leu11)SP impaired the ability of BB, vasopressin, or bradykinin to increase cytosolic Ca2+ and ERK activity. (D-Arg1, D-Trp5,7,9, Leu11)SP decreased SCLC growth in vitro, and (D-Arg1, D-Trp5,7,9, Leu11)SP has a unique tertiary structure in with two type IV non-standard turns, which juxtapose the N- and C-terminal adjacent to one another (29). Due to this unique structure (D-Arg1, D-Trp5,7,9, Leu11)SP may be able to interact with multiple GPCR. In contrast, AM-37 and ST-36 are small molecules that have a different structure from that of (D-Arg1, D-Trp5,7,9, Leu11)SP.
AM-37 and ST-36 inhibited the proliferation of NSCLC cells such as NCI-H1299 and H727, which have BB1R, BB2R, or BRS-3. In contrast, AM-37 and ST-36 have little effect on NSCLC cell line NCI-H1975, which lacks BB1R, BB2R, and BRS-3. It remains to be determined if AM-37 or ST-36 are synergistic with gefitinib at inhibiting the growth of NSCLC. A goal is to identify GPCR antagonists, which potentiate the action of TKI in NSCLC patients.
Conclusion
AM-37 and ST-36 are small molecules, which bind to the BB1R, BB2R, and BRS-3. Because AM-37 and ST-36 inhibit the ability of BA1 to increase cytosolic Ca2+ as well as increase EGFR and ERK tyrosine phosphorylation, they function as BB1R, BB2R, and BRS-3 antagonists. A particular advantage of AM-37 and ST-36 is that they will inhibit the growth of NSCLC cells if they have BB1R, BB2R, or BRS-3.
Author Contributions
TM and SM were responsible for the receptor binding studies. TM and NT were responsible for the cell culture and calcium experiments. TM, PM, and IR-A were responsible for the transactivation and growth experiments. ML was responsible for the synthesis of the small molecules. TM and RJ were responsible for the writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Acknowledgments
The authors thank Drs. M. Nicklaus, E. Lacivita, D. Venzon, and M. Peach for helpful discussions.
Funding
This research was supported by the intramural programs of the NCI and NIDDK of the NIH.
References
1. Moreno P, Ramos-Alvarez I, Moody TW, Jensen RT. Bombesin related peptides/receptors and their promising therapeutic roles in cancer imaging, targeting and treatment. Expert Opin Ther Targets (2016) 20:1055–73. doi:10.1517/14728222.2016.1164694
2. Ramos-Alvarez I, Moreno P, Mantey SA, Nakamura T, Nuche-Berenguer B, Moody TW, et al. Insights into bombesin receptors and ligands: highlighting recent advances. Peptides (2015) 72:128–44. doi:10.1016/j.peptides.2015.04.026
3. Mantey SA, Weber HC, Sainz E, Akeson M, Ryan RR, Pradhan TK, et al. Discovery of a high affinity radioligand for the human orphan receptor, bombesin receptor subtype3, which demonstrates it has a unique pharmacology compared to other mammalin bombesin receptors. J Biol Chem (1997) 272:26062–71. doi:10.1074/jbc.272.41.26062
4. Majumdar ID, Weber HC. Appetite-modifying effects of bombesin receptor subtype-3 agonists. Handb Exp Pharmacol (2012) 209:405–32. doi:10.1007/978-3-642-24716-3_19
5. Moody TW, Merali Z. Bombesin-like peptides and associated receptors within the brain: distribution and behavioral implications. Peptides (2004) 25:511–20. doi:10.1016/j.peptides.2004.02.012
6. Moody TW, Pert CB, Gazdar AF, Carney DN, Minna JD. High levels of intracellular bombesin characterize human small-cell lung carcinoma. Science (1981) 214:1246–8. doi:10.1126/science.6272398
7. Wood SM, Wood JR, Ghatei MA, Lee YC, O’Shaughnessy D, Bloom SR. Bombesin, somatostatin and neurotensin-like immunoreactivity in bronchial carcinoma. J Clin Endocrinol Metab (1981) 53:1310–2. doi:10.1210/jcem-53-6-1310
8. Cuttitta F, Carney DN, Mulshine J, Moody TW, Fedorko J, Fischler A, et al. Bombesin-like peptides can function as autocrine growth factors in human small cell lung cancer. Nature (1985) 316:823–6. doi:10.1038/316823a0
9. Giaccone G, Battey J, Gazdar AF, Oie H, Draoui M, Moody TW. Neuromedin B is present in lung cancer cell lines. Cancer Res (1993) 52:2732s–6s.
10. Moody TW, Moreno P, Jensen RT. Neuropeptides as lung cancer growth factors. Peptides (2015) 72:106–11. doi:10.1016/j.peptides.2015.03.018
11. Moody TW, Nuche-Berenguer B, Nakamura T, Jensen RT. EGFR transactivation by peptide G protein-coupled receptors in cancer. Curr Drug Targets (2016) 17:520–8. doi:10.2174/1389450116666150107153609
12. Jensen RT, Battey JF, Spindel ER, Benya RV. Mammalian bombesin receptors: nomenclature, distribution, pharmacology, signaling and functions in normal and disease states. Pharmacol Rev (2008) 60:1–42. doi:10.1124/pr.107.07108
13. Moody TW, Sancho V, Di Florio A, Nuche-Berenguer B, Mantey SA, Jensen RT. Bombesin receptor subtype-3 agonists stimulate the growth of lung cancer cells and increase EGF receptor tyrosine phosphorylation. Peptides (2011) 32:1677–84. doi:10.1016/j.peptides.2011.06.011
14. Lacivita E, Schepetkin IA, Stama ML, Kirpotina LN, Colabufo NA, Perrone R, et al. Novel 3-(1H-indoyl-3-yl)-2-[3-(4-methoxyphenyl)ureido]propanamides as selective agonist of human formyl-peptide receptor2. Bioorg Med Chem (2015) 23:3913–24. doi:10.1016/j.bmc.2014.12.007
15. Chang Y, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem Pharmacol (1973) 22:3099–108. doi:10.1016/0006-2952(73)90196-2
16. Moody TW, Berna MJ, Mantey S, Sancho V, Ridnour L, Wink DA, et al. Neuromedin B receptors regulate EGF receptor tyrosine phosphorylation in lung cancer cells. Eur J Pharmacol (2010) 637:38–45. doi:10.1016/j.ejphar.2010.03.057
17. Kaufman J, Horn L, Carbone D. Molecular biology of lung cancer. In: DeVita W Jr, Lawrence T, Rosenberg SA, editors. Cancer: Principles and Practice of Oncology. Philadelphia: Lippincott, Williams & Wilkens (2011). p. 789–98.
18. Lopes GL, Vattimo EF, Castro Junior GD. Identifying activating mutations in the EGFR gene: prognostic and therapeutic implications in non-small cell lung cancer. J Bras Pneumol (2015) 41:365–75. doi:10.1590/S1806-37132015000004531
19. Corjay M, Dobrzanski DJ, Way JM, Viallet J, Shapira H, Worland P, et al. Two distinct bombesin receptor subtypes are expressed and functional in human lung carcinoma cells. J Biol Chem (1991) 266:18771–9.
20. Siegfried JM, Krishnamachary N, Gaither Davis A, Gubish C, Hunt JD, Shriver SP. Evidence for autocrine actions of neuromedin B and gastrin-releasing peptide in non-small cell lung cancer. Pulm Pharmacol Ther (1999) 12:291–302. doi:10.1006/pupt.1999.0210
21. Reubi JC, Wenger S, Schmuckli-Mauer J, Schaer JC, Gugger M. Bombesin receptor subtypes in human cancers: detection with the universal radioligand 125I-[D-Tyr6, β-Ala11, Phe13, Nle14]bombesin(6-14). Clin Cancer Res (2002) 8:11139–46.
22. Moody TW, Lee M, Kris RM, Bellot F, Bepler G, Oie H, et al. Lung carcinoid cell lines have bombesin-like peptides and EGF receptors. J Cell Biochem (1990) 43:139–47. doi:10.1002/jcb.240430205
23. Thomas SM, Grandis JR, Wentzel AL, Gooding WE, Lui VWY, Siegfried JM. Gastrin-releasing peptide receptor mediates activation of the epidermal growth factor receptor in lung cancer cells. Neoplasia (2006) 7:426–31. doi:10.1593/neo.04454
24. Moody TW, Mantey SA, Moreno P, Nakamura T, Lacivita E, Leopoldo M, et al. ML-18 is a non-peptide bombesin receptor subtype-3 antagonist which inhibits lung cancer growth. Peptides (2015) 64:55–61. doi:10.1016/j.peptides.2014.12.005
25. Nakamura T, Ramos-Alvarez I, Iordanskaia T, Moreno P, Mantey SA, Jensen RT. Molecular basis for high affinity and selectivity of peptide antagonists, Bantag-1, for the orphan BB3 receptor. Biochem Pharmacol (2016) 115:64–76. doi:10.1016/j.bcp.2016.06.013
26. Carrieri A, Lacivita E, Danilo Belviso B, Caliandro R, Mastrorilli P, Gallo V, et al. Structural determinants in the binding of BB2 receptor ligands: in silico, x-ray and NMR studies in PD176252 analogues. Curr Top Med Chem (2017) 17:1599–610. doi:10.2174/1568026617666161104102459
27. Ashwood V, Brownhill V, Higginbottom M, Horwell D, Hughes J, Lewthwaite A, et al. PD176252-the first high affinity non-peptide gastrin-releasing peptide (BB2) receptor antagonist. Bioorg Med Chem Lett (1996) 8:2589–94. doi:10.1016/S0960-894X(98)00462-4
28. Seckl MJ, Higgins T, Widner F, Rozengurt E. [D-Arg1, D-Trp5,7,9, Leu11] substance P, A novel potent inhibitor of signal transduction and growth in vitro and in vivo in small cell lung cancer cells. Cancer Res (1997) 57:51–4.
Keywords: small molecule antagonists, GRPR, NMBR, bombesin receptor subtype-3, lung cancer
Citation: Moody TW, Tashakkori N, Mantey SA, Moreno P, Ramos-Alvarez I, Leopoldo M and Jensen RT (2017) AM-37 and ST-36 Are Small Molecule Bombesin Receptor Antagonists. Front. Endocrinol. 8:176. doi: 10.3389/fendo.2017.00176
Received: 30 November 2016; Accepted: 05 July 2017;
Published: 21 July 2017
Edited by:
Hubert Vaudry, University of Rouen, FranceReviewed by:
Miriam Goebel-Stengel, HELIOS Klinik Zerbst, GermanyJana Sopkova-de Oliveira Santos, Centre d’Etudes et de Recherche sur le Médicament de Normandie (CERMN), France
Copyright: © 2017 Moody, Tashakkori, Mantey, Moreno, Ramos-Alvarez, Leopoldo and Jensen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Terry W. Moody, bW9vZHl0QG1haWwubmloLmdvdg==