 Stefanie Fruhwürth
Stefanie Fruhwürth Heike Vogel
Heike Vogel Annette Schürmann
Annette Schürmann Kevin Jon Williams
Kevin Jon Williams- 1Department of Molecular and Clinical Medicine, Wallenberg Laboratory, Sahlgrenska University Hospital, Gothenburg, Sweden
- 2Department of Experimental Diabetology, German Institute of Human Nutrition Potsdam-Rehbruecke, Nuthetal, Germany
- 3German Center for Diabetes Research (DZD), München-Neuherberg, Germany
- 4Department of Medicine, Section of Endocrinology, Diabetes, and Metabolism, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, United States
Obesity has become a worldwide health problem, but we still do not understand the molecular mechanisms that contribute to overeating and low expenditure of energy. Leptin has emerged as a major regulator of energy balance through its actions in the hypothalamus. Importantly, obese people exhibit high circulating levels of leptin, yet the hypothalamus no longer responds normally to this hormone to suppress appetite or to increase energy expenditure. Several well-known hypotheses have been proposed to explain impaired central responsiveness to the effects of leptin in obesity, including defective transit across the blood–brain barrier at the arcuate nucleus, hypothalamic endoplasmic reticulum stress, maladaptive sterile inflammation in the hypothalamus, and overexpression of molecules that may inhibit leptin signaling. We also discuss a new explanation that is based on our group’s recent discovery of a signaling pathway that we named “NSAPP” after its five main protein components. The NSAPP pathway consists of an oxide transport chain that causes a transient, targeted burst in intracellular hydrogen peroxide (H2O2) to inactivate redox-sensitive members of the protein tyrosine phosphatase gene family. The NSAPP oxide transport chain is required for full activation of canonical leptin signaling in neurons but fails to function normally in states of overnutrition. Remarkably, leptin and insulin both require the NSAPP oxide transport chain, suggesting that a defect in this pathway could explain simultaneous resistance to the appetite-suppressing effects of both hormones in obesity.
Introduction
In just the past few decades, obesity has become a worldwide health problem. The underlying cause is excessive food intake and a sedentary lifestyle, resulting in a chronic positive energy imbalance. The maintenance of a healthy energy balance is essential for the prevention of obesity. Successful strategies to achieve sustained weight loss to reverse obesity must address appetite and food intake as well as energy expenditure. Why has keeping a healthy energy balance become so difficult recently?
A major regulator of appetite and hence food intake is the adipocyte-derived hormone leptin (1, 2). Because blood levels of leptin rise chronically in proportion to body fat mass (3), this hormone indicates caloric prosperity. In addition, blood concentrations of leptin concentrations rise acutely after a carbohydrate-containing meal, apparently stimulated by insulin (4, 5). Accordingly, leptin enters the central nervous system (CNS) where it provokes specific neuronal signals in the hypothalamus that mediate leptin’s appetite-suppressing effects (6, 7). In addition to these direct homeostatic actions in the hypothalamus, leptin has gained recognition as a modulator of neural circuits governing motivation and reward (8–10). Leptin acts via the mesolimbic dopaminergic “reward system” to suppress the motivational drive to seek and consume food (11, 12). Leptin also triggers responses in the CNS that can increase energy expenditure, e.g., via activation of non-shivering thermogenesis in brown adipose tissue (BAT) and the induction of BAT-like thermogenesis in white adipose tissue (WAT). This latter process is known colloquially as “browning” and is discussed in more detail below.
Obese individuals typically exhibit high circulating levels of leptin because overall fat mass is increased, yet the hypothalamus no longer responds normally to leptin to suppress appetite (13–15). The exact molecular mechanisms responsible for the poor response to leptin in the brain remain unknown. Here, we review several well-known hypotheses that have been proposed to explain impaired central responsiveness to the appetite-suppressing effects of leptin in obesity. We also discuss a new explanation that is based on our group’s recent discovery of a signaling pathway—the NSAPP oxide transport chain—that is required for full activation of canonical leptin signaling in neurons but fails to function normally in states of overnutrition.
Central Regulation of Energy Balance—Normal Canonical Effects of Leptin
The first studies showing that a hormone from the circulation regulates feeding centrally took advantage of a pair of spontaneous mutant mice with severe heritable hyperphagia and obesity (16). One of these obese mice, ob/ob, was later used to identify the leptin gene, where the obesogenic mutation resides (2). The other obese mouse, db/db, carries two copies of a defective allele in the gene encoding the long form of the leptin receptor, LepRb (Leprdb/db). The db/db mouse is a key animal model of overnutrition, obesity, and type 2 diabetes mellitus. Restoration of CNS expression of functional LepRb in db/db mice is sufficient to normalize food intake and reverse their obesity (17).
The primary CNS site involved in the regulation of appetite by leptin is the arcuate nucleus of the hypothalamus (ARC). Within the ARC, leptin acts on LepRb, to inhibit neurons that express the orexigenic (appetite-stimulating) neuropeptide agouti-related peptide (AgRP), while simultaneously stimulating nearby neurons that express the anorexigenic (appetite-suppressing) neuropeptide proopiomelanocortin (POMC). Both of these actions work together to reduce food intake (6, 7).
Although leptin drives AgRP and POMC expression in opposite directions, the hormone acts in those two types of ARC neurons primarily via the same signaling cascades. Binding of leptin to the LepRb, which has no intrinsic kinase activity, causes the receptor to recruit intracellular Janus kinase 2 (JAK2). The recruitment of JAK2 in turn activates diverse signal transduction cascades through autophosphorylation and phosphorylation of LepRb and signal transducer and activator of transcription 3 (STAT3) (blue in Figure 1). Phosphorylation of STAT3 activates it, to silence the AgRP gene and drive expression of the Pomc gene. Leptin–LepRb binding also activates intracellular phosphoinositide-3 kinase (PI3K) (18) and causes phosphorylation and hence nuclear exclusion of the transcription factor FOXO1 (19). Consistent with this model, unphosphorylated FOXO1 and phosphorylated STAT3 appear to act on the Agrp and Pomc promoters through squelching, meaning that the two proteins compete for binding to overlapping sites within the two promoters (19). Both JAK2–STAT3 and PI3K have been implicated in leptin’s anorexigenic effect [reviewed in Ref. (20, 21)].
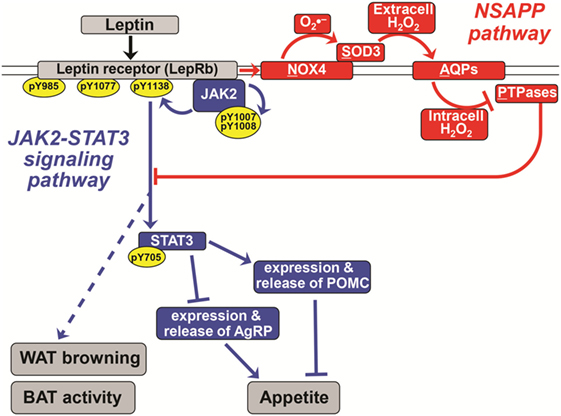
Figure 1. A novel pathway essential for canonical leptin signaling in hypothalamic neurons. Leptin activates signaling pathways to control the expression of appetite-regulating neuropeptides [proopiomelanocortin (POMC) and agouti-related peptide (AgRP)] in hypothalamic neurons. The canonical Janus kinase 2 (JAK2)–signal transducer and activator of transcription 3 (STAT3) signaling pathway is shown in blue. In red is the newly discovered NSAPP pathway that we showed to be essential for producing an intracellular burst of H2O2 that inhibits key protein tyrosine phosphatases (PTPases), thereby allowing canonical leptin signaling to propagate. Normal leptin signaling is required to suppress appetite and to stimulate white adipose tissue (WAT) browning and brown adipose tissue (BAT) thermogenic activity. Pointed arrowheads indicate stimulation of the immediately downstream molecule or process; flat arrowheads indicate inhibition. Specific phosphorylation sites are indicated within yellow ovals. The double line indicates the plasma membrane; all molecules above it are extracellular, and all named molecules below it in the schematic are intracellular. The leptin receptor (LepRb), NADPH oxidase-4 (NOX4), and aquaporins are transmembrane molecules. Solid lines represent experimentally demonstrated pathways; dotted lines are less well-characterized or putative. Adapted from Ref. (21–23).
Regarding energy expenditure, leptin activates additional pathways in the brain that trigger efferent outflow to adipose tissue to alter its metabolism. Thus, abundant adipose tissue produces leptin, a prosperity signal to the brain, and then the brain sends neuronal signals back to adipose tissue to increase energy expenditure. Importantly, there are two general types of adipose tissue in humans and other mammals. WAT stores chemical energy as triacylglycerols for use during periods of fasting or starvation. BAT also contains triacylglycerols but is enriched in mitochondria and adrenergic (sympathetic) innervation, both of which contribute to its brown color. BAT expresses uncoupling protein-1 (UCP1), which causes its mitochondria to dissipate the energy from oxidation of macronutrients as heat rather than harnessing this energy to make ATP. In human infants and in small rodents, which have high surface area-to-volume ratios, a major function of BAT is to maintain body temperature during cold exposure. Research during recent years provided unequivocal evidence for the existence of metabolically active BAT in normal human adults as well (24–26).
Leptin action in the hypothalamus, in part through local activation of the extracellular signal-regulated kinase (ERK) (27), increases sympathetic outflow to β3-adrenergic receptors on BAT adipocytes, thereby stimulating UCP1 expression and thermogenic activity (28). Several hypothalamic nuclei have been shown to be involved in activation of BAT and browning of WAT by leptin and other factors [reviewed in (29)]. Leptin acts in the mediobasal hypothalamus also to suppress WAT lipogenesis in an STAT3-independent manner (30). The fact that db/db mice (17) and leptin-deficient ob/ob mice (Lepob/ob) (31) are cold intolerant supports a key role for leptin in heat production. Restoration of CNS expression of functional LepRb in db/db mice also restores cold tolerance (17). Abundance and activity of BAT or brown-like fat is reduced or absent in obese humans (25, 32), suggesting that energy expenditure could be substantially increased in these individuals if therapies could be found to stimulate BAT thermogenic activity and the browning of WAT (33). Owing to issues of safety and efficacy, however, no pharmacologic agents (34, 35) or devices (36) that were designed primarily to stimulate BAT have yet achieved regulatory approval for weight loss. Some data indicate pharmacological activation of the glucagon-like peptide-1 (GLP-1) system might increase BAT thermogenesis. For example, central administration of GLP-1 and GLP-1 receptor agonists has been shown to increase BAT thermogenesis in mice (37, 38).
Stimulation of WAT to increase their content of mitochondria and UCP1, a process called browning, has been shown to increase energy expenditure and suppress high-fat diet-induced obesity in rodents (39–41). Recently, Dodd and colleagues demonstrated that leptin acts on POMC neurons to promote the browning of WAT (42). WAT browning has been demonstrated in humans as well (43). Per gram of tissue, WAT after browning shows a lower thermogenic capacity compared with BAT (44). Nevertheless, there is much more WAT mass than BAT mass, suggesting that browning of WAT could substantially increase overall energy expenditure.
Defective Central Responses to Leptin in Obesity
As noted earlier, obese individuals typically exhibit hyperleptinema, yet this overabundant leptin loses its normal appetite-suppressing effects (13–15). Strikingly, the ARC becomes selectively leptin resistant in mice with diet-induced obesity, whereas other hypothalamic and extrahypothalamic nuclei remain leptin responsive (15, 45). Also, there is evidence in diet-induced obese mice that exogenous leptin still activates BAT, even though leptin no longer suppresses food intake (45). Stellate cells in the liver also appear to retain their responsiveness to leptin in obesity; the hormone stimulates those cells to synthesize collagen and may thereby contribute to hepatic fibrosis and cirrhosis (46).
Several different mechanisms have been hypothesized to impair leptin responsiveness in the ARC in obesity [reviewed in Ref. (47)], including defective transit across the blood–brain barrier (BBB) that lines the ARC, hypothalamic endoplasmic reticulum (ER) stress, maladaptive sterile inflammation in the hypothalamus, and inhibited LepRb signaling owing to abnormal overexpression of suppressor of cytokine signaling 3 and protein tyrosine phosphatases (PTPases). Leptin transport across the BBB has been reported to be gradually impaired during high-fat feeding (48). In humans, the ratio of leptin concentrations in cerebrospinal fluid versus serum was found to be 4.3-fold higher in lean individuals than in obese individuals (49). Tanycytes, which are specialized glia in the BBB, have been reported to bring circulating leptin into the hypothalamus (50, 51), and there is evidence that leptin transport requires ERK activation in these cells (50). Recently, it was reported that ER stress in the ventromedial hypothalamus in obese Zucker rats leads to reduced BAT thermogenesis and weight gain, which could be rescued by overexpression of a chemical chaperone (52). Furthermore, it has been shown that histone deacetylase 5 activity is a regulator of leptin signaling (53). This picture is further complicated by evidence of discrepancies between endogenous and exogenous leptin sensitivity (54). Using a leptin receptor antagonist, Ottaway and colleagues concluded that diet-induced obese mice have essentially normal sensitivity to their endogenous leptin, despite other work indicating impaired sensitivity to exogenous leptin and the fact that these mice continue to overeat. Different animal models of obesity can give different results, and we recently reviewed problems with high-fat-fed rodent models of obesity (21).
Hypothalamic neurons and other cell types contain PTPases that dephosphorylate specific tyrosyl residues in canonical leptin and insulin signaling cascades, thereby attenuating or terminating the action of these hormones [Figure 1; Ref. (55)]. Accordingly, abnormal increases in the abundance (and possibly activity) of hypothalamic PTPases have been implicated in high-fat diet-induced obesity and central leptin resistance (56, 57). Conversely, genetic deletion of specific PTPases in the brain promotes leptin signaling in the ARC in association with decreased food intake, increased energy expenditure, and hence reduced adiposity. For example, deletion of protein tyrosine phosphatase 1B and T-cell protein tyrosine phosphatase enhances leptin signaling in POMC neurons and prevents diet-induced obesity by increasing WAT browning and energy expenditure (42).
Levels of PTPase activity in fasted, lean animals are sufficiently high to quickly undo the phosphorylation of key tyrosyl residues in LepRb, JAK2, and STAT3. Thus, for normal leptin signaling to propagate, hypothalamic neurons require a robust system to transiently inactivate PTPases that otherwise interfere with phosphorylation and activation of LepRb, JAK2, and STAT3. The enzymatic activity of PTPases depends on the presence of a reduced cysteine in a conserved motif, CX5R(S/T), within the active site (58, 59). Thus, certain members of the PTPase gene family are among the most redox-sensitive molecules in the cell. PTPase activity is regulated by reversible oxidation of that key active-site cysteine, and any disturbance in this process will affect leptin signaling.
We and others have shown that leptin normally induces a transient burst in reactive oxygen species (ROS) in neurons and other cell types (22, 60–62). Leptin seems to induce an increase in an ROS species with a long half-life—namely, hydrogen peroxide (H2O2; see below). Importantly, Diano et al. reported lower hypothalamic content of ROS in obesity and that central injection of low, non-toxic amounts of H2O2 mimicked the appetite-suppressing effects of leptin and restored leptin sensitivity in diet-induced obese mice (61). Nevertheless, the molecular mechanism by which leptin normally stimulates ROS production had remained uncharacterized.
The Newly Discovered NSAPP Signaling Pathway is Essential for Canonical Leptin Signaling in Hypothalamic Neurons
Recently, our group discovered a new signaling pathway that we named “NSAPP” after its major protein components [red in Figure 1; (21, 23)]. The pathway consists of an oxide transport chain, in which certain hormones stimulate NADPH oxidase-4 (NOX4) to generate the superoxide ion . NOX4 hands to the nearby superoxide dismutase-3 (SOD3) for efficient conversion into H2O2. This H2O2 is generated extracellularly and requires aquaporins to cross the plasma membrane to enter the cell. Inside the cell, the H2O2 is targeted to inactivate redox-sensitive PTPases and the closely related enzyme PTEN. We initially showed that the NSAPP oxide transport chain is required for normal, balanced insulin signaling through the PI3K–AKT pathway in liver and in endothelium (21, 23). The NSAPP pathway fails to function normally in states of overnutrition, thereby providing a molecular explanation for pathway-selective insulin resistance, also known as imbalanced insulin action (21, 23).
In recent work (22), we found that all proteins of the NSAPP oxide transport chain are present in rat hypothalamus. In murine hypothalamic cell lines, leptin induced a burst in intracellular staining by a fluorogenic probe for ROS that we definitively identified as H2O2 by its quenching by catalase. Inhibition of NOX4 with diphenyliodonium abolished the leptin-induced H2O2 burst and blocked leptin signaling to key tyrosine phosphorylation sites on JAK2 and STAT3. Strikingly, knockdown of Sod3 also blocked leptin signaling to JAK2 and STAT3. Consistent with our findings, NOX4-deficient mice are unusually susceptible to diet-induced obesity and early-onset insulin resistance for handling glucose (63). Moreover, the anorexigenic effect of insulin requires an increase in hypothalamic ROS mediated through NADPH oxidases that is blunted in high-fat diet-fed mice (64).
The NSAPP oxide transport chain finally provides a molecular explanation for how leptin normally provokes an ROS burst in hypothalamic neurons (22). Moreover, the NSAPP pathway is essential for canonical leptin signaling to JAK2 and then STAT3, which in turn regulates the expression of key neuropeptides, such as POMC and AgRP, that control appetite (Figure 1).
Outlook
Remarkably, leptin and insulin both require the NSAPP oxide transport chain, suggesting that a defect in this pathway could explain simultaneous resistance to the appetite-suppressing effects of both hormones in obesity. Thus, we hypothesize that interference with the NSAPP signaling pathway in the hypothalami of lean animals will produce defects in central control of food intake and energy expenditure, causing overeating, positive caloric imbalance, and weight gain. In the other direction, unraveling the molecular basis for NSAPP dysfunction in overnutrition has now become a top priority. At this point, restoration of normal hypothalamic NSAPP function in obesity should be considered as an attractive, but entirely unexplored, strategy to promote weight loss.
Author Contributions
All the authors reviewed the literature and approved the final version of manuscript. SF wrote the first draft. HV, AS, and KJW finalized the manuscript.
Conflict of Interest Statement
KJW has an ownership interest in Hygieia, Inc., which provides insulin management services in Northern Ireland and in Michigan, USA. The other authors declare that they have no conflicts of interest.
The reviewer RN and handling Editor declared their shared affiliation.
Funding
This work was supported by grants from the Swedish Heart-Lung Foundation (Hjärt-Lungfonden), ALF-medel Västra Götalandsregionen, and the Ruth and Yonatan Ben-Avraham Fund (KJW); and the German Ministry of Education and Research and the State of Brandenburg (DZD grant 82DZD00302, AS).
References
1. Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science (1995) 269:543–6. doi:10.1126/science.7624777
2. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature (1994) 372:425–32. doi:10.1038/372425a0
3. Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med (1996) 334:292–5. doi:10.1056/NEJM199602013340503
4. Evans K, Clark ML, Frayn KN. Carbohydrate and fat have different effects on plasma leptin concentrations and adipose tissue leptin production. Clin Sci (Lond) (2001) 100:493–8. doi:10.1042/CS20000197
5. Saladin R, De Vos P, Guerre-Millo M, Leturque A, Girard J, Staels B, et al. Transient increase in obese gene expression after food intake or insulin administration. Nature (1995) 377:527–9. doi:10.1038/377527a0
6. Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature (2001) 411:480–4. doi:10.1038/35078085
7. Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjørbaeek C, et al. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron (1999) 23:775–86. doi:10.1016/S0896-6273(01)80035-0
8. Fernandes MF, Matthys D, Hryhorczuk C, Sharma S, Mogra S, Alquier T, et al. Leptin suppresses the rewarding effects of running via STAT3 signaling in dopamine neurons. Cell Metab (2015) 22:741–9. doi:10.1016/j.cmet.2015.08.003
9. Khanh DV, Choi YH, Moh SH, Kinyua AW, Kim KW. Leptin and insulin signaling in dopaminergic neurons: relationship between energy balance and reward system. Front Psychol (2014) 5:846. doi:10.3389/fpsyg.2014.00846
10. Murray S, Tulloch A, Gold MS, Avena NM. Hormonal and neural mechanisms of food reward, eating behaviour and obesity. Nat Rev Endocrinol (2014) 10:540–52. doi:10.1038/nrendo.2014.91
11. Evans MC, Anderson GM. Dopamine neuron-restricted leptin receptor signaling reduces some aspects of food reward but exacerbates the obesity of leptin receptor-deficient male mice. Endocrinology (2017) 158:4246–56. doi:10.1210/en.2017-00513
12. Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, et al. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron (2006) 51:801–10. doi:10.1016/j.neuron.2006.08.023
13. Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, et al. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab (2007) 5:181–94. doi:10.1016/j.cmet.2007.02.004
14. Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, et al. Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA (1999) 282:1568–75. doi:10.1001/jama.282.16.1568
15. Münzberg H, Flier JS, Bjørbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology (2004) 145:4880–9. doi:10.1210/en.2004-0726
16. Coleman DL. A historical perspective on leptin. Nat Med (2010) 16:1097–9. doi:10.1038/nm1010-1097
17. de Luca C, Kowalski TJ, Zhang Y, Elmquist JK, Lee C, Kilimann MW, et al. Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. J Clin Invest (2005) 115:3484–93. doi:10.1172/JCI24059
18. Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG Jr, Schwartz MW. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature (2001) 413:794–5. doi:10.1038/35101657
19. Kitamura T, Feng Y, Kitamura YI, Chua SC Jr, Xu AW, Barsh GS, et al. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med (2006) 12:534–40. doi:10.1038/nm1392
20. Shan X, Yeo GS. Central leptin and ghrelin signalling: comparing and contrasting their mechanisms of action in the brain. Rev Endocr Metab Disord (2011) 12:197–209. doi:10.1007/s11154-011-9171-7
21. Williams KJ, Wu X. Imbalanced insulin action in chronic over nutrition: clinical harm, molecular mechanisms, and a way forward. Atherosclerosis (2016) 247:225–82. doi:10.1016/j.atherosclerosis.2016.02.004
22. Fruhwürth S, Wu X, Williams KJ. A novel pathway essential for canonical leptin signaling in hypothalamic neurons. Diabetes (2017) 66(Suppl 1A):LB73.
23. Wu X, Williams KJ. NOX4 pathway as a source of selective insulin resistance and responsiveness. Arterioscler Thromb Vasc Biol (2012) 32:1236–45. doi:10.1161/ATVBAHA.111.244525
24. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med (2009) 360:1509–17. doi:10.1056/NEJMoa0810780
25. van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med (2009) 360:1500–8. doi:10.1056/NEJMoa0808718
26. Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, et al. Functional brown adipose tissue in healthy adults. N Engl J Med (2009) 360:1518–25. doi:10.1056/NEJMoa0808949
27. Rahmouni K, Sigmund CD, Haynes WG, Mark AL. Hypothalamic ERK mediates the anorectic and thermogenic sympathetic effects of leptin. Diabetes (2009) 58:536–42. doi:10.2337/db08-0822
28. Commins SP, Watson PM, Levin N, Beiler RJ, Gettys TW. Central leptin regulates the UCP1 and ob genes in brown and white adipose tissue via different ß-adrenoceptor subtypes. J Biol Chem (2000) 275:33059–67. doi:10.1074/jbc.M006328200
29. Contreras C, Nogueiras R, Dieguez C, Rahmouni K, Lopez M. Traveling from the hypothalamus to the adipose tissue: the thermogenic pathway. Redox Biol (2017) 12:854–63. doi:10.1016/j.redox.2017.04.019
30. Buettner C, Muse ED, Cheng A, Chen L, Scherer T, Pocai A, et al. Leptin controls adipose tissue lipogenesis via central, STAT3-independent mechanisms. Nat Med (2008) 14:667–75. doi:10.1038/nm1775
31. Trayhurn P, Thurlby PL, James WP. Thermogenic defect in pre-obese ob/ob mice. Nature (1977) 266:60–2. doi:10.1038/266060a0
32. Ouellet V, Routhier-Labadie A, Bellemare W, Lakhal-Chaieb L, Turcotte E, Carpentier AC, et al. Outdoor temperature, age, sex, body mass index, and diabetic status determine the prevalence, mass, and glucose-uptake activity of 18F-FDG-detected BAT in humans. J Clin Endocrinol Metab (2011) 96:192–9. doi:10.1210/jc.2010-0989
33. Yoneshiro T, Aita S, Matsushita M, Kayahara T, Kameya T, Kawai Y, et al. Recruited brown adipose tissue as an antiobesity agent in humans. J Clin Invest (2013) 123:3404–8. doi:10.1172/JCI67803
34. Arch JR. Challenges in ß3-adrenoceptor agonist drug development. Ther Adv Endocrinol Metab (2011) 2:59–64. doi:10.1177/2042018811398517
35. Cypess AM, Weiner LS, Roberts-Toler C, Franquet Elia E, Kessler SH, Kahn PA, et al. Activation of human brown adipose tissue by a ß3-adrenergic receptor agonist. Cell Metab (2015) 21:33–8. doi:10.1016/j.cmet.2014.12.009
36. Martinez-Tellez B, Sanchez-Delgado G, Garcia-Rivero Y, Alcantara JMA, Martinez-Avila WD, Munoz-Hernandez MV, et al. A new personalized cooling protocol to activate brown adipose tissue in young adults. Front Physiol (2017) 8:863. doi:10.3389/fphys.2017.00863
37. Beiroa D, Imbernon M, Gallego R, Senra A, Herranz D, Villarroya F, et al. GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes (2014) 63:3346–58. doi:10.2337/db14-0302
38. Lockie SH, Heppner KM, Chaudhary N, Chabenne JR, Morgan DA, Veyrat-Durebex C, et al. Direct control of brown adipose tissue thermogenesis by central nervous system glucagon-like peptide-1 receptor signaling. Diabetes (2012) 61:2753–62. doi:10.2337/db11-1556
39. Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, et al. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell (2014) 156:304–16. doi:10.1016/j.cell.2013.12.021
40. Shao M, Ishibashi J, Kusminski CM, Wang QA, Hepler C, Vishvanath L, et al. Zfp423 maintains white adipocyte identity through suppression of the beige cell thermogenic gene program. Cell Metab (2016) 23:1167–84. doi:10.1016/j.cmet.2016.04.023
41. Stadion M, Schwerbel K, Graja A, Baumeier C, Rödiger M, Jonas W, et al. Increased Ifi202b/IFI16 expression stimulates adipogenesis in mice and humans. Diabetologia (2018). doi:10.1007/s00125-018-4571-9
42. Dodd GT, Decherf S, Loh K, Simonds SE, Wiede F, Balland E, et al. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell (2015) 160:88–104. doi:10.1016/j.cell.2014.12.022
43. Sidossis LS, Porter C, Saraf MK, Borsheim E, Radhakrishnan RS, Chao T, et al. Browning of subcutaneous white adipose tissue in humans. Cell Metab (2015) 22:219–27. doi:10.1016/j.cmet.2015.06.022
44. Shabalina IG, Petrovic N, de Jong JM, Kalinovich AV, Cannon B, Nedergaard J. UCP1 in brite/beige adipose tissue mitochondria is functionally thermogenic. Cell Rep (2013) 5:1196–203. doi:10.1016/j.celrep.2013.10.044
45. Enriori PJ, Sinnayah P, Simonds SE, Garcia Rudaz C, Cowley MA. Leptin action in the dorsomedial hypothalamus increases sympathetic tone to brown adipose tissue in spite of systemic leptin resistance. J Neurosci (2011) 31:12189–97. doi:10.1523/JNEUROSCI.2336-11.2011
46. Saxena NK, Anania FA. Adipocytokines and hepatic fibrosis. Trends Endocrinol Metab (2015) 26:153–61. doi:10.1016/j.tem.2015.01.002
47. Quarta C, Sánchez-Garrido MA, Tschöp MH, Clemmensen C. Renaissance of leptin for obesity therapy. Diabetologia (2016) 59:920–7. doi:10.1007/s00125-016-3906-7
48. El-Haschimi K, Pierroz DD, Hileman SM, Bjørbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest (2000) 105:1827–32. doi:10.1172/JCI9842
49. Caro JF, Kolaczynski JW, Nyce MR, Ohannesian JP, Opentanova I, Goldman WH, et al. Decreased cerebrospinal-fluid/serum leptin ratio in obesity: a possible mechanism for leptin resistance. Lancet (1996) 348:159–61. doi:10.1016/S0140-6736(96)03173-X
50. Balland E, Dam J, Langlet F, Caron E, Steculorum S, Messina A, et al. Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab (2014) 19:293–301. doi:10.1016/j.cmet.2013.12.015
51. Mullier A, Bouret SG, Prevot V, Dehouck B. Differential distribution of tight junction proteins suggests a role for tanycytes in blood-hypothalamus barrier regulation in the adult mouse brain. J Comp Neurol (2010) 518:943–62. doi:10.1002/cne.22273
52. Contreras C, Gonzalez-Garcia I, Martinez-Sanchez N, Seoane-Collazo P, Jacas J, Morgan DA, et al. Central ceramide-induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell Rep (2014) 9:366–77. doi:10.1016/j.celrep.2014.08.057
53. Kabra DG, Pfuhlmann K, Garcia-Caceres C, Schriever SC, Casquero Garcia V, Kebede AF, et al. Hypothalamic leptin action is mediated by histone deacetylase 5. Nat Commun (2016) 7:10782. doi:10.1038/ncomms10782
54. Ottaway N, Mahbod P, Rivero B, Norman LA, Gertler A, D’Alessio DA, et al. Diet-induced obese mice retain endogenous leptin action. Cell Metab (2015) 21:877–82. doi:10.1016/j.cmet.2015.04.015
55. Zhang ZY, Dodd GT, Tiganis T. Protein tyrosine phosphatases in hypothalamic insulin and leptin signaling. Trends Pharmacol Sci (2015) 36:661–74. doi:10.1016/j.tips.2015.07.003
56. Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med (2006) 12:917–24. doi:10.1038/nm1435
57. Loh K, Fukushima A, Zhang X, Galic S, Briggs D, Enriori PJ, et al. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab (2011) 14:684–99. doi:10.1016/j.cmet.2011.09.011
58. Ross SH, Lindsay Y, Safrany ST, Lorenzo O, Villa F, Toth R, et al. Differential redox regulation within the PTP superfamily. Cell Signal (2007) 19:1521–30. doi:10.1016/j.cellsig.2007.01.026
59. Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol (2006) 7:833–46. doi:10.1038/nrm2039
60. Chetboun M, Abitbol G, Rozenberg K, Rozenfeld H, Deutsch A, Sampson SR, et al. Maintenance of redox state and pancreatic ß-cell function: role of leptin and adiponectin. J Cell Biochem (2012) 113:1966–76. doi:10.1002/jcb.24065
61. Diano S, Liu ZW, Jeong JK, Dietrich MO, Ruan HB, Kim E, et al. Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nat Med (2011) 17:1121–7. doi:10.1038/nm.2421
62. Palomba L, Silvestri C, Imperatore R, Morello G, Piscitelli F, Martella A, et al. Negative regulation of leptin-induced reactive oxygen species (ROS) formation by cannabinoid CB1 receptor activation in hypothalamic neurons. J Biol Chem (2015) 290:13669–77. doi:10.1074/jbc.M115.646885
63. Li Y, Mouche S, Sajic T, Veyrat-Durebex C, Supale R, Pierroz D, et al. Deficiency in the NADPH oxidase 4 predisposes towards diet-induced obesity. Int J Obes (Lond) (2012) 36:1503–13. doi:10.1038/ijo.2011.279
Keywords: leptin, hypothalamus, obesity, energy balance, signaling
Citation: Fruhwürth S, Vogel H, Schürmann A and Williams KJ (2018) Novel Insights into How Overnutrition Disrupts the Hypothalamic Actions of Leptin. Front. Endocrinol. 9:89. doi: 10.3389/fendo.2018.00089
Received: 06 December 2017; Accepted: 23 February 2018;
Published: 26 March 2018
Edited by:
Sulay Tovar, Universidade de Santiago de Compostela, SpainReviewed by:
Cristina Contreras, Complutense University of Madrid, SpainRuben Nogueiras, Universidade de Santiago de Compostela, Spain
Copyright: © 2018 Fruhwürth, Vogel, Schürmann and Williams. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevin Jon Williams, a2V2aW4uam9uLndpbGxpYW1zQGd1LnNl