 Ariel Tarasiuk1,2*
Ariel Tarasiuk1,2* Yael Segev3
Yael Segev3
- 1Sleep-Wake Disorders Unit, Soroka University Medical Center, Beer-Sheva, Israel
- 2Department of Physiology, Faculty of Health Sciences, Ben-Gurion University of the Negev, Beer-Sheva, Israel
- 3Shraga Segal Department of Microbiology and Immunology, Ben-Gurion University of the Negev, Beer-Sheva, Israel
Pediatric obstructive sleep apnea (OSA) is a syndrome manifesting with snoring and increased respiratory effort due to increased upper airway resistance. In addition to cause the abnormal sleep, this syndrome has been shown to elicit either growth retardation or metabolic syndrome and obesity. Treating OSA by adenotonsillectomy is usually associated with increased risk for obesity, despite near complete restoration of breathing and sleep. However, the underlying mechanism linking upper airways obstruction (AO) to persistent change in food intake, metabolism, and growth remains unclear. Rodent models have examined the impact of intermittent hypoxia on metabolism. However, an additional defining feature of OSA that is not related to intermittent hypoxia is enhanced respiratory loading leading to increased respiratory effort and abnormal sleep. The focus of this mini review is on recent evidence indicating the persistent abnormalities in endocrine regulation of feeding and growth that are not fully restored by the chronic upper AO removal in rats. Here, we highlight important aspects related to abnormal regulation of metabolism that are not related to intermittent hypoxia per se, in an animal model that mimics many of the clinical features of pediatric OSA. Our evidence from the AO model indicates that obstruction removal may not be sufficient to prevent the post-removal tendency for abnormal growth.
Introduction
Obstructive sleep-disordered breathing includes a spectrum of clinical entities with variable severity ranging from primary snoring to obstructive sleep apnea (OSA) (1, 2). Children with OSA suffer from upper airway obstruction (AO) during sleep that is manifested by increased respiratory efforts, large variations in intrathoracic pressure (up to −50 mmHg during peak inspiration), intermittent hypoxia, ultimately leading to sleep fragmentation and nonrestorative sleep. OSA is relatively common in children, and it may have serious consequences on longitudinal growth, body weight, energy metabolism, cardiovascular and neurobehavioral abnormalities, and increased health-care utilization (1–10). Estimates of OSA prevalence range between 1 and 5.7% depending mainly on the populations studied (1, 2, 11, 12). It is estimated that 5–56% of OSA cases develop growth retardation, with the lower prevalence probably reflecting increased awareness and earlier diagnosis and treatment (13–17). The mechanisms underlying the development of growth retardation in OSA continue to be highly controversial. Three main possibilities have been put forward to explain growth retardation in OSA. First, it is possible that dysphagia (18) is due to enlarged tonsils and adenoids, and decreased appetite due to changes in olfactory acuity in some cases. Second, it has been postulated that dysregulation of energy supply/energy expenditure balance (3, 18–20), due to the increased respiratory efforts (work of breathing) during sleep, will lead to increased metabolic expenditure and contribute to slow weight gain in these children (3). However, this mechanism has been disputed, as total energy expenditure was not affected by OSA (21). Third, more recently, impaired homeostasis of hormones such as growth hormone (GH), ghrelin, and leptin has been reported (4, 5, 22–25). The GH homeostasis is recognized as a key mechanism underlying impaired longitudinal growth (1, 4, 5). GH secretion occurs in pulses from the anterior pituitary somatotropic cells mainly during deep slow wave sleep onset (26, 27). Deep slow wave sleep is initiated in the preoptic area of the hypothalamus and consists of delta electroencephalogram activity, i.e., high-amplitude brain waves with a frequency of oscillation between 0.5 and 4 Hz (23, 28–31). OSA has also been shown to cause growth failure in some young children, and metabolic syndrome and obesity were reported in other cases (1, 3, 5, 6, 13, 15, 16, 18). OSA is most prevalent in 2- to 8-year-old children, when tonsil and adenoid volume is largest relative to the upper airway diameter; these children are usually referred to adenotonsillectomy as the first-line treatment (1, 3, 6, 15, 32). A currently poorly understood phenomenon is the fact that treatment of OSA can lead to accelerated weight gain in children, i.e., it normalizes weight in children who have failure to thrive, but increases the risk for obesity in overweight patients (1, 2, 5, 6, 15, 32–35). Regulation of energy expenditure is multifactorial and includes factors such as metabolic rate at rest, physical activity, and thermic effect of food intake (19, 20, 36, 37). Whole-body energy balance to promote weight gain may be altered following treatment of OSA (19, 20). However, study design and the between-group variability make a conclusion on the effect of treatment difficult. Although adenotonsillectomy in children and positive airway pressure (in adults) treatments predispose humans to a positive energy balance and accelerate body weight gain, sedentary lifestyles, dietary intake, and selection of high caloric/glycemic index foods may have greater impacts on weight change (6, 19, 20, 38–40). However, the majority of clinical studies concentrated on elucidating the endocrine consequences of the surgical treatment while data on normal healthy controls barely exist.
Experimental models of sleep apnea provide mechanistic insight into the apnea generation as well as into its impact on cardiovascular, metabolic, and psychological consequences (41, 42). The commonly used model to study OSA involves implementation of intermittent hypoxia, i.e., the repetitive brief hypoxic episodes like those that occur in OSA (43) or specifically dusting sleep (44, 45) to explore the impact of intermittent hypoxia on cardiovascular (43, 46), sleep (47, 48), and neurocognitive parameters (42). Another defining feature of OSA, however, is the mechanical changes in work of breathing that are not associated with intermittent hypoxia. To elucidate the role of the mechanical load, we first developed a model of mechanical obstruction in rodents in 1991 (49). We used this model 2 years later to elucidate the impact of chronic upper AO on cardiac function in rats (50). Here, we highlight important aspects related to abnormal regulation of metabolism that are not related to intermittent hypoxia per se, in an animal model that features many of the clinical signs of pediatric OSA.
The Upper AO Model
In the AO model, respiratory load is surgically induced in 22-day-old rats by tracheal narrowing and animals were followed up to 7 weeks; this period is comparable to half a year up to 20 years in humans (25). AO induces adaptive response in the respiratory system including alterations in respiratory muscles (49, 51), control of ventilation (52, 53), sleep, growth, and metabolism (23–25, 31, 54–57). Following surgery trachea diameter was reduced by 45%, its resistance increased by 46–100%, and respiratory effort more than doubled (23, 52, 53, 55); following obstruction removal, trachea diameter was normalized to control values (25). One of the limitations of this model is the fact that respiratory loading was both inspiratory and expiratory and not sleep related, resembling pediatric subglottic stenosis. In clinical OSA, however, the upper AO is predominately inspiratory and manifested mainly during sleep (58). The similarity of this model and sleep apnea are striking as obstructed animals exhibit sleep fragmentation and abnormal growth similar to OSA (1, 2, 23, 54, 55). The AO elicits audible wheezing, especially following activity, whereas no sign of respiratory distress was observed at rest (49, 50, 53). Under these conditions, animals maintain PO2 in the normal range with no evidence for gas exchange abnormalities (24, 25, 50, 52, 54, 57), hemoglobin and lactate levels (23, 25, 52, 54, 57), and daily food intake were higher (25, 53, 54). By contrast, intermittent hypoxia can lead to decreased food intake, erythropoiesis, and liver injury (59–65). In children with OSA, oxygen saturation may decrease with airway loading during sleep (66). However, in AO, no changes were found in liver enzymes, liver histology, prolyl-hydroxylase 2, or hypoxia-inducible factors (25). These findings support the notion that intermittent hypoxia does not play a role in this abnormal energy metabolism and growth observed in the AO animal model.
Orexin and Breathing
Airways obstruction leads to adaptive changes in the respiratory mechanics including large inspiratory swings in pleural pressure and increased diaphragmatic contractile force (49, 50) to maintain respiratory homeostasis (Figure 1) (25, 52–54). These adaptations are critical especially during sleep, a condition where respiratory muscle force may not be sufficient to support obstructed ventilation. AO leads to increased hypothalamic orexin level, wakefulness, and hunger (23–25). The orexins (orexin A and orexin B) are novel hypothalamic peptides derived from the common precursor prepro-orexin that acts through two subtypes of receptors [orexin receptor 1 (OX1R) and OX2R]. Orexin neurons have emerged as a key orchestrator of sleep–wake activity, and breathing and feeding centers receive stimulatory inputs from orexin neurons (67–70). Orexin neurons are active during wakefulness but show little or no activity during paradoxical sleep (rapid eye movement sleep in humans) and slow wave sleep (70–72). Orexin-containing neurons located in the lateral hypothalamus contribute to carbon dioxide (CO2) chemoreception (73–77) in a vigilance-state- and diurnal-cycle-dependent manner (73, 76). Prepro-orexin knockout mice exhibit a large decrease in the ventilatory response CO2 during wakefulness that contributes to a higher arterial PCO2 (78). Hypothalamic orexin cell firing can be stimulated by extracellular pH levels, at least partial closure of TASK-like channels (74), and possibly via acid-sensing ion channels (77, 79). Respiratory acidosis produced by OSA causes arousal and stimulates breathing, which normalizes extracellular levels of protons (79). In the AO model, increase in hypothalamic orexin plays an important role in maintaining respiration (23, 24, 54) and increased food intake (24, 25). Administration of almorexant (dual orexin receptor antagonist) normalized sleep (54) but induced severe breathing difficulties in the AO group, while it affected neither sleep nor breathing in control animals. Similar breathing difficulties were found during recovery sleep from 4-h sleep deprivation protocol when sleep was stimulated and orexin level is minimal. Orexin plays a role in maintaining breathing homeostasis in AO, probably via its primary role in CO2 chemoreception (76). Further studies are needed to quantify the role of TASK-like channels (74) and acid-sensing ion channels in regulation of breathing in AO model. Leptin has an important role in central chemoreception; leptin deficiency in ob/ob mice produces marked depression of the hypercapnic ventilatory response (62). In AO rats, however, leptin decreases and does not play a role in respiratory regulation (25, 54).
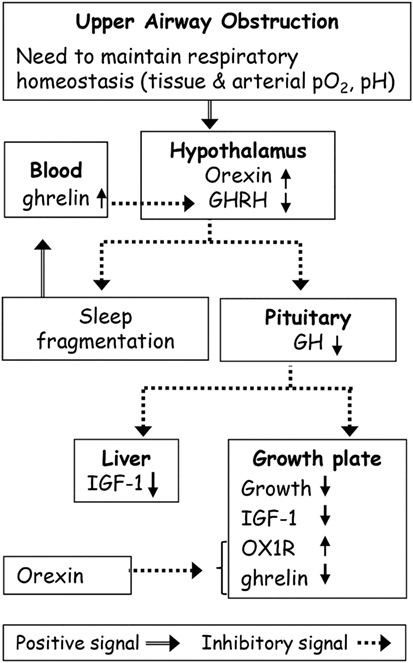
Figure 1. Possible effects of upper airway obstruction (AO) on sleep and growth. Upper AO leads to adaptive changes in the respiratory mechanics to maintain respiratory homeostasis. Orexin plays a role in maintaining breathing homeostasis in AO via its primary role in carbon dioxide chemoreception. Orexin inhibits growth hormone (GH) release from the pituitary gland through inhibition of hypothalamic hormone-releasing hormone (GHRH) neurons. Abnormalities in GHRH underlie both growth and sleep disorders in AO. The continuous ghrelin elevation in AO possibly due to partial sleep loss and increased orexin was sufficient to desensitize the hypothalamic–pituitary–GH axis. AO causes suppression of the GH axis, and of the global and local growth plate IGF-1 levels leading to growth retardation. Orexin receptor 1 (OX1R) plays a role in growth retardation by modulation of local ghrelin levels.
Orexin–GH Axis and Feeding
The need to maintain ventilation in AO is associated with upregulation of hypothalamic orexin, which causes suppression of the growth hormone-releasing hormone/growth hormone axis (GHRH/GH) and decreased sleep (Figure 1) (23, 31, 54). AO disruption of sleep leads to adverse health outcomes in rodents, i.e., increased appetite hormones (orexin, ghrelin) and food intake and GH suppression (23–25). Orexin serves as an important link between peripheral metabolism and homeostatic challenges including sleep, respiration, feeding, and neuroendocrine homeostasis (67–70, 80–82). Orexin-A inhibits GH release from the pituitary gland through inhibition of GHRHergic neurons of the periventricular nucleus and of the arcuate nucleus and stimulation of somatostatin in the hypothalamus (82–85). Enhanced GH secretion and induction of sleep are two parallel and closely interrelated consequences of hypothalamic GHRHergic neurons activation (29, 86, 87). Abnormalities in GHRH levels could underlie both growth and sleep disorders in AO (23). The decreased GHRH content in AO was related to increased hypothalamic orexin and somatostatin levels, and in obstruction removal group to increased somatostatin (25, 54). Administration of ritanserin (selective 5HT2 antagonist) normalized both GHRH content and slow wave sleep (23, 31, 54).
Interestingly, AO elicits sustained ghrelin elevation, sufficient to desensitize the hypothalamic–pituitary–GH axis while still causing hyperphagia (25). This hyperphagia was also attributed to upregulation of orexin and mediators that are activated by ghrelin, such as neuropeptide Y and agouti-related peptide, and to decreased circulatory leptin levels. Gut-derived ghrelin both stimulates feeding behavior and causes release of GHRH from the hypothalamus in response to fasting (88–90) by activation of the GH-secretagogue receptors (91). At least three types of neurons, such as GHRHergic, neuropeptide Y, and agouti-related peptidergic, are well-defined targets for ghrelin action on feeding (92). Acceleration of growth by ghrelin depends on its level and pattern of secretion (93). Short exposure to ghrelin causes augmentation of GH release and increases appetite via the hypothalamic GHRH receptor, while continuous exposure increases feeding but suppresses the GH release. The most prevalent explanation of the impaired somatic growth in OSA patients is that their GH homeostasis is disturbed (4). A meta-analysis study (5) found the evidence of improved endocrine homeostasis of the GH axis that is associated with improved somatic growth following adenotonsillectomy, supporting the concept that GH homeostasis is impaired in pediatric OSA. However, the limitation of these studies was that the comparison has been done to hormone levels in the short time windows before and after the surgical intervention, not including healthy controls. Levels of IGF-1 and of its main binding protein, IGF-binding protein 3, are well-established predictors of the mean GH levels across the day (94–96). One of the interesting findings in the AO model is that following the obstruction removal, the GH/IGF-1 axis does not normalize and these animals still exhibit substantial growth retardation (25). Both GHRH and IGF-1 levels do not reach control values and obstruction removal animals continue to exhibit shorter body length despite normalized tracheal diameter, indicating persistent deregulation of GH axis (25). Increased food intake in AO was probably a physiological adaptation required to provide the extra energy needed for increased additional wakefulness (23, 31, 36, 54) and possibly elevated energy expenditure due to increased work of breathing (49, 51). Increased feeding behavior was associated with persistent remodeling of the appetite homeostasis long after the successful removal of the upper AO (25). It is possible that AO (treated or untreated) leads to persistent alterations of appetite homeostasis and increases the preference for a carbohydrate-rich diet. Further studies are needed to explore the whole-body energy balance using an open-circuit indirect calorimeter (36) and to find out whether AO obstruction removal affects nutritional preference for carbohydrate-rich vs. high-fat diet that in turn may contribute to abnormal body weight gain. In children, despite resuming normal sleep and ventilation by adenotonsillectomy, the risk for obesity remains high (3, 6, 32–35). This has been attributed to a shift toward less active lifestyle and, possibly, to unhealthy food choices, highlighting the importance of changing the lifestyle following surgery (6, 19, 32, 38–40).
Metabolic Consequences of AO
Airways obstruction leads to partial sleep loss, i.e., 30–45% elevation of wake duration, from early life to adulthood (23, 24, 31, 54). Low weight gain and growth were observed in AO animals despite increased energy intake and increased intestine surface area to absorb water and nutrients (25, 54), similar to the effect observed during chronic partial sleep loss in rats (97). Sleep fragmentation, regardless of the methodology, including the AO in rats, leads to considerable pathophysiological abnormalities including decreased body temperature (98), decreased GHRH/GH axis (27, 99), and increased feeding behavior (61, 97). Similar sleep abnormalities in humans may increase risk for type 2 diabetes, obesity, and cardiovascular diseases (100, 101). The difference in metabolic response to sleep abnormalities between humans and rats may simply reflect an inter-species difference (102), although other contributing factors such as intermittent hypoxia cannot be excluded in the case of sleep apnea (1, 2, 22). Moreover, the different levels of physical activity in human (6) and AO animals may also play a role. A striking reduction of adipose tissue content and distribution was the main factor contributing to the slow body weight gain in AO rats (54). Slower weight gain (or even weight loss) was also reported in earlier studies of sleep deprivation in rodents, but these reports were not consistent. Thus, whereas most studies showed an increase in food intake (97, 103), some groups reported no change in energy intake following sleep loss (102). Short sleep duration augments gut-derived ghrelin and appetite (99). Ghrelin drives motivated behavior such as motivated movement, feeding, and increased arousal (88, 104). The essential need to provide the energy required to support loaded breathing in AO (49) accompanied by enhanced wakefulness (36) makes the increased feeding essential for physiological adaptation to the chronic respiratory condition.
Bone Metabolism and Architecture
Growth retardation is highly prevalent in children suffering from OSA. It aging adults, OSA has been shown to affect bone metabolism and bone mass via decreased sleep quality, nocturnal hypoxia, inflammation, etc. (105). The endocrine effect of GHRH/GH is induction of IGF-1 in various organs, including the epiphyseal growth plate (EGP) in bones (106). AO causes a significant suppression of the GH axis, and of the global and local EGP IGF-1 levels (Figure 1) (55, 57). The global IGF-1 levels are primarily determined by liver synthesis; other peripheral tissues contribute somewhat to circulating global IGF-1 levels but their contributions are less significant (107). Local IGF-1 mediates the GH effect on the EGP by binding to the specific receptors (94, 108). Reduced local IGF-1 release in the EGP will lead to growth retardation (109).
We assessed somatic growth by measuring long bones length and by microCT scanning (24, 55, 110). Longitudinal growth is a well-orchestrated process that occurs via endochondral ossification at the growth plates (111). In AO animals, growth gain was inversely correlated with enhanced upper airways resistance (55). Both proliferative and hypertrophic zones were narrowed in the AO animals and these animals exhibited reduced bone mass. These pathophysiological bone abnormalities were related to EGP failure and dysregulation of the levels of endochondral ossification markers (e.g., collagen type II, X, Sox9, and RankL) (24). Pharmacological stimulation of GH release restored local EGP IGF-1. The growth parameters were only partially restored (55), however, indicating that other pathways are involved. Seeking the identity of these pathways, we focused our attention on orexin and ghrelin peptides in AO animals. OX1R has been shown to play an important role in bone remodeling and metabolism by modulation of local ghrelin levels (112). Several ghrelin related signaling pathways, including phosphatidylinositol 3-kinase/AKT, apparently are involved in bone metabolism and development (113, 114). Orexins activate transcription factor peroxisome proliferator-activated receptor gamma (PPARγ) in bone marrow adipose tissue, causing bone mass loss and leading to fatty bone marrow (115–117). Obstruction removal and administration of almorexant normalized sleep but did not reverse the phenomena (24); these finding indicate persistent bone remodeling that is not related to loss of developmental time per se. Increased expression of orexin and OX1R, and suppression of GH-secretagogue receptor and local ghrelin in the EGP are the main mechanisms contributing to increased adipocyte differentiation and fatty bone marrow found in AO. Dual orexin receptor antagonist considerably improved EGP growth and restored sleep in AO animals. Our findings support the possibility that OX1R plays a significant role in bone development in AO animals (Figure 1) (24). The robust increase of PPARγ and reduction in Sox9 levels strongly suggest that the course of early bone development has been impaired (118). Orexins have been shown capable to stimulate marrow PPARγ that in return upregulates marrow adipogenesis and causes bone mass loss (112, 115, 116). Downregulation of Sox9 also promotes adipocyte differentiation, a process that is activated by PPARγ (119).
Conclusion
The AO model provides mechanistic insight about functional interactions between orexin and the hypothalamic–pituitary axis in regulation of sleep, breathing, and growth. AO leads to adaptive changes in the respiratory mechanics to maintain respiratory homeostasis. The available information supports the notion that AO leads to increased hypothalamic orexin release, which plays an important role in maintaining respiration, but elicits sleep, energy metabolism, and growth abnormalities.
Author Contributions
AT and YS wrote the manuscript and recruited funds.
Conflict of Interest Statement
The authors have declared that no conflict of interest exists and none of the material in this manuscript has previously been published.
The reviewer JJ and handling Editor declared their shared affiliation.
Funding
This study was supported by the Israel Science Foundation grant No. 31/14.
References
1. Marcus CL, Brooks LJ, Draper KA, Gozal D, Halbower AC, Jones J, et al. Diagnosis and management of childhood obstructive sleep apnea syndrome. Pediatrics (2012) 130:e714–55. doi:10.1542/peds.2012-1672
2. Kaditis AG, Alonso Alvarez ML, Boudewyns A, Alexopoulos EI, Ersu R, Joosten K, et al. Obstructive sleep disordered breathing in 2- to 18-year-old children: diagnosis and management. Eur Respir J (2016) 4:69–94. doi:10.1183/13993003.00385-2015
3. Marcus CL, Carroll JL, Koerner CB, Hamer A, Lutz J, Loughlin GM. Determinants of growth in children with the obstructive sleep apnea syndrome. J Pediatr (1994) 125:556–62. doi:10.1016/S0022-3476(94)70007-9
4. Bar A, Tarasiuk A, Segev Y, Phillip M, Tal A. The effect of adenotonsillectomy on serum insulin-like growth factor-I and growth in children with obstructive sleep apnea syndrome. J Pediatr (1999) 135:76–80. doi:10.1016/S0022-3476(99)70331-8
5. Bonuck KA, Freeman K, Henderson J. Growth and growth biomarker changes after adenotonsillectomy: systematic review and meta-analysis. Arch Dis Child (2009) 94:83–91. doi:10.1136/adc.2008.141192
6. Katz ES, Moore RH, Rosen CL, Mitchell RB, Amin R, Arens R, et al. Growth after adenotonsillectomy for obstructive sleep apnea: an RCT. Pediatrics (2014) 134:282–9. doi:10.1542/peds.2014-0591
7. Marcus CL, Moore RH, Rosen CL, Giordani B, Garetz SL, Taylor HG, et al. Childhood adenotonsillectomy trial (CHAT). A randomized trial of adenotonsillectomy for childhood sleep apnea. N Engl J Med (2013) 368:2366–76. doi:10.1056/NEJMoa1215881
8. Israel LP, Benharoch D, Gopas J, Goldbart AD. A pro-inflammatory role for nuclear factor kappa B in childhood obstructive sleep apnea syndrome. Sleep (2013) 36:1947–55. doi:10.5665/sleep.3236
9. Tarasiuk A, Reuveni H. The economic impact of obstructive sleep apnea. Curr Opin Pulm Med (2013) 19:639–44. doi:10.1097/MCP.0b013e3283659e1e
10. Tarasiuk A, Greenberg-Dotan S, Simon-Tuval T, Freidman B, Goldbart AD, Tal A, et al. Elevated morbidity and health care use in children with obstructive sleep apnea syndrome. Am J Respir Crit Care Med (2007) 175:55–61. doi:10.1164/rccm.200604-577OC
11. Bixler EO, Vgontzas AN, Lin HM, Liao D, Calhoun S, Vela-Bueno A, et al. Sleep disordered breathing in children in a general population sample: prevalence and risk factors. Sleep (2009) 32:731–6. doi:10.1093/sleep/32.6.731
12. Rosen CL, Larkin EK, Kirchner HL, Emancipator JL, Bivins SF, Surovec SA, et al. Prevalence and risk factors for sleep-disordered breathing in 8- to 11-year-old children: association with race and prematurity. J Pediatr (2003) 142:383–9. doi:10.1067/mpd.2003.28
13. Brouillette RT, Fernbach SK, Hunt CE. Obstructive sleep apnea in infants and children. J Pediatr (1982) 100:31–40. doi:10.1016/S0022-3476(82)80231-X
14. Bonuck K, Parikh S, Bassila M. Growth failure and sleep disordered breathing: a review of the literature. Int J Pediatr Otorhinolaryngol (2006) 70:769–78. doi:10.1016/j.ijporl.2005.11.012
15. Williams EF III, Woo P, Miller R, Kellman RM. The effects of adenotonsillectomy on growth in young children. Otolaryngol Head Neck Surg (1991) 104:509–16. doi:10.1177/019459989110400415
16. Guilleminault C, Korobkin R, Winkle R. A review of 50 children with obstructive sleep apnea syndrome. Lung (1981) 159:275–87. doi:10.1007/BF02713925
17. O’Brien LM, Gozal D. Behavioural and neurocognitive implications of snoring and obstructive sleep apnoea in children: facts and theory. Paediatr Respir Rev (2002) 3:3–9. doi:10.1053/prrv.2002.0177
18. Stradling JR, Thomas G, Warley HRA, Williams P, Freeland A. Effect of adenotonsillectomy on nocturnal hypoxaemia, sleep disturbance, and symptoms in snoring children. Lancet (1990) 335:249–53. doi:10.1016/0140-6736(90)90068-G
19. Tachikawa R, Ikeda K, Minami T, Matsumoto T, Hamada S, Murase K, et al. Changes in energy metabolism after continuous positive airway pressure for obstructive sleep apnea. Am J Respir Crit Care Med (2016) 194:729–38. doi:10.1164/rccm.201511-2314OC
20. Shechter A. Effects of continuous positive airway pressure on energy balance regulation: a systematic review. Eur Respir J (2016) 48:1640–57. doi:10.1183/13993003.00689-2016
21. Bland RM, Bulgarelli S, Ventham JC, Jackson D, Reilly JJ, Paton JY. Total energy expenditure in children with obstructive sleep apnoea syndrome. Eur Respir J (2001) 18:164–9. doi:10.1183/09031936.01.99104401
22. Pillar G, Shehadeh N. Abdominal fat and sleep apnea: the chicken or the egg? Diabetes Care (2008) 31:S303–9. doi:10.2337/dc08-s272
23. Tarasiuk A, Berdugo-Boura N, Troib A, Segev Y. Role of growth hormone-releasing hormone in sleep and growth impairments induced by upper airway obstruction in rats. Eur Respir J (2011) 38:870–7. doi:10.1183/09031936.00197610
24. Tarasiuk A, Levi A, Assadi MH, Troib A, Segev Y. Orexin plays a role in growth impediment induced by obstructive sleep breathing in rats. Sleep (2016) 39:887–97. doi:10.5665/sleep.5648
25. Assadi MH, Shknevsky E, Segev Y, Tarasiuk A. Abnormal growth and feeding behavior persist after removal of upper airway obstruction in juvenile rats. Sci Rep (2017) 7(1):2730. doi:10.1038/s41598-017-02843-5
26. Brandenberger G, Weibel L. The 24-h growth hormone rhythm in men: sleep and circadian influences questioned. J Sleep Res (2004) 13:251–5. doi:10.1111/j.1365-2869.2004.00415.x
27. Everson CA, Crowley WR. Reductions in circulating anabolic hormones induced by sustained sleep deprivation in rats. Am J Physiol Endocrinol Metab (2004) 286:E1060–70. doi:10.1152/ajpendo.00553.2003
28. Zhang J, Obál F Jr, Zheng T, Fang J, Taishi P, Krueger JM. Intrapreoptic microinjection of GHRH or its antagonist alters sleep in rats. J Neurosci (1999) 19:2187–94. doi:10.1523/JNEUROSCI.19-06-02187.1999
29. Obal F Jr, Krueger JM. GHRH and sleep. Sleep Med Rev (2004) 8:367–77. doi:10.1016/j.smrv.2004.03.005
30. Ben-Israel N, Zigel Y, Tal A, Segev Y, Tarasiuk A. Adenotonsillectomy improves slow-wave activity in children with obstructive sleep apnoea. Eur Respir J (2011) 37:1144–50. doi:10.1183/09031936.00106710
31. Gradwohl G, Berdugo-Boura N, Segev Y, Tarasiuk A. Chronic upper airway obstruction induces abnormal sleep/wake dynamics in juvenile rats. PLoS One (2014) 9:e97111. doi:10.1371/journal.pone.0097111
32. Amin R, Anthony L, Somers V, Fenchel M, McConnell K, Jefferies J, et al. Growth velocity predicts recurrence of sleep-disordered breathing 1 year after adenotonsillectomy. Am J Respir Crit Care Med (2008) 177:654–9. doi:10.1164/rccm.200710-1610OC
33. Jeyakumar A, Fettman N, Armbrecht ES, Mitchell R. A systematic review of adenotonsillectomy as a risk factor for childhood obesity. Otolaryngol Head Neck Surg (2011) 144:154–8. doi:10.1177/0194599810392328
34. Czechowicz JA, Chang KW. Analysis of growth curves in children after adenotonsillectomy. JAMA Otolaryngol Head Neck Surg (2014) 140:491–6. doi:10.1001/jamaoto.2014.411
35. Levi J, Leoniak S, Schmidt R. Evaluating tonsillectomy as a risk factor for childhood obesity. Arch Otolaryngol Head Neck Surg (2012) 138:897–901. doi:10.1001/2013.jamaoto.252
36. Markwald RR, Melanson EL, Smith MR, Higgins J, Perreault L, Eckel RH, et al. Impact of insufficient sleep on total daily energy expenditure, food intake, and weight gain. Proc Natl Acad Sci U S A (2013) 110:5695–700. doi:10.1073/pnas.1216951110
37. Hill JO, Wyatt HR, Peters JC. Energy balance and obesity. Circulation (2012) 126:126–32. doi:10.1161/CIRCULATIONAHA.111.087213
38. Smurra M, Philip P, Taillard J, Guilleminault C, Bioulac B, Gin H. CPAP treatment does not affect glucose–insulin metabolism in sleep apneic patients. Sleep Med (2001) 2:207–13. doi:10.1016/S1389-9457(00)00079-4
39. Beebe DW, Miller N, Kirk S, Daniels SR, Amin R. The association between obstructive sleep apnea and dietary choices among obese individuals during middle to late childhood. Sleep Med (2011) 12:797–9. doi:10.1016/j.sleep.2010.12.020
40. Beebe DW, Simon S, Summer S, Hemmer S, Strotman D, Dolan LM. Dietary intake following experimentally restricted sleep in adolescents. Sleep (2013) 36:827–34. doi:10.5665/sleep.2704
41. Davis EM, O’Donnell CP. Rodent models of sleep apnea. Respir Physiol Neurobiol (2013) 188:355–61. doi:10.1016/j.resp.2013.05.022
42. Gozal D, Daniel JM, Dohanich GP. Behavioral and anatomical correlates of chronic episodic hypoxia during sleep in the rat. J Neurosci (2001) 21:2442–50. doi:10.1523/JNEUROSCI.21-07-02442.2001
43. Fletcher EC, Lesske J, Qian W, Miller CC III, Unger T. Repetitive, episodic hypoxia causes diurnal elevation of blood pressure in rats. Hypertension (1992) 19:555–61. doi:10.1161/01.HYP.19.6.555
44. Rubin AE, Polotsky VY, Balbir A, Krishnan JA, Schwartz AR, Smith PL, et al. Differences in sleep-induced hypoxia between A/J and DBA/2J mouse strains. Am J Respir Crit Care Med (2003) 168:1520–7. doi:10.1164/rccm.200304-462OC
45. Tagaito Y, Polotsky VY, Campen MJ, Wilson JA, Balbir A, Smith PL, et al. A model of sleep-disordered breathing in the C57BL/6J mouse. J Appl Physiol (2001) 91:2758–66. doi:10.1152/jappl.2001.91.6.2758
46. Chen L, Einbinder E, Zhang Q, Hasday J, Balke CW, Scharf SM. Oxidative stress and left ventricular function with chronic intermittent hypoxia in rats. Am J Respir Crit Care Med (2005) 172:915–20. doi:10.1164/rccm.200504-560OC
47. Polotsky VY, Rubin AE, Balbir A, Dean T, Smith PL, Schwartz AR, et al. Intermittent hypoxia causes REM sleep deficits and decreases EEG delta power in NREM sleep in the C57BL/6J mouse. Sleep Med (2006) 7:7–16. doi:10.1016/j.sleep.2005.06.006
48. Veasey SC, Davis CW, Fenik P, Zhan G, Hsu YJ, Pratico D, et al. Long-term intermittent hypoxia in mice: protracted hypersomnolence with oxidative injury to sleep-wake brain regions. Sleep (2004) 27:194–201. doi:10.1093/sleep/27.2.194
49. Tarasiuk A, Scharf SM, Miller MJ. Effect of chronic resistive loading on inspiratory muscles in rats. J Appl Physiol (1991) 70:216–22. doi:10.1152/jappl.1991.70.1.216
50. Salejee I, Tarasiuk A, Reder I, Scharf SM. Chronic upper airway obstruction produces right but not left ventricular hypertrophy in rats. Am Rev Respir Dis (1993) 148:1346–50. doi:10.1164/ajrccm/148.5.1346
51. Prezant DJ, Aldrich TK, Richner B, Gentry EI, Valentine DE, Nagashima H, et al. Effects of long-term continuous respiratory resistive loading on rat diaphragm function and structure. J Appl Physiol (1993) 74:1212–9. doi:10.1152/jappl.1993.74.3.1212
52. Greenberg HE, Tarasiuk A, Rao RS, Kupferman M, Kane N, Scharf SM. Effect of chronic resistive loading on ventilatory control in a rat model. Am J Respir Crit Care Med (1995) 152:666–76. doi:10.1164/ajrccm.152.2.7633724
53. Greenberg HE, Rao RS, Sica AL, Scharf SM. Effect of chronic resistive loading on hypoxic ventilatory responsiveness. J Appl Physiol (1997) 82:500–7. doi:10.1152/jappl.1997.82.2.500
54. Tarasiuk A, Levi A, Berdugo-Boura N, Yahalom A, Segev Y. Role of orexin in respiratory and sleep homeostasis during upper airway obstruction in rats. Sleep (2014) 37:987–98. doi:10.5665/sleep.3676
55. Segev Y, Berdugo-Boura N, Porati O, Tarasiuk A. Upper airway loading induces growth retardation and change in local chondrocyte IGF-I expression is reversed by stimulation of GH release in juvenile rats. J Appl Physiol (2008) 105:1602–9. doi:10.1152/japplphysiol.90772.2008
56. Tarasiuk A, Segev Y. Chronic resistive airway loading reduces weight due to low serum IGF-1 in rats. Resp Physiol Neurobiol (2005) 145:177–82. doi:10.1016/j.resp.2004.10.007
57. Tarasiuk A, Segev Y. Chronic upper airway resistive loading induces growth retardation via the GH/IGF-1 axis in pre-pubescent rats. J Appl Physiol (2007) 102:913–8. doi:10.1152/japplphysiol.00838.2006
58. Arens R, Marcus CL. Pathophysiology of upper airway obstruction: a developmental perspective. Sleep (2004) 27:997–1019. doi:10.1093/sleep/27.5.997
59. Savransky V, Reinke C, Jun J, Bevans-Fonti S, Nanayakkara A, Li J, et al. Chronic intermittent hypoxia and acetaminophen induce synergistic liver injury in mice. Exp Physiol (2009) 94:228–39. doi:10.1113/expphysiol.2008.044883
60. Polotsky VY, Patil SP, Savransky V, Laffan A, Fonti S, Frame LA, et al. Obstructive sleep apnea, insulin resistance, and steatohepatitis in severe obesity. Am J Respir Crit Care Med (2009) 179:228–34. doi:10.1164/rccm.200804-608OC
61. Nath B, Szabo G. Hypoxia and hypoxia inducible factors: diverse roles in liver diseases. Hepatology (2012) 55:622–33. doi:10.1002/hep.25497
62. O’Donnell CP, Schaub CD, Haines AS, Berkowitz DE, Tankersley CG, Schwartz AR, et al. Leptin prevents respiratory depression in obesity. Am J Respir Crit Care Med (1999) 159:1477–84. doi:10.1164/ajrccm.159.5.9809025
63. Iioka Y, Tatsumi K, Sugito K, Moriya T, Kuriyama T. Effects of insulin-like growth factor on nitrogen balance during hypoxic exposure. Eur Respir J (2002) 20:293–9. doi:10.1183/09031936.02.00234302
64. Thomas A, Belaidi E, Moulin S, Horman S, van der Zon GC, Viollet B, et al. Chronic intermittent hypoxia impairs insulin sensitivity but improves whole-body glucose tolerance by activating skeletal muscle AMPK. Diabetes (2017) 66:2942–51. doi:10.2337/db17-0186
65. Alvarez-Martins I, Remédio L, Matias I, Diogo LN, Monteiro EC, Dias S. The impact of chronic intermittent hypoxia on hematopoiesis and the bone marrow microenvironment. Pflugers Arch (2016) 468:919–32. doi:10.1007/s00424-016-1797-6
66. Pillar G, Schnall RP, Peled N, Oliven A, Lavie P. Impaired respiratory response to resistive loading during sleep in healthy offspring of patients with obstructive sleep apnea. Am J Respir Crit Care Med (1997) 155:1602–8. doi:10.1164/ajrccm.155.5.9154864
67. de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, et al. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci U S A (1998) 95:322–7. doi:10.1073/pnas.95.1.322
68. Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell (1999) 98:437–51. doi:10.1016/S0092-8674(00)81973-X
69. Sakurai T. Roles of orexins in regulation of feeding and wakefulness. Neuroreport (2002) 13:987–95. doi:10.1097/00001756-200206120-00001
70. Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci (2005) 25:6716–20. doi:10.1523/JNEUROSCI.1887-05.2005
71. Tsunematsu T, Kilduff TS, Boyden ES, Takahashi S, Tominaga M, Yamanaka A. Acute optogenetic silencing of orexin/hypocretin neurons induces slow-wave sleep in mice. J Neurosci (2011) 31:10529–39. doi:10.1523/JNEUROSCI.0784-11.2011
72. Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron (2005) 46:787–98. doi:10.1016/j.neuron.2005.04.035
73. Williams RH, Jensen LT, Verkhratsky A, Fugger L, Burdakov D. Control of hypothalamic orexin neurons by acid and CO2. Proc Natl Acad Sci U S A (2007) 104:10685–90. doi:10.1073/pnas.0702676104
74. Gestreau C, Bevengut M, Dutschmann M. The dual role of the orexin/hypocretin system in modulating wakefulness and respiratory drive. Curr Opin Pulm Med (2008) 14:512–8. doi:10.1097/MCP.0b013e32831311d3
75. Nakamura A, Zhang W, Yanagisawa M, Fukuda Y, Kuwaki T. Vigilance state-dependent attenuation of hypercapnic chemoreflex and exaggerated sleep apnea in orexin knockout mice. J Appl Physiol (2007) 102:241–8. doi:10.1152/japplphysiol.00679.2006
76. Li A, Nattie E. Antagonism of rat orexin receptors by almorexant attenuates central chemoreception in wakefulness in the active period of the diurnal cycle. J Physiol (2010) 588:2935–44. doi:10.1113/jphysiol.2010.191288
77. Song N, Zhang G, Geng W, Liu Z, Jin W, Li L, et al. Acid sensing ion channel 1 in lateral hypothalamus contributes to breathing control. PLoS One (2012) 7:e39982. doi:10.1371/journal.pone.0039982
78. Kuwaki T. Hypothalamic modulation of breathing. Adv Exp Med Biol (2010) 669:243–7. doi:10.1007/978-1-4419-5692-7_49
79. Kernder A, De Luca R, Yanovsky Y, Haas HL, Sergeeva OA. Acid-sensing hypothalamic neurons controlling arousal. Cell Mol Neurobiol (2014) 34:777–89. doi:10.1007/s10571-014-0065-6
80. Fujiki N, Yoshida Y, Ripley B, Honda K, Mignot E, Nishino S. Changes in CSF hypocretin-1 (orexin A) levels in rats across 24 hours and in response to food deprivation. Neuroreport (2001) 12:993–7. doi:10.1097/00001756-200104170-00026
81. Messina G, Dalia C, Tafuri D, Monda V, Palmieri F, Dato A, et al. Orexin-A controls sympathetic activity and eating behavior. Front Psychol (2014) 5:997. doi:10.3389/fpsyg.2014.00997
82. López M, Tena-Sempere M, Diéguez C. Cross-talk between orexins (hypocretins) and the neuroendocrine axes (hypothalamic-pituitary axes). Front Neuroendocrinol (2010) 31:113–27. doi:10.1016/j.yfrne.2009.07.001
83. Seoane LM, Tovar SA, Perez D, Mallo F, Lopez M, Senaris R, et al. Orexin A suppresses in vivo GH secretion. Eur J Endocrinol (2004) 150:731–6. doi:10.1530/eje.0.1500731
84. Horvath TL, Diano S, van den Pol AN. Synaptic interaction between hypocretin (orexin) and neuropeptide Y cells in the rodent and primate hypothalamus: a novel circuit implicated in metabolic and endocrine regulations. J Neurosci (1999) 19:1072–87. doi:10.1523/JNEUROSCI.19-03-01072.1999
85. López M, Seoane LM, Tovar S, Nogueiras R, Diéguez C, Señarís R. Orexin-A regulates growth hormone-releasing hormone mRNA content in a nucleus-specific manner and somatostatin mRNA content in a growth hormone-dependent fashion in the rat hypothalamus. Eur J Neurosci (2004) 19:2080–8. doi:10.1111/j.0953-816X.2004.03318.x
86. Gardi J, Obal F Jr, Fang J, Zhang J, Krueger JM. Diurnal variations and sleep deprivation-induced changes in rat hypothalamic GHRH and somatostatin contents. Am J Physiol Regul Integr Comp Physiol (1999) 277:R1339–44. doi:10.1152/ajpregu.1999.277.5.R1339
87. Obál F Jr, Payne L, Kapás L, Opp M, Krueger JM. Inhibition of growth hormone-releasing factor suppresses both sleep and growth hormone secretion in the rat. Brain Res (1991) 557:149–53. doi:10.1016/0006-8993(91)90128-I
88. Wren AM, Small CJ, Abbott CR, Dhillo WS, Seal LJ, Cohen MA, et al. Ghrelin causes hyperphagia and obesity in rats. Diabetes (2001) 50:2540–7. doi:10.2337/diabetes.50.11.2540
89. Kageyama H, Takenoya F, Shiba K, Shioda S. Neuronal circuits involving ghrelin in the hypothalamus-mediated regulation of feeding. Neuropeptides (2010) 44:133–8. doi:10.1016/j.npep.2009.11.010
90. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature (1999) 402:656–60. doi:10.1038/45230
91. Howard AD, Feighner SD, Cully DF, Arena JP, Liberator PA, Rosenblum CI, et al. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science (1996) 273:974–7. doi:10.1126/science.273.5277.974
92. Dickson SL, Luckman SM. Induction of c-fos messenger ribonucleic acid in neuropeptide Y and growth hormone (GH)-releasing factor neurons in the rat arcuate nucleus following systemic injection of the GH secretagogue, GH-releasing peptide-6. Endocrinology (1997) 138:771–7. doi:10.1210/endo.138.2.4907
93. Thompson NM, Davies JS, Mode A, Houston PA, Wells T. Pattern-dependent suppression of growth hormone (GH) pulsatility by ghrelin and GH-releasing peptide-6 in moderately GH-deficient rats. Endocrinology (2003) 144:4859–67. doi:10.1210/en.2003-0423
94. Barkan AL, Beitins IZ, Kelch RP. Plasma insulin-like growth factor-I/somatomedin-C in acromegaly: correlation with the degree of growth hormone hypersecretion. J Clin Endocrinol Metab (1988) 67:69–73. doi:10.1210/jcem-67-1-69
96. Werner H, Weinstein D, Bentov I. Similarities and differences between insulin and IGF-I: structures, receptors, and signalling pathways. Arch Physiol Biochem (2008) 114:17–22. doi:10.1080/13813450801900694
97. Everson CA, Szabo A. Recurrent restriction of sleep and inadequate recuperation induce both adaptive changes and pathological outcomes. Am J Physiol Regul Integr Comp Physiol (2009) 297:R1430–40. doi:10.1152/ajpregu.00230.2009
98. Rechtschaffen A, Bergmann BM. Sleep deprivation in the rat: an update of the 1989 paper. Sleep (2002) 25:18–24. doi:10.1093/sleep/25.1.18
99. Bodosi B, Gardi J, Hajdu I, Szentirmai E, Obal F Jr, Krueger JM. Rhythms of ghrelin, leptin, and sleep in rats: effects of the normal diurnal cycle, restricted feeding, and sleep deprivation. Am J Physiol Regul Integr Comp Physiol (2004) 287:R1071–9. doi:10.1152/ajpregu.00294.2004
100. Spiegel K, Knutson K, Leproult R, Tasali E, Van Cauter E. Sleep loss: a novel risk factor for insulin resistance and type 2 diabetes. J Appl Physiol (2005) 99:2008–19. doi:10.1152/japplphysiol.00660.2005
101. Mullington JM, Haack M, Toth M, Serrador JM, Meier-Ewert HK. Cardiovascular, inflammatory, and metabolic consequences of sleep deprivation. Prog Cardiovasc Dis (2009) 51:294–302. doi:10.1016/j.pcad.2008.10.003
102. Vetrivelan R, Fuller PM, Yokota S, Lu J, Saper CB. Metabolic effects of chronic sleep restriction in rats. Sleep (2012) 35:1511–20. doi:10.5665/sleep.2200
103. Everson CA, Bergmann BM, Rechtschaffen A. Sleep deprivation in the rat. III. Total sleep deprivation. Sleep (1989) 12:13–21. doi:10.1093/sleep/12.1.13
104. Jerlhag E, Egecioglu E, Dickson SL, Andersson M, Svensson L, Engel JA. Ghrelin stimulates locomotor activity and accumbal dopamine-overflow via central cholinergic systems in mice: implications for its involvement in brain reward. Addict Biol (2006) 11:45–54. doi:10.1111/j.1369-1600.2006.00002.x
105. Swanson CM, Shea SA, Stone KL, Cauley JA, Rosen CJ, Redline S, et al. Obstructive sleep apnea and metabolic bone disease: insights into the relationship between bone and sleep. J Bone Miner Res (2015) 30:199–211. doi:10.1002/jbmr.2446
106. Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev (1995) 16:3–34. doi:10.1210/er.16.1.3
107. Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL, et al. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest (2002) 110:771–81. doi:10.1172/JCI15463
108. Nilsson O, Marino R, De Luca F, Phillip M, Baron J. Endocrine regulation of the growth plate. Horm Res (2005) 64:157–65. doi:10.1159/000088791
109. Smink JJ, Gresnigt MG, Hamers N, Koedam JA, Berger R, Van Buul-Offers SC. Short-term glucocorticoid treatment of prepubertal mice decreases growth and IGF-I expression in the growth plate. J Endocrinol (2003) 177:381–8. doi:10.1677/joe.0.1770381
110. Segev Y, Landau D, Davidoff-Friedman S, Weinreb M, Phillip M. Involvement of the skeletal GH/IGF system in an experimental model of diabetes-induced growth retardation. Acta Diabetol (2002) 39:61–7. doi:10.1007/s005920200015
111. Hamrick MW, Skedros JG, Pennington C, McNeil PL. Increased osteogenic response to exercise in metaphyseal versus diaphyseal cortical bone. J Musculoskelet Neuronal Interact (2006) 6:258–63.
112. Wei W, Motoike T, Krzeszinski JY, Jin Z, Xie XJ, Dechow PC, et al. Orexin regulates bone remodeling via a dominant positive central action and a subordinate negative peripheral action. Cell Metab (2014) 19:927–40. doi:10.1016/j.cmet.2014.03.016
113. Zhang R, Murakami S, Coustry F, Wang Y, de Crombrugghe B. Constitutive activation of MKK6 in chondrocytes of transgenic mice inhibits proliferation and delays endochondral bone formation. Proc Natl Acad Sci U S A (2006) 103:365–70. doi:10.1073/pnas.0507979103
114. Monemdjou R, Vasheghani F, Fahmi H, Perez G, Blati M, Taniguchi N, et al. Association of cartilage-specific deletion of peroxisome proliferator-activated receptor γ with abnormal endochondral ossification and impaired cartilage growth and development in a murine model. Arthritis Rheum (2012) 64:1551–61. doi:10.1002/art.33490
115. Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med (2013) 19:557–66. doi:10.1038/nm.3159
116. Sugii S, Olson P, Sears DD, Saberi M, Atkins AR, Barish GD, et al. PPARgamma activation in adipocytes is sufficient for systemic insulin sensitization. Proc Natl Acad Sci U S A (2009) 106:22504–9. doi:10.1073/pnas.0912487106
117. Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung UI, Kubota N, et al. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J Clin Invest (2004) 113:846–55. doi:10.1172/JCI19900
118. Mackie EJ, Ahmed YA, Tatarczuch L, Chen KS, Mirams M. Endochondral ossification: how cartilage is converted into bone in the developing skeleton. Int J Biochem Cell Biol (2008) 40:46–62. doi:10.1016/j.biocel.2007.06.009
Keywords: sleep-disordered breathing, upper airway obstruction, sleep, growth, metabolism, rats
Citation: Tarasiuk A and Segev Y (2018) Abnormal Growth and Feeding Behavior in Upper Airway Obstruction in Rats. Front. Endocrinol. 9:298. doi: 10.3389/fendo.2018.00298
Received: 22 March 2018; Accepted: 18 May 2018;
Published: 04 June 2018
Edited by:
Slava Berger, Johns Hopkins Medicine, United StatesReviewed by:
Jonathan C. Jun, Johns Hopkins University, United StatesZhichao Feng, Albert Einstein College of Medicine, United States
Copyright: © 2018 Tarasiuk and Segev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ariel Tarasiuk, dGFyYXNpdWtAYmd1LmFjLmls